User login
Safe Opioid Prescribing for Acute Noncancer Pain in Hospitalized Adults: A Systematic Review of Existing Guidelines
Pain is prevalent among hospitalized patients, occurring in 52%-71% of patients in cross-sectional surveys.1-3 Opioid administration is also common, with more than half of nonsurgical patients in United States (US) hospitals receiving at least one dose of opioid during hospitalization.4 Studies have also begun to define the degree to which hospital prescribing contributes to long-term use. Among opioid-naïve patients admitted to the hospital, 15%-25% fill an opioid prescription in the week after hospital discharge,5,6 43% of such patients fill another opioid prescription 90 days postdischarge,6 and 15% meet the criteria for long-term use at one year.7 With about 37 million discharges from US hospitals each year,8 these estimates suggest that hospitalization contributes to initiation of long-term opioid use in millions of adults each year.
Additionally, studies in the emergency department and hospital settings demonstrate large variations in prescribing of opioids between providers and hospitals.4,9 Variation unrelated to patient characteristics highlights areas of clinical uncertainty and the corresponding need for prescribing standards and guidance. To our knowledge, there are no existing guidelines on safe prescribing of opioids in hospitalized patients, aside from guidelines specifically focused on the perioperative, palliative care, or end-of-life settings.
Thus, in the context of the current opioid epidemic, the Society of Hospital Medicine (SHM) sought to develop a consensus statement to assist clinicians practicing medicine in the inpatient setting in safe prescribing of opioids for acute, noncancer pain on the medical services. We define “safe” prescribing as proposed by Aronson: “a process that recommends a medicine appropriate to the patient’s condition and minimizes the risk of undue harm from it.”10 To inform development of the consensus statement, SHM convened a working group to systematically review existing guidelines on the more general management of acute pain. This article describes the methods and results of our systematic review of existing guidelines for managing acute pain. The Consensus Statement derived from these existing guidelines, applied to the hospital setting, appears in a companion article.
METHODS
Steps in the systematic review process included: 1) searching for relevant guidelines, 2) applying exclusion criteria, 3) assessing the quality of the guidelines, and 4) synthesizing guideline recommendations to identify issues potentially relevant to medical inpatients with acute pain. Details of the protocol for this systematic review were registered on PROSPERO and can be accessed at https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=71846.
Data Sources and Search Terms

Guideline Inclusion/Exclusion Criteria
We defined guidelines as statements that include recommendations intended to optimize patient care that are informed by a systematic review of evidence and an assessment of the benefits and harm of alternative care options, consistent with the National Academies’ definition.11 To be eligible, guidelines had to be published in English and include recommendations on prescribing opioids for acute, noncancer pain. We excluded guidelines focused on chronic pain or palliative care, guidelines derived entirely from another guideline, and guidelines published before 2010, since such guidelines may contain outdated information.12 Because we were interested in general principles regarding safe use of opioids for managing acute pain, we excluded guidelines that focused exclusively on specific disease processes (eg, cancer, low-back pain, and sickle cell anemia). As we were specifically interested in the management of acute pain in the hospital setting, we also excluded guidelines that focused exclusively on specific nonhospital settings of care (eg, outpatient care clinics and nursing homes). We included guidelines related to care in the emergency department (ED) given the hospital-based location of care and the high degree of similarity in scope of practice and patient population, as most hospitalized adults are admitted through the ED. Finally, we excluded guidelines focusing on management in the intensive care setting (including the post-anesthesia care unit) given the inherent differences in patient population and management options between the intensive and nonintensive care areas of the hospital.
Guideline Quality Assessment
Guideline Synthesis and Analysis
We extracted recommendations from each guideline related to the following topics: 1) deciding when to use opioids, nonopioid medications, and nonmedication-based pain management modalities, 2) best practices in screening/monitoring/education prior to prescribing an opioid and/or during treatment, 3) opioid selection considerations, including selection of dose, duration, and route of administration, 4) strategies to minimize the risk of opioid-related adverse events, and 5) safe practices on discharge.
Role of the Funding Source
The Society of Hospital Medicine provided administrative and material support for the project, but had no role in the design or execution of the scientific evaluation.
RESULTS
Guideline Quality Assessment
See Table 1 for the AGREE II scaled domain scores, and Appendix Table 1 for the ratings on each individual item within a domain. The range of scaled scores for each of the AGREE II domains were as follows: Scope and purpose 52%-89%, stakeholder involvement 30%-81%, rigor of development 46%-81%, clarity of presentation 59%-72%, applicability 10%-57%, and editorial independence 42%-78%. Overall guideline assessment scores ranged from 4 to 5.33 on a scale from 1 to 7. Three of the guidelines (NICE, ACOEM, and WSAMDG)16,17,19 were recommended for use without modification by 2 out of 3 guideline appraisers, and one of the guidelines (ACEP)18 was recommended for use with modification by all 3 appraisers. The guideline by NICE19 was rated the highest both overall (5.33), and on 4 of the 6 AGREE II domains.
Although the guidelines each included a systematic review of the literature, the NICE19 and WSAMDG17 guidelines did not include the strength of recommendations or provide clear links between each recommendation and the underlying evidence base. When citations were present, we reviewed them to determine the type of data upon which the recommendations were based and included this information in Table 2. The majority of the recommendations in Table 2 are based on expert opinion alone, or other guidelines.
Guideline Synthesis and Analysis
Table 2 contains a synthesis of the recommendations related to each of our 5 prespecified content areas. Despite the generally low quality of the evidence supporting the recommendations, there were many areas of concordance across guidelines.
Deciding When to Use Opioids, Nonopioid Medications, and Nonmedication-Based Pain Management Modalities
Three out of 4 guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy,16-18 2 guidelines recommended treating mild to moderate pain with nonopioid medications, including acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs),16,17 and 2 guidelines recommended co-prescribing opioids with nonopioid analgesic medications to reduce total opioid requirements and improve pain control.16,17 Each of these recommendations was supported by at least one randomized controlled trial.
Best Practices in Screening/Monitoring/Education to Occur Prior to Prescribing an Opioid and/or During Treatment
Three guidelines recommended checking prescription drug monitoring programs (PDMPs), all based on expert consensus.16-18 Only the WSAMDG guideline offered guidance as to the optimal timing to check the PDMP in this setting, specifically recommending to check before prescribing opioids.17 Two guidelines also recommended helping patients set reasonable expectations about their recovery and educating patients about the risks/side effects of opioid therapy, all based on expert consensus or other guidelines.17,19
Opioid Selection Considerations, Including Selection of Dose, Duration, and Route of Administration
Three guidelines recommended using the lowest effective dose, supported by expert consensus and observational data in the outpatient setting demonstrating that overdose risk increases with opioid dose.16-18 Three guidelines recommended using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain based on expert consensus.16-18 Two guidelines recommended using as-needed rather than scheduled dosing of opioids based on expert recommendation.16, 17
Strategies to Minimize the Risk of Opioid-Related Adverse Events
Several strategies to minimize the risk of opioid-related adverse events were identified, but most were only recommended by a single guideline. Strategies recommended by more than one guideline included using a recognized opioid dose conversion guide when prescribing, reviewing, or changing opioid prescriptions (based on expert consensus);16,19 avoiding co-administration of parenteral and oral as-needed opioids, and if as-needed opioids from different routes are necessary, providing a clear indication for use of each (based on expert consensus and other guidelines);17,19 and avoiding/using caution when co-prescribing opioids with other central nervous system depressant medications16,17 (supported by observational studies demonstrating increased risk in the outpatient setting).
Safe Practices on Discharge
All 4 of the guidelines recommended prescribing a limited duration of opioids for the acute pain episode; however the maximum recommended duration varied widely from one week to 30 days.16-19 It is important to note that because these guidelines were not focused on hospitalization specifically, these maximum recommended durations of use reflect the entire acute pain episode (ie, not prescribing on discharge specifically). The guideline with the longest maximum recommended duration was from NICE, based in the United Kingdom, while the US-based guideline development groups uniformly recommended 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
DISCUSSION
This systematic review identified only 4 existing guidelines that included recommendations on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions, specific nonhospital settings, or the intensive care setting. Although 2 of the identified guidelines offered sparse recommendations specific to the hospital setting, we found no guidelines that focused exclusively on the period of hospitalization specifically outside of the perioperative period. Furthermore, the guideline recommendations were largely based on expert opinion. Although these factors limit the confidence with which the recommendations can be applied to the hospital setting, they nonetheless represent the best guidance currently available to standardize and improve the safety of prescribing opioids in the hospital setting.
This paucity of guidance specific to patients hospitalized in general, nonintensive care areas of the hospital is important because pain management in this setting differs in a number of ways from pain management in the ambulatory or intensive care unit settings (including the post-anesthesia care unit). First, there are differences in the monitoring strategies that are available in each of these settings (eg, variability in nurse-to-patient ratios, frequency of measuring vital signs, and availability of continuous pulse oximetry/capnography). Second, there are differences in available/feasible routes of medication administration depending on the setting of care. Finally, there are differences in the patients themselves, including severity of illness, baseline and expected functional status, pain severity, and ability to communicate.
Accordingly, to avoid substantial heterogeneity in recommendations obtained from this review, we chose to focus on guidelines most relevant to clinicians practicing medicine in nonintensive care areas of the hospital. This resulted in the exclusion of 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management and included use of advanced management procedures beyond the scope of practice for general internists,20,21 and one guideline that focused on management in the intensive care unit.22 Within the set of guidelines included in this review, we did include recommendations designated for the postoperative period that we felt were relevant to the care of hospitalized patients more generally. In fact, the ACOEM guideline, which includes postoperative recommendations, specifically noted that these recommendations are mostly comparable to those for treating acute pain more generally.16
In addition to the lack of guidance specific to the setting in which most hospitalists practice, most of the recommendations in the existing guidelines are based on expert consensus. Guidelines based on expert opinion typically carry a lower strength of recommendation, and, accordingly, should be applied with some caution and accompanied by diligent tracking of outcome metrics, as these recommendations are applied to local health systems. Recommendations may have unintended consequences that are not necessarily apparent at the outset, and the specific circumstances of each patient must be considered when deciding how best to apply recommendations. Additional research will be necessary to track the impact of the recommended prescribing practices on patient outcomes, particularly given that many states have already begun instituting regulations on safe opioid prescribing despite the limited nature of the evidence. Furthermore, although several studies have identified patient- and prescribing-related risk factors for opioid-related adverse events in surgical patient populations, given the differences in patient characteristics and prescribing patterns in these settings, research to understand the risk factors in hospitalized medical patients specifically is important to inform evidence-based, safe prescribing recommendations in this setting.
Despite the largely expert consensus-based nature of the recommendations, we found substantial overlap in the recommendations between the guidelines, spanning our prespecified topics of interest related to safe prescribing. Most guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy, checking PDMPs, using the lowest effective dose, and using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain. There was less consensus on risk mitigation strategies, where the majority of recommendations were endorsed by only 1 or 2 guidelines. Finally, all 4 guidelines recommended prescribing a limited duration of opioids for the acute pain episode, with US-based guidelines recommending 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
There are limitations to our evaluation. As previously noted, in order to avoid substantial heterogeneity in management recommendations, we excluded 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management,20,21 and one guideline focused on management in the intensive care unit.22 Accordingly, recommendations contained in this review may or may not be applicable to those settings, and readers interested in those settings specifically are directed to those guidelines. Additionally, we decided to exclude guidelines that focused on managing acute pain in specific conditions (eg, sickle cell disease and pancreatitis) because our goal was to identify generalizable principles of safe prescribing of opioids that apply regardless of clinical condition. Despite this goal, it is important to recognize that not all of the recommendations are generalizable to all types of pain; clinicians interested in management principles specific to certain disease states are encouraged to review disease-specific informational material. Finally, although we used rigorous, pre-defined search criteria and registered our protocol on PROSPERO, it is possible that our search strategy missed relevant guidelines.
In conclusion, we identified few guidelines on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions or nonhospital settings, and no guidelines focused on acute pain management in general, nonintensive care areas of the hospital specifically. Nevertheless, the guidelines that we identified make consistent recommendations related to our prespecified topic areas of relevance to the hospital setting, although most recommendations are based exclusively on expert opinion. Our systematic review nonetheless provides guidance in an area where guidance has thus far been limited. Future research should investigate risk factors for opioid-related adverse events in hospitalized, nonsurgical patients, and the effectiveness of interventions designed to reduce their occurrence.
ACKNOWLEDGMENTS
Dr. Herzig had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors would like to acknowledge and thank Kevin Vuernick, Jenna Goldstein, Meghan Mallouk, and Chris Frost, MD, from SHM for their facilitation of this project and dedication to this purpose.
Disclosures: Dr. Herzig received compensation from the Society of Hospital Medicine for her editorial role at the Journal of Hospital Medicine (unrelated to the present work). Dr. Jena received consulting fees from Pfizer, Inc., Hill Rom Services, Inc., Bristol Myers Squibb, Novartis Pharmaceuticals, Vertex Pharmaceuticals, and Precision Health Economics (all unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding: The Society of Hospital Medicine (SHM) provided administrative assistance and material support, but had no role in or influence on the scientific conduct of the study. Dr. Herzig was funded by grant number K23AG042459 from the National Institute on Aging. Dr. Mosher was supported, in part, by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). None of the funding agencies had involvement in any aspect of the study, including design, conduct, or reporting of the study
1. Melotti RM, Samolsky-Dekel BG, Ricchi E, et al. Pain prevalence and predictors among inpatients in a major Italian teaching hospital. A baseline survey towards a pain free hospital. Eur J Pain. 2005;9(5):485-495. PubMed
2. Sawyer J, Haslam L, Robinson S, Daines P, Stilos K. Pain prevalence study in a large Canadian teaching hospital. Pain Manag Nurs. 2008;9(3):104-112. PubMed
3. Strohbuecker B, Mayer H, Evers GC, Sabatowski R. Pain prevalence in hospitalized patients in a German university teaching hospital. J Pain Symptom Manage. 2005;29(5):498-506. PubMed
4. Herzig SJ, Rothberg MB, Cheung M, Ngo LH, Marcantonio ER. Opioid utilization and opioid-related adverse events in nonsurgical patients in US hospitals. J Hosp Med. 2014;9(2):73-81. PubMed
5. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2015;31(5):478-485. PubMed
6. Jena AB, Goldman D, Karaca-Mandic P. Hospital prescribing of opioids to medicare neneficiaries. JAMA Intern Med. 2016;176(7):990-997. PubMed
7. Mosher HJ, Hofmeyer B, Hadlandsmyth K, Richardson KK, Lund BC. Predictors of long-term opioid use after opioid initiation at discharge from medical and surgical hospitalizations. JHM. Accepted for Publication November 11, 2017. PubMed
8. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. HCUP Statistical Brief #180. 2014. Agency for Healthcare Research and Quality, Rockville, MD. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb180-Hospitalizations-United-States-2012.pdf. Accessed June 29, 2015. PubMed
9. Barnett ML, Olenski AR, Jena AB. Opioid-prescribing patterns of emergency physicians and risk of long-term use. N Engl J Med. 2017;376(7):663-673. PubMed
10. Aronson JK. Balanced prescribing. Br J Clin Pharmacol. 2006;62(6):629-632. PubMed
11. IOM (Institute of Medicine). 2011. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press.
12. Shekelle PG, Ortiz E, Rhodes S, et al. Validity of the agency for healthcare research and quality clinical practice guidelines: How quickly do guidelines become outdated? JAMA. 2001;286(12):1461-1467. PubMed
13. Brouwers MC, Kho ME, Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842. PubMed
14. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 1: performance, usefulness and areas for improvement. CMAJ. 2010;182(10):1045-1052. PubMed
15. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. Hegmann KT, Weiss MS, Bowden K, et al. ACOEM practice guidelines: opioids for treatment of acute, subacute, chronic, and postoperative pain. J Occup Environ Med. 2014;56(12):e143-e159. PubMed
17. Washington State Agency Medical Directors’ Group. Interagency Guideline on Prescribing Opioids for Pain. http://www.agencymeddirectors.wa.gov/Files/2015AMDGOpioidGuideline.pdf. Accessed December 5, 2017.
18. Cantrill SV, Brown MD, Carlisle RJ, et al. Clinical policy: critical issues in the prescribing of opioids for adult patients in the emergency department. Ann Emerg Med. 2012;60(4):499-525. PubMed
19. National Institute for Healthcare Excellence. Controlled drugs: Safe use and management. https://www.nice.org.uk/guidance/ng46/chapter/Recommendations. Accessed December 5, 2017.
20. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2012;116(2):248-273. PubMed
21. Apfelbaum JL, Silverstein JH, Chung FF, et al. Practice guidelines for postanesthetic care: an updated report by the American Society of Anesthesiologists Task Force on Postanesthetic Care. Anesthesiology. 2013;118(2):291-307. PubMed
22. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
Pain is prevalent among hospitalized patients, occurring in 52%-71% of patients in cross-sectional surveys.1-3 Opioid administration is also common, with more than half of nonsurgical patients in United States (US) hospitals receiving at least one dose of opioid during hospitalization.4 Studies have also begun to define the degree to which hospital prescribing contributes to long-term use. Among opioid-naïve patients admitted to the hospital, 15%-25% fill an opioid prescription in the week after hospital discharge,5,6 43% of such patients fill another opioid prescription 90 days postdischarge,6 and 15% meet the criteria for long-term use at one year.7 With about 37 million discharges from US hospitals each year,8 these estimates suggest that hospitalization contributes to initiation of long-term opioid use in millions of adults each year.
Additionally, studies in the emergency department and hospital settings demonstrate large variations in prescribing of opioids between providers and hospitals.4,9 Variation unrelated to patient characteristics highlights areas of clinical uncertainty and the corresponding need for prescribing standards and guidance. To our knowledge, there are no existing guidelines on safe prescribing of opioids in hospitalized patients, aside from guidelines specifically focused on the perioperative, palliative care, or end-of-life settings.
Thus, in the context of the current opioid epidemic, the Society of Hospital Medicine (SHM) sought to develop a consensus statement to assist clinicians practicing medicine in the inpatient setting in safe prescribing of opioids for acute, noncancer pain on the medical services. We define “safe” prescribing as proposed by Aronson: “a process that recommends a medicine appropriate to the patient’s condition and minimizes the risk of undue harm from it.”10 To inform development of the consensus statement, SHM convened a working group to systematically review existing guidelines on the more general management of acute pain. This article describes the methods and results of our systematic review of existing guidelines for managing acute pain. The Consensus Statement derived from these existing guidelines, applied to the hospital setting, appears in a companion article.
METHODS
Steps in the systematic review process included: 1) searching for relevant guidelines, 2) applying exclusion criteria, 3) assessing the quality of the guidelines, and 4) synthesizing guideline recommendations to identify issues potentially relevant to medical inpatients with acute pain. Details of the protocol for this systematic review were registered on PROSPERO and can be accessed at https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=71846.
Data Sources and Search Terms
Guideline Inclusion/Exclusion Criteria
We defined guidelines as statements that include recommendations intended to optimize patient care that are informed by a systematic review of evidence and an assessment of the benefits and harm of alternative care options, consistent with the National Academies’ definition.11 To be eligible, guidelines had to be published in English and include recommendations on prescribing opioids for acute, noncancer pain. We excluded guidelines focused on chronic pain or palliative care, guidelines derived entirely from another guideline, and guidelines published before 2010, since such guidelines may contain outdated information.12 Because we were interested in general principles regarding safe use of opioids for managing acute pain, we excluded guidelines that focused exclusively on specific disease processes (eg, cancer, low-back pain, and sickle cell anemia). As we were specifically interested in the management of acute pain in the hospital setting, we also excluded guidelines that focused exclusively on specific nonhospital settings of care (eg, outpatient care clinics and nursing homes). We included guidelines related to care in the emergency department (ED) given the hospital-based location of care and the high degree of similarity in scope of practice and patient population, as most hospitalized adults are admitted through the ED. Finally, we excluded guidelines focusing on management in the intensive care setting (including the post-anesthesia care unit) given the inherent differences in patient population and management options between the intensive and nonintensive care areas of the hospital.
Guideline Quality Assessment
Guideline Synthesis and Analysis
We extracted recommendations from each guideline related to the following topics: 1) deciding when to use opioids, nonopioid medications, and nonmedication-based pain management modalities, 2) best practices in screening/monitoring/education prior to prescribing an opioid and/or during treatment, 3) opioid selection considerations, including selection of dose, duration, and route of administration, 4) strategies to minimize the risk of opioid-related adverse events, and 5) safe practices on discharge.
Role of the Funding Source
The Society of Hospital Medicine provided administrative and material support for the project, but had no role in the design or execution of the scientific evaluation.
RESULTS
Guideline Quality Assessment
See Table 1 for the AGREE II scaled domain scores, and Appendix Table 1 for the ratings on each individual item within a domain. The range of scaled scores for each of the AGREE II domains were as follows: Scope and purpose 52%-89%, stakeholder involvement 30%-81%, rigor of development 46%-81%, clarity of presentation 59%-72%, applicability 10%-57%, and editorial independence 42%-78%. Overall guideline assessment scores ranged from 4 to 5.33 on a scale from 1 to 7. Three of the guidelines (NICE, ACOEM, and WSAMDG)16,17,19 were recommended for use without modification by 2 out of 3 guideline appraisers, and one of the guidelines (ACEP)18 was recommended for use with modification by all 3 appraisers. The guideline by NICE19 was rated the highest both overall (5.33), and on 4 of the 6 AGREE II domains.
Although the guidelines each included a systematic review of the literature, the NICE19 and WSAMDG17 guidelines did not include the strength of recommendations or provide clear links between each recommendation and the underlying evidence base. When citations were present, we reviewed them to determine the type of data upon which the recommendations were based and included this information in Table 2. The majority of the recommendations in Table 2 are based on expert opinion alone, or other guidelines.
Guideline Synthesis and Analysis
Table 2 contains a synthesis of the recommendations related to each of our 5 prespecified content areas. Despite the generally low quality of the evidence supporting the recommendations, there were many areas of concordance across guidelines.
Deciding When to Use Opioids, Nonopioid Medications, and Nonmedication-Based Pain Management Modalities
Three out of 4 guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy,16-18 2 guidelines recommended treating mild to moderate pain with nonopioid medications, including acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs),16,17 and 2 guidelines recommended co-prescribing opioids with nonopioid analgesic medications to reduce total opioid requirements and improve pain control.16,17 Each of these recommendations was supported by at least one randomized controlled trial.
Best Practices in Screening/Monitoring/Education to Occur Prior to Prescribing an Opioid and/or During Treatment
Three guidelines recommended checking prescription drug monitoring programs (PDMPs), all based on expert consensus.16-18 Only the WSAMDG guideline offered guidance as to the optimal timing to check the PDMP in this setting, specifically recommending to check before prescribing opioids.17 Two guidelines also recommended helping patients set reasonable expectations about their recovery and educating patients about the risks/side effects of opioid therapy, all based on expert consensus or other guidelines.17,19
Opioid Selection Considerations, Including Selection of Dose, Duration, and Route of Administration
Three guidelines recommended using the lowest effective dose, supported by expert consensus and observational data in the outpatient setting demonstrating that overdose risk increases with opioid dose.16-18 Three guidelines recommended using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain based on expert consensus.16-18 Two guidelines recommended using as-needed rather than scheduled dosing of opioids based on expert recommendation.16, 17
Strategies to Minimize the Risk of Opioid-Related Adverse Events
Several strategies to minimize the risk of opioid-related adverse events were identified, but most were only recommended by a single guideline. Strategies recommended by more than one guideline included using a recognized opioid dose conversion guide when prescribing, reviewing, or changing opioid prescriptions (based on expert consensus);16,19 avoiding co-administration of parenteral and oral as-needed opioids, and if as-needed opioids from different routes are necessary, providing a clear indication for use of each (based on expert consensus and other guidelines);17,19 and avoiding/using caution when co-prescribing opioids with other central nervous system depressant medications16,17 (supported by observational studies demonstrating increased risk in the outpatient setting).
Safe Practices on Discharge
All 4 of the guidelines recommended prescribing a limited duration of opioids for the acute pain episode; however the maximum recommended duration varied widely from one week to 30 days.16-19 It is important to note that because these guidelines were not focused on hospitalization specifically, these maximum recommended durations of use reflect the entire acute pain episode (ie, not prescribing on discharge specifically). The guideline with the longest maximum recommended duration was from NICE, based in the United Kingdom, while the US-based guideline development groups uniformly recommended 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
DISCUSSION
This systematic review identified only 4 existing guidelines that included recommendations on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions, specific nonhospital settings, or the intensive care setting. Although 2 of the identified guidelines offered sparse recommendations specific to the hospital setting, we found no guidelines that focused exclusively on the period of hospitalization specifically outside of the perioperative period. Furthermore, the guideline recommendations were largely based on expert opinion. Although these factors limit the confidence with which the recommendations can be applied to the hospital setting, they nonetheless represent the best guidance currently available to standardize and improve the safety of prescribing opioids in the hospital setting.
This paucity of guidance specific to patients hospitalized in general, nonintensive care areas of the hospital is important because pain management in this setting differs in a number of ways from pain management in the ambulatory or intensive care unit settings (including the post-anesthesia care unit). First, there are differences in the monitoring strategies that are available in each of these settings (eg, variability in nurse-to-patient ratios, frequency of measuring vital signs, and availability of continuous pulse oximetry/capnography). Second, there are differences in available/feasible routes of medication administration depending on the setting of care. Finally, there are differences in the patients themselves, including severity of illness, baseline and expected functional status, pain severity, and ability to communicate.
Accordingly, to avoid substantial heterogeneity in recommendations obtained from this review, we chose to focus on guidelines most relevant to clinicians practicing medicine in nonintensive care areas of the hospital. This resulted in the exclusion of 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management and included use of advanced management procedures beyond the scope of practice for general internists,20,21 and one guideline that focused on management in the intensive care unit.22 Within the set of guidelines included in this review, we did include recommendations designated for the postoperative period that we felt were relevant to the care of hospitalized patients more generally. In fact, the ACOEM guideline, which includes postoperative recommendations, specifically noted that these recommendations are mostly comparable to those for treating acute pain more generally.16
In addition to the lack of guidance specific to the setting in which most hospitalists practice, most of the recommendations in the existing guidelines are based on expert consensus. Guidelines based on expert opinion typically carry a lower strength of recommendation, and, accordingly, should be applied with some caution and accompanied by diligent tracking of outcome metrics, as these recommendations are applied to local health systems. Recommendations may have unintended consequences that are not necessarily apparent at the outset, and the specific circumstances of each patient must be considered when deciding how best to apply recommendations. Additional research will be necessary to track the impact of the recommended prescribing practices on patient outcomes, particularly given that many states have already begun instituting regulations on safe opioid prescribing despite the limited nature of the evidence. Furthermore, although several studies have identified patient- and prescribing-related risk factors for opioid-related adverse events in surgical patient populations, given the differences in patient characteristics and prescribing patterns in these settings, research to understand the risk factors in hospitalized medical patients specifically is important to inform evidence-based, safe prescribing recommendations in this setting.
Despite the largely expert consensus-based nature of the recommendations, we found substantial overlap in the recommendations between the guidelines, spanning our prespecified topics of interest related to safe prescribing. Most guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy, checking PDMPs, using the lowest effective dose, and using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain. There was less consensus on risk mitigation strategies, where the majority of recommendations were endorsed by only 1 or 2 guidelines. Finally, all 4 guidelines recommended prescribing a limited duration of opioids for the acute pain episode, with US-based guidelines recommending 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
There are limitations to our evaluation. As previously noted, in order to avoid substantial heterogeneity in management recommendations, we excluded 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management,20,21 and one guideline focused on management in the intensive care unit.22 Accordingly, recommendations contained in this review may or may not be applicable to those settings, and readers interested in those settings specifically are directed to those guidelines. Additionally, we decided to exclude guidelines that focused on managing acute pain in specific conditions (eg, sickle cell disease and pancreatitis) because our goal was to identify generalizable principles of safe prescribing of opioids that apply regardless of clinical condition. Despite this goal, it is important to recognize that not all of the recommendations are generalizable to all types of pain; clinicians interested in management principles specific to certain disease states are encouraged to review disease-specific informational material. Finally, although we used rigorous, pre-defined search criteria and registered our protocol on PROSPERO, it is possible that our search strategy missed relevant guidelines.
In conclusion, we identified few guidelines on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions or nonhospital settings, and no guidelines focused on acute pain management in general, nonintensive care areas of the hospital specifically. Nevertheless, the guidelines that we identified make consistent recommendations related to our prespecified topic areas of relevance to the hospital setting, although most recommendations are based exclusively on expert opinion. Our systematic review nonetheless provides guidance in an area where guidance has thus far been limited. Future research should investigate risk factors for opioid-related adverse events in hospitalized, nonsurgical patients, and the effectiveness of interventions designed to reduce their occurrence.
ACKNOWLEDGMENTS
Dr. Herzig had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors would like to acknowledge and thank Kevin Vuernick, Jenna Goldstein, Meghan Mallouk, and Chris Frost, MD, from SHM for their facilitation of this project and dedication to this purpose.
Disclosures: Dr. Herzig received compensation from the Society of Hospital Medicine for her editorial role at the Journal of Hospital Medicine (unrelated to the present work). Dr. Jena received consulting fees from Pfizer, Inc., Hill Rom Services, Inc., Bristol Myers Squibb, Novartis Pharmaceuticals, Vertex Pharmaceuticals, and Precision Health Economics (all unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding: The Society of Hospital Medicine (SHM) provided administrative assistance and material support, but had no role in or influence on the scientific conduct of the study. Dr. Herzig was funded by grant number K23AG042459 from the National Institute on Aging. Dr. Mosher was supported, in part, by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). None of the funding agencies had involvement in any aspect of the study, including design, conduct, or reporting of the study
Pain is prevalent among hospitalized patients, occurring in 52%-71% of patients in cross-sectional surveys.1-3 Opioid administration is also common, with more than half of nonsurgical patients in United States (US) hospitals receiving at least one dose of opioid during hospitalization.4 Studies have also begun to define the degree to which hospital prescribing contributes to long-term use. Among opioid-naïve patients admitted to the hospital, 15%-25% fill an opioid prescription in the week after hospital discharge,5,6 43% of such patients fill another opioid prescription 90 days postdischarge,6 and 15% meet the criteria for long-term use at one year.7 With about 37 million discharges from US hospitals each year,8 these estimates suggest that hospitalization contributes to initiation of long-term opioid use in millions of adults each year.
Additionally, studies in the emergency department and hospital settings demonstrate large variations in prescribing of opioids between providers and hospitals.4,9 Variation unrelated to patient characteristics highlights areas of clinical uncertainty and the corresponding need for prescribing standards and guidance. To our knowledge, there are no existing guidelines on safe prescribing of opioids in hospitalized patients, aside from guidelines specifically focused on the perioperative, palliative care, or end-of-life settings.
Thus, in the context of the current opioid epidemic, the Society of Hospital Medicine (SHM) sought to develop a consensus statement to assist clinicians practicing medicine in the inpatient setting in safe prescribing of opioids for acute, noncancer pain on the medical services. We define “safe” prescribing as proposed by Aronson: “a process that recommends a medicine appropriate to the patient’s condition and minimizes the risk of undue harm from it.”10 To inform development of the consensus statement, SHM convened a working group to systematically review existing guidelines on the more general management of acute pain. This article describes the methods and results of our systematic review of existing guidelines for managing acute pain. The Consensus Statement derived from these existing guidelines, applied to the hospital setting, appears in a companion article.
METHODS
Steps in the systematic review process included: 1) searching for relevant guidelines, 2) applying exclusion criteria, 3) assessing the quality of the guidelines, and 4) synthesizing guideline recommendations to identify issues potentially relevant to medical inpatients with acute pain. Details of the protocol for this systematic review were registered on PROSPERO and can be accessed at https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=71846.
Data Sources and Search Terms
Guideline Inclusion/Exclusion Criteria
We defined guidelines as statements that include recommendations intended to optimize patient care that are informed by a systematic review of evidence and an assessment of the benefits and harm of alternative care options, consistent with the National Academies’ definition.11 To be eligible, guidelines had to be published in English and include recommendations on prescribing opioids for acute, noncancer pain. We excluded guidelines focused on chronic pain or palliative care, guidelines derived entirely from another guideline, and guidelines published before 2010, since such guidelines may contain outdated information.12 Because we were interested in general principles regarding safe use of opioids for managing acute pain, we excluded guidelines that focused exclusively on specific disease processes (eg, cancer, low-back pain, and sickle cell anemia). As we were specifically interested in the management of acute pain in the hospital setting, we also excluded guidelines that focused exclusively on specific nonhospital settings of care (eg, outpatient care clinics and nursing homes). We included guidelines related to care in the emergency department (ED) given the hospital-based location of care and the high degree of similarity in scope of practice and patient population, as most hospitalized adults are admitted through the ED. Finally, we excluded guidelines focusing on management in the intensive care setting (including the post-anesthesia care unit) given the inherent differences in patient population and management options between the intensive and nonintensive care areas of the hospital.
Guideline Quality Assessment
Guideline Synthesis and Analysis
We extracted recommendations from each guideline related to the following topics: 1) deciding when to use opioids, nonopioid medications, and nonmedication-based pain management modalities, 2) best practices in screening/monitoring/education prior to prescribing an opioid and/or during treatment, 3) opioid selection considerations, including selection of dose, duration, and route of administration, 4) strategies to minimize the risk of opioid-related adverse events, and 5) safe practices on discharge.
Role of the Funding Source
The Society of Hospital Medicine provided administrative and material support for the project, but had no role in the design or execution of the scientific evaluation.
RESULTS
Guideline Quality Assessment
See Table 1 for the AGREE II scaled domain scores, and Appendix Table 1 for the ratings on each individual item within a domain. The range of scaled scores for each of the AGREE II domains were as follows: Scope and purpose 52%-89%, stakeholder involvement 30%-81%, rigor of development 46%-81%, clarity of presentation 59%-72%, applicability 10%-57%, and editorial independence 42%-78%. Overall guideline assessment scores ranged from 4 to 5.33 on a scale from 1 to 7. Three of the guidelines (NICE, ACOEM, and WSAMDG)16,17,19 were recommended for use without modification by 2 out of 3 guideline appraisers, and one of the guidelines (ACEP)18 was recommended for use with modification by all 3 appraisers. The guideline by NICE19 was rated the highest both overall (5.33), and on 4 of the 6 AGREE II domains.
Although the guidelines each included a systematic review of the literature, the NICE19 and WSAMDG17 guidelines did not include the strength of recommendations or provide clear links between each recommendation and the underlying evidence base. When citations were present, we reviewed them to determine the type of data upon which the recommendations were based and included this information in Table 2. The majority of the recommendations in Table 2 are based on expert opinion alone, or other guidelines.
Guideline Synthesis and Analysis
Table 2 contains a synthesis of the recommendations related to each of our 5 prespecified content areas. Despite the generally low quality of the evidence supporting the recommendations, there were many areas of concordance across guidelines.
Deciding When to Use Opioids, Nonopioid Medications, and Nonmedication-Based Pain Management Modalities
Three out of 4 guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy,16-18 2 guidelines recommended treating mild to moderate pain with nonopioid medications, including acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs),16,17 and 2 guidelines recommended co-prescribing opioids with nonopioid analgesic medications to reduce total opioid requirements and improve pain control.16,17 Each of these recommendations was supported by at least one randomized controlled trial.
Best Practices in Screening/Monitoring/Education to Occur Prior to Prescribing an Opioid and/or During Treatment
Three guidelines recommended checking prescription drug monitoring programs (PDMPs), all based on expert consensus.16-18 Only the WSAMDG guideline offered guidance as to the optimal timing to check the PDMP in this setting, specifically recommending to check before prescribing opioids.17 Two guidelines also recommended helping patients set reasonable expectations about their recovery and educating patients about the risks/side effects of opioid therapy, all based on expert consensus or other guidelines.17,19
Opioid Selection Considerations, Including Selection of Dose, Duration, and Route of Administration
Three guidelines recommended using the lowest effective dose, supported by expert consensus and observational data in the outpatient setting demonstrating that overdose risk increases with opioid dose.16-18 Three guidelines recommended using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain based on expert consensus.16-18 Two guidelines recommended using as-needed rather than scheduled dosing of opioids based on expert recommendation.16, 17
Strategies to Minimize the Risk of Opioid-Related Adverse Events
Several strategies to minimize the risk of opioid-related adverse events were identified, but most were only recommended by a single guideline. Strategies recommended by more than one guideline included using a recognized opioid dose conversion guide when prescribing, reviewing, or changing opioid prescriptions (based on expert consensus);16,19 avoiding co-administration of parenteral and oral as-needed opioids, and if as-needed opioids from different routes are necessary, providing a clear indication for use of each (based on expert consensus and other guidelines);17,19 and avoiding/using caution when co-prescribing opioids with other central nervous system depressant medications16,17 (supported by observational studies demonstrating increased risk in the outpatient setting).
Safe Practices on Discharge
All 4 of the guidelines recommended prescribing a limited duration of opioids for the acute pain episode; however the maximum recommended duration varied widely from one week to 30 days.16-19 It is important to note that because these guidelines were not focused on hospitalization specifically, these maximum recommended durations of use reflect the entire acute pain episode (ie, not prescribing on discharge specifically). The guideline with the longest maximum recommended duration was from NICE, based in the United Kingdom, while the US-based guideline development groups uniformly recommended 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
DISCUSSION
This systematic review identified only 4 existing guidelines that included recommendations on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions, specific nonhospital settings, or the intensive care setting. Although 2 of the identified guidelines offered sparse recommendations specific to the hospital setting, we found no guidelines that focused exclusively on the period of hospitalization specifically outside of the perioperative period. Furthermore, the guideline recommendations were largely based on expert opinion. Although these factors limit the confidence with which the recommendations can be applied to the hospital setting, they nonetheless represent the best guidance currently available to standardize and improve the safety of prescribing opioids in the hospital setting.
This paucity of guidance specific to patients hospitalized in general, nonintensive care areas of the hospital is important because pain management in this setting differs in a number of ways from pain management in the ambulatory or intensive care unit settings (including the post-anesthesia care unit). First, there are differences in the monitoring strategies that are available in each of these settings (eg, variability in nurse-to-patient ratios, frequency of measuring vital signs, and availability of continuous pulse oximetry/capnography). Second, there are differences in available/feasible routes of medication administration depending on the setting of care. Finally, there are differences in the patients themselves, including severity of illness, baseline and expected functional status, pain severity, and ability to communicate.
Accordingly, to avoid substantial heterogeneity in recommendations obtained from this review, we chose to focus on guidelines most relevant to clinicians practicing medicine in nonintensive care areas of the hospital. This resulted in the exclusion of 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management and included use of advanced management procedures beyond the scope of practice for general internists,20,21 and one guideline that focused on management in the intensive care unit.22 Within the set of guidelines included in this review, we did include recommendations designated for the postoperative period that we felt were relevant to the care of hospitalized patients more generally. In fact, the ACOEM guideline, which includes postoperative recommendations, specifically noted that these recommendations are mostly comparable to those for treating acute pain more generally.16
In addition to the lack of guidance specific to the setting in which most hospitalists practice, most of the recommendations in the existing guidelines are based on expert consensus. Guidelines based on expert opinion typically carry a lower strength of recommendation, and, accordingly, should be applied with some caution and accompanied by diligent tracking of outcome metrics, as these recommendations are applied to local health systems. Recommendations may have unintended consequences that are not necessarily apparent at the outset, and the specific circumstances of each patient must be considered when deciding how best to apply recommendations. Additional research will be necessary to track the impact of the recommended prescribing practices on patient outcomes, particularly given that many states have already begun instituting regulations on safe opioid prescribing despite the limited nature of the evidence. Furthermore, although several studies have identified patient- and prescribing-related risk factors for opioid-related adverse events in surgical patient populations, given the differences in patient characteristics and prescribing patterns in these settings, research to understand the risk factors in hospitalized medical patients specifically is important to inform evidence-based, safe prescribing recommendations in this setting.
Despite the largely expert consensus-based nature of the recommendations, we found substantial overlap in the recommendations between the guidelines, spanning our prespecified topics of interest related to safe prescribing. Most guidelines recommended restricting opioid use to severe pain or pain that has not responded to nonopioid therapy, checking PDMPs, using the lowest effective dose, and using short-acting opioids and/or avoiding use of long-acting/extended-release opioids for acute pain. There was less consensus on risk mitigation strategies, where the majority of recommendations were endorsed by only 1 or 2 guidelines. Finally, all 4 guidelines recommended prescribing a limited duration of opioids for the acute pain episode, with US-based guidelines recommending 1 to 2 weeks as the maximum duration of opioid use, including the period of hospitalization.
There are limitations to our evaluation. As previously noted, in order to avoid substantial heterogeneity in management recommendations, we excluded 2 guidelines intended for anesthesiologists that focused exclusively on perioperative management,20,21 and one guideline focused on management in the intensive care unit.22 Accordingly, recommendations contained in this review may or may not be applicable to those settings, and readers interested in those settings specifically are directed to those guidelines. Additionally, we decided to exclude guidelines that focused on managing acute pain in specific conditions (eg, sickle cell disease and pancreatitis) because our goal was to identify generalizable principles of safe prescribing of opioids that apply regardless of clinical condition. Despite this goal, it is important to recognize that not all of the recommendations are generalizable to all types of pain; clinicians interested in management principles specific to certain disease states are encouraged to review disease-specific informational material. Finally, although we used rigorous, pre-defined search criteria and registered our protocol on PROSPERO, it is possible that our search strategy missed relevant guidelines.
In conclusion, we identified few guidelines on safe opioid prescribing practices for managing acute, noncancer pain, outside of the context of specific conditions or nonhospital settings, and no guidelines focused on acute pain management in general, nonintensive care areas of the hospital specifically. Nevertheless, the guidelines that we identified make consistent recommendations related to our prespecified topic areas of relevance to the hospital setting, although most recommendations are based exclusively on expert opinion. Our systematic review nonetheless provides guidance in an area where guidance has thus far been limited. Future research should investigate risk factors for opioid-related adverse events in hospitalized, nonsurgical patients, and the effectiveness of interventions designed to reduce their occurrence.
ACKNOWLEDGMENTS
Dr. Herzig had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors would like to acknowledge and thank Kevin Vuernick, Jenna Goldstein, Meghan Mallouk, and Chris Frost, MD, from SHM for their facilitation of this project and dedication to this purpose.
Disclosures: Dr. Herzig received compensation from the Society of Hospital Medicine for her editorial role at the Journal of Hospital Medicine (unrelated to the present work). Dr. Jena received consulting fees from Pfizer, Inc., Hill Rom Services, Inc., Bristol Myers Squibb, Novartis Pharmaceuticals, Vertex Pharmaceuticals, and Precision Health Economics (all unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding: The Society of Hospital Medicine (SHM) provided administrative assistance and material support, but had no role in or influence on the scientific conduct of the study. Dr. Herzig was funded by grant number K23AG042459 from the National Institute on Aging. Dr. Mosher was supported, in part, by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). None of the funding agencies had involvement in any aspect of the study, including design, conduct, or reporting of the study
1. Melotti RM, Samolsky-Dekel BG, Ricchi E, et al. Pain prevalence and predictors among inpatients in a major Italian teaching hospital. A baseline survey towards a pain free hospital. Eur J Pain. 2005;9(5):485-495. PubMed
2. Sawyer J, Haslam L, Robinson S, Daines P, Stilos K. Pain prevalence study in a large Canadian teaching hospital. Pain Manag Nurs. 2008;9(3):104-112. PubMed
3. Strohbuecker B, Mayer H, Evers GC, Sabatowski R. Pain prevalence in hospitalized patients in a German university teaching hospital. J Pain Symptom Manage. 2005;29(5):498-506. PubMed
4. Herzig SJ, Rothberg MB, Cheung M, Ngo LH, Marcantonio ER. Opioid utilization and opioid-related adverse events in nonsurgical patients in US hospitals. J Hosp Med. 2014;9(2):73-81. PubMed
5. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2015;31(5):478-485. PubMed
6. Jena AB, Goldman D, Karaca-Mandic P. Hospital prescribing of opioids to medicare neneficiaries. JAMA Intern Med. 2016;176(7):990-997. PubMed
7. Mosher HJ, Hofmeyer B, Hadlandsmyth K, Richardson KK, Lund BC. Predictors of long-term opioid use after opioid initiation at discharge from medical and surgical hospitalizations. JHM. Accepted for Publication November 11, 2017. PubMed
8. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. HCUP Statistical Brief #180. 2014. Agency for Healthcare Research and Quality, Rockville, MD. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb180-Hospitalizations-United-States-2012.pdf. Accessed June 29, 2015. PubMed
9. Barnett ML, Olenski AR, Jena AB. Opioid-prescribing patterns of emergency physicians and risk of long-term use. N Engl J Med. 2017;376(7):663-673. PubMed
10. Aronson JK. Balanced prescribing. Br J Clin Pharmacol. 2006;62(6):629-632. PubMed
11. IOM (Institute of Medicine). 2011. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press.
12. Shekelle PG, Ortiz E, Rhodes S, et al. Validity of the agency for healthcare research and quality clinical practice guidelines: How quickly do guidelines become outdated? JAMA. 2001;286(12):1461-1467. PubMed
13. Brouwers MC, Kho ME, Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842. PubMed
14. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 1: performance, usefulness and areas for improvement. CMAJ. 2010;182(10):1045-1052. PubMed
15. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. Hegmann KT, Weiss MS, Bowden K, et al. ACOEM practice guidelines: opioids for treatment of acute, subacute, chronic, and postoperative pain. J Occup Environ Med. 2014;56(12):e143-e159. PubMed
17. Washington State Agency Medical Directors’ Group. Interagency Guideline on Prescribing Opioids for Pain. http://www.agencymeddirectors.wa.gov/Files/2015AMDGOpioidGuideline.pdf. Accessed December 5, 2017.
18. Cantrill SV, Brown MD, Carlisle RJ, et al. Clinical policy: critical issues in the prescribing of opioids for adult patients in the emergency department. Ann Emerg Med. 2012;60(4):499-525. PubMed
19. National Institute for Healthcare Excellence. Controlled drugs: Safe use and management. https://www.nice.org.uk/guidance/ng46/chapter/Recommendations. Accessed December 5, 2017.
20. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2012;116(2):248-273. PubMed
21. Apfelbaum JL, Silverstein JH, Chung FF, et al. Practice guidelines for postanesthetic care: an updated report by the American Society of Anesthesiologists Task Force on Postanesthetic Care. Anesthesiology. 2013;118(2):291-307. PubMed
22. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
1. Melotti RM, Samolsky-Dekel BG, Ricchi E, et al. Pain prevalence and predictors among inpatients in a major Italian teaching hospital. A baseline survey towards a pain free hospital. Eur J Pain. 2005;9(5):485-495. PubMed
2. Sawyer J, Haslam L, Robinson S, Daines P, Stilos K. Pain prevalence study in a large Canadian teaching hospital. Pain Manag Nurs. 2008;9(3):104-112. PubMed
3. Strohbuecker B, Mayer H, Evers GC, Sabatowski R. Pain prevalence in hospitalized patients in a German university teaching hospital. J Pain Symptom Manage. 2005;29(5):498-506. PubMed
4. Herzig SJ, Rothberg MB, Cheung M, Ngo LH, Marcantonio ER. Opioid utilization and opioid-related adverse events in nonsurgical patients in US hospitals. J Hosp Med. 2014;9(2):73-81. PubMed
5. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2015;31(5):478-485. PubMed
6. Jena AB, Goldman D, Karaca-Mandic P. Hospital prescribing of opioids to medicare neneficiaries. JAMA Intern Med. 2016;176(7):990-997. PubMed
7. Mosher HJ, Hofmeyer B, Hadlandsmyth K, Richardson KK, Lund BC. Predictors of long-term opioid use after opioid initiation at discharge from medical and surgical hospitalizations. JHM. Accepted for Publication November 11, 2017. PubMed
8. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. HCUP Statistical Brief #180. 2014. Agency for Healthcare Research and Quality, Rockville, MD. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb180-Hospitalizations-United-States-2012.pdf. Accessed June 29, 2015. PubMed
9. Barnett ML, Olenski AR, Jena AB. Opioid-prescribing patterns of emergency physicians and risk of long-term use. N Engl J Med. 2017;376(7):663-673. PubMed
10. Aronson JK. Balanced prescribing. Br J Clin Pharmacol. 2006;62(6):629-632. PubMed
11. IOM (Institute of Medicine). 2011. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press.
12. Shekelle PG, Ortiz E, Rhodes S, et al. Validity of the agency for healthcare research and quality clinical practice guidelines: How quickly do guidelines become outdated? JAMA. 2001;286(12):1461-1467. PubMed
13. Brouwers MC, Kho ME, Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-E842. PubMed
14. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 1: performance, usefulness and areas for improvement. CMAJ. 2010;182(10):1045-1052. PubMed
15. Brouwers MC, Kho ME, Browman GP, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. Hegmann KT, Weiss MS, Bowden K, et al. ACOEM practice guidelines: opioids for treatment of acute, subacute, chronic, and postoperative pain. J Occup Environ Med. 2014;56(12):e143-e159. PubMed
17. Washington State Agency Medical Directors’ Group. Interagency Guideline on Prescribing Opioids for Pain. http://www.agencymeddirectors.wa.gov/Files/2015AMDGOpioidGuideline.pdf. Accessed December 5, 2017.
18. Cantrill SV, Brown MD, Carlisle RJ, et al. Clinical policy: critical issues in the prescribing of opioids for adult patients in the emergency department. Ann Emerg Med. 2012;60(4):499-525. PubMed
19. National Institute for Healthcare Excellence. Controlled drugs: Safe use and management. https://www.nice.org.uk/guidance/ng46/chapter/Recommendations. Accessed December 5, 2017.
20. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology. 2012;116(2):248-273. PubMed
21. Apfelbaum JL, Silverstein JH, Chung FF, et al. Practice guidelines for postanesthetic care: an updated report by the American Society of Anesthesiologists Task Force on Postanesthetic Care. Anesthesiology. 2013;118(2):291-307. PubMed
22. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
© 2018 Society of Hospital Medicine
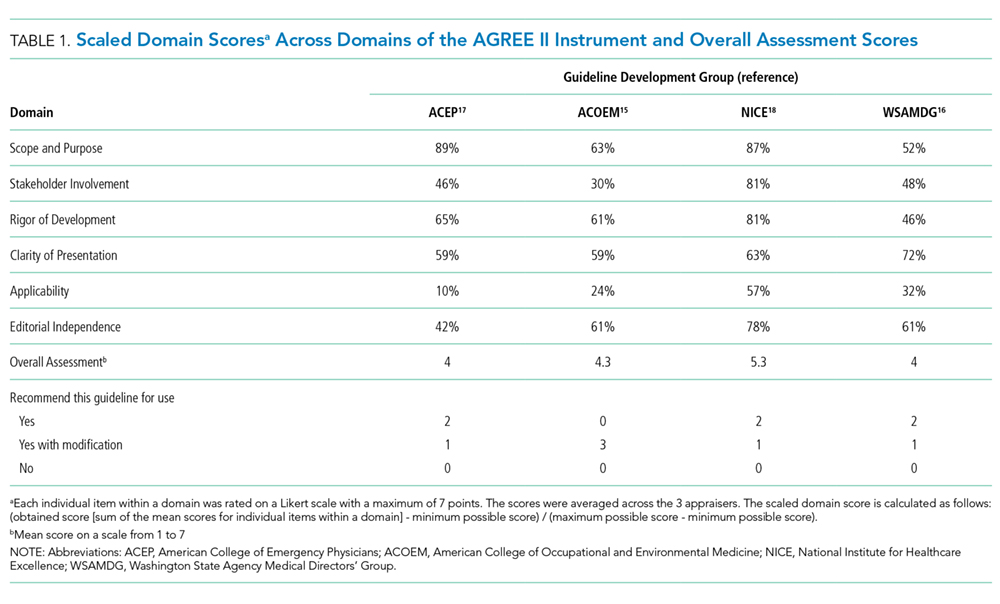
Gallstones: Watch and wait, or intervene?
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES

Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS

Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
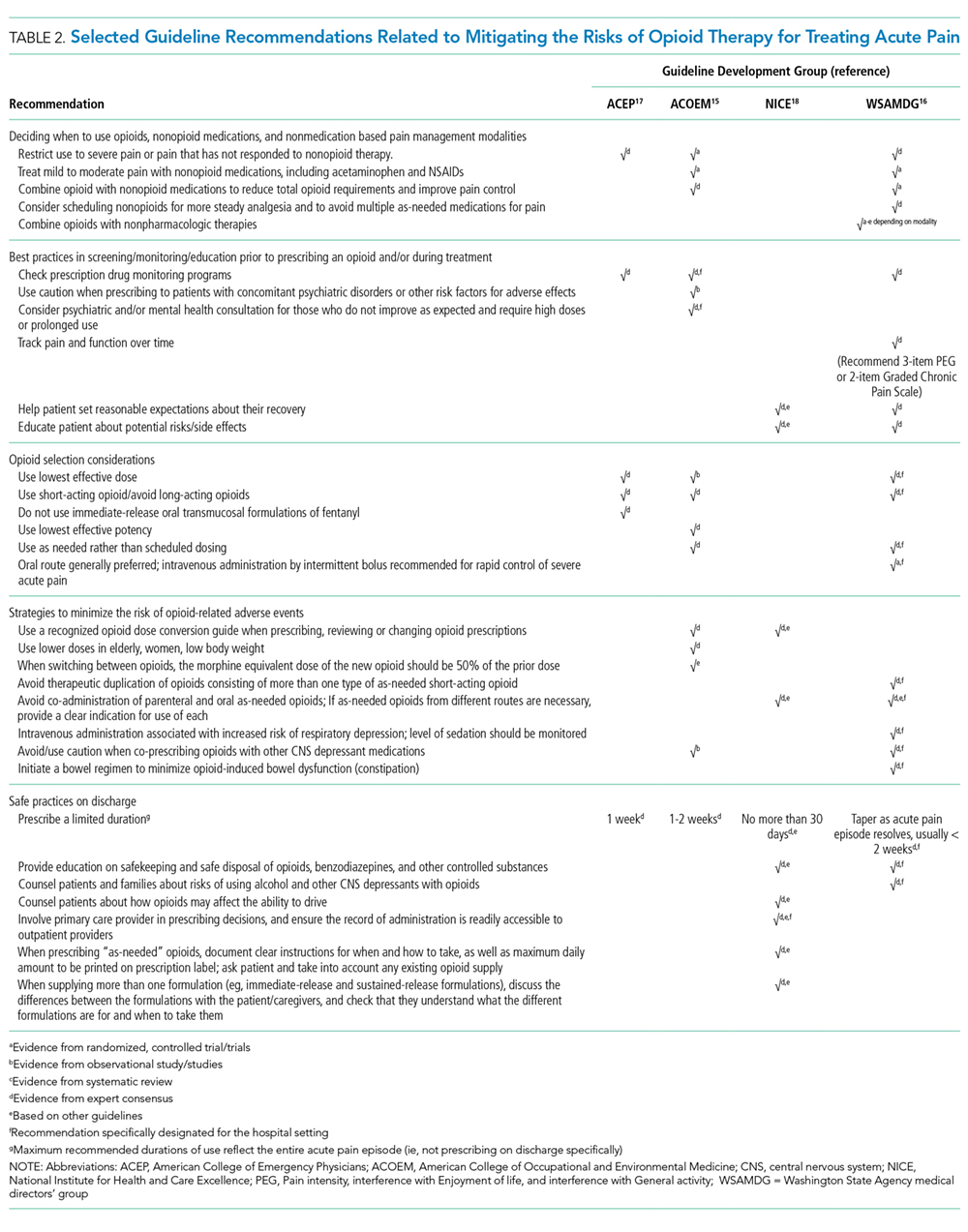
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones

Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones

For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
.")
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:

- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.

Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES
Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS
Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones
Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones
For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:
- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.
Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
The prevalence of gallstones is approximately 10% to 15% of the adult US population.1,2 Most cases are asymptomatic, as gallstones are usually discovered incidentally during routine imaging for other abdominal conditions, and only about 20% of patients with asymptomatic gallstones develop clinically significant complications.2,3
Nevertheless, gallstones carry significant healthcare costs. In 2004, the median inpatient cost for any gallstone-related disease was $11,584, with an overall annual cost of $6.2 billion.4,5
Laparoscopic cholecystectomy is the standard treatment for symptomatic cholelithiasis. For asymptomatic cholelithasis, the usual approach is expectant management (“watch and wait”), but prophylactic cholecystectomy may be an option in certain patients at high risk.
CHEMICAL COMPOSITION
Gallstones can be classified into 2 main categories based on their predominant chemical composition: cholesterol or pigment.
Cholesterol gallstones
About 75% of gallstones are composed of cholesterol.3,4 In the past, this type of stone was thought to be caused by gallbladder inflammation, bile stasis, and absorption of bile salts from damaged mucosa. However, it is now known that cholesterol gallstones are the result of biliary supersaturation caused by cholesterol hypersecretion into the gallbladder, gallbladder hypomotility, accelerated cholesterol nucleation and crystallization, and mucin gel accumulation.
Pigment gallstones
Black pigment gallstones account for 10% to 15% of all gallstones.6 They are caused by chronic hemolysis in association with supersaturation of bile with calcium hydrogen bilirubinate, along with deposition of calcium carbonate, phosphate, and inorganic salts.7
Brown pigment stones, accounting for 5% to 10% of all gallstones,6 are caused by infection in the obstructed bile ducts, where bacteria that produce beta-glucuronidase, phospholipase, and slime contribute to formation of the stone.8,9
RISK FACTORS FOR GALLSTONES
Age. After age 40, the risk increases dramatically, with an incidence 4 times higher for those ages 40 to 69 than in younger people.10
Female sex. Women of reproductive age are 4 times more likely to develop gallstones than men, but this gap narrows after menopause.11 The higher risk is attributed to female sex hormones, pregnancy, and oral contraceptive use. Estrogen decreases secretion of bile salts and increases secretion of cholesterol into the gallbladder, which leads to cholesterol supersaturation. Progesterone acts synergistically by causing hypomobility of the gallbladder, which in turn leads to bile stasis.12,13
Ethnicity. The risk is higher in Mexican Americans and Native Americans than in other ethnic groups.14
Rapid weight loss, such as after bariatric surgery, occurs from decreased caloric intake and promotes bile stasis, while lipolysis increases cholesterol mobilization and secretion into the gallbladder. This creates an environment conducive to bile supersaturation with cholesterol, leading to gallstone formation.
Chronic hemolytic disorders carry an increased risk of developing calcium bilirubinate stones due to increased excretion of bilirubin during hemolysis.
Obesity and diabetes mellitus are both attributed to insulin resistance. Obesity also increases bile stasis and cholesterol saturation.
CLINICAL PRESENTATION OF GALLSTONES (CHOLELITHIASIS)
Most patients with gallstones (cholelithiasis) experience no symptoms. Their gallstones are often discovered incidentally during imaging tests for unrelated or unexplained abdominal symptoms. Most patients with asymptomatic gallstones remain symptom-free, while about 20% develop gallstone-related symptoms.2,3
Abdominal pain is the most common symptom. The phrase biliary colic—suggesting pain that is fluctuating in nature—appears ubiquitously in the medical literature, but it does not correctly characterize the pain associated with gallstones.
Most patients with gallstone symptoms describe a constant and often severe pain in the right upper abdomen, epigastrium, or both, often persisting for 30 to 120 minutes. Symptoms are frequently reported in the epigastrium when only visceral pain fibers are stimulated due to gallbladder distention. This is usually called midline pain; however, pain occurs in the back and right shoulder in up to 60% of patients, with involvement of somatic fibers.15,16 Gallstone pain is not relieved by change of position or passage of stool or gas.
Onset of symptoms more than an hour after eating or in the late evening or at night also very strongly suggests biliary pain. Patients with a history of biliary pain are more likely to experience it again, with a 69% chance of developing recurrent pain within 2 years.17
GALLSTONE-RELATED COMPLICATIONS
Acute gallbladder inflammation (cholecystitis)
Gallbladder inflammation (cholecystitis) is the most common complication, occurring in up to 10% of symptomatic cases. Many patients with acute cholecystitis present with right upper quadrant pain that may be accompanied by anorexia, nausea, or vomiting. Inspiratory arrest on deep palpation of the right upper quadrant (Murphy sign) has a specificity of 79% to 96% for acute cholecystitis.20 Markers of systemic inflammation such as fever, elevated white blood cell count, and elevated C-reactive protein are highly suggestive of acute cholecystitis.20,21
Bile duct stones (choledocholithiasis)
Bile duct stones (choledocholithiasis) are detected in 3.4% to 12% of patients with gallstones.22,23 Most stones in the common bile duct migrate there from the gallbladder via the cystic duct. Less commonly, primary duct stones form in the duct due to biliary stasis. Removing the gallbladder does not completely eliminate the risk of bile duct stones, as stones can remain or recur after surgery.
Bile duct stones can obstruct the common bile duct, which disrupts normal bile flow and leads to jaundice. Other symptoms may include pruritus, right upper quadrant pain, nausea, and vomiting. Serum levels of bilirubin, aspartate aminotransferase, alanine aminotransferase (ALT), and alkaline phosphatase are usually high.24
Acute bacterial infection (cholangitis)
Acute bacterial infection of the biliary system (cholangitis) is usually associated with obstruction of the common bile duct. Common symptoms of acute cholangitis include right upper quadrant pain, fever, and jaundice (Charcot triad), and these are present in about 50% to 75% of cases.21 In severe cases, patients can develop altered mental status and septicemic shock in addition to the Charcot triad, a condition called the Reynold pentad. White blood cell counts and serum levels of C-reactive protein, bilirubin, aminotransferases, and alkaline phosphatase are usually elevated.21
Pancreatitis
Approximately 4% to 8% of patients with gallstones develop inflammation of the pancreas (pancreatitis).25 The diagnosis of acute pancreatitis requires at least 2 of the following:26,27
- Abdominal pain (typically epigastric, often radiating to the back)
- Amylase or lipase levels at least 3 times above the normal limit
- Imaging findings that suggest acute pancreatitis.
Gallstone-related pancreatitis should be considered if the ALT level is greater than 150 U/mL, which has a 97% specificity for gallstone-related pancreatitis.28
ABDOMINAL ULTRASONOGRAPHY FOR DIAGNOSIS
Transabdominal ultrasonography, with a sensitivity of 84% to 89% and a specificity of up to 99%, is the test of choice for detecting gallstones.29 The characteristic findings of acute cholecystitis on ultrasonography include enlargement of the gallbladder, thickening of the gallbladder wall, presence of pericholecystic fluid, and tenderness elicited by the ultrasound probe over the gallbladder (sonographic Murphy sign).
Scintigraphy as a second test
Acute cholecystitis is primarily a clinical diagnosis and typically does not require additional imaging beyond ultrasonography. When there is discordance between clinical and ultrasonographic findings, the most accurate second imaging test is scintigraphy of the biliary tract, usually performed with technetium-labeled hydroxy iminodiacetic acid. Given intravenously, the radionuclide is rapidly taken up by the liver and then secreted into the bile. In acute cholecystitis, the cystic duct is functionally occluded and the isotope does not enter the gallbladder, creating an imaging void compared with a normal appearance.
Scintigraphy is more sensitive than abdominal ultrasonography, with a sensitivity of up to 97% vs 81% to 88%, respectively.29,30 The tests have about equal specificity.
Even though scintigraphy is more sensitive, abdominal ultrasonography is often the initial test for patients with suspected acute cholecystitis because it is more widely available, takes less time, does not involve radiation exposure, and can assess for the presence or absence of gallstones and dilation of the intra- and extrahepatic bile ducts.
Looking for stones in the common bile duct
When acute cholangitis due to choledocholithiasis is suspected, abdominal ultrasonography is a prudent initial test to look for gallstones or biliary dilation suggesting obstruction by stones in the common bile duct. Abdominal ultrasonography has only a 22% to 55% sensitivity for visualizing stones in the common bile duct, but it has a 77% to 87% sensitivity for detecting common bile duct dilation, a surrogate marker of stones.31
The normal bile duct diameter ranges from 3 to 6 mm, although mild dilation is often seen in older patients or after cholecystectomy or Roux-en-Y gastric bypass surgery.32,33 Bile duct dilation of up to 10 mm can be considered normal in patients after cholecystectomy.34 A normal-appearing bile duct on ultrasonography has a negative predictive value of 95% for excluding common bile duct stones.31
Endoscopic ultrasonography (EUS), magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP) have similar sensitivity (89%–94%, 85%–92%, and 89%–93%, respectively) and specificity (94%–95%, 93%–97%, and 100%, respectively) for detecting common bile duct stones.35–37 EUS is superior to MRCP in detecting stones smaller than 6 mm.38
ERCP should be reserved for managing rather than diagnosing common bile duct stones because of the risk of pancreatitis and perforation. Patients undergoing cholecystectomy who are suspected of having choledocholithiasis may undergo intraoperative cholangiography or laparoscopic common bile duct ultrasonography.
WATCH AND WAIT, OR INTERVENE?
Asymptomatic gallstones
Standard treatment for these patients is expectant management. Cholecystectomy is not recommended for patients with asymptomatic gallstones.47 Nevertheless, some patients may benefit from prophylactic cholecystectomy. We and others48 suggest considering cholecystectomy in the following patients.
Patients with chronic hemolytic anemia (including children with sickle cell anemia and spherocytosis). These patients have a higher risk of developing calcium bilirubinate stones, and cholecystectomy has improved outcomes.49 It should be noted that most of these data come from pediatric populations and have been extrapolated to adults.
Native Americans, who have a higher risk of gallbladder cancer if they have gallstones.2,50
Conversely, calcification of the gallbladder wall (“porcelain gallbladder”) is no longer considered an absolute indication for cholecystectomy. This condition was thought to be associated with a high rate of gallbladder carcinoma, but analyses of larger, more recent data sets found much smaller risks.51,52 Further, cholecystectomy in these patients was found to be associated with high rates of postoperative complications. Thus, prophylactic cholecystectomy is no longer recommended in asymptomatic cases of porcelain gallbladder.
In addition, concomitant cholecystectomy in patients undergoing bariatric surgery is no longer considered the therapeutic standard. Historically, cholecystectomy was performed in these patients because of the increased risk of gallstones associated with rapid weight loss after surgery. However, research now weighs against concomitant cholecystectomy with bariatric surgery and most other abdominal surgeries for asymptomatic gallstones.53
Laparoscopic surgery for symptomatic gallstones
For patients experiencing acute cholecystitis, laparoscopic cholecystectomy within 72 hours is recommended.48 There were safety concerns regarding higher rates of morbidity and conversion from laparoscopic to open cholecystectomy in patients who underwent surgery before the acute cholecystitis episode had settled. However, a large meta-analysis found no significant difference between early and delayed laparoscopic cholecystectomy in bile duct injury or conversion rates.54 Further, early cholecystectomy—defined as within 1 week of symptom onset—has been found to reduce gallstone-related complications, shorten hospital stays, and lower costs.55–57 If the patient cannot undergo surgery, percutaneous cholecystotomy or novel endoscopic gallbladder drainage interventions can be used.
Several variables predict the presence of bile duct stones in patients who have symptoms (Table 4). Based on these predictors, the ASGE classifies the probabilities as low (< 10%), intermediate (10% to 50%), and high (> 50%)31:
- High-risk patients should undergo preoperative ERCP and stone extraction if needed
- Intermediate-risk patients should undergo preoperative imaging with EUS or MRCP or intraoperative bile duct evaluation, depending on the availability, costs, and local expertise.
Patients with associated cholangitis should be given intravenous fluids and broad-spectrum antibiotics. Biliary decompression should be done as early as possible to decrease the risk of morbidity and mortality. For acute cholangitis, ERCP is the treatment of choice.25
Patients with acute gallstone pancreatitis should receive conservative management with intravenous isotonic solutions and pain control, followed by laparoscopic cholecystectomy.48
The timing of laparoscopic cholecystectomy in acute gallstone pancreatitis has been debated. Studies conducted during the era of open cholecystectomy reported similar or worse outcomes if cholecystectomy was done sooner rather than later.
However, in 1999, Uhl et al58 reported that 48 of 77 patients admitted with acute gallstone pancreatitis were able to undergo laparoscopic cholecystectomy during the same admission. Success rates were 85% (30 of 35 patients) in those with mild disease and 62% (8 of 13 patients) in those with severe disease. They concluded laparoscopic cholecystectomy could be safely performed within 7 days in patients with mild disease, whereas in severe disease at least 3 weeks should elapse because of the risk of infection.
In a randomized trial published in 2010, Aboulian et al59 reported that hospital length of stay (the primary end point) was shorter in 25 patients who underwent laparoscopic cholecystectomy early (within 48 hours of admission) than in 25 patients who underwent surgery after abdominal pain had resolved and laboratory enzymes showed a normalizing trend, 3.5 vs 5.8 days (P = .0016). Rates of perioperative complications and need for conversion to open surgery were similar between the 2 groups.
If there is associated cholangitis, patients should also be given broad-spectrum antibiotics and should undergo ERCP within 24 hours of admission.25–27
SUMMARY
Gallstones are common in US adults. Abdominal ultrasonography is the diagnostic imaging test of choice to detect gallbladder stones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct. Fortunately, most gallstones are asymptomatic and can usually be managed expectantly. In patients who have symptoms or have gallstone complications, laparoscopic cholecystectomy is the standard of care.
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
- Schirmer BD, Winters KL, Edlich RF. Cholelithiasis and cholecystitis. J Long Term Eff Med Implants 2005; 15(3):329–338. doi:10.1615/JLongTermEffMedImplants.v15.i3.90
- Stinton LM, Shaffer EA. Epidemiology of gallbladder disease: cholelithiasis and cancer. Gut Liver 2012; 6(2):172–187. doi:10.5009/gnl.2012.6.2.172
- Lee JY, Keane MG, Pereira S. Diagnosis and treatment of gallstone disease. Practitioner 2015; 259(1783):15–19.
- Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology 2004; 126(5):1448–1453. doi:10.1053/j.gastro.2004.01.025
- Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part I: overall and upper gastrointestinal diseases. Gastroenterology 2009; 136(2):376–386. doi:10.1053/j.gastro.2008.12.015
- Cariati A. Gallstone classification in Western countries. Indian J Surg 2015; 77(suppl 2):376–380. doi.org/10.1007/s12262-013-0847-y
- Carey MC. Pathogenesis of gallstones. Am J Surg 1993; 165(4):410–419. doi:10.1016/S0002-9610(05)80932-8
- Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers 2016; 2:16024. doi:10.1038/nrdp.2016.24
- Stewart L, Oesterle AL, Erdan I, Griffiss JM, Way LW. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg 2002; 6(6):891–904.
- Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione Study. Hepatology 1987; 7(5):913–917. doi:10.1002/hep.1840070520
- Sood S, Winn T, Ibrahim S, et al. Natural history of asymptomatic gallstones: differential behaviour in male and female subjects. Med J Malaysia 2015; 70(6):341–345.
- Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med 1993; 119(2):116–120. doi:10.7326/0003-4819-119-2-199307150-00004
- Etminan M, Delaney JA, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011; 183(8):899–904. doi:10.1503/cmaj.110161
- Everhart JE, Khare M, Hill M, Maurer KR. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology 1999; 117(3):632–639.
- Festi D, Sottili S, Colecchia A, et al. Clinical manifestations of gallstone disease: evidence from the multicenter Italian study on cholelithiasis (MICOL). Hepatology 1999; 30(4):839–846. doi:10.1002/hep.510300401
- Berhane T, Vetrhus M, Hausken T, Olafsson S, Sondenaa K. Pain attacks in non-complicated and complicated gallstone disease have a characteristic pattern and are accompanied by dyspepsia in most patients: the results of a prospective study. Scand J Gastroenterol 2006; 41(1):93–101. doi:10.1080/00365520510023990
- Thistle JL, Cleary PA, Lachin JM, Tyor MP, Hersh T. The natural history of cholelithiasis: the National Cooperative Gallstone Study. Ann Intern Med 1984; 101(2):171–175. doi:10.7326/0003-4819-101-2-171
- Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg 1993; 165(4):399–404. doi:0.1016/S0002-9610(05)80930-4
- Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol 1989; 42(2):127–136. doi:10.1016/0895-4356(89)90086-3
- Hirota M, Takada T, Kawarada Y, et al. Diagnostic criteria and severity assessment of acute cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):78–82. doi:10.1007/s00534-006-1159-4
- Miura F, Takada T, Kawarada Y, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1):27–34. doi:10.1007/s00534-006-1153-x
- Koo KP, Traverso LW. Do preoperative indicators predict the presence of common bile duct stones during laparoscopic cholecystectomy? Am J Surg 1996; 171(5):495–499. doi:10.1016/S0002-9610(97)89611-0
- Collins C, Maguire D, Ireland A, Fitzgerald E, O’Sullivan GC. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: natural history of choledocholithiasis revisited. Ann Surg 2004; 239(1):28–33. doi:10.1097/01.sla.0000103069.00170.9c
- Costi R, Gnocchi A, Di Mario F, Sarli L. Diagnosis and management of choledocholithiasis in the golden age of imaging, endoscopy and laparoscopy. World J Gastroenterol 2014; 20(37):13382–13401. doi:10.3748/wjg.v20.i37.13382
- European Association for the Study of the Liver (EASL). EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol 2016; 65(1):146–181. doi:10.1016/j.jhep.2016.03.005
- Greenberg JA, Hsu J, Bawazeer M, et al. Clinical practice guideline: management of acute pancreatitis. Can J Surg 2016; 59 (2):128–140. doi:10.1503/cjs.015015
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108(9):1400–1416. doi:10.1038/ajg.2013.218
- Moolla Z, Anderson F, Thomson SR. Use of amylase and alanine transaminase to predict acute gallstone pancreatitis in a population with high HIV prevalence. World J Surg 2013; 37(1):156–161. doi:10.1007/s00268-012-1801-z
- Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med 1994; 154(22):2573–2581. doi:10.1001/archinte.1994.00420220069008
- Kiewiet JJ, Leeuwenburgh MM, Bipat S, et al. A systematic review and meta-analysis of diagnostic performance of imaging in acute cholecystitis. Radiology 2012; 264(3):708–720. doi:10.1148/radiol.12111561
- ASGE Standards of Practice Committee; Maple JT, Ben-Menachem T, Anderson MA, et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71(1):1–9. doi:10.1016/j.gie.2009.09.041
- Bachar GN, Cohen M, Belenky A, Atar E, Gideon S. Effect of aging on the adult extrahepatic bile duct: a sonographic study. J Ultrasound Med 2003; 22(9):879–885. doi:10.7863/jum.2003.22.9.879
- El-Hayek K, Timratana P, Meranda J, Shimizu H, Eldar S, Chand B. Post Roux-en-Y gastric bypass biliary dilation: natural process or significant entity? J Gastrointest Surg 2012; 16(12):2185–2189. doi:10.1007/s11605-012-2058-4
- Park SM, Kim WS, Bae IH, et al. Common bile duct dilatation after cholecystectomy: a one-year prospective study. J Korean Surg Soc 2012; 83(2):97–101. doi:10.4174/jkss.2012.83.2.97
- Tse F, Liu L, Barkun AN, Armstrong D, Moayyedi P. EUS: a meta-analysis of test performance in suspected choledocholithiasis. Gastrointest Endosc 2008; 67(2):235–244. doi:10.1016/j.gie.2007.09.047
- Verma D, Kapadia A, Eisen GM, Adler DG. EUS vs MRCP for detection of choledocholithiasis. Gastrointest Endosc 2006; 64(2):248–254. doi:10.1016/j.gie.2005.12.038
- Tseng LJ, Jao YT, Mo LR, Lin RC. Over-the-wire US catheter probe as an adjunct to ERCP in the detection of choledocholithiasis. Gastrointest Endosc 2001; 54(6):720–723. doi:10.1067/mge.2001.119255
- Kondo S, Isayama H, Akahane M, et al. Detection of common bile duct stones: comparison between endoscopic ultrasonography, magnetic resonance cholangiography, and helical-computed-tomographic cholangiography. Eur J Radiol 2005; 54(2):271–275. doi:10.1016/j.ejrad.2004.07.007
- Attili AF, De Santis A, Capri R, Repice AM, Maselli S. The natural history of gallstones: the GREPCO experience. The GREPCO Group. Hepatology 1995; 21(3):656–660. doi:10.1016/0270-9139(95)90514-6
- Sakorafas GH, Milingos D, Peros G. Asymptomatic cholelithiasis: is cholecystectomy really needed? A critical reappraisal 15 years after the introduction of laparoscopic cholecystectomy. Dig Dis Sci 2007; 52(5):1313–1325. doi:10.1007/s10620-006-9107-3
- Gracie WA, Ransohoff DF. The natural history of silent gallstones: the innocent gallstone is not a myth. N Engl J Med 1982; 307(13):798–800. doi:10.1056/NEJM198209233071305
- McSherry CK, Ferstenberg H, Calhoun WF, Lahman E, Virshup M. The natural history of diagnosed gallstone disease in symptomatic and asymptomatic patients. Ann Surg 1985; 202(1):59–63. doi:10.1097/00000658-198507000-00009
- Wada K, Wada K, Imamura T. Natural course of asymptomatic gallstone disease. Nihon Rinsho 1993; 51(7):1737–1743. Japanese.
- Halldestam I, Enell EL, Kullman E, Borch K. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg 2004; 91(6):734–738. doi:10.1002/bjs.4547
- Festi D, Reggiani ML, Attili AF, et al. Natural history of gallstone disease: expectant management or active treatment? Results from a population-based cohort study. J Gastroenterol Hepatol 2010; 25(4):719–724. doi:10.1111/j.1440-1746.2009.06146.x
- Shabanzadeh DM, Sorensen LT, Jorgensen T. A prediction rule for risk stratification of incidentally discovered gallstones: results from a large cohort study. Gastroenterology 2016; 150(1):156–167e1. doi:10.1053/j.gastro.2015.09.002
- Overby DW, Apelgren KN, Richardson W, Fanelli R; Society of American Gastrointestinal and Endoscopic Surgeons. SAGES guidelines for the clinical application of laparoscopic biliary tract surgery. Surg Endosc 2010; 24(10):2368–2386. doi:10.1007/s00464-010-1268-7
- Abraham S, Rivero HG, Erlikh IV, Griffith LF, Kondamudi VK. Surgical and nonsurgical management of gallstones. Am Fam Physician 2014; 89(10):795–802.
- Currò G,, Iapichino G, Lorenzini C, Palmeri R, Cucinotta E. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc 2006; 20(2):252–255. doi:10.1007/s00464-005-0318-z
- Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome. Clin Epidemiol 2014; 6:99–109. doi:10.2147/CLEP.S37357
- Chen GL, Akmal Y, DiFronzo AL, Vuong B, O’Connor V. Porcelain gallbladder: no longer an indication for prophylactic cholecystectomy. Am Surg 2015; 81(10):936–940.
- Schnelldorfer T. Porcelain gallbladder: a benign process or concern for malignancy? J Gastrointest Surg 2013; 17(6):1161–1168. doi:10.1007/s11605-013-2170-0
- Warschkow R, Tarantino I, Ukegjini K, et al. Concomitant cholecystectomy during laparoscopic Roux-en-Y gastric bypass in obese patients is not justified: a meta-analysis. Obes Surg 2013; 23(3)3979–408. doi:10.1007/s11695-012-0852-4
- Gurusamy K, Samraj K, Gluud C, Wilson E, Davidson BR. Meta-analysis of randomized controlled trials on the safety and effectiveness of early versus delayed laparoscopic cholecystectomy for acute cholecystitis. Br J Surg 2010; 97(2):141–150. doi:10.1002/bjs.6870
- Papi C, Catarci M, D’Ambrosio L, et al. Timing of cholecystectomy for acute calculous cholecystitis: a meta-analysis. Am J Gastroenterol 2004; 99(1):147–155. doi:10.1046/j.1572-0241.2003.04002.x
- Gurusamy KS, Davidson C, Gluud C, Davidson BR. Early versus delayed laparoscopic cholecystectomy for people with acute cholecystitis. Cochrane Database Syst Rev 2013; 6:CD005440. doi:10.1002/14651858
- Menahem B, Mulliri A, Fohlen A, Guittet L, Alves A, Lubrano J. Delayed laparoscopic cholecystectomy increases the total hospital stay compared to an early laparoscopic cholecystectomy after acute cholecystitis: an updated meta-analysis of randomized controlled trials. HPB (Oxford) 2015; 17(10):857–862. doi:10.1111/hpb.12449
- Uhl W, Müller CA, Krähenbühl L, Schmid SW, Schölzel S, Büchler MW. Acute gallstone pancreatitis: timing of laparoscopic cholecystectomy in mild and severe disease. Surg Endosc 1999; 13(11):1070–1076. doi:10.1007/s004649901175
- Aboulian A, Chan T, Yaghoubian A, et al. Early cholecystectomy safely decreases hospital stay in patients with mild gallstone pancreatitis: a randomized prospective study. Ann Surg 2010(4): 251:615–619. doi:10.1097/SLA.0b013e3181c38f1f
KEY POINTS
- Abdominal pain is the primary symptom associated with gallstones.
- Abdominal ultrasonography is the diagnostic test of choice to detect gallstones and assess for findings suggestive of acute cholecystitis and dilation of the common bile duct.
- First-line therapy for asymptomatic gallstones is expectant management.
- First-line therapy for symptomatic gallstones is cholecystectomy.
Musculoskeletal ultrasonography basics
Ultrasonography has been used to evaluate musculoskeletal problems for decades but has only recently become more widely available in the United States. Advances in technology and physician familiarity are increasing its role in orthopedic imaging.
No single imaging method can yield all musculoskeletal diagnoses. Like any imaging technique, ultrasonography has strengths and weaknesses specific to orthopedics. Radiography, computed tomography (CT), and magnetic resonance imaging (MRI) play important roles for investigating musculoskeletal problems and are complementary to each other and to ultrasonography.
To help clinicians make informed decisions about ordering musculoskeletal ultrasonography, this article reviews the basic physics underlying ultrasonography, its advantages and disadvantages compared with other imaging methods, and common clinical applications.
CLASSIC TECHNOLOGY MAKING A RESURGENCE
The first reports of the use of musculoskeletal ultrasonography appeared in the 1970s for investigating the rotator cuff,1–3 actually preceding reports of its use in obstetrics and gynecology.4 In the 1980s, reports emerged for evaluating the Achilles tendon.5,6 After that, its popularity in the United States plateaued, likely because of the advent of MRI, lower reimbursement and greater variability in interpretation compared with MRI, as well as a lack of physicians and sonographers trained in its use.7,8
Musculoskeletal ultrasonography is currently experiencing a resurgence. Although it remains a specialized service more commonly available in large hospitals, its use is increasing rapidly, and it will likely become more widely available.
SPECIAL TRAINING REQUIRED
Musculoskeletal ultrasonography is simply an ultrasonographic examination of part of the musculoskeletal system. But because not all ultrasonographic transducers offer sufficient resolution for musculoskeletal evaluation and not all sonographers and imaging physicians are familiar with the specialized techniques, musculoskeletal ultrasonography often has a separate designation (eg, “MSKUS,” “MSUS”). At Cleveland Clinic, it is offered through the department of musculoskeletal imaging by subspecialty-trained musculoskeletal radiologists and specially trained musculoskeletal ultrasonographers with 4 to 5 years of training in the technique.
Musculoskeletal ultrasonography is also performed by physician groups with specialized training, including sports medicine physicians, rheumatologists, physiatrists, neurologists, and orthopedic surgeons. The American Institute of Ultrasound in Medicine offers voluntary accreditation for practice groups using musculoskeletal ultrasonography. Certification in musculoskeletal radiology is offered to sonographers through the American Registry for Diagnostic Medical Sonography.
SONOGRAPHY HAS UNIQUE QUALITIES
Ultrasonography uses high-frequency sound waves to generate images. The transducer (or probe) emits sound from the many piezoelectric elements at its surface, and the sound waves travel through and react with tissues. Sound reflected by tissues is detected by the transducer and converted to an image. Objects that reflect sound appear hyperechoic (brighter), whereas tissues that reflect little or no sound appear hypoechoic.
High-resolution imaging of superficial structures

(B, arrow).
Ultrasonography involves a fundamental trade-off between image resolution and imaging depth. Higher-frequency sound waves do not penetrate far into tissues but generate a higher-resolution image; lower-frequency sound waves can penetrate much further but yield a lower-resolution image. Although high-resolution imaging of deep structures with ultrasonography is not possible (Figure 1), many musculoskeletal structures are located superficially and are amenable to ultrasonographic evaluation.
Be aware of artifacts

Some materials attenuate sound very little, such as simple fluid. Low attenuation results in artifacts on ultrasonography, making tissues behind the simple fluid appear brighter than neighboring tissues. These artifacts may be reported as “increased through transmission” or “posterior acoustic enhancement.” Conversely, metal and bone reflect all sound waves that reach them, rendering any structures beyond them invisible. This “shadowing” creates a problem for imaging of structures in or near bone. Subcutaneous fat also attenuates sound waves, limiting the use of ultrasonography for patients with obesity (Figure 2).

High-frequency linear transducer sharpens images
High-frequency linear transducers reduce anisotropy because their flat surface keeps sound waves more uniformly perpendicular to the structure of interest.4,7 Their development has allowed imaging of superficial structures that is superior to that of MRI. A high-frequency linear transducer offers more than twice the spatial resolution of a typical 1.5T MRI examination of superficial tissue.12,13
Operator experience is critical
Ultrasonography examinations, more than other imaging tests, are dependent on operator experience. A solid understanding of musculoskeletal anatomy is imperative. Because the probe images only a thin section of tissue (about the thickness of a credit card), referencing adjacent structures for orientation is more difficult with ultrasonography than with CT or MRI.
The accuracy of ultrasonography is highly dependent on acquiring and interpreting images, whereas the accuracy of MRI is dependent primarily on image interpretation.7 Interpreting physicians must check that sonographers capture relevant targets.
STRENGTHS OF MUSCULOSKELETAL ULTRASONOGRAPHY
Ultrasonography has multiple advantages:
No ionizing radiation exposure.
Portability. Unlike CT or MRI, ultrasonography equipment is portable.
Increased patient comfort. Patient positioning for an ultrasonography examination is more flexible than for MRI or CT,14 and the examination does not induce claustrophobia.8
High-resolution imaging. Ultrasonography provides very-high-resolution imaging of superficial soft tissues—in some cases, higher than MRI or CT.
Real-time dynamic examinations are possible with ultrasonography, unlike with CT or MRI, and may increase test sensitivity.4,15–18
Implanted hardware is less of a problem. Although ultrasonography cannot image beyond implanted orthopedic metallic hardware, the hardware does not obscure surrounding soft tissues as it does on CT and MRI.6,19,20 Also, ultrasonography is safe for patients with a pacemaker.8
WEAKNESSES

The main disadvantages of musculoskeletal ultrasonography are inherent to its limited field of view, making it inappropriate for a survey examination (eg, for ankle pain, knee pain, hip pain).4 Unlike CT and MRI, ultrasonography does not provide a “bird’s-eye view,” and important abnormalities can be missed during evaluation of large areas (Figure 4).
Ultrasonography also cannot evaluate bone or intra-articular structures such as cartilage, bone marrow, labrum, and intra-articular ligaments; MRI is the standard for evaluating these structures.21
Ultrasonography is time-consuming. To perform a detailed examination of the anterior, posterior, medial, and lateral aspects of the hip, knee, or ankle would require 1.5 to 2 hours of scanning time and an additional 10 to 25 minutes of image checking and interpretation.
CURRENT CLINICAL INDICATIONS
Musculoskeletal ultrasonography is best used for clinical questions regarding limited, superficial musculoskeletal problems.
Fluid collections
Ultrasonography can help evaluate small fluid collections in soft tissue. As is true for a lung opacity on chest radiography, soft-tissue fluid detected on ultrasonography is nonspecific, and results must be correlated with the clinical picture to narrow the differential diagnosis.
Fluid collections can be classified as loculated or nonloculated.
Nonloculated fluid involves more fluid than is simply interposed between tissue planes and has no wall or defined margins. It can be simple or complex in appearance: simple fluid is anechoic, and complex fluid appears more heterogeneous and may contain septations or debris.
Subcutaneous edema, which may occur postoperatively or from trauma, venous insufficiency, or inflammatory or infectious processes, appears on ultrasonography as nonloculated fluid interspersed between subcutaneous fat lobules.
Loculated fluid collections have well-defined margins or a discrete wall that does not follow normal tissue planes. They can also be simple or complex and can be caused by hematoma, abscess, or ganglion. Less commonly, neoplasms can mimic a loculated fluid collection (Figure 4).
A ganglion is a specific type of loculated fluid collection containing synovial fluid arising from a joint or tendon sheath. It tends to occur in specific locations, most commonly around the wrist, most often arising from the dorsal scapholunate ligament and volar wrist between the radial artery and flexor carpi radialis.22 On MRI, it can be difficult to distinguish between small vascular structures and a small ganglion, especially in the hands and feet.23
Ultrasonography can also help identify a Baker cyst, a specific fluid collection arising from the semimembranosus bursa between the medial head of the gastrocnemius tendon and the semimembranosus tendon. Ultrasonography can also detect inflammation, rupture, or leaking associated with a Baker cyst.24
Power Doppler is an ultrasonographic examination that can detect increased blood flow surrounding a fluid collection and determine the likelihood of an acute inflammatory or infectious cause.25
Joint effusion and synovitis
Musculoskeletal ultrasonography can help evaluate joints for effusion and synovitis. It is highly sensitive (94%) and specific (95%) for synovitis, making it superior to contrast-enhanced MRI.26,27 The area of concern should be limited to 1 quadrant of a joint (anterior, posterior, medial, or lateral); for problems beyond that, MRI should be considered.
A joint effusion appears as a distended joint capsule containing hypoechoic (complex) or anechoic (simple) joint fluid.
Complex joint fluid may contain debris and occurs with hemarthrosis, infection, and inflammation.23 Hypertrophied synovium is hypoechoic and can mimic complex joint fluid.
Power Doppler evaluation can help distinguish synovitis from joint fluid by demonstrating blood flow, a feature of synovitis but not of simple joint fluid. Power Doppler is the most sensitive means of detecting blood flow, although it does not show direction of flow.28
Using ultrasonography can help to improve disease control and minimize disabling changes by monitoring synovitis therapy. In addition, subclinical synovitis and enthesitis (inflammation of insertion sites of tendons or ligaments into bone) detected by ultrasonography may predict future disease and disease flares.29–31
Ultrasonographic guidance for a wide range of procedures is increasing rapidly.32–36 Multiple studies have shown the advantage of ultrasonography-guided aspiration and injection compared with techniques without imaging guidance.37,38
Soft-tissue masses
Accurately diagnosing soft-tissue masses can be difficult. A mass may remain indeterminate even after multiple imaging studies, requiring biopsy or surgical referral. However, for a few specific masses, ultrasonography is highly accurate and can eliminate the need for further imaging.

Ultrasonography can help evaluate soft- tissue masses no larger than 5 cm in diameter and no deeper than superficial muscular fascia. If the mass is larger or deeper than that, ultrasonography is less reliable for showing the margins of the mass and its relationship to adjacent structures (Figure 5). Further imaging by MRI may be recommended in such cases.
Fortunately, many of the most common soft-tissue masses can be accurately diagnosed with ultrasonography, including lipomas, ganglion cysts, foreign bodies, and simple fluid collections.4,39 Nerve-sheath tumors can also be diagnosed with ultrasonography if the lesion clearly arises from a nerve. Other soft-tissue masses are likely to be indeterminate with ultrasonography, requiring follow-up with MRI with contrast.
Tendons
Musculoskeletal ultrasonography can be effective for evaluating tendons around joints, especially 1 or a small number of nearby superficial tendons. Tendons particularly well suited for ultrasonographic examination include:
- Upper-extremity tendons located in the rotator cuff or around the elbow, and flexor and extensor tendons of the hands; ultrasonographic evaluation of the rotator cuff is highly accurate, equivalent to that of MRI for partial-thickness and full-thickness tearing40–43
- Lower-extremity tendons of the extensor mechanism of the knee, distal hamstring tendons, tendons around the ankle,44–46 and flexor and extensor tendons of the foot.

