User login
Treatment-resistant OCD: There’s more we can do
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”

Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
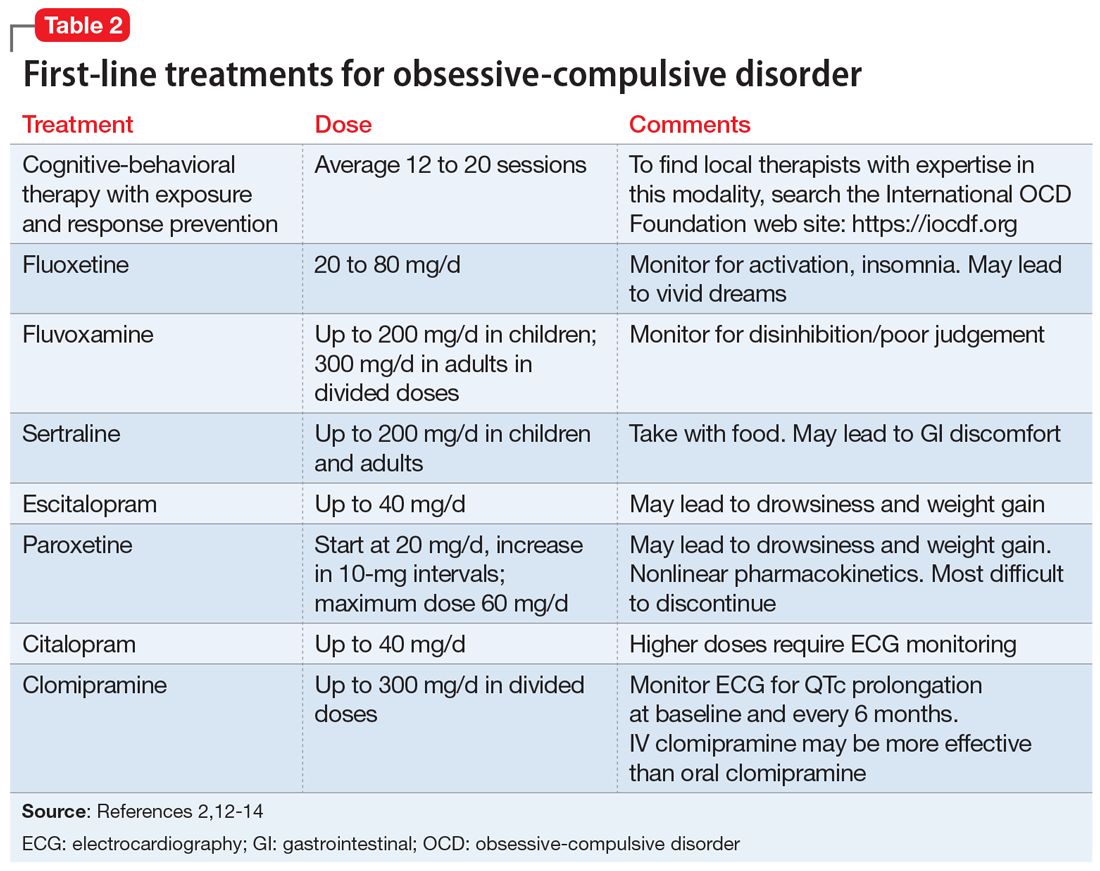
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
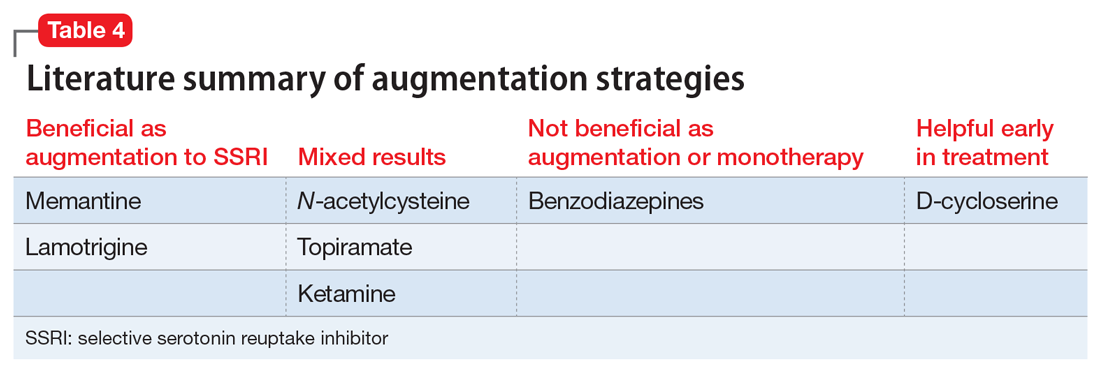
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.

Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.

Disruptive mood dysregulation disorder: A better understanding
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
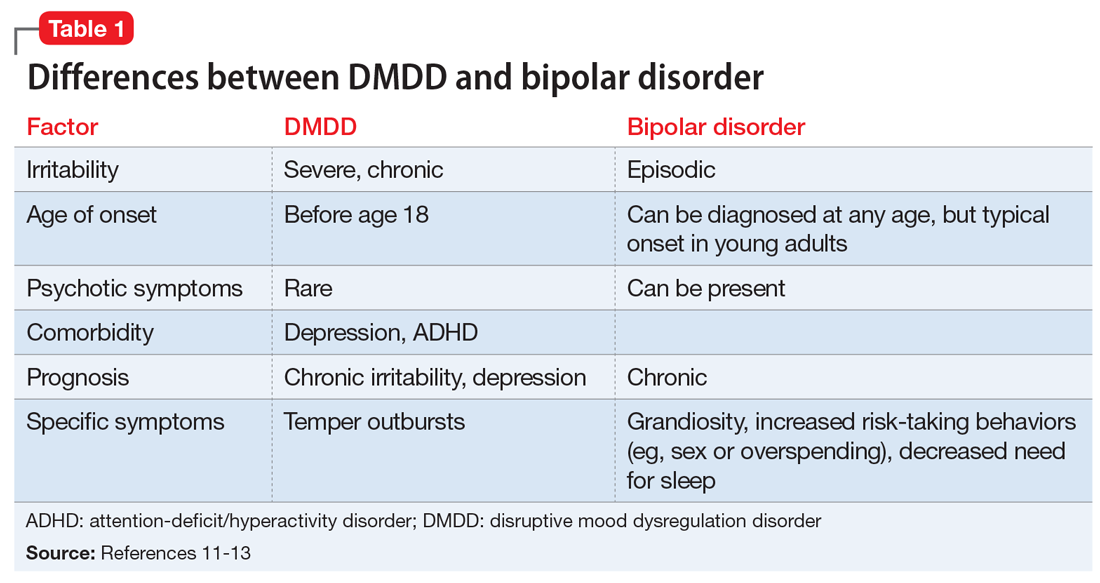
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
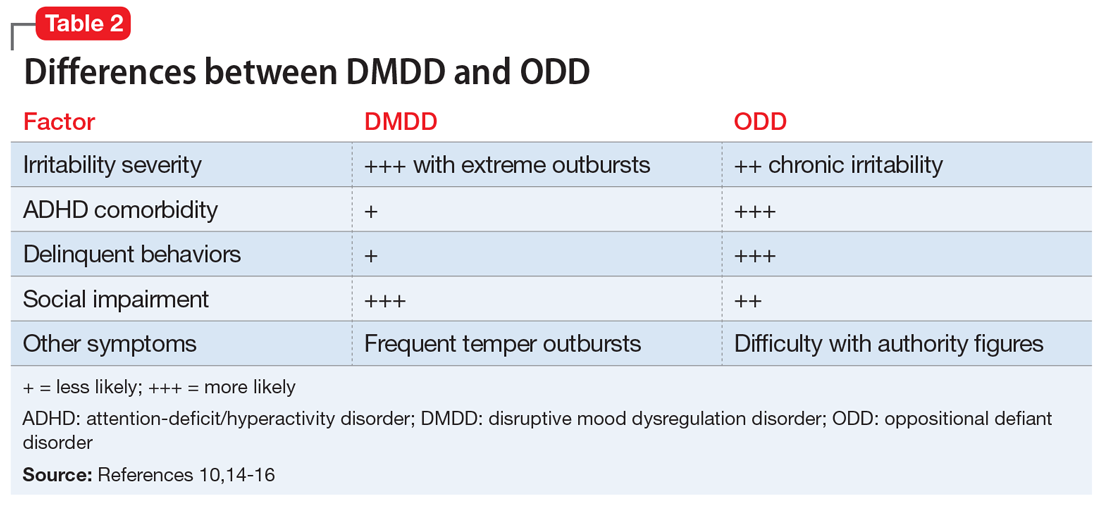
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.

Bright light therapy for bipolar depression
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.

COMPRESS: Key questions to ask during shift changes in a psychiatric ER
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
Clinical errors are common during shift changes in a hospital setting.1-3 Clinicians on the outgoing shift may forget to communicate important details, such as medication dosages, critical laboratory orders, or other interventions, to the clinicians in the next shift. To help myself formally structure the sign-out process for each patient during a shift change in a psychiatric emergency room, I came up with the acronym COMPRESS for key questions to ask the outgoing provider:
Communicate. Did you communicate with this patient in any way at any time during your shift?
Orders. Did you write any orders for this patient? If not, had another clinician already written orders for this patient?
Medications. Did you review and reconcile the medication list for this patient? If not, had another clinician already reviewed and reconciled the medication list for this patient?
PRogrESs. Did you write a progress note for this patient? If not, had the attending clinician written a progress note for this patient within the last 24 hours?
Sign. Did you sign all of your orders and progress notes for this patient?
In my experience in the psychiatric emergency room, COMPRESS has helped me efficiently structure the outgoing clinicians’ reports about my patients by having them provide vital clinical sign-out information before they leave. I hope that other clinicians working in this setting also find these questions useful.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.
1. Dubosh NM, Carney D, Fisher J, et al. Implementation of an emergency department sign-out checklist improves transfer of information at shift change. J Emerg Med. 2014;47(5):580-585.
2. Estryn-Behar MR, Milanini-Magny G, Chaumon E, et al. Shift change handovers and subsequent interruptions: potential impacts on quality of care. J Patient Saf. 2014;10(1):29-44.
3. Mardis T, Mardis M, Davis J, et al. Bedside shift-to-shift handoffs: a systematic review of the literature. J Nurs Care Qual. 2016;31(1):54-60.
Looking up patients online: Why it’s a bad idea
Searching for someone on the Internet and viewing his or her social media profile is an effective way to obtain information about people, including patients. Following our patients’ “digital footprint” may help us understand the context of their lives, reconcile discrepancies in what they have told us, or allow us to confront denial and address incomplete reporting.1 However, perusing our patients’ online profiles could negatively impact treatment and adherence. Consider these factors before looking up your patients’ online profiles1-3:
Inaccurate information. Information on the Internet, especially what you can find on user-generated forums, is largely unregulated; as a result, the veracity of that information cannot be guaranteed.1 Patients may choose to portray themselves inaccurately on their online profiles, and their identities often cannot be confirmed. Even if some information is accurate, you might discover things that you did not expect to learn about your patients, including important information that they did not share, or even something they lied about. This can create the conundrums of what to do with such information and how to discuss it at the next visit.
Impact on treatment. Despite patients’ online activities being displayed for the world to see, many patients do not expect their clinicians to access their online information. They might perceive such perusal as a breach of trust, which might lead some to view the doctor–patient relationship as adversarial. Accessing this information also could create a more intimate relationship than intended. Even if a clinician acquires consent to perform a search, patients may still feel coerced into allowing it because they might feel that declining to grant permission would make the clinician suspect that they have something to hide, or that the clinician would search without consent.2
In addition, if patients are aware that their psychiatrists are monitoring them, they might change their behavior. For example, they may delete certain data, add additional information that may not be accurate, or censor future social media posts. Knowing that their clinicians could be paying attention to them around the clock also might motivate certain patients to act out more or become withdrawn.
Possible medicolegal repercussions. If clinicians are able to access their patients’ electronic profiles, are they then legally obligated to monitor them? For example, if a patient who posts a picture with a noose around his neck later completes suicide, does the clinician who intermittently monitored this patient’s online profile face legal ramifications for not seeing the post? Do clinicians have to call 911 for vaguely suicidal tweets? What responsibilities does a clinician have at the first sign of an innocuous “sad” emoji? The sheer volume of online content that patients can create over different outlets is staggering. It can be overwhelming and ineffective to attempt to monitor patients’ online activities in addition to attending to one’s usual clinical duties, and the medicolegal repercussions of doing so are largely unknown.
Before searching the Internet to learn more about your patients, first consider the ramifications of doing so. While such searches could be helpful, they may lead to poor adherence, a lack of trust, or legal quagmires.
1. Fisher CE, Appelbaum PS. Beyond Googling: the ethics of using patients’ electronic footprints in psychiatric practice. Harv Rev Psychiatry. 2017;25(4):170-179.
2. Ashby GA, O’Brien A, Bowman
3. Cox-George C. The changing face(book) of psychiatry: can we justify ‘following’ patients’ social media activity? BJPsych Bulletin. 2015;39(6):284-285.
Searching for someone on the Internet and viewing his or her social media profile is an effective way to obtain information about people, including patients. Following our patients’ “digital footprint” may help us understand the context of their lives, reconcile discrepancies in what they have told us, or allow us to confront denial and address incomplete reporting.1 However, perusing our patients’ online profiles could negatively impact treatment and adherence. Consider these factors before looking up your patients’ online profiles1-3:
Inaccurate information. Information on the Internet, especially what you can find on user-generated forums, is largely unregulated; as a result, the veracity of that information cannot be guaranteed.1 Patients may choose to portray themselves inaccurately on their online profiles, and their identities often cannot be confirmed. Even if some information is accurate, you might discover things that you did not expect to learn about your patients, including important information that they did not share, or even something they lied about. This can create the conundrums of what to do with such information and how to discuss it at the next visit.
Impact on treatment. Despite patients’ online activities being displayed for the world to see, many patients do not expect their clinicians to access their online information. They might perceive such perusal as a breach of trust, which might lead some to view the doctor–patient relationship as adversarial. Accessing this information also could create a more intimate relationship than intended. Even if a clinician acquires consent to perform a search, patients may still feel coerced into allowing it because they might feel that declining to grant permission would make the clinician suspect that they have something to hide, or that the clinician would search without consent.2
In addition, if patients are aware that their psychiatrists are monitoring them, they might change their behavior. For example, they may delete certain data, add additional information that may not be accurate, or censor future social media posts. Knowing that their clinicians could be paying attention to them around the clock also might motivate certain patients to act out more or become withdrawn.
Possible medicolegal repercussions. If clinicians are able to access their patients’ electronic profiles, are they then legally obligated to monitor them? For example, if a patient who posts a picture with a noose around his neck later completes suicide, does the clinician who intermittently monitored this patient’s online profile face legal ramifications for not seeing the post? Do clinicians have to call 911 for vaguely suicidal tweets? What responsibilities does a clinician have at the first sign of an innocuous “sad” emoji? The sheer volume of online content that patients can create over different outlets is staggering. It can be overwhelming and ineffective to attempt to monitor patients’ online activities in addition to attending to one’s usual clinical duties, and the medicolegal repercussions of doing so are largely unknown.
Before searching the Internet to learn more about your patients, first consider the ramifications of doing so. While such searches could be helpful, they may lead to poor adherence, a lack of trust, or legal quagmires.
Searching for someone on the Internet and viewing his or her social media profile is an effective way to obtain information about people, including patients. Following our patients’ “digital footprint” may help us understand the context of their lives, reconcile discrepancies in what they have told us, or allow us to confront denial and address incomplete reporting.1 However, perusing our patients’ online profiles could negatively impact treatment and adherence. Consider these factors before looking up your patients’ online profiles1-3:
Inaccurate information. Information on the Internet, especially what you can find on user-generated forums, is largely unregulated; as a result, the veracity of that information cannot be guaranteed.1 Patients may choose to portray themselves inaccurately on their online profiles, and their identities often cannot be confirmed. Even if some information is accurate, you might discover things that you did not expect to learn about your patients, including important information that they did not share, or even something they lied about. This can create the conundrums of what to do with such information and how to discuss it at the next visit.
Impact on treatment. Despite patients’ online activities being displayed for the world to see, many patients do not expect their clinicians to access their online information. They might perceive such perusal as a breach of trust, which might lead some to view the doctor–patient relationship as adversarial. Accessing this information also could create a more intimate relationship than intended. Even if a clinician acquires consent to perform a search, patients may still feel coerced into allowing it because they might feel that declining to grant permission would make the clinician suspect that they have something to hide, or that the clinician would search without consent.2
In addition, if patients are aware that their psychiatrists are monitoring them, they might change their behavior. For example, they may delete certain data, add additional information that may not be accurate, or censor future social media posts. Knowing that their clinicians could be paying attention to them around the clock also might motivate certain patients to act out more or become withdrawn.
Possible medicolegal repercussions. If clinicians are able to access their patients’ electronic profiles, are they then legally obligated to monitor them? For example, if a patient who posts a picture with a noose around his neck later completes suicide, does the clinician who intermittently monitored this patient’s online profile face legal ramifications for not seeing the post? Do clinicians have to call 911 for vaguely suicidal tweets? What responsibilities does a clinician have at the first sign of an innocuous “sad” emoji? The sheer volume of online content that patients can create over different outlets is staggering. It can be overwhelming and ineffective to attempt to monitor patients’ online activities in addition to attending to one’s usual clinical duties, and the medicolegal repercussions of doing so are largely unknown.
Before searching the Internet to learn more about your patients, first consider the ramifications of doing so. While such searches could be helpful, they may lead to poor adherence, a lack of trust, or legal quagmires.
1. Fisher CE, Appelbaum PS. Beyond Googling: the ethics of using patients’ electronic footprints in psychiatric practice. Harv Rev Psychiatry. 2017;25(4):170-179.
2. Ashby GA, O’Brien A, Bowman
3. Cox-George C. The changing face(book) of psychiatry: can we justify ‘following’ patients’ social media activity? BJPsych Bulletin. 2015;39(6):284-285.
1. Fisher CE, Appelbaum PS. Beyond Googling: the ethics of using patients’ electronic footprints in psychiatric practice. Harv Rev Psychiatry. 2017;25(4):170-179.
2. Ashby GA, O’Brien A, Bowman
3. Cox-George C. The changing face(book) of psychiatry: can we justify ‘following’ patients’ social media activity? BJPsych Bulletin. 2015;39(6):284-285.
Aripiprazole lauroxil nanocrystal suspension

Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL

Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
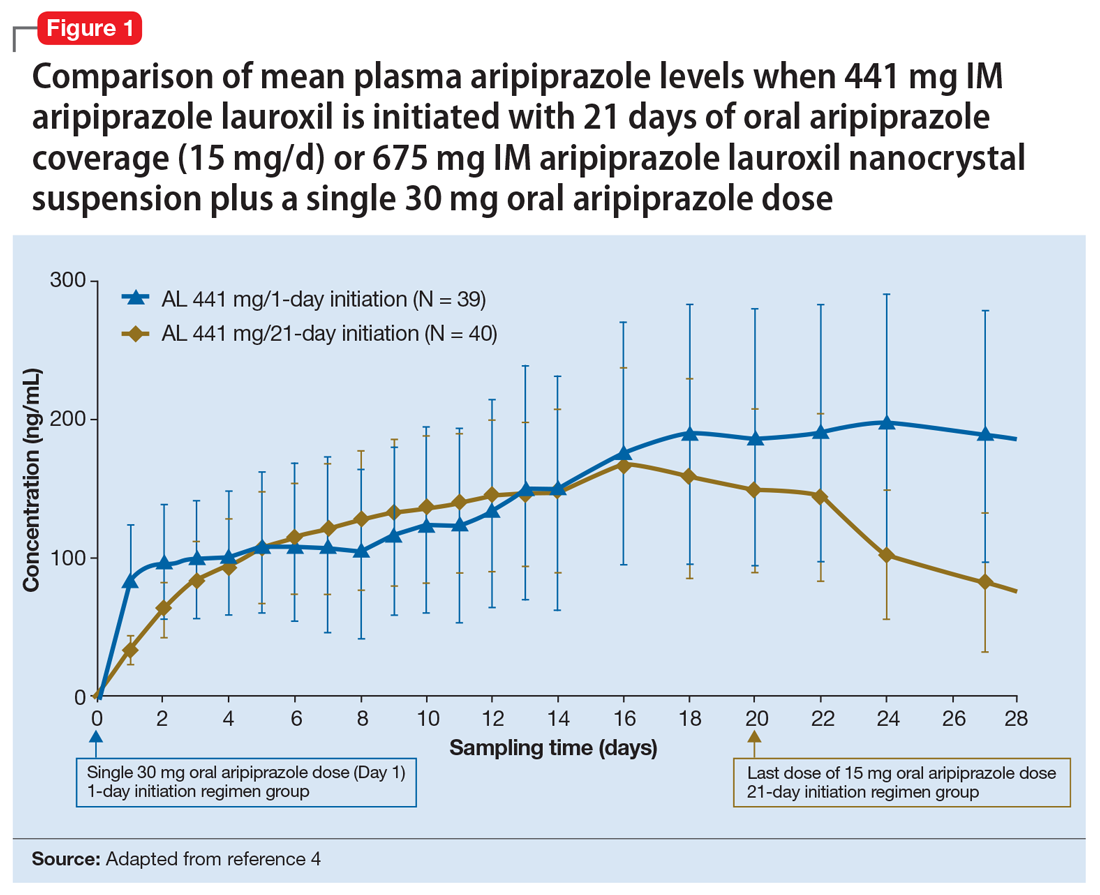
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
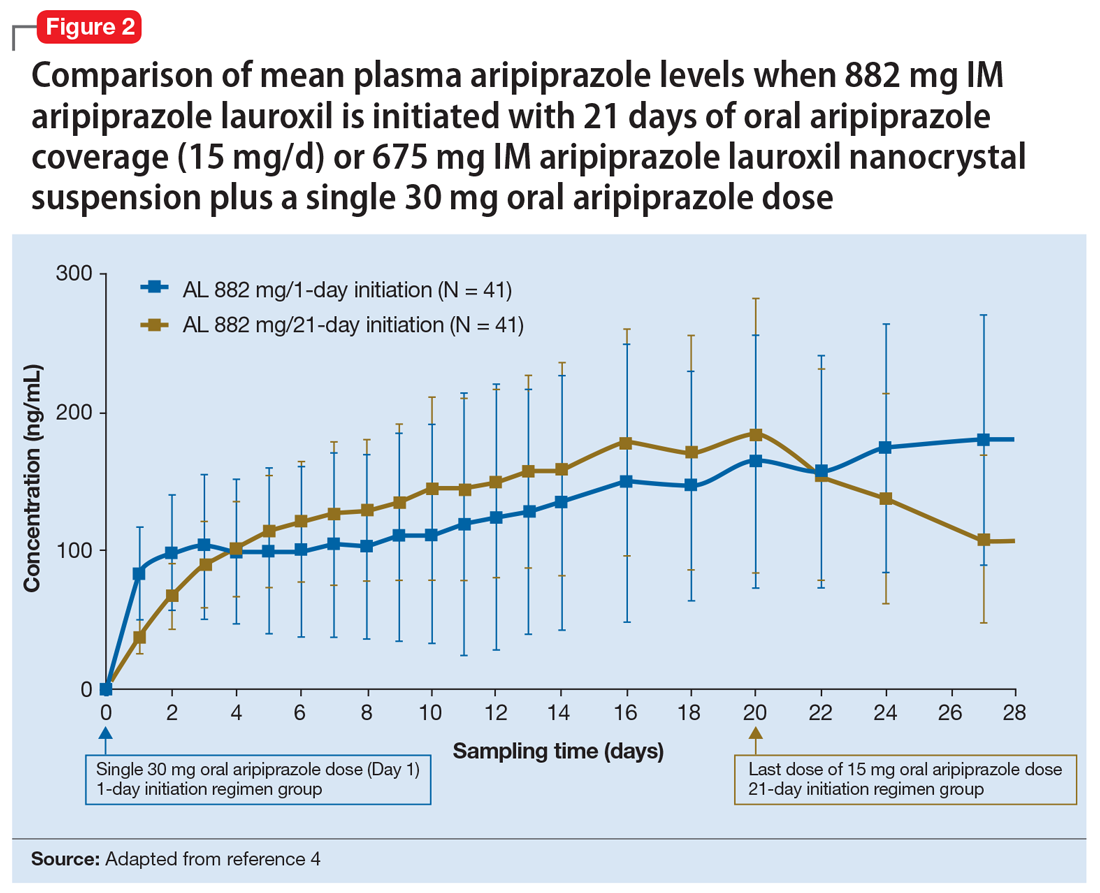
Continue to: Clinical considerations
Clinical considerations
AL
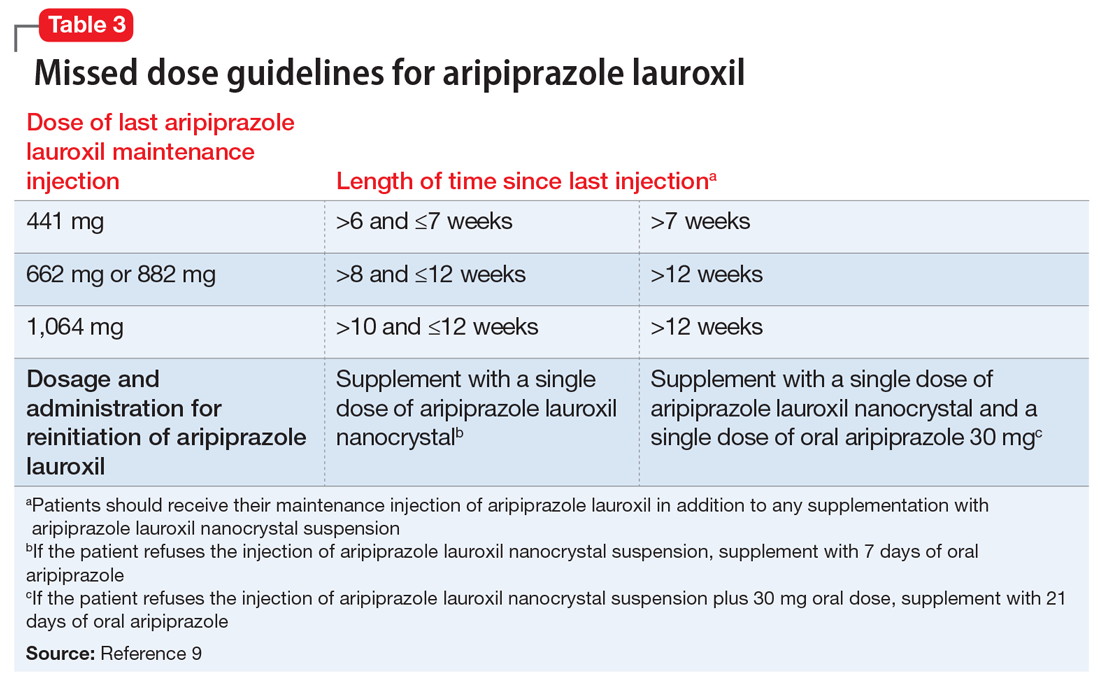
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
Office approach to small fiber neuropathy
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES

Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.

Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.

INITIAL TESTING FOR AN UNDERLYING CAUSE

Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control

Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies

PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES
Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.
Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.
INITIAL TESTING FOR AN UNDERLYING CAUSE
Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control
Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies
PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
Peripheral neuropathy is the most common reason for an outpatient neurology visit in the United States and accounts for over $10 billion in healthcare spending each year.1,2 When the disorder affects only small, thinly myelinated or unmyelinated nerve fibers, it is referred to as small fiber neuropathy, which commonly presents as numbness and burning pain in the feet.
This article details the manifestations and evaluation of small fiber neuropathy, with an eye toward diagnosing an underlying cause amenable to treatment.
OLDER PATIENTS MOST AFFECTED
The epidemiology of small fiber neuropathy is not well established. It occurs more commonly in older patients, but data are mixed on prevalence by sex.3–6 In a Dutch study,3 the overall prevalence was at least 53 cases per 100,000, with the highest rate in men over age 65.
CHARACTERISTIC SENSORY DISTURBANCES
Sensations vary in quality and time
Patients with small fiber neuropathy typically present with a symmetric length-dependent (“stocking-glove”) distribution of sensory changes, starting in the feet and gradually ascending up the legs and then to the hands.
Commonly reported neuropathic symptoms include various combinations of burning, numbness, tingling, itching, sunburn-like, and frostbite-like sensations. Nonneuropathic symptoms may include tightness, a vise-like squeezing of the feet, and the sensation of a sock rolled up at the end of the shoe. Cramps or spasms may also be reported but rarely occur in isolation.7
Symptoms are typically worse at the end of the day and while sitting or lying down at night. They can arise spontaneously but may also be triggered by something as minor as the touch of clothing or cool air against the skin. Bedsheet sensitivity of the feet is reported so often that it is used as an outcome measure in clinical trials. Symptoms can also be exacerbated by extremes in ambient temperature and are especially worse in cold weather.
Random patterns suggest an immune cause
Symptoms may also have a non–length-dependent distribution that is asymmetric, patchy, intermittent, and migratory, and can involve the face, proximal limbs, and trunk. Symptoms may vary throughout the day, eg, starting with electric-shock sensations on one side of the face, followed by perineal numbness and then tingling in the arms lasting for a few minutes to several hours. While such patterns may be seen with diabetes and other common etiologies, they often suggest an underlying immune-mediated disorder such as Sjögren syndrome or sarcoidosis.8–10 Although large fiber polyneuropathy may also be non–length-dependent, the deficits are usually fixed, with no migratory component.
Autonomic features may be prominent
Autonomic symptoms occur in nearly half of patients and can be as troublesome as neuropathic pain.3 Small nerve fibers mediate somatic and autonomic functions, an evolutionary link that may reflect visceral defense mechanisms responding to pain as a signal of danger.11 This may help explain the multisystemic nature of symptoms, which can include sweating abnormalities, bowel and bladder disturbances, dry eyes, dry mouth, gastrointestinal dysmotility, skin changes (eg, discoloration, loss of hair, shiny skin), sexual dysfunction, orthostatic hypotension, and palpitations. In some cases, isolated dysautonomia may be seen.
TARGETED EXAMINATION
History: Medications, alcohol, infections
When a patient presents with neuropathic pain in the feet, a detailed history should be obtained, including alcohol use, family history of neuropathy, and use of neurotoxic medications such as metronidazole, colchicine, and chemotherapeutic agents.
Human immunodeficiency virus (HIV) and hepatitis C infection are well known to be associated with small fiber neuropathy, so relevant risk factors (eg, blood transfusions, sexual history, intravenous drug use) should be asked about. Recent illnesses and vaccinations are another important line of questioning, as a small-fiber variant of Guillain-Barré syndrome has been described.12
Assess reflexes, strength, sensation
On physical examination, particular attention should be focused on searching for abnormalities indicating large nerve fiber involvement (eg, absent deep tendon reflexes, weakness of the toes). However, absent ankle deep tendon reflexes and reduced vibratory sense may also occur in healthy elderly people.
Similarly, proprioception, motor strength, balance, and vibratory sensation are functions of large myelinated nerve fibers, and thus remain unaffected in patients with only small fiber neuropathy.
Evidence of a systemic disorder should also be sought, as it may indicate an underlying etiology.
DIAGNOSTIC TESTING
Although patients with either large or small fiber neuropathy may have subjective hyperesthesia or numbness of the distal lower extremities, the absence of significant abnormalities on neurologic examination should prompt consideration of small fiber neuropathy.
Electromyography worthwhile
Nerve conduction studies and needle electrode examination evaluate only large nerve fiber conditions. While electromyographic results are normal in patients with isolated small fiber neuropathy, the test can help evaluate subclinical large nerve fiber involvement and alternative diagnoses such as bilateral S1 radiculopathy. Nerve conduction studies may be less useful in patients over age 75, as they may lack sural sensory responses because of aging changes.13
Skin biopsy easy to do
Skin biopsy for evaluating intraepidermal nerve fiber density is one of the most widely used tests for small fiber neuropathy. This minimally invasive procedure can now be performed in a primary care office using readily available tools or prepackaged kits and analyzed by several commercial laboratories.
Reduced intraepidermal nerve fiber density on skin biopsy has been described in various other conditions such as fibromyalgia and chronic pain syndromes.16,17 The clinical significance of these findings remains uncertain.
Quantitative sudomotor axon reflex testing
Quantitative sudomotor axon reflex testing (QSART) is a noninvasive autonomic study that assesses the volume of sweat produced by the limbs in response to acetylcholine. A measure of postganglionic sympathetic sudomotor nerve function, QSART has a sensitivity of up to 80% and can be used to diagnose small fiber neuropathy.18 In a series of 115 patients with sarcoidosis small fiber neuropathy,9 the QSART and skin biopsy findings were concordant in 17 cases and complementary in 29, allowing for confirmation of small fiber neuropathy in patients whose condition would have remained undiagnosed had only one test been performed. QSART can also be considered in cases where skin biopsy may be contraindicated (eg, patient use of anticoagulation). Of note, the study may be affected by a number of external factors, including caffeine, tobacco, antihistamines, and tricyclic antidepressants; these should be held before testing.
Other diagnostic studies
Other tests may be helpful, as follows:
Tilt-table and cardiovagal testing may be useful for patients with orthostasis and palpitations.
Thermoregulatory sweat testing can be used to evaluate patients with abnormal patterns of sweating, eg, hyperhidrosis of the face and head.
INITIAL TESTING FOR AN UNDERLYING CAUSE
Glucose tolerance test for diabetes
Diabetes is the most common identifiable cause of small fiber neuropathy and accounts for about a third of all cases.5 Impaired glucose tolerance is also thought to be a risk factor and has been found in up to 50% of idiopathic cases, but the association is still being debated.21
While testing for hemoglobin A1c is more convenient for the patient, especially because it does not require fasting, a 2-hour oral glucose tolerance test is more sensitive for detecting glucose dysmetabolism.22
Lipid panel for metabolic syndrome
Small fiber neuropathy is associated with individual components of the metabolic syndrome, which include obesity, hyperglycemia, and dyslipidemia. Of these, dyslipidemia has emerged as the primary factor involved in the development of small fiber neuropathy, via an inflammatory pathway or oxidative stress mechanism.23,24
Vitamin B12 deficiency testing
Vitamin B12 deficiency, a potentially correctable cause of small fiber neuropathy, may be underdiagnosed, especially as values obtained by blood testing may not reflect tissue uptake. Causes of vitamin B12 deficiency include reduced intake, pernicious anemia, and medications that can affect absorption of vitamin B12 (eg, proton pump inhibitors, histamine 2 receptor antagonists, metformin).
Testing should include:
- Complete blood cell count to evaluate for vitamin B12-related macrocytic anemia and other hematologic abnormalities
- Serum vitamin B12 level
- Methylmalonic acid or homocysteine level in patients with subclinical or mild vitamin B12 deficiency, manifested as low to normal vitamin B12 levels (< 400 pg/mL); methylmalonic acid and homocysteine require vitamin B12 as a cofactor for enzymatic conversion, and either or both may be elevated in early vitamin B12 deficiency.
Celiac antibody panel
Celiac disease, a T-cell mediated enteropathy characterized by gluten intolerance and a herpetiform-like rash, can be associated with small fiber neuropathy.25 In some cases, neuropathy symptoms are preceded by the onset of gastrointestinal symptoms, or they may occur in isolation.25
Inflammatory disease testing
Sjögren syndrome accounts for nearly 10% of cases of small fiber neuropathy. Associated neuropathic symptoms are often non–length-dependent, can precede sicca symptoms for up to 6 years, and in some cases are the sole manifestation of the disease.10 Small fiber neuropathy may also be associated with vasculitis, systemic lupus erythematosus, and other connective tissue disorders.
Testing should include:
- Erythrocyte sedimentation rate, C-reactive protein, and antinuclear antibodies: though these are nonspecific markers of inflammation, they may support an immune-mediated etiology if positive
- Extractable nuclear antigen panel: Sjögren syndrome A and B autoantibodies are the most important components in this setting5,11
- The Schirmer test or salivary gland biopsy should be considered for seronegative patients with sicca or a suspected immune-mediated etiology, as the sensitivity of antibody testing ranges from only 10% to 55%.10
Thyroid function testing
Hypothyroidism, and less commonly hyperthyroidism, are associated with small fiber neuropathy.
Metabolic tests for liver and kidney disease
Renal insufficiency and liver impairment are well-known causes of small nerve fiber dysfunction. Testing should include:
- Comprehensive metabolic panel
- Gamma-glutamyltransferase if alcohol abuse is suspected, since heavy alcohol use is one of the most common causes of both large and small fiber neuropathy.
HIV and hepatitis C testing
For patients with relevant risk factors, HIV and hepatitis C testing should be part of the initial workup (and as second-tier testing for others). Patients who test positive for hepatitis C should undergo further testing for cryoglobulinemia, which can present with painful small fiber neuropathy.26
Serum and urine immunoelectrophoresis
Paraproteinemia, with causes ranging from monoclonal gammopathy of uncertain significance to multiple myeloma, has been associated with small fiber neuropathy. An abnormal serum or urine immunoelectrophoresis test warrants further investigation and possibly referral to a hematology-oncology specialist.
SECOND-TIER TESTING
Less common treatable causes of small fiber neuropathy may also be evaluated.
Copper, vitamin B1 (thiamine), or vitamin B6 (pyridoxine) deficiency testing. Although vitamin B6 toxicity may also result in neuropathy due to its toxic effect on the dorsal root ganglia, the mildly elevated vitamin B6 levels often found in patients being evaluated for neuropathy are unlikely to be the primary cause of symptoms. Many laboratories require fasting samples for accurate vitamin B6 levels.
Angiotensin-converting enzyme levels for sarcoidosis. Small fiber neuropathy is common in sarcoidosis, occurring in more than 30% of patients with systemic disease.27 However, screening for sarcoidosis by measuring serum levels is often falsely positive and is not cost-effective. In a study of 195 patients with idiopathic small fiber neuropathy,11 44% had an elevated serum level, but no evidence of sarcoidosis was seen on further testing, which included computed tomography of the chest in 29 patients.12 Thus, this test is best used for patients with evidence of systemic disease.
Amyloid testing for amyloidosis. Fat pad or bone marrow biopsy should be considered in the appropriate clinical setting.
Paraneoplastic autoantibody panel for occult cancer. Such testing may also be considered if clinically warranted. However, if a patient is found to have low positive titers of paraneoplastic antibodies and suspicion is low for an occult cancer (eg, no weight loss or early satiety), repeat confirmatory testing at another laboratory should be done before embarking on an extensive search for malignancy.
Ganglionic acetylcholine receptor antibody testing for autoimmune autonomic ganglionopathy. This should be ordered for patients with prominent autonomic dysfunction. The antibody test can be ordered separately or as part of an autoantibody panel. The antibody may indicate a primary immune-mediated process or a paraneoplastic disease.28
Genetic mutation testing. Recent discoveries of gene mutations leading to peripheral nerve hyperexcitability of voltage-gated sodium channels have elucidated a hereditary cause of small fiber neuropathy in nearly 30% of cases that were once thought to be idiopathic.29,30 Genetic testing for mutations in SCN9A and SCN10 (which code for the Nav1.7 and Nav1.8 sodium channels, respectively) is commercially available and may be considered for those with a family history of neuropathic pain in the feet or for young, otherwise healthy patients.
Fabry disease is an X-linked lysosomal disorder characterized by angiokeratomas, cardiac and renal impairment, and small fiber neuropathy. Treatment is now available, but screening is not cost-efficient and should only be pursued in patients with other symptoms of the disease.31,32
OTHER POSSIBLE CAUSES
Guillain-Barré syndrome
A Guillain-Barré syndrome variant has been reported that is characterized by ascending limb paresthesias and cerebrospinal fluid albuminocytologic dissociation in the setting of preserved deep tendon reflexes and normal findings on EMG.12 The clinical course is similar to that of typical Guillain-Barré syndrome, in that symptoms follow an upper respiratory or gastrointestinal tract infection, reach their nadir at 4 weeks, and then gradually improve. Some patients respond to intravenous immune globulin.
Vaccine-associated
Postvaccination small fiber neuropathy has also been reported. The nature of the association is unclear.33
Parkinson disease
Small fiber neuropathy is associated with Parkinson disease. It is attributed to a number of proposed factors, including neurodegeneration that occurs parallel to central nervous system decline, as well as intestinal malabsorption with resultant vitamin deficiency.34,35
Rapid glycemic lowering
Aggressive treatment of diabetes, defined as at least a 2-point reduction of serum hemoglobin A1c level over 3 months, may result in acute small fiber neuropathy. It manifests as severe distal extremity pain and dysautonomia.
In a retrospective study,36 104 (10.9%) of 954 patients presenting to a tertiary diabetic clinic developed treatment-induced diabetic neuropathy with symptoms occurring within 8 weeks of rapid glycemic control. The severity of neuropathy correlated with the degree and rate of glycemic lowering. The condition was reversible in some cases.
TREATING SPECIFIC DISORDERS
For patients with an identified cause of neuropathy, targeted treatment offers the best chance of halting progression and possibly improving symptoms. Below are recommendations for addressing neuropathy associated with the common diagnoses.
Diabetes, impaired glucose tolerance, and metabolic syndrome. In addition to glycemic- and lipid-lowering therapies, lifestyle modifications with a specific focus on exercise and nutrition are integral to treating diabetes and related disorders.
In the Look AHEAD (Action for Health in Diabetes) study,37 which evaluated the effects of intensive lifestyle intervention on neuropathy in 5,145 overweight patients with type 2 diabetes, patients in the intervention group had lower pain scores and better touch sensation in the toes compared with controls at 1 year. Differences correlated with the degree of weight loss and reduction of hemoglobin A1c and lipid levels.
As running and walking may not be feasible for many patients owing to pain, stationary cycling, aqua therapy, and swimming are other options. A stationary recumbent bike may be useful for older patients with balance issues.
Vitamin B12 deficiency. As reduced absorption rather than low dietary intake is the primary cause of vitamin B12 deficiency for many patients, parenteral rather than oral supplementation may be best. A suggested regimen is subcutaneous or intramuscular methylcobalamin injection of 1,000 µg given daily for 1 week, then once weekly for 1 month, followed by a maintenance dose once a month for at least 6 to 12 months. Alternatively, a daily dose of vitamin B12 1,000 µg can be taken sublingually.
Sjögren syndrome. According to anecdotal case reports, intravenous immune globulin, corticosteroids, and other immunosuppressants help painful small fiber neuropathy and dysautonomia associated with Sjögren syndrome.10
Sarcoidosis. Sarcoidosis-associated small fiber neuropathy may also respond to intravenous immune globulin, as well as infliximab and combination therapy.9 Culver et al38 found that cibinetide, an experimental erythropoetin agonist, resulted in improved corneal nerve fiber measures in patients with small fiber neuropathy associated with sarcoidosis.
Celiac disease. A gluten-free diet is the treatment for celiac disease and can help some patients.
GENERAL MANAGEMENT
For all patients, regardless of whether the cause of small fiber neuropathy has been identified, managing symptoms remains key, as pain and autonomic dysfunction can markedly impair quality of life. A multidisciplinary approach that incorporates pain medications, physical therapy, and lifestyle modifications is ideal. Integrative holistic treatments such as natural supplements, yoga, and other mind-body therapies may also help.
Pain control
Mexiletine, a voltage-gated sodium channel blocker used as an antiarrhythmic, may help refractory pain or hereditary small fiber neuropathy related to sodium channel dysfunction. However, it is not recommended for diabetic neuropathy.39
Combination regimens that use drugs with different mechanisms of action can be effective. In one study, combined gabapentin and nortriptyline were more effective than either drug alone for neuropathic pain.40
Inhaled cannabis reduced pain in patients with HIV and diabetic neuropathy in a number of studies. Side effects included euphoria, somnolence, and cognitive impairment.41,42 The use of medical marijuana is not yet legal nationwide and may affect employability even in states in which it has been legalized.
Owing to the opioid epidemic and high addiction potential, opioids are no longer a preferred recommendation for chronic treatment of noncancer-related neuropathy. A population-based study of 2,892 patients with neuropathy found that those on chronic opioid therapy (≥ 90 days) had worse functional outcomes and higher rates of addiction and overdose than those on short-term therapy.43 However, the opioid agonist tramadol was found to be effective in reducing neuropathic pain and may be a safer option for patients with chronic small fiber neuropathy.44
Integrative, holistic therapies
PROGNOSIS
For many patients, small fiber neuropathy is a slowly progressive disorder that reaches a clinical plateau lasting for years, with progression to large fiber involvement reported in 13% to 36% of cases; over half of patients in one series either improved or remained stable over a period of 2 years.5,57 Long-term studies are needed to fully understand the natural disease course. In the meantime, treating underlying disease and managing symptoms are imperative to patient care.
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
- Burke JF, Skolarus LE, Callaghan BC, Kerber KA. Choosing Wisely: highest-cost tests in outpatient neurology. Ann Neurol 2013; 73(5):679–683. doi:10.1002/ana.23865
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes Care 2003; 26(6):1790–1795. pmid:12766111
- Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. Incidence and prevalence of small-fiber neuropathy: a survey in the Netherlands. Neurology 2013; 81(15):1356–1360. doi:10.1212/WNL.0b013e3182a8236e
- Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 1999; 53(8):1641–1647. pmid:10563606
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131(pt 7):1912–1925. doi:10.1093/brain/awn093
- Lacomis D. Small-fiber neuropathy. Muscle Nerve 2002; 26(2):173–188. doi:10.1002/mus.10181
- Lopate G, Streif E, Harms M, Weihl C, Pestronk A. Cramps and small-fiber neuropathy. Muscle Nerve 2013; 48(2):252–255. doi:10.1002/mus.23757
- Khan S, Zhou L. Characterization of non-length-dependent small-fiber sensory neuropathy. Muscle Nerve 2012; 45(1):86–91. doi:10.1002/mus.22255
- Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017; 126:135–138. doi:10.1016/j.rmed.2017.03.011
- Berkowitz AL, Samuels MA. The neurology of Sjogren’s syndrome and the rheumatology of peripheral neuropathy and myelitis. Pract Neurol 2014; 14(1):14–22. doi:10.1136/practneurol-2013-000651
- Lang M, Treister R, Oaklander AL. Diagnostic value of blood tests for occult causes of initially idiopathic small-fiber polyneuropathy. J Neurol 2016; 263(12):2515–2527. doi:10.1007/s00415-016-8270-5
- Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002; 72(4):540–542. pmid:11909922
- Tavee JO, Polston D, Zhou L, Shields RW, Butler RS, Levin KH. Sural sensory nerve action potential, epidermal nerve fiber density, and quantitative sudomotor axon reflex in the healthy elderly. Muscle Nerve 2014; 49(4):564–569. doi:10.1002/mus.23971
- Tavee J, Zhou L. Small fiber neuropathy: a burning problem. Cleve Clin J Med 2009; 76(5):297–305. doi:10.3949/ccjm.76a.08070
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53(8):1634–1640. pmid:10563605
- Oaklander AL, Herzog ZD, Downs HM, Klein MM. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia. Pain 2013; 154(11):2310–2316. doi:10.1016/j.pain.2013.06.001
- Üçeyler N, Zeller D, Kahn AK, et al. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013; 136(pt 6):1857–1867. doi:10.1093/brain/awt053
- Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15(6):661–665. doi:10.1002/mus.880150605
- Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: a non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia 2003; 46(5):683–688. doi:10.1007/s00125-003-1086-8
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 2018; 25(2):348–355. doi:10.1111/ene.13508
- Smith AG. Impaired glucose tolerance and metabolic syndrome in idiopathic neuropathy. J Peripher Nerv Syst 2012; 17(suppl 2):15–21. doi:10.1111/j.1529-8027.2012.00390.x
- Hoffman-Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol 2006; 63(8):1075–1079. doi:10.1001/archneur.63.8.noc50336
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst 2009; 14(4):257–267. doi:10.1111/j.1529-8027.2009.00237.x
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes 2009; 58(7):1634–1640. doi:10.2337/db08-1771
- Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003; 60(10):1581–1585. pmid:12771245
- Gemignani F, Brindani F, Alfieri S, et al. Clinical spectrum of cryoglobulinaemic neuropathy. J Neurol Neurosurg Psychiatry 2005; 76(10):1410–1414. doi:10.1136/jnnp.2004.057620
- Bakkers M, Merkies IS, Lauria G, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 2009; 73(14):1142–1148. doi:10.1212/WNL.0b013e3181bacf05
- Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med 2000; 343(12):847–855. doi:10.1056/NEJM200009213431204
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71(1):26–39. doi:10.1002/ana.22485
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19(2):53–65. doi:10.1111/jns5.12071
- Samuelsson K, Kostulas K, Vrethem M, Rolfs A, Press R. Idiopathic small fiber neuropathy: phenotype, etiologies, and the search for Fabry disease. J Clin Neurol 2014; 10(2):108–118. doi:10.3988/jcn.2014.10.2.108
- de Greef BT, Hoeijmakers JG, Wolters EE, et al. No Fabry disease in patients presenting with isolated small fiber neuropathy. PLoS One 2016; 11(2):e0148316. doi:10.1371/journal.pone.0148316
- Souayah N, Ajroud-Driss S, Sander HW, Brannagan TH, Hays AP, Chin RL. Small fiber neuropathy following vaccination for rabies, varicella or Lyme disease. Vaccine 2009; 27(52):7322–7325. doi:10.1016/j.vaccine.2009.09.077
- Nolano M, Provitera V, Manganelli F, et al. Loss of cutaneous large and small fibers in naive and l-dopa–treated PD patients. Neurology 2017; 89(8):776–784. doi:10.1212/WNL.0000000000004274
- Zis P, Grünewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral neuropathy in idiopathic Parkinson’s disease: a systematic review. J Neurol Sci 2017; 378:204–209. doi:10.1016/j.jns.2017.05.023
- Gibbons CH, Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 2015; 138(pt 1):43–52. doi:10.1093/brain/awu307
- Look AHEAD Research Group. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia 2017; 60(6):980–988. doi:10.1007/s00125-017-4253-z
- Culver DA, Dahan A, Bajorunas D, et al. Cibinetide improves corneal nerve fiber abundance in patients with sarcoidosis-associated small nerve fiber loss and neuropathic pain. Invest Ophthalmol Vis Sci 2017; 58(6):BIO52–BIO60. doi:10.1167/iovs.16-21291
- Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R 2011; 3(4):345–352.e21. doi:10.1016/j.pmrj.2011.03.008
- Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374(9697):1252–1261. doi:10.1016/S0140-6736(09)61081-3
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology 2009; 34(3):672–680. doi:10.1038/npp.2008.120
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, Atkinson JH. Efficacy of inhaled cannabis on painful diabetic neuropathy. J Pain 2015; 16(7):616–627. doi:10.1016/j.jpain.2015.03.008
- Hoffman EM, Watson JC, St Sauver J, Staff NP, Klein CJ. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol 2017; 74(7):773–779. doi:10.1001/jamaneurol.2017.0486
- Harati Y, Gooch C, Swenson M, et al. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50(6):1842–1846. pmid:9633738
- Sima AA, Calvani M, Mehra M, Amato A; Acetyl-L-Carnitine Study Group. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28(1):89–94. pmid:15616239
- Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 1995; 38(12):1425–1433. pmid:8786016
- Scarpini E, Sacilotto G, Baron P, Cusini M, Scarlato G. Effect of acetyl-L-carnitine in the treatment of painful peripheral neuropathies in HIV+ patients. J Peripher Nerv Syst 1997; 2(3):250-252. pmid: 10975731
- Hershman DL, Unger JM, Crew KD, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol 2013; 31(20):2627-2633. doi:10.1200/JCO.2012.44.8738
- Amara S. Oral glutamine for the prevention of chemotherapy-induced peripheral neuropathy. Ann Pharmacother 2008; 42(10):1481-1485. doi:10.1345/aph.1L179
- Huang JS, Wu CL, Fan CW, Chen WH, Yeh KY, Chang PH. Intravenous glutamine appears to reduce the severity of symptomatic platinum-induced neuropathy: a prospective randomized study. J Chemother 2015; 27(4):235-240. doi:10.1179/1973947815Y.0000000011
- Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: possible involvement of opioid system. Eur J Pharmacol 2014; 723:202-206. doi:10.1016/j.ejphar.2013.11.033
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MD, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205-211. doi:10.1016/j.neuro.2012.09.011
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain 2017; 21(3):456-465. doi:10.1002/ejp.939
- Lewis EJ, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology 2017; 88(24):2294–2301. doi:10.1212/WNL.0000000000004033
- Hu D, Wang C, Li F, et al. A combined water extract of frankincense and myrrh alleviates neuropathic pain in mice via modulation of TRPV1. Neural Plast 2017; 2017:3710821. doi:10.1155/2017/3710821
- Tavee J, Rensel M, Planchon SM, Butler RS, Stone L. Effects of meditation on pain and quality of life in multiple sclerosis and peripheral neuropathy: a pilot study. Int J MS Care 2011; 13(4):163–168. doi:10.7224/1537-2073-13.4.163
- Khoshnoodi MA, Truelove S, Burakgazi A, Hoke A, Mammen AL, Polydefkis M. Longitudinal assessment of small fiber neuropathy: evidence of a non-length-dependent distal axonopathy. JAMA Neurol 2016; 73(6):684–690. doi:10.1001/jamaneurol.2016.0057
KEY POINTS
- Patients typically develop a symmetric “stocking-glove” pattern of sensory loss in the feet and hands.
- The diagnosis may be confirmed with skin biopsy for nerve fiber density, which can easily be done in a clinic setting with commercially available kits.
- Diabetes is the most common identifiable cause of small fiber neuropathy.
- Serologic testing can help uncover a vitamin deficiency or other potentially treatable condition.
- Antiepileptics, antidepressants, and topical agents are first-line drugs for managing pain.
Genitourinary syndrome of menopause in breast cancer survivors: Treatments are available
Many breast cancer survivors and women at high risk of breast cancer suffer from genitourinary syndrome of menopause (GSM), a term that encompasses any urinary, genital, or sexual dysfunction related to a hypoestrogenic state. Although GSM is usually caused by postmenopausal estrogen loss, it can also be caused by cancer treatments such as chemotherapy, radiation, and systemic endocrine therapy (eg, tamoxifen, aromatase inhibitors). These treatments can substantially decrease systemic estrogen levels, causing GSM symptoms that can profoundly worsen quality of life.
Managing GSM in these women poses a dilemma because systemic estrogen-containing therapies can increase the risk of breast cancer, and nonhormonal vaginal lubricants and moisturizers provide only minimal benefit. Fortunately, there are hormonal options, including locally applied estrogen, intravaginal dehydroepiandrosterone (DHEA), and estrogen receptor agonists/antagonists (ospemifene and bazedoxifene).
Here, we review the clinical management of GSM in breast cancer survivors and women at high risk of breast cancer and the efficacy and safety of available treatments, including their impact on breast cancer risk.
DRYNESS, IRRITATION, ATROPHY
The term GSM describes vulvovaginal and genitourinary symptoms associated with estrogen loss after menopause. Common symptoms are vaginal dryness, dyspareunia, irritation of genital skin, and pruritus.

