User login
The Role of Hospital Medicine in Emergency Preparedness: A Framework for Hospitalist Leadership in Disaster Preparedness, Response, and Recovery
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23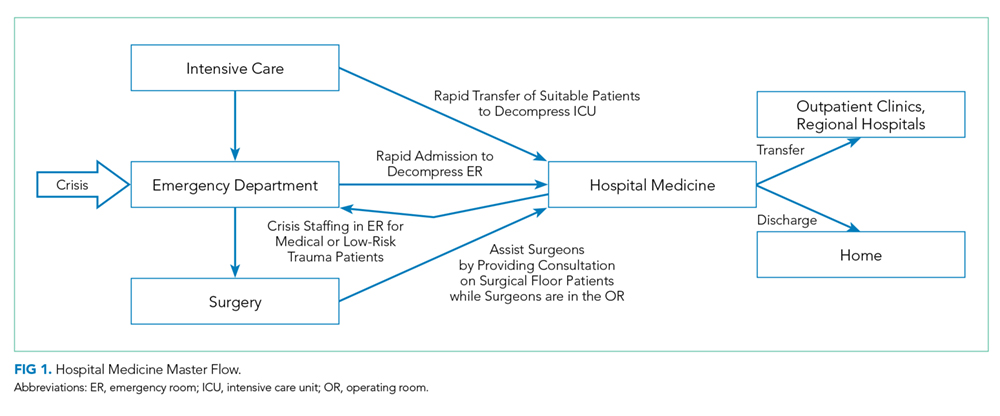
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders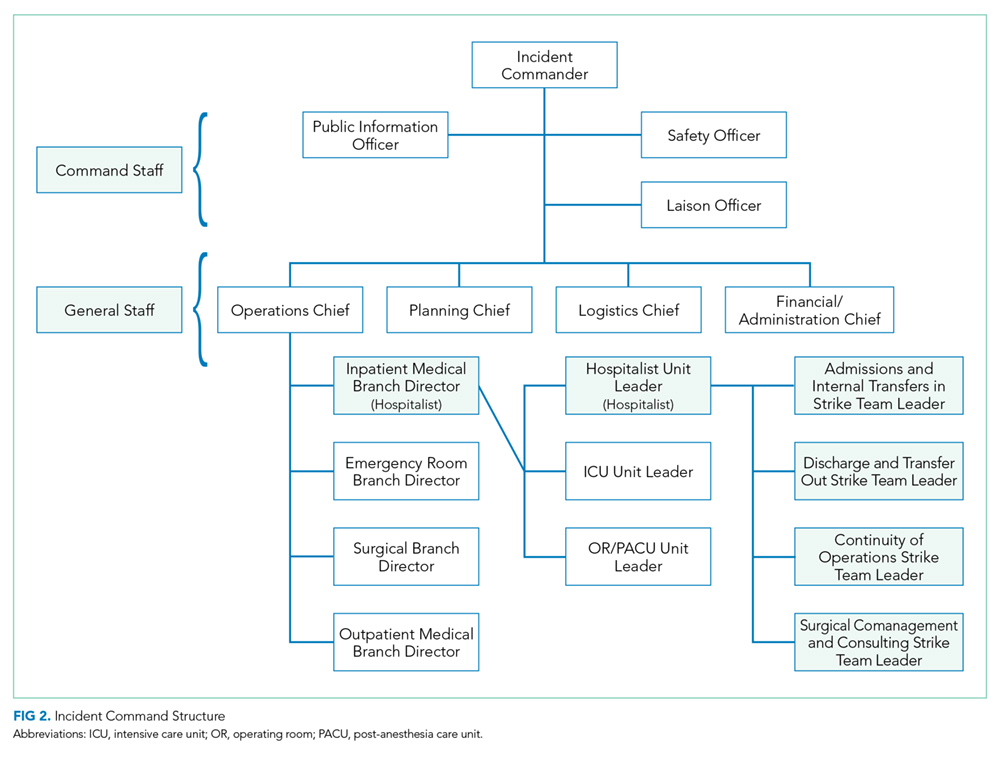
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
© 2018 Society of Hospital Medicine
Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
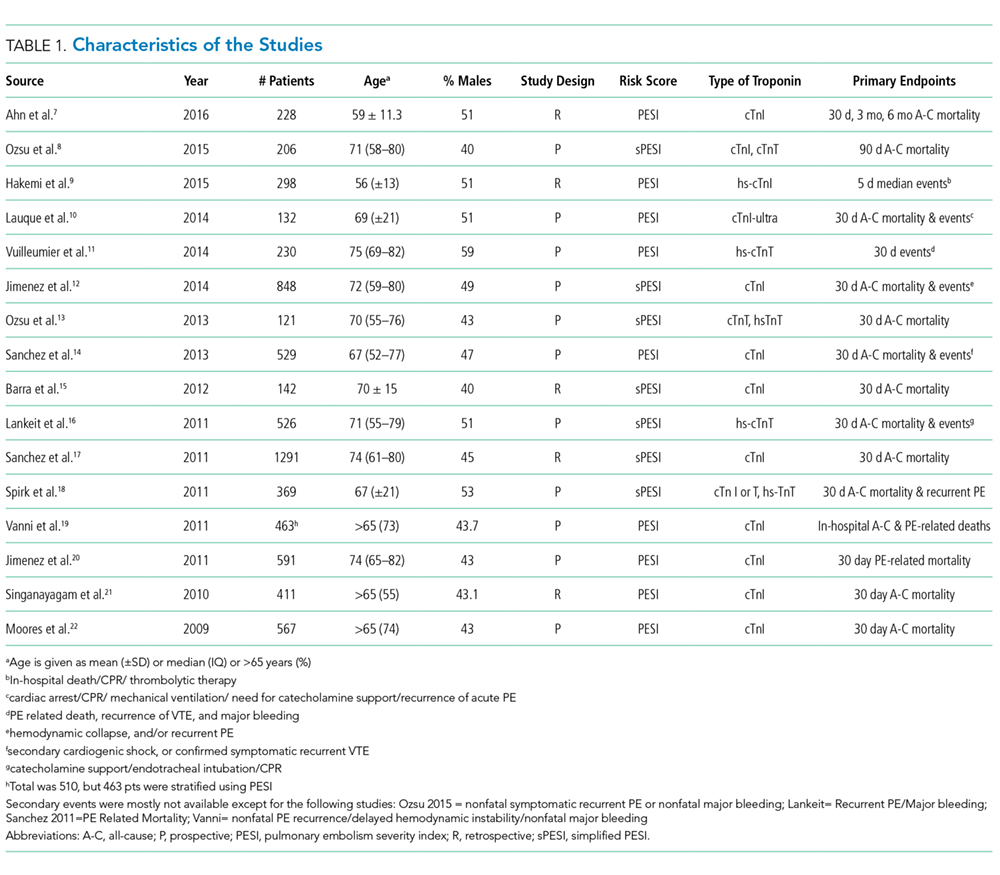
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
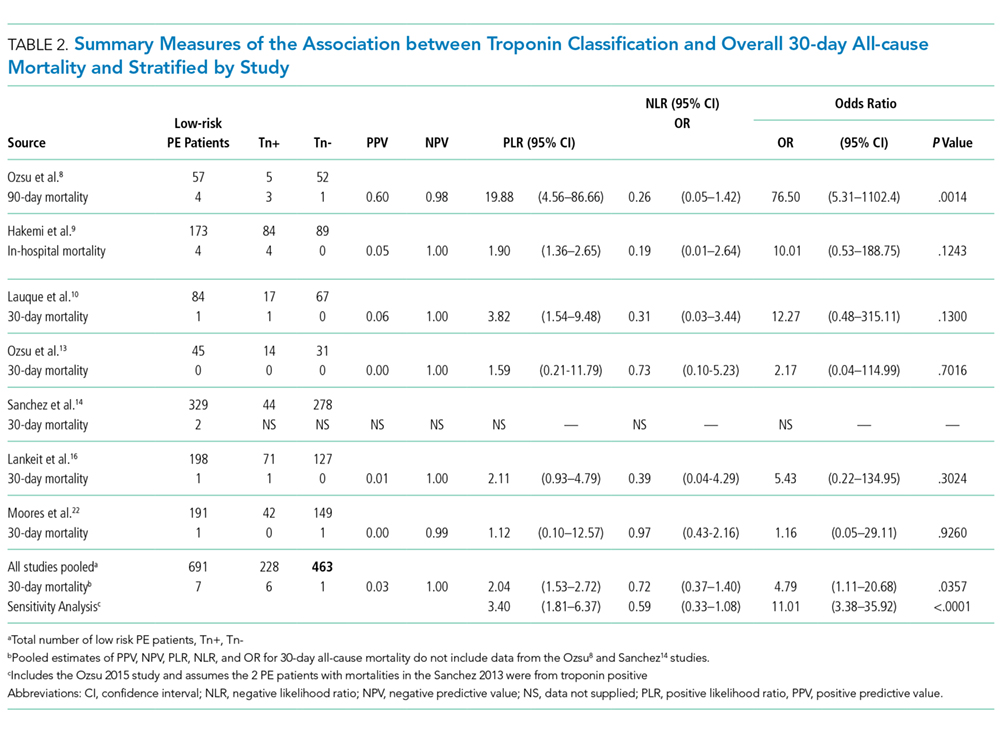
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).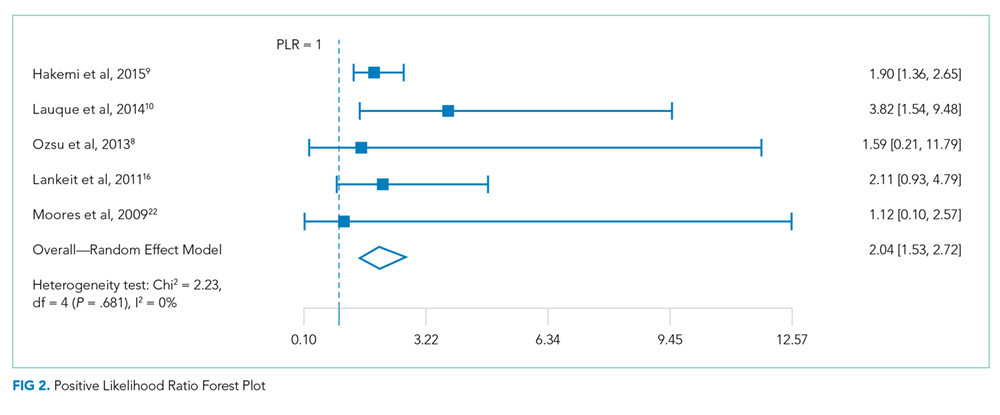
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
© 2018 Society of Hospital Medicine
Antipsychotics for patients with dementia: The road less traveled
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
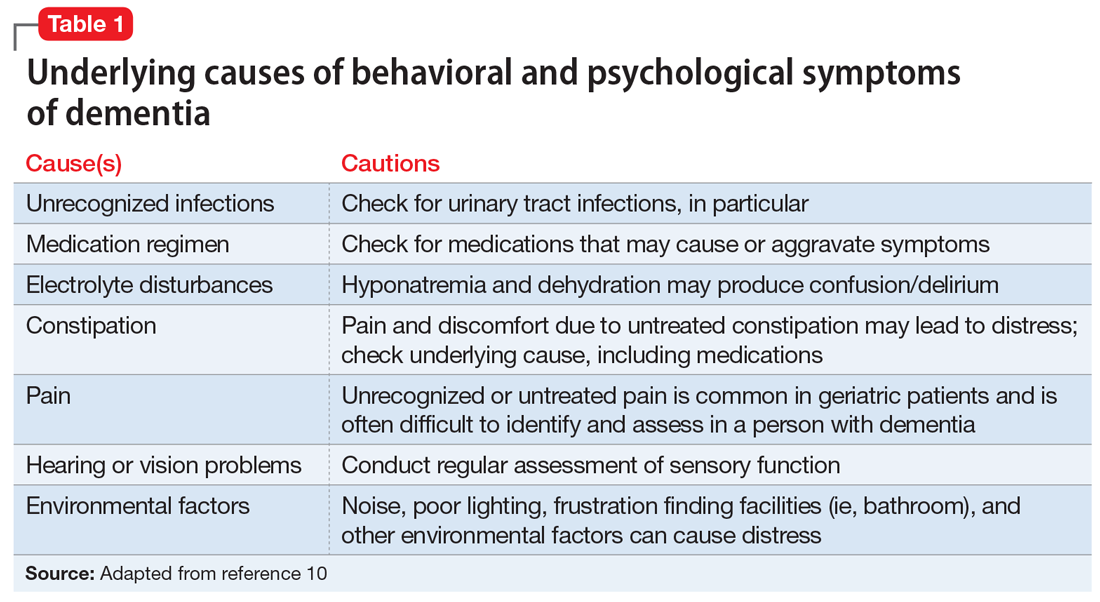
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.

Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10

Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing

When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.
Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10
Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing
When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.
Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10
Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing
When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.

5 Strategies for managing antipsychotic-induced hyperprolactinemia
There is a well-established relationship between antipsychotic treatment and hyperprolactinemia. Most antipsychotics have been linked to increased prolactin levels, and the risk appears to be dose-related.1 Antipsychotic-induced hyperprolactinemia can be asymptomatic, but it also has been associated with several adverse effects, including menstrual irregularity, osteoporosis, gynecomastia, and sexual dysfunction. Here I discuss what to do before starting a patient on an antipsychotic, and 5 treatment strategies for addressing antipsychotic-induced hyperprolactinemia.
Get a baseline prolactin level
Before starting a patient on an antipsychotic, obtain a baseline prolactin level measurement. If the patient later develops hyperprolactinemia, having a baseline measurement will make it easier to determine if the antipsychotic is a potential cause. Also, it is helpful to gather additional information regarding baseline psychosexual function and menstruation before starting an antipsychotic.

It is critical to determine if a temporal relationship exists between exposure to an antipsychotic and increase in prolactin levels.3 If the time course is unclear, laboratory tests need to be performed, including assessing liver, renal, and thyroid function or imaging of the pituitary gland. Also, hyperprolactinemia should not be diagnosed based on a single blood test result, because emotional and physical stress can elevate prolactin levels.
Continued to: 5 strategies for addressing hyperprolactinemia
5 strategies for addressing hyperprolactinemia
1. Reduce the antipsychotic dose. Because the risk of hyperprolactinemia is dose-dependent, reducing the antipsychotic dose could be helpful for some patients.
2. Switch to a prolactin-sparing antipsychotic, such as clozapine, quetiapine, olanzapine, or ziprasidone. However, it is often difficult to predict positive outcomes because switching antipsychotics may cause new adverse effects or trigger a psychotic relapse.
3. Consider sex hormone replacement therapy. A combined oral contraceptive could prevent osteoporosis and help estrogen deficiency symptoms in women who require antipsychotic medication. However, this treatment approach may worsen galactorrhea.