Ultrasonography can help diagnose a variety of tendon abnormalities (Table 1),48,49 including tearing, for which a dynamic examination can be performed.
Many tendons have a tendon sheath containing tenosynovium, while others have surrounding peritenon only; either can become thickened and inflamed. Tenosynovitis is a nonspecific finding and may be inflammatory, infectious, or posttraumatic. The presence of tendon sheath fluid alone on ultrasonography can be a normal finding, and some tendon sheaths that communicate with adjacent joints (eg, the long head biceps tendon, the flexor hallucis longus tendon) commonly contain simple fluid.6 A dynamic examination with ultrasonography can help diagnose snapping related to abnormal tendon movement, for example, in the case of intra-sheath and extra-sheath subluxation of the peroneal tendons.45,50,51
Ligaments
Ultrasonography can detect abnormalities in many superficial ligaments (Table 1).
Ankle. Ankle ligaments are superficial and can be clearly visualized. The diagnostic accuracy of ultrasonography for tearing of the anterior talofibular ligament may be as high as 100%.50,52,53
Elbow and thumb. The larger of the collateral ligaments of the elbow, especially the ulnar collateral ligament, and the ulnar collateral ligament of the thumb can be effectively evaluated with ultrasonography.54,55
Knee. The collateral ligaments of the knee can be seen with ultrasonography, but injuries of the external ligaments of the knee are often associated with intrinsic derangements that cannot be evaluated with ultrasonography.56,57 Intra-articular ligaments such as the anterior cruciate ligament are also not amenable to ultrasonography.
Dynamic examination of a ligament with ultrasonography can help determine the grade of the injury.
Deeply located ligaments (eg, around the hip) and ligaments surrounded by bone, such as the Lisfranc ligament, cannot be completely seen on ultrasonography.
Muscle
Musculoskeletal ultrasonography is useful for small areas of concern within a muscle (Table 1). It can detect muscle strains and tears, intramuscular collections or lesions, and fascial scarring or fascial injuries such as superficial muscle herniation. Although ultrasonography may yield a definitive diagnosis for a muscle problem, further imaging may be needed.
Nerves
Ultrasonography is useful for peripheral nerve investigation but requires a steep learning curve for sonographers and interpreting physicians.58,59 It is best suited for directed questions regarding focal abnormal nerve findings on physical examination.
Ultrasonography can help identify areas of nerve entrapment caused by a mass or dynamic compression. It can detect neuritis (Table 1), lesions of peripheral nerves (eg, nerve-sheath tumors), and neuromas (eg, Morton neuroma of the intermetatarsal space). In a large meta-analysis, ultrasonography and MRI were found to be equally accurate for detecting Morton neuroma.60 Even for nerve-sheath tumors located deep to the muscular fascia, ultrasonography can confirm the diagnosis because of the characteristic appearance of the nerves. Ultrasonography can also demonstrate a large extent of the course of superficial peripheral nerves while keeping the imaging plane appropriately oriented to the nerves.
Acknowledgment: We would like to sincerely thank Megan Griffiths, MA, for her help in the preparation and submission of this manuscript.
- Hamilton JV, Flinn G Jr, Haynie CC, Cefalo RC. Diagnosis of rectus sheath hematoma by B-mode ultrasound: a case report. Am J Obstet Gynecol 1976; 125(4):562–565. doi:10.1016/0002-9378(76)90379-3
- Zweymüller VK, Kratochwil A. Ultrasound diagnosis of bone and soft tissue tumours. Wien Klin Wochenschr 1975; 87(12):397–398. German.
- Mayer V. Ultrasonography of the rotator cuff. J Ultrasound Med 1985; 4(11):608, 607. doi:10.7863/jum.1985.4.11.608
- McNally EG. The development and clinical applications of musculoskeletal ultrasound. Skeletal Radiol 2011; 40(9):1223–1231. doi:10.1007/s00256-011-1220-5
- Ignashin NS, Girshin SG, Tsypin IS. Ultrasonic scanning in subcutaneous rupture of the Achilles tendon. Vestn Khir Im I I Grek 1981; 127(9):82–85. Russian.
- Robinson P. Sonography of common tendon injuries. AJR Am J Roentgenol 2009; 193(3):607–618. doi:10.2214/AJR.09.2808
- Jacobson JA. Musculoskeletal ultrasound: focused impact on MRI. AJR Am J Roentgenol 2009; 193(3):619–627. doi:10.2214/AJR.09.2841
- Nazarian LN. The top 10 reasons musculoskeletal sonography is an important complementary or alternative technique to MRI. AJR Am J Roentgenol 2008; 190(6):1621–1626. doi:10.2214/AJR.07.3385
- AIUM technical bulletin. Transducer manipulation. American Institute of Ultrasound in Medicine. J Ultrasound Med 1999; 18(2):169–175. doi:10.7863/jum.1999.18.2.169
- Connolly DJ, Berman L, McNally EG. The use of beam angulation to overcome anisotropy when viewing human tendon with high frequency linear array ultrasound. Br J Radiol 2001; 74 (878):183–185. doi:10.1259/bjr.74.878.740183
- Crass JR, van de Vegte GL, Harkavy LA. Tendon echogenicity: ex vivo study. Radiology 1988; 167(2):499–501. doi:10.1148/radiology.167.2.3282264
- Erickson SJ. High-resolution imaging of the musculoskeletal system. Radiology 1997; 205(3):593–618. doi:10.1148/radiology.205.3.9393511
- Link TM, Majumdar S, Peterfy C, et al. High resolution MRI of small joints: impact of spatial resolution on diagnostic performance and SNR. Magn Reson Imaging 1998; 16(2):147–155. doi:10.1016/S0730-725X(97)00244-0
- Middleton WD, Payne WT, Teefey SA, Hildebolt CF, Rubin DA, Yamaguchi K. Sonography and MRI of the shoulder: comparison of patient satisfaction. AJR Am J Roentgenol 2004; 183(5):1449–1452. doi:10.2214/ajr.183.5.1831449
- Khoury V, Cardinal E, Bureau NJ. Musculoskeletal sonography: a dynamic tool for usual and unusual disorders. AJR Am J Roentgenol 2007; 188(1):W63–W73. doi:10.2214/AJR.06.0579
- Farin PU, Jaroma H, Harju A, Soimakallio S. Medial displacement of the biceps brachii tendon: evaluation with dynamic sonography during maximal external shoulder rotation. Radiology 1995; 195(3):845–848. doi:10.1148/radiology.195.3.7754019
- Miller TT, Adler RS, Friedman L. Sonography of injury of the ulnar collateral ligament of the elbow-initial experience. Skeletal Radiol 2004; 33(7):386–391. doi:10.1007/s00256-004-0788-4
- Nazarian LN, McShane JM, Ciccotti MG, O’Kane PL, Harwood MI. Dynamic US of the anterior band of the ulnar collateral ligament of the elbow in asymptomatic major league baseball pitchers. Radiology 2003; 227(1):149–154. doi:10.1148/radiol.2271020288
- Jacobson JA, Lax MJ. Musculoskeletal sonography of the postoperative orthopedic patient. Semin Musculoskelet Radiol 2002; 6(1):67–77. doi:10.1055/s-2002-23165
- Sofka CM, Adler RS. Original report. Sonographic evaluation of shoulder arthroplasty. AJR Am J Roentgenol 2003; 180(4):1117–1120. doi:10.2214/ajr.180.4.1801117
- Silvestri E, Martinoli C, Derchi LE, Bertolotto M, Chiaramondia M, Rosenberg I. Echotexture of peripheral nerves: correlation between US and histologic findings and criteria to differentiate tendons. Radiology 1995; 197(1):291–296. doi:10.1148/radiology.197.1.7568840
- Cardinal E, Buckwalter KA, Braunstein EM, Mih AD. Occult dorsal carpal ganglion: comparison of US and MR imaging. Radiology 1994; 193(1):259–262. doi:10.1148/radiology.193.1.8090903
- Jacobson JA. Musculoskeletal ultrasound and MRI: which do I choose? Semin Musculoskelet Radiol 2005; 9(2):135–149. doi:10.1055/s-2005-872339
- Ward EE, Jacobson JA, Fessell DP, Hayes CW, van Holsbeeck M. Sonographic detection of Baker’s cysts: comparison with MR imaging. AJR Am J Roentgenol 2001; 176(2):373–380. doi:10.2214/ajr.176.2.1760373
- Bhasin S, Cheung PP. The role of power Doppler ultrasonography as disease activity marker in rheumatoid arthritis. Dis Markers 2015; 2015:325909. doi:10.1155/2015/325909
- Fukuba E, Yoshizako T, Kitagaki H, Murakawa Y, Kondo M, Uchida N. Power Doppler ultrasonography for assessment of rheumatoid synovitis: comparison with dynamic magnetic resonance imaging. Clin Imaging 2013; 37(1):134–137. doi:10.1016/j.clinimag.2012.02.008
- Takase-Minegishi K, Horita N, Kobayashi K, et al. Diagnostic test accuracy of ultrasound for synovitis in rheumatoid arthritis: systematic review and meta-analysis. Rheumatology (Oxford) 2018; 57(1):49–58. doi:10.1093/rheumatology/kex036
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet 2009; 373(9664):659–672. doi:10.1016/S0140-6736(09)60008-8
- Ash ZR, Tinazzi I, Gallego CC, et al. Psoriasis patients with nail disease have a greater magnitude of underlying systemic subclinical enthesopathy than those with normal nails. Ann Rheum Dis 2012; 71(4):553–556. doi:10.1136/annrheumdis-2011-200478
- Han J, Geng Y, Deng X, Zhang Z. Subclinical synovitis assessed by ultrasound predicts flare and progressive bone erosion in rheumatoid arthritis patients with clinical remission: a systematic review and metaanalysis. J Rheumatol 2016; 43(11):2010–2018. doi.org/10.3899/jrheum.160193
- Iagnocco A, Finucci A, Ceccarelli F, Perricone C, Iorgoveanu V, Valesini G. Power Doppler ultrasound monitoring of response to anti-tumour necrosis factor alpha treatment in patients with rheumatoid arthritis. Rheumatology (Oxford) 2015; 54(10):1890–1896. doi:10.1093/rheumatology/kev211
- Henning PT. Ultrasound-guided foot and ankle procedures. Phys Med Rehabil Clin N Am 2016; 27(3):649–671. doi:10.1016/j.pmr.2016.04.005
- Lueders DR, Smith J, Sellon JL. Ultrasound-guided knee procedures. Phys Med Rehabil Clin North Am 2016; 27(3):631–648. doi:10.1016/j.pmr.2016.04.010
- Payne JM. Ultrasound-guided hip procedures. Phys Med Rehabil Clin North Am 2016; 27(3):607–629. doi:10.1016/j.pmr.2016.04.004
- Strakowski JA. Ultrasound-guided peripheral nerve procedures. Phys Med Rehabil Clin North Am 2016; 27(3):687–715. doi:10.1016/j.pmr.2016.04.006
- Sussman WI, Williams CJ, Mautner K. Ultrasound-guided elbow procedures. Phys Med Rehabil Clin North Am 2016; 27(3):573–587. doi:10.1016/j.pmr.2016.04.002
- Finnoff JT. The evolution of diagnostic and interventional ultrasound in sports medicine. PM R 2016; 8(suppl 3):S133–S138. doi:10.1016/j.pmrj.2015.09.022
- Wu T, Dong Y, Song H, Fu Y, Li JH. Ultrasound-guided versus landmark in knee arthrocentesis: a systematic review. Semin Arthritis Rheum 2016; 45(5):627–632. doi:10.1016/j.semarthrit.2015.10.011
- Failla JM, van Holsbeeck M, Vanderschueren G. Detection of a 0.5-mm-thick thorn using ultrasound: a case report. J Hand Surg Am 1995; 20(3):456–457.
- Teefey SA, Hasan SA, Middleton WD, Patel M, Wright RW, Yamaguchi K. Ultrasonography of the rotator cuff. A comparison of ultrasonographic and arthroscopic findings in one hundred consecutive cases. J Bone Joint Surg Am 2000; 82(4):498–504.
- van Holsbeeck MT, Kolowich PA, Eyler WR, et al. US depiction of partial-thickness tear of the rotator cuff. Radiology 1995; 197(2):443–446. doi:10.1148/radiology.197.2.7480690
- Balich SM, Sheley RC, Brown TR, Sauser DD, Quinn SF. MR imaging of the rotator cuff tendon: interobserver agreement and analysis of interpretive errors. Radiology 1997; 204(1):191–194. doi:10.1148/radiology.204.1.9205245
- Dinnes J, Loveman E, McIntyre L, Waugh N. The effectiveness of diagnostic tests for the assessment of shoulder pain due to soft tissue disorders: a systematic review. Health Technol Assess 2003; 7(29):1–166. doi:10.3310/hta7290
- Rockett MS, Waitches G, Sudakoff G, Brage M. Use of ultrasonography versus magnetic resonance imaging for tendon abnormalities around the ankle. Foot Ankle Int 1998; 19(9):604–612.
- Grant TH, Kelikian AS, Jereb SE, McCarthy RJ. Ultrasound diagnosis of peroneal tendon tears. A surgical correlation. J Bone Joint Surg Am 2005; 87(8):1788–1794. doi:10.2106/JBJS.D.02450
- Hartgerink P, Fessell DP, Jacobson JA, van Holsbeeck MT. Full- versus partial-thickness Achilles tendon tears: sonographic accuracy and characterization in 26 cases with surgical correlation. Radiology 2001; 220(2):406–412. doi:10.1148/radiology.220.2.r01au41406
- Cho KH, Park BH, Yeon KM. Ultrasound of the adult hip. Semin Ultrasound CT MR 2000; 21(3):214–230.
- Adler RS, Finzel KC. The complementary roles of MR imaging and ultrasound of tendons. Radiol Clin North Am 2005; 43(4):771–807. doi:10.1016/j.rcl.2005.02.011
- Martinoli C, Bianchi S, Derchi LE. Tendon and nerve sonography. Radiol Clin North Am 1999; 37(4):691–711. doi:10.1016/S0033-8389(05)70124-X
- Fessell DP, Vanderschueren GM, Jacobson JA, et al. US of the ankle: technique, anatomy, and diagnosis of pathologic conditions. Radiographics 1998; 18(2):325–340. doi:10.1148/radiographics.18.2.9536481
- Neustadter J, Raikin SM, Nazarian LN. Dynamic sonographic evaluation of peroneal tendon subluxation. AJR Am J Roentgenol 2004; 183(4):985–988. doi:10.2214/ajr.183.4.1830985
- Verhaven EF, Shahabpour M, Handelberg FW, Vaes PH, Opdecam PJ. The accuracy of three-dimensional magnetic resonance imaging in the diagnosis of ruptures of the lateral ligaments of the ankle. Am J Sports Med 1991; 19(6):583–587. doi:10.1177/036354659101900605
- Milz P, Milz S, Steinborn M, Mittlmeier T, Putz R, Reiser M. Lateral ankle ligaments and tibiofibular syndesmosis. 13-MHz high-frequency sonography and MRI compared in 20 patients. Acta Orthop Scand 1998; 69(1):51–55.
- De Smet AA, Winter TC, Best TM, Bernhardt DT. Dynamic sonography with valgus stress to assess elbow ulnar collateral ligament injury in baseball pitchers. Skeletal Radiol 2002; 31(11):671–676. doi:10.1007/s00256-002-0558-0
- Melville DM, Jacobson JA, Fessell DP. Ultrasound of the thumb ulnar collateral ligament: technique and pathology. AJR Am J Roentgenol 2014; 202(2):W168. doi:10.2214/AJR.13.11335
- Court-Payen M. Sonography of the knee: intra-articular pathology. J Clin Ultrasound 2004; 32(9):481–490. doi:10.1002/jcu.20069
- Azzoni R, Cabitza P. Is there a role for sonography in the diagnosis of tears of the knee menisci? J Clin Ultrasound 2002; 30(8):472–476. doi:10.1002/jcu.10106
- Jacobson JA, Wilson TJ, Yang LJ. Sonography of common peripheral nerve disorders with clinical correlation. J Ultrasound Med 2016; 35(4):683–693. doi:10.7863/ultra.15.05061
- Ali ZS, Pisapia JM, Ma TS, Zager EL, Heuer GG, Khoury V. Ultrasonographic evaluation of peripheral nerves. World Neurosurg 2016; 85(1):333–339. doi:10.1016/j.wneu.2015.10.005
- Bignotti B, Signori A, Sormani MP, Molfetta L, Martinoli C, Tagliafico A. Ultrasound versus magnetic resonance imaging for Morton neuroma: systematic review and meta-analysis. Eur Radiol 2015; 25(8):2254–2262. doi:10.1007/s00330-015-3633-3
Ultrasonography has been used to evaluate musculoskeletal problems for decades but has only recently become more widely available in the United States. Advances in technology and physician familiarity are increasing its role in orthopedic imaging.
No single imaging method can yield all musculoskeletal diagnoses. Like any imaging technique, ultrasonography has strengths and weaknesses specific to orthopedics. Radiography, computed tomography (CT), and magnetic resonance imaging (MRI) play important roles for investigating musculoskeletal problems and are complementary to each other and to ultrasonography.
To help clinicians make informed decisions about ordering musculoskeletal ultrasonography, this article reviews the basic physics underlying ultrasonography, its advantages and disadvantages compared with other imaging methods, and common clinical applications.
CLASSIC TECHNOLOGY MAKING A RESURGENCE
The first reports of the use of musculoskeletal ultrasonography appeared in the 1970s for investigating the rotator cuff,1–3 actually preceding reports of its use in obstetrics and gynecology.4 In the 1980s, reports emerged for evaluating the Achilles tendon.5,6 After that, its popularity in the United States plateaued, likely because of the advent of MRI, lower reimbursement and greater variability in interpretation compared with MRI, as well as a lack of physicians and sonographers trained in its use.7,8
Musculoskeletal ultrasonography is currently experiencing a resurgence. Although it remains a specialized service more commonly available in large hospitals, its use is increasing rapidly, and it will likely become more widely available.
SPECIAL TRAINING REQUIRED
Musculoskeletal ultrasonography is simply an ultrasonographic examination of part of the musculoskeletal system. But because not all ultrasonographic transducers offer sufficient resolution for musculoskeletal evaluation and not all sonographers and imaging physicians are familiar with the specialized techniques, musculoskeletal ultrasonography often has a separate designation (eg, “MSKUS,” “MSUS”). At Cleveland Clinic, it is offered through the department of musculoskeletal imaging by subspecialty-trained musculoskeletal radiologists and specially trained musculoskeletal ultrasonographers with 4 to 5 years of training in the technique.
Musculoskeletal ultrasonography is also performed by physician groups with specialized training, including sports medicine physicians, rheumatologists, physiatrists, neurologists, and orthopedic surgeons. The American Institute of Ultrasound in Medicine offers voluntary accreditation for practice groups using musculoskeletal ultrasonography. Certification in musculoskeletal radiology is offered to sonographers through the American Registry for Diagnostic Medical Sonography.
SONOGRAPHY HAS UNIQUE QUALITIES
Ultrasonography uses high-frequency sound waves to generate images. The transducer (or probe) emits sound from the many piezoelectric elements at its surface, and the sound waves travel through and react with tissues. Sound reflected by tissues is detected by the transducer and converted to an image. Objects that reflect sound appear hyperechoic (brighter), whereas tissues that reflect little or no sound appear hypoechoic.
High-resolution imaging of superficial structures
(B, arrow).
Ultrasonography involves a fundamental trade-off between image resolution and imaging depth. Higher-frequency sound waves do not penetrate far into tissues but generate a higher-resolution image; lower-frequency sound waves can penetrate much further but yield a lower-resolution image. Although high-resolution imaging of deep structures with ultrasonography is not possible (Figure 1), many musculoskeletal structures are located superficially and are amenable to ultrasonographic evaluation.
Be aware of artifacts
Some materials attenuate sound very little, such as simple fluid. Low attenuation results in artifacts on ultrasonography, making tissues behind the simple fluid appear brighter than neighboring tissues. These artifacts may be reported as “increased through transmission” or “posterior acoustic enhancement.” Conversely, metal and bone reflect all sound waves that reach them, rendering any structures beyond them invisible. This “shadowing” creates a problem for imaging of structures in or near bone. Subcutaneous fat also attenuates sound waves, limiting the use of ultrasonography for patients with obesity (Figure 2).
High-frequency linear transducer sharpens images
High-frequency linear transducers reduce anisotropy because their flat surface keeps sound waves more uniformly perpendicular to the structure of interest.4,7 Their development has allowed imaging of superficial structures that is superior to that of MRI. A high-frequency linear transducer offers more than twice the spatial resolution of a typical 1.5T MRI examination of superficial tissue.12,13
Operator experience is critical
Ultrasonography examinations, more than other imaging tests, are dependent on operator experience. A solid understanding of musculoskeletal anatomy is imperative. Because the probe images only a thin section of tissue (about the thickness of a credit card), referencing adjacent structures for orientation is more difficult with ultrasonography than with CT or MRI.
The accuracy of ultrasonography is highly dependent on acquiring and interpreting images, whereas the accuracy of MRI is dependent primarily on image interpretation.7 Interpreting physicians must check that sonographers capture relevant targets.
STRENGTHS OF MUSCULOSKELETAL ULTRASONOGRAPHY
Ultrasonography has multiple advantages:
No ionizing radiation exposure.
Portability. Unlike CT or MRI, ultrasonography equipment is portable.
Increased patient comfort. Patient positioning for an ultrasonography examination is more flexible than for MRI or CT,14 and the examination does not induce claustrophobia.8
High-resolution imaging. Ultrasonography provides very-high-resolution imaging of superficial soft tissues—in some cases, higher than MRI or CT.
Real-time dynamic examinations are possible with ultrasonography, unlike with CT or MRI, and may increase test sensitivity.4,15–18
Implanted hardware is less of a problem. Although ultrasonography cannot image beyond implanted orthopedic metallic hardware, the hardware does not obscure surrounding soft tissues as it does on CT and MRI.6,19,20 Also, ultrasonography is safe for patients with a pacemaker.8
WEAKNESSES
The main disadvantages of musculoskeletal ultrasonography are inherent to its limited field of view, making it inappropriate for a survey examination (eg, for ankle pain, knee pain, hip pain).4 Unlike CT and MRI, ultrasonography does not provide a “bird’s-eye view,” and important abnormalities can be missed during evaluation of large areas (Figure 4).
Ultrasonography also cannot evaluate bone or intra-articular structures such as cartilage, bone marrow, labrum, and intra-articular ligaments; MRI is the standard for evaluating these structures.21
Ultrasonography is time-consuming. To perform a detailed examination of the anterior, posterior, medial, and lateral aspects of the hip, knee, or ankle would require 1.5 to 2 hours of scanning time and an additional 10 to 25 minutes of image checking and interpretation.
CURRENT CLINICAL INDICATIONS
Musculoskeletal ultrasonography is best used for clinical questions regarding limited, superficial musculoskeletal problems.
Fluid collections
Ultrasonography can help evaluate small fluid collections in soft tissue. As is true for a lung opacity on chest radiography, soft-tissue fluid detected on ultrasonography is nonspecific, and results must be correlated with the clinical picture to narrow the differential diagnosis.
Fluid collections can be classified as loculated or nonloculated.
Nonloculated fluid involves more fluid than is simply interposed between tissue planes and has no wall or defined margins. It can be simple or complex in appearance: simple fluid is anechoic, and complex fluid appears more heterogeneous and may contain septations or debris.
Subcutaneous edema, which may occur postoperatively or from trauma, venous insufficiency, or inflammatory or infectious processes, appears on ultrasonography as nonloculated fluid interspersed between subcutaneous fat lobules.
Loculated fluid collections have well-defined margins or a discrete wall that does not follow normal tissue planes. They can also be simple or complex and can be caused by hematoma, abscess, or ganglion. Less commonly, neoplasms can mimic a loculated fluid collection (Figure 4).
A ganglion is a specific type of loculated fluid collection containing synovial fluid arising from a joint or tendon sheath. It tends to occur in specific locations, most commonly around the wrist, most often arising from the dorsal scapholunate ligament and volar wrist between the radial artery and flexor carpi radialis.22 On MRI, it can be difficult to distinguish between small vascular structures and a small ganglion, especially in the hands and feet.23
Ultrasonography can also help identify a Baker cyst, a specific fluid collection arising from the semimembranosus bursa between the medial head of the gastrocnemius tendon and the semimembranosus tendon. Ultrasonography can also detect inflammation, rupture, or leaking associated with a Baker cyst.24
Power Doppler is an ultrasonographic examination that can detect increased blood flow surrounding a fluid collection and determine the likelihood of an acute inflammatory or infectious cause.25
Joint effusion and synovitis
Musculoskeletal ultrasonography can help evaluate joints for effusion and synovitis. It is highly sensitive (94%) and specific (95%) for synovitis, making it superior to contrast-enhanced MRI.26,27 The area of concern should be limited to 1 quadrant of a joint (anterior, posterior, medial, or lateral); for problems beyond that, MRI should be considered.
A joint effusion appears as a distended joint capsule containing hypoechoic (complex) or anechoic (simple) joint fluid.
Complex joint fluid may contain debris and occurs with hemarthrosis, infection, and inflammation.23 Hypertrophied synovium is hypoechoic and can mimic complex joint fluid.
Power Doppler evaluation can help distinguish synovitis from joint fluid by demonstrating blood flow, a feature of synovitis but not of simple joint fluid. Power Doppler is the most sensitive means of detecting blood flow, although it does not show direction of flow.28
Using ultrasonography can help to improve disease control and minimize disabling changes by monitoring synovitis therapy. In addition, subclinical synovitis and enthesitis (inflammation of insertion sites of tendons or ligaments into bone) detected by ultrasonography may predict future disease and disease flares.29–31
Ultrasonographic guidance for a wide range of procedures is increasing rapidly.32–36 Multiple studies have shown the advantage of ultrasonography-guided aspiration and injection compared with techniques without imaging guidance.37,38
Soft-tissue masses
Accurately diagnosing soft-tissue masses can be difficult. A mass may remain indeterminate even after multiple imaging studies, requiring biopsy or surgical referral. However, for a few specific masses, ultrasonography is highly accurate and can eliminate the need for further imaging.
Ultrasonography can help evaluate soft- tissue masses no larger than 5 cm in diameter and no deeper than superficial muscular fascia. If the mass is larger or deeper than that, ultrasonography is less reliable for showing the margins of the mass and its relationship to adjacent structures (Figure 5). Further imaging by MRI may be recommended in such cases.
Fortunately, many of the most common soft-tissue masses can be accurately diagnosed with ultrasonography, including lipomas, ganglion cysts, foreign bodies, and simple fluid collections.4,39 Nerve-sheath tumors can also be diagnosed with ultrasonography if the lesion clearly arises from a nerve. Other soft-tissue masses are likely to be indeterminate with ultrasonography, requiring follow-up with MRI with contrast.
Tendons
Musculoskeletal ultrasonography can be effective for evaluating tendons around joints, especially 1 or a small number of nearby superficial tendons. Tendons particularly well suited for ultrasonographic examination include:
- Upper-extremity tendons located in the rotator cuff or around the elbow, and flexor and extensor tendons of the hands; ultrasonographic evaluation of the rotator cuff is highly accurate, equivalent to that of MRI for partial-thickness and full-thickness tearing40–43
- Lower-extremity tendons of the extensor mechanism of the knee, distal hamstring tendons, tendons around the ankle,44–46 and flexor and extensor tendons of the foot.
Ultrasonography can help diagnose a variety of tendon abnormalities (Table 1),48,49 including tearing, for which a dynamic examination can be performed.
Many tendons have a tendon sheath containing tenosynovium, while others have surrounding peritenon only; either can become thickened and inflamed. Tenosynovitis is a nonspecific finding and may be inflammatory, infectious, or posttraumatic. The presence of tendon sheath fluid alone on ultrasonography can be a normal finding, and some tendon sheaths that communicate with adjacent joints (eg, the long head biceps tendon, the flexor hallucis longus tendon) commonly contain simple fluid.6 A dynamic examination with ultrasonography can help diagnose snapping related to abnormal tendon movement, for example, in the case of intra-sheath and extra-sheath subluxation of the peroneal tendons.45,50,51
Ligaments
Ultrasonography can detect abnormalities in many superficial ligaments (Table 1).
Ankle. Ankle ligaments are superficial and can be clearly visualized. The diagnostic accuracy of ultrasonography for tearing of the anterior talofibular ligament may be as high as 100%.50,52,53
Elbow and thumb. The larger of the collateral ligaments of the elbow, especially the ulnar collateral ligament, and the ulnar collateral ligament of the thumb can be effectively evaluated with ultrasonography.54,55
Knee. The collateral ligaments of the knee can be seen with ultrasonography, but injuries of the external ligaments of the knee are often associated with intrinsic derangements that cannot be evaluated with ultrasonography.56,57 Intra-articular ligaments such as the anterior cruciate ligament are also not amenable to ultrasonography.
Dynamic examination of a ligament with ultrasonography can help determine the grade of the injury.
Deeply located ligaments (eg, around the hip) and ligaments surrounded by bone, such as the Lisfranc ligament, cannot be completely seen on ultrasonography.
Muscle
Musculoskeletal ultrasonography is useful for small areas of concern within a muscle (Table 1). It can detect muscle strains and tears, intramuscular collections or lesions, and fascial scarring or fascial injuries such as superficial muscle herniation. Although ultrasonography may yield a definitive diagnosis for a muscle problem, further imaging may be needed.
Nerves
Ultrasonography is useful for peripheral nerve investigation but requires a steep learning curve for sonographers and interpreting physicians.58,59 It is best suited for directed questions regarding focal abnormal nerve findings on physical examination.
Ultrasonography can help identify areas of nerve entrapment caused by a mass or dynamic compression. It can detect neuritis (Table 1), lesions of peripheral nerves (eg, nerve-sheath tumors), and neuromas (eg, Morton neuroma of the intermetatarsal space). In a large meta-analysis, ultrasonography and MRI were found to be equally accurate for detecting Morton neuroma.60 Even for nerve-sheath tumors located deep to the muscular fascia, ultrasonography can confirm the diagnosis because of the characteristic appearance of the nerves. Ultrasonography can also demonstrate a large extent of the course of superficial peripheral nerves while keeping the imaging plane appropriately oriented to the nerves.
Acknowledgment: We would like to sincerely thank Megan Griffiths, MA, for her help in the preparation and submission of this manuscript.
Ultrasonography has been used to evaluate musculoskeletal problems for decades but has only recently become more widely available in the United States. Advances in technology and physician familiarity are increasing its role in orthopedic imaging.
No single imaging method can yield all musculoskeletal diagnoses. Like any imaging technique, ultrasonography has strengths and weaknesses specific to orthopedics. Radiography, computed tomography (CT), and magnetic resonance imaging (MRI) play important roles for investigating musculoskeletal problems and are complementary to each other and to ultrasonography.
To help clinicians make informed decisions about ordering musculoskeletal ultrasonography, this article reviews the basic physics underlying ultrasonography, its advantages and disadvantages compared with other imaging methods, and common clinical applications.
CLASSIC TECHNOLOGY MAKING A RESURGENCE
The first reports of the use of musculoskeletal ultrasonography appeared in the 1970s for investigating the rotator cuff,1–3 actually preceding reports of its use in obstetrics and gynecology.4 In the 1980s, reports emerged for evaluating the Achilles tendon.5,6 After that, its popularity in the United States plateaued, likely because of the advent of MRI, lower reimbursement and greater variability in interpretation compared with MRI, as well as a lack of physicians and sonographers trained in its use.7,8
Musculoskeletal ultrasonography is currently experiencing a resurgence. Although it remains a specialized service more commonly available in large hospitals, its use is increasing rapidly, and it will likely become more widely available.
SPECIAL TRAINING REQUIRED
Musculoskeletal ultrasonography is simply an ultrasonographic examination of part of the musculoskeletal system. But because not all ultrasonographic transducers offer sufficient resolution for musculoskeletal evaluation and not all sonographers and imaging physicians are familiar with the specialized techniques, musculoskeletal ultrasonography often has a separate designation (eg, “MSKUS,” “MSUS”). At Cleveland Clinic, it is offered through the department of musculoskeletal imaging by subspecialty-trained musculoskeletal radiologists and specially trained musculoskeletal ultrasonographers with 4 to 5 years of training in the technique.
Musculoskeletal ultrasonography is also performed by physician groups with specialized training, including sports medicine physicians, rheumatologists, physiatrists, neurologists, and orthopedic surgeons. The American Institute of Ultrasound in Medicine offers voluntary accreditation for practice groups using musculoskeletal ultrasonography. Certification in musculoskeletal radiology is offered to sonographers through the American Registry for Diagnostic Medical Sonography.
SONOGRAPHY HAS UNIQUE QUALITIES
Ultrasonography uses high-frequency sound waves to generate images. The transducer (or probe) emits sound from the many piezoelectric elements at its surface, and the sound waves travel through and react with tissues. Sound reflected by tissues is detected by the transducer and converted to an image. Objects that reflect sound appear hyperechoic (brighter), whereas tissues that reflect little or no sound appear hypoechoic.
High-resolution imaging of superficial structures
(B, arrow).
Ultrasonography involves a fundamental trade-off between image resolution and imaging depth. Higher-frequency sound waves do not penetrate far into tissues but generate a higher-resolution image; lower-frequency sound waves can penetrate much further but yield a lower-resolution image. Although high-resolution imaging of deep structures with ultrasonography is not possible (Figure 1), many musculoskeletal structures are located superficially and are amenable to ultrasonographic evaluation.
Be aware of artifacts
Some materials attenuate sound very little, such as simple fluid. Low attenuation results in artifacts on ultrasonography, making tissues behind the simple fluid appear brighter than neighboring tissues. These artifacts may be reported as “increased through transmission” or “posterior acoustic enhancement.” Conversely, metal and bone reflect all sound waves that reach them, rendering any structures beyond them invisible. This “shadowing” creates a problem for imaging of structures in or near bone. Subcutaneous fat also attenuates sound waves, limiting the use of ultrasonography for patients with obesity (Figure 2).
High-frequency linear transducer sharpens images
High-frequency linear transducers reduce anisotropy because their flat surface keeps sound waves more uniformly perpendicular to the structure of interest.4,7 Their development has allowed imaging of superficial structures that is superior to that of MRI. A high-frequency linear transducer offers more than twice the spatial resolution of a typical 1.5T MRI examination of superficial tissue.12,13
Operator experience is critical
Ultrasonography examinations, more than other imaging tests, are dependent on operator experience. A solid understanding of musculoskeletal anatomy is imperative. Because the probe images only a thin section of tissue (about the thickness of a credit card), referencing adjacent structures for orientation is more difficult with ultrasonography than with CT or MRI.
The accuracy of ultrasonography is highly dependent on acquiring and interpreting images, whereas the accuracy of MRI is dependent primarily on image interpretation.7 Interpreting physicians must check that sonographers capture relevant targets.
STRENGTHS OF MUSCULOSKELETAL ULTRASONOGRAPHY
Ultrasonography has multiple advantages:
No ionizing radiation exposure.
Portability. Unlike CT or MRI, ultrasonography equipment is portable.
Increased patient comfort. Patient positioning for an ultrasonography examination is more flexible than for MRI or CT,14 and the examination does not induce claustrophobia.8
High-resolution imaging. Ultrasonography provides very-high-resolution imaging of superficial soft tissues—in some cases, higher than MRI or CT.
Real-time dynamic examinations are possible with ultrasonography, unlike with CT or MRI, and may increase test sensitivity.4,15–18
Implanted hardware is less of a problem. Although ultrasonography cannot image beyond implanted orthopedic metallic hardware, the hardware does not obscure surrounding soft tissues as it does on CT and MRI.6,19,20 Also, ultrasonography is safe for patients with a pacemaker.8
WEAKNESSES
The main disadvantages of musculoskeletal ultrasonography are inherent to its limited field of view, making it inappropriate for a survey examination (eg, for ankle pain, knee pain, hip pain).4 Unlike CT and MRI, ultrasonography does not provide a “bird’s-eye view,” and important abnormalities can be missed during evaluation of large areas (Figure 4).
Ultrasonography also cannot evaluate bone or intra-articular structures such as cartilage, bone marrow, labrum, and intra-articular ligaments; MRI is the standard for evaluating these structures.21
Ultrasonography is time-consuming. To perform a detailed examination of the anterior, posterior, medial, and lateral aspects of the hip, knee, or ankle would require 1.5 to 2 hours of scanning time and an additional 10 to 25 minutes of image checking and interpretation.
CURRENT CLINICAL INDICATIONS
Musculoskeletal ultrasonography is best used for clinical questions regarding limited, superficial musculoskeletal problems.
Fluid collections
Ultrasonography can help evaluate small fluid collections in soft tissue. As is true for a lung opacity on chest radiography, soft-tissue fluid detected on ultrasonography is nonspecific, and results must be correlated with the clinical picture to narrow the differential diagnosis.
Fluid collections can be classified as loculated or nonloculated.
Nonloculated fluid involves more fluid than is simply interposed between tissue planes and has no wall or defined margins. It can be simple or complex in appearance: simple fluid is anechoic, and complex fluid appears more heterogeneous and may contain septations or debris.
Subcutaneous edema, which may occur postoperatively or from trauma, venous insufficiency, or inflammatory or infectious processes, appears on ultrasonography as nonloculated fluid interspersed between subcutaneous fat lobules.
Loculated fluid collections have well-defined margins or a discrete wall that does not follow normal tissue planes. They can also be simple or complex and can be caused by hematoma, abscess, or ganglion. Less commonly, neoplasms can mimic a loculated fluid collection (Figure 4).
A ganglion is a specific type of loculated fluid collection containing synovial fluid arising from a joint or tendon sheath. It tends to occur in specific locations, most commonly around the wrist, most often arising from the dorsal scapholunate ligament and volar wrist between the radial artery and flexor carpi radialis.22 On MRI, it can be difficult to distinguish between small vascular structures and a small ganglion, especially in the hands and feet.23
Ultrasonography can also help identify a Baker cyst, a specific fluid collection arising from the semimembranosus bursa between the medial head of the gastrocnemius tendon and the semimembranosus tendon. Ultrasonography can also detect inflammation, rupture, or leaking associated with a Baker cyst.24
Power Doppler is an ultrasonographic examination that can detect increased blood flow surrounding a fluid collection and determine the likelihood of an acute inflammatory or infectious cause.25
Joint effusion and synovitis
Musculoskeletal ultrasonography can help evaluate joints for effusion and synovitis. It is highly sensitive (94%) and specific (95%) for synovitis, making it superior to contrast-enhanced MRI.26,27 The area of concern should be limited to 1 quadrant of a joint (anterior, posterior, medial, or lateral); for problems beyond that, MRI should be considered.
A joint effusion appears as a distended joint capsule containing hypoechoic (complex) or anechoic (simple) joint fluid.
Complex joint fluid may contain debris and occurs with hemarthrosis, infection, and inflammation.23 Hypertrophied synovium is hypoechoic and can mimic complex joint fluid.
Power Doppler evaluation can help distinguish synovitis from joint fluid by demonstrating blood flow, a feature of synovitis but not of simple joint fluid. Power Doppler is the most sensitive means of detecting blood flow, although it does not show direction of flow.28
Using ultrasonography can help to improve disease control and minimize disabling changes by monitoring synovitis therapy. In addition, subclinical synovitis and enthesitis (inflammation of insertion sites of tendons or ligaments into bone) detected by ultrasonography may predict future disease and disease flares.29–31
Ultrasonographic guidance for a wide range of procedures is increasing rapidly.32–36 Multiple studies have shown the advantage of ultrasonography-guided aspiration and injection compared with techniques without imaging guidance.37,38
Soft-tissue masses
Accurately diagnosing soft-tissue masses can be difficult. A mass may remain indeterminate even after multiple imaging studies, requiring biopsy or surgical referral. However, for a few specific masses, ultrasonography is highly accurate and can eliminate the need for further imaging.
Ultrasonography can help evaluate soft- tissue masses no larger than 5 cm in diameter and no deeper than superficial muscular fascia. If the mass is larger or deeper than that, ultrasonography is less reliable for showing the margins of the mass and its relationship to adjacent structures (Figure 5). Further imaging by MRI may be recommended in such cases.
Fortunately, many of the most common soft-tissue masses can be accurately diagnosed with ultrasonography, including lipomas, ganglion cysts, foreign bodies, and simple fluid collections.4,39 Nerve-sheath tumors can also be diagnosed with ultrasonography if the lesion clearly arises from a nerve. Other soft-tissue masses are likely to be indeterminate with ultrasonography, requiring follow-up with MRI with contrast.
Tendons
Musculoskeletal ultrasonography can be effective for evaluating tendons around joints, especially 1 or a small number of nearby superficial tendons. Tendons particularly well suited for ultrasonographic examination include:
- Upper-extremity tendons located in the rotator cuff or around the elbow, and flexor and extensor tendons of the hands; ultrasonographic evaluation of the rotator cuff is highly accurate, equivalent to that of MRI for partial-thickness and full-thickness tearing40–43
- Lower-extremity tendons of the extensor mechanism of the knee, distal hamstring tendons, tendons around the ankle,44–46 and flexor and extensor tendons of the foot.
Ultrasonography can help diagnose a variety of tendon abnormalities (Table 1),48,49 including tearing, for which a dynamic examination can be performed.
Many tendons have a tendon sheath containing tenosynovium, while others have surrounding peritenon only; either can become thickened and inflamed. Tenosynovitis is a nonspecific finding and may be inflammatory, infectious, or posttraumatic. The presence of tendon sheath fluid alone on ultrasonography can be a normal finding, and some tendon sheaths that communicate with adjacent joints (eg, the long head biceps tendon, the flexor hallucis longus tendon) commonly contain simple fluid.6 A dynamic examination with ultrasonography can help diagnose snapping related to abnormal tendon movement, for example, in the case of intra-sheath and extra-sheath subluxation of the peroneal tendons.45,50,51
Ligaments
Ultrasonography can detect abnormalities in many superficial ligaments (Table 1).
Ankle. Ankle ligaments are superficial and can be clearly visualized. The diagnostic accuracy of ultrasonography for tearing of the anterior talofibular ligament may be as high as 100%.50,52,53
Elbow and thumb. The larger of the collateral ligaments of the elbow, especially the ulnar collateral ligament, and the ulnar collateral ligament of the thumb can be effectively evaluated with ultrasonography.54,55
Knee. The collateral ligaments of the knee can be seen with ultrasonography, but injuries of the external ligaments of the knee are often associated with intrinsic derangements that cannot be evaluated with ultrasonography.56,57 Intra-articular ligaments such as the anterior cruciate ligament are also not amenable to ultrasonography.
Dynamic examination of a ligament with ultrasonography can help determine the grade of the injury.
Deeply located ligaments (eg, around the hip) and ligaments surrounded by bone, such as the Lisfranc ligament, cannot be completely seen on ultrasonography.
Muscle
Musculoskeletal ultrasonography is useful for small areas of concern within a muscle (Table 1). It can detect muscle strains and tears, intramuscular collections or lesions, and fascial scarring or fascial injuries such as superficial muscle herniation. Although ultrasonography may yield a definitive diagnosis for a muscle problem, further imaging may be needed.
Nerves
Ultrasonography is useful for peripheral nerve investigation but requires a steep learning curve for sonographers and interpreting physicians.58,59 It is best suited for directed questions regarding focal abnormal nerve findings on physical examination.
Ultrasonography can help identify areas of nerve entrapment caused by a mass or dynamic compression. It can detect neuritis (Table 1), lesions of peripheral nerves (eg, nerve-sheath tumors), and neuromas (eg, Morton neuroma of the intermetatarsal space). In a large meta-analysis, ultrasonography and MRI were found to be equally accurate for detecting Morton neuroma.60 Even for nerve-sheath tumors located deep to the muscular fascia, ultrasonography can confirm the diagnosis because of the characteristic appearance of the nerves. Ultrasonography can also demonstrate a large extent of the course of superficial peripheral nerves while keeping the imaging plane appropriately oriented to the nerves.
Acknowledgment: We would like to sincerely thank Megan Griffiths, MA, for her help in the preparation and submission of this manuscript.
- Hamilton JV, Flinn G Jr, Haynie CC, Cefalo RC. Diagnosis of rectus sheath hematoma by B-mode ultrasound: a case report. Am J Obstet Gynecol 1976; 125(4):562–565. doi:10.1016/0002-9378(76)90379-3
- Zweymüller VK, Kratochwil A. Ultrasound diagnosis of bone and soft tissue tumours. Wien Klin Wochenschr 1975; 87(12):397–398. German.
- Mayer V. Ultrasonography of the rotator cuff. J Ultrasound Med 1985; 4(11):608, 607. doi:10.7863/jum.1985.4.11.608
- McNally EG. The development and clinical applications of musculoskeletal ultrasound. Skeletal Radiol 2011; 40(9):1223–1231. doi:10.1007/s00256-011-1220-5
- Ignashin NS, Girshin SG, Tsypin IS. Ultrasonic scanning in subcutaneous rupture of the Achilles tendon. Vestn Khir Im I I Grek 1981; 127(9):82–85. Russian.
- Robinson P. Sonography of common tendon injuries. AJR Am J Roentgenol 2009; 193(3):607–618. doi:10.2214/AJR.09.2808
- Jacobson JA. Musculoskeletal ultrasound: focused impact on MRI. AJR Am J Roentgenol 2009; 193(3):619–627. doi:10.2214/AJR.09.2841
- Nazarian LN. The top 10 reasons musculoskeletal sonography is an important complementary or alternative technique to MRI. AJR Am J Roentgenol 2008; 190(6):1621–1626. doi:10.2214/AJR.07.3385
- AIUM technical bulletin. Transducer manipulation. American Institute of Ultrasound in Medicine. J Ultrasound Med 1999; 18(2):169–175. doi:10.7863/jum.1999.18.2.169
- Connolly DJ, Berman L, McNally EG. The use of beam angulation to overcome anisotropy when viewing human tendon with high frequency linear array ultrasound. Br J Radiol 2001; 74 (878):183–185. doi:10.1259/bjr.74.878.740183
- Crass JR, van de Vegte GL, Harkavy LA. Tendon echogenicity: ex vivo study. Radiology 1988; 167(2):499–501. doi:10.1148/radiology.167.2.3282264
- Erickson SJ. High-resolution imaging of the musculoskeletal system. Radiology 1997; 205(3):593–618. doi:10.1148/radiology.205.3.9393511
- Link TM, Majumdar S, Peterfy C, et al. High resolution MRI of small joints: impact of spatial resolution on diagnostic performance and SNR. Magn Reson Imaging 1998; 16(2):147–155. doi:10.1016/S0730-725X(97)00244-0
- Middleton WD, Payne WT, Teefey SA, Hildebolt CF, Rubin DA, Yamaguchi K. Sonography and MRI of the shoulder: comparison of patient satisfaction. AJR Am J Roentgenol 2004; 183(5):1449–1452. doi:10.2214/ajr.183.5.1831449
- Khoury V, Cardinal E, Bureau NJ. Musculoskeletal sonography: a dynamic tool for usual and unusual disorders. AJR Am J Roentgenol 2007; 188(1):W63–W73. doi:10.2214/AJR.06.0579
- Farin PU, Jaroma H, Harju A, Soimakallio S. Medial displacement of the biceps brachii tendon: evaluation with dynamic sonography during maximal external shoulder rotation. Radiology 1995; 195(3):845–848. doi:10.1148/radiology.195.3.7754019
- Miller TT, Adler RS, Friedman L. Sonography of injury of the ulnar collateral ligament of the elbow-initial experience. Skeletal Radiol 2004; 33(7):386–391. doi:10.1007/s00256-004-0788-4
- Nazarian LN, McShane JM, Ciccotti MG, O’Kane PL, Harwood MI. Dynamic US of the anterior band of the ulnar collateral ligament of the elbow in asymptomatic major league baseball pitchers. Radiology 2003; 227(1):149–154. doi:10.1148/radiol.2271020288
- Jacobson JA, Lax MJ. Musculoskeletal sonography of the postoperative orthopedic patient. Semin Musculoskelet Radiol 2002; 6(1):67–77. doi:10.1055/s-2002-23165
- Sofka CM, Adler RS. Original report. Sonographic evaluation of shoulder arthroplasty. AJR Am J Roentgenol 2003; 180(4):1117–1120. doi:10.2214/ajr.180.4.1801117
- Silvestri E, Martinoli C, Derchi LE, Bertolotto M, Chiaramondia M, Rosenberg I. Echotexture of peripheral nerves: correlation between US and histologic findings and criteria to differentiate tendons. Radiology 1995; 197(1):291–296. doi:10.1148/radiology.197.1.7568840
- Cardinal E, Buckwalter KA, Braunstein EM, Mih AD. Occult dorsal carpal ganglion: comparison of US and MR imaging. Radiology 1994; 193(1):259–262. doi:10.1148/radiology.193.1.8090903
- Jacobson JA. Musculoskeletal ultrasound and MRI: which do I choose? Semin Musculoskelet Radiol 2005; 9(2):135–149. doi:10.1055/s-2005-872339
- Ward EE, Jacobson JA, Fessell DP, Hayes CW, van Holsbeeck M. Sonographic detection of Baker’s cysts: comparison with MR imaging. AJR Am J Roentgenol 2001; 176(2):373–380. doi:10.2214/ajr.176.2.1760373
- Bhasin S, Cheung PP. The role of power Doppler ultrasonography as disease activity marker in rheumatoid arthritis. Dis Markers 2015; 2015:325909. doi:10.1155/2015/325909
- Fukuba E, Yoshizako T, Kitagaki H, Murakawa Y, Kondo M, Uchida N. Power Doppler ultrasonography for assessment of rheumatoid synovitis: comparison with dynamic magnetic resonance imaging. Clin Imaging 2013; 37(1):134–137. doi:10.1016/j.clinimag.2012.02.008
- Takase-Minegishi K, Horita N, Kobayashi K, et al. Diagnostic test accuracy of ultrasound for synovitis in rheumatoid arthritis: systematic review and meta-analysis. Rheumatology (Oxford) 2018; 57(1):49–58. doi:10.1093/rheumatology/kex036
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet 2009; 373(9664):659–672. doi:10.1016/S0140-6736(09)60008-8
- Ash ZR, Tinazzi I, Gallego CC, et al. Psoriasis patients with nail disease have a greater magnitude of underlying systemic subclinical enthesopathy than those with normal nails. Ann Rheum Dis 2012; 71(4):553–556. doi:10.1136/annrheumdis-2011-200478
- Han J, Geng Y, Deng X, Zhang Z. Subclinical synovitis assessed by ultrasound predicts flare and progressive bone erosion in rheumatoid arthritis patients with clinical remission: a systematic review and metaanalysis. J Rheumatol 2016; 43(11):2010–2018. doi.org/10.3899/jrheum.160193
- Iagnocco A, Finucci A, Ceccarelli F, Perricone C, Iorgoveanu V, Valesini G. Power Doppler ultrasound monitoring of response to anti-tumour necrosis factor alpha treatment in patients with rheumatoid arthritis. Rheumatology (Oxford) 2015; 54(10):1890–1896. doi:10.1093/rheumatology/kev211
- Henning PT. Ultrasound-guided foot and ankle procedures. Phys Med Rehabil Clin N Am 2016; 27(3):649–671. doi:10.1016/j.pmr.2016.04.005
- Lueders DR, Smith J, Sellon JL. Ultrasound-guided knee procedures. Phys Med Rehabil Clin North Am 2016; 27(3):631–648. doi:10.1016/j.pmr.2016.04.010
- Payne JM. Ultrasound-guided hip procedures. Phys Med Rehabil Clin North Am 2016; 27(3):607–629. doi:10.1016/j.pmr.2016.04.004
- Strakowski JA. Ultrasound-guided peripheral nerve procedures. Phys Med Rehabil Clin North Am 2016; 27(3):687–715. doi:10.1016/j.pmr.2016.04.006
- Sussman WI, Williams CJ, Mautner K. Ultrasound-guided elbow procedures. Phys Med Rehabil Clin North Am 2016; 27(3):573–587. doi:10.1016/j.pmr.2016.04.002
- Finnoff JT. The evolution of diagnostic and interventional ultrasound in sports medicine. PM R 2016; 8(suppl 3):S133–S138. doi:10.1016/j.pmrj.2015.09.022
- Wu T, Dong Y, Song H, Fu Y, Li JH. Ultrasound-guided versus landmark in knee arthrocentesis: a systematic review. Semin Arthritis Rheum 2016; 45(5):627–632. doi:10.1016/j.semarthrit.2015.10.011
- Failla JM, van Holsbeeck M, Vanderschueren G. Detection of a 0.5-mm-thick thorn using ultrasound: a case report. J Hand Surg Am 1995; 20(3):456–457.
- Teefey SA, Hasan SA, Middleton WD, Patel M, Wright RW, Yamaguchi K. Ultrasonography of the rotator cuff. A comparison of ultrasonographic and arthroscopic findings in one hundred consecutive cases. J Bone Joint Surg Am 2000; 82(4):498–504.
- van Holsbeeck MT, Kolowich PA, Eyler WR, et al. US depiction of partial-thickness tear of the rotator cuff. Radiology 1995; 197(2):443–446. doi:10.1148/radiology.197.2.7480690
- Balich SM, Sheley RC, Brown TR, Sauser DD, Quinn SF. MR imaging of the rotator cuff tendon: interobserver agreement and analysis of interpretive errors. Radiology 1997; 204(1):191–194. doi:10.1148/radiology.204.1.9205245
- Dinnes J, Loveman E, McIntyre L, Waugh N. The effectiveness of diagnostic tests for the assessment of shoulder pain due to soft tissue disorders: a systematic review. Health Technol Assess 2003; 7(29):1–166. doi:10.3310/hta7290
- Rockett MS, Waitches G, Sudakoff G, Brage M. Use of ultrasonography versus magnetic resonance imaging for tendon abnormalities around the ankle. Foot Ankle Int 1998; 19(9):604–612.
- Grant TH, Kelikian AS, Jereb SE, McCarthy RJ. Ultrasound diagnosis of peroneal tendon tears. A surgical correlation. J Bone Joint Surg Am 2005; 87(8):1788–1794. doi:10.2106/JBJS.D.02450
- Hartgerink P, Fessell DP, Jacobson JA, van Holsbeeck MT. Full- versus partial-thickness Achilles tendon tears: sonographic accuracy and characterization in 26 cases with surgical correlation. Radiology 2001; 220(2):406–412. doi:10.1148/radiology.220.2.r01au41406
- Cho KH, Park BH, Yeon KM. Ultrasound of the adult hip. Semin Ultrasound CT MR 2000; 21(3):214–230.
- Adler RS, Finzel KC. The complementary roles of MR imaging and ultrasound of tendons. Radiol Clin North Am 2005; 43(4):771–807. doi:10.1016/j.rcl.2005.02.011
- Martinoli C, Bianchi S, Derchi LE. Tendon and nerve sonography. Radiol Clin North Am 1999; 37(4):691–711. doi:10.1016/S0033-8389(05)70124-X
- Fessell DP, Vanderschueren GM, Jacobson JA, et al. US of the ankle: technique, anatomy, and diagnosis of pathologic conditions. Radiographics 1998; 18(2):325–340. doi:10.1148/radiographics.18.2.9536481
- Neustadter J, Raikin SM, Nazarian LN. Dynamic sonographic evaluation of peroneal tendon subluxation. AJR Am J Roentgenol 2004; 183(4):985–988. doi:10.2214/ajr.183.4.1830985
- Verhaven EF, Shahabpour M, Handelberg FW, Vaes PH, Opdecam PJ. The accuracy of three-dimensional magnetic resonance imaging in the diagnosis of ruptures of the lateral ligaments of the ankle. Am J Sports Med 1991; 19(6):583–587. doi:10.1177/036354659101900605
- Milz P, Milz S, Steinborn M, Mittlmeier T, Putz R, Reiser M. Lateral ankle ligaments and tibiofibular syndesmosis. 13-MHz high-frequency sonography and MRI compared in 20 patients. Acta Orthop Scand 1998; 69(1):51–55.
- De Smet AA, Winter TC, Best TM, Bernhardt DT. Dynamic sonography with valgus stress to assess elbow ulnar collateral ligament injury in baseball pitchers. Skeletal Radiol 2002; 31(11):671–676. doi:10.1007/s00256-002-0558-0
- Melville DM, Jacobson JA, Fessell DP. Ultrasound of the thumb ulnar collateral ligament: technique and pathology. AJR Am J Roentgenol 2014; 202(2):W168. doi:10.2214/AJR.13.11335
- Court-Payen M. Sonography of the knee: intra-articular pathology. J Clin Ultrasound 2004; 32(9):481–490. doi:10.1002/jcu.20069
- Azzoni R, Cabitza P. Is there a role for sonography in the diagnosis of tears of the knee menisci? J Clin Ultrasound 2002; 30(8):472–476. doi:10.1002/jcu.10106
- Jacobson JA, Wilson TJ, Yang LJ. Sonography of common peripheral nerve disorders with clinical correlation. J Ultrasound Med 2016; 35(4):683–693. doi:10.7863/ultra.15.05061
- Ali ZS, Pisapia JM, Ma TS, Zager EL, Heuer GG, Khoury V. Ultrasonographic evaluation of peripheral nerves. World Neurosurg 2016; 85(1):333–339. doi:10.1016/j.wneu.2015.10.005
- Bignotti B, Signori A, Sormani MP, Molfetta L, Martinoli C, Tagliafico A. Ultrasound versus magnetic resonance imaging for Morton neuroma: systematic review and meta-analysis. Eur Radiol 2015; 25(8):2254–2262. doi:10.1007/s00330-015-3633-3
- Hamilton JV, Flinn G Jr, Haynie CC, Cefalo RC. Diagnosis of rectus sheath hematoma by B-mode ultrasound: a case report. Am J Obstet Gynecol 1976; 125(4):562–565. doi:10.1016/0002-9378(76)90379-3
- Zweymüller VK, Kratochwil A. Ultrasound diagnosis of bone and soft tissue tumours. Wien Klin Wochenschr 1975; 87(12):397–398. German.
- Mayer V. Ultrasonography of the rotator cuff. J Ultrasound Med 1985; 4(11):608, 607. doi:10.7863/jum.1985.4.11.608
- McNally EG. The development and clinical applications of musculoskeletal ultrasound. Skeletal Radiol 2011; 40(9):1223–1231. doi:10.1007/s00256-011-1220-5
- Ignashin NS, Girshin SG, Tsypin IS. Ultrasonic scanning in subcutaneous rupture of the Achilles tendon. Vestn Khir Im I I Grek 1981; 127(9):82–85. Russian.
- Robinson P. Sonography of common tendon injuries. AJR Am J Roentgenol 2009; 193(3):607–618. doi:10.2214/AJR.09.2808
- Jacobson JA. Musculoskeletal ultrasound: focused impact on MRI. AJR Am J Roentgenol 2009; 193(3):619–627. doi:10.2214/AJR.09.2841
- Nazarian LN. The top 10 reasons musculoskeletal sonography is an important complementary or alternative technique to MRI. AJR Am J Roentgenol 2008; 190(6):1621–1626. doi:10.2214/AJR.07.3385
- AIUM technical bulletin. Transducer manipulation. American Institute of Ultrasound in Medicine. J Ultrasound Med 1999; 18(2):169–175. doi:10.7863/jum.1999.18.2.169
- Connolly DJ, Berman L, McNally EG. The use of beam angulation to overcome anisotropy when viewing human tendon with high frequency linear array ultrasound. Br J Radiol 2001; 74 (878):183–185. doi:10.1259/bjr.74.878.740183
- Crass JR, van de Vegte GL, Harkavy LA. Tendon echogenicity: ex vivo study. Radiology 1988; 167(2):499–501. doi:10.1148/radiology.167.2.3282264
- Erickson SJ. High-resolution imaging of the musculoskeletal system. Radiology 1997; 205(3):593–618. doi:10.1148/radiology.205.3.9393511
- Link TM, Majumdar S, Peterfy C, et al. High resolution MRI of small joints: impact of spatial resolution on diagnostic performance and SNR. Magn Reson Imaging 1998; 16(2):147–155. doi:10.1016/S0730-725X(97)00244-0
- Middleton WD, Payne WT, Teefey SA, Hildebolt CF, Rubin DA, Yamaguchi K. Sonography and MRI of the shoulder: comparison of patient satisfaction. AJR Am J Roentgenol 2004; 183(5):1449–1452. doi:10.2214/ajr.183.5.1831449
- Khoury V, Cardinal E, Bureau NJ. Musculoskeletal sonography: a dynamic tool for usual and unusual disorders. AJR Am J Roentgenol 2007; 188(1):W63–W73. doi:10.2214/AJR.06.0579
- Farin PU, Jaroma H, Harju A, Soimakallio S. Medial displacement of the biceps brachii tendon: evaluation with dynamic sonography during maximal external shoulder rotation. Radiology 1995; 195(3):845–848. doi:10.1148/radiology.195.3.7754019
- Miller TT, Adler RS, Friedman L. Sonography of injury of the ulnar collateral ligament of the elbow-initial experience. Skeletal Radiol 2004; 33(7):386–391. doi:10.1007/s00256-004-0788-4
- Nazarian LN, McShane JM, Ciccotti MG, O’Kane PL, Harwood MI. Dynamic US of the anterior band of the ulnar collateral ligament of the elbow in asymptomatic major league baseball pitchers. Radiology 2003; 227(1):149–154. doi:10.1148/radiol.2271020288
- Jacobson JA, Lax MJ. Musculoskeletal sonography of the postoperative orthopedic patient. Semin Musculoskelet Radiol 2002; 6(1):67–77. doi:10.1055/s-2002-23165
- Sofka CM, Adler RS. Original report. Sonographic evaluation of shoulder arthroplasty. AJR Am J Roentgenol 2003; 180(4):1117–1120. doi:10.2214/ajr.180.4.1801117
- Silvestri E, Martinoli C, Derchi LE, Bertolotto M, Chiaramondia M, Rosenberg I. Echotexture of peripheral nerves: correlation between US and histologic findings and criteria to differentiate tendons. Radiology 1995; 197(1):291–296. doi:10.1148/radiology.197.1.7568840
- Cardinal E, Buckwalter KA, Braunstein EM, Mih AD. Occult dorsal carpal ganglion: comparison of US and MR imaging. Radiology 1994; 193(1):259–262. doi:10.1148/radiology.193.1.8090903
- Jacobson JA. Musculoskeletal ultrasound and MRI: which do I choose? Semin Musculoskelet Radiol 2005; 9(2):135–149. doi:10.1055/s-2005-872339
- Ward EE, Jacobson JA, Fessell DP, Hayes CW, van Holsbeeck M. Sonographic detection of Baker’s cysts: comparison with MR imaging. AJR Am J Roentgenol 2001; 176(2):373–380. doi:10.2214/ajr.176.2.1760373
- Bhasin S, Cheung PP. The role of power Doppler ultrasonography as disease activity marker in rheumatoid arthritis. Dis Markers 2015; 2015:325909. doi:10.1155/2015/325909
- Fukuba E, Yoshizako T, Kitagaki H, Murakawa Y, Kondo M, Uchida N. Power Doppler ultrasonography for assessment of rheumatoid synovitis: comparison with dynamic magnetic resonance imaging. Clin Imaging 2013; 37(1):134–137. doi:10.1016/j.clinimag.2012.02.008
- Takase-Minegishi K, Horita N, Kobayashi K, et al. Diagnostic test accuracy of ultrasound for synovitis in rheumatoid arthritis: systematic review and meta-analysis. Rheumatology (Oxford) 2018; 57(1):49–58. doi:10.1093/rheumatology/kex036
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet 2009; 373(9664):659–672. doi:10.1016/S0140-6736(09)60008-8
- Ash ZR, Tinazzi I, Gallego CC, et al. Psoriasis patients with nail disease have a greater magnitude of underlying systemic subclinical enthesopathy than those with normal nails. Ann Rheum Dis 2012; 71(4):553–556. doi:10.1136/annrheumdis-2011-200478
- Han J, Geng Y, Deng X, Zhang Z. Subclinical synovitis assessed by ultrasound predicts flare and progressive bone erosion in rheumatoid arthritis patients with clinical remission: a systematic review and metaanalysis. J Rheumatol 2016; 43(11):2010–2018. doi.org/10.3899/jrheum.160193
- Iagnocco A, Finucci A, Ceccarelli F, Perricone C, Iorgoveanu V, Valesini G. Power Doppler ultrasound monitoring of response to anti-tumour necrosis factor alpha treatment in patients with rheumatoid arthritis. Rheumatology (Oxford) 2015; 54(10):1890–1896. doi:10.1093/rheumatology/kev211
- Henning PT. Ultrasound-guided foot and ankle procedures. Phys Med Rehabil Clin N Am 2016; 27(3):649–671. doi:10.1016/j.pmr.2016.04.005
- Lueders DR, Smith J, Sellon JL. Ultrasound-guided knee procedures. Phys Med Rehabil Clin North Am 2016; 27(3):631–648. doi:10.1016/j.pmr.2016.04.010
- Payne JM. Ultrasound-guided hip procedures. Phys Med Rehabil Clin North Am 2016; 27(3):607–629. doi:10.1016/j.pmr.2016.04.004
- Strakowski JA. Ultrasound-guided peripheral nerve procedures. Phys Med Rehabil Clin North Am 2016; 27(3):687–715. doi:10.1016/j.pmr.2016.04.006
- Sussman WI, Williams CJ, Mautner K. Ultrasound-guided elbow procedures. Phys Med Rehabil Clin North Am 2016; 27(3):573–587. doi:10.1016/j.pmr.2016.04.002
- Finnoff JT. The evolution of diagnostic and interventional ultrasound in sports medicine. PM R 2016; 8(suppl 3):S133–S138. doi:10.1016/j.pmrj.2015.09.022
- Wu T, Dong Y, Song H, Fu Y, Li JH. Ultrasound-guided versus landmark in knee arthrocentesis: a systematic review. Semin Arthritis Rheum 2016; 45(5):627–632. doi:10.1016/j.semarthrit.2015.10.011
- Failla JM, van Holsbeeck M, Vanderschueren G. Detection of a 0.5-mm-thick thorn using ultrasound: a case report. J Hand Surg Am 1995; 20(3):456–457.
- Teefey SA, Hasan SA, Middleton WD, Patel M, Wright RW, Yamaguchi K. Ultrasonography of the rotator cuff. A comparison of ultrasonographic and arthroscopic findings in one hundred consecutive cases. J Bone Joint Surg Am 2000; 82(4):498–504.
- van Holsbeeck MT, Kolowich PA, Eyler WR, et al. US depiction of partial-thickness tear of the rotator cuff. Radiology 1995; 197(2):443–446. doi:10.1148/radiology.197.2.7480690
- Balich SM, Sheley RC, Brown TR, Sauser DD, Quinn SF. MR imaging of the rotator cuff tendon: interobserver agreement and analysis of interpretive errors. Radiology 1997; 204(1):191–194. doi:10.1148/radiology.204.1.9205245
- Dinnes J, Loveman E, McIntyre L, Waugh N. The effectiveness of diagnostic tests for the assessment of shoulder pain due to soft tissue disorders: a systematic review. Health Technol Assess 2003; 7(29):1–166. doi:10.3310/hta7290
- Rockett MS, Waitches G, Sudakoff G, Brage M. Use of ultrasonography versus magnetic resonance imaging for tendon abnormalities around the ankle. Foot Ankle Int 1998; 19(9):604–612.
- Grant TH, Kelikian AS, Jereb SE, McCarthy RJ. Ultrasound diagnosis of peroneal tendon tears. A surgical correlation. J Bone Joint Surg Am 2005; 87(8):1788–1794. doi:10.2106/JBJS.D.02450
- Hartgerink P, Fessell DP, Jacobson JA, van Holsbeeck MT. Full- versus partial-thickness Achilles tendon tears: sonographic accuracy and characterization in 26 cases with surgical correlation. Radiology 2001; 220(2):406–412. doi:10.1148/radiology.220.2.r01au41406
- Cho KH, Park BH, Yeon KM. Ultrasound of the adult hip. Semin Ultrasound CT MR 2000; 21(3):214–230.
- Adler RS, Finzel KC. The complementary roles of MR imaging and ultrasound of tendons. Radiol Clin North Am 2005; 43(4):771–807. doi:10.1016/j.rcl.2005.02.011
- Martinoli C, Bianchi S, Derchi LE. Tendon and nerve sonography. Radiol Clin North Am 1999; 37(4):691–711. doi:10.1016/S0033-8389(05)70124-X
- Fessell DP, Vanderschueren GM, Jacobson JA, et al. US of the ankle: technique, anatomy, and diagnosis of pathologic conditions. Radiographics 1998; 18(2):325–340. doi:10.1148/radiographics.18.2.9536481
- Neustadter J, Raikin SM, Nazarian LN. Dynamic sonographic evaluation of peroneal tendon subluxation. AJR Am J Roentgenol 2004; 183(4):985–988. doi:10.2214/ajr.183.4.1830985
- Verhaven EF, Shahabpour M, Handelberg FW, Vaes PH, Opdecam PJ. The accuracy of three-dimensional magnetic resonance imaging in the diagnosis of ruptures of the lateral ligaments of the ankle. Am J Sports Med 1991; 19(6):583–587. doi:10.1177/036354659101900605
- Milz P, Milz S, Steinborn M, Mittlmeier T, Putz R, Reiser M. Lateral ankle ligaments and tibiofibular syndesmosis. 13-MHz high-frequency sonography and MRI compared in 20 patients. Acta Orthop Scand 1998; 69(1):51–55.
- De Smet AA, Winter TC, Best TM, Bernhardt DT. Dynamic sonography with valgus stress to assess elbow ulnar collateral ligament injury in baseball pitchers. Skeletal Radiol 2002; 31(11):671–676. doi:10.1007/s00256-002-0558-0
- Melville DM, Jacobson JA, Fessell DP. Ultrasound of the thumb ulnar collateral ligament: technique and pathology. AJR Am J Roentgenol 2014; 202(2):W168. doi:10.2214/AJR.13.11335
- Court-Payen M. Sonography of the knee: intra-articular pathology. J Clin Ultrasound 2004; 32(9):481–490. doi:10.1002/jcu.20069
- Azzoni R, Cabitza P. Is there a role for sonography in the diagnosis of tears of the knee menisci? J Clin Ultrasound 2002; 30(8):472–476. doi:10.1002/jcu.10106
- Jacobson JA, Wilson TJ, Yang LJ. Sonography of common peripheral nerve disorders with clinical correlation. J Ultrasound Med 2016; 35(4):683–693. doi:10.7863/ultra.15.05061
- Ali ZS, Pisapia JM, Ma TS, Zager EL, Heuer GG, Khoury V. Ultrasonographic evaluation of peripheral nerves. World Neurosurg 2016; 85(1):333–339. doi:10.1016/j.wneu.2015.10.005
- Bignotti B, Signori A, Sormani MP, Molfetta L, Martinoli C, Tagliafico A. Ultrasound versus magnetic resonance imaging for Morton neuroma: systematic review and meta-analysis. Eur Radiol 2015; 25(8):2254–2262. doi:10.1007/s00330-015-3633-3
KEY POINTS
- Ultrasonography can be used to evaluate small fluid collections in soft tissue; joint effusions and synovitis; soft tissue masses (≤ 5 cm in diameter); tendon, ligament and muscle injuries; and peripheral nerve entrapment and lesions.
- Ultrasonography is not appropriate for survey examinations of vague or diffuse symptoms or for evaluating soft-tissue areas more than a few centimeters in diameter or more than a few centimeters deep.
- Musculoskeletal ultrasonography requires specially trained sonographers and interpreting physicians.
The female athlete triad: It takes a team
Striving for athletic excellence, many young women—and some young men—create an energy deficit from increased exercise, decreased intake, or both. In women, the resulting energy deficit can suppress the menstrual cycle and in turn lead to bone demineralization in a syndrome called the female athlete triad.
Primary care physicians should be aware of this syndrome because it can lead to short-term and long-term health complications, and they are in a good position to screen for, diagnose, and treat it. However, a study of 931 US physicians in 2015 found that only 37% had heard of it.1
DEFINITION HAS CHANGED: ONLY 1 OF 3 COMPONENTS NEEDED
In 1972, Title IX of the Education Amendment Act was passed, prohibiting sex discrimination in any higher education program or activity receiving federal financial aid. Since then, female athletic participation in the United States has increased more than 10-fold.2
Also increasing has been awareness of the link between athletics, eating disorders, and amenorrhea. The American College of Sports Medicine coined the term female athlete triad in 1992, describing it as the constellation of disordered eating, amenorrhea, and osteoporosis (all 3 needed to be present).3 They broadened the definition in 2007 so that the syndrome can be diagnosed if any of the following is present4:
- Low energy availability (with or without an eating disorder)
- Menstrual dysfunction
- Decreased bone mineral density.
Recognizing that low energy availability can affect athletes of either sex and have consequences beyond the female reproductive system and skeleton, in 2014 the International Olympic Committee introduced a broader term called relative energy deficiency in sport.5,6 Like the triad, this condition occurs when energy intake falls below energy output to the point that it negatively affects an athlete’s physical and mental health.
THE COMPONENTS ARE COMMON
The female athlete triad can be seen in high school, collegiate, and elite athletes7 and is especially common in sports with subjective judging (gymnastics, figure skating) or endurance sports that emphasize leanness (eg, running).8
In a review of 65 studies, Gibbs et al9 found that the prevalence of any one of the triad conditions in exercising women and female athletes ranged from 16.0% to 60.0%, the prevalence of any 2 ranged from 2.7% to 27.0%, and the prevalence of all 3 ranged from 0% to 15.9%.
Low energy availability is categorized as either intentional (ie, due to disordered eating) or unintentional (ie, due to activities not associated with eating). Sustained low energy availability is often associated with eating disorders and subsequent low self-esteem, depression, and anxiety disorders.4
The prevalence of eating disorders is high in female athletes—31% and 20% in 2 large studies of elite female athletes, compared with 5.5% and 9%, respectively, in the general population.10,11 Another study found that the prevalence of disordered eating was 46.7% in sports that emphasize leanness, such as track and gymnastics, compared with 19.8% in sports that did not, such as basketball and soccer.12
Calorie restriction is common. In a study of 15 elite ballet dancers and 15 matched controls, the dancers were found to consume only about 3/4 as many calories per day as the controls (1,577 vs 2,075 kcal/day, P ≤ .01).13
Menstrual dysfunction. In small studies, the prevalence of secondary amenorrhea was as high as 69% in dancers and 65% in long-distance runners.4,14–16
Decreased bone mineral density. According to a systematic review, the prevalence of osteopenia in amenorrheic athletes ranged between 22% and 50% and the prevalence of osteoporosis was 0% to 13%, compared with 12% and 2.3%, respectively, in the general population.17
THE COMPONENTS ARE LINKED