LOCAL ESTROGEN THERAPY
Systemic estrogen therapy is widely used and effective for GSM, but there are concerns that it could increase the risk of breast cancer. After the Women’s Health Initiative in 2002 showed higher rates of cardiovascular disease and breast cancer with systemic estrogen-progestin use,5 the use of this hormone therapy declined by approximately 80%.6 Since then, healthcare providers have turned to local (ie, vaginal) estrogen therapies to manage GSM. These therapies have several advantages over systemic hormone therapy:
- Lower risk of adverse effects on the breast and cardiovascular system
- Greater efficacy in treating GSM
- In general, no need for progesterone when low-dose local estrogen is given to a woman with a uterus.7
Is locally applied estrogen systemically absorbed?
Despite these advantages, concerns remain as to whether vaginal estrogen therapy has adverse consequences associated with systemic absorption, particularly from atrophic vaginal tissues.
Santen,8 in a 2015 review of 33 studies, concluded that systemic absorption from low-dose vaginal estrogen is minimal, which provides some rationale for using it to treat vulvovaginal atrophy in postmenopausal women. This finding also suggests that the US Food and Drug Administration (FDA) “black box” warning of possible toxicities with vaginal estrogen is likely overstated, given that serum estrogen levels remained within normal postmenopausal levels.
Nevertheless, many providers are apprehensive about prescribing vaginal estrogen in women with a history of breast cancer because the threshold for systemic estrogen levels associated with breast cancer recurrence has not been established.
ACOG statement. In 2016, a committee of the American College of Obstetricians and Gynecologists cited data showing that low-dose vaginal estrogens do not result in sustained serum estrogen levels exceeding the normal postmenopausal range, and that the use of vaginal estrogens does not increase the risk of cancer recurrence.9 However, they recommend caution with vaginal estrogen use, especially in women with a history of estrogen-dependent breast cancer, reserving it for patients with GSM symptoms nonresponsive to nonhormonal treatment and specifying that it be used in low doses.
Vaginal estrogen formulations