4. Use a dopamine receptor agonist. Dopamine receptor agonists, such as cabergoline or bromocriptine, have been shown to suppress prolactin secretion. Clinicians should always proceed cautiously because these medications can potentially increase the risk of psychosis.
5. Examine the potential benefits of adding aripiprazole because it can be used for augmentation to reduce prolactin levels in patients receiving other antipsychotics. In some cases, dopamine receptors can be exposed to competition between a partial agonist (aripiprazole) and an antagonist (the current antipsychotic). This competition may decrease the effectiveness of the current antipsychotic.1 Also, adding another antipsychotic could increase overall adverse effects.
1. Montejo ÁL, Arango C, Bernardo M, et al. Multidisciplinary consensus on the therapeutic recommendations for iatrogenic hyperprolactinemia secondary to antipsychotics. Front Neuroendocrinol. 2017;45:25-34.
2. Taylor D, Paton C, Kapur S. Schizophrenia. In: Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines in psychiatry. 12th ed. Chichester, UK: Wiley Blackwell; 2015:133-134.
3. Miyamoto BE, Galecki M, Francois D. Guidelines for antipsychotic-induced hyperprolactinemia. Psychiatr Ann. 2015;45(5):266,268,270-272.
There is a well-established relationship between antipsychotic treatment and hyperprolactinemia. Most antipsychotics have been linked to increased prolactin levels, and the risk appears to be dose-related.1 Antipsychotic-induced hyperprolactinemia can be asymptomatic, but it also has been associated with several adverse effects, including menstrual irregularity, osteoporosis, gynecomastia, and sexual dysfunction. Here I discuss what to do before starting a patient on an antipsychotic, and 5 treatment strategies for addressing antipsychotic-induced hyperprolactinemia.
Get a baseline prolactin level
Before starting a patient on an antipsychotic, obtain a baseline prolactin level measurement. If the patient later develops hyperprolactinemia, having a baseline measurement will make it easier to determine if the antipsychotic is a potential cause. Also, it is helpful to gather additional information regarding baseline psychosexual function and menstruation before starting an antipsychotic.
It is critical to determine if a temporal relationship exists between exposure to an antipsychotic and increase in prolactin levels.3 If the time course is unclear, laboratory tests need to be performed, including assessing liver, renal, and thyroid function or imaging of the pituitary gland. Also, hyperprolactinemia should not be diagnosed based on a single blood test result, because emotional and physical stress can elevate prolactin levels.
Continued to: 5 strategies for addressing hyperprolactinemia
5 strategies for addressing hyperprolactinemia
1. Reduce the antipsychotic dose. Because the risk of hyperprolactinemia is dose-dependent, reducing the antipsychotic dose could be helpful for some patients.
2. Switch to a prolactin-sparing antipsychotic, such as clozapine, quetiapine, olanzapine, or ziprasidone. However, it is often difficult to predict positive outcomes because switching antipsychotics may cause new adverse effects or trigger a psychotic relapse.
3. Consider sex hormone replacement therapy. A combined oral contraceptive could prevent osteoporosis and help estrogen deficiency symptoms in women who require antipsychotic medication. However, this treatment approach may worsen galactorrhea.
4. Use a dopamine receptor agonist. Dopamine receptor agonists, such as cabergoline or bromocriptine, have been shown to suppress prolactin secretion. Clinicians should always proceed cautiously because these medications can potentially increase the risk of psychosis.