dysfunction play causative roles in bone mineral density pathology. Within each component of the triad a spectrum of dysfunction exists, with all 3 components exhibiting serious health end points including low energy availability, functional hypothalamic amenorrhea, and osteoporosis.
The 3 components of the female athlete triad—low energy availability, menstrual dysfunction, and decreased bone mineral density—are linked, and each exists on a spectrum (Figure 1). The long-term consequences are far-reaching and can affect the cardiovascular, endocrine, reproductive, skeletal, gastrointestinal, renal, and central nervous systems.
Low energy availability is the driving force of the triad, causing menstrual irregularity and subsequent low bone mineral density.
Menstrual dysfunction. Low energy availability can contribute to menstrual disturbances because the body suppresses reproductive function to prevent pregnancy. Functional hypothalamic amenorrhea results from decreased gonadotropin-releasing hormone leading to decreased gonadotropin release from the pituitary gland and, ultimately, to low circulating estrogen levels.18 Menstrual irregularities related to the triad include:
- Primary amenorrhea (a delay in menarche)
- Oligomenorrhea (menstrual cycles occurring at intervals greater than every 35 days)
- Secondary amenorrhea (cessation of menstruation for 3 consecutive months).
(Primary amenorrhea is defined as no menses by age 15 in the presence of normal secondary sexual development or within 5 years after breast development if that occurs before the age of 10. Secondary amenorrhea is defined as the loss of menses for 90 or more days after menarche.19)
In animal studies, reducing dietary intake by more than 30% resulted in infertility.4 Menstrual abnormalities can present as early as 5 days after a patient enters a state of low energy availability.20 Symptoms of menstrual dysfunction are largely indicative of hypogonadism and include vaginal dryness, infertility, and impaired bone health.
Bone health in women with the female athlete triad can range from optimal to osteoporosis.
Low bone mineral density is a result of low energy availability and menstrual dysfunction leading to estrogen deficiency.21,22 Specifically, menstrual abnormalities can result in low estrogen and overactivity of osteoclasts, while low energy availability alters the metabolic environment, inducing changes in insulinlike growth factor 1, leptin, and peptide YY, resulting in deficiencies in vitamin D and calcium—nutrients necessary for bone mineralization. In turn, bone health and density are compromised.21,22
Ninety percent of peak bone mass is attained by age 18. Those who have low bone mineral density as part of the female athlete triad can suffer from long-lasting effects on their bone health.
SCREENING
Untreated, the triad can lead to fatigue, poor sports performance, and a number of serious comorbid conditions such as osteopenia and osteoporosis (leading to stress fractures) anemia, heart arrhythmias, and amenorrhea. Therefore, it is important for primary care providers to screen female athletes for the triad during routine office visits.
In 2014, the Triad Consensus Panel recommended screening female athletes at the high school and collegiate levels during a preparticipation physical evaluation and then every year by a primary care physician, athletic trainer, team physician, or coach.23
Risk factors include signs of dietary restriction, low body mass index, delayed menarche, oligomenorrhea or amenorrhea, and bone stress reactions or fractures.23 Athletes should be questioned about their menstrual history (age of menarche, frequency, and duration of menstrual cycles), history of stress fractures, medication history, family history (osteoporosis, eating disorders, and fractures),24,25 and dietary habits.

Physical findings include low body mass index, recent weight loss, orthostatic hypotension, lanugo, hypercarotenemia, and signs of eating disorders (restrictive, binging, purging) (Table 1).25–27
Additionally, it is important to ascertain if the patient receives critical comments regarding performance or body image from coaches, parents, or teammates and if sport-specific training began early in life.
Certain personality factors and behaviors are clues, such as perfectionism, obsessiveness, frequent weight cycling, and overtraining.4,25 If any of the triad components are apparent, a deeper evaluation can be completed.
Specific screening questions
The Female Athlete Triad Coalition recommends asking 11 screening questions and having prompt discussions regarding the athlete’s nutritional status and body image.23 If the patient gives a worrisome response to a screening question, further workup for a formal diagnosis should be initiated.
Questions about nutritional status.
- Do you worry about your weight?
- Are you trying to gain or lose weight, or has anyone recommended that you do so?
- Are you on a special diet or do you avoid certain types of foods or food groups?
- Have you ever had an eating disorder?
Questions about menstrual function.
- Have you ever had a menstrual period?
- How old were you when you had your first menstrual period?
- When was your most recent menstrual period?
- How many periods have you had in the last 12 months?
- Are you presently taking any female hormones (estrogen, progesterone, birth control pills)?
Questions about bone health.
- Have you ever had a stress fracture?
- Have you ever been told you have low bone density (osteopenia or osteoporosis)?
Along similar lines, the American Academy of Pediatrics, American Academy of Family Physicians, and American College of Sports Medicine28 have a list of 7 questions:
- Do you worry about your weight?
- Do you limit the foods you eat?
- Do you lose weight to meet image requirements for sports?
- Have you ever suffered from an eating disorder?
- How old were you when you had your first menstrual period?
- How many menstrual cycles have you had in the past 12 months?
- Have you ever had a stress fracture?
These questions are not being widely used. A study of the National Collegiate Athletic Association Division I universities found that only 9% of universities included 9 or more of the recommended 12 questions that the Female Athlete Triad Coalition was recommending at that time, and 22% asked only 1 or 2 of the questions. None of the universities included all 12.29 These findings are not surprising, given that screening for the triad is not state-mandated. Screening discrepancies among providers largely stem from knowledge gaps, nonstandardized questionnaires, lack of time at appointments, and the sensitive nature of the questioning (eg, disordered eating).30
DIAGNOSING THE TRIAD
Given that the signs of low energy availability and menstrual dysfunction are often subtle, the diagnosis of the triad for those at risk requires input from a multidisciplinary team including a physician, sports dietitian, mental health professional, exercise physiologist, and other medical consultants.