Several types of locally applied estrogens are available, each with different properties and affinity for estrogen receptors. These include conjugated estrogens, 17-beta estradiol, estradiol acetate, and estradiol hemihydrate. Three delivery systems are FDA-approved: creams, rings, and tablets (Table 2).
Vaginal creams. Two vaginal creams are available, one (Estrace) containing 17-beta estradiol and the other (Premarin) containing conjugated estrogens.
The FDA-approved dosage for 17-beta estradiol is 2 to 4 g/day for 1 or 2 weeks, then gradually reduced to half the dose for a similar time. Maintenance dosing is 1 g 1 to 3 times per week. However, the ACOG statement notes that the FDA-approved dosages are higher than those proven to be effective and currently used in clinical practice, eg, 0.5 g twice a week.9
The FDA-approved dosage of conjugated estrogen cream for moderate to severe dyspareunia is 0.5 g daily for 21 days, then off for 7 days, or 0.5 g twice a week.
Vaginal tablets. The vaginal tablet Vagifem and its generic equivalent Yuvafem contain 10 µg of estradiol hemihydrate. The FDA-approved dosage is 10 µg daily for 2 weeks, followed by 10 µg twice a week, inserted into the lower third of the vagina. This dosage is significantly lower than that of estrogen creams.
Vaginal insert. A newly approved vaginal insert (Imvexxy) contains estradiol 4 µg (the lowest dose of vaginal estradiol available) or 10 µg, in a coconut oil vehicle. Its indications are for moderate to severe dyspareunia due to menopause and atrophic vaginitis due to menopause. A study cited in its package insert (www.accessdata.fda.gov/drugsatfda_docs/label/2018/208564s000lbl.pdf) showed that, in patients who used this product, systemic absorption of estradiol remained within the postmenopausal range. Its effects on breast cancer have not yet been studied.
Vaginal rings. Two vaginal rings are marketed. One (Estring) contains 17-beta estradiol, and the other (Femring) contains estradiol acetate. Only the 17-beta estradiol ring delivers a low dose to vaginal tissues, releasing 7.5 µg/day for 90 days. The estradiol acetate ring releases 0.05 mg/day or 0.10 mg/day and is a systemic treatment meant to be used with a progestin, not for local therapy.
VAGINAL ANDROGEN THERAPY: DHEA
After menopause, as the ovaries stop making estrogen from androstenedione, some production continues in other tissues, with DHEA as the primary precursor of androgens that are ultimately converted to estrogen. This has led to the theory that the cause of GSM is not estrogen deficiency but androgen deficiency. Evidence reviewed by Labrie et al11 shows that vulvovaginal atrophy is caused by decreased DHEA availability, which in turn causes sex steroid deficiency-related menopausal symptoms.11 Thus, it is reasonable to conclude that menopausal symptoms can be relieved by giving DHEA.
This theory has been borne out in clinical trials, in which DHEA in a vaginal tablet formulation increased the maturation of vaginal cells and lowered vaginal pH, leading to relief of GSM symptoms.12
The only DHEA product FDA-approved for treating GSM-related symptoms is prasterone (Intrarosa), indicated for moderate to severe dyspareunia due to vulvovaginal atrophy. The recommended dosing is a single 6.5-mg intravaginal tablet (0.5% prasterone) inserted nightly at bedtime. Its efficacy for treating hypoactive sexual desire disorder in postmenopausal women is being investigated.
Breast cancer implications
Because DHEA is converted to estrogen by aromatization, healthcare providers might hesitate to use it in women who have a history of hormone-sensitive cancer. Data on the safety of intravaginal DHEA use in breast cancer survivors are limited. However, studies have found that prasterone has highly beneficial effects on dyspareunia, vaginal dryness, and objective signs of vulvovaginal atrophy without significant drug-related adverse effects.12,13 In these studies, serum estrogen levels in women treated with DHEA were within the values observed in normal postmenopausal women. In addition, there are no aromatase enzymes in the endometrium, so even high doses of vaginal DHEA (in contrast to high doses of vaginal estrogen) will not stimulate the endometrium.
Clinically, this evidence indicates that DHEA exerts both estrogenic and androgenic activity in the vagina without increasing serum estrogen levels, making it a good alternative to topical estrogen therapy.
OSPEMIFENE: AN ESTROGEN RECEPTOR AGONIST/ANTAGONIST
Ospemifene (Osphena) is an estrogen receptor agonist/antagonist, a class of drugs previously called selective estrogen receptor modulators (SERMs). It is FDA-approved to treat moderate to severe dyspareunia secondary to vulvar and vaginal atrophy.
Ospemifene has unique estrogenic effects on the vaginal mucosa, having been shown to increase the number of epithelial cells, lower the vaginal pH, and decrease the percentage of parabasal cells seen on Papanicolaou smears after 12 weeks of use.14
Unlike tamoxifen, another drug of this class, ospemifene does not change the endometrial lining.14 Similarly, ospemifene acts as an estrogenic agonist in bone and, thus, has the potential for use in preventing and managing osteoporosis or for use in women at risk of fractures.
Breast cancer impact
In preclinical trials, ospemifene was found to have antiestrogenic effects on breast tissue, similar to those seen with tamoxifen.
In a model using human tumor grafts, ospemifene decreased tumor growth in mice implanted with estrogen receptor-positive breast cancer cells.15
In a mouse model using breast cancer cells that were biologically and histologically similar to those of humans, ospemifene had strong antiestrogenic effects on the breast tissue.16 The evidence suggests that ospemifene has a favorable effect on vulvar and vaginal atrophy.17
Ospemifene is FDA-approved to treat moderate to severe dyspareunia secondary to menopause. Recommended dosing is 60 mg/day orally with food.
Its antiestrogenic effects on breast tissue make it a promising option for women with a history of estrogen-receptor positive breast cancer. However, further study is needed to fully understand its effects on human breast tissue. According to the manufacturer’s package insert (www.osphena.com/files/pdf/osphena_prescribing_information.pdf), because the drug has not been adequately studied in women with breast cancer, it should not be used in women with known or suspected breast cancer or a history of breast cancer.
CONJUGATED ESTROGENS PLUS BAZEDOXIFENE
The combination of conjugated estrogens and bazedoxifene (Duavee) is a progesterone-free alternative for treating various menopausal symptoms. Bazedoxifene is another estrogen receptor agonist/antagonist, and it was added to counteract estrogen’s effects on the endometrium, thus replacing progesterone. This protective effect has been validated in clinical trials, which also found a favorable safety profile in breast tissue.18,19
SMART trials. The efficacy of this combination was studied in a series of large phase 3 multicenter trials called the SMART (Selective Estrogens, Menopause, and Response to Therapy) trials.20–23 Treated patients had markedly fewer vasomotor symptoms at 1 year, along with an increase in superficial cells and intermediate cells of the vaginal epithelium and a decrease in parabasal cells. They also had a substantial decrease in the incidence of dyspareunia.
Its effects on breast tissue were evaluated in the SMART-5 trial. Therapy had no net impact on breast density, suggesting that it has an estrogen-neutral effect on the breast.23
These results suggest that combined conjugated estrogens and bazedoxifene could be a noteworthy treatment option for GSM in women with a history of estrogen receptor-positive breast cancer, particularly in those with vasomotor symptoms and bone loss. However, the combination has not been studied specifically in breast cancer survivors.
Dosage. The FDA-approved dosing is 20 mg/0.45 mg per day orally to treat vasomotor symptoms, GSM, and osteoporosis in postmenopausal women with a uterus.
LASER THERAPY AND RADIOFREQUENCY HEAT: AN OFF-LABEL OPTION
Low-dose radiofrequency thermal therapy, delivered by carbon dioxide laser or by radiofrequency heat, has been used with some success to treat urinary stress incontinence and vaginal laxity in postpartum women. It may be an option for GSM, although it is not FDA-approved for this indication, and the FDA has recently issued a warning about it.24
Marketing literature promotes laser therapy as an effective option that stimulates vaginal connective tissue to produce new collagen, which then promotes improved blood flow and tissue regeneration for vaginal lubrication and elasticity.
A study comparing fractional carbon dioxide vaginal laser treatment and local estrogen therapy in postmenopausal women with vulvovaginal atrophy found that laser therapy was an effective treatment for vulvovaginal atrophy (dyspareunia, dryness, and burning), both alone and with local estrogen.25
Despite the promising effects of laser therapy for treating vulvovaginal atrophy in GSM, studies have not determined its short-term or long-term safety profile. Furthermore, laser therapy does not improve impaired sexual function, ie, decreased libido, arousal, and sexual satisfaction. Another important consideration is that the cost of laser therapy in 2017 was estimated to be $2,000 to $3,000 per treatment, not covered by healthcare insurance.
CLINICAL APPROACH
Symptoms of GSM are common in breast cancer survivors, both pre- and postmenopausal, especially those treated with tamoxifen or an aromatase inhibitor. Estimates are that 60% of postmenopausal breast cancer survivors and 40% of premenopausal breast cancer survivors suffer from GSM.26 Unfortunately, many women do not seek medical attention for their symptoms.
A variety of hormonal and nonhormonal options are available for these patients. We recommend an interdisciplinary approach to treatment, with the decision to use hormonal options made in collaboration with the patient’s oncologist and the patient herself, in an informed, shared decision-making process that takes into consideration the risks and possible benefits depending on the symptoms.
The first step in selecting a management plan for GSM symptoms in women with breast cancer is to conduct a thorough assessment to provide data for individualizing the care plan. The decision to use a hormonal option should be made in collaboration with a woman’s oncologist and should include an informed decision-making process during which the potential risks and benefits, including the breast cancer impact, are fully disclosed.
Alternatives to systemic estrogen
Vaginal estrogen is an effective and safe option to treat GSM in women with either estrogen receptor-negative or estrogen receptor-positive breast cancer. It often completely cures the symptoms without any noticeable increase in serum estrogen levels.
Vaginal DHEA therapy is a nonestrogen option shown to effectively treat GSM without increasing systemic levels of estrogen or testosterone. This profile makes vaginal DHEA therapy a particularly attractive treatment for symptoms of GSM in women at risk for breast cancer.
Use of an estrogen receptor agonist/antagonist in breast cancer survivors needs careful consideration. Ospemifene has antiestrogenic effects that make it a good option for women with bone loss and those at high risk for breast cancer, but it should not be used concurrently with tamoxifen or raloxifene. Additionally, ospemifene does not cause uterine hyperplasia, so it can be used in women with a uterus.
Although more study is needed, we do have options to improve the overall quality of life in breast cancer survivors with GSM.
- Lester J, Pahouja G, Andersen B, Lustberg M. Atrophic vaginitis in breast cancer survivors: a difficult survivorship issue. J Pers Med 2015; 5(2):50–66. doi:10.3390/jpm5020050
- Chin SN, Trinkaus M, Simmons C, et al. Prevalence and severity of urogenital symptoms in postmenopausal women receiving endocrine therapy for breast cancer. Clin Breast Cancer 2009; 9(2):108–117. doi:10.3816/CBC.2009.n.020
- Fallowfield L, Cella D, Cuzick J, Francis S, Locker G, Howell A. Quality of life of postmenopausal women in the Arimidex, Tamoxifen, Alone or in Combination (ATAC) adjuvant breast cancer trial. J Clin Oncol 2004; 22(21):4261–4271. doi:10.1200/JCO.2004.08.029
- Cella D, Fallowfield LJ. Recognition and management of treatment-related side effects for breast cancer patients receiving adjuvant endocrine therapy. Breast Cancer Res Treat 2008; 107(2):167–180. doi:10.1007/s10549-007-9548-1
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288(3):321–333. pmid:12117397
- Tsai SA, Stefanick ML, Stafford RS. Trends in menopausal hormone therapy use of US office-based physicians, 2000–2009. Menopause 2011; 18(4):385–392. doi:10.1097/gme.0b013e3181f43404
- North American Menopause Society. Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–902. doi:10.1097/GME.0b013e3182a122c2
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–134. doi:10.3109/13697137.2014.947254
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice, Farrell R. ACOG Committee Opinion No. 659: the use of vaginal estrogen in women with a history of estrogen-dependent breast cancer. Obstet Gynecol 2016; 127(3):e93–e96. doi:10.1097/AOG.0000000000001351
- Santoro N, Epperson CN, Mathews SB. Menopausal symptoms and their management. Endocrinol Metab Clin North Am 2015; 44(3):497–515. doi:10.1016/j.ecl.2015.05.001
- Labrie F, Archer DF, Martel C, Vaillancourt M, Montesino M. Combined data of intravaginal prasterone against vulvovaginal atrophy of menopause. Menopause 2017; 24(11):1246–1256. doi:10.1097/GME.0000000000000910
- Labrie F, Archer D, Bouchard C, et al. Serum steroid levels during 12-week intravaginal dehydroepiandrosterone administration. Menopause 2009; 16(5):897–906. doi:10.1097/gme.0b013e31819e8930
- Labrie F, Cusan L, Gomez JL, et al. Effect of intravaginal DHEA on serum DHEA and eleven of its metabolites in postmenopausal women. J Steroid Biochem Mol Biol 2008; 111(3-5):178–194. doi:10.1016/j.jsbmb.2008.06.003
- Soe LH, Wurz GT, Kao CJ, Degregorio MW. Ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy: potential benefits in bone and breast. Int J Womens Health 2013; 5:605–611. doi:10.2147/IJWH.S39146
- Taras TL, Wurz GT, DeGregorio MW. In vitro and in vivo biologic effects of ospemifene (FC-1271a) in breast cancer. J Steroid Biochem Mol Biol 2001; 77(4–5):271–279. pmid:11457665
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97(3):230–240. doi:10.1016/j.jsbmb.2005.06.027
- Archer DF, Carr BR, Pinkerton JV, Taylor HS, Constantine GD. Effects of ospemifene on the female reproductive and urinary tracts: translation from preclinical models into clinical evidence. Menopause 2015; 22(7):786–796. doi:10.1097/GME.0000000000000365
- Mirkin S, Pickar JH. Management of osteoporosis and menopausal symptoms: focus on bazedoxifene/conjugated estrogen combination. Int J Womens Health 2013; 5:465–475. doi:10.2147/IJWH.S39455
- Kagan R, Goldstein SR, Pickar JH, Komm BS. Patient considerations in the management of menopausal symptoms: role of conjugated estrogens with bazedoxifene. Ther Clin Risk Manag 2016; 12:549–562. doi:10.2147/TCRM.S63833
- Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril 2009; 92(3):1025–1038. doi:10.1016/j.fertnstert.2009.03.113
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16(6):1116–1124. doi:10.1097/gme.0b013e3181a7df0d
- Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause 2010; 17(2):281–289. doi:10.1097/GME.0b013e3181b7c65f
- Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens: a randomized controlled trial. Obstet Gynecol 2013; 121(5):959–968. doi:10.1097/AOG.0b013e31828c5974
- FDA. U.S. Food & Drug Administration. FDA Statement. Statement from FDA Commissioner Scott Gottlieb, M.D., on efforts to safeguard women’s health from deceptive health claims and significant risks related to devices marketed for use in medical procedures for “vaginal rejuvenation.” www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm615130.htm. Accessed August 20, 2018.
- Cruz VL, Steiner ML, Pompei LM, et al. Randomized, double-blind, placebo-controlled clinical trial for evaluating the efficacy of fractional CO2 laser compared with topical estriol in the treatment of vaginal atrophy in postmenopausal women. Menopause 2018; 25(1):21–28. doi:10.1097/GME.0000000000000955
- Biglia N, Bounous VE, D’Alonzo M, et al. Vaginal atrophy in breast cancer survivors: attitude and approaches among oncologists. Clin Breast Cancer 2017; 17(8):611–617. doi:10.1016/j.clbc.2017.05.008
Many breast cancer survivors and women at high risk of breast cancer suffer from genitourinary syndrome of menopause (GSM), a term that encompasses any urinary, genital, or sexual dysfunction related to a hypoestrogenic state. Although GSM is usually caused by postmenopausal estrogen loss, it can also be caused by cancer treatments such as chemotherapy, radiation, and systemic endocrine therapy (eg, tamoxifen, aromatase inhibitors). These treatments can substantially decrease systemic estrogen levels, causing GSM symptoms that can profoundly worsen quality of life.
Managing GSM in these women poses a dilemma because systemic estrogen-containing therapies can increase the risk of breast cancer, and nonhormonal vaginal lubricants and moisturizers provide only minimal benefit. Fortunately, there are hormonal options, including locally applied estrogen, intravaginal dehydroepiandrosterone (DHEA), and estrogen receptor agonists/antagonists (ospemifene and bazedoxifene).
Here, we review the clinical management of GSM in breast cancer survivors and women at high risk of breast cancer and the efficacy and safety of available treatments, including their impact on breast cancer risk.
DRYNESS, IRRITATION, ATROPHY
The term GSM describes vulvovaginal and genitourinary symptoms associated with estrogen loss after menopause. Common symptoms are vaginal dryness, dyspareunia, irritation of genital skin, and pruritus.
LOCAL ESTROGEN THERAPY
Systemic estrogen therapy is widely used and effective for GSM, but there are concerns that it could increase the risk of breast cancer. After the Women’s Health Initiative in 2002 showed higher rates of cardiovascular disease and breast cancer with systemic estrogen-progestin use,5 the use of this hormone therapy declined by approximately 80%.6 Since then, healthcare providers have turned to local (ie, vaginal) estrogen therapies to manage GSM. These therapies have several advantages over systemic hormone therapy:
- Lower risk of adverse effects on the breast and cardiovascular system
- Greater efficacy in treating GSM
- In general, no need for progesterone when low-dose local estrogen is given to a woman with a uterus.7
Is locally applied estrogen systemically absorbed?
Despite these advantages, concerns remain as to whether vaginal estrogen therapy has adverse consequences associated with systemic absorption, particularly from atrophic vaginal tissues.
Santen,8 in a 2015 review of 33 studies, concluded that systemic absorption from low-dose vaginal estrogen is minimal, which provides some rationale for using it to treat vulvovaginal atrophy in postmenopausal women. This finding also suggests that the US Food and Drug Administration (FDA) “black box” warning of possible toxicities with vaginal estrogen is likely overstated, given that serum estrogen levels remained within normal postmenopausal levels.
Nevertheless, many providers are apprehensive about prescribing vaginal estrogen in women with a history of breast cancer because the threshold for systemic estrogen levels associated with breast cancer recurrence has not been established.
ACOG statement. In 2016, a committee of the American College of Obstetricians and Gynecologists cited data showing that low-dose vaginal estrogens do not result in sustained serum estrogen levels exceeding the normal postmenopausal range, and that the use of vaginal estrogens does not increase the risk of cancer recurrence.9 However, they recommend caution with vaginal estrogen use, especially in women with a history of estrogen-dependent breast cancer, reserving it for patients with GSM symptoms nonresponsive to nonhormonal treatment and specifying that it be used in low doses.
Vaginal estrogen formulations
Several types of locally applied estrogens are available, each with different properties and affinity for estrogen receptors. These include conjugated estrogens, 17-beta estradiol, estradiol acetate, and estradiol hemihydrate. Three delivery systems are FDA-approved: creams, rings, and tablets (Table 2).
Vaginal creams. Two vaginal creams are available, one (Estrace) containing 17-beta estradiol and the other (Premarin) containing conjugated estrogens.
The FDA-approved dosage for 17-beta estradiol is 2 to 4 g/day for 1 or 2 weeks, then gradually reduced to half the dose for a similar time. Maintenance dosing is 1 g 1 to 3 times per week. However, the ACOG statement notes that the FDA-approved dosages are higher than those proven to be effective and currently used in clinical practice, eg, 0.5 g twice a week.9
The FDA-approved dosage of conjugated estrogen cream for moderate to severe dyspareunia is 0.5 g daily for 21 days, then off for 7 days, or 0.5 g twice a week.
Vaginal tablets. The vaginal tablet Vagifem and its generic equivalent Yuvafem contain 10 µg of estradiol hemihydrate. The FDA-approved dosage is 10 µg daily for 2 weeks, followed by 10 µg twice a week, inserted into the lower third of the vagina. This dosage is significantly lower than that of estrogen creams.
Vaginal insert. A newly approved vaginal insert (Imvexxy) contains estradiol 4 µg (the lowest dose of vaginal estradiol available) or 10 µg, in a coconut oil vehicle. Its indications are for moderate to severe dyspareunia due to menopause and atrophic vaginitis due to menopause. A study cited in its package insert (www.accessdata.fda.gov/drugsatfda_docs/label/2018/208564s000lbl.pdf) showed that, in patients who used this product, systemic absorption of estradiol remained within the postmenopausal range. Its effects on breast cancer have not yet been studied.
Vaginal rings. Two vaginal rings are marketed. One (Estring) contains 17-beta estradiol, and the other (Femring) contains estradiol acetate. Only the 17-beta estradiol ring delivers a low dose to vaginal tissues, releasing 7.5 µg/day for 90 days. The estradiol acetate ring releases 0.05 mg/day or 0.10 mg/day and is a systemic treatment meant to be used with a progestin, not for local therapy.
VAGINAL ANDROGEN THERAPY: DHEA
After menopause, as the ovaries stop making estrogen from androstenedione, some production continues in other tissues, with DHEA as the primary precursor of androgens that are ultimately converted to estrogen. This has led to the theory that the cause of GSM is not estrogen deficiency but androgen deficiency. Evidence reviewed by Labrie et al11 shows that vulvovaginal atrophy is caused by decreased DHEA availability, which in turn causes sex steroid deficiency-related menopausal symptoms.11 Thus, it is reasonable to conclude that menopausal symptoms can be relieved by giving DHEA.
This theory has been borne out in clinical trials, in which DHEA in a vaginal tablet formulation increased the maturation of vaginal cells and lowered vaginal pH, leading to relief of GSM symptoms.12
The only DHEA product FDA-approved for treating GSM-related symptoms is prasterone (Intrarosa), indicated for moderate to severe dyspareunia due to vulvovaginal atrophy. The recommended dosing is a single 6.5-mg intravaginal tablet (0.5% prasterone) inserted nightly at bedtime. Its efficacy for treating hypoactive sexual desire disorder in postmenopausal women is being investigated.
Breast cancer implications
Because DHEA is converted to estrogen by aromatization, healthcare providers might hesitate to use it in women who have a history of hormone-sensitive cancer. Data on the safety of intravaginal DHEA use in breast cancer survivors are limited. However, studies have found that prasterone has highly beneficial effects on dyspareunia, vaginal dryness, and objective signs of vulvovaginal atrophy without significant drug-related adverse effects.12,13 In these studies, serum estrogen levels in women treated with DHEA were within the values observed in normal postmenopausal women. In addition, there are no aromatase enzymes in the endometrium, so even high doses of vaginal DHEA (in contrast to high doses of vaginal estrogen) will not stimulate the endometrium.
Clinically, this evidence indicates that DHEA exerts both estrogenic and androgenic activity in the vagina without increasing serum estrogen levels, making it a good alternative to topical estrogen therapy.
OSPEMIFENE: AN ESTROGEN RECEPTOR AGONIST/ANTAGONIST
Ospemifene (Osphena) is an estrogen receptor agonist/antagonist, a class of drugs previously called selective estrogen receptor modulators (SERMs). It is FDA-approved to treat moderate to severe dyspareunia secondary to vulvar and vaginal atrophy.
Ospemifene has unique estrogenic effects on the vaginal mucosa, having been shown to increase the number of epithelial cells, lower the vaginal pH, and decrease the percentage of parabasal cells seen on Papanicolaou smears after 12 weeks of use.14
Unlike tamoxifen, another drug of this class, ospemifene does not change the endometrial lining.14 Similarly, ospemifene acts as an estrogenic agonist in bone and, thus, has the potential for use in preventing and managing osteoporosis or for use in women at risk of fractures.
Breast cancer impact
In preclinical trials, ospemifene was found to have antiestrogenic effects on breast tissue, similar to those seen with tamoxifen.
In a model using human tumor grafts, ospemifene decreased tumor growth in mice implanted with estrogen receptor-positive breast cancer cells.15
In a mouse model using breast cancer cells that were biologically and histologically similar to those of humans, ospemifene had strong antiestrogenic effects on the breast tissue.16 The evidence suggests that ospemifene has a favorable effect on vulvar and vaginal atrophy.17
Ospemifene is FDA-approved to treat moderate to severe dyspareunia secondary to menopause. Recommended dosing is 60 mg/day orally with food.
Its antiestrogenic effects on breast tissue make it a promising option for women with a history of estrogen-receptor positive breast cancer. However, further study is needed to fully understand its effects on human breast tissue. According to the manufacturer’s package insert (www.osphena.com/files/pdf/osphena_prescribing_information.pdf), because the drug has not been adequately studied in women with breast cancer, it should not be used in women with known or suspected breast cancer or a history of breast cancer.
CONJUGATED ESTROGENS PLUS BAZEDOXIFENE
The combination of conjugated estrogens and bazedoxifene (Duavee) is a progesterone-free alternative for treating various menopausal symptoms. Bazedoxifene is another estrogen receptor agonist/antagonist, and it was added to counteract estrogen’s effects on the endometrium, thus replacing progesterone. This protective effect has been validated in clinical trials, which also found a favorable safety profile in breast tissue.18,19
SMART trials. The efficacy of this combination was studied in a series of large phase 3 multicenter trials called the SMART (Selective Estrogens, Menopause, and Response to Therapy) trials.20–23 Treated patients had markedly fewer vasomotor symptoms at 1 year, along with an increase in superficial cells and intermediate cells of the vaginal epithelium and a decrease in parabasal cells. They also had a substantial decrease in the incidence of dyspareunia.
Its effects on breast tissue were evaluated in the SMART-5 trial. Therapy had no net impact on breast density, suggesting that it has an estrogen-neutral effect on the breast.23
These results suggest that combined conjugated estrogens and bazedoxifene could be a noteworthy treatment option for GSM in women with a history of estrogen receptor-positive breast cancer, particularly in those with vasomotor symptoms and bone loss. However, the combination has not been studied specifically in breast cancer survivors.
Dosage. The FDA-approved dosing is 20 mg/0.45 mg per day orally to treat vasomotor symptoms, GSM, and osteoporosis in postmenopausal women with a uterus.
LASER THERAPY AND RADIOFREQUENCY HEAT: AN OFF-LABEL OPTION
Low-dose radiofrequency thermal therapy, delivered by carbon dioxide laser or by radiofrequency heat, has been used with some success to treat urinary stress incontinence and vaginal laxity in postpartum women. It may be an option for GSM, although it is not FDA-approved for this indication, and the FDA has recently issued a warning about it.24
Marketing literature promotes laser therapy as an effective option that stimulates vaginal connective tissue to produce new collagen, which then promotes improved blood flow and tissue regeneration for vaginal lubrication and elasticity.