5. Examine the potential benefits of adding aripiprazole because it can be used for augmentation to reduce prolactin levels in patients receiving other antipsychotics. In some cases, dopamine receptors can be exposed to competition between a partial agonist (aripiprazole) and an antagonist (the current antipsychotic). This competition may decrease the effectiveness of the current antipsychotic.1 Also, adding another antipsychotic could increase overall adverse effects.
There is a well-established relationship between antipsychotic treatment and hyperprolactinemia. Most antipsychotics have been linked to increased prolactin levels, and the risk appears to be dose-related.1 Antipsychotic-induced hyperprolactinemia can be asymptomatic, but it also has been associated with several adverse effects, including menstrual irregularity, osteoporosis, gynecomastia, and sexual dysfunction. Here I discuss what to do before starting a patient on an antipsychotic, and 5 treatment strategies for addressing antipsychotic-induced hyperprolactinemia.
Get a baseline prolactin level
Before starting a patient on an antipsychotic, obtain a baseline prolactin level measurement. If the patient later develops hyperprolactinemia, having a baseline measurement will make it easier to determine if the antipsychotic is a potential cause. Also, it is helpful to gather additional information regarding baseline psychosexual function and menstruation before starting an antipsychotic.
It is critical to determine if a temporal relationship exists between exposure to an antipsychotic and increase in prolactin levels.3 If the time course is unclear, laboratory tests need to be performed, including assessing liver, renal, and thyroid function or imaging of the pituitary gland. Also, hyperprolactinemia should not be diagnosed based on a single blood test result, because emotional and physical stress can elevate prolactin levels.
Continued to: 5 strategies for addressing hyperprolactinemia
5 strategies for addressing hyperprolactinemia
1. Reduce the antipsychotic dose. Because the risk of hyperprolactinemia is dose-dependent, reducing the antipsychotic dose could be helpful for some patients.
2. Switch to a prolactin-sparing antipsychotic, such as clozapine, quetiapine, olanzapine, or ziprasidone. However, it is often difficult to predict positive outcomes because switching antipsychotics may cause new adverse effects or trigger a psychotic relapse.
3. Consider sex hormone replacement therapy. A combined oral contraceptive could prevent osteoporosis and help estrogen deficiency symptoms in women who require antipsychotic medication. However, this treatment approach may worsen galactorrhea.
4. Use a dopamine receptor agonist. Dopamine receptor agonists, such as cabergoline or bromocriptine, have been shown to suppress prolactin secretion. Clinicians should always proceed cautiously because these medications can potentially increase the risk of psychosis.
5. Examine the potential benefits of adding aripiprazole because it can be used for augmentation to reduce prolactin levels in patients receiving other antipsychotics. In some cases, dopamine receptors can be exposed to competition between a partial agonist (aripiprazole) and an antagonist (the current antipsychotic). This competition may decrease the effectiveness of the current antipsychotic.1 Also, adding another antipsychotic could increase overall adverse effects.
1. Montejo ÁL, Arango C, Bernardo M, et al. Multidisciplinary consensus on the therapeutic recommendations for iatrogenic hyperprolactinemia secondary to antipsychotics. Front Neuroendocrinol. 2017;45:25-34.
2. Taylor D, Paton C, Kapur S. Schizophrenia. In: Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines in psychiatry. 12th ed. Chichester, UK: Wiley Blackwell; 2015:133-134.
3. Miyamoto BE, Galecki M, Francois D. Guidelines for antipsychotic-induced hyperprolactinemia. Psychiatr Ann. 2015;45(5):266,268,270-272.
1. Montejo ÁL, Arango C, Bernardo M, et al. Multidisciplinary consensus on the therapeutic recommendations for iatrogenic hyperprolactinemia secondary to antipsychotics. Front Neuroendocrinol. 2017;45:25-34.
2. Taylor D, Paton C, Kapur S. Schizophrenia. In: Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines in psychiatry. 12th ed. Chichester, UK: Wiley Blackwell; 2015:133-134.
3. Miyamoto BE, Galecki M, Francois D. Guidelines for antipsychotic-induced hyperprolactinemia. Psychiatr Ann. 2015;45(5):266,268,270-272.
Psychiatric considerations in menopause
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan

Continued to: Why menopause?
Why menopause?

Perimenopausal mood disorders

However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.

Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan
Continued to: Why menopause?
Why menopause?
Perimenopausal mood disorders
However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.
Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan
Continued to: Why menopause?
Why menopause?
Perimenopausal mood disorders
However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.
Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.

Caring for patients with autism spectrum disorder
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.

Vitamin B6 for tardive dyskinesia?
Although antipsychotics have revolutionized the treatment of severe mental illnesses, adverse effects often present a substantial obstacle to adherence. One of the most tenacious and difficult-to-treat adverse effects is tardive dyskinesia (TD), a neuromotor syndrome with characteristic involuntary repetitive movements, typically of the muscles of the jaw, lips, and tongue. In addition to spasms and grimacing, patients can have choreoathetoid movements of the neck. In more extreme presentations, some patients can have difficulty breathing. TD is a largely irreversible condition. It is often a disfiguring lifelong disability that can further stigmatize patients who already suffer scorn and derision. TD usually has a delayed onset after a patient is started on an antipsychotic.1 The syndrome is more commonly associated with first-generation antipsychotics, but affects up to 20% of patients who are treated with second-generation antipsychotics.1 In the United States, TD affects as many as 500,000 patients.1
There are several palliative interventions for TD, but the evidence for a consistently reliable treatment is weak. Branched-chain amino acids, ginkgo biloba, melatonin, and vitamin E have been investigated as interventions. Other approaches include switching to an alternate antipsychotic such as clozapine, adjusting the antipsychotic dose, using anticholinergic medications, adjunctive amantadine, gamma aminobutyric acid agonists, or adding tetrabenazine.
The FDA recently approved two vesicular monoamine transporter 2 (VMAT2) inhibitors, deutetrabenazine and valbenazine, for addressing symptoms of TD. However, these medications can cost tens of thousands of dollars per year, and also carry the risk of adverse effects such as sedation, akathisia, urinary retention, constipation, and muscle pain.2 When treating a patient who develops TD, one might consider other potentially effective therapies with low adverse effect profiles that may be more cost-effective than existing treatments. The bioactive form of vitamin B6 (pyridoxine), pyridoxal-5-phosphate, has been used to treat various antipsychotic-induced movement disorders. Preliminary evidence suggests that vitamin B6 may help reduce the symptoms of TD.
A recent Cochrane Database Review (2015)3 of pyridoxal-5-phosphate treatment for TD found a significant improvement in symptoms compared with placebo. Although the studies included in this review were limited by modest sample sizes and short follow-up periods, 2 of the investigations revealed improvements of >40% in extrapyramidal symptoms with vitamin B6 compared with placebo. Lerner et al (2001)4 conducted a randomized, double-blind, placebo-controlled crossover trial in which 15 inpatients with schizophrenia who met the criteria for TD were assigned to vitamin B6, 400 mg/d, or placebo for 4 weeks. After a 2-week washout period, the placebo group was given vitamin B6 and vice versa. Compared with placebo, mean scores on the parkinsonism and dyskinetic movement subscales of the Extrapyramidal Symptom Rating Scale were significantly better in the third week of treatment with vitamin B6.
Lerner et al (2007)5 later conducted a separate crossover study using the same design with a washout period. This trial included a larger sample size (50 inpatients with DSM-IV diagnoses of schizophrenia or schizoaffective disorder and TD) and the dosage of vitamin B6 was increased to 1,200 mg/d over 26 weeks. Patients who received vitamin B6 experienced a significantly greater decrease in Extrapyramidal Symptom Rating Scale scores compared with those in the placebo group.
Continued to: A 29-year-old woman with treatment-resistant schizophrenia...
Umar et al (2016)6 published a case review of a 29-year-old woman with treatment-resistant schizophrenia with TD who was treated with clozapine, 400 mg/d. She was started on vitamin B6, 450 mg/d, for 4 weeks, and then her dose was increased to 600 mg/d. At 6 months, she experienced a 78% reduction in the severity of her TD symptoms, as measured by the Abnormal Involuntary Movement Scale. The authors reported that this improvement was maintained for 1 year after vitamin B6 was stopped.
Miodownik et al (2008)7 reported in a study of 89 patients with schizophrenia that those with TD (n = 40) had diminished amounts of vitamin B6 in their plasma compared with patients without symptoms of motor disturbances (n = 49).
Vitamin B6 has been known to improve other psychotropic-induced movement disorders. In a study of lithium-induced tremors, treatment with pyridoxine, 900 to 1,200 mg/d, resulted in “impressive improvement until total disappearance of tremor.”8 Lerner et al (2004)9 also reported significant improvement for patients with neuroleptic-induced akathisia who were treated with vitamin B6.
Some proposed mechanisms of action
Pyridoxal-5-phosphate is a coenzyme in the synthesis of dopamine and other neurotransmitters. This might explain in part the biochemical mechanism of vitamin B6 in attenuating motor symptoms following long-term dopamine blockade. Chronic neurotransmitter antagonism may result in an upregulation of dopamine receptors in response. This compensatory reaction might create a dopamine receptor super-sensitivity in the nigrostriatal pathways.10
Another potential mechanism of action might be vitamin B6’s potent antioxidant properties and its scavenging of free radicals. The neurotoxicity of oxidative stress has been implicated in various movement disorders and psychiatric conditions.