Table 2 lists diagnostic tests the primary care provider should consider.
Diagnosing low energy availability
Energy availability is the dietary energy remaining after exercise energy expenditure; it is normalized to fat-free (lean) mass to account for resting energy expenditure. It is a product of energy intake, energy expenditure, and stored energy, and is calculated as:
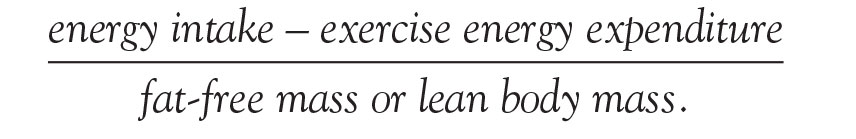
An optimal value is at least 45 kcal/kg/day, while physiologic changes start to occur at less than 30 kcal/kg/day.4,31 Low energy is often seen in adult patients with a body mass index less than 17.5 kg/m2 and adolescent patients who are less than 80% of expected body weight.
Energy availability is hard to calculate, but certain assessments can be performed in a primary care setting to approximate it. To assess dietary intake, patients can bring in a 3-, 4-, or 7-day dietary log or complete a 24-hour food recall or food-frequency questionnaire in the office. To objectively document energy expenditure, patients can use heart rate monitors, accelerometers, an exercise diary, and web-based calculators. The fat-free mass can be calculated using a bioelectric impedance scale and skinfold caliper measurements.26
Those with chronic energy deficiency states may have reduced resting metabolic rates, with measured rates less than 90% of predicted and low triiodothyronine (T3) levels.31
Diagnosing menstrual dysfunction
When evaluating patients with menstrual dysfunction, it is important to first rule out pregnancy and endocrinopathies. These include thyroid dysfunction, hyperprolactinemia, primary ovarian insufficiency, other hypothalamic and pituitary disorders, and hyperandrogenic conditions such as polycystic ovarian syndrome, ovarian tumor, adrenal tumor, nonclassic congenital adrenal hyperplasia, and Cushing syndrome.
Depending on the patient’s age, laboratory tests can include follicle-stimulating hormone, luteinizing hormone, prolactin, serum estradiol, and a progesterone challenge.32 For hyperandrogenic symptoms, measuring total and free testosterone, dehydroepiandrosterone sulfate, 24-hour urine cortisol, and 17-hydroxyprogesterone levels may be helpful.
An endocrinologist should be consulted to evaluate the underlying cause of amenorrhea and address any associated hormonal imbalances. Attributing menstrual dysfunction to low energy availability is generally a diagnosis of exclusion. Additionally, outflow tract obstruction should be considered and ruled out with transvaginal ultrasonography in patients with primary amenorrhea.
A patient with hypoestrogenemia and amenorrhea may have the same steroid hormone profile as that of a menopausal woman. Lack of estrogen results in impaired endothelial cell function and arterial dilation, with accelerated development of atherosclerosis and subsequent cardiovascular events.33,34 Further, low energy availability has been linked to negative cardiovascular effects such as decreased vessel dilation leading to decreased tissue perfusion and hastened development of atherosclerosis.33 Female athletes with hypoestrogenism may show reduced perfusion of working muscle, impaired aerobic metabolism in skeletal muscle, elevated low-density lipoprotein cholesterol, and vaginal dryness.4
Diagnosing low bone mineral density
The most common clinical manifestations of low bone mineral density in female athletes are bone stress reactions such as stress fractures. In a study of 311 female high school athletes, 65.6% suffered from musculoskeletal injury from trauma or overuse including stress fractures and the patellofemoral syndrome.35 Many athletes seek medical attention from their primary care physician for stress reactions, providing an opportunity for triad screening.36
In postmenopausal women, osteopenia and osteoporosis are defined using the T score. However, in premenopausal women and adolescents, the International Society for Clinical Densitometry recommends using the Z score. A Z score less than –2.0 is described as “low bone density for chronological age.”14 For the diagnosis of osteoporosis in children and premenopausal women, the Society recommends using a Z score less than –2.0 along with the presence of a secondary risk factor for fracture such as undernutrition, hypogonadism, or a history of fracture.
Table 2 summarizes the diagnosis of low bone mineral density and osteoporosis in premenopausal women, adolescents, and children as well as when to order dual-energy x-ray absorptiometry (DEXA).37,38
Adolescents with low bone mineral density should have an annual DEXA scan of the total hip and lumbar spine.22 Amenorrheic athletes typically present with low areal density at the lumbar spine, reduced trabecular volumetric bone mineral density and bone strength index at the distal radius, and deterioration of the distal tibia.39
EARLY INTERVENTION IS ESSENTIAL
Early intervention is essential in patients with any component of the female athlete triad to prevent long-term adverse health effects. Successful treatment is strongly correlated with a trusting relationship between the athlete and the multidisciplinary team involved in her treatment.40
If needed, selective serotonin reuptake inhibitors and other psychotropic medications can be prescribed for comorbid conditions including bulimia nervosa, anxiety, depression, and obsessive-compulsive disorder. Primary providers can identify risk factors that prompt the evaluation and diagnosis of the triad, as well as support the goals of treatment and help manage comorbid conditions.
Eat more, exercise less
The primary goal is to restore body weight, maximizing nutritional and energy status by modifying the diet and adjusting exercise behavior to increase energy availability.41 Creating an energy-positive state by increasing intake, decreasing energy expenditure, or both increases energy availability, subsequently improves bone mineral density, and normalizes menstrual function.40
To sustain normal physiologic function, an energy availability of at least 45 kcal/kg/day is recommended.42 Patients should consume a minimum of 2,000 kcal/day, although energy needs may far exceed that, depending on energy expenditure. Olympic athletes participating in women’s crew and other sports have been anecdotally known to require over 12,000 kcal a day to maintain weight and performance. Goals include a body mass index of at least 18.5 kg/m2 in adults and a body weight of at least 90% of predicted in adolescents.
Involving a dietitian in the care team can help ensure that the patient consumes an adequate amount of macronutrients and micronutrients necessary for bone growth; these include calcium, vitamin D, iron, zinc, and vitamin K.4,32 For patients with disordered eating, referral to a mental health professional is important to help them avoid pathologic eating behaviors, reduce dieting attempts, and alter negative emotions associated with food and body image.
Once treatment begins, patients must undergo standardized periodic monitoring of their body weight. Although positive effects such as normalization of metabolic hormones (eg, insulinlike growth factor 1) may be seen in days to weeks by reversing low energy availability, it may take several months for menstrual function to improve and years for measurable improvement in bone mineral density to occur.23
Menstrual function should improve with weight gain
Normalizing menses in patients with the female athlete triad depends on improving the low energy availability and inducing weight gain.
Pharmacotherapy such as combined oral contraceptives can treat symptoms of hypogonadism.25 However, combined oral contraceptives do not restore spontaneous menses but rather induce withdrawal bleeding, which can lead to a false sense of security.23 While there are some benefits to prescribing combined oral contraceptives to treat hypogonadism, nonpharmacologic methods should be tried initially to restore menses, including increasing caloric intake and body weight. Golden et al showed that hormone replacement with combined oral contraceptives did not improve bone density in women with low estrogen states (eg, anorexia nervosa, osteopenia).43 Further, combined oral contraceptives may worsen bone health, as oral estrogen suppresses hepatic production of insulinlike growth factor 1, a bone trophic hormone.23
Treating low bone mineral density
Improving energy availability and menstrual function can help improve bone mineral density. Nutritional enhancement is recommended for mineralization of trabecular bone and growth of cortical bone. Supplemental calcium (1,000–1,500 mg daily) and vitamin D (600–1,000 IU daily) should be incorporated into the treatment of low bone mineral density.15,19
Additionally, weight gain has a positive effect on bone mineral density independent of its effect on the resumption of menses. However, weight gain alone does not normalize bone mineral density. Resuming normal physiologic production of hormones with estrogen-dependent effects on bone health is integral to normalization as well.23
Resistance training is encouraged to increase lean mass, although caution must be used to prevent fractures during high-impact activity. Bone mineral density may take up to several years to improve and may not be fully reversible.4,25,39
Pharmacologic therapy for low bone mineral density has unclear outcomes in women under age 50. The decision to treat should be based on bone mineral density along with fracture history. Given their unknown effects on the human fetal skeleton, bisphosphonates and denosumab should be used with caution in women of childbearing potential.24 No studies have used denosumab or teriparatide in women with the female athlete triad. Despite concerns regarding use of these drugs in premenopausal women, drug therapy should be strongly considered in women with a history of fracture and those with a high risk of subsequent fracture. The decision to treat can be made in conjunction with the athlete’s endocrinologist.
RETURN TO PLAY
If an athlete is noted to be at risk for or diagnosed with the female athlete triad, it is important to formulate a plan for her to return to play once her health improves.
De Souza et al provided a cumulative risk assessment for risk stratification and made recommendations on when an athlete should return to play depending on her level of risk.23 Using this grading system, primary care physicians can risk-stratify their patients. Those at low risk may be fully cleared to return to play, while those at moderate to high risk must first follow up with a multidisciplinary team to develop treatment strategies for improving their health.
Once a patient reaches her established goals, she may provisionally return to play under the close supervision of a team physician or primary care physician. A written treatment contract, including the goals set by the multidisciplinary team, should be followed closely as the athlete continues to participate in the sport.23;
- Curry EJ, Logan C, Ackerman K, McInnis KC, Matzkin EG. Female athlete triad awareness among multispecialty physicians. Sports Med Open 2015; 1(1):38. doi:10.1186/s40798-015-0037-5
- National Federation of State High School Associations. 2012–13 high school athletics participation survey. http://old.nfhs.org/content.aspx?id=3282. Accessed February 1, 2018.
- Otis CL, Drinkwater B, Johnson M, Loucks A, Wilmore J. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 1997; 29(5):i–ix.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39(10):1867–1882. doi:10.1249/mss.0b013e318149f111
- Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med 2014; 48(7):491–497. doi:10.1136/bjsports-2014-093502
- Tenforde AS, Barrack MT, Nattiv A, Fredericson M. Parallels with the female athlete triad in male athletes. Sports Med 2016; 46(2):171–182. doi:10.1007/s40279-015-0411-y
- Thein-Nissenbaum JM, Carr KE. Female athlete triad syndrome in the high school athlete. Phys Ther Sport 2011; 12(3):108–116. doi:10.1016/j.ptsp.2011.04.002
- Matzkin E, Curry EJ, Whitlock K. Female athlete triad: past, present, and future. J Am Acad Orthop Surg 2015; 23(7):424–432. doi:10.5435/JAAOS-D-14-00168
- Gibbs JC, Williams NI, De Souza MJ. Prevalence of individual and combined components of the female athlete triad. Med Sci Sports Exerc 2013; 45(5):985–996. doi:10.1249/MSS.0b013e31827e1bdc
- Byrne S, McLean N. Elite athletes: effects of the pressure to be thin. J Sci Med Sport 2002; 5(2):80–94. doi:10.1016/S1440-2440(02)80029-9
- Sundgot-Borgen J, Torstveit MK. Prevalence of eating disorders in elite athletes is higher than in the general population. Clin J Sport Med 2004; 14(1):25–32.
- Lynch SL, Hoch AZ. The female runner: gender specifics. Clin Sports Med 2010; 29(3):477–498. doi:10.1016/j.csm.2010.03.003
- Doyle-Lucas AF, Akers JD, Davy BM. Energetic efficiency, menstrual irregularity, and bone mineral density in elite professional female ballet dancers. J Dance Med Sci 2010; 14(4):146–154.
- Thein-Nissenbaum J. Long term consequences of the female athlete triad. Maturitas 2013; 75(2):107–112. doi:10.1016/j.maturitas.2013.02.010
- Hilibrand MJ, Hammoud S, Bishop M, Woods D, Fredrick RW, Dodson CC. Common injuries and ailments of the female athlete; pathophysiology, treatment and prevention. Phys Sportsmed 2015; 43(4):403–411. doi:10.1080/00913847.2015.1092856
- Demorest RA, Hergenroeder AC. Preface. Sports medicine and sports injuries. Adolesc Med State Art Rev 2015; 26(1):xv–xvi.
- Khan KM, Liu-Ambrose T, Sran MM, Ashe MC, Donaldson MG, Wark JD. New criteria for female athlete triad syndrome? Br J Sports Med 2002; 36(1):10–13. doi:10.1136/bjsm.36.1.10
- Fontana R, Della Torre S. The deep correlation between energy metabolism and reproduction: a view of the effects of nutrition for women fertility. Nutrients 2016; 8:87. www.ncbi.nlm.nih.gov/pmc/articles/PMC4772050/. Accessed February 2, 2018.
- Nazem TG, Ackerman K. The female athlete triad. Sports Health 2012; 4(4):302–311. doi:10.1177/1941738112439685
- Pantano KJ. Coaching concerns in physically active girls and young women—part 1: the female athlete triad. Strength Conditioning J 2009; 31(6):38–43. doi:10.1519/SSC.0b013e3181c105dd
- Micklesfield LK, Hugo J, Johnson C, Noakes TD, Lambert EV. Factors associated with menstrual dysfunction and self-reported bone stress injuries in female runners in the ultra- and half-marathons of the Two Oceans. Br J Sports Med 2007; 41(10):679–683. doi:10.1136/bjsm.2007.037077
- Lambrinoudaki I, Papadimitriou D. Pathophysiology of bone loss in the female athlete. Ann N Y Acad Sci 2010; 1205:45–50. doi:10.1111/j.1749-6632.2010.05681.x
- De Souza MJ, Nattiv A, Joy E, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad: 1st International Conference held in San Francisco, CA, May 2012, and 2nd International Conference held in Indianapolis, IN, May 2013. Clin J Sport Med 2014; 24(2):96–119. doi:10.1136/bjsports-2013-093218
- Horn E, Gergen N, McGarry KA. The female athlete triad. R I Med J (2013) 2014; 97(11):18–21.
- Joy E, De Souza MJ, Nattiv A, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad. Curr Sports Med Rep 2014; 13(4):219–232. doi:10.1249/JSR.0000000000000077
- Temme KE, Hoch AZ. Recognition and rehabilitation of the female athlete triad/tetrad: a multidisciplinary approach. Curr Sports Med Rep 2013; 12(3):190–199. doi:10.1249/JSR.0b013e318296190b
- Pritts SD, Susman J. Diagnosis of eating disorders in primary care. Am Fam Physician 2003; 67(2):297–304.
- Berhardt DR, Roberts WO, editors. Preparticipation Physical Evaluation, 4th Ed. American Academy of Pediatrics, Elk Grove Village, IL, 2010.
- Mencias T, Noon M, Hoch AZ. Female athlete triad screening in National Collegiate Athletic Association Division I athletes: is the preparticipation evaluation form effective? Clin J Sport Med 2012; 22(2):122–125. doi:10.1097/JSM.0b013e3182425aee
- Javed A, Tebben PJ, Fischer PR, Lteif AN. Female athlete triad and its components: toward improved screening and management. Mayo Clin Proc 2013; 88(9):996–1009. doi:10.1016/j.mayocp.2013.07.001
- Melin A, Tornberg AB, Skouby S, et al. Energy availability and the female athlete triad in elite endurance athletes. Scand J Med Sci Sports 2015; 25(5):610–622. doi:10.1111/sms.12261
- Warr BJ, Woolf K. The female athlete triad: patients do best with a team approach to care. JAAPA 2011; 24(4):50–55.
- Hoch AZ, Lal S, Jurva JW, Gutterman DD. The female athlete triad and cardiovascular dysfunction. Phys Med Rehabil Clin North Am 2007; 18(3):385–400. doi:10.1016/j.pmr.2007.05.001
- Lanser EM, Zach KN, Hoch AZ. The female athlete triad and endothelial dysfunction. PM R 2011; 3(5):458–465. doi:10.1016/j.pmrj.2010.12.024
- Thein-Nissenbaum JM, Rauh MJ, Carr KE, Loud KJ, McGuine TA. Associations between disordered eating, menstrual dysfunction, and musculoskeletal injury among high school athletes. J Orthop Sports Phys Ther 2011; 41(2):60–69. doi:10.2519/jospt.2011.3312
- Ducher G, Turner AI, Kukuljan S, et al. Obstacles in the optimization of bone health outcomes in the female athlete triad. Sports Med 2011; 41(7):587–607. doi:10.2165/11588770-000000000-00000
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39(1):155–167. doi:10.1016/j.ecl.2009.11.002
- House S, Loud K, Shubkin C. Female athlete triad for the primary care pediatrician. Curr Opin Pediatr 2013; 25(6):755–761. doi:10.1097/MOP.0000000000000033
- Mallinson RJ, De Souza MJ. Current perspectives on the etiology and manifestation of the “silent” component of the female athlete triad. Int J Womens Health 2014; 6:451–467. doi:10.2147/IJWH.S38603
- Deimel JF, Dunlap BJ. The female athlete triad. Clin Sports Med 2012; 31(2):247–254. doi:10.1016/j.csm.2011.09.007
- Manore MM, Kam LC, Loucks AB; International Association of Athletics Federations. The female athlete triad: components, nutrition issues, and health consequences. J Sports Sci 2007; 25(suppl 1):S61–S71. doi:10.1080/02640410701607320
- Witkop CT, Warren MP. Understanding the spectrum of the female athlete triad. Obstet Gynecol 2010; 116(6):1444–1448. doi:10.1097/AOG.0b013e3181fbed40
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15(3):135–143. doi:10.1016/S1083-3188(02)00145-6
Striving for athletic excellence, many young women—and some young men—create an energy deficit from increased exercise, decreased intake, or both. In women, the resulting energy deficit can suppress the menstrual cycle and in turn lead to bone demineralization in a syndrome called the female athlete triad.
Primary care physicians should be aware of this syndrome because it can lead to short-term and long-term health complications, and they are in a good position to screen for, diagnose, and treat it. However, a study of 931 US physicians in 2015 found that only 37% had heard of it.1
DEFINITION HAS CHANGED: ONLY 1 OF 3 COMPONENTS NEEDED
In 1972, Title IX of the Education Amendment Act was passed, prohibiting sex discrimination in any higher education program or activity receiving federal financial aid. Since then, female athletic participation in the United States has increased more than 10-fold.2
Also increasing has been awareness of the link between athletics, eating disorders, and amenorrhea. The American College of Sports Medicine coined the term female athlete triad in 1992, describing it as the constellation of disordered eating, amenorrhea, and osteoporosis (all 3 needed to be present).3 They broadened the definition in 2007 so that the syndrome can be diagnosed if any of the following is present4:
- Low energy availability (with or without an eating disorder)
- Menstrual dysfunction
- Decreased bone mineral density.
Recognizing that low energy availability can affect athletes of either sex and have consequences beyond the female reproductive system and skeleton, in 2014 the International Olympic Committee introduced a broader term called relative energy deficiency in sport.5,6 Like the triad, this condition occurs when energy intake falls below energy output to the point that it negatively affects an athlete’s physical and mental health.
THE COMPONENTS ARE COMMON
The female athlete triad can be seen in high school, collegiate, and elite athletes7 and is especially common in sports with subjective judging (gymnastics, figure skating) or endurance sports that emphasize leanness (eg, running).8
In a review of 65 studies, Gibbs et al9 found that the prevalence of any one of the triad conditions in exercising women and female athletes ranged from 16.0% to 60.0%, the prevalence of any 2 ranged from 2.7% to 27.0%, and the prevalence of all 3 ranged from 0% to 15.9%.
Low energy availability is categorized as either intentional (ie, due to disordered eating) or unintentional (ie, due to activities not associated with eating). Sustained low energy availability is often associated with eating disorders and subsequent low self-esteem, depression, and anxiety disorders.4
The prevalence of eating disorders is high in female athletes—31% and 20% in 2 large studies of elite female athletes, compared with 5.5% and 9%, respectively, in the general population.10,11 Another study found that the prevalence of disordered eating was 46.7% in sports that emphasize leanness, such as track and gymnastics, compared with 19.8% in sports that did not, such as basketball and soccer.12
Calorie restriction is common. In a study of 15 elite ballet dancers and 15 matched controls, the dancers were found to consume only about 3/4 as many calories per day as the controls (1,577 vs 2,075 kcal/day, P ≤ .01).13
Menstrual dysfunction. In small studies, the prevalence of secondary amenorrhea was as high as 69% in dancers and 65% in long-distance runners.4,14–16
Decreased bone mineral density. According to a systematic review, the prevalence of osteopenia in amenorrheic athletes ranged between 22% and 50% and the prevalence of osteoporosis was 0% to 13%, compared with 12% and 2.3%, respectively, in the general population.17
THE COMPONENTS ARE LINKED
dysfunction play causative roles in bone mineral density pathology. Within each component of the triad a spectrum of dysfunction exists, with all 3 components exhibiting serious health end points including low energy availability, functional hypothalamic amenorrhea, and osteoporosis.
The 3 components of the female athlete triad—low energy availability, menstrual dysfunction, and decreased bone mineral density—are linked, and each exists on a spectrum (Figure 1). The long-term consequences are far-reaching and can affect the cardiovascular, endocrine, reproductive, skeletal, gastrointestinal, renal, and central nervous systems.
Low energy availability is the driving force of the triad, causing menstrual irregularity and subsequent low bone mineral density.
Menstrual dysfunction. Low energy availability can contribute to menstrual disturbances because the body suppresses reproductive function to prevent pregnancy. Functional hypothalamic amenorrhea results from decreased gonadotropin-releasing hormone leading to decreased gonadotropin release from the pituitary gland and, ultimately, to low circulating estrogen levels.18 Menstrual irregularities related to the triad include:
- Primary amenorrhea (a delay in menarche)
- Oligomenorrhea (menstrual cycles occurring at intervals greater than every 35 days)
- Secondary amenorrhea (cessation of menstruation for 3 consecutive months).
(Primary amenorrhea is defined as no menses by age 15 in the presence of normal secondary sexual development or within 5 years after breast development if that occurs before the age of 10. Secondary amenorrhea is defined as the loss of menses for 90 or more days after menarche.19)
In animal studies, reducing dietary intake by more than 30% resulted in infertility.4 Menstrual abnormalities can present as early as 5 days after a patient enters a state of low energy availability.20 Symptoms of menstrual dysfunction are largely indicative of hypogonadism and include vaginal dryness, infertility, and impaired bone health.
Bone health in women with the female athlete triad can range from optimal to osteoporosis.
Low bone mineral density is a result of low energy availability and menstrual dysfunction leading to estrogen deficiency.21,22 Specifically, menstrual abnormalities can result in low estrogen and overactivity of osteoclasts, while low energy availability alters the metabolic environment, inducing changes in insulinlike growth factor 1, leptin, and peptide YY, resulting in deficiencies in vitamin D and calcium—nutrients necessary for bone mineralization. In turn, bone health and density are compromised.21,22
Ninety percent of peak bone mass is attained by age 18. Those who have low bone mineral density as part of the female athlete triad can suffer from long-lasting effects on their bone health.
SCREENING
Untreated, the triad can lead to fatigue, poor sports performance, and a number of serious comorbid conditions such as osteopenia and osteoporosis (leading to stress fractures) anemia, heart arrhythmias, and amenorrhea. Therefore, it is important for primary care providers to screen female athletes for the triad during routine office visits.
In 2014, the Triad Consensus Panel recommended screening female athletes at the high school and collegiate levels during a preparticipation physical evaluation and then every year by a primary care physician, athletic trainer, team physician, or coach.23
Risk factors include signs of dietary restriction, low body mass index, delayed menarche, oligomenorrhea or amenorrhea, and bone stress reactions or fractures.23 Athletes should be questioned about their menstrual history (age of menarche, frequency, and duration of menstrual cycles), history of stress fractures, medication history, family history (osteoporosis, eating disorders, and fractures),24,25 and dietary habits.
Physical findings include low body mass index, recent weight loss, orthostatic hypotension, lanugo, hypercarotenemia, and signs of eating disorders (restrictive, binging, purging) (Table 1).25–27
Additionally, it is important to ascertain if the patient receives critical comments regarding performance or body image from coaches, parents, or teammates and if sport-specific training began early in life.
Certain personality factors and behaviors are clues, such as perfectionism, obsessiveness, frequent weight cycling, and overtraining.4,25 If any of the triad components are apparent, a deeper evaluation can be completed.
Specific screening questions
The Female Athlete Triad Coalition recommends asking 11 screening questions and having prompt discussions regarding the athlete’s nutritional status and body image.23 If the patient gives a worrisome response to a screening question, further workup for a formal diagnosis should be initiated.
Questions about nutritional status.
- Do you worry about your weight?
- Are you trying to gain or lose weight, or has anyone recommended that you do so?
- Are you on a special diet or do you avoid certain types of foods or food groups?
- Have you ever had an eating disorder?
Questions about menstrual function.
- Have you ever had a menstrual period?
- How old were you when you had your first menstrual period?
- When was your most recent menstrual period?
- How many periods have you had in the last 12 months?
- Are you presently taking any female hormones (estrogen, progesterone, birth control pills)?
Questions about bone health.
- Have you ever had a stress fracture?
- Have you ever been told you have low bone density (osteopenia or osteoporosis)?
Along similar lines, the American Academy of Pediatrics, American Academy of Family Physicians, and American College of Sports Medicine28 have a list of 7 questions:
- Do you worry about your weight?
- Do you limit the foods you eat?
- Do you lose weight to meet image requirements for sports?
- Have you ever suffered from an eating disorder?
- How old were you when you had your first menstrual period?
- How many menstrual cycles have you had in the past 12 months?
- Have you ever had a stress fracture?
These questions are not being widely used. A study of the National Collegiate Athletic Association Division I universities found that only 9% of universities included 9 or more of the recommended 12 questions that the Female Athlete Triad Coalition was recommending at that time, and 22% asked only 1 or 2 of the questions. None of the universities included all 12.29 These findings are not surprising, given that screening for the triad is not state-mandated. Screening discrepancies among providers largely stem from knowledge gaps, nonstandardized questionnaires, lack of time at appointments, and the sensitive nature of the questioning (eg, disordered eating).30
DIAGNOSING THE TRIAD
Given that the signs of low energy availability and menstrual dysfunction are often subtle, the diagnosis of the triad for those at risk requires input from a multidisciplinary team including a physician, sports dietitian, mental health professional, exercise physiologist, and other medical consultants.
Table 2 lists diagnostic tests the primary care provider should consider.
Diagnosing low energy availability
Energy availability is the dietary energy remaining after exercise energy expenditure; it is normalized to fat-free (lean) mass to account for resting energy expenditure. It is a product of energy intake, energy expenditure, and stored energy, and is calculated as:
An optimal value is at least 45 kcal/kg/day, while physiologic changes start to occur at less than 30 kcal/kg/day.4,31 Low energy is often seen in adult patients with a body mass index less than 17.5 kg/m2 and adolescent patients who are less than 80% of expected body weight.
Energy availability is hard to calculate, but certain assessments can be performed in a primary care setting to approximate it. To assess dietary intake, patients can bring in a 3-, 4-, or 7-day dietary log or complete a 24-hour food recall or food-frequency questionnaire in the office. To objectively document energy expenditure, patients can use heart rate monitors, accelerometers, an exercise diary, and web-based calculators. The fat-free mass can be calculated using a bioelectric impedance scale and skinfold caliper measurements.26
Those with chronic energy deficiency states may have reduced resting metabolic rates, with measured rates less than 90% of predicted and low triiodothyronine (T3) levels.31
Diagnosing menstrual dysfunction
When evaluating patients with menstrual dysfunction, it is important to first rule out pregnancy and endocrinopathies. These include thyroid dysfunction, hyperprolactinemia, primary ovarian insufficiency, other hypothalamic and pituitary disorders, and hyperandrogenic conditions such as polycystic ovarian syndrome, ovarian tumor, adrenal tumor, nonclassic congenital adrenal hyperplasia, and Cushing syndrome.
Depending on the patient’s age, laboratory tests can include follicle-stimulating hormone, luteinizing hormone, prolactin, serum estradiol, and a progesterone challenge.32 For hyperandrogenic symptoms, measuring total and free testosterone, dehydroepiandrosterone sulfate, 24-hour urine cortisol, and 17-hydroxyprogesterone levels may be helpful.
An endocrinologist should be consulted to evaluate the underlying cause of amenorrhea and address any associated hormonal imbalances. Attributing menstrual dysfunction to low energy availability is generally a diagnosis of exclusion. Additionally, outflow tract obstruction should be considered and ruled out with transvaginal ultrasonography in patients with primary amenorrhea.
A patient with hypoestrogenemia and amenorrhea may have the same steroid hormone profile as that of a menopausal woman. Lack of estrogen results in impaired endothelial cell function and arterial dilation, with accelerated development of atherosclerosis and subsequent cardiovascular events.33,34 Further, low energy availability has been linked to negative cardiovascular effects such as decreased vessel dilation leading to decreased tissue perfusion and hastened development of atherosclerosis.33 Female athletes with hypoestrogenism may show reduced perfusion of working muscle, impaired aerobic metabolism in skeletal muscle, elevated low-density lipoprotein cholesterol, and vaginal dryness.4
Diagnosing low bone mineral density
The most common clinical manifestations of low bone mineral density in female athletes are bone stress reactions such as stress fractures. In a study of 311 female high school athletes, 65.6% suffered from musculoskeletal injury from trauma or overuse including stress fractures and the patellofemoral syndrome.35 Many athletes seek medical attention from their primary care physician for stress reactions, providing an opportunity for triad screening.36
In postmenopausal women, osteopenia and osteoporosis are defined using the T score. However, in premenopausal women and adolescents, the International Society for Clinical Densitometry recommends using the Z score. A Z score less than –2.0 is described as “low bone density for chronological age.”14 For the diagnosis of osteoporosis in children and premenopausal women, the Society recommends using a Z score less than –2.0 along with the presence of a secondary risk factor for fracture such as undernutrition, hypogonadism, or a history of fracture.
Table 2 summarizes the diagnosis of low bone mineral density and osteoporosis in premenopausal women, adolescents, and children as well as when to order dual-energy x-ray absorptiometry (DEXA).37,38
Adolescents with low bone mineral density should have an annual DEXA scan of the total hip and lumbar spine.22 Amenorrheic athletes typically present with low areal density at the lumbar spine, reduced trabecular volumetric bone mineral density and bone strength index at the distal radius, and deterioration of the distal tibia.39
EARLY INTERVENTION IS ESSENTIAL
Early intervention is essential in patients with any component of the female athlete triad to prevent long-term adverse health effects. Successful treatment is strongly correlated with a trusting relationship between the athlete and the multidisciplinary team involved in her treatment.40
If needed, selective serotonin reuptake inhibitors and other psychotropic medications can be prescribed for comorbid conditions including bulimia nervosa, anxiety, depression, and obsessive-compulsive disorder. Primary providers can identify risk factors that prompt the evaluation and diagnosis of the triad, as well as support the goals of treatment and help manage comorbid conditions.
Eat more, exercise less
The primary goal is to restore body weight, maximizing nutritional and energy status by modifying the diet and adjusting exercise behavior to increase energy availability.41 Creating an energy-positive state by increasing intake, decreasing energy expenditure, or both increases energy availability, subsequently improves bone mineral density, and normalizes menstrual function.40
To sustain normal physiologic function, an energy availability of at least 45 kcal/kg/day is recommended.42 Patients should consume a minimum of 2,000 kcal/day, although energy needs may far exceed that, depending on energy expenditure. Olympic athletes participating in women’s crew and other sports have been anecdotally known to require over 12,000 kcal a day to maintain weight and performance. Goals include a body mass index of at least 18.5 kg/m2 in adults and a body weight of at least 90% of predicted in adolescents.
Involving a dietitian in the care team can help ensure that the patient consumes an adequate amount of macronutrients and micronutrients necessary for bone growth; these include calcium, vitamin D, iron, zinc, and vitamin K.4,32 For patients with disordered eating, referral to a mental health professional is important to help them avoid pathologic eating behaviors, reduce dieting attempts, and alter negative emotions associated with food and body image.
Once treatment begins, patients must undergo standardized periodic monitoring of their body weight. Although positive effects such as normalization of metabolic hormones (eg, insulinlike growth factor 1) may be seen in days to weeks by reversing low energy availability, it may take several months for menstrual function to improve and years for measurable improvement in bone mineral density to occur.23
Menstrual function should improve with weight gain
Normalizing menses in patients with the female athlete triad depends on improving the low energy availability and inducing weight gain.
Pharmacotherapy such as combined oral contraceptives can treat symptoms of hypogonadism.25 However, combined oral contraceptives do not restore spontaneous menses but rather induce withdrawal bleeding, which can lead to a false sense of security.23 While there are some benefits to prescribing combined oral contraceptives to treat hypogonadism, nonpharmacologic methods should be tried initially to restore menses, including increasing caloric intake and body weight. Golden et al showed that hormone replacement with combined oral contraceptives did not improve bone density in women with low estrogen states (eg, anorexia nervosa, osteopenia).43 Further, combined oral contraceptives may worsen bone health, as oral estrogen suppresses hepatic production of insulinlike growth factor 1, a bone trophic hormone.23
Treating low bone mineral density
Improving energy availability and menstrual function can help improve bone mineral density. Nutritional enhancement is recommended for mineralization of trabecular bone and growth of cortical bone. Supplemental calcium (1,000–1,500 mg daily) and vitamin D (600–1,000 IU daily) should be incorporated into the treatment of low bone mineral density.15,19
Additionally, weight gain has a positive effect on bone mineral density independent of its effect on the resumption of menses. However, weight gain alone does not normalize bone mineral density. Resuming normal physiologic production of hormones with estrogen-dependent effects on bone health is integral to normalization as well.23
Resistance training is encouraged to increase lean mass, although caution must be used to prevent fractures during high-impact activity. Bone mineral density may take up to several years to improve and may not be fully reversible.4,25,39
Pharmacologic therapy for low bone mineral density has unclear outcomes in women under age 50. The decision to treat should be based on bone mineral density along with fracture history. Given their unknown effects on the human fetal skeleton, bisphosphonates and denosumab should be used with caution in women of childbearing potential.24 No studies have used denosumab or teriparatide in women with the female athlete triad. Despite concerns regarding use of these drugs in premenopausal women, drug therapy should be strongly considered in women with a history of fracture and those with a high risk of subsequent fracture. The decision to treat can be made in conjunction with the athlete’s endocrinologist.
RETURN TO PLAY
If an athlete is noted to be at risk for or diagnosed with the female athlete triad, it is important to formulate a plan for her to return to play once her health improves.
De Souza et al provided a cumulative risk assessment for risk stratification and made recommendations on when an athlete should return to play depending on her level of risk.23 Using this grading system, primary care physicians can risk-stratify their patients. Those at low risk may be fully cleared to return to play, while those at moderate to high risk must first follow up with a multidisciplinary team to develop treatment strategies for improving their health.
Once a patient reaches her established goals, she may provisionally return to play under the close supervision of a team physician or primary care physician. A written treatment contract, including the goals set by the multidisciplinary team, should be followed closely as the athlete continues to participate in the sport.23;
Striving for athletic excellence, many young women—and some young men—create an energy deficit from increased exercise, decreased intake, or both. In women, the resulting energy deficit can suppress the menstrual cycle and in turn lead to bone demineralization in a syndrome called the female athlete triad.
Primary care physicians should be aware of this syndrome because it can lead to short-term and long-term health complications, and they are in a good position to screen for, diagnose, and treat it. However, a study of 931 US physicians in 2015 found that only 37% had heard of it.1
DEFINITION HAS CHANGED: ONLY 1 OF 3 COMPONENTS NEEDED
In 1972, Title IX of the Education Amendment Act was passed, prohibiting sex discrimination in any higher education program or activity receiving federal financial aid. Since then, female athletic participation in the United States has increased more than 10-fold.2
Also increasing has been awareness of the link between athletics, eating disorders, and amenorrhea. The American College of Sports Medicine coined the term female athlete triad in 1992, describing it as the constellation of disordered eating, amenorrhea, and osteoporosis (all 3 needed to be present).3 They broadened the definition in 2007 so that the syndrome can be diagnosed if any of the following is present4:
- Low energy availability (with or without an eating disorder)
- Menstrual dysfunction
- Decreased bone mineral density.
Recognizing that low energy availability can affect athletes of either sex and have consequences beyond the female reproductive system and skeleton, in 2014 the International Olympic Committee introduced a broader term called relative energy deficiency in sport.5,6 Like the triad, this condition occurs when energy intake falls below energy output to the point that it negatively affects an athlete’s physical and mental health.
THE COMPONENTS ARE COMMON
The female athlete triad can be seen in high school, collegiate, and elite athletes7 and is especially common in sports with subjective judging (gymnastics, figure skating) or endurance sports that emphasize leanness (eg, running).8
In a review of 65 studies, Gibbs et al9 found that the prevalence of any one of the triad conditions in exercising women and female athletes ranged from 16.0% to 60.0%, the prevalence of any 2 ranged from 2.7% to 27.0%, and the prevalence of all 3 ranged from 0% to 15.9%.
Low energy availability is categorized as either intentional (ie, due to disordered eating) or unintentional (ie, due to activities not associated with eating). Sustained low energy availability is often associated with eating disorders and subsequent low self-esteem, depression, and anxiety disorders.4
The prevalence of eating disorders is high in female athletes—31% and 20% in 2 large studies of elite female athletes, compared with 5.5% and 9%, respectively, in the general population.10,11 Another study found that the prevalence of disordered eating was 46.7% in sports that emphasize leanness, such as track and gymnastics, compared with 19.8% in sports that did not, such as basketball and soccer.12
Calorie restriction is common. In a study of 15 elite ballet dancers and 15 matched controls, the dancers were found to consume only about 3/4 as many calories per day as the controls (1,577 vs 2,075 kcal/day, P ≤ .01).13
Menstrual dysfunction. In small studies, the prevalence of secondary amenorrhea was as high as 69% in dancers and 65% in long-distance runners.4,14–16
Decreased bone mineral density. According to a systematic review, the prevalence of osteopenia in amenorrheic athletes ranged between 22% and 50% and the prevalence of osteoporosis was 0% to 13%, compared with 12% and 2.3%, respectively, in the general population.17
THE COMPONENTS ARE LINKED
dysfunction play causative roles in bone mineral density pathology. Within each component of the triad a spectrum of dysfunction exists, with all 3 components exhibiting serious health end points including low energy availability, functional hypothalamic amenorrhea, and osteoporosis.
The 3 components of the female athlete triad—low energy availability, menstrual dysfunction, and decreased bone mineral density—are linked, and each exists on a spectrum (Figure 1). The long-term consequences are far-reaching and can affect the cardiovascular, endocrine, reproductive, skeletal, gastrointestinal, renal, and central nervous systems.
Low energy availability is the driving force of the triad, causing menstrual irregularity and subsequent low bone mineral density.
Menstrual dysfunction. Low energy availability can contribute to menstrual disturbances because the body suppresses reproductive function to prevent pregnancy. Functional hypothalamic amenorrhea results from decreased gonadotropin-releasing hormone leading to decreased gonadotropin release from the pituitary gland and, ultimately, to low circulating estrogen levels.18 Menstrual irregularities related to the triad include:
- Primary amenorrhea (a delay in menarche)
- Oligomenorrhea (menstrual cycles occurring at intervals greater than every 35 days)
- Secondary amenorrhea (cessation of menstruation for 3 consecutive months).
(Primary amenorrhea is defined as no menses by age 15 in the presence of normal secondary sexual development or within 5 years after breast development if that occurs before the age of 10. Secondary amenorrhea is defined as the loss of menses for 90 or more days after menarche.19)
In animal studies, reducing dietary intake by more than 30% resulted in infertility.4 Menstrual abnormalities can present as early as 5 days after a patient enters a state of low energy availability.20 Symptoms of menstrual dysfunction are largely indicative of hypogonadism and include vaginal dryness, infertility, and impaired bone health.
Bone health in women with the female athlete triad can range from optimal to osteoporosis.
Low bone mineral density is a result of low energy availability and menstrual dysfunction leading to estrogen deficiency.21,22 Specifically, menstrual abnormalities can result in low estrogen and overactivity of osteoclasts, while low energy availability alters the metabolic environment, inducing changes in insulinlike growth factor 1, leptin, and peptide YY, resulting in deficiencies in vitamin D and calcium—nutrients necessary for bone mineralization. In turn, bone health and density are compromised.21,22
Ninety percent of peak bone mass is attained by age 18. Those who have low bone mineral density as part of the female athlete triad can suffer from long-lasting effects on their bone health.
SCREENING
Untreated, the triad can lead to fatigue, poor sports performance, and a number of serious comorbid conditions such as osteopenia and osteoporosis (leading to stress fractures) anemia, heart arrhythmias, and amenorrhea. Therefore, it is important for primary care providers to screen female athletes for the triad during routine office visits.
In 2014, the Triad Consensus Panel recommended screening female athletes at the high school and collegiate levels during a preparticipation physical evaluation and then every year by a primary care physician, athletic trainer, team physician, or coach.23
Risk factors include signs of dietary restriction, low body mass index, delayed menarche, oligomenorrhea or amenorrhea, and bone stress reactions or fractures.23 Athletes should be questioned about their menstrual history (age of menarche, frequency, and duration of menstrual cycles), history of stress fractures, medication history, family history (osteoporosis, eating disorders, and fractures),24,25 and dietary habits.
Physical findings include low body mass index, recent weight loss, orthostatic hypotension, lanugo, hypercarotenemia, and signs of eating disorders (restrictive, binging, purging) (Table 1).25–27
Additionally, it is important to ascertain if the patient receives critical comments regarding performance or body image from coaches, parents, or teammates and if sport-specific training began early in life.
Certain personality factors and behaviors are clues, such as perfectionism, obsessiveness, frequent weight cycling, and overtraining.4,25 If any of the triad components are apparent, a deeper evaluation can be completed.
Specific screening questions
The Female Athlete Triad Coalition recommends asking 11 screening questions and having prompt discussions regarding the athlete’s nutritional status and body image.23 If the patient gives a worrisome response to a screening question, further workup for a formal diagnosis should be initiated.
Questions about nutritional status.
- Do you worry about your weight?
- Are you trying to gain or lose weight, or has anyone recommended that you do so?
- Are you on a special diet or do you avoid certain types of foods or food groups?
- Have you ever had an eating disorder?
Questions about menstrual function.
- Have you ever had a menstrual period?
- How old were you when you had your first menstrual period?
- When was your most recent menstrual period?
- How many periods have you had in the last 12 months?
- Are you presently taking any female hormones (estrogen, progesterone, birth control pills)?
Questions about bone health.
- Have you ever had a stress fracture?
- Have you ever been told you have low bone density (osteopenia or osteoporosis)?
Along similar lines, the American Academy of Pediatrics, American Academy of Family Physicians, and American College of Sports Medicine28 have a list of 7 questions:
- Do you worry about your weight?
- Do you limit the foods you eat?
- Do you lose weight to meet image requirements for sports?
- Have you ever suffered from an eating disorder?
- How old were you when you had your first menstrual period?
- How many menstrual cycles have you had in the past 12 months?
- Have you ever had a stress fracture?
These questions are not being widely used. A study of the National Collegiate Athletic Association Division I universities found that only 9% of universities included 9 or more of the recommended 12 questions that the Female Athlete Triad Coalition was recommending at that time, and 22% asked only 1 or 2 of the questions. None of the universities included all 12.29 These findings are not surprising, given that screening for the triad is not state-mandated. Screening discrepancies among providers largely stem from knowledge gaps, nonstandardized questionnaires, lack of time at appointments, and the sensitive nature of the questioning (eg, disordered eating).30
DIAGNOSING THE TRIAD
Given that the signs of low energy availability and menstrual dysfunction are often subtle, the diagnosis of the triad for those at risk requires input from a multidisciplinary team including a physician, sports dietitian, mental health professional, exercise physiologist, and other medical consultants.
Table 2 lists diagnostic tests the primary care provider should consider.
Diagnosing low energy availability
Energy availability is the dietary energy remaining after exercise energy expenditure; it is normalized to fat-free (lean) mass to account for resting energy expenditure. It is a product of energy intake, energy expenditure, and stored energy, and is calculated as:
An optimal value is at least 45 kcal/kg/day, while physiologic changes start to occur at less than 30 kcal/kg/day.4,31 Low energy is often seen in adult patients with a body mass index less than 17.5 kg/m2 and adolescent patients who are less than 80% of expected body weight.
Energy availability is hard to calculate, but certain assessments can be performed in a primary care setting to approximate it. To assess dietary intake, patients can bring in a 3-, 4-, or 7-day dietary log or complete a 24-hour food recall or food-frequency questionnaire in the office. To objectively document energy expenditure, patients can use heart rate monitors, accelerometers, an exercise diary, and web-based calculators. The fat-free mass can be calculated using a bioelectric impedance scale and skinfold caliper measurements.26
Those with chronic energy deficiency states may have reduced resting metabolic rates, with measured rates less than 90% of predicted and low triiodothyronine (T3) levels.31
Diagnosing menstrual dysfunction
When evaluating patients with menstrual dysfunction, it is important to first rule out pregnancy and endocrinopathies. These include thyroid dysfunction, hyperprolactinemia, primary ovarian insufficiency, other hypothalamic and pituitary disorders, and hyperandrogenic conditions such as polycystic ovarian syndrome, ovarian tumor, adrenal tumor, nonclassic congenital adrenal hyperplasia, and Cushing syndrome.
Depending on the patient’s age, laboratory tests can include follicle-stimulating hormone, luteinizing hormone, prolactin, serum estradiol, and a progesterone challenge.32 For hyperandrogenic symptoms, measuring total and free testosterone, dehydroepiandrosterone sulfate, 24-hour urine cortisol, and 17-hydroxyprogesterone levels may be helpful.
An endocrinologist should be consulted to evaluate the underlying cause of amenorrhea and address any associated hormonal imbalances. Attributing menstrual dysfunction to low energy availability is generally a diagnosis of exclusion. Additionally, outflow tract obstruction should be considered and ruled out with transvaginal ultrasonography in patients with primary amenorrhea.
A patient with hypoestrogenemia and amenorrhea may have the same steroid hormone profile as that of a menopausal woman. Lack of estrogen results in impaired endothelial cell function and arterial dilation, with accelerated development of atherosclerosis and subsequent cardiovascular events.33,34 Further, low energy availability has been linked to negative cardiovascular effects such as decreased vessel dilation leading to decreased tissue perfusion and hastened development of atherosclerosis.33 Female athletes with hypoestrogenism may show reduced perfusion of working muscle, impaired aerobic metabolism in skeletal muscle, elevated low-density lipoprotein cholesterol, and vaginal dryness.4
Diagnosing low bone mineral density
The most common clinical manifestations of low bone mineral density in female athletes are bone stress reactions such as stress fractures. In a study of 311 female high school athletes, 65.6% suffered from musculoskeletal injury from trauma or overuse including stress fractures and the patellofemoral syndrome.35 Many athletes seek medical attention from their primary care physician for stress reactions, providing an opportunity for triad screening.36
In postmenopausal women, osteopenia and osteoporosis are defined using the T score. However, in premenopausal women and adolescents, the International Society for Clinical Densitometry recommends using the Z score. A Z score less than –2.0 is described as “low bone density for chronological age.”14 For the diagnosis of osteoporosis in children and premenopausal women, the Society recommends using a Z score less than –2.0 along with the presence of a secondary risk factor for fracture such as undernutrition, hypogonadism, or a history of fracture.
Table 2 summarizes the diagnosis of low bone mineral density and osteoporosis in premenopausal women, adolescents, and children as well as when to order dual-energy x-ray absorptiometry (DEXA).37,38
Adolescents with low bone mineral density should have an annual DEXA scan of the total hip and lumbar spine.22 Amenorrheic athletes typically present with low areal density at the lumbar spine, reduced trabecular volumetric bone mineral density and bone strength index at the distal radius, and deterioration of the distal tibia.39
EARLY INTERVENTION IS ESSENTIAL
Early intervention is essential in patients with any component of the female athlete triad to prevent long-term adverse health effects. Successful treatment is strongly correlated with a trusting relationship between the athlete and the multidisciplinary team involved in her treatment.40
If needed, selective serotonin reuptake inhibitors and other psychotropic medications can be prescribed for comorbid conditions including bulimia nervosa, anxiety, depression, and obsessive-compulsive disorder. Primary providers can identify risk factors that prompt the evaluation and diagnosis of the triad, as well as support the goals of treatment and help manage comorbid conditions.
Eat more, exercise less
The primary goal is to restore body weight, maximizing nutritional and energy status by modifying the diet and adjusting exercise behavior to increase energy availability.41 Creating an energy-positive state by increasing intake, decreasing energy expenditure, or both increases energy availability, subsequently improves bone mineral density, and normalizes menstrual function.40
To sustain normal physiologic function, an energy availability of at least 45 kcal/kg/day is recommended.42 Patients should consume a minimum of 2,000 kcal/day, although energy needs may far exceed that, depending on energy expenditure. Olympic athletes participating in women’s crew and other sports have been anecdotally known to require over 12,000 kcal a day to maintain weight and performance. Goals include a body mass index of at least 18.5 kg/m2 in adults and a body weight of at least 90% of predicted in adolescents.
Involving a dietitian in the care team can help ensure that the patient consumes an adequate amount of macronutrients and micronutrients necessary for bone growth; these include calcium, vitamin D, iron, zinc, and vitamin K.4,32 For patients with disordered eating, referral to a mental health professional is important to help them avoid pathologic eating behaviors, reduce dieting attempts, and alter negative emotions associated with food and body image.
Once treatment begins, patients must undergo standardized periodic monitoring of their body weight. Although positive effects such as normalization of metabolic hormones (eg, insulinlike growth factor 1) may be seen in days to weeks by reversing low energy availability, it may take several months for menstrual function to improve and years for measurable improvement in bone mineral density to occur.23
Menstrual function should improve with weight gain
Normalizing menses in patients with the female athlete triad depends on improving the low energy availability and inducing weight gain.
Pharmacotherapy such as combined oral contraceptives can treat symptoms of hypogonadism.25 However, combined oral contraceptives do not restore spontaneous menses but rather induce withdrawal bleeding, which can lead to a false sense of security.23 While there are some benefits to prescribing combined oral contraceptives to treat hypogonadism, nonpharmacologic methods should be tried initially to restore menses, including increasing caloric intake and body weight. Golden et al showed that hormone replacement with combined oral contraceptives did not improve bone density in women with low estrogen states (eg, anorexia nervosa, osteopenia).43 Further, combined oral contraceptives may worsen bone health, as oral estrogen suppresses hepatic production of insulinlike growth factor 1, a bone trophic hormone.23
Treating low bone mineral density
Improving energy availability and menstrual function can help improve bone mineral density. Nutritional enhancement is recommended for mineralization of trabecular bone and growth of cortical bone. Supplemental calcium (1,000–1,500 mg daily) and vitamin D (600–1,000 IU daily) should be incorporated into the treatment of low bone mineral density.15,19
Additionally, weight gain has a positive effect on bone mineral density independent of its effect on the resumption of menses. However, weight gain alone does not normalize bone mineral density. Resuming normal physiologic production of hormones with estrogen-dependent effects on bone health is integral to normalization as well.23
Resistance training is encouraged to increase lean mass, although caution must be used to prevent fractures during high-impact activity. Bone mineral density may take up to several years to improve and may not be fully reversible.4,25,39
Pharmacologic therapy for low bone mineral density has unclear outcomes in women under age 50. The decision to treat should be based on bone mineral density along with fracture history. Given their unknown effects on the human fetal skeleton, bisphosphonates and denosumab should be used with caution in women of childbearing potential.24 No studies have used denosumab or teriparatide in women with the female athlete triad. Despite concerns regarding use of these drugs in premenopausal women, drug therapy should be strongly considered in women with a history of fracture and those with a high risk of subsequent fracture. The decision to treat can be made in conjunction with the athlete’s endocrinologist.
RETURN TO PLAY
If an athlete is noted to be at risk for or diagnosed with the female athlete triad, it is important to formulate a plan for her to return to play once her health improves.
De Souza et al provided a cumulative risk assessment for risk stratification and made recommendations on when an athlete should return to play depending on her level of risk.23 Using this grading system, primary care physicians can risk-stratify their patients. Those at low risk may be fully cleared to return to play, while those at moderate to high risk must first follow up with a multidisciplinary team to develop treatment strategies for improving their health.
Once a patient reaches her established goals, she may provisionally return to play under the close supervision of a team physician or primary care physician. A written treatment contract, including the goals set by the multidisciplinary team, should be followed closely as the athlete continues to participate in the sport.23;
- Curry EJ, Logan C, Ackerman K, McInnis KC, Matzkin EG. Female athlete triad awareness among multispecialty physicians. Sports Med Open 2015; 1(1):38. doi:10.1186/s40798-015-0037-5
- National Federation of State High School Associations. 2012–13 high school athletics participation survey. http://old.nfhs.org/content.aspx?id=3282. Accessed February 1, 2018.
- Otis CL, Drinkwater B, Johnson M, Loucks A, Wilmore J. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 1997; 29(5):i–ix.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39(10):1867–1882. doi:10.1249/mss.0b013e318149f111
- Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med 2014; 48(7):491–497. doi:10.1136/bjsports-2014-093502
- Tenforde AS, Barrack MT, Nattiv A, Fredericson M. Parallels with the female athlete triad in male athletes. Sports Med 2016; 46(2):171–182. doi:10.1007/s40279-015-0411-y
- Thein-Nissenbaum JM, Carr KE. Female athlete triad syndrome in the high school athlete. Phys Ther Sport 2011; 12(3):108–116. doi:10.1016/j.ptsp.2011.04.002
- Matzkin E, Curry EJ, Whitlock K. Female athlete triad: past, present, and future. J Am Acad Orthop Surg 2015; 23(7):424–432. doi:10.5435/JAAOS-D-14-00168
- Gibbs JC, Williams NI, De Souza MJ. Prevalence of individual and combined components of the female athlete triad. Med Sci Sports Exerc 2013; 45(5):985–996. doi:10.1249/MSS.0b013e31827e1bdc
- Byrne S, McLean N. Elite athletes: effects of the pressure to be thin. J Sci Med Sport 2002; 5(2):80–94. doi:10.1016/S1440-2440(02)80029-9
- Sundgot-Borgen J, Torstveit MK. Prevalence of eating disorders in elite athletes is higher than in the general population. Clin J Sport Med 2004; 14(1):25–32.
- Lynch SL, Hoch AZ. The female runner: gender specifics. Clin Sports Med 2010; 29(3):477–498. doi:10.1016/j.csm.2010.03.003
- Doyle-Lucas AF, Akers JD, Davy BM. Energetic efficiency, menstrual irregularity, and bone mineral density in elite professional female ballet dancers. J Dance Med Sci 2010; 14(4):146–154.
- Thein-Nissenbaum J. Long term consequences of the female athlete triad. Maturitas 2013; 75(2):107–112. doi:10.1016/j.maturitas.2013.02.010
- Hilibrand MJ, Hammoud S, Bishop M, Woods D, Fredrick RW, Dodson CC. Common injuries and ailments of the female athlete; pathophysiology, treatment and prevention. Phys Sportsmed 2015; 43(4):403–411. doi:10.1080/00913847.2015.1092856
- Demorest RA, Hergenroeder AC. Preface. Sports medicine and sports injuries. Adolesc Med State Art Rev 2015; 26(1):xv–xvi.
- Khan KM, Liu-Ambrose T, Sran MM, Ashe MC, Donaldson MG, Wark JD. New criteria for female athlete triad syndrome? Br J Sports Med 2002; 36(1):10–13. doi:10.1136/bjsm.36.1.10
- Fontana R, Della Torre S. The deep correlation between energy metabolism and reproduction: a view of the effects of nutrition for women fertility. Nutrients 2016; 8:87. www.ncbi.nlm.nih.gov/pmc/articles/PMC4772050/. Accessed February 2, 2018.
- Nazem TG, Ackerman K. The female athlete triad. Sports Health 2012; 4(4):302–311. doi:10.1177/1941738112439685
- Pantano KJ. Coaching concerns in physically active girls and young women—part 1: the female athlete triad. Strength Conditioning J 2009; 31(6):38–43. doi:10.1519/SSC.0b013e3181c105dd
- Micklesfield LK, Hugo J, Johnson C, Noakes TD, Lambert EV. Factors associated with menstrual dysfunction and self-reported bone stress injuries in female runners in the ultra- and half-marathons of the Two Oceans. Br J Sports Med 2007; 41(10):679–683. doi:10.1136/bjsm.2007.037077
- Lambrinoudaki I, Papadimitriou D. Pathophysiology of bone loss in the female athlete. Ann N Y Acad Sci 2010; 1205:45–50. doi:10.1111/j.1749-6632.2010.05681.x
- De Souza MJ, Nattiv A, Joy E, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad: 1st International Conference held in San Francisco, CA, May 2012, and 2nd International Conference held in Indianapolis, IN, May 2013. Clin J Sport Med 2014; 24(2):96–119. doi:10.1136/bjsports-2013-093218
- Horn E, Gergen N, McGarry KA. The female athlete triad. R I Med J (2013) 2014; 97(11):18–21.
- Joy E, De Souza MJ, Nattiv A, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad. Curr Sports Med Rep 2014; 13(4):219–232. doi:10.1249/JSR.0000000000000077
- Temme KE, Hoch AZ. Recognition and rehabilitation of the female athlete triad/tetrad: a multidisciplinary approach. Curr Sports Med Rep 2013; 12(3):190–199. doi:10.1249/JSR.0b013e318296190b
- Pritts SD, Susman J. Diagnosis of eating disorders in primary care. Am Fam Physician 2003; 67(2):297–304.
- Berhardt DR, Roberts WO, editors. Preparticipation Physical Evaluation, 4th Ed. American Academy of Pediatrics, Elk Grove Village, IL, 2010.
- Mencias T, Noon M, Hoch AZ. Female athlete triad screening in National Collegiate Athletic Association Division I athletes: is the preparticipation evaluation form effective? Clin J Sport Med 2012; 22(2):122–125. doi:10.1097/JSM.0b013e3182425aee
- Javed A, Tebben PJ, Fischer PR, Lteif AN. Female athlete triad and its components: toward improved screening and management. Mayo Clin Proc 2013; 88(9):996–1009. doi:10.1016/j.mayocp.2013.07.001
- Melin A, Tornberg AB, Skouby S, et al. Energy availability and the female athlete triad in elite endurance athletes. Scand J Med Sci Sports 2015; 25(5):610–622. doi:10.1111/sms.12261
- Warr BJ, Woolf K. The female athlete triad: patients do best with a team approach to care. JAAPA 2011; 24(4):50–55.
- Hoch AZ, Lal S, Jurva JW, Gutterman DD. The female athlete triad and cardiovascular dysfunction. Phys Med Rehabil Clin North Am 2007; 18(3):385–400. doi:10.1016/j.pmr.2007.05.001
- Lanser EM, Zach KN, Hoch AZ. The female athlete triad and endothelial dysfunction. PM R 2011; 3(5):458–465. doi:10.1016/j.pmrj.2010.12.024
- Thein-Nissenbaum JM, Rauh MJ, Carr KE, Loud KJ, McGuine TA. Associations between disordered eating, menstrual dysfunction, and musculoskeletal injury among high school athletes. J Orthop Sports Phys Ther 2011; 41(2):60–69. doi:10.2519/jospt.2011.3312
- Ducher G, Turner AI, Kukuljan S, et al. Obstacles in the optimization of bone health outcomes in the female athlete triad. Sports Med 2011; 41(7):587–607. doi:10.2165/11588770-000000000-00000
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39(1):155–167. doi:10.1016/j.ecl.2009.11.002
- House S, Loud K, Shubkin C. Female athlete triad for the primary care pediatrician. Curr Opin Pediatr 2013; 25(6):755–761. doi:10.1097/MOP.0000000000000033
- Mallinson RJ, De Souza MJ. Current perspectives on the etiology and manifestation of the “silent” component of the female athlete triad. Int J Womens Health 2014; 6:451–467. doi:10.2147/IJWH.S38603
- Deimel JF, Dunlap BJ. The female athlete triad. Clin Sports Med 2012; 31(2):247–254. doi:10.1016/j.csm.2011.09.007
- Manore MM, Kam LC, Loucks AB; International Association of Athletics Federations. The female athlete triad: components, nutrition issues, and health consequences. J Sports Sci 2007; 25(suppl 1):S61–S71. doi:10.1080/02640410701607320
- Witkop CT, Warren MP. Understanding the spectrum of the female athlete triad. Obstet Gynecol 2010; 116(6):1444–1448. doi:10.1097/AOG.0b013e3181fbed40
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15(3):135–143. doi:10.1016/S1083-3188(02)00145-6
- Curry EJ, Logan C, Ackerman K, McInnis KC, Matzkin EG. Female athlete triad awareness among multispecialty physicians. Sports Med Open 2015; 1(1):38. doi:10.1186/s40798-015-0037-5
- National Federation of State High School Associations. 2012–13 high school athletics participation survey. http://old.nfhs.org/content.aspx?id=3282. Accessed February 1, 2018.
- Otis CL, Drinkwater B, Johnson M, Loucks A, Wilmore J. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 1997; 29(5):i–ix.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39(10):1867–1882. doi:10.1249/mss.0b013e318149f111
- Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med 2014; 48(7):491–497. doi:10.1136/bjsports-2014-093502
- Tenforde AS, Barrack MT, Nattiv A, Fredericson M. Parallels with the female athlete triad in male athletes. Sports Med 2016; 46(2):171–182. doi:10.1007/s40279-015-0411-y
- Thein-Nissenbaum JM, Carr KE. Female athlete triad syndrome in the high school athlete. Phys Ther Sport 2011; 12(3):108–116. doi:10.1016/j.ptsp.2011.04.002
- Matzkin E, Curry EJ, Whitlock K. Female athlete triad: past, present, and future. J Am Acad Orthop Surg 2015; 23(7):424–432. doi:10.5435/JAAOS-D-14-00168
- Gibbs JC, Williams NI, De Souza MJ. Prevalence of individual and combined components of the female athlete triad. Med Sci Sports Exerc 2013; 45(5):985–996. doi:10.1249/MSS.0b013e31827e1bdc
- Byrne S, McLean N. Elite athletes: effects of the pressure to be thin. J Sci Med Sport 2002; 5(2):80–94. doi:10.1016/S1440-2440(02)80029-9
- Sundgot-Borgen J, Torstveit MK. Prevalence of eating disorders in elite athletes is higher than in the general population. Clin J Sport Med 2004; 14(1):25–32.
- Lynch SL, Hoch AZ. The female runner: gender specifics. Clin Sports Med 2010; 29(3):477–498. doi:10.1016/j.csm.2010.03.003
- Doyle-Lucas AF, Akers JD, Davy BM. Energetic efficiency, menstrual irregularity, and bone mineral density in elite professional female ballet dancers. J Dance Med Sci 2010; 14(4):146–154.
- Thein-Nissenbaum J. Long term consequences of the female athlete triad. Maturitas 2013; 75(2):107–112. doi:10.1016/j.maturitas.2013.02.010
- Hilibrand MJ, Hammoud S, Bishop M, Woods D, Fredrick RW, Dodson CC. Common injuries and ailments of the female athlete; pathophysiology, treatment and prevention. Phys Sportsmed 2015; 43(4):403–411. doi:10.1080/00913847.2015.1092856
- Demorest RA, Hergenroeder AC. Preface. Sports medicine and sports injuries. Adolesc Med State Art Rev 2015; 26(1):xv–xvi.
- Khan KM, Liu-Ambrose T, Sran MM, Ashe MC, Donaldson MG, Wark JD. New criteria for female athlete triad syndrome? Br J Sports Med 2002; 36(1):10–13. doi:10.1136/bjsm.36.1.10
- Fontana R, Della Torre S. The deep correlation between energy metabolism and reproduction: a view of the effects of nutrition for women fertility. Nutrients 2016; 8:87. www.ncbi.nlm.nih.gov/pmc/articles/PMC4772050/. Accessed February 2, 2018.
- Nazem TG, Ackerman K. The female athlete triad. Sports Health 2012; 4(4):302–311. doi:10.1177/1941738112439685
- Pantano KJ. Coaching concerns in physically active girls and young women—part 1: the female athlete triad. Strength Conditioning J 2009; 31(6):38–43. doi:10.1519/SSC.0b013e3181c105dd
- Micklesfield LK, Hugo J, Johnson C, Noakes TD, Lambert EV. Factors associated with menstrual dysfunction and self-reported bone stress injuries in female runners in the ultra- and half-marathons of the Two Oceans. Br J Sports Med 2007; 41(10):679–683. doi:10.1136/bjsm.2007.037077
- Lambrinoudaki I, Papadimitriou D. Pathophysiology of bone loss in the female athlete. Ann N Y Acad Sci 2010; 1205:45–50. doi:10.1111/j.1749-6632.2010.05681.x
- De Souza MJ, Nattiv A, Joy E, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad: 1st International Conference held in San Francisco, CA, May 2012, and 2nd International Conference held in Indianapolis, IN, May 2013. Clin J Sport Med 2014; 24(2):96–119. doi:10.1136/bjsports-2013-093218
- Horn E, Gergen N, McGarry KA. The female athlete triad. R I Med J (2013) 2014; 97(11):18–21.
- Joy E, De Souza MJ, Nattiv A, et al. 2014 Female Athlete Triad Coalition consensus statement on treatment and return to play of the female athlete triad. Curr Sports Med Rep 2014; 13(4):219–232. doi:10.1249/JSR.0000000000000077
- Temme KE, Hoch AZ. Recognition and rehabilitation of the female athlete triad/tetrad: a multidisciplinary approach. Curr Sports Med Rep 2013; 12(3):190–199. doi:10.1249/JSR.0b013e318296190b
- Pritts SD, Susman J. Diagnosis of eating disorders in primary care. Am Fam Physician 2003; 67(2):297–304.
- Berhardt DR, Roberts WO, editors. Preparticipation Physical Evaluation, 4th Ed. American Academy of Pediatrics, Elk Grove Village, IL, 2010.
- Mencias T, Noon M, Hoch AZ. Female athlete triad screening in National Collegiate Athletic Association Division I athletes: is the preparticipation evaluation form effective? Clin J Sport Med 2012; 22(2):122–125. doi:10.1097/JSM.0b013e3182425aee
- Javed A, Tebben PJ, Fischer PR, Lteif AN. Female athlete triad and its components: toward improved screening and management. Mayo Clin Proc 2013; 88(9):996–1009. doi:10.1016/j.mayocp.2013.07.001
- Melin A, Tornberg AB, Skouby S, et al. Energy availability and the female athlete triad in elite endurance athletes. Scand J Med Sci Sports 2015; 25(5):610–622. doi:10.1111/sms.12261
- Warr BJ, Woolf K. The female athlete triad: patients do best with a team approach to care. JAAPA 2011; 24(4):50–55.
- Hoch AZ, Lal S, Jurva JW, Gutterman DD. The female athlete triad and cardiovascular dysfunction. Phys Med Rehabil Clin North Am 2007; 18(3):385–400. doi:10.1016/j.pmr.2007.05.001
- Lanser EM, Zach KN, Hoch AZ. The female athlete triad and endothelial dysfunction. PM R 2011; 3(5):458–465. doi:10.1016/j.pmrj.2010.12.024
- Thein-Nissenbaum JM, Rauh MJ, Carr KE, Loud KJ, McGuine TA. Associations between disordered eating, menstrual dysfunction, and musculoskeletal injury among high school athletes. J Orthop Sports Phys Ther 2011; 41(2):60–69. doi:10.2519/jospt.2011.3312
- Ducher G, Turner AI, Kukuljan S, et al. Obstacles in the optimization of bone health outcomes in the female athlete triad. Sports Med 2011; 41(7):587–607. doi:10.2165/11588770-000000000-00000
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39(1):155–167. doi:10.1016/j.ecl.2009.11.002
- House S, Loud K, Shubkin C. Female athlete triad for the primary care pediatrician. Curr Opin Pediatr 2013; 25(6):755–761. doi:10.1097/MOP.0000000000000033
- Mallinson RJ, De Souza MJ. Current perspectives on the etiology and manifestation of the “silent” component of the female athlete triad. Int J Womens Health 2014; 6:451–467. doi:10.2147/IJWH.S38603
- Deimel JF, Dunlap BJ. The female athlete triad. Clin Sports Med 2012; 31(2):247–254. doi:10.1016/j.csm.2011.09.007
- Manore MM, Kam LC, Loucks AB; International Association of Athletics Federations. The female athlete triad: components, nutrition issues, and health consequences. J Sports Sci 2007; 25(suppl 1):S61–S71. doi:10.1080/02640410701607320
- Witkop CT, Warren MP. Understanding the spectrum of the female athlete triad. Obstet Gynecol 2010; 116(6):1444–1448. doi:10.1097/AOG.0b013e3181fbed40
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15(3):135–143. doi:10.1016/S1083-3188(02)00145-6
KEY POINTS
- Low energy availability is the driving force of the triad, causing menstrual irregularity and subsequent low bone mineral density.
- Recognizing that men as well as women can suffer from energy deficiency and that it can affect more than the female reproductive system and skeleton, the International Olympic Committee has proposed calling the disorder relative energy deficiency in sport.
- Screening for the triad with a specific set of questions is recommended during the preparticipation assessment.
- Early intervention and treatment prevents serious health consequences including life-threatening arrhythmias, amenorrhea, and osteoporosis.