A study comparing fractional carbon dioxide vaginal laser treatment and local estrogen therapy in postmenopausal women with vulvovaginal atrophy found that laser therapy was an effective treatment for vulvovaginal atrophy (dyspareunia, dryness, and burning), both alone and with local estrogen.25
Despite the promising effects of laser therapy for treating vulvovaginal atrophy in GSM, studies have not determined its short-term or long-term safety profile. Furthermore, laser therapy does not improve impaired sexual function, ie, decreased libido, arousal, and sexual satisfaction. Another important consideration is that the cost of laser therapy in 2017 was estimated to be $2,000 to $3,000 per treatment, not covered by healthcare insurance.
CLINICAL APPROACH
Symptoms of GSM are common in breast cancer survivors, both pre- and postmenopausal, especially those treated with tamoxifen or an aromatase inhibitor. Estimates are that 60% of postmenopausal breast cancer survivors and 40% of premenopausal breast cancer survivors suffer from GSM.26 Unfortunately, many women do not seek medical attention for their symptoms.
A variety of hormonal and nonhormonal options are available for these patients. We recommend an interdisciplinary approach to treatment, with the decision to use hormonal options made in collaboration with the patient’s oncologist and the patient herself, in an informed, shared decision-making process that takes into consideration the risks and possible benefits depending on the symptoms.
The first step in selecting a management plan for GSM symptoms in women with breast cancer is to conduct a thorough assessment to provide data for individualizing the care plan. The decision to use a hormonal option should be made in collaboration with a woman’s oncologist and should include an informed decision-making process during which the potential risks and benefits, including the breast cancer impact, are fully disclosed.
Alternatives to systemic estrogen
Vaginal estrogen is an effective and safe option to treat GSM in women with either estrogen receptor-negative or estrogen receptor-positive breast cancer. It often completely cures the symptoms without any noticeable increase in serum estrogen levels.
Vaginal DHEA therapy is a nonestrogen option shown to effectively treat GSM without increasing systemic levels of estrogen or testosterone. This profile makes vaginal DHEA therapy a particularly attractive treatment for symptoms of GSM in women at risk for breast cancer.
Use of an estrogen receptor agonist/antagonist in breast cancer survivors needs careful consideration. Ospemifene has antiestrogenic effects that make it a good option for women with bone loss and those at high risk for breast cancer, but it should not be used concurrently with tamoxifen or raloxifene. Additionally, ospemifene does not cause uterine hyperplasia, so it can be used in women with a uterus.
Although more study is needed, we do have options to improve the overall quality of life in breast cancer survivors with GSM.
Many breast cancer survivors and women at high risk of breast cancer suffer from genitourinary syndrome of menopause (GSM), a term that encompasses any urinary, genital, or sexual dysfunction related to a hypoestrogenic state. Although GSM is usually caused by postmenopausal estrogen loss, it can also be caused by cancer treatments such as chemotherapy, radiation, and systemic endocrine therapy (eg, tamoxifen, aromatase inhibitors). These treatments can substantially decrease systemic estrogen levels, causing GSM symptoms that can profoundly worsen quality of life.
Managing GSM in these women poses a dilemma because systemic estrogen-containing therapies can increase the risk of breast cancer, and nonhormonal vaginal lubricants and moisturizers provide only minimal benefit. Fortunately, there are hormonal options, including locally applied estrogen, intravaginal dehydroepiandrosterone (DHEA), and estrogen receptor agonists/antagonists (ospemifene and bazedoxifene).
Here, we review the clinical management of GSM in breast cancer survivors and women at high risk of breast cancer and the efficacy and safety of available treatments, including their impact on breast cancer risk.
DRYNESS, IRRITATION, ATROPHY
The term GSM describes vulvovaginal and genitourinary symptoms associated with estrogen loss after menopause. Common symptoms are vaginal dryness, dyspareunia, irritation of genital skin, and pruritus.
LOCAL ESTROGEN THERAPY
Systemic estrogen therapy is widely used and effective for GSM, but there are concerns that it could increase the risk of breast cancer. After the Women’s Health Initiative in 2002 showed higher rates of cardiovascular disease and breast cancer with systemic estrogen-progestin use,5 the use of this hormone therapy declined by approximately 80%.6 Since then, healthcare providers have turned to local (ie, vaginal) estrogen therapies to manage GSM. These therapies have several advantages over systemic hormone therapy:
- Lower risk of adverse effects on the breast and cardiovascular system
- Greater efficacy in treating GSM
- In general, no need for progesterone when low-dose local estrogen is given to a woman with a uterus.7
Is locally applied estrogen systemically absorbed?
Despite these advantages, concerns remain as to whether vaginal estrogen therapy has adverse consequences associated with systemic absorption, particularly from atrophic vaginal tissues.
Santen,8 in a 2015 review of 33 studies, concluded that systemic absorption from low-dose vaginal estrogen is minimal, which provides some rationale for using it to treat vulvovaginal atrophy in postmenopausal women. This finding also suggests that the US Food and Drug Administration (FDA) “black box” warning of possible toxicities with vaginal estrogen is likely overstated, given that serum estrogen levels remained within normal postmenopausal levels.
Nevertheless, many providers are apprehensive about prescribing vaginal estrogen in women with a history of breast cancer because the threshold for systemic estrogen levels associated with breast cancer recurrence has not been established.
ACOG statement. In 2016, a committee of the American College of Obstetricians and Gynecologists cited data showing that low-dose vaginal estrogens do not result in sustained serum estrogen levels exceeding the normal postmenopausal range, and that the use of vaginal estrogens does not increase the risk of cancer recurrence.9 However, they recommend caution with vaginal estrogen use, especially in women with a history of estrogen-dependent breast cancer, reserving it for patients with GSM symptoms nonresponsive to nonhormonal treatment and specifying that it be used in low doses.
Vaginal estrogen formulations
Several types of locally applied estrogens are available, each with different properties and affinity for estrogen receptors. These include conjugated estrogens, 17-beta estradiol, estradiol acetate, and estradiol hemihydrate. Three delivery systems are FDA-approved: creams, rings, and tablets (Table 2).
Vaginal creams. Two vaginal creams are available, one (Estrace) containing 17-beta estradiol and the other (Premarin) containing conjugated estrogens.
The FDA-approved dosage for 17-beta estradiol is 2 to 4 g/day for 1 or 2 weeks, then gradually reduced to half the dose for a similar time. Maintenance dosing is 1 g 1 to 3 times per week. However, the ACOG statement notes that the FDA-approved dosages are higher than those proven to be effective and currently used in clinical practice, eg, 0.5 g twice a week.9
The FDA-approved dosage of conjugated estrogen cream for moderate to severe dyspareunia is 0.5 g daily for 21 days, then off for 7 days, or 0.5 g twice a week.
Vaginal tablets. The vaginal tablet Vagifem and its generic equivalent Yuvafem contain 10 µg of estradiol hemihydrate. The FDA-approved dosage is 10 µg daily for 2 weeks, followed by 10 µg twice a week, inserted into the lower third of the vagina. This dosage is significantly lower than that of estrogen creams.
Vaginal insert. A newly approved vaginal insert (Imvexxy) contains estradiol 4 µg (the lowest dose of vaginal estradiol available) or 10 µg, in a coconut oil vehicle. Its indications are for moderate to severe dyspareunia due to menopause and atrophic vaginitis due to menopause. A study cited in its package insert (www.accessdata.fda.gov/drugsatfda_docs/label/2018/208564s000lbl.pdf) showed that, in patients who used this product, systemic absorption of estradiol remained within the postmenopausal range. Its effects on breast cancer have not yet been studied.
Vaginal rings. Two vaginal rings are marketed. One (Estring) contains 17-beta estradiol, and the other (Femring) contains estradiol acetate. Only the 17-beta estradiol ring delivers a low dose to vaginal tissues, releasing 7.5 µg/day for 90 days. The estradiol acetate ring releases 0.05 mg/day or 0.10 mg/day and is a systemic treatment meant to be used with a progestin, not for local therapy.
VAGINAL ANDROGEN THERAPY: DHEA
After menopause, as the ovaries stop making estrogen from androstenedione, some production continues in other tissues, with DHEA as the primary precursor of androgens that are ultimately converted to estrogen. This has led to the theory that the cause of GSM is not estrogen deficiency but androgen deficiency. Evidence reviewed by Labrie et al11 shows that vulvovaginal atrophy is caused by decreased DHEA availability, which in turn causes sex steroid deficiency-related menopausal symptoms.11 Thus, it is reasonable to conclude that menopausal symptoms can be relieved by giving DHEA.
This theory has been borne out in clinical trials, in which DHEA in a vaginal tablet formulation increased the maturation of vaginal cells and lowered vaginal pH, leading to relief of GSM symptoms.12
The only DHEA product FDA-approved for treating GSM-related symptoms is prasterone (Intrarosa), indicated for moderate to severe dyspareunia due to vulvovaginal atrophy. The recommended dosing is a single 6.5-mg intravaginal tablet (0.5% prasterone) inserted nightly at bedtime. Its efficacy for treating hypoactive sexual desire disorder in postmenopausal women is being investigated.
Breast cancer implications
Because DHEA is converted to estrogen by aromatization, healthcare providers might hesitate to use it in women who have a history of hormone-sensitive cancer. Data on the safety of intravaginal DHEA use in breast cancer survivors are limited. However, studies have found that prasterone has highly beneficial effects on dyspareunia, vaginal dryness, and objective signs of vulvovaginal atrophy without significant drug-related adverse effects.12,13 In these studies, serum estrogen levels in women treated with DHEA were within the values observed in normal postmenopausal women. In addition, there are no aromatase enzymes in the endometrium, so even high doses of vaginal DHEA (in contrast to high doses of vaginal estrogen) will not stimulate the endometrium.
Clinically, this evidence indicates that DHEA exerts both estrogenic and androgenic activity in the vagina without increasing serum estrogen levels, making it a good alternative to topical estrogen therapy.
OSPEMIFENE: AN ESTROGEN RECEPTOR AGONIST/ANTAGONIST
Ospemifene (Osphena) is an estrogen receptor agonist/antagonist, a class of drugs previously called selective estrogen receptor modulators (SERMs). It is FDA-approved to treat moderate to severe dyspareunia secondary to vulvar and vaginal atrophy.
Ospemifene has unique estrogenic effects on the vaginal mucosa, having been shown to increase the number of epithelial cells, lower the vaginal pH, and decrease the percentage of parabasal cells seen on Papanicolaou smears after 12 weeks of use.14
Unlike tamoxifen, another drug of this class, ospemifene does not change the endometrial lining.14 Similarly, ospemifene acts as an estrogenic agonist in bone and, thus, has the potential for use in preventing and managing osteoporosis or for use in women at risk of fractures.
Breast cancer impact
In preclinical trials, ospemifene was found to have antiestrogenic effects on breast tissue, similar to those seen with tamoxifen.
In a model using human tumor grafts, ospemifene decreased tumor growth in mice implanted with estrogen receptor-positive breast cancer cells.15
In a mouse model using breast cancer cells that were biologically and histologically similar to those of humans, ospemifene had strong antiestrogenic effects on the breast tissue.16 The evidence suggests that ospemifene has a favorable effect on vulvar and vaginal atrophy.17
Ospemifene is FDA-approved to treat moderate to severe dyspareunia secondary to menopause. Recommended dosing is 60 mg/day orally with food.
Its antiestrogenic effects on breast tissue make it a promising option for women with a history of estrogen-receptor positive breast cancer. However, further study is needed to fully understand its effects on human breast tissue. According to the manufacturer’s package insert (www.osphena.com/files/pdf/osphena_prescribing_information.pdf), because the drug has not been adequately studied in women with breast cancer, it should not be used in women with known or suspected breast cancer or a history of breast cancer.
CONJUGATED ESTROGENS PLUS BAZEDOXIFENE
The combination of conjugated estrogens and bazedoxifene (Duavee) is a progesterone-free alternative for treating various menopausal symptoms. Bazedoxifene is another estrogen receptor agonist/antagonist, and it was added to counteract estrogen’s effects on the endometrium, thus replacing progesterone. This protective effect has been validated in clinical trials, which also found a favorable safety profile in breast tissue.18,19
SMART trials. The efficacy of this combination was studied in a series of large phase 3 multicenter trials called the SMART (Selective Estrogens, Menopause, and Response to Therapy) trials.20–23 Treated patients had markedly fewer vasomotor symptoms at 1 year, along with an increase in superficial cells and intermediate cells of the vaginal epithelium and a decrease in parabasal cells. They also had a substantial decrease in the incidence of dyspareunia.
Its effects on breast tissue were evaluated in the SMART-5 trial. Therapy had no net impact on breast density, suggesting that it has an estrogen-neutral effect on the breast.23
These results suggest that combined conjugated estrogens and bazedoxifene could be a noteworthy treatment option for GSM in women with a history of estrogen receptor-positive breast cancer, particularly in those with vasomotor symptoms and bone loss. However, the combination has not been studied specifically in breast cancer survivors.
Dosage. The FDA-approved dosing is 20 mg/0.45 mg per day orally to treat vasomotor symptoms, GSM, and osteoporosis in postmenopausal women with a uterus.
LASER THERAPY AND RADIOFREQUENCY HEAT: AN OFF-LABEL OPTION
Low-dose radiofrequency thermal therapy, delivered by carbon dioxide laser or by radiofrequency heat, has been used with some success to treat urinary stress incontinence and vaginal laxity in postpartum women. It may be an option for GSM, although it is not FDA-approved for this indication, and the FDA has recently issued a warning about it.24
Marketing literature promotes laser therapy as an effective option that stimulates vaginal connective tissue to produce new collagen, which then promotes improved blood flow and tissue regeneration for vaginal lubrication and elasticity.
A study comparing fractional carbon dioxide vaginal laser treatment and local estrogen therapy in postmenopausal women with vulvovaginal atrophy found that laser therapy was an effective treatment for vulvovaginal atrophy (dyspareunia, dryness, and burning), both alone and with local estrogen.25
Despite the promising effects of laser therapy for treating vulvovaginal atrophy in GSM, studies have not determined its short-term or long-term safety profile. Furthermore, laser therapy does not improve impaired sexual function, ie, decreased libido, arousal, and sexual satisfaction. Another important consideration is that the cost of laser therapy in 2017 was estimated to be $2,000 to $3,000 per treatment, not covered by healthcare insurance.
CLINICAL APPROACH
Symptoms of GSM are common in breast cancer survivors, both pre- and postmenopausal, especially those treated with tamoxifen or an aromatase inhibitor. Estimates are that 60% of postmenopausal breast cancer survivors and 40% of premenopausal breast cancer survivors suffer from GSM.26 Unfortunately, many women do not seek medical attention for their symptoms.
A variety of hormonal and nonhormonal options are available for these patients. We recommend an interdisciplinary approach to treatment, with the decision to use hormonal options made in collaboration with the patient’s oncologist and the patient herself, in an informed, shared decision-making process that takes into consideration the risks and possible benefits depending on the symptoms.
The first step in selecting a management plan for GSM symptoms in women with breast cancer is to conduct a thorough assessment to provide data for individualizing the care plan. The decision to use a hormonal option should be made in collaboration with a woman’s oncologist and should include an informed decision-making process during which the potential risks and benefits, including the breast cancer impact, are fully disclosed.
Alternatives to systemic estrogen
Vaginal estrogen is an effective and safe option to treat GSM in women with either estrogen receptor-negative or estrogen receptor-positive breast cancer. It often completely cures the symptoms without any noticeable increase in serum estrogen levels.
Vaginal DHEA therapy is a nonestrogen option shown to effectively treat GSM without increasing systemic levels of estrogen or testosterone. This profile makes vaginal DHEA therapy a particularly attractive treatment for symptoms of GSM in women at risk for breast cancer.
Use of an estrogen receptor agonist/antagonist in breast cancer survivors needs careful consideration. Ospemifene has antiestrogenic effects that make it a good option for women with bone loss and those at high risk for breast cancer, but it should not be used concurrently with tamoxifen or raloxifene. Additionally, ospemifene does not cause uterine hyperplasia, so it can be used in women with a uterus.
Although more study is needed, we do have options to improve the overall quality of life in breast cancer survivors with GSM.
- Lester J, Pahouja G, Andersen B, Lustberg M. Atrophic vaginitis in breast cancer survivors: a difficult survivorship issue. J Pers Med 2015; 5(2):50–66. doi:10.3390/jpm5020050
- Chin SN, Trinkaus M, Simmons C, et al. Prevalence and severity of urogenital symptoms in postmenopausal women receiving endocrine therapy for breast cancer. Clin Breast Cancer 2009; 9(2):108–117. doi:10.3816/CBC.2009.n.020
- Fallowfield L, Cella D, Cuzick J, Francis S, Locker G, Howell A. Quality of life of postmenopausal women in the Arimidex, Tamoxifen, Alone or in Combination (ATAC) adjuvant breast cancer trial. J Clin Oncol 2004; 22(21):4261–4271. doi:10.1200/JCO.2004.08.029
- Cella D, Fallowfield LJ. Recognition and management of treatment-related side effects for breast cancer patients receiving adjuvant endocrine therapy. Breast Cancer Res Treat 2008; 107(2):167–180. doi:10.1007/s10549-007-9548-1
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288(3):321–333. pmid:12117397
- Tsai SA, Stefanick ML, Stafford RS. Trends in menopausal hormone therapy use of US office-based physicians, 2000–2009. Menopause 2011; 18(4):385–392. doi:10.1097/gme.0b013e3181f43404
- North American Menopause Society. Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–902. doi:10.1097/GME.0b013e3182a122c2
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–134. doi:10.3109/13697137.2014.947254
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice, Farrell R. ACOG Committee Opinion No. 659: the use of vaginal estrogen in women with a history of estrogen-dependent breast cancer. Obstet Gynecol 2016; 127(3):e93–e96. doi:10.1097/AOG.0000000000001351
- Santoro N, Epperson CN, Mathews SB. Menopausal symptoms and their management. Endocrinol Metab Clin North Am 2015; 44(3):497–515. doi:10.1016/j.ecl.2015.05.001
- Labrie F, Archer DF, Martel C, Vaillancourt M, Montesino M. Combined data of intravaginal prasterone against vulvovaginal atrophy of menopause. Menopause 2017; 24(11):1246–1256. doi:10.1097/GME.0000000000000910
- Labrie F, Archer D, Bouchard C, et al. Serum steroid levels during 12-week intravaginal dehydroepiandrosterone administration. Menopause 2009; 16(5):897–906. doi:10.1097/gme.0b013e31819e8930
- Labrie F, Cusan L, Gomez JL, et al. Effect of intravaginal DHEA on serum DHEA and eleven of its metabolites in postmenopausal women. J Steroid Biochem Mol Biol 2008; 111(3-5):178–194. doi:10.1016/j.jsbmb.2008.06.003
- Soe LH, Wurz GT, Kao CJ, Degregorio MW. Ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy: potential benefits in bone and breast. Int J Womens Health 2013; 5:605–611. doi:10.2147/IJWH.S39146
- Taras TL, Wurz GT, DeGregorio MW. In vitro and in vivo biologic effects of ospemifene (FC-1271a) in breast cancer. J Steroid Biochem Mol Biol 2001; 77(4–5):271–279. pmid:11457665
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97(3):230–240. doi:10.1016/j.jsbmb.2005.06.027
- Archer DF, Carr BR, Pinkerton JV, Taylor HS, Constantine GD. Effects of ospemifene on the female reproductive and urinary tracts: translation from preclinical models into clinical evidence. Menopause 2015; 22(7):786–796. doi:10.1097/GME.0000000000000365
- Mirkin S, Pickar JH. Management of osteoporosis and menopausal symptoms: focus on bazedoxifene/conjugated estrogen combination. Int J Womens Health 2013; 5:465–475. doi:10.2147/IJWH.S39455
- Kagan R, Goldstein SR, Pickar JH, Komm BS. Patient considerations in the management of menopausal symptoms: role of conjugated estrogens with bazedoxifene. Ther Clin Risk Manag 2016; 12:549–562. doi:10.2147/TCRM.S63833
- Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril 2009; 92(3):1025–1038. doi:10.1016/j.fertnstert.2009.03.113
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16(6):1116–1124. doi:10.1097/gme.0b013e3181a7df0d
- Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause 2010; 17(2):281–289. doi:10.1097/GME.0b013e3181b7c65f
- Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens: a randomized controlled trial. Obstet Gynecol 2013; 121(5):959–968. doi:10.1097/AOG.0b013e31828c5974
- FDA. U.S. Food & Drug Administration. FDA Statement. Statement from FDA Commissioner Scott Gottlieb, M.D., on efforts to safeguard women’s health from deceptive health claims and significant risks related to devices marketed for use in medical procedures for “vaginal rejuvenation.” www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm615130.htm. Accessed August 20, 2018.
- Cruz VL, Steiner ML, Pompei LM, et al. Randomized, double-blind, placebo-controlled clinical trial for evaluating the efficacy of fractional CO2 laser compared with topical estriol in the treatment of vaginal atrophy in postmenopausal women. Menopause 2018; 25(1):21–28. doi:10.1097/GME.0000000000000955
- Biglia N, Bounous VE, D’Alonzo M, et al. Vaginal atrophy in breast cancer survivors: attitude and approaches among oncologists. Clin Breast Cancer 2017; 17(8):611–617. doi:10.1016/j.clbc.2017.05.008
- Lester J, Pahouja G, Andersen B, Lustberg M. Atrophic vaginitis in breast cancer survivors: a difficult survivorship issue. J Pers Med 2015; 5(2):50–66. doi:10.3390/jpm5020050
- Chin SN, Trinkaus M, Simmons C, et al. Prevalence and severity of urogenital symptoms in postmenopausal women receiving endocrine therapy for breast cancer. Clin Breast Cancer 2009; 9(2):108–117. doi:10.3816/CBC.2009.n.020
- Fallowfield L, Cella D, Cuzick J, Francis S, Locker G, Howell A. Quality of life of postmenopausal women in the Arimidex, Tamoxifen, Alone or in Combination (ATAC) adjuvant breast cancer trial. J Clin Oncol 2004; 22(21):4261–4271. doi:10.1200/JCO.2004.08.029
- Cella D, Fallowfield LJ. Recognition and management of treatment-related side effects for breast cancer patients receiving adjuvant endocrine therapy. Breast Cancer Res Treat 2008; 107(2):167–180. doi:10.1007/s10549-007-9548-1
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288(3):321–333. pmid:12117397
- Tsai SA, Stefanick ML, Stafford RS. Trends in menopausal hormone therapy use of US office-based physicians, 2000–2009. Menopause 2011; 18(4):385–392. doi:10.1097/gme.0b013e3181f43404
- North American Menopause Society. Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause 2013; 20(9):888–902. doi:10.1097/GME.0b013e3182a122c2
- Santen RJ. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric 2015; 18(2):121–134. doi:10.3109/13697137.2014.947254
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice, Farrell R. ACOG Committee Opinion No. 659: the use of vaginal estrogen in women with a history of estrogen-dependent breast cancer. Obstet Gynecol 2016; 127(3):e93–e96. doi:10.1097/AOG.0000000000001351
- Santoro N, Epperson CN, Mathews SB. Menopausal symptoms and their management. Endocrinol Metab Clin North Am 2015; 44(3):497–515. doi:10.1016/j.ecl.2015.05.001
- Labrie F, Archer DF, Martel C, Vaillancourt M, Montesino M. Combined data of intravaginal prasterone against vulvovaginal atrophy of menopause. Menopause 2017; 24(11):1246–1256. doi:10.1097/GME.0000000000000910
- Labrie F, Archer D, Bouchard C, et al. Serum steroid levels during 12-week intravaginal dehydroepiandrosterone administration. Menopause 2009; 16(5):897–906. doi:10.1097/gme.0b013e31819e8930
- Labrie F, Cusan L, Gomez JL, et al. Effect of intravaginal DHEA on serum DHEA and eleven of its metabolites in postmenopausal women. J Steroid Biochem Mol Biol 2008; 111(3-5):178–194. doi:10.1016/j.jsbmb.2008.06.003
- Soe LH, Wurz GT, Kao CJ, Degregorio MW. Ospemifene for the treatment of dyspareunia associated with vulvar and vaginal atrophy: potential benefits in bone and breast. Int J Womens Health 2013; 5:605–611. doi:10.2147/IJWH.S39146
- Taras TL, Wurz GT, DeGregorio MW. In vitro and in vivo biologic effects of ospemifene (FC-1271a) in breast cancer. J Steroid Biochem Mol Biol 2001; 77(4–5):271–279. pmid:11457665
- Wurz GT, Read KC, Marchisano-Karpman C, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol 2005; 97(3):230–240. doi:10.1016/j.jsbmb.2005.06.027
- Archer DF, Carr BR, Pinkerton JV, Taylor HS, Constantine GD. Effects of ospemifene on the female reproductive and urinary tracts: translation from preclinical models into clinical evidence. Menopause 2015; 22(7):786–796. doi:10.1097/GME.0000000000000365
- Mirkin S, Pickar JH. Management of osteoporosis and menopausal symptoms: focus on bazedoxifene/conjugated estrogen combination. Int J Womens Health 2013; 5:465–475. doi:10.2147/IJWH.S39455
- Kagan R, Goldstein SR, Pickar JH, Komm BS. Patient considerations in the management of menopausal symptoms: role of conjugated estrogens with bazedoxifene. Ther Clin Risk Manag 2016; 12:549–562. doi:10.2147/TCRM.S63833
- Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril 2009; 92(3):1025–1038. doi:10.1016/j.fertnstert.2009.03.113
- Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause 2009; 16(6):1116–1124. doi:10.1097/gme.0b013e3181a7df0d
- Kagan R, Williams RS, Pan K, Mirkin S, Pickar JH. A randomized, placebo- and active-controlled trial of bazedoxifene/conjugated estrogens for treatment of moderate to severe vulvar/vaginal atrophy in postmenopausal women. Menopause 2010; 17(2):281–289. doi:10.1097/GME.0b013e3181b7c65f
- Pinkerton JV, Harvey JA, Pan K, et al. Breast effects of bazedoxifene-conjugated estrogens: a randomized controlled trial. Obstet Gynecol 2013; 121(5):959–968. doi:10.1097/AOG.0b013e31828c5974
- FDA. U.S. Food & Drug Administration. FDA Statement. Statement from FDA Commissioner Scott Gottlieb, M.D., on efforts to safeguard women’s health from deceptive health claims and significant risks related to devices marketed for use in medical procedures for “vaginal rejuvenation.” www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm615130.htm. Accessed August 20, 2018.
- Cruz VL, Steiner ML, Pompei LM, et al. Randomized, double-blind, placebo-controlled clinical trial for evaluating the efficacy of fractional CO2 laser compared with topical estriol in the treatment of vaginal atrophy in postmenopausal women. Menopause 2018; 25(1):21–28. doi:10.1097/GME.0000000000000955
- Biglia N, Bounous VE, D’Alonzo M, et al. Vaginal atrophy in breast cancer survivors: attitude and approaches among oncologists. Clin Breast Cancer 2017; 17(8):611–617. doi:10.1016/j.clbc.2017.05.008
KEY POINTS
- In general, locally applied hormonal therapies relieve GSM symptoms without increasing breast cancer risk.
- DHEA relieves vaginal symptoms without increasing serum estrogen levels.
- Ospemifene has antiestrogenic effects on breast tissue that make it an attractive option for women with breast cancer.
- The combination of conjugated estrogens and bazedoxifene offers a progesterone-free treatment for GSM symptoms in women desiring systemic hormone therapy.