In all of the studies described here, patients continued to receive daily antipsychotic treatment. In these trials, the adverse effects of vitamin B6 were minimal or negligible. In one study, vitamin B6 was reported to have had a better adverse effect profile than placebo.4
1. Carbon M, Hsieh CH, Kane JM, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Smith Mosley LL, Mosely II JF, Fleischfresser JR, et al. Vesicular monoamine transporter type 2 (VMAT2) inhibitors in the management of tardive dyskinesia. Clin Med Rev Case Rep. 2017;4(12):1-5.
3. Adelufosi AO, Abayomi O, Ojo M. Pyridoxal 5 phosphate for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2015;(4):CD010501.
4. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B(6) in the treatment of tardive dyskinesia: a double-blind, placebo-controlled, crossover study. Am J Psychiatry. 2001;158(9):1511-1514.
5. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B6 treatment for tardive dyskinesia: a randomized, double-blind, placebo-controlled, crossover study. J Clin Psychiatry. 2007;68(11):1648-1654.
6. Umar MU, Isa AA, Abba AH. High dose pyridoxine for the treatment of tardive dyskinesia: clinical case and review of literature. Ther Adv Psychopharmacol. 2016;6(2):152-156.
7. Miodownik C, Meoded A, Libov I, et al. Pyridoxal plasma level in schizophrenic and schizoaffective patients with and without tardive dyskinesia. Clin Neuropharmacol. 2008;31(4):197-203.
8. Miodownik C, Witztum E, Lerner V. Lithium-induced tremor treated with vitamin B6: a preliminary case series. Int J Psychiatry Med. 2002;32(1):103-108.
9. Lerner V, Bergman J, Statsenko N, et al. Vitamin B6 treatment in acute neuroleptic-induced akathisia: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2004;65(11):1550-1554.
10. Miller, BJ. Tardive dyskinesia: a review of the literature. Psychiatric Times. http://www.psychiatrictimes.com/articles/tardive-dyskinesia-review-literature. Published June 27, 2017. Accessed July 31, 2018.
Although antipsychotics have revolutionized the treatment of severe mental illnesses, adverse effects often present a substantial obstacle to adherence. One of the most tenacious and difficult-to-treat adverse effects is tardive dyskinesia (TD), a neuromotor syndrome with characteristic involuntary repetitive movements, typically of the muscles of the jaw, lips, and tongue. In addition to spasms and grimacing, patients can have choreoathetoid movements of the neck. In more extreme presentations, some patients can have difficulty breathing. TD is a largely irreversible condition. It is often a disfiguring lifelong disability that can further stigmatize patients who already suffer scorn and derision. TD usually has a delayed onset after a patient is started on an antipsychotic.1 The syndrome is more commonly associated with first-generation antipsychotics, but affects up to 20% of patients who are treated with second-generation antipsychotics.1 In the United States, TD affects as many as 500,000 patients.1
There are several palliative interventions for TD, but the evidence for a consistently reliable treatment is weak. Branched-chain amino acids, ginkgo biloba, melatonin, and vitamin E have been investigated as interventions. Other approaches include switching to an alternate antipsychotic such as clozapine, adjusting the antipsychotic dose, using anticholinergic medications, adjunctive amantadine, gamma aminobutyric acid agonists, or adding tetrabenazine.
The FDA recently approved two vesicular monoamine transporter 2 (VMAT2) inhibitors, deutetrabenazine and valbenazine, for addressing symptoms of TD. However, these medications can cost tens of thousands of dollars per year, and also carry the risk of adverse effects such as sedation, akathisia, urinary retention, constipation, and muscle pain.2 When treating a patient who develops TD, one might consider other potentially effective therapies with low adverse effect profiles that may be more cost-effective than existing treatments. The bioactive form of vitamin B6 (pyridoxine), pyridoxal-5-phosphate, has been used to treat various antipsychotic-induced movement disorders. Preliminary evidence suggests that vitamin B6 may help reduce the symptoms of TD.
A recent Cochrane Database Review (2015)3 of pyridoxal-5-phosphate treatment for TD found a significant improvement in symptoms compared with placebo. Although the studies included in this review were limited by modest sample sizes and short follow-up periods, 2 of the investigations revealed improvements of >40% in extrapyramidal symptoms with vitamin B6 compared with placebo. Lerner et al (2001)4 conducted a randomized, double-blind, placebo-controlled crossover trial in which 15 inpatients with schizophrenia who met the criteria for TD were assigned to vitamin B6, 400 mg/d, or placebo for 4 weeks. After a 2-week washout period, the placebo group was given vitamin B6 and vice versa. Compared with placebo, mean scores on the parkinsonism and dyskinetic movement subscales of the Extrapyramidal Symptom Rating Scale were significantly better in the third week of treatment with vitamin B6.
Lerner et al (2007)5 later conducted a separate crossover study using the same design with a washout period. This trial included a larger sample size (50 inpatients with DSM-IV diagnoses of schizophrenia or schizoaffective disorder and TD) and the dosage of vitamin B6 was increased to 1,200 mg/d over 26 weeks. Patients who received vitamin B6 experienced a significantly greater decrease in Extrapyramidal Symptom Rating Scale scores compared with those in the placebo group.
Continued to: A 29-year-old woman with treatment-resistant schizophrenia...
Umar et al (2016)6 published a case review of a 29-year-old woman with treatment-resistant schizophrenia with TD who was treated with clozapine, 400 mg/d. She was started on vitamin B6, 450 mg/d, for 4 weeks, and then her dose was increased to 600 mg/d. At 6 months, she experienced a 78% reduction in the severity of her TD symptoms, as measured by the Abnormal Involuntary Movement Scale. The authors reported that this improvement was maintained for 1 year after vitamin B6 was stopped.
Miodownik et al (2008)7 reported in a study of 89 patients with schizophrenia that those with TD (n = 40) had diminished amounts of vitamin B6 in their plasma compared with patients without symptoms of motor disturbances (n = 49).
Vitamin B6 has been known to improve other psychotropic-induced movement disorders. In a study of lithium-induced tremors, treatment with pyridoxine, 900 to 1,200 mg/d, resulted in “impressive improvement until total disappearance of tremor.”8 Lerner et al (2004)9 also reported significant improvement for patients with neuroleptic-induced akathisia who were treated with vitamin B6.
Some proposed mechanisms of action
Pyridoxal-5-phosphate is a coenzyme in the synthesis of dopamine and other neurotransmitters. This might explain in part the biochemical mechanism of vitamin B6 in attenuating motor symptoms following long-term dopamine blockade. Chronic neurotransmitter antagonism may result in an upregulation of dopamine receptors in response. This compensatory reaction might create a dopamine receptor super-sensitivity in the nigrostriatal pathways.10
Another potential mechanism of action might be vitamin B6’s potent antioxidant properties and its scavenging of free radicals. The neurotoxicity of oxidative stress has been implicated in various movement disorders and psychiatric conditions.
In all of the studies described here, patients continued to receive daily antipsychotic treatment. In these trials, the adverse effects of vitamin B6 were minimal or negligible. In one study, vitamin B6 was reported to have had a better adverse effect profile than placebo.4
Although antipsychotics have revolutionized the treatment of severe mental illnesses, adverse effects often present a substantial obstacle to adherence. One of the most tenacious and difficult-to-treat adverse effects is tardive dyskinesia (TD), a neuromotor syndrome with characteristic involuntary repetitive movements, typically of the muscles of the jaw, lips, and tongue. In addition to spasms and grimacing, patients can have choreoathetoid movements of the neck. In more extreme presentations, some patients can have difficulty breathing. TD is a largely irreversible condition. It is often a disfiguring lifelong disability that can further stigmatize patients who already suffer scorn and derision. TD usually has a delayed onset after a patient is started on an antipsychotic.1 The syndrome is more commonly associated with first-generation antipsychotics, but affects up to 20% of patients who are treated with second-generation antipsychotics.1 In the United States, TD affects as many as 500,000 patients.1
There are several palliative interventions for TD, but the evidence for a consistently reliable treatment is weak. Branched-chain amino acids, ginkgo biloba, melatonin, and vitamin E have been investigated as interventions. Other approaches include switching to an alternate antipsychotic such as clozapine, adjusting the antipsychotic dose, using anticholinergic medications, adjunctive amantadine, gamma aminobutyric acid agonists, or adding tetrabenazine.
The FDA recently approved two vesicular monoamine transporter 2 (VMAT2) inhibitors, deutetrabenazine and valbenazine, for addressing symptoms of TD. However, these medications can cost tens of thousands of dollars per year, and also carry the risk of adverse effects such as sedation, akathisia, urinary retention, constipation, and muscle pain.2 When treating a patient who develops TD, one might consider other potentially effective therapies with low adverse effect profiles that may be more cost-effective than existing treatments. The bioactive form of vitamin B6 (pyridoxine), pyridoxal-5-phosphate, has been used to treat various antipsychotic-induced movement disorders. Preliminary evidence suggests that vitamin B6 may help reduce the symptoms of TD.