Aripiprazole, brexpiprazole, and cariprazine: Not all the same
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
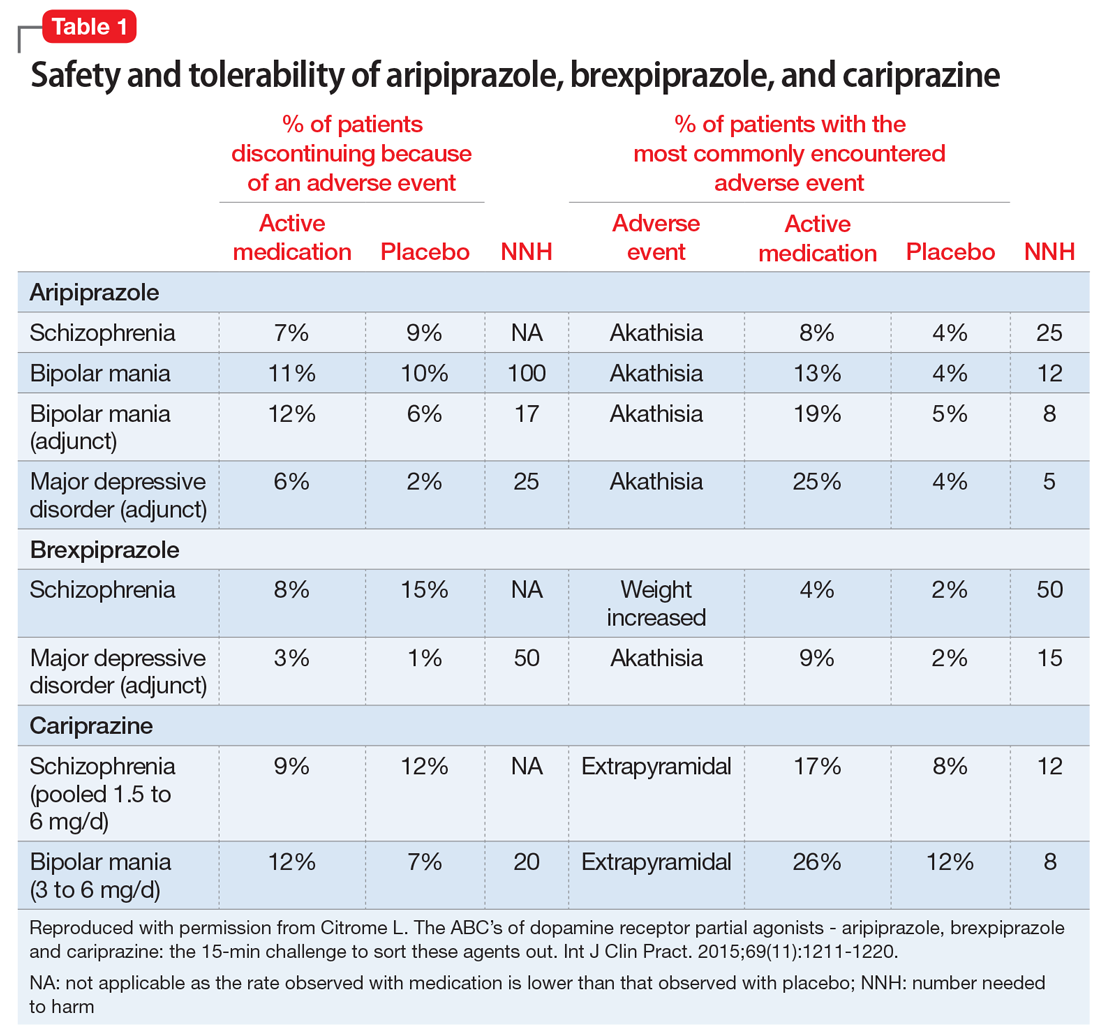
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.

Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
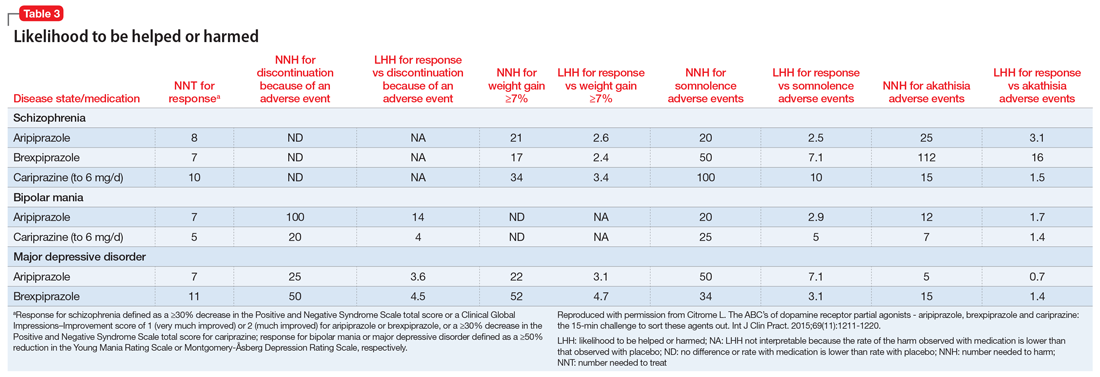
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.

Anxiety and joint hypermobility: An unexpected association
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.

The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
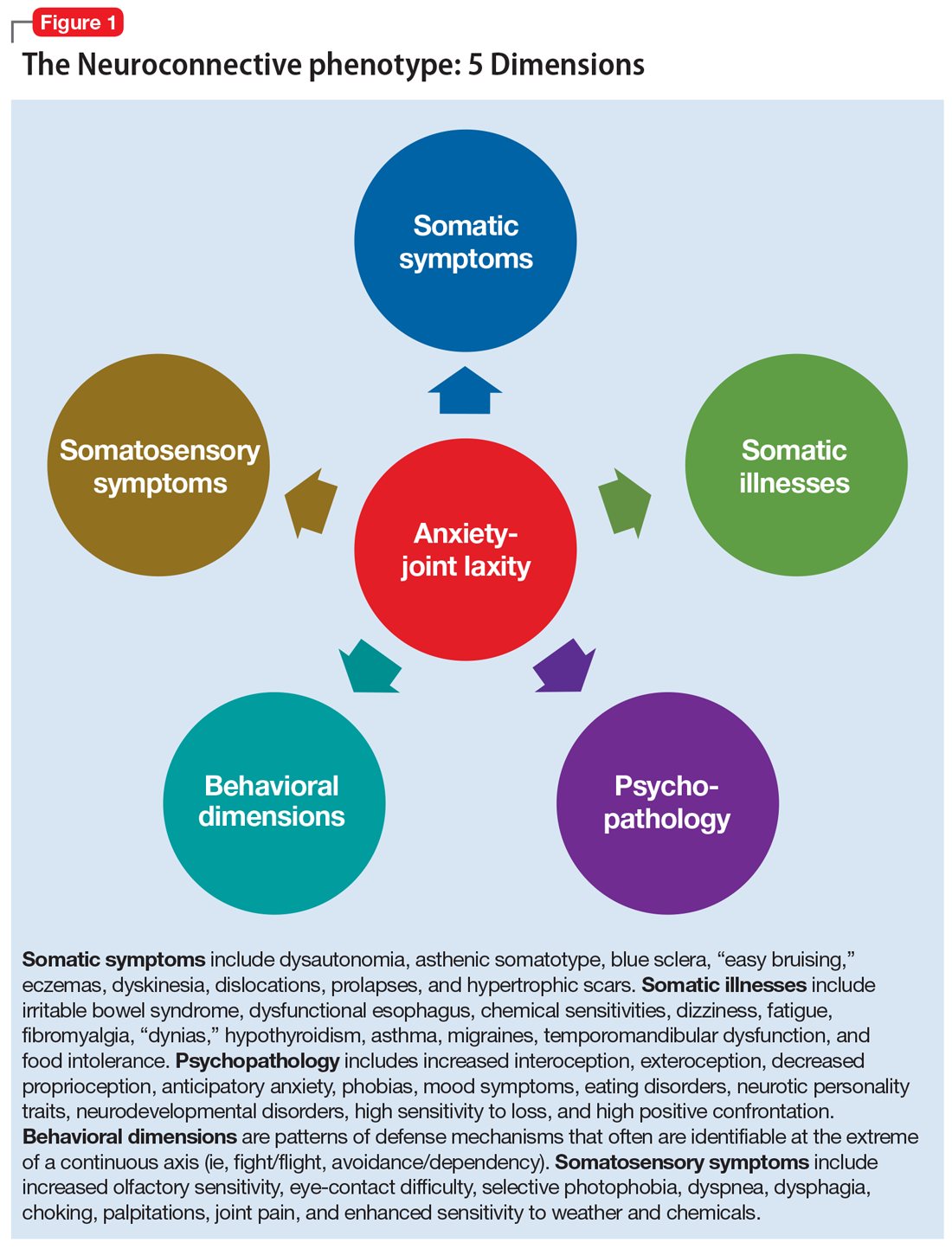
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
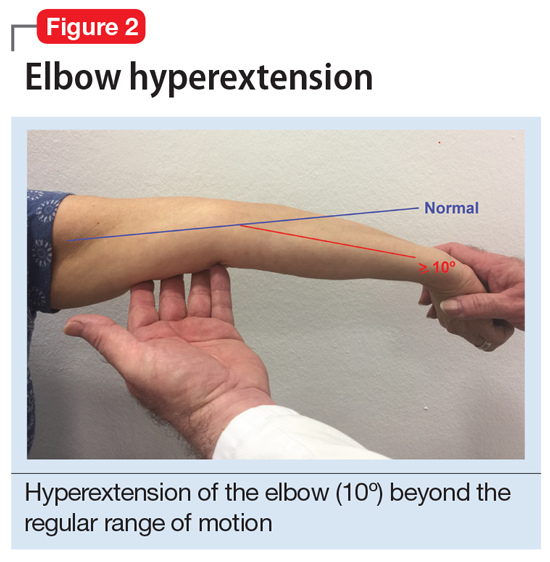
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
PTSD: A systematic approach to diagnosis and treatment
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.

Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
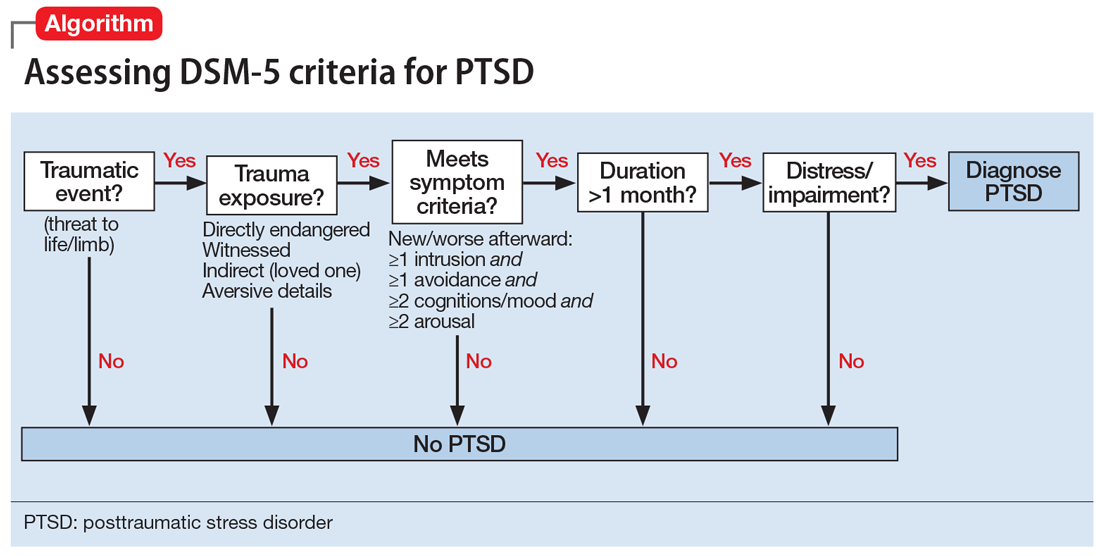
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
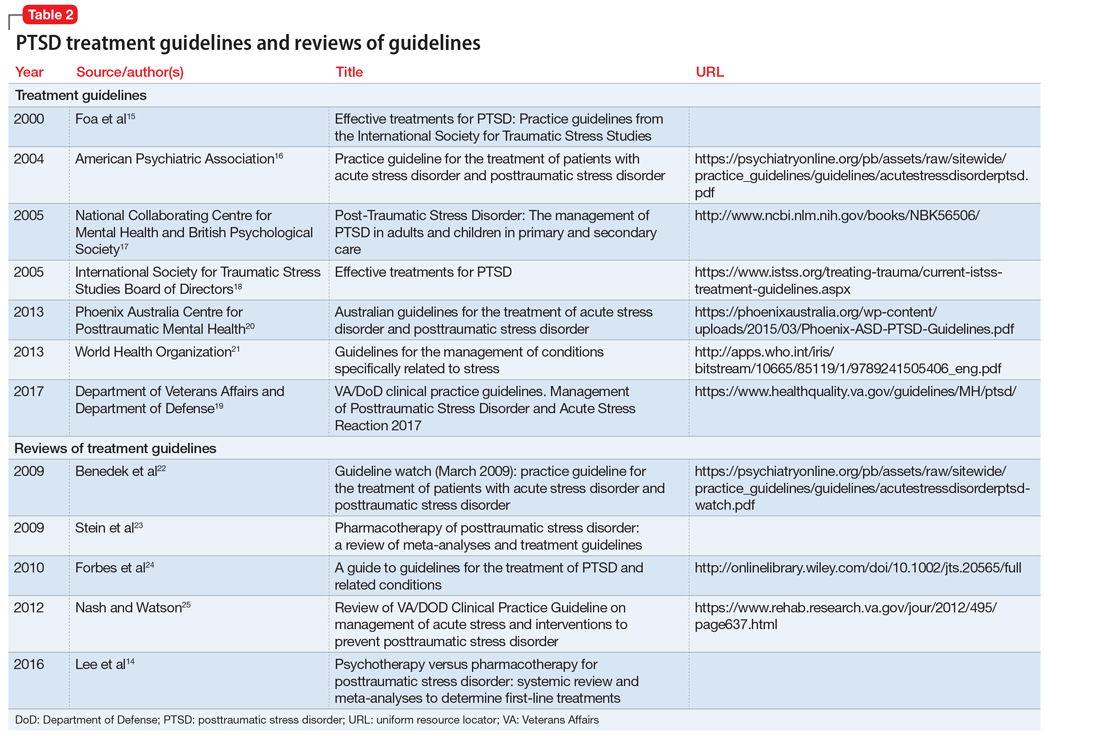
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.

Psychiatric consults: Documenting 6 essential elements
Written communication is an essential skill for a consultation-liaison (C-L) psychiatrist, but unfortunately, how to write a consultation note is not part of formal didactics in medical school or residency training.1 Documentation of a consultation note is a permanent medical record entry that conveys current physician-to-physician information. While considerable literature describes the consultation process, little has been published about composing a consultation note.1,2 Residents and clinicians who do not have frequent consultations may be unfamiliar with the consultation environment and their role as an expert consultant. Therefore, more explicit guidance on documentation and optimal formatting of the consultation note is needed.
The Box provides an outline for completing the Recommendations/Treatment Plan section of psychiatric consultation notes. When providing your recommendations, it is best to use bullet points, numbering, or bold text; do not bury the information in a dense paragraph.3 Be sure to address each of the following 6 elements.

1. Primary consult concern. The first section of the Recommendations section should include the reason for the consult, which may be the most important part of the consultation process.1,2 It is important to recognize that an unclear consult question may be a sign of the primary team’s knowledge gap in psychiatry. The role of the C-L psychiatrist is to help the primary team organize their thoughts and concerns regarding their patient to decide on the final consult question.1 The active consult question may change as clinical issues evolve.
2. Safety and critical issues. Include an assessment of or recommendation on safety and critical issues. An important consideration is whether to recommend a patient sitter and to provide a reason for this recommendation. Occasionally, critical issues are more pressing than the primary consult concern. For example, there are several situations in which abnormal laboratory values and acute medical issues manifest as psychiatric symptoms, including hyponatremia, hypoglycemia, hypotension, low oxygen saturation, or infection. The connection between the 2 may not be clear to the primary treatment team; thus, include a statement to draw their attention to this.
3. Nonpharmacologic recommendations.
4. Psychopharmacology. In this section, the C-L psychiatrist should provide information on the use of any psychotropic medications and an explanation of their indications. If there are discrepancies between a patient’s home and hospital-ordered medications, clarify which medications the patient should be taking while hospitalized. If the C-L treatment team recommends initiating a new medication, provide details regarding the specific medication, dose, route, administration time, and titration schedule, as well as the specific situation for any as-needed medications. It is important to include the indication for any recommended medications, as well as any potential adverse effects. If psychotropic medications are not indicated, add a statement to emphasize this.
5. Social work support. Document any issues that need to be clarified by social work. This might include clarification of a patient’s insurance coverage, current living situation, or durable power of attorney. Also, document how the treatment team would prefer social work to assist with the patient’s care by (for example) providing the patient with resources for outpatient mental health and/or substance abuse treatment or housing options.
Continue to: Disposition
6. Disposition. Finally, include a recommendation regarding disposition. If transfer to a psychiatric facility is not indicated, provide a statement to affirm this. If transfer to a psychiatric facility is recommended, include a discussion of the patient’s appropriateness in the assessment and recommendations. It is helpful to inform the primary team of criteria that may or may not allow the patient to transfer to or be accepted by a psychiatry unit (eg, the patient will need to be off IV medications and able to tolerate oral intake prior to transfer). When transfer is not possible, communicate the reason to the primary treatment team and other ancillary staff.
Communicating responsibilities and expectations
After concluding the Recommendations section, end the consultation note with a brief sentence of gratitude (eg, “Thank you for this consultation and allowing us to assist in the care of your patient.”) and a comment regarding the C-L treatment team’s plan for follow-up. Also, include your contact information in case the primary treatment team has any questions or concerns.
The Recommendations section of a psychiatric consultation note is vital to convey current physician-to-physician recommendations. With the potential complexities in assessing and caring for a medically ill patient with comorbid psychiatric diagnoses, psychiatrists with less C-L experience may be unfamiliar with the essential elements of a consultation note. It is helpful to use a Template to ensure that the consultation and documentation are complete.
1. Garrick TR, Stotland, NL. How to write a psychiatric consultation. Am J Psychiatry. 1982;139(7):849-855.
2. Alexander T, Bloch S. The written report in consultation-liaison psychiatry: a proposed schema. Aust N Z J Psychiatry. 2002;36(2):251-258.
3. von Gunten CF, Weissman DE. Writing the consultation note #267. J Palliat Med. 2013;16(5):579-580.
Written communication is an essential skill for a consultation-liaison (C-L) psychiatrist, but unfortunately, how to write a consultation note is not part of formal didactics in medical school or residency training.1 Documentation of a consultation note is a permanent medical record entry that conveys current physician-to-physician information. While considerable literature describes the consultation process, little has been published about composing a consultation note.1,2 Residents and clinicians who do not have frequent consultations may be unfamiliar with the consultation environment and their role as an expert consultant. Therefore, more explicit guidance on documentation and optimal formatting of the consultation note is needed.
The Box provides an outline for completing the Recommendations/Treatment Plan section of psychiatric consultation notes. When providing your recommendations, it is best to use bullet points, numbering, or bold text; do not bury the information in a dense paragraph.3 Be sure to address each of the following 6 elements.
1. Primary consult concern. The first section of the Recommendations section should include the reason for the consult, which may be the most important part of the consultation process.1,2 It is important to recognize that an unclear consult question may be a sign of the primary team’s knowledge gap in psychiatry. The role of the C-L psychiatrist is to help the primary team organize their thoughts and concerns regarding their patient to decide on the final consult question.1 The active consult question may change as clinical issues evolve.
2. Safety and critical issues. Include an assessment of or recommendation on safety and critical issues. An important consideration is whether to recommend a patient sitter and to provide a reason for this recommendation. Occasionally, critical issues are more pressing than the primary consult concern. For example, there are several situations in which abnormal laboratory values and acute medical issues manifest as psychiatric symptoms, including hyponatremia, hypoglycemia, hypotension, low oxygen saturation, or infection. The connection between the 2 may not be clear to the primary treatment team; thus, include a statement to draw their attention to this.
3. Nonpharmacologic recommendations.
4. Psychopharmacology. In this section, the C-L psychiatrist should provide information on the use of any psychotropic medications and an explanation of their indications. If there are discrepancies between a patient’s home and hospital-ordered medications, clarify which medications the patient should be taking while hospitalized. If the C-L treatment team recommends initiating a new medication, provide details regarding the specific medication, dose, route, administration time, and titration schedule, as well as the specific situation for any as-needed medications. It is important to include the indication for any recommended medications, as well as any potential adverse effects. If psychotropic medications are not indicated, add a statement to emphasize this.
5. Social work support. Document any issues that need to be clarified by social work. This might include clarification of a patient’s insurance coverage, current living situation, or durable power of attorney. Also, document how the treatment team would prefer social work to assist with the patient’s care by (for example) providing the patient with resources for outpatient mental health and/or substance abuse treatment or housing options.
Continue to: Disposition
6. Disposition. Finally, include a recommendation regarding disposition. If transfer to a psychiatric facility is not indicated, provide a statement to affirm this. If transfer to a psychiatric facility is recommended, include a discussion of the patient’s appropriateness in the assessment and recommendations. It is helpful to inform the primary team of criteria that may or may not allow the patient to transfer to or be accepted by a psychiatry unit (eg, the patient will need to be off IV medications and able to tolerate oral intake prior to transfer). When transfer is not possible, communicate the reason to the primary treatment team and other ancillary staff.
Communicating responsibilities and expectations
After concluding the Recommendations section, end the consultation note with a brief sentence of gratitude (eg, “Thank you for this consultation and allowing us to assist in the care of your patient.”) and a comment regarding the C-L treatment team’s plan for follow-up. Also, include your contact information in case the primary treatment team has any questions or concerns.
The Recommendations section of a psychiatric consultation note is vital to convey current physician-to-physician recommendations. With the potential complexities in assessing and caring for a medically ill patient with comorbid psychiatric diagnoses, psychiatrists with less C-L experience may be unfamiliar with the essential elements of a consultation note. It is helpful to use a Template to ensure that the consultation and documentation are complete.
Written communication is an essential skill for a consultation-liaison (C-L) psychiatrist, but unfortunately, how to write a consultation note is not part of formal didactics in medical school or residency training.1 Documentation of a consultation note is a permanent medical record entry that conveys current physician-to-physician information. While considerable literature describes the consultation process, little has been published about composing a consultation note.1,2 Residents and clinicians who do not have frequent consultations may be unfamiliar with the consultation environment and their role as an expert consultant. Therefore, more explicit guidance on documentation and optimal formatting of the consultation note is needed.
The Box provides an outline for completing the Recommendations/Treatment Plan section of psychiatric consultation notes. When providing your recommendations, it is best to use bullet points, numbering, or bold text; do not bury the information in a dense paragraph.3 Be sure to address each of the following 6 elements.
1. Primary consult concern. The first section of the Recommendations section should include the reason for the consult, which may be the most important part of the consultation process.1,2 It is important to recognize that an unclear consult question may be a sign of the primary team’s knowledge gap in psychiatry. The role of the C-L psychiatrist is to help the primary team organize their thoughts and concerns regarding their patient to decide on the final consult question.1 The active consult question may change as clinical issues evolve.
2. Safety and critical issues. Include an assessment of or recommendation on safety and critical issues. An important consideration is whether to recommend a patient sitter and to provide a reason for this recommendation. Occasionally, critical issues are more pressing than the primary consult concern. For example, there are several situations in which abnormal laboratory values and acute medical issues manifest as psychiatric symptoms, including hyponatremia, hypoglycemia, hypotension, low oxygen saturation, or infection. The connection between the 2 may not be clear to the primary treatment team; thus, include a statement to draw their attention to this.
3. Nonpharmacologic recommendations.
4. Psychopharmacology. In this section, the C-L psychiatrist should provide information on the use of any psychotropic medications and an explanation of their indications. If there are discrepancies between a patient’s home and hospital-ordered medications, clarify which medications the patient should be taking while hospitalized. If the C-L treatment team recommends initiating a new medication, provide details regarding the specific medication, dose, route, administration time, and titration schedule, as well as the specific situation for any as-needed medications. It is important to include the indication for any recommended medications, as well as any potential adverse effects. If psychotropic medications are not indicated, add a statement to emphasize this.
5. Social work support. Document any issues that need to be clarified by social work. This might include clarification of a patient’s insurance coverage, current living situation, or durable power of attorney. Also, document how the treatment team would prefer social work to assist with the patient’s care by (for example) providing the patient with resources for outpatient mental health and/or substance abuse treatment or housing options.
Continue to: Disposition
6. Disposition. Finally, include a recommendation regarding disposition. If transfer to a psychiatric facility is not indicated, provide a statement to affirm this. If transfer to a psychiatric facility is recommended, include a discussion of the patient’s appropriateness in the assessment and recommendations. It is helpful to inform the primary team of criteria that may or may not allow the patient to transfer to or be accepted by a psychiatry unit (eg, the patient will need to be off IV medications and able to tolerate oral intake prior to transfer). When transfer is not possible, communicate the reason to the primary treatment team and other ancillary staff.
Communicating responsibilities and expectations
After concluding the Recommendations section, end the consultation note with a brief sentence of gratitude (eg, “Thank you for this consultation and allowing us to assist in the care of your patient.”) and a comment regarding the C-L treatment team’s plan for follow-up. Also, include your contact information in case the primary treatment team has any questions or concerns.
The Recommendations section of a psychiatric consultation note is vital to convey current physician-to-physician recommendations. With the potential complexities in assessing and caring for a medically ill patient with comorbid psychiatric diagnoses, psychiatrists with less C-L experience may be unfamiliar with the essential elements of a consultation note. It is helpful to use a Template to ensure that the consultation and documentation are complete.
1. Garrick TR, Stotland, NL. How to write a psychiatric consultation. Am J Psychiatry. 1982;139(7):849-855.
2. Alexander T, Bloch S. The written report in consultation-liaison psychiatry: a proposed schema. Aust N Z J Psychiatry. 2002;36(2):251-258.
3. von Gunten CF, Weissman DE. Writing the consultation note #267. J Palliat Med. 2013;16(5):579-580.
1. Garrick TR, Stotland, NL. How to write a psychiatric consultation. Am J Psychiatry. 1982;139(7):849-855.
2. Alexander T, Bloch S. The written report in consultation-liaison psychiatry: a proposed schema. Aust N Z J Psychiatry. 2002;36(2):251-258.
3. von Gunten CF, Weissman DE. Writing the consultation note #267. J Palliat Med. 2013;16(5):579-580.
Tardive dyskinesia: 5 Steps for prevention
Tardive dyskinesia (TD) is an elusive-to-treat adverse effect of antipsychotics that has caused extreme discomfort (in a literal and figurative sense) for patients and their psychiatrists. In 2017, the prevalence of TD as a result of exposure to dopamine antagonists was approximately 30% with first-generation antipsychotics and 20% with second-generation antipsychotics.1 There have been several effective attempts at reducing rates of TD, including lowering the dosing, shifting to second-generation antipsychotics, and using recently introduced pharmacologic treatments for TD. The past 2 years have seen increased efforts at treating this often-irreversible adverse effect with pharmacotherapy, such as the recently marketed vesicular monoamine transporter-2 (VMAT2) inhibitors valbenazine and deutetrabenazine, as well as the supplement Ginkgo biloba,2 although issues with cost, adverse effects, or drug–drug interactions could limit the benefits of these agents.
Despite these strategies, one approach has been largely overlooked: prevention. Although it is included in many guidelines and literature reports, prevention has become less of a standard of practice and more of a cliché. Prevention is the key strategy for lowering the rate of TD, and it should be the assumed responsibility of each clinician in every prescription they write throughout the entire continuum of care. Here, we provide steps to take to help prevent TD, and what to consider when TD occurs.
1. Realize that we are all responsible for TD. We know TD exists, but we often feel that this adverse effect is not our fault. Avoid adapting a philosophy of “someone else caused it,” “they didn’t cause it yet,” or “it’s going to happen anyway.” We must remember that every unnecessary exposure to a dopamine antagonist increases the risk of TD, even if we don’t see the adverse effect firsthand.
2. Treat first-episode psychosis early and aggressively. Doing so may prevent chronicity of the illness, which would save a patient from long-term, high-dose exposure to antipsychotics. Lower the risk of TD with atypical antipsychotics and offer long-acting injectables when possible to improve medication adherence.
3. Treat both acute and chronic symptoms of psychosis throughout the continuum of care. The choice of medication and dose should be reevaluated at each interaction to enhance improvement of acute symptoms and to minimize chronic adverse effects. Always recognize the differences in aggressive treatment of an acute episode of psychosis vs maintenance treatment of baseline symptoms. Also, assess for TD by conducting abnormal involuntary movement scale (AIMS) examinations at baseline and at least biannually.
4. Use clozapine instead of 2 antipsychotics in chronic, refractory patients when possible. Clozapine is largely underutilize
5. Consider pharmacotherapy if TD has already occurred. Psychiatrists have been waiting for pharmacologic options for treating TD for quite some time. Explore using VMAT2 inhibitors and other agents when it is too late to implement prevention or when a patient’s symptoms are refractory to other treatments. However, avoid anticholinergic medications; there is insufficient data to support the use of these agents in the treatment of TD.5
1. Carbon M, Hsieh C, Kane J, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Zheng W, Xiang Y, Ng H, et al. Extract of ginkgo biloba for tardive dyskinesia: meta-analysis of randomized controlled trials. Pharmacopsychiatry. 2016;49(3):107-111.
3. Tiihonen J, Mittendorfer-Rutz E, Majak M, et al. Real-world effectiveness of antipsychotic treatments in a nationwide cohort of 29 823 patients with schizophrenia. JAMA Psychiatry. 2017;74(7):686-693.
4. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
5. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
Tardive dyskinesia (TD) is an elusive-to-treat adverse effect of antipsychotics that has caused extreme discomfort (in a literal and figurative sense) for patients and their psychiatrists. In 2017, the prevalence of TD as a result of exposure to dopamine antagonists was approximately 30% with first-generation antipsychotics and 20% with second-generation antipsychotics.1 There have been several effective attempts at reducing rates of TD, including lowering the dosing, shifting to second-generation antipsychotics, and using recently introduced pharmacologic treatments for TD. The past 2 years have seen increased efforts at treating this often-irreversible adverse effect with pharmacotherapy, such as the recently marketed vesicular monoamine transporter-2 (VMAT2) inhibitors valbenazine and deutetrabenazine, as well as the supplement Ginkgo biloba,2 although issues with cost, adverse effects, or drug–drug interactions could limit the benefits of these agents.
Despite these strategies, one approach has been largely overlooked: prevention. Although it is included in many guidelines and literature reports, prevention has become less of a standard of practice and more of a cliché. Prevention is the key strategy for lowering the rate of TD, and it should be the assumed responsibility of each clinician in every prescription they write throughout the entire continuum of care. Here, we provide steps to take to help prevent TD, and what to consider when TD occurs.
1. Realize that we are all responsible for TD. We know TD exists, but we often feel that this adverse effect is not our fault. Avoid adapting a philosophy of “someone else caused it,” “they didn’t cause it yet,” or “it’s going to happen anyway.” We must remember that every unnecessary exposure to a dopamine antagonist increases the risk of TD, even if we don’t see the adverse effect firsthand.
2. Treat first-episode psychosis early and aggressively. Doing so may prevent chronicity of the illness, which would save a patient from long-term, high-dose exposure to antipsychotics. Lower the risk of TD with atypical antipsychotics and offer long-acting injectables when possible to improve medication adherence.
3. Treat both acute and chronic symptoms of psychosis throughout the continuum of care. The choice of medication and dose should be reevaluated at each interaction to enhance improvement of acute symptoms and to minimize chronic adverse effects. Always recognize the differences in aggressive treatment of an acute episode of psychosis vs maintenance treatment of baseline symptoms. Also, assess for TD by conducting abnormal involuntary movement scale (AIMS) examinations at baseline and at least biannually.
4. Use clozapine instead of 2 antipsychotics in chronic, refractory patients when possible. Clozapine is largely underutilize
5. Consider pharmacotherapy if TD has already occurred. Psychiatrists have been waiting for pharmacologic options for treating TD for quite some time. Explore using VMAT2 inhibitors and other agents when it is too late to implement prevention or when a patient’s symptoms are refractory to other treatments. However, avoid anticholinergic medications; there is insufficient data to support the use of these agents in the treatment of TD.5
Tardive dyskinesia (TD) is an elusive-to-treat adverse effect of antipsychotics that has caused extreme discomfort (in a literal and figurative sense) for patients and their psychiatrists. In 2017, the prevalence of TD as a result of exposure to dopamine antagonists was approximately 30% with first-generation antipsychotics and 20% with second-generation antipsychotics.1 There have been several effective attempts at reducing rates of TD, including lowering the dosing, shifting to second-generation antipsychotics, and using recently introduced pharmacologic treatments for TD. The past 2 years have seen increased efforts at treating this often-irreversible adverse effect with pharmacotherapy, such as the recently marketed vesicular monoamine transporter-2 (VMAT2) inhibitors valbenazine and deutetrabenazine, as well as the supplement Ginkgo biloba,2 although issues with cost, adverse effects, or drug–drug interactions could limit the benefits of these agents.
Despite these strategies, one approach has been largely overlooked: prevention. Although it is included in many guidelines and literature reports, prevention has become less of a standard of practice and more of a cliché. Prevention is the key strategy for lowering the rate of TD, and it should be the assumed responsibility of each clinician in every prescription they write throughout the entire continuum of care. Here, we provide steps to take to help prevent TD, and what to consider when TD occurs.
1. Realize that we are all responsible for TD. We know TD exists, but we often feel that this adverse effect is not our fault. Avoid adapting a philosophy of “someone else caused it,” “they didn’t cause it yet,” or “it’s going to happen anyway.” We must remember that every unnecessary exposure to a dopamine antagonist increases the risk of TD, even if we don’t see the adverse effect firsthand.
2. Treat first-episode psychosis early and aggressively. Doing so may prevent chronicity of the illness, which would save a patient from long-term, high-dose exposure to antipsychotics. Lower the risk of TD with atypical antipsychotics and offer long-acting injectables when possible to improve medication adherence.
3. Treat both acute and chronic symptoms of psychosis throughout the continuum of care. The choice of medication and dose should be reevaluated at each interaction to enhance improvement of acute symptoms and to minimize chronic adverse effects. Always recognize the differences in aggressive treatment of an acute episode of psychosis vs maintenance treatment of baseline symptoms. Also, assess for TD by conducting abnormal involuntary movement scale (AIMS) examinations at baseline and at least biannually.
4. Use clozapine instead of 2 antipsychotics in chronic, refractory patients when possible. Clozapine is largely underutilize
5. Consider pharmacotherapy if TD has already occurred. Psychiatrists have been waiting for pharmacologic options for treating TD for quite some time. Explore using VMAT2 inhibitors and other agents when it is too late to implement prevention or when a patient’s symptoms are refractory to other treatments. However, avoid anticholinergic medications; there is insufficient data to support the use of these agents in the treatment of TD.5
1. Carbon M, Hsieh C, Kane J, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Zheng W, Xiang Y, Ng H, et al. Extract of ginkgo biloba for tardive dyskinesia: meta-analysis of randomized controlled trials. Pharmacopsychiatry. 2016;49(3):107-111.
3. Tiihonen J, Mittendorfer-Rutz E, Majak M, et al. Real-world effectiveness of antipsychotic treatments in a nationwide cohort of 29 823 patients with schizophrenia. JAMA Psychiatry. 2017;74(7):686-693.
4. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
5. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
1. Carbon M, Hsieh C, Kane J, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Zheng W, Xiang Y, Ng H, et al. Extract of ginkgo biloba for tardive dyskinesia: meta-analysis of randomized controlled trials. Pharmacopsychiatry. 2016;49(3):107-111.
3. Tiihonen J, Mittendorfer-Rutz E, Majak M, et al. Real-world effectiveness of antipsychotic treatments in a nationwide cohort of 29 823 patients with schizophrenia. JAMA Psychiatry. 2017;74(7):686-693.
4. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
5. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.