Bicuspid aortic valve: Basics and beyond
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22


Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22
Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22
Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
KEY POINTS
- Associated aortopathies such as aortic root dilation, aneurysm, dissection, and coarctation may initially be asymptomatic.
- Regular surveillance with transthoracic echocardiography (TTE) is required.
- Transesophageal echocardiography should be performed if TTE does not clearly show the aorta and aortic root. Magnetic resonance imaging or computed tomographic angiography may also be needed to measure the aortic root and ascending thoracic aorta.
- If initial imaging is normal and there is no aortic dilation, repeat imaging should be done every 5 to 10 years. If any abnormality is found, annual surveillance is needed.
- Women with a bicuspid aortic valve who are contemplating pregnancy should undergo echocardiography first, and some may need to undergo surgery.
Ablation of atrial fibrillation: Facts for the referring physician
A 64-year-old man with hypertension but without known structural heart disease presents for a second opinion on management of his atrial fibrillation. The condition was first diagnosed at age 38, when he experienced palpitations and shortness of breath on exertion; at times he also experienced decreased endurance and fatigue without overt palpitations. At first, these episodes occurred about twice a year, and the patient was managed with a beta-blocker for rate control and an oral anticoagulant.
Over the past 10 years, the episodes have become more frequent and longer-lasting and have required frequent cardioversions. He was given flecainide for rhythm control but continued to have frequent episodes, and so about 1 year ago he was switched to amiodarone, which controlled his rhythm better. However, after reading about side effects of amiodarone, he decided to seek a second opinion.
He was evaluated by our team and eventually underwent radiofrequency ablation. During the procedure, he was noted to have diffuse scarring and fibrosis of his left atrium, and afterward he continued to require antiarrhythmic drugs to maintain sinus rhythm.
Should he have been referred sooner? What factors should primary care physicians consider when referring a patient with atrial fibrillation for ablation?
THE EPIDEMIC OF ATRIAL FIBRILLATION
Atrial fibrillation is a large and growing public health problem. In 2010, it was estimated to affect 2.7 to 6.1 million people in the United States, and with the rapid aging of our population, its prevalence is expected to rise to between 5.6 and 12 million by 2050.1–3 It is associated with significant morbidity, poor quality of life, and increased risk of death, heart failure, stroke, and cognitive impairment.
The number of new cases per year has increased over the years despite research and preventive measures, which may reflect aging of the population and increased survival rates in patients with cardiovascular or comorbid conditions.1,4
Thus, atrial fibrillation is one of the most common cardiovascular conditions encountered by primary care physicians and cardiologists, putting them at the forefront of its management. Proper treatment in its early stages and referral to a specialist for advanced management may alter its natural history and improve clinical outcomes.
HOW DOES ATRIAL FIBRILLATION ARISE AND PERSIST?
Much is still unknown about the pathogenesis of atrial fibrillation, but considerable progress has been made in the past few decades, opening the door for clinical ablative strategies.
Multiple wavelet hypothesis
Until the late 1980s, the most widely accepted conceptual mechanism of atrial fibrillation was the multiple wavelet hypothesis developed by Moe et al.5 According to this hypothesis, atrial fibrillation begins with multiple independent wavelets occurring simultaneously and spreading randomly throughout both atria, and it persists if there are a minimum number of coexisting wavelets, increased atrial mass, and heterogeneous conduction delays across the atrial tissue.
The surgical maze procedure, in which a series of incisions arranged in a maze-like pattern is created in the left atrium, was predicated on this model. The theory was that these surgical lesions would compartmentalize the atria into discrete electrical segments and thereby reduce the number of circulating random wavelets.6,7
However, experimental and clinical studies suggest that although randomly propagating wavelets can contribute to maintaining atrial fibrillation, focal triggers are noted in most cases.
Focal triggers
In 1997, Jaïs et al8 observed that atrial fibrillation is often triggered by a rapidly firing ectopic focus and that ablation of that focus can eliminate it. These ectopic foci are often found at or near the ostia of the pulmonary veins or near the superior vena cava.8,9 It is now well established that ectopic foci in the pulmonary veins are crucial triggers that initiate atrial fibrillation.
Trigger-and-substrate theory

The substrate for maintaining atrial fibrillation consists of an abnormal left atrium with heterogeneous fibrosis (scarring) and conduction delays. Any heart disease that increases left atrial pressure could lead to atrial dilation and remodeling, which could be substrates for atrial fibrillation. Extensive atrial remodeling and scarring are associated with progression and persistence of atrial fibrillation and make rhythm control more challenging.
Atrial fibrillation begets atrial fibrillation
As shown in the case above, over time, paroxysmal atrial fibrillation often progresses to persistent and long-standing atrial fibrillation if not aggressively managed initially.
In 1972, Davies and Pomerance10 performed 100 autopsies and found that the people who had had atrial fibrillation for longer than 1 month had lost muscle mass in the sinus node and internodal tract, and their atria were dilated. The study introduced the concept that atrial fibrillation itself causes pathologic changes in the atrium.
Wijffels et al,11 in an experiment in goats, showed that atrial fibrillation produced by rapid bursts of atrial pacing was initially paroxysmal. However, as they continued to induce atrial fibrillation over and over again, it lasted progressively longer until it would persist for more than 24 hours. Thus, in a relatively short time, the atria went from supporting paroxysmal fibrillation to supporting persistent fibrillation.
Atrial fibrillation leads to electrophysiologic and anatomic remodeling in the atrium, which leads to a shorter action potential duration and a shorter refractory period. This in turn makes it easier for atrial fibrillation to persist.12
Because atrial fibrillation tends to progress, intervening early may improve its outcomes. Early ablation has been shown to improve the chances of staying in sinus rhythm in both paroxysmal and persistent atrial fibrillation.13–15
CATHETER ABLATION OF ATRIAL FIBRILLATION
The goal of ablation is to prevent atrial fibrillation by eliminating the trigger that initiates it, altering the arrhythmogenic substrate, or both.
Pulmonary vein isolation
The most common ablation strategy is to electrically isolate the pulmonary veins by creating circumferential lesions around their antra. This creates a nonconducting rim of scar tissue, electrically disconnecting the pulmonary veins from the atrium.
Ablation outside of the pulmonary veins
Because recurrence rates are high in patients with persistent atrial fibrillation who undergo pulmonary vein ablation alone, the search continues for adjunctive strategies to improve outcomes. Although these strategies have a sound rationale based on experimental data and anecdotal evidence in humans, they have not yet been convincingly shown to be helpful in large clinical studies. Nonetheless, it is possible that more extensive substrate ablation—atrial “debulking”—could improve outcomes by reducing the amount of tissue that can fibrillate.
Linear ablation. Creating lines of ablation (as in the maze procedure) isolates different segments of the left atrium. Often, these lines are created along the roof of the left atrium between the right and left upper pulmonary veins and from the mitral valve to the left inferior pulmonary vein. The benefit of linear ablation has not been proven, and gaps in such lines may introduce atrial flutter.
Triggers not in the pulmonary veins. Common sites of nonpulmonary vein triggers include the posterior wall of the left atrium, the superior vena cava, the coronary sinus, and along the ligament of Marshall. Provocative maneuvers such as isoproterenol infusion can help find those triggers, which can then be ablated. A limitation is that there is no protocol proven to reproducibly elicit triggers.
Complex fractionated atrial electrograms are areas in the atrium with highly fractionated, low voltage potentials. They may be critical sites of substrate for atrial fibrillation, and many electrophysiologists target them in patients with persistent atrial fibrillation. But despite initial enthusiasm, doing so has not resulted in better outcomes in persistent atrial fibrillation.
Rotors. Animal studies have shown that atrial fibrillation can be triggered or maintained by localized sources of organized reentrant circuits (rotors) or focal impulses. Recent studies have shown that these electrical rotors and focal sources could potentially be mapped and ablated in humans. But positive results in initial reports have not been reproduced, and this remains an area of controversy.
Our practice. We isolate the pulmonary veins with antral ablations, ablate the posterior wall, and extend the ablation toward the septum and inferior to the right pulmonary veins, with good long-term outcomes.14 The rationale behind ablating the posterior wall is that it shares embryologic origins with the pulmonary veins and may be a common source of triggers in atrial fibrillation.
We do not routinely create empiric ablation lines in the left or right atrium unless the patient has atrial flutter. Empiric ablation lines have not been convincingly shown to provide additional benefit compared with our extensive ablation approach, which involves the posterior wall. Empiric ablation of the appendage or coronary sinus is typically reserved for repeat ablation in patients with recurrent persistent atrial fibrillation.
RATIONALE FOR TREATING ATRIAL FIBRILLATION WITH ABLATION
To control symptoms
At this time, the primary aim of atrial fibrillation ablation is to reduce symptoms and improve quality of life. In theory, ablation could also decrease the risk of stroke, heart failure, and death. However, these outcomes have not been systematically evaluated in any large randomized controlled trial.
To control rhythm and improve survival
Randomized controlled trials of rhythm vs rate control of atrial fibrillation16–18 have failed to demonstrate that restoring sinus rhythm is associated with better survival. All of these trials used antiarrhythmic drugs for rhythm control. However, nonrandomized studies19,20 showed that maintaining sinus rhythm is associated with a significant reduction in mortality rates, whereas the use of antiarrhythmic drugs increased mortality risk.
This suggests that the beneficial effect of restoring sinus rhythm may be offset by adverse effects of antiarrhythmic drugs, and if rhythm control could be achieved by a method other than antiarrhythmic drug therapy, it may be superior to rate control. On the other hand, these data may be affected by residual confounding. This topic deserves further research, but maintaining sinus rhythm is typically preferred whenever possible.
Discontinuing anticoagulation is not a goal at this time
Retrospective studies have reported a low risk of stroke in patients who discontinue anticoagulation several months after undergoing atrial fibrillation ablation.21–23 However, atrial fibrillation can recur, and risk of stroke increases with age.
Therefore, guidelines24 still recommend continuing anticoagulation after ablation. Generally, we do not offer ablation with a goal of discontinuing anticoagulation. That said, stopping anticoagulation may be considered after long-term suppression of paroxysmal atrial fibrillation on a case-by-case basis in patients deemed to be at low risk. Left atrial appendage closure devices may eventually allow concomitant atrial fibrillation ablation and closure of the appendage, so that anticoagulation could then be stopped. This remains a topic of investigation.
Who should be considered for ablation?
There are no absolute age or comorbidity contraindications to ablation. Everyone who has atrial fibrillation deserves, in our opinion, a referral to the electrophysiology clinic.

PROCEDURAL CONSIDERATIONS
Atrial fibrillation ablation is most often performed by electrophysiologists using a minimally invasive endovascular approach. The patient can be under either moderate sedation or general anesthesia; we prefer general anesthesia for patient comfort, safety, and efficacy.


We use an electrogram-based technique to target and eliminate electrical potentials and ensure continuity of ablation sets, with additional guidance by 3-dimensional cardiac mapping systems and intracardiac echocardiography. We also use contact force-sensing catheters to ensure catheter-tissue contact during ablation and to avoid excessive contact, which may enhance the safety of the procedure.
Energy: Hot or cold
Two types of energy can be used for ablation:
Radiofrequency energy (low voltage, high frequency—30 kHz to 1.5 mHz) is delivered to the endocardial surface via a point-source catheter. The radiofrequency energy produces controlled, focal thermal ablation.