A recent Cochrane Database Review (2015)3 of pyridoxal-5-phosphate treatment for TD found a significant improvement in symptoms compared with placebo. Although the studies included in this review were limited by modest sample sizes and short follow-up periods, 2 of the investigations revealed improvements of >40% in extrapyramidal symptoms with vitamin B6 compared with placebo. Lerner et al (2001)4 conducted a randomized, double-blind, placebo-controlled crossover trial in which 15 inpatients with schizophrenia who met the criteria for TD were assigned to vitamin B6, 400 mg/d, or placebo for 4 weeks. After a 2-week washout period, the placebo group was given vitamin B6 and vice versa. Compared with placebo, mean scores on the parkinsonism and dyskinetic movement subscales of the Extrapyramidal Symptom Rating Scale were significantly better in the third week of treatment with vitamin B6.
Lerner et al (2007)5 later conducted a separate crossover study using the same design with a washout period. This trial included a larger sample size (50 inpatients with DSM-IV diagnoses of schizophrenia or schizoaffective disorder and TD) and the dosage of vitamin B6 was increased to 1,200 mg/d over 26 weeks. Patients who received vitamin B6 experienced a significantly greater decrease in Extrapyramidal Symptom Rating Scale scores compared with those in the placebo group.
Continued to: A 29-year-old woman with treatment-resistant schizophrenia...
Umar et al (2016)6 published a case review of a 29-year-old woman with treatment-resistant schizophrenia with TD who was treated with clozapine, 400 mg/d. She was started on vitamin B6, 450 mg/d, for 4 weeks, and then her dose was increased to 600 mg/d. At 6 months, she experienced a 78% reduction in the severity of her TD symptoms, as measured by the Abnormal Involuntary Movement Scale. The authors reported that this improvement was maintained for 1 year after vitamin B6 was stopped.
Miodownik et al (2008)7 reported in a study of 89 patients with schizophrenia that those with TD (n = 40) had diminished amounts of vitamin B6 in their plasma compared with patients without symptoms of motor disturbances (n = 49).
Vitamin B6 has been known to improve other psychotropic-induced movement disorders. In a study of lithium-induced tremors, treatment with pyridoxine, 900 to 1,200 mg/d, resulted in “impressive improvement until total disappearance of tremor.”8 Lerner et al (2004)9 also reported significant improvement for patients with neuroleptic-induced akathisia who were treated with vitamin B6.
Some proposed mechanisms of action
Pyridoxal-5-phosphate is a coenzyme in the synthesis of dopamine and other neurotransmitters. This might explain in part the biochemical mechanism of vitamin B6 in attenuating motor symptoms following long-term dopamine blockade. Chronic neurotransmitter antagonism may result in an upregulation of dopamine receptors in response. This compensatory reaction might create a dopamine receptor super-sensitivity in the nigrostriatal pathways.10
Another potential mechanism of action might be vitamin B6’s potent antioxidant properties and its scavenging of free radicals. The neurotoxicity of oxidative stress has been implicated in various movement disorders and psychiatric conditions.
In all of the studies described here, patients continued to receive daily antipsychotic treatment. In these trials, the adverse effects of vitamin B6 were minimal or negligible. In one study, vitamin B6 was reported to have had a better adverse effect profile than placebo.4
1. Carbon M, Hsieh CH, Kane JM, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Smith Mosley LL, Mosely II JF, Fleischfresser JR, et al. Vesicular monoamine transporter type 2 (VMAT2) inhibitors in the management of tardive dyskinesia. Clin Med Rev Case Rep. 2017;4(12):1-5.
3. Adelufosi AO, Abayomi O, Ojo M. Pyridoxal 5 phosphate for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2015;(4):CD010501.
4. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B(6) in the treatment of tardive dyskinesia: a double-blind, placebo-controlled, crossover study. Am J Psychiatry. 2001;158(9):1511-1514.
5. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B6 treatment for tardive dyskinesia: a randomized, double-blind, placebo-controlled, crossover study. J Clin Psychiatry. 2007;68(11):1648-1654.
6. Umar MU, Isa AA, Abba AH. High dose pyridoxine for the treatment of tardive dyskinesia: clinical case and review of literature. Ther Adv Psychopharmacol. 2016;6(2):152-156.
7. Miodownik C, Meoded A, Libov I, et al. Pyridoxal plasma level in schizophrenic and schizoaffective patients with and without tardive dyskinesia. Clin Neuropharmacol. 2008;31(4):197-203.
8. Miodownik C, Witztum E, Lerner V. Lithium-induced tremor treated with vitamin B6: a preliminary case series. Int J Psychiatry Med. 2002;32(1):103-108.
9. Lerner V, Bergman J, Statsenko N, et al. Vitamin B6 treatment in acute neuroleptic-induced akathisia: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2004;65(11):1550-1554.
10. Miller, BJ. Tardive dyskinesia: a review of the literature. Psychiatric Times. http://www.psychiatrictimes.com/articles/tardive-dyskinesia-review-literature. Published June 27, 2017. Accessed July 31, 2018.
1. Carbon M, Hsieh CH, Kane JM, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Smith Mosley LL, Mosely II JF, Fleischfresser JR, et al. Vesicular monoamine transporter type 2 (VMAT2) inhibitors in the management of tardive dyskinesia. Clin Med Rev Case Rep. 2017;4(12):1-5.
3. Adelufosi AO, Abayomi O, Ojo M. Pyridoxal 5 phosphate for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2015;(4):CD010501.
4. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B(6) in the treatment of tardive dyskinesia: a double-blind, placebo-controlled, crossover study. Am J Psychiatry. 2001;158(9):1511-1514.
5. Lerner V, Miodownik C, Kapstan A, et al. Vitamin B6 treatment for tardive dyskinesia: a randomized, double-blind, placebo-controlled, crossover study. J Clin Psychiatry. 2007;68(11):1648-1654.
6. Umar MU, Isa AA, Abba AH. High dose pyridoxine for the treatment of tardive dyskinesia: clinical case and review of literature. Ther Adv Psychopharmacol. 2016;6(2):152-156.
7. Miodownik C, Meoded A, Libov I, et al. Pyridoxal plasma level in schizophrenic and schizoaffective patients with and without tardive dyskinesia. Clin Neuropharmacol. 2008;31(4):197-203.
8. Miodownik C, Witztum E, Lerner V. Lithium-induced tremor treated with vitamin B6: a preliminary case series. Int J Psychiatry Med. 2002;32(1):103-108.
9. Lerner V, Bergman J, Statsenko N, et al. Vitamin B6 treatment in acute neuroleptic-induced akathisia: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2004;65(11):1550-1554.
10. Miller, BJ. Tardive dyskinesia: a review of the literature. Psychiatric Times. http://www.psychiatrictimes.com/articles/tardive-dyskinesia-review-literature. Published June 27, 2017. Accessed July 31, 2018.
Data-driven prescribing
Computational psychiatry is an emerging field in which artificial intelligence and machine learning are used to find hidden patterns in big data to better understand, predict, and treat mental illness. The field uses various mathematical models to predict the dependent variable y based on the independent variable x. One application of analytics in medicine was the Framingham Heart Study, which used multivariate logistic regression to predict heart disease.1
Analytics could be used to predict the number of bad outcomes associated with different psychiatric medications over time. To demonstrate this, I examined a select data set of 8 psychiatric medications (aripiprazole, ziprasidone, risperidone, olanzapine, sertraline, trazodone, amitriptyline, and lithium) accounting for 59,827 bad outcomes during a 15-year period as reported by U.S. poison control centers,2 and plotted these on the y-axis.
When considering the independent variable to use as a predictor for bad outcomes, I used a composite index derived with the relative lethality (RL) equation, f(x) = 310x /LD50, where x is the daily dose of a medication prescribed for 30 days, and LD50 is the rat oral lethal dose 50.3 I plotted the RL of the 8 medications on the x-axis. Then I attempted to find a mathematical function that would best fit the x and y intersection points (Figure 1). I used the Excel data analysis pack to run a logarithmic regression model (Figure 2).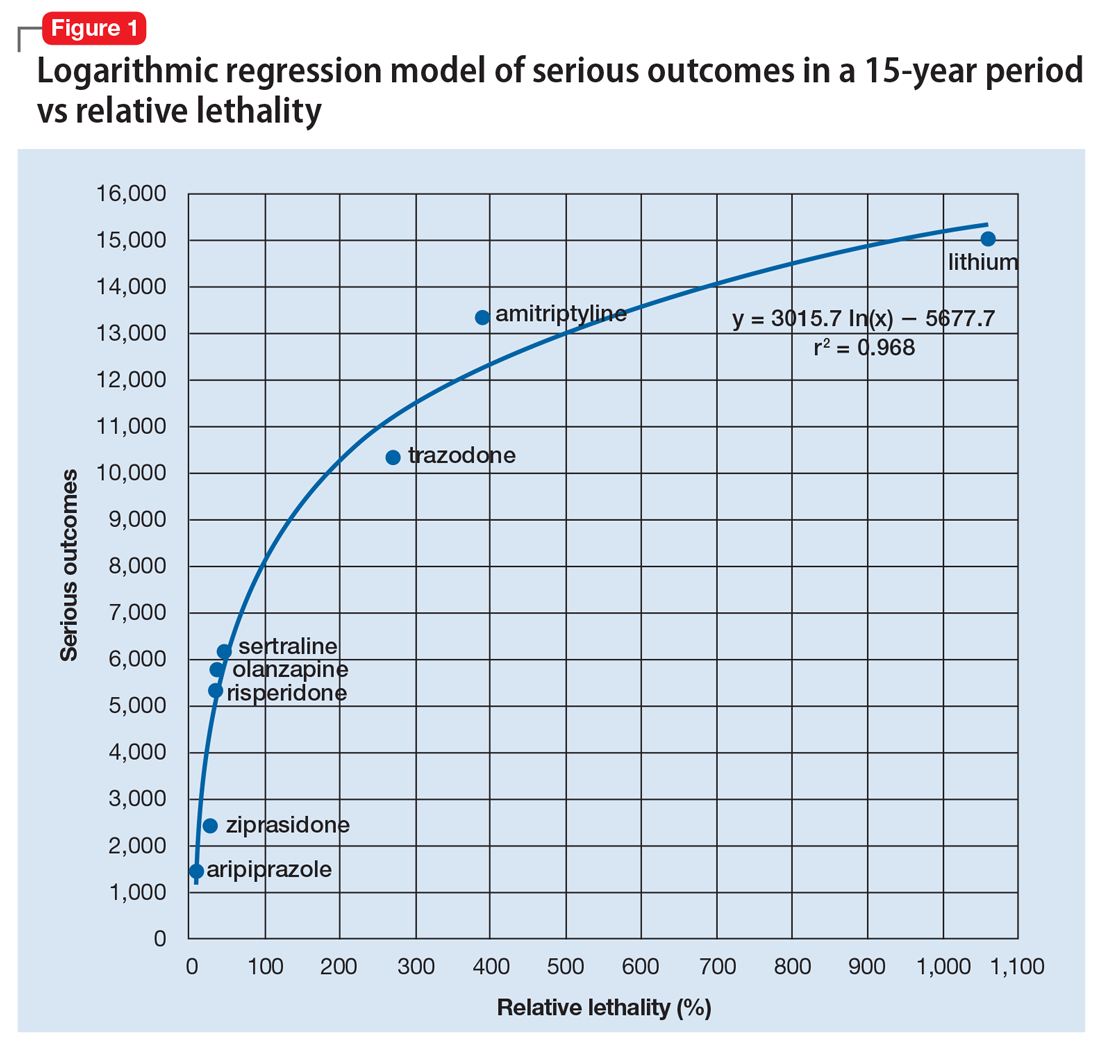
The model predicts that medications with a lower RL will have fewer serious outcomes, including mortality. The coefficient of determination r2 = 0.968, which indicates that 97% of the variation in serious outcomes is attributed to variation in RL, and 3% may be due to other factors, such as the poor quality of U.S. poison control data. This is a very significant correlation, and the causality is self-evident.
Continued to: The distribution of bad outcomes in the model was...
The distribution of bad outcomes in the model was: 1,446 for aripiprazole (RL = 9.76%), 2,387 for ziprasidone (RL = 24.80%), 5,352 for risperidone (RL = 32.63%), 5,798 for olanzapine (RL = 35.03%), 6,120 for sertraline (RL = 46.72%), 10,343 for trazodone (RL = 269.57%), 13,345 for amitriptyline (RL = 387.50%), and 15,036 for lithium (RL = 1,062.86%). The regression equation is: serious outcomes = –5,677.7 + 3,015.7 × ln (RL).
Some doctors may argue that such a data set is too small to make a meaningful model. However, the number of possible ways of ranking the drugs by bad outcomes is 8! = 40,320, so the probability of guessing the right sequence is P = .000024801. To appreciate how small this probability is, imagine trying to find a person of interest in half a football stadium on Superbowl Sunday.
The RL composite index correctly predicted the ranking order of serious outcomes for the 8 medications and may be useful for finding such outcomes in any drug class. For example, with angiotensin-converting enzyme inhibitors (n = 11) the number of possible combinations is 11! = 39,916,800. The probability of guessing the right sequence is like finding a person of interest in Poland. The model predicts the following decreasing sequence: 1) captopril, 2) fosinopril, 3) quinapril, 4) benazepril, 5) enalapril, 6) lisinopril, 7) moexipril, 8) perindopril, 9) cilazapril, 10) ramipril, 11) trandolapril. The predicted number of bad outcomes is highest for captopril, and lowest for trandolapril. The usefulness of the machine learning algorithm becomes immediately apparent.
Data can inform prescribing
Analytics can expose a critical flaw in the academic psychiatry paradigm for prescribing medications. For example, some doctors may regard lithium as the “gold standard” for treating certain mood disorders, but there is evidence that olanzapine is “significantly more effective than lithium in preventing recurrence of manic and mixed episodes.”4 Olanzapine is also 30 times safer than lithium based on its RL index, and had 9,238 fewer bad outcomes based on the 15-year data from U.S. poison control centers.2 A patient who intends to attempt suicide would easily be able to find the lethal dose of lithium from a “suicide” web site, and would quickly be able to figure out that the monthly amount of lithium his or her psychiatrist prescribed, would exceed the lethal dose.
When academia and reality collide, the use of analytics will have the final word by preventing suicide in the short term and reducing the number of bad outcomes in the long term. The irony of data science is that mathematical models can find optimal solutions to complex problems in a fraction of a second, but it may take years for a paradigm shift.
1. Bertsimas D, O’Hair AK, Pulleyblank WR. The analytics edge. Belmont, MA: Dynamic Ideas LLC; 2016.
2. Nelson JC, Spyker DA. Morbidity and mortality associated with medications used in the treatment of depression: an analysis of cases reported to U.S. poison control centers, 2000-2014. Am J Psychiatry. 2017;174(5):438-450.
3. Giurca D. Decreasing suicide risk with math. Current Psychiatry. 2018;17(2):57-59,A,B.
4. Tohen M, Greil W, Calabrese JR, et al. Olanzapine versus lithium in the maintenance treatment of bipolar disorder: a 12-month, randomized, double-blind, controlled clinical trial. Am J Psychiatry. 2005;162(7):1281-1290.
Computational psychiatry is an emerging field in which artificial intelligence and machine learning are used to find hidden patterns in big data to better understand, predict, and treat mental illness. The field uses various mathematical models to predict the dependent variable y based on the independent variable x. One application of analytics in medicine was the Framingham Heart Study, which used multivariate logistic regression to predict heart disease.1
Analytics could be used to predict the number of bad outcomes associated with different psychiatric medications over time. To demonstrate this, I examined a select data set of 8 psychiatric medications (aripiprazole, ziprasidone, risperidone, olanzapine, sertraline, trazodone, amitriptyline, and lithium) accounting for 59,827 bad outcomes during a 15-year period as reported by U.S. poison control centers,2 and plotted these on the y-axis.
When considering the independent variable to use as a predictor for bad outcomes, I used a composite index derived with the relative lethality (RL) equation, f(x) = 310x /LD50, where x is the daily dose of a medication prescribed for 30 days, and LD50 is the rat oral lethal dose 50.3 I plotted the RL of the 8 medications on the x-axis. Then I attempted to find a mathematical function that would best fit the x and y intersection points (Figure 1). I used the Excel data analysis pack to run a logarithmic regression model (Figure 2).
The model predicts that medications with a lower RL will have fewer serious outcomes, including mortality. The coefficient of determination r2 = 0.968, which indicates that 97% of the variation in serious outcomes is attributed to variation in RL, and 3% may be due to other factors, such as the poor quality of U.S. poison control data. This is a very significant correlation, and the causality is self-evident.
Continued to: The distribution of bad outcomes in the model was...
The distribution of bad outcomes in the model was: 1,446 for aripiprazole (RL = 9.76%), 2,387 for ziprasidone (RL = 24.80%), 5,352 for risperidone (RL = 32.63%), 5,798 for olanzapine (RL = 35.03%), 6,120 for sertraline (RL = 46.72%), 10,343 for trazodone (RL = 269.57%), 13,345 for amitriptyline (RL = 387.50%), and 15,036 for lithium (RL = 1,062.86%). The regression equation is: serious outcomes = –5,677.7 + 3,015.7 × ln (RL).
Some doctors may argue that such a data set is too small to make a meaningful model. However, the number of possible ways of ranking the drugs by bad outcomes is 8! = 40,320, so the probability of guessing the right sequence is P = .000024801. To appreciate how small this probability is, imagine trying to find a person of interest in half a football stadium on Superbowl Sunday.
The RL composite index correctly predicted the ranking order of serious outcomes for the 8 medications and may be useful for finding such outcomes in any drug class. For example, with angiotensin-converting enzyme inhibitors (n = 11) the number of possible combinations is 11! = 39,916,800. The probability of guessing the right sequence is like finding a person of interest in Poland. The model predicts the following decreasing sequence: 1) captopril, 2) fosinopril, 3) quinapril, 4) benazepril, 5) enalapril, 6) lisinopril, 7) moexipril, 8) perindopril, 9) cilazapril, 10) ramipril, 11) trandolapril. The predicted number of bad outcomes is highest for captopril, and lowest for trandolapril. The usefulness of the machine learning algorithm becomes immediately apparent.
Data can inform prescribing
Analytics can expose a critical flaw in the academic psychiatry paradigm for prescribing medications. For example, some doctors may regard lithium as the “gold standard” for treating certain mood disorders, but there is evidence that olanzapine is “significantly more effective than lithium in preventing recurrence of manic and mixed episodes.”4 Olanzapine is also 30 times safer than lithium based on its RL index, and had 9,238 fewer bad outcomes based on the 15-year data from U.S. poison control centers.2 A patient who intends to attempt suicide would easily be able to find the lethal dose of lithium from a “suicide” web site, and would quickly be able to figure out that the monthly amount of lithium his or her psychiatrist prescribed, would exceed the lethal dose.