In a randomized trial,25 these ablation technologies were shown to be equivalent for preventing recurrences of atrial fibrillation. We use both in our practice. The choice depends primarily on the planned ablation set, given that balloon cryoablation can achieve antral isolation of the pulmonary veins but allows little or no substrate modification.
Improved ablation technology

Contact force-sensing catheters. Radiofrequency ablation catheters are now equipped with a pressure sensor at the tip that measures how hard the catheter is pressing on the heart wall.26,27 In our experience, this has improved the outcomes of ablation procedures, primarily in persistent atrial fibrillation.28

Complications of ablation
Although catheter ablation for atrial fibrillation is safe, it is still one of the most complex electrophysiologic procedures. Improvements in technology and techniques and accumulated experience over the past 15 years have made ablation safer, especially in tertiary care centers. But adverse outcomes are more frequent in low-volume centers.29
Minor procedural complications include pericarditis, complications at the site of vascular access, and anesthesia-related complications. While they do not affect the long-term outcome for the patient, they may increase hospital length of stay and cause temporary inconvenience.
Major complications include cardiac perforation and tamponade, periprocedural stroke, pulmonary vein stenosis, atrioesophageal fistula, phrenic nerve paralysis, major bleeding, myocardial infarction, and death. In a worldwide survey published in 2005, when atrial fibrillation ablation was still novel, the rate of major complications was 6%.30 By 2010, this had declined to 4.5%,31 and the rates of major complications may be significantly lower in more experienced centers.29 In our practice, in 2015, the rate of major complications was 1.3% (unpublished data).
Outcomes of catheter ablation
Clinical outcomes depend on many factors including the type of atrial fibrillation (paroxysmal vs nonparoxysmal), overall health of the atria (atrial size and scarring), patient age and comorbidities, and most importantly, the center’s and operator’s experience.
In randomized controlled trials comparing ablation and antiarrhythmic drug therapy, the efficacy of ablation in maintaining sinus rhythm has been in the range of 66% to 86% vs 16% to 22% for drug therapy,32,33 but these trials have been predominantly in middle-aged white men with paroxysmal atrial fibrillation. These trials also showed that catheter ablation reduced symptoms and improved quality of life. Ablation is less effective in persistent than in paroxysmal atrial fibrillation.34
In a long-term study from our group,14 660 (79.4%) of 831 patients who underwent ablation in 2005 were arrhythmia-free and not on antiarrhythmic drug therapy after a total of 1,019 ablations (an average of 1.2 ablations per patient) at a median of 55 months; 125 patients (15%, 41 with more than 1 ablation) continued to have atrial arrhythmia, controlled with drugs in 87 patients (69.6%). Only 38 patients (4.6%) continued to have drug-resistant atrial fibrillation and were treated with rate control with negative dromotropic agents.
Recent evidence
The largest randomized controlled trial of catheter ablation vs drug therapy for atrial fibrillation (Catheter Ablation Versus Antiarrhythmic Drug Therapy for Atrial Fibrillation [CABANA]) was completed recently, and the results were presented at a national meeting, although they have not yet been published in a peer-reviewed journal.35
A total of 2,204 patients with atrial fibrillation (42.4% paroxysmal, 47.3% persistent, and 10.3% long-standing persistent) were randomized to either ablation or drug therapy. Median follow-up was 4 years. The crossover rate was high—9.2% of those randomized to ablation did not undergo it, and 27.5% of those randomized to drug therapy underwent ablation.
The incidence of the primary end point (a composite of death, disabling stroke, serious bleeding, and cardiac arrest) was not significantly different between the 2 groups in the intention-to-treat analysis; however, given the high crossover rates, the as-treated and per-protocol analyses become important, and as-treated and per-protocol analyses revealed a significant benefit of ablation compared with drug therapy. The hazard ratio (HR) for the primary composite outcome was 0.67 (P = .006) on as-treated analysis and 0.73 (P = .05) on per-protocol analysis. The HR for all-cause mortality was 0.60 (P = .005) on as-treated analysis.
PERIPROCEDURAL CONSIDERATIONS
Periprocedural anticoagulation
The risk of thromboembolism is increased during, immediately following, and for several weeks to months after atrial fibrillation ablation.36,37
During the procedure, the risk is related to transseptal sheath placement, electrode catheters in the left atrium, and char formation on ablation catheters. These risks are mitigated with proper and careful sheath and catheter manipulation, maintenance of bubble-free irrigation through lines and sheaths, use of irrigated catheters, and initiation of heparin before transseptal access. Heparin is also infused during the procedure, with close monitoring of activated clotting time.
Postprocedurally, the transiently increased clotting risk could be due to damaged endothelium from the ablation itself and stunning of atrial tissue, which results in impaired contraction. Damaged endothelium improves as the tissue heals, and the stunning resolves by electrical reverse remodeling with sinus rhythm maintenance.
In view of these risks, the referring physician and electrophysiologist must pay careful attention to anticoagulation before and after ablation.
Before the procedure. It is safe to continue anticoagulation uninterrupted through the procedure.38,39 If the patient is on warfarin, we want the international normalized ratio to be in the therapeutic range when we perform atrial fibrillation ablation, and the patient takes his or her usual dose on the day of the procedure. If taking a direct oral anticoagulant, patients typically skip a dose the day before ablation and again on the morning of the procedure, and resume taking it immediately afterward while in the anesthesia recovery room.
During the procedure, we start heparin before transseptal puncture, adjust it to achieve an activated clotting time of 300 to 400 seconds, and keep it in this range as long as there are sheaths or catheters in the left atrium.
After the procedure. The current guidelines24 recommend that oral anticoagulation be continued without interruption for at least 2 months after the procedure, and in most cases indefinitely, depending on age and comorbidities. The decision to stop anticoagulation after 2 months is typically based on the stroke risk as assessed by the CHA2DS2-VASc score (www.chadsvasc.org) and not on the success of the ablation procedure.
ANTIARRHYTHMIC DRUGS AFTER THE PROCEDURE
Some patients actually experience more atrial fibrillation in the first weeks to months after the procedure. The mechanism in this setting may be different from that causing the arrhythmia in the first place. The causes of early recurrence of atrial arrhythmias include postablation inflammation, temporary autonomic imbalance, and delay of atrial radiofrequency lesion formation.40,41 These arrhythmias may completely resolve as the ablation lesions heal and scars mature.
It has been hypothesized that short-term use of antiarrhythmic drugs after atrial fibrillation ablation is effective in preventing arrhythmias because it alters atrial electrophysiologic characteristics induced by the above transient factors. A recent systematic review of 6 clinical trials showed that short-term use of antiarrhythmic drugs reduces the risk of early arrhythmia recurrence but does not reduce recurrence in the long term.42
In terms of outcomes, any arrhythmias that occur in the first 3 months do not necessarily affect long-term success. This is referred to as the “blanking period.” However, generally speaking, it is preferable to maintain sinus rhythm during that time to avoid further anatomic or electrical left atrial adverse remodeling. In many situations, patients continue taking the same antiarrhythmic agent or start on antiarrhythmic therapy in the first few months after ablation.43,44
The mechanisms of late recurrence of atrial arrhythmias after ablation are thought to be different from those in early recurrence. Late recurrence has been ascribed to incomplete pulmonary vein isolation, recovery of pulmonary vein-left atrium connections, or recovery of any other lines of ablation created in the procedure.45,46 For late recurrence of atrial arrhythmia, studies and guidelines suggest that repeat ablation may be an option.24,47
PRACTICAL CONSIDERATIONS FOR PROCEDURAL PLANNING
Before the procedure, some electrophysiologists use cardiac computed tomography or magnetic resonance imaging to evaluate the pulmonary vein anatomy. This helps in planning and in selecting the appropriate tools for the procedure.
The patient is asked to fast on the day of the procedure. The procedure can take 3 to 6 hours, depending on the patient’s anatomy and the operator’s technique and experience. It can be performed with the patient under general anesthesia or conscious sedation. Currently, we use general anesthesia most of the time to maximize patient comfort.
After the procedure, our patients must stay in bed for 4 hours and stay overnight for observation. If no complications arise, they are discharged the next day.
- Go AS. The epidemiology of atrial fibrillation in elderly persons: the tip of the iceberg. Am J Geriatr Cardiol 2005; 14(2):56–61. pmid:15785146
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285(18):2370–2375. pmid:11343485
- Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006; 114(2):119–125. doi:10.1161/CIRCULATIONAHA.105.595140
- Piccini JP, Hammill BG, Sinner MF, et al. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ Cardiovasc Qual Outcomes 2012; 5(1):85–93. doi:10.1161/CIRCOUTCOMES.111.962688
- Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J 1964; 67:200–220. pmid:14118488
- Cox JL, Schuessler RB, Boineau JP. The surgical treatment of atrial fibrillation. I. Summary of the current concepts of the mechanisms of atrial flutter and atrial fibrillation. J Thorac Cardiovasc Surg 1991; 101(3):402–405. pmid:1999933
- Cox JL, Schuessler RB, D’Agostino HJ Jr, et al. The surgical treatment of atrial fibrillation. III. Development of a definitive surgical procedure. J Thorac Cardiovasc Surg 1991; 101(4):569–583. pmid:2008095
- Jaïs P, Haïssaguerre M, Shah DC, et al. A focal source of atrial fibrillation treated by discrete radiofrequency ablation. Circulation 1997; 95(3):572–576. pmid:9024141
- Haïssaguerre M, Jaïs P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998; 339(10):659–666. doi:10.1056/NEJM199809033391003
- Davies MJ, Pomerance A. Pathology of atrial fibrillation in man. Br Heart J 1972; 34(5):520–525. pmid:5031645
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995; 92(7):1954–1968. pmid:7671380
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002; 415(6868):219–226. doi:10.1038/415219a
- Medi C, Sparks PB, Morton JB, et al. Pulmonary vein antral isolation for paroxysmal atrial fibrillation: results from long-term follow-up. J Cardiovasc Electrophysiol 2011; 22(2):137–141. doi:10.1111/j.1540-8167.2010.01885.x
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4(3):271–278. doi:10.1161/CIRCEP.111.962100
- Hussein AA, Saliba WI, Barakat A, et al. Radiofrequency ablation of persistent atrial fibrillation: diagnosis-to-ablation time, markers of pathways of atrial remodeling, and outcomes. Circ Arrhythm Electrophysiol 2016; 9(1):e003669. doi:10.1161/CIRCEP.115.003669
- Carlsson J, Miketic S, Windeler J, et al. Randomized trial of rate-control versus rhythm-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol 2003; 41(10):1690–1696. pmid:12767648
- Van Gelder IC, Hagens VE, Bosker HA, et al; Rate Control versus Electrical Cardioversion for Persistent Atrial Fibrillation Study Group. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med 2002; 347(23):1834–1840. doi:10.1056/NEJMoa021375
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347(23):1825–1833. doi:10.1056/NEJMoa021328
- Hagens VE, Crijns HJ, Van Veldhuisen DJ, et al; RAte Control versus Electrical cardioversion for persistent atrial fibrillation study group. Rate control versus rhythm control for patients with persistent atrial fibrillation with mild to moderate heart failure: results from the RAte Control versus Electrical cardioversion (RACE) study. Am Heart J 2005; 149(6):1106–111. doi:10.1016/j.ahj.2004.11.030
- Pedersen OD, Bagger H, Keller N, Marchant B, Køber L, Torp-Pedersen C. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: a Danish investigations of arrhythmia and mortality on dofetilide (diamond) substudy. Circulation 2001; 104(3):292–296. pmid:11457747
- Guiot A, Jongnarangsin K, Chugh A, et al. Anticoagulant therapy and risk of cerebrovascular events after catheter ablation of atrial fibrillation in the elderly. J Cardiovasc Electrophysiol 2012; 23(1):36–43. doi:10.1111/j.1540-8167.2011.02141.x
- Oral H, Chugh A, Ozaydin M, et al. Risk of thromboembolic events after percutaneous left atrial radiofrequency ablation of atrial fibrillation. Circulation 2006; 114(8):759–765. doi:10.1161/CIRCULATIONAHA.106.641225
- Themistoclakis S, Corrado A, Marchlinski FE, et al. The risk of thromboembolism and need for oral anticoagulation after successful atrial fibrillation ablation. J Am Coll Cardiol 2010; 55(8):735–743. doi:10.1016/j.jacc.2009.11.039
- Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. J Arrhythm 2017; 33(5):369–409. doi:10.1016/j.joa.2017.08.001
- Kuck KH, Brugada J, Fürnkranz A, et al; FIRE AND ICE Investigators. Cryoballoon or radiofrequency ablation for paroxysmal atrial fibrillation. N Engl J Med 2016; 374(23):2235–2245. doi:10.1056/NEJMoa1602014
- Reddy VY, Dukkipati SR, Neuzil P, et al. Randomized, controlled trial of the safety and effectiveness of a contact force-sensing irrigated catheter for ablation of paroxysmal atrial fibrillation: results of the TactiCath Contact Force Ablation Catheter Study for Atrial Fibrillation (TOCCASTAR) Study. Circulation 2015; 132(10):907–915. doi:10.1161/CIRCULATIONAHA.114.014092
- Natale A, Reddy VY, Monir G, et al. Paroxysmal AF catheter ablation with a contact force sensing catheter: results of the prospective, multicenter SMART-AF trial. J Am Coll Cardiol 2014; 64(7):647–656. doi:10.1016/j.jacc.2014.04.072
- Hussein AA, Barakat AF, Saliba WI, et al. Persistent atrial fibrillation ablation with or without contact force sensing. J Cardiovasc Electrophysiol 2017; 28(5):483–488. doi:10.1111/jce.13179
- Deshmukh A, Patel NJ, Pant I, et al. In-hospital complications associated with catheter ablation of atrial fibrillation in the United States between 2000 and 2010: analysis of 93,801 procedures. Circulation 2013; 128(19):2104–2112. doi:10.1161/CIRCULATIONAHA.113.003862
- Cappato R, Calkins H, Chen SA, et al. Worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circulation 2005; 111(9):1100–1105. doi:10.1161/01.CIR.0000157153.30978.67
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3(1):32–38. doi:10.1161/CIRCEP.109.859116
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293(21):2634–2640. doi:10.1001/jama.293.21.2634
- Jaïs P, Cauchemez B, Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 study. Circulation 2008; 118(24):2498–2505. doi:10.1161/CIRCULATIONAHA.108.772582
- Brooks AG, Stiles MK, Laborderie J, et al. Outcomes of long-standing persistent atrial fibrillation ablation: a systematic review. Heart Rhythm 2010; 7(6):835–846. doi:10.1016/j.hrthm.2010.01.017
- Packer DL, Lee KL, Mark DB, Robb RA. Catheter Ablation versus Antiarrhythmic Drug Therapy for Atrial Fibrillation Trial, CABANA. http://cabanatrial.org/. Accessed September 10, 2018.
- Scherr D, Sharma K, Dalal D, et al. Incidence and predictors of periprocedural cerebrovascular accident in patients undergoing catheter ablation of atrial fibrillation. J Cardiovasc Electrophysiol 2009; 20(12):1357–1363. doi:10.1111/j.1540-8167.2009.01540.x
- Wazni OM, Rossillo A, Marrouche NF, et al. Embolic events and char formation during pulmonary vein isolation in patients with atrial fibrillation: impact of different anticoagulation regimens and importance of intracardiac echo imaging. J Cardiovasc Electrophysiol 2005; 16(6):576–581. doi:10.1111/j.1540-8167.2005.40480.x
- Hussein AA, Martin DO, Saliba W, et al. Radiofrequency ablation of atrial fibrillation under therapeutic international normalized ratio: a safe and efficacious periprocedural anticoagulation strategy. Heart Rhythm 2009; 6(10):1425–1429. doi:10.1016/j.hrthm.2009.07.007
- Bassiouny M, Saliba W, Rickard J, et al. Use of dabigatran for periprocedural anticoagulation in patients undergoing catheter ablation for atrial fibrillation. Circ Arrhythm Electrophysiol 2013; 6(3):460–466. doi:10.1161/CIRCEP.113.000320
- Koyama T, Tada H, Sekiguchi Y, et al. Prevention of atrial fibrillation recurrence with corticosteroids after radiofrequency catheter ablation: a randomized controlled trial. J Am Coll Cardiol 2010; 56(18):1463–1472. doi:10.1016/j.jacc.2010.04.057
- Oral H, Knight BP, Ozaydin M, et al. Clinical significance of early recurrences of atrial fibrillation after pulmonary vein isolation. J Am Coll Cardiol 2002; 40(1):100–104. pmid:12103262
- Chen W, Liu H, Ling Z, et al. Efficacy of short-term antiarrhythmic drugs use after catheter ablation of atrial fibrillation—a systematic review with meta-analyses and trial sequential analyses of randomized controlled trials. PLoS One 2016; 11(5):e0156121. doi:10.1371/journal.pone.0156121
- Leong-Sit P, Roux JF, Zado E, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study): six-month follow-up study. Circ Arrhythm Electrophysiol 2011; 4(1):11–14. doi:10.1161/CIRCEP.110.955393
- Roux JF, Zado E, Callans DJ, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study). Circulation 2009; 120(12):1036–1040. doi:10.1161/CIRCULATIONAHA.108.839639
- Sotomi Y, Inoue K, Ito N, et al. Cause of very late recurrence of atrial fibrillation or flutter after catheter ablation for atrial fibrillation. Am J Cardiol 2013; 111(4):552–556. doi:10.1016/j.amjcard.2012.10.040
- Lee SH, Tai CT, Hsieh MH, et al. Predictors of early and late recurrence of atrial fibrillation after catheter ablation of paroxysmal atrial fibrillation. J Interv Card Electrophysiol. 2004 Jun;10(3):221-6. doi:10.1023/B:JICE.0000026915.02503.92
- Zhang XD, Gu J, Jiang WF, et al. Optimal rhythm-control strategy for recurrent atrial tachycardia after catheter ablation of persistent atrial fibrillation: a randomized clinical trial. Eur Heart J 2014; 35(20):1327–1334. doi:10.1093/eurheartj/ehu017
A 64-year-old man with hypertension but without known structural heart disease presents for a second opinion on management of his atrial fibrillation. The condition was first diagnosed at age 38, when he experienced palpitations and shortness of breath on exertion; at times he also experienced decreased endurance and fatigue without overt palpitations. At first, these episodes occurred about twice a year, and the patient was managed with a beta-blocker for rate control and an oral anticoagulant.
Over the past 10 years, the episodes have become more frequent and longer-lasting and have required frequent cardioversions. He was given flecainide for rhythm control but continued to have frequent episodes, and so about 1 year ago he was switched to amiodarone, which controlled his rhythm better. However, after reading about side effects of amiodarone, he decided to seek a second opinion.
He was evaluated by our team and eventually underwent radiofrequency ablation. During the procedure, he was noted to have diffuse scarring and fibrosis of his left atrium, and afterward he continued to require antiarrhythmic drugs to maintain sinus rhythm.
Should he have been referred sooner? What factors should primary care physicians consider when referring a patient with atrial fibrillation for ablation?
THE EPIDEMIC OF ATRIAL FIBRILLATION
Atrial fibrillation is a large and growing public health problem. In 2010, it was estimated to affect 2.7 to 6.1 million people in the United States, and with the rapid aging of our population, its prevalence is expected to rise to between 5.6 and 12 million by 2050.1–3 It is associated with significant morbidity, poor quality of life, and increased risk of death, heart failure, stroke, and cognitive impairment.
The number of new cases per year has increased over the years despite research and preventive measures, which may reflect aging of the population and increased survival rates in patients with cardiovascular or comorbid conditions.1,4
Thus, atrial fibrillation is one of the most common cardiovascular conditions encountered by primary care physicians and cardiologists, putting them at the forefront of its management. Proper treatment in its early stages and referral to a specialist for advanced management may alter its natural history and improve clinical outcomes.
HOW DOES ATRIAL FIBRILLATION ARISE AND PERSIST?
Much is still unknown about the pathogenesis of atrial fibrillation, but considerable progress has been made in the past few decades, opening the door for clinical ablative strategies.
Multiple wavelet hypothesis
Until the late 1980s, the most widely accepted conceptual mechanism of atrial fibrillation was the multiple wavelet hypothesis developed by Moe et al.5 According to this hypothesis, atrial fibrillation begins with multiple independent wavelets occurring simultaneously and spreading randomly throughout both atria, and it persists if there are a minimum number of coexisting wavelets, increased atrial mass, and heterogeneous conduction delays across the atrial tissue.
The surgical maze procedure, in which a series of incisions arranged in a maze-like pattern is created in the left atrium, was predicated on this model. The theory was that these surgical lesions would compartmentalize the atria into discrete electrical segments and thereby reduce the number of circulating random wavelets.6,7
However, experimental and clinical studies suggest that although randomly propagating wavelets can contribute to maintaining atrial fibrillation, focal triggers are noted in most cases.
Focal triggers
In 1997, Jaïs et al8 observed that atrial fibrillation is often triggered by a rapidly firing ectopic focus and that ablation of that focus can eliminate it. These ectopic foci are often found at or near the ostia of the pulmonary veins or near the superior vena cava.8,9 It is now well established that ectopic foci in the pulmonary veins are crucial triggers that initiate atrial fibrillation.
Trigger-and-substrate theory
The substrate for maintaining atrial fibrillation consists of an abnormal left atrium with heterogeneous fibrosis (scarring) and conduction delays. Any heart disease that increases left atrial pressure could lead to atrial dilation and remodeling, which could be substrates for atrial fibrillation. Extensive atrial remodeling and scarring are associated with progression and persistence of atrial fibrillation and make rhythm control more challenging.
Atrial fibrillation begets atrial fibrillation
As shown in the case above, over time, paroxysmal atrial fibrillation often progresses to persistent and long-standing atrial fibrillation if not aggressively managed initially.
In 1972, Davies and Pomerance10 performed 100 autopsies and found that the people who had had atrial fibrillation for longer than 1 month had lost muscle mass in the sinus node and internodal tract, and their atria were dilated. The study introduced the concept that atrial fibrillation itself causes pathologic changes in the atrium.
Wijffels et al,11 in an experiment in goats, showed that atrial fibrillation produced by rapid bursts of atrial pacing was initially paroxysmal. However, as they continued to induce atrial fibrillation over and over again, it lasted progressively longer until it would persist for more than 24 hours. Thus, in a relatively short time, the atria went from supporting paroxysmal fibrillation to supporting persistent fibrillation.
Atrial fibrillation leads to electrophysiologic and anatomic remodeling in the atrium, which leads to a shorter action potential duration and a shorter refractory period. This in turn makes it easier for atrial fibrillation to persist.12
Because atrial fibrillation tends to progress, intervening early may improve its outcomes. Early ablation has been shown to improve the chances of staying in sinus rhythm in both paroxysmal and persistent atrial fibrillation.13–15
CATHETER ABLATION OF ATRIAL FIBRILLATION
The goal of ablation is to prevent atrial fibrillation by eliminating the trigger that initiates it, altering the arrhythmogenic substrate, or both.
Pulmonary vein isolation
The most common ablation strategy is to electrically isolate the pulmonary veins by creating circumferential lesions around their antra. This creates a nonconducting rim of scar tissue, electrically disconnecting the pulmonary veins from the atrium.
Ablation outside of the pulmonary veins
Because recurrence rates are high in patients with persistent atrial fibrillation who undergo pulmonary vein ablation alone, the search continues for adjunctive strategies to improve outcomes. Although these strategies have a sound rationale based on experimental data and anecdotal evidence in humans, they have not yet been convincingly shown to be helpful in large clinical studies. Nonetheless, it is possible that more extensive substrate ablation—atrial “debulking”—could improve outcomes by reducing the amount of tissue that can fibrillate.
Linear ablation. Creating lines of ablation (as in the maze procedure) isolates different segments of the left atrium. Often, these lines are created along the roof of the left atrium between the right and left upper pulmonary veins and from the mitral valve to the left inferior pulmonary vein. The benefit of linear ablation has not been proven, and gaps in such lines may introduce atrial flutter.
Triggers not in the pulmonary veins. Common sites of nonpulmonary vein triggers include the posterior wall of the left atrium, the superior vena cava, the coronary sinus, and along the ligament of Marshall. Provocative maneuvers such as isoproterenol infusion can help find those triggers, which can then be ablated. A limitation is that there is no protocol proven to reproducibly elicit triggers.
Complex fractionated atrial electrograms are areas in the atrium with highly fractionated, low voltage potentials. They may be critical sites of substrate for atrial fibrillation, and many electrophysiologists target them in patients with persistent atrial fibrillation. But despite initial enthusiasm, doing so has not resulted in better outcomes in persistent atrial fibrillation.
Rotors. Animal studies have shown that atrial fibrillation can be triggered or maintained by localized sources of organized reentrant circuits (rotors) or focal impulses. Recent studies have shown that these electrical rotors and focal sources could potentially be mapped and ablated in humans. But positive results in initial reports have not been reproduced, and this remains an area of controversy.
Our practice. We isolate the pulmonary veins with antral ablations, ablate the posterior wall, and extend the ablation toward the septum and inferior to the right pulmonary veins, with good long-term outcomes.14 The rationale behind ablating the posterior wall is that it shares embryologic origins with the pulmonary veins and may be a common source of triggers in atrial fibrillation.
We do not routinely create empiric ablation lines in the left or right atrium unless the patient has atrial flutter. Empiric ablation lines have not been convincingly shown to provide additional benefit compared with our extensive ablation approach, which involves the posterior wall. Empiric ablation of the appendage or coronary sinus is typically reserved for repeat ablation in patients with recurrent persistent atrial fibrillation.
RATIONALE FOR TREATING ATRIAL FIBRILLATION WITH ABLATION
To control symptoms
At this time, the primary aim of atrial fibrillation ablation is to reduce symptoms and improve quality of life. In theory, ablation could also decrease the risk of stroke, heart failure, and death. However, these outcomes have not been systematically evaluated in any large randomized controlled trial.
To control rhythm and improve survival
Randomized controlled trials of rhythm vs rate control of atrial fibrillation16–18 have failed to demonstrate that restoring sinus rhythm is associated with better survival. All of these trials used antiarrhythmic drugs for rhythm control. However, nonrandomized studies19,20 showed that maintaining sinus rhythm is associated with a significant reduction in mortality rates, whereas the use of antiarrhythmic drugs increased mortality risk.
This suggests that the beneficial effect of restoring sinus rhythm may be offset by adverse effects of antiarrhythmic drugs, and if rhythm control could be achieved by a method other than antiarrhythmic drug therapy, it may be superior to rate control. On the other hand, these data may be affected by residual confounding. This topic deserves further research, but maintaining sinus rhythm is typically preferred whenever possible.
Discontinuing anticoagulation is not a goal at this time
Retrospective studies have reported a low risk of stroke in patients who discontinue anticoagulation several months after undergoing atrial fibrillation ablation.21–23 However, atrial fibrillation can recur, and risk of stroke increases with age.
Therefore, guidelines24 still recommend continuing anticoagulation after ablation. Generally, we do not offer ablation with a goal of discontinuing anticoagulation. That said, stopping anticoagulation may be considered after long-term suppression of paroxysmal atrial fibrillation on a case-by-case basis in patients deemed to be at low risk. Left atrial appendage closure devices may eventually allow concomitant atrial fibrillation ablation and closure of the appendage, so that anticoagulation could then be stopped. This remains a topic of investigation.
Who should be considered for ablation?
There are no absolute age or comorbidity contraindications to ablation. Everyone who has atrial fibrillation deserves, in our opinion, a referral to the electrophysiology clinic.
PROCEDURAL CONSIDERATIONS
Atrial fibrillation ablation is most often performed by electrophysiologists using a minimally invasive endovascular approach. The patient can be under either moderate sedation or general anesthesia; we prefer general anesthesia for patient comfort, safety, and efficacy.
We use an electrogram-based technique to target and eliminate electrical potentials and ensure continuity of ablation sets, with additional guidance by 3-dimensional cardiac mapping systems and intracardiac echocardiography. We also use contact force-sensing catheters to ensure catheter-tissue contact during ablation and to avoid excessive contact, which may enhance the safety of the procedure.
Energy: Hot or cold
Two types of energy can be used for ablation:
Radiofrequency energy (low voltage, high frequency—30 kHz to 1.5 mHz) is delivered to the endocardial surface via a point-source catheter. The radiofrequency energy produces controlled, focal thermal ablation.
In a randomized trial,25 these ablation technologies were shown to be equivalent for preventing recurrences of atrial fibrillation. We use both in our practice. The choice depends primarily on the planned ablation set, given that balloon cryoablation can achieve antral isolation of the pulmonary veins but allows little or no substrate modification.
Improved ablation technology
Contact force-sensing catheters. Radiofrequency ablation catheters are now equipped with a pressure sensor at the tip that measures how hard the catheter is pressing on the heart wall.26,27 In our experience, this has improved the outcomes of ablation procedures, primarily in persistent atrial fibrillation.28
Complications of ablation
Although catheter ablation for atrial fibrillation is safe, it is still one of the most complex electrophysiologic procedures. Improvements in technology and techniques and accumulated experience over the past 15 years have made ablation safer, especially in tertiary care centers. But adverse outcomes are more frequent in low-volume centers.29
Minor procedural complications include pericarditis, complications at the site of vascular access, and anesthesia-related complications. While they do not affect the long-term outcome for the patient, they may increase hospital length of stay and cause temporary inconvenience.
Major complications include cardiac perforation and tamponade, periprocedural stroke, pulmonary vein stenosis, atrioesophageal fistula, phrenic nerve paralysis, major bleeding, myocardial infarction, and death. In a worldwide survey published in 2005, when atrial fibrillation ablation was still novel, the rate of major complications was 6%.30 By 2010, this had declined to 4.5%,31 and the rates of major complications may be significantly lower in more experienced centers.29 In our practice, in 2015, the rate of major complications was 1.3% (unpublished data).
Outcomes of catheter ablation
Clinical outcomes depend on many factors including the type of atrial fibrillation (paroxysmal vs nonparoxysmal), overall health of the atria (atrial size and scarring), patient age and comorbidities, and most importantly, the center’s and operator’s experience.
In randomized controlled trials comparing ablation and antiarrhythmic drug therapy, the efficacy of ablation in maintaining sinus rhythm has been in the range of 66% to 86% vs 16% to 22% for drug therapy,32,33 but these trials have been predominantly in middle-aged white men with paroxysmal atrial fibrillation. These trials also showed that catheter ablation reduced symptoms and improved quality of life. Ablation is less effective in persistent than in paroxysmal atrial fibrillation.34
In a long-term study from our group,14 660 (79.4%) of 831 patients who underwent ablation in 2005 were arrhythmia-free and not on antiarrhythmic drug therapy after a total of 1,019 ablations (an average of 1.2 ablations per patient) at a median of 55 months; 125 patients (15%, 41 with more than 1 ablation) continued to have atrial arrhythmia, controlled with drugs in 87 patients (69.6%). Only 38 patients (4.6%) continued to have drug-resistant atrial fibrillation and were treated with rate control with negative dromotropic agents.
Recent evidence
The largest randomized controlled trial of catheter ablation vs drug therapy for atrial fibrillation (Catheter Ablation Versus Antiarrhythmic Drug Therapy for Atrial Fibrillation [CABANA]) was completed recently, and the results were presented at a national meeting, although they have not yet been published in a peer-reviewed journal.35
A total of 2,204 patients with atrial fibrillation (42.4% paroxysmal, 47.3% persistent, and 10.3% long-standing persistent) were randomized to either ablation or drug therapy. Median follow-up was 4 years. The crossover rate was high—9.2% of those randomized to ablation did not undergo it, and 27.5% of those randomized to drug therapy underwent ablation.
The incidence of the primary end point (a composite of death, disabling stroke, serious bleeding, and cardiac arrest) was not significantly different between the 2 groups in the intention-to-treat analysis; however, given the high crossover rates, the as-treated and per-protocol analyses become important, and as-treated and per-protocol analyses revealed a significant benefit of ablation compared with drug therapy. The hazard ratio (HR) for the primary composite outcome was 0.67 (P = .006) on as-treated analysis and 0.73 (P = .05) on per-protocol analysis. The HR for all-cause mortality was 0.60 (P = .005) on as-treated analysis.
PERIPROCEDURAL CONSIDERATIONS
Periprocedural anticoagulation
The risk of thromboembolism is increased during, immediately following, and for several weeks to months after atrial fibrillation ablation.36,37
During the procedure, the risk is related to transseptal sheath placement, electrode catheters in the left atrium, and char formation on ablation catheters. These risks are mitigated with proper and careful sheath and catheter manipulation, maintenance of bubble-free irrigation through lines and sheaths, use of irrigated catheters, and initiation of heparin before transseptal access. Heparin is also infused during the procedure, with close monitoring of activated clotting time.
Postprocedurally, the transiently increased clotting risk could be due to damaged endothelium from the ablation itself and stunning of atrial tissue, which results in impaired contraction. Damaged endothelium improves as the tissue heals, and the stunning resolves by electrical reverse remodeling with sinus rhythm maintenance.
In view of these risks, the referring physician and electrophysiologist must pay careful attention to anticoagulation before and after ablation.
Before the procedure. It is safe to continue anticoagulation uninterrupted through the procedure.38,39 If the patient is on warfarin, we want the international normalized ratio to be in the therapeutic range when we perform atrial fibrillation ablation, and the patient takes his or her usual dose on the day of the procedure. If taking a direct oral anticoagulant, patients typically skip a dose the day before ablation and again on the morning of the procedure, and resume taking it immediately afterward while in the anesthesia recovery room.
During the procedure, we start heparin before transseptal puncture, adjust it to achieve an activated clotting time of 300 to 400 seconds, and keep it in this range as long as there are sheaths or catheters in the left atrium.
After the procedure. The current guidelines24 recommend that oral anticoagulation be continued without interruption for at least 2 months after the procedure, and in most cases indefinitely, depending on age and comorbidities. The decision to stop anticoagulation after 2 months is typically based on the stroke risk as assessed by the CHA2DS2-VASc score (www.chadsvasc.org) and not on the success of the ablation procedure.
ANTIARRHYTHMIC DRUGS AFTER THE PROCEDURE
Some patients actually experience more atrial fibrillation in the first weeks to months after the procedure. The mechanism in this setting may be different from that causing the arrhythmia in the first place. The causes of early recurrence of atrial arrhythmias include postablation inflammation, temporary autonomic imbalance, and delay of atrial radiofrequency lesion formation.40,41 These arrhythmias may completely resolve as the ablation lesions heal and scars mature.
It has been hypothesized that short-term use of antiarrhythmic drugs after atrial fibrillation ablation is effective in preventing arrhythmias because it alters atrial electrophysiologic characteristics induced by the above transient factors. A recent systematic review of 6 clinical trials showed that short-term use of antiarrhythmic drugs reduces the risk of early arrhythmia recurrence but does not reduce recurrence in the long term.42
In terms of outcomes, any arrhythmias that occur in the first 3 months do not necessarily affect long-term success. This is referred to as the “blanking period.” However, generally speaking, it is preferable to maintain sinus rhythm during that time to avoid further anatomic or electrical left atrial adverse remodeling. In many situations, patients continue taking the same antiarrhythmic agent or start on antiarrhythmic therapy in the first few months after ablation.43,44
The mechanisms of late recurrence of atrial arrhythmias after ablation are thought to be different from those in early recurrence. Late recurrence has been ascribed to incomplete pulmonary vein isolation, recovery of pulmonary vein-left atrium connections, or recovery of any other lines of ablation created in the procedure.45,46 For late recurrence of atrial arrhythmia, studies and guidelines suggest that repeat ablation may be an option.24,47
PRACTICAL CONSIDERATIONS FOR PROCEDURAL PLANNING
Before the procedure, some electrophysiologists use cardiac computed tomography or magnetic resonance imaging to evaluate the pulmonary vein anatomy. This helps in planning and in selecting the appropriate tools for the procedure.
The patient is asked to fast on the day of the procedure. The procedure can take 3 to 6 hours, depending on the patient’s anatomy and the operator’s technique and experience. It can be performed with the patient under general anesthesia or conscious sedation. Currently, we use general anesthesia most of the time to maximize patient comfort.
After the procedure, our patients must stay in bed for 4 hours and stay overnight for observation. If no complications arise, they are discharged the next day.
A 64-year-old man with hypertension but without known structural heart disease presents for a second opinion on management of his atrial fibrillation. The condition was first diagnosed at age 38, when he experienced palpitations and shortness of breath on exertion; at times he also experienced decreased endurance and fatigue without overt palpitations. At first, these episodes occurred about twice a year, and the patient was managed with a beta-blocker for rate control and an oral anticoagulant.