When academia and reality collide, the use of analytics will have the final word by preventing suicide in the short term and reducing the number of bad outcomes in the long term. The irony of data science is that mathematical models can find optimal solutions to complex problems in a fraction of a second, but it may take years for a paradigm shift.
Computational psychiatry is an emerging field in which artificial intelligence and machine learning are used to find hidden patterns in big data to better understand, predict, and treat mental illness. The field uses various mathematical models to predict the dependent variable y based on the independent variable x. One application of analytics in medicine was the Framingham Heart Study, which used multivariate logistic regression to predict heart disease.1
Analytics could be used to predict the number of bad outcomes associated with different psychiatric medications over time. To demonstrate this, I examined a select data set of 8 psychiatric medications (aripiprazole, ziprasidone, risperidone, olanzapine, sertraline, trazodone, amitriptyline, and lithium) accounting for 59,827 bad outcomes during a 15-year period as reported by U.S. poison control centers,2 and plotted these on the y-axis.
When considering the independent variable to use as a predictor for bad outcomes, I used a composite index derived with the relative lethality (RL) equation, f(x) = 310x /LD50, where x is the daily dose of a medication prescribed for 30 days, and LD50 is the rat oral lethal dose 50.3 I plotted the RL of the 8 medications on the x-axis. Then I attempted to find a mathematical function that would best fit the x and y intersection points (Figure 1). I used the Excel data analysis pack to run a logarithmic regression model (Figure 2).
The model predicts that medications with a lower RL will have fewer serious outcomes, including mortality. The coefficient of determination r2 = 0.968, which indicates that 97% of the variation in serious outcomes is attributed to variation in RL, and 3% may be due to other factors, such as the poor quality of U.S. poison control data. This is a very significant correlation, and the causality is self-evident.
Continued to: The distribution of bad outcomes in the model was...
The distribution of bad outcomes in the model was: 1,446 for aripiprazole (RL = 9.76%), 2,387 for ziprasidone (RL = 24.80%), 5,352 for risperidone (RL = 32.63%), 5,798 for olanzapine (RL = 35.03%), 6,120 for sertraline (RL = 46.72%), 10,343 for trazodone (RL = 269.57%), 13,345 for amitriptyline (RL = 387.50%), and 15,036 for lithium (RL = 1,062.86%). The regression equation is: serious outcomes = –5,677.7 + 3,015.7 × ln (RL).
Some doctors may argue that such a data set is too small to make a meaningful model. However, the number of possible ways of ranking the drugs by bad outcomes is 8! = 40,320, so the probability of guessing the right sequence is P = .000024801. To appreciate how small this probability is, imagine trying to find a person of interest in half a football stadium on Superbowl Sunday.
The RL composite index correctly predicted the ranking order of serious outcomes for the 8 medications and may be useful for finding such outcomes in any drug class. For example, with angiotensin-converting enzyme inhibitors (n = 11) the number of possible combinations is 11! = 39,916,800. The probability of guessing the right sequence is like finding a person of interest in Poland. The model predicts the following decreasing sequence: 1) captopril, 2) fosinopril, 3) quinapril, 4) benazepril, 5) enalapril, 6) lisinopril, 7) moexipril, 8) perindopril, 9) cilazapril, 10) ramipril, 11) trandolapril. The predicted number of bad outcomes is highest for captopril, and lowest for trandolapril. The usefulness of the machine learning algorithm becomes immediately apparent.
Data can inform prescribing
Analytics can expose a critical flaw in the academic psychiatry paradigm for prescribing medications. For example, some doctors may regard lithium as the “gold standard” for treating certain mood disorders, but there is evidence that olanzapine is “significantly more effective than lithium in preventing recurrence of manic and mixed episodes.”4 Olanzapine is also 30 times safer than lithium based on its RL index, and had 9,238 fewer bad outcomes based on the 15-year data from U.S. poison control centers.2 A patient who intends to attempt suicide would easily be able to find the lethal dose of lithium from a “suicide” web site, and would quickly be able to figure out that the monthly amount of lithium his or her psychiatrist prescribed, would exceed the lethal dose.
When academia and reality collide, the use of analytics will have the final word by preventing suicide in the short term and reducing the number of bad outcomes in the long term. The irony of data science is that mathematical models can find optimal solutions to complex problems in a fraction of a second, but it may take years for a paradigm shift.
1. Bertsimas D, O’Hair AK, Pulleyblank WR. The analytics edge. Belmont, MA: Dynamic Ideas LLC; 2016.
2. Nelson JC, Spyker DA. Morbidity and mortality associated with medications used in the treatment of depression: an analysis of cases reported to U.S. poison control centers, 2000-2014. Am J Psychiatry. 2017;174(5):438-450.
3. Giurca D. Decreasing suicide risk with math. Current Psychiatry. 2018;17(2):57-59,A,B.
4. Tohen M, Greil W, Calabrese JR, et al. Olanzapine versus lithium in the maintenance treatment of bipolar disorder: a 12-month, randomized, double-blind, controlled clinical trial. Am J Psychiatry. 2005;162(7):1281-1290.
1. Bertsimas D, O’Hair AK, Pulleyblank WR. The analytics edge. Belmont, MA: Dynamic Ideas LLC; 2016.
2. Nelson JC, Spyker DA. Morbidity and mortality associated with medications used in the treatment of depression: an analysis of cases reported to U.S. poison control centers, 2000-2014. Am J Psychiatry. 2017;174(5):438-450.
3. Giurca D. Decreasing suicide risk with math. Current Psychiatry. 2018;17(2):57-59,A,B.
4. Tohen M, Greil W, Calabrese JR, et al. Olanzapine versus lithium in the maintenance treatment of bipolar disorder: a 12-month, randomized, double-blind, controlled clinical trial. Am J Psychiatry. 2005;162(7):1281-1290.
The benzodiazepine dilemma
As clinicians, we are faced with a conflict when deciding whether or not to prescribe a benzodiazepine. If we prescribe one of these agents, we might be putting our patients at risk for dependence and abuse. However, if we do not prescribe them, we risk providing inadequate treatment, especially for patients with panic disorder.
Benzodiazepine dependence and abuse can take many forms. Dependence can be psychological as well as physiologic. While many patients will adhere to their prescribing regimen, some may sell their benzodiazepines, falsely claim that they have “panic attacks,” or take a fatal overdose of an opioid and benzodiazepine combination.
Here I discuss the pros and cons of restricting benzodiazepines use to low doses and/or combination therapy with antidepressants.
_
Weighing the benefits of restricted prescribing
Some double-blind studies referenced in the American Psychiatric Association (APA) 2010 Practice Guideline for the Treatment of Patients with Panic Disorder1 suggest that benzodiazepine duration of treatment and dosages should be severely restricted. These studies found that:
- Although the combination of a selective serotonin reuptake inhibitor (SSRI) and a benzodiazepine initially decreased the number of panic attacks more quickly than SSRI monotherapy, the 2 treatments are equally effective after 4 or 5 weeks.2,3
- For the treatment of panic disorder, a low dosage of a benzodiazepine (clonazepam 1 mg/d or alprazolam 2 mg/d) was as effective as a higher dosage (clonazepam 2 mg/d or alprazolam 6 mg/d).4,5
However, these studies could be misleading. They all excluded patients with a comorbid condition, such as bipolar disorder or depression, that was more severe than their panic disorder. Severe comorbidity is associated with more severe panic symptoms,6,7 which might require an SSRI/benzodiazepine combination or a higher benzodiazepine dosage.
The APA Practice Guideline suggests the following possible options:
- benzodiazepine augmentation if there is a partial response to an SSRI
- substitution with a different SSRI or a serotonin-norepinephrine reuptake inhibitor (SNRI) if there is no response to an SSRI
- benzodiazepine augmentation or substitution if there is still no therapeutic response.
Continue to: The APA Practice Guideline also states...
The APA Practice Guideline also states that although the highest “usual therapeutic dose” for panic disorder is clonazepam 2 mg/d or alprazolam 4 mg/d, “higher doses are sometimes used for patients who do not respond to the usual therapeutic dose.”1
Presumably, an SSRI/benzodiazepine combination should be considered if an SSRI alleviates major depressive disorder but does not alleviate a comorbid panic disorder. However, the APA Practice Guideline does not include studies that investigated this clinical scenario.
Monitor carefully for dependency/abuse
Restricting benzodiazepine use to low doses over a short period of time may decrease the risk of dependence and abuse. However, this practice may also limit or prevent effective treatment for adherent patients with panic disorder who do not adequately respond to SSRI or SNRI monotherapy.
Therefore, clinicians need to carefully differentiate between patients who are adherent to their prescribed dosages and those who may be at risk for benzodiazepine dependence and abuse. Consider using prescription drug monitoring programs and drug screens to help detect patients who “doctor shop” for benzodiazepines, or who could be abusing opioids, alcohol, marijuana, or other substances while taking a benzodiazepine.
1. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, 2nd edition. Washington DC: American Psychiatric Association. 2010. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed March 7, 2018.
2. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
3. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
4. Lydiard RB, Lesser IM, Ballenger JC, et al. A fixed-dose study of alprazolam 2 mg, alprazolam 6 mg, and placebo in panic disorder. J Clin Psychopharmacol. 1992;12(2):966-103.
5. Rosenbaum JF, Moroz G, Bowden CL. Clonazepam in the treatment of panic disorder with or without agoraphobia: a dose-response study of efficacy, safety, and discontinuance. Clonazepam Panic Disorder Dose-Response Study Group. J Clin Psychopharmacol. 1997;17(5):390-400.
6. Goodwin RD, Hoven CW. Bipolar-panic comorbidity in the general population: prevalence and associated morbidity. J Affect Disord. 2002;70(1):27-33.
7. Roy-Byrne PP, Stang P, Wittchen HU, et al. Lifetime panic-depression comorbidity in the National Comorbidity Survey. Association with symptoms, impairment, course and help-seeking. Br J Psychiatry. 2000;176:229-235.
As clinicians, we are faced with a conflict when deciding whether or not to prescribe a benzodiazepine. If we prescribe one of these agents, we might be putting our patients at risk for dependence and abuse. However, if we do not prescribe them, we risk providing inadequate treatment, especially for patients with panic disorder.
Benzodiazepine dependence and abuse can take many forms. Dependence can be psychological as well as physiologic. While many patients will adhere to their prescribing regimen, some may sell their benzodiazepines, falsely claim that they have “panic attacks,” or take a fatal overdose of an opioid and benzodiazepine combination.
Here I discuss the pros and cons of restricting benzodiazepines use to low doses and/or combination therapy with antidepressants.
_
Weighing the benefits of restricted prescribing
Some double-blind studies referenced in the American Psychiatric Association (APA) 2010 Practice Guideline for the Treatment of Patients with Panic Disorder1 suggest that benzodiazepine duration of treatment and dosages should be severely restricted. These studies found that:
- Although the combination of a selective serotonin reuptake inhibitor (SSRI) and a benzodiazepine initially decreased the number of panic attacks more quickly than SSRI monotherapy, the 2 treatments are equally effective after 4 or 5 weeks.2,3
- For the treatment of panic disorder, a low dosage of a benzodiazepine (clonazepam 1 mg/d or alprazolam 2 mg/d) was as effective as a higher dosage (clonazepam 2 mg/d or alprazolam 6 mg/d).4,5
However, these studies could be misleading. They all excluded patients with a comorbid condition, such as bipolar disorder or depression, that was more severe than their panic disorder. Severe comorbidity is associated with more severe panic symptoms,6,7 which might require an SSRI/benzodiazepine combination or a higher benzodiazepine dosage.
The APA Practice Guideline suggests the following possible options:
- benzodiazepine augmentation if there is a partial response to an SSRI
- substitution with a different SSRI or a serotonin-norepinephrine reuptake inhibitor (SNRI) if there is no response to an SSRI
- benzodiazepine augmentation or substitution if there is still no therapeutic response.
Continue to: The APA Practice Guideline also states...
The APA Practice Guideline also states that although the highest “usual therapeutic dose” for panic disorder is clonazepam 2 mg/d or alprazolam 4 mg/d, “higher doses are sometimes used for patients who do not respond to the usual therapeutic dose.”1
Presumably, an SSRI/benzodiazepine combination should be considered if an SSRI alleviates major depressive disorder but does not alleviate a comorbid panic disorder. However, the APA Practice Guideline does not include studies that investigated this clinical scenario.
Monitor carefully for dependency/abuse
Restricting benzodiazepine use to low doses over a short period of time may decrease the risk of dependence and abuse. However, this practice may also limit or prevent effective treatment for adherent patients with panic disorder who do not adequately respond to SSRI or SNRI monotherapy.
Therefore, clinicians need to carefully differentiate between patients who are adherent to their prescribed dosages and those who may be at risk for benzodiazepine dependence and abuse. Consider using prescription drug monitoring programs and drug screens to help detect patients who “doctor shop” for benzodiazepines, or who could be abusing opioids, alcohol, marijuana, or other substances while taking a benzodiazepine.
As clinicians, we are faced with a conflict when deciding whether or not to prescribe a benzodiazepine. If we prescribe one of these agents, we might be putting our patients at risk for dependence and abuse. However, if we do not prescribe them, we risk providing inadequate treatment, especially for patients with panic disorder.
Benzodiazepine dependence and abuse can take many forms. Dependence can be psychological as well as physiologic. While many patients will adhere to their prescribing regimen, some may sell their benzodiazepines, falsely claim that they have “panic attacks,” or take a fatal overdose of an opioid and benzodiazepine combination.
Here I discuss the pros and cons of restricting benzodiazepines use to low doses and/or combination therapy with antidepressants.
_
Weighing the benefits of restricted prescribing
Some double-blind studies referenced in the American Psychiatric Association (APA) 2010 Practice Guideline for the Treatment of Patients with Panic Disorder1 suggest that benzodiazepine duration of treatment and dosages should be severely restricted. These studies found that:
- Although the combination of a selective serotonin reuptake inhibitor (SSRI) and a benzodiazepine initially decreased the number of panic attacks more quickly than SSRI monotherapy, the 2 treatments are equally effective after 4 or 5 weeks.2,3
- For the treatment of panic disorder, a low dosage of a benzodiazepine (clonazepam 1 mg/d or alprazolam 2 mg/d) was as effective as a higher dosage (clonazepam 2 mg/d or alprazolam 6 mg/d).4,5
However, these studies could be misleading. They all excluded patients with a comorbid condition, such as bipolar disorder or depression, that was more severe than their panic disorder. Severe comorbidity is associated with more severe panic symptoms,6,7 which might require an SSRI/benzodiazepine combination or a higher benzodiazepine dosage.
The APA Practice Guideline suggests the following possible options:
- benzodiazepine augmentation if there is a partial response to an SSRI
- substitution with a different SSRI or a serotonin-norepinephrine reuptake inhibitor (SNRI) if there is no response to an SSRI
- benzodiazepine augmentation or substitution if there is still no therapeutic response.
Continue to: The APA Practice Guideline also states...
The APA Practice Guideline also states that although the highest “usual therapeutic dose” for panic disorder is clonazepam 2 mg/d or alprazolam 4 mg/d, “higher doses are sometimes used for patients who do not respond to the usual therapeutic dose.”1
Presumably, an SSRI/benzodiazepine combination should be considered if an SSRI alleviates major depressive disorder but does not alleviate a comorbid panic disorder. However, the APA Practice Guideline does not include studies that investigated this clinical scenario.
Monitor carefully for dependency/abuse
Restricting benzodiazepine use to low doses over a short period of time may decrease the risk of dependence and abuse. However, this practice may also limit or prevent effective treatment for adherent patients with panic disorder who do not adequately respond to SSRI or SNRI monotherapy.
Therefore, clinicians need to carefully differentiate between patients who are adherent to their prescribed dosages and those who may be at risk for benzodiazepine dependence and abuse. Consider using prescription drug monitoring programs and drug screens to help detect patients who “doctor shop” for benzodiazepines, or who could be abusing opioids, alcohol, marijuana, or other substances while taking a benzodiazepine.
1. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, 2nd edition. Washington DC: American Psychiatric Association. 2010. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed March 7, 2018.
2. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
3. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
4. Lydiard RB, Lesser IM, Ballenger JC, et al. A fixed-dose study of alprazolam 2 mg, alprazolam 6 mg, and placebo in panic disorder. J Clin Psychopharmacol. 1992;12(2):966-103.
5. Rosenbaum JF, Moroz G, Bowden CL. Clonazepam in the treatment of panic disorder with or without agoraphobia: a dose-response study of efficacy, safety, and discontinuance. Clonazepam Panic Disorder Dose-Response Study Group. J Clin Psychopharmacol. 1997;17(5):390-400.
6. Goodwin RD, Hoven CW. Bipolar-panic comorbidity in the general population: prevalence and associated morbidity. J Affect Disord. 2002;70(1):27-33.
7. Roy-Byrne PP, Stang P, Wittchen HU, et al. Lifetime panic-depression comorbidity in the National Comorbidity Survey. Association with symptoms, impairment, course and help-seeking. Br J Psychiatry. 2000;176:229-235.
1. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder, 2nd edition. Washington DC: American Psychiatric Association. 2010. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed March 7, 2018.
2. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
3. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
4. Lydiard RB, Lesser IM, Ballenger JC, et al. A fixed-dose study of alprazolam 2 mg, alprazolam 6 mg, and placebo in panic disorder. J Clin Psychopharmacol. 1992;12(2):966-103.
5. Rosenbaum JF, Moroz G, Bowden CL. Clonazepam in the treatment of panic disorder with or without agoraphobia: a dose-response study of efficacy, safety, and discontinuance. Clonazepam Panic Disorder Dose-Response Study Group. J Clin Psychopharmacol. 1997;17(5):390-400.
6. Goodwin RD, Hoven CW. Bipolar-panic comorbidity in the general population: prevalence and associated morbidity. J Affect Disord. 2002;70(1):27-33.
7. Roy-Byrne PP, Stang P, Wittchen HU, et al. Lifetime panic-depression comorbidity in the National Comorbidity Survey. Association with symptoms, impairment, course and help-seeking. Br J Psychiatry. 2000;176:229-235.