Over the past 10 years, the episodes have become more frequent and longer-lasting and have required frequent cardioversions. He was given flecainide for rhythm control but continued to have frequent episodes, and so about 1 year ago he was switched to amiodarone, which controlled his rhythm better. However, after reading about side effects of amiodarone, he decided to seek a second opinion.
He was evaluated by our team and eventually underwent radiofrequency ablation. During the procedure, he was noted to have diffuse scarring and fibrosis of his left atrium, and afterward he continued to require antiarrhythmic drugs to maintain sinus rhythm.
Should he have been referred sooner? What factors should primary care physicians consider when referring a patient with atrial fibrillation for ablation?
THE EPIDEMIC OF ATRIAL FIBRILLATION
Atrial fibrillation is a large and growing public health problem. In 2010, it was estimated to affect 2.7 to 6.1 million people in the United States, and with the rapid aging of our population, its prevalence is expected to rise to between 5.6 and 12 million by 2050.1–3 It is associated with significant morbidity, poor quality of life, and increased risk of death, heart failure, stroke, and cognitive impairment.
The number of new cases per year has increased over the years despite research and preventive measures, which may reflect aging of the population and increased survival rates in patients with cardiovascular or comorbid conditions.1,4
Thus, atrial fibrillation is one of the most common cardiovascular conditions encountered by primary care physicians and cardiologists, putting them at the forefront of its management. Proper treatment in its early stages and referral to a specialist for advanced management may alter its natural history and improve clinical outcomes.
HOW DOES ATRIAL FIBRILLATION ARISE AND PERSIST?
Much is still unknown about the pathogenesis of atrial fibrillation, but considerable progress has been made in the past few decades, opening the door for clinical ablative strategies.
Multiple wavelet hypothesis
Until the late 1980s, the most widely accepted conceptual mechanism of atrial fibrillation was the multiple wavelet hypothesis developed by Moe et al.5 According to this hypothesis, atrial fibrillation begins with multiple independent wavelets occurring simultaneously and spreading randomly throughout both atria, and it persists if there are a minimum number of coexisting wavelets, increased atrial mass, and heterogeneous conduction delays across the atrial tissue.
The surgical maze procedure, in which a series of incisions arranged in a maze-like pattern is created in the left atrium, was predicated on this model. The theory was that these surgical lesions would compartmentalize the atria into discrete electrical segments and thereby reduce the number of circulating random wavelets.6,7
However, experimental and clinical studies suggest that although randomly propagating wavelets can contribute to maintaining atrial fibrillation, focal triggers are noted in most cases.
Focal triggers
In 1997, Jaïs et al8 observed that atrial fibrillation is often triggered by a rapidly firing ectopic focus and that ablation of that focus can eliminate it. These ectopic foci are often found at or near the ostia of the pulmonary veins or near the superior vena cava.8,9 It is now well established that ectopic foci in the pulmonary veins are crucial triggers that initiate atrial fibrillation.
Trigger-and-substrate theory
The substrate for maintaining atrial fibrillation consists of an abnormal left atrium with heterogeneous fibrosis (scarring) and conduction delays. Any heart disease that increases left atrial pressure could lead to atrial dilation and remodeling, which could be substrates for atrial fibrillation. Extensive atrial remodeling and scarring are associated with progression and persistence of atrial fibrillation and make rhythm control more challenging.
Atrial fibrillation begets atrial fibrillation
As shown in the case above, over time, paroxysmal atrial fibrillation often progresses to persistent and long-standing atrial fibrillation if not aggressively managed initially.
In 1972, Davies and Pomerance10 performed 100 autopsies and found that the people who had had atrial fibrillation for longer than 1 month had lost muscle mass in the sinus node and internodal tract, and their atria were dilated. The study introduced the concept that atrial fibrillation itself causes pathologic changes in the atrium.
Wijffels et al,11 in an experiment in goats, showed that atrial fibrillation produced by rapid bursts of atrial pacing was initially paroxysmal. However, as they continued to induce atrial fibrillation over and over again, it lasted progressively longer until it would persist for more than 24 hours. Thus, in a relatively short time, the atria went from supporting paroxysmal fibrillation to supporting persistent fibrillation.
Atrial fibrillation leads to electrophysiologic and anatomic remodeling in the atrium, which leads to a shorter action potential duration and a shorter refractory period. This in turn makes it easier for atrial fibrillation to persist.12
Because atrial fibrillation tends to progress, intervening early may improve its outcomes. Early ablation has been shown to improve the chances of staying in sinus rhythm in both paroxysmal and persistent atrial fibrillation.13–15
CATHETER ABLATION OF ATRIAL FIBRILLATION
The goal of ablation is to prevent atrial fibrillation by eliminating the trigger that initiates it, altering the arrhythmogenic substrate, or both.
Pulmonary vein isolation
The most common ablation strategy is to electrically isolate the pulmonary veins by creating circumferential lesions around their antra. This creates a nonconducting rim of scar tissue, electrically disconnecting the pulmonary veins from the atrium.
Ablation outside of the pulmonary veins
Because recurrence rates are high in patients with persistent atrial fibrillation who undergo pulmonary vein ablation alone, the search continues for adjunctive strategies to improve outcomes. Although these strategies have a sound rationale based on experimental data and anecdotal evidence in humans, they have not yet been convincingly shown to be helpful in large clinical studies. Nonetheless, it is possible that more extensive substrate ablation—atrial “debulking”—could improve outcomes by reducing the amount of tissue that can fibrillate.
Linear ablation. Creating lines of ablation (as in the maze procedure) isolates different segments of the left atrium. Often, these lines are created along the roof of the left atrium between the right and left upper pulmonary veins and from the mitral valve to the left inferior pulmonary vein. The benefit of linear ablation has not been proven, and gaps in such lines may introduce atrial flutter.
Triggers not in the pulmonary veins. Common sites of nonpulmonary vein triggers include the posterior wall of the left atrium, the superior vena cava, the coronary sinus, and along the ligament of Marshall. Provocative maneuvers such as isoproterenol infusion can help find those triggers, which can then be ablated. A limitation is that there is no protocol proven to reproducibly elicit triggers.
Complex fractionated atrial electrograms are areas in the atrium with highly fractionated, low voltage potentials. They may be critical sites of substrate for atrial fibrillation, and many electrophysiologists target them in patients with persistent atrial fibrillation. But despite initial enthusiasm, doing so has not resulted in better outcomes in persistent atrial fibrillation.
Rotors. Animal studies have shown that atrial fibrillation can be triggered or maintained by localized sources of organized reentrant circuits (rotors) or focal impulses. Recent studies have shown that these electrical rotors and focal sources could potentially be mapped and ablated in humans. But positive results in initial reports have not been reproduced, and this remains an area of controversy.
Our practice. We isolate the pulmonary veins with antral ablations, ablate the posterior wall, and extend the ablation toward the septum and inferior to the right pulmonary veins, with good long-term outcomes.14 The rationale behind ablating the posterior wall is that it shares embryologic origins with the pulmonary veins and may be a common source of triggers in atrial fibrillation.
We do not routinely create empiric ablation lines in the left or right atrium unless the patient has atrial flutter. Empiric ablation lines have not been convincingly shown to provide additional benefit compared with our extensive ablation approach, which involves the posterior wall. Empiric ablation of the appendage or coronary sinus is typically reserved for repeat ablation in patients with recurrent persistent atrial fibrillation.
RATIONALE FOR TREATING ATRIAL FIBRILLATION WITH ABLATION
To control symptoms
At this time, the primary aim of atrial fibrillation ablation is to reduce symptoms and improve quality of life. In theory, ablation could also decrease the risk of stroke, heart failure, and death. However, these outcomes have not been systematically evaluated in any large randomized controlled trial.
To control rhythm and improve survival
Randomized controlled trials of rhythm vs rate control of atrial fibrillation16–18 have failed to demonstrate that restoring sinus rhythm is associated with better survival. All of these trials used antiarrhythmic drugs for rhythm control. However, nonrandomized studies19,20 showed that maintaining sinus rhythm is associated with a significant reduction in mortality rates, whereas the use of antiarrhythmic drugs increased mortality risk.
This suggests that the beneficial effect of restoring sinus rhythm may be offset by adverse effects of antiarrhythmic drugs, and if rhythm control could be achieved by a method other than antiarrhythmic drug therapy, it may be superior to rate control. On the other hand, these data may be affected by residual confounding. This topic deserves further research, but maintaining sinus rhythm is typically preferred whenever possible.
Discontinuing anticoagulation is not a goal at this time
Retrospective studies have reported a low risk of stroke in patients who discontinue anticoagulation several months after undergoing atrial fibrillation ablation.21–23 However, atrial fibrillation can recur, and risk of stroke increases with age.
Therefore, guidelines24 still recommend continuing anticoagulation after ablation. Generally, we do not offer ablation with a goal of discontinuing anticoagulation. That said, stopping anticoagulation may be considered after long-term suppression of paroxysmal atrial fibrillation on a case-by-case basis in patients deemed to be at low risk. Left atrial appendage closure devices may eventually allow concomitant atrial fibrillation ablation and closure of the appendage, so that anticoagulation could then be stopped. This remains a topic of investigation.
Who should be considered for ablation?
There are no absolute age or comorbidity contraindications to ablation. Everyone who has atrial fibrillation deserves, in our opinion, a referral to the electrophysiology clinic.
PROCEDURAL CONSIDERATIONS
Atrial fibrillation ablation is most often performed by electrophysiologists using a minimally invasive endovascular approach. The patient can be under either moderate sedation or general anesthesia; we prefer general anesthesia for patient comfort, safety, and efficacy.
We use an electrogram-based technique to target and eliminate electrical potentials and ensure continuity of ablation sets, with additional guidance by 3-dimensional cardiac mapping systems and intracardiac echocardiography. We also use contact force-sensing catheters to ensure catheter-tissue contact during ablation and to avoid excessive contact, which may enhance the safety of the procedure.
Energy: Hot or cold
Two types of energy can be used for ablation:
Radiofrequency energy (low voltage, high frequency—30 kHz to 1.5 mHz) is delivered to the endocardial surface via a point-source catheter. The radiofrequency energy produces controlled, focal thermal ablation.
In a randomized trial,25 these ablation technologies were shown to be equivalent for preventing recurrences of atrial fibrillation. We use both in our practice. The choice depends primarily on the planned ablation set, given that balloon cryoablation can achieve antral isolation of the pulmonary veins but allows little or no substrate modification.
Improved ablation technology
Contact force-sensing catheters. Radiofrequency ablation catheters are now equipped with a pressure sensor at the tip that measures how hard the catheter is pressing on the heart wall.26,27 In our experience, this has improved the outcomes of ablation procedures, primarily in persistent atrial fibrillation.28
Complications of ablation
Although catheter ablation for atrial fibrillation is safe, it is still one of the most complex electrophysiologic procedures. Improvements in technology and techniques and accumulated experience over the past 15 years have made ablation safer, especially in tertiary care centers. But adverse outcomes are more frequent in low-volume centers.29
Minor procedural complications include pericarditis, complications at the site of vascular access, and anesthesia-related complications. While they do not affect the long-term outcome for the patient, they may increase hospital length of stay and cause temporary inconvenience.
Major complications include cardiac perforation and tamponade, periprocedural stroke, pulmonary vein stenosis, atrioesophageal fistula, phrenic nerve paralysis, major bleeding, myocardial infarction, and death. In a worldwide survey published in 2005, when atrial fibrillation ablation was still novel, the rate of major complications was 6%.30 By 2010, this had declined to 4.5%,31 and the rates of major complications may be significantly lower in more experienced centers.29 In our practice, in 2015, the rate of major complications was 1.3% (unpublished data).
Outcomes of catheter ablation
Clinical outcomes depend on many factors including the type of atrial fibrillation (paroxysmal vs nonparoxysmal), overall health of the atria (atrial size and scarring), patient age and comorbidities, and most importantly, the center’s and operator’s experience.
In randomized controlled trials comparing ablation and antiarrhythmic drug therapy, the efficacy of ablation in maintaining sinus rhythm has been in the range of 66% to 86% vs 16% to 22% for drug therapy,32,33 but these trials have been predominantly in middle-aged white men with paroxysmal atrial fibrillation. These trials also showed that catheter ablation reduced symptoms and improved quality of life. Ablation is less effective in persistent than in paroxysmal atrial fibrillation.34
In a long-term study from our group,14 660 (79.4%) of 831 patients who underwent ablation in 2005 were arrhythmia-free and not on antiarrhythmic drug therapy after a total of 1,019 ablations (an average of 1.2 ablations per patient) at a median of 55 months; 125 patients (15%, 41 with more than 1 ablation) continued to have atrial arrhythmia, controlled with drugs in 87 patients (69.6%). Only 38 patients (4.6%) continued to have drug-resistant atrial fibrillation and were treated with rate control with negative dromotropic agents.
Recent evidence
The largest randomized controlled trial of catheter ablation vs drug therapy for atrial fibrillation (Catheter Ablation Versus Antiarrhythmic Drug Therapy for Atrial Fibrillation [CABANA]) was completed recently, and the results were presented at a national meeting, although they have not yet been published in a peer-reviewed journal.35
A total of 2,204 patients with atrial fibrillation (42.4% paroxysmal, 47.3% persistent, and 10.3% long-standing persistent) were randomized to either ablation or drug therapy. Median follow-up was 4 years. The crossover rate was high—9.2% of those randomized to ablation did not undergo it, and 27.5% of those randomized to drug therapy underwent ablation.
The incidence of the primary end point (a composite of death, disabling stroke, serious bleeding, and cardiac arrest) was not significantly different between the 2 groups in the intention-to-treat analysis; however, given the high crossover rates, the as-treated and per-protocol analyses become important, and as-treated and per-protocol analyses revealed a significant benefit of ablation compared with drug therapy. The hazard ratio (HR) for the primary composite outcome was 0.67 (P = .006) on as-treated analysis and 0.73 (P = .05) on per-protocol analysis. The HR for all-cause mortality was 0.60 (P = .005) on as-treated analysis.
PERIPROCEDURAL CONSIDERATIONS
Periprocedural anticoagulation
The risk of thromboembolism is increased during, immediately following, and for several weeks to months after atrial fibrillation ablation.36,37
During the procedure, the risk is related to transseptal sheath placement, electrode catheters in the left atrium, and char formation on ablation catheters. These risks are mitigated with proper and careful sheath and catheter manipulation, maintenance of bubble-free irrigation through lines and sheaths, use of irrigated catheters, and initiation of heparin before transseptal access. Heparin is also infused during the procedure, with close monitoring of activated clotting time.
Postprocedurally, the transiently increased clotting risk could be due to damaged endothelium from the ablation itself and stunning of atrial tissue, which results in impaired contraction. Damaged endothelium improves as the tissue heals, and the stunning resolves by electrical reverse remodeling with sinus rhythm maintenance.
In view of these risks, the referring physician and electrophysiologist must pay careful attention to anticoagulation before and after ablation.
Before the procedure. It is safe to continue anticoagulation uninterrupted through the procedure.38,39 If the patient is on warfarin, we want the international normalized ratio to be in the therapeutic range when we perform atrial fibrillation ablation, and the patient takes his or her usual dose on the day of the procedure. If taking a direct oral anticoagulant, patients typically skip a dose the day before ablation and again on the morning of the procedure, and resume taking it immediately afterward while in the anesthesia recovery room.
During the procedure, we start heparin before transseptal puncture, adjust it to achieve an activated clotting time of 300 to 400 seconds, and keep it in this range as long as there are sheaths or catheters in the left atrium.
After the procedure. The current guidelines24 recommend that oral anticoagulation be continued without interruption for at least 2 months after the procedure, and in most cases indefinitely, depending on age and comorbidities. The decision to stop anticoagulation after 2 months is typically based on the stroke risk as assessed by the CHA2DS2-VASc score (www.chadsvasc.org) and not on the success of the ablation procedure.
ANTIARRHYTHMIC DRUGS AFTER THE PROCEDURE
Some patients actually experience more atrial fibrillation in the first weeks to months after the procedure. The mechanism in this setting may be different from that causing the arrhythmia in the first place. The causes of early recurrence of atrial arrhythmias include postablation inflammation, temporary autonomic imbalance, and delay of atrial radiofrequency lesion formation.40,41 These arrhythmias may completely resolve as the ablation lesions heal and scars mature.
It has been hypothesized that short-term use of antiarrhythmic drugs after atrial fibrillation ablation is effective in preventing arrhythmias because it alters atrial electrophysiologic characteristics induced by the above transient factors. A recent systematic review of 6 clinical trials showed that short-term use of antiarrhythmic drugs reduces the risk of early arrhythmia recurrence but does not reduce recurrence in the long term.42
In terms of outcomes, any arrhythmias that occur in the first 3 months do not necessarily affect long-term success. This is referred to as the “blanking period.” However, generally speaking, it is preferable to maintain sinus rhythm during that time to avoid further anatomic or electrical left atrial adverse remodeling. In many situations, patients continue taking the same antiarrhythmic agent or start on antiarrhythmic therapy in the first few months after ablation.43,44
The mechanisms of late recurrence of atrial arrhythmias after ablation are thought to be different from those in early recurrence. Late recurrence has been ascribed to incomplete pulmonary vein isolation, recovery of pulmonary vein-left atrium connections, or recovery of any other lines of ablation created in the procedure.45,46 For late recurrence of atrial arrhythmia, studies and guidelines suggest that repeat ablation may be an option.24,47
PRACTICAL CONSIDERATIONS FOR PROCEDURAL PLANNING
Before the procedure, some electrophysiologists use cardiac computed tomography or magnetic resonance imaging to evaluate the pulmonary vein anatomy. This helps in planning and in selecting the appropriate tools for the procedure.
The patient is asked to fast on the day of the procedure. The procedure can take 3 to 6 hours, depending on the patient’s anatomy and the operator’s technique and experience. It can be performed with the patient under general anesthesia or conscious sedation. Currently, we use general anesthesia most of the time to maximize patient comfort.
After the procedure, our patients must stay in bed for 4 hours and stay overnight for observation. If no complications arise, they are discharged the next day.
- Go AS. The epidemiology of atrial fibrillation in elderly persons: the tip of the iceberg. Am J Geriatr Cardiol 2005; 14(2):56–61. pmid:15785146
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285(18):2370–2375. pmid:11343485
- Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006; 114(2):119–125. doi:10.1161/CIRCULATIONAHA.105.595140
- Piccini JP, Hammill BG, Sinner MF, et al. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ Cardiovasc Qual Outcomes 2012; 5(1):85–93. doi:10.1161/CIRCOUTCOMES.111.962688
- Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J 1964; 67:200–220. pmid:14118488
- Cox JL, Schuessler RB, Boineau JP. The surgical treatment of atrial fibrillation. I. Summary of the current concepts of the mechanisms of atrial flutter and atrial fibrillation. J Thorac Cardiovasc Surg 1991; 101(3):402–405. pmid:1999933
- Cox JL, Schuessler RB, D’Agostino HJ Jr, et al. The surgical treatment of atrial fibrillation. III. Development of a definitive surgical procedure. J Thorac Cardiovasc Surg 1991; 101(4):569–583. pmid:2008095
- Jaïs P, Haïssaguerre M, Shah DC, et al. A focal source of atrial fibrillation treated by discrete radiofrequency ablation. Circulation 1997; 95(3):572–576. pmid:9024141
- Haïssaguerre M, Jaïs P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998; 339(10):659–666. doi:10.1056/NEJM199809033391003
- Davies MJ, Pomerance A. Pathology of atrial fibrillation in man. Br Heart J 1972; 34(5):520–525. pmid:5031645
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995; 92(7):1954–1968. pmid:7671380
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002; 415(6868):219–226. doi:10.1038/415219a
- Medi C, Sparks PB, Morton JB, et al. Pulmonary vein antral isolation for paroxysmal atrial fibrillation: results from long-term follow-up. J Cardiovasc Electrophysiol 2011; 22(2):137–141. doi:10.1111/j.1540-8167.2010.01885.x
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4(3):271–278. doi:10.1161/CIRCEP.111.962100
- Hussein AA, Saliba WI, Barakat A, et al. Radiofrequency ablation of persistent atrial fibrillation: diagnosis-to-ablation time, markers of pathways of atrial remodeling, and outcomes. Circ Arrhythm Electrophysiol 2016; 9(1):e003669. doi:10.1161/CIRCEP.115.003669
- Carlsson J, Miketic S, Windeler J, et al. Randomized trial of rate-control versus rhythm-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol 2003; 41(10):1690–1696. pmid:12767648
- Van Gelder IC, Hagens VE, Bosker HA, et al; Rate Control versus Electrical Cardioversion for Persistent Atrial Fibrillation Study Group. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med 2002; 347(23):1834–1840. doi:10.1056/NEJMoa021375
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347(23):1825–1833. doi:10.1056/NEJMoa021328
- Hagens VE, Crijns HJ, Van Veldhuisen DJ, et al; RAte Control versus Electrical cardioversion for persistent atrial fibrillation study group. Rate control versus rhythm control for patients with persistent atrial fibrillation with mild to moderate heart failure: results from the RAte Control versus Electrical cardioversion (RACE) study. Am Heart J 2005; 149(6):1106–111. doi:10.1016/j.ahj.2004.11.030
- Pedersen OD, Bagger H, Keller N, Marchant B, Køber L, Torp-Pedersen C. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: a Danish investigations of arrhythmia and mortality on dofetilide (diamond) substudy. Circulation 2001; 104(3):292–296. pmid:11457747
- Guiot A, Jongnarangsin K, Chugh A, et al. Anticoagulant therapy and risk of cerebrovascular events after catheter ablation of atrial fibrillation in the elderly. J Cardiovasc Electrophysiol 2012; 23(1):36–43. doi:10.1111/j.1540-8167.2011.02141.x
- Oral H, Chugh A, Ozaydin M, et al. Risk of thromboembolic events after percutaneous left atrial radiofrequency ablation of atrial fibrillation. Circulation 2006; 114(8):759–765. doi:10.1161/CIRCULATIONAHA.106.641225
- Themistoclakis S, Corrado A, Marchlinski FE, et al. The risk of thromboembolism and need for oral anticoagulation after successful atrial fibrillation ablation. J Am Coll Cardiol 2010; 55(8):735–743. doi:10.1016/j.jacc.2009.11.039
- Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. J Arrhythm 2017; 33(5):369–409. doi:10.1016/j.joa.2017.08.001
- Kuck KH, Brugada J, Fürnkranz A, et al; FIRE AND ICE Investigators. Cryoballoon or radiofrequency ablation for paroxysmal atrial fibrillation. N Engl J Med 2016; 374(23):2235–2245. doi:10.1056/NEJMoa1602014
- Reddy VY, Dukkipati SR, Neuzil P, et al. Randomized, controlled trial of the safety and effectiveness of a contact force-sensing irrigated catheter for ablation of paroxysmal atrial fibrillation: results of the TactiCath Contact Force Ablation Catheter Study for Atrial Fibrillation (TOCCASTAR) Study. Circulation 2015; 132(10):907–915. doi:10.1161/CIRCULATIONAHA.114.014092
- Natale A, Reddy VY, Monir G, et al. Paroxysmal AF catheter ablation with a contact force sensing catheter: results of the prospective, multicenter SMART-AF trial. J Am Coll Cardiol 2014; 64(7):647–656. doi:10.1016/j.jacc.2014.04.072
- Hussein AA, Barakat AF, Saliba WI, et al. Persistent atrial fibrillation ablation with or without contact force sensing. J Cardiovasc Electrophysiol 2017; 28(5):483–488. doi:10.1111/jce.13179
- Deshmukh A, Patel NJ, Pant I, et al. In-hospital complications associated with catheter ablation of atrial fibrillation in the United States between 2000 and 2010: analysis of 93,801 procedures. Circulation 2013; 128(19):2104–2112. doi:10.1161/CIRCULATIONAHA.113.003862
- Cappato R, Calkins H, Chen SA, et al. Worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circulation 2005; 111(9):1100–1105. doi:10.1161/01.CIR.0000157153.30978.67
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3(1):32–38. doi:10.1161/CIRCEP.109.859116
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293(21):2634–2640. doi:10.1001/jama.293.21.2634
- Jaïs P, Cauchemez B, Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 study. Circulation 2008; 118(24):2498–2505. doi:10.1161/CIRCULATIONAHA.108.772582
- Brooks AG, Stiles MK, Laborderie J, et al. Outcomes of long-standing persistent atrial fibrillation ablation: a systematic review. Heart Rhythm 2010; 7(6):835–846. doi:10.1016/j.hrthm.2010.01.017
- Packer DL, Lee KL, Mark DB, Robb RA. Catheter Ablation versus Antiarrhythmic Drug Therapy for Atrial Fibrillation Trial, CABANA. http://cabanatrial.org/. Accessed September 10, 2018.
- Scherr D, Sharma K, Dalal D, et al. Incidence and predictors of periprocedural cerebrovascular accident in patients undergoing catheter ablation of atrial fibrillation. J Cardiovasc Electrophysiol 2009; 20(12):1357–1363. doi:10.1111/j.1540-8167.2009.01540.x
- Wazni OM, Rossillo A, Marrouche NF, et al. Embolic events and char formation during pulmonary vein isolation in patients with atrial fibrillation: impact of different anticoagulation regimens and importance of intracardiac echo imaging. J Cardiovasc Electrophysiol 2005; 16(6):576–581. doi:10.1111/j.1540-8167.2005.40480.x
- Hussein AA, Martin DO, Saliba W, et al. Radiofrequency ablation of atrial fibrillation under therapeutic international normalized ratio: a safe and efficacious periprocedural anticoagulation strategy. Heart Rhythm 2009; 6(10):1425–1429. doi:10.1016/j.hrthm.2009.07.007
- Bassiouny M, Saliba W, Rickard J, et al. Use of dabigatran for periprocedural anticoagulation in patients undergoing catheter ablation for atrial fibrillation. Circ Arrhythm Electrophysiol 2013; 6(3):460–466. doi:10.1161/CIRCEP.113.000320
- Koyama T, Tada H, Sekiguchi Y, et al. Prevention of atrial fibrillation recurrence with corticosteroids after radiofrequency catheter ablation: a randomized controlled trial. J Am Coll Cardiol 2010; 56(18):1463–1472. doi:10.1016/j.jacc.2010.04.057
- Oral H, Knight BP, Ozaydin M, et al. Clinical significance of early recurrences of atrial fibrillation after pulmonary vein isolation. J Am Coll Cardiol 2002; 40(1):100–104. pmid:12103262
- Chen W, Liu H, Ling Z, et al. Efficacy of short-term antiarrhythmic drugs use after catheter ablation of atrial fibrillation—a systematic review with meta-analyses and trial sequential analyses of randomized controlled trials. PLoS One 2016; 11(5):e0156121. doi:10.1371/journal.pone.0156121
- Leong-Sit P, Roux JF, Zado E, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study): six-month follow-up study. Circ Arrhythm Electrophysiol 2011; 4(1):11–14. doi:10.1161/CIRCEP.110.955393
- Roux JF, Zado E, Callans DJ, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study). Circulation 2009; 120(12):1036–1040. doi:10.1161/CIRCULATIONAHA.108.839639
- Sotomi Y, Inoue K, Ito N, et al. Cause of very late recurrence of atrial fibrillation or flutter after catheter ablation for atrial fibrillation. Am J Cardiol 2013; 111(4):552–556. doi:10.1016/j.amjcard.2012.10.040
- Lee SH, Tai CT, Hsieh MH, et al. Predictors of early and late recurrence of atrial fibrillation after catheter ablation of paroxysmal atrial fibrillation. J Interv Card Electrophysiol. 2004 Jun;10(3):221-6. doi:10.1023/B:JICE.0000026915.02503.92
- Zhang XD, Gu J, Jiang WF, et al. Optimal rhythm-control strategy for recurrent atrial tachycardia after catheter ablation of persistent atrial fibrillation: a randomized clinical trial. Eur Heart J 2014; 35(20):1327–1334. doi:10.1093/eurheartj/ehu017
- Go AS. The epidemiology of atrial fibrillation in elderly persons: the tip of the iceberg. Am J Geriatr Cardiol 2005; 14(2):56–61. pmid:15785146
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285(18):2370–2375. pmid:11343485
- Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006; 114(2):119–125. doi:10.1161/CIRCULATIONAHA.105.595140
- Piccini JP, Hammill BG, Sinner MF, et al. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ Cardiovasc Qual Outcomes 2012; 5(1):85–93. doi:10.1161/CIRCOUTCOMES.111.962688
- Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. Am Heart J 1964; 67:200–220. pmid:14118488
- Cox JL, Schuessler RB, Boineau JP. The surgical treatment of atrial fibrillation. I. Summary of the current concepts of the mechanisms of atrial flutter and atrial fibrillation. J Thorac Cardiovasc Surg 1991; 101(3):402–405. pmid:1999933
- Cox JL, Schuessler RB, D’Agostino HJ Jr, et al. The surgical treatment of atrial fibrillation. III. Development of a definitive surgical procedure. J Thorac Cardiovasc Surg 1991; 101(4):569–583. pmid:2008095
- Jaïs P, Haïssaguerre M, Shah DC, et al. A focal source of atrial fibrillation treated by discrete radiofrequency ablation. Circulation 1997; 95(3):572–576. pmid:9024141
- Haïssaguerre M, Jaïs P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998; 339(10):659–666. doi:10.1056/NEJM199809033391003
- Davies MJ, Pomerance A. Pathology of atrial fibrillation in man. Br Heart J 1972; 34(5):520–525. pmid:5031645
- Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995; 92(7):1954–1968. pmid:7671380
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002; 415(6868):219–226. doi:10.1038/415219a
- Medi C, Sparks PB, Morton JB, et al. Pulmonary vein antral isolation for paroxysmal atrial fibrillation: results from long-term follow-up. J Cardiovasc Electrophysiol 2011; 22(2):137–141. doi:10.1111/j.1540-8167.2010.01885.x
- Hussein AA, Saliba WI, Martin DO, et al. Natural history and long-term outcomes of ablated atrial fibrillation. Circ Arrhythm Electrophysiol 2011; 4(3):271–278. doi:10.1161/CIRCEP.111.962100
- Hussein AA, Saliba WI, Barakat A, et al. Radiofrequency ablation of persistent atrial fibrillation: diagnosis-to-ablation time, markers of pathways of atrial remodeling, and outcomes. Circ Arrhythm Electrophysiol 2016; 9(1):e003669. doi:10.1161/CIRCEP.115.003669
- Carlsson J, Miketic S, Windeler J, et al. Randomized trial of rate-control versus rhythm-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol 2003; 41(10):1690–1696. pmid:12767648
- Van Gelder IC, Hagens VE, Bosker HA, et al; Rate Control versus Electrical Cardioversion for Persistent Atrial Fibrillation Study Group. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med 2002; 347(23):1834–1840. doi:10.1056/NEJMoa021375
- Wyse DG, Waldo AL, DiMarco JP, et al; Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347(23):1825–1833. doi:10.1056/NEJMoa021328
- Hagens VE, Crijns HJ, Van Veldhuisen DJ, et al; RAte Control versus Electrical cardioversion for persistent atrial fibrillation study group. Rate control versus rhythm control for patients with persistent atrial fibrillation with mild to moderate heart failure: results from the RAte Control versus Electrical cardioversion (RACE) study. Am Heart J 2005; 149(6):1106–111. doi:10.1016/j.ahj.2004.11.030
- Pedersen OD, Bagger H, Keller N, Marchant B, Køber L, Torp-Pedersen C. Efficacy of dofetilide in the treatment of atrial fibrillation-flutter in patients with reduced left ventricular function: a Danish investigations of arrhythmia and mortality on dofetilide (diamond) substudy. Circulation 2001; 104(3):292–296. pmid:11457747
- Guiot A, Jongnarangsin K, Chugh A, et al. Anticoagulant therapy and risk of cerebrovascular events after catheter ablation of atrial fibrillation in the elderly. J Cardiovasc Electrophysiol 2012; 23(1):36–43. doi:10.1111/j.1540-8167.2011.02141.x
- Oral H, Chugh A, Ozaydin M, et al. Risk of thromboembolic events after percutaneous left atrial radiofrequency ablation of atrial fibrillation. Circulation 2006; 114(8):759–765. doi:10.1161/CIRCULATIONAHA.106.641225
- Themistoclakis S, Corrado A, Marchlinski FE, et al. The risk of thromboembolism and need for oral anticoagulation after successful atrial fibrillation ablation. J Am Coll Cardiol 2010; 55(8):735–743. doi:10.1016/j.jacc.2009.11.039
- Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. J Arrhythm 2017; 33(5):369–409. doi:10.1016/j.joa.2017.08.001
- Kuck KH, Brugada J, Fürnkranz A, et al; FIRE AND ICE Investigators. Cryoballoon or radiofrequency ablation for paroxysmal atrial fibrillation. N Engl J Med 2016; 374(23):2235–2245. doi:10.1056/NEJMoa1602014
- Reddy VY, Dukkipati SR, Neuzil P, et al. Randomized, controlled trial of the safety and effectiveness of a contact force-sensing irrigated catheter for ablation of paroxysmal atrial fibrillation: results of the TactiCath Contact Force Ablation Catheter Study for Atrial Fibrillation (TOCCASTAR) Study. Circulation 2015; 132(10):907–915. doi:10.1161/CIRCULATIONAHA.114.014092
- Natale A, Reddy VY, Monir G, et al. Paroxysmal AF catheter ablation with a contact force sensing catheter: results of the prospective, multicenter SMART-AF trial. J Am Coll Cardiol 2014; 64(7):647–656. doi:10.1016/j.jacc.2014.04.072
- Hussein AA, Barakat AF, Saliba WI, et al. Persistent atrial fibrillation ablation with or without contact force sensing. J Cardiovasc Electrophysiol 2017; 28(5):483–488. doi:10.1111/jce.13179
- Deshmukh A, Patel NJ, Pant I, et al. In-hospital complications associated with catheter ablation of atrial fibrillation in the United States between 2000 and 2010: analysis of 93,801 procedures. Circulation 2013; 128(19):2104–2112. doi:10.1161/CIRCULATIONAHA.113.003862
- Cappato R, Calkins H, Chen SA, et al. Worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circulation 2005; 111(9):1100–1105. doi:10.1161/01.CIR.0000157153.30978.67
- Cappato R, Calkins H, Chen SA, et al. Updated worldwide survey on the methods, efficacy, and safety of catheter ablation for human atrial fibrillation. Circ Arrhythm Electrophysiol 2010; 3(1):32–38. doi:10.1161/CIRCEP.109.859116
- Wazni OM, Marrouche NF, Martin DO, et al. Radiofrequency ablation vs antiarrhythmic drugs as first-line treatment of symptomatic atrial fibrillation: a randomized trial. JAMA 2005; 293(21):2634–2640. doi:10.1001/jama.293.21.2634
- Jaïs P, Cauchemez B, Macle L, et al. Catheter ablation versus antiarrhythmic drugs for atrial fibrillation: the A4 study. Circulation 2008; 118(24):2498–2505. doi:10.1161/CIRCULATIONAHA.108.772582
- Brooks AG, Stiles MK, Laborderie J, et al. Outcomes of long-standing persistent atrial fibrillation ablation: a systematic review. Heart Rhythm 2010; 7(6):835–846. doi:10.1016/j.hrthm.2010.01.017
- Packer DL, Lee KL, Mark DB, Robb RA. Catheter Ablation versus Antiarrhythmic Drug Therapy for Atrial Fibrillation Trial, CABANA. http://cabanatrial.org/. Accessed September 10, 2018.
- Scherr D, Sharma K, Dalal D, et al. Incidence and predictors of periprocedural cerebrovascular accident in patients undergoing catheter ablation of atrial fibrillation. J Cardiovasc Electrophysiol 2009; 20(12):1357–1363. doi:10.1111/j.1540-8167.2009.01540.x
- Wazni OM, Rossillo A, Marrouche NF, et al. Embolic events and char formation during pulmonary vein isolation in patients with atrial fibrillation: impact of different anticoagulation regimens and importance of intracardiac echo imaging. J Cardiovasc Electrophysiol 2005; 16(6):576–581. doi:10.1111/j.1540-8167.2005.40480.x
- Hussein AA, Martin DO, Saliba W, et al. Radiofrequency ablation of atrial fibrillation under therapeutic international normalized ratio: a safe and efficacious periprocedural anticoagulation strategy. Heart Rhythm 2009; 6(10):1425–1429. doi:10.1016/j.hrthm.2009.07.007
- Bassiouny M, Saliba W, Rickard J, et al. Use of dabigatran for periprocedural anticoagulation in patients undergoing catheter ablation for atrial fibrillation. Circ Arrhythm Electrophysiol 2013; 6(3):460–466. doi:10.1161/CIRCEP.113.000320
- Koyama T, Tada H, Sekiguchi Y, et al. Prevention of atrial fibrillation recurrence with corticosteroids after radiofrequency catheter ablation: a randomized controlled trial. J Am Coll Cardiol 2010; 56(18):1463–1472. doi:10.1016/j.jacc.2010.04.057
- Oral H, Knight BP, Ozaydin M, et al. Clinical significance of early recurrences of atrial fibrillation after pulmonary vein isolation. J Am Coll Cardiol 2002; 40(1):100–104. pmid:12103262
- Chen W, Liu H, Ling Z, et al. Efficacy of short-term antiarrhythmic drugs use after catheter ablation of atrial fibrillation—a systematic review with meta-analyses and trial sequential analyses of randomized controlled trials. PLoS One 2016; 11(5):e0156121. doi:10.1371/journal.pone.0156121
- Leong-Sit P, Roux JF, Zado E, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study): six-month follow-up study. Circ Arrhythm Electrophysiol 2011; 4(1):11–14. doi:10.1161/CIRCEP.110.955393
- Roux JF, Zado E, Callans DJ, et al. Antiarrhythmics after ablation of atrial fibrillation (5A Study). Circulation 2009; 120(12):1036–1040. doi:10.1161/CIRCULATIONAHA.108.839639
- Sotomi Y, Inoue K, Ito N, et al. Cause of very late recurrence of atrial fibrillation or flutter after catheter ablation for atrial fibrillation. Am J Cardiol 2013; 111(4):552–556. doi:10.1016/j.amjcard.2012.10.040
- Lee SH, Tai CT, Hsieh MH, et al. Predictors of early and late recurrence of atrial fibrillation after catheter ablation of paroxysmal atrial fibrillation. J Interv Card Electrophysiol. 2004 Jun;10(3):221-6. doi:10.1023/B:JICE.0000026915.02503.92
- Zhang XD, Gu J, Jiang WF, et al. Optimal rhythm-control strategy for recurrent atrial tachycardia after catheter ablation of persistent atrial fibrillation: a randomized clinical trial. Eur Heart J 2014; 35(20):1327–1334. doi:10.1093/eurheartj/ehu017
KEY POINTS
- Atrial fibrillation is increasing in prevalence with the aging of the US population and is associated with worsening quality of life and increased risk of stroke, heart failure, and death.
- Atrial fibrillation results in adverse atrial remodeling and fibrosis, eventually leading to persistence of the arrhythmia and making rhythm control difficult.
- Catheter ablation has evolved to be a safe procedure with technologic advancements, especially in experienced tertiary care centers.
- The primary aim of atrial fibrillation ablation is to reduce symptoms and improve quality of life. In theory, it could also decrease the risk of stroke, heart failure, and death, but these outcomes have not been systematically evaluated in a large randomized controlled trial.