User login
Can taming inflammation help reduce aggression?
Several psychiatric disorders, including depression, schizophrenia, bipolar disorder, Alzheimer’s disease, traumatic brain injury, autism, and posttraumatic stress disorder, are associated with a dysregulated immune response and elevated levels of inflammatory biomarkers. Inflammation has long been associated with an increased risk of aggressive behavior.1,2 By taming immune system dysregulation, we might be able to more effectively reduce inflammation, and thus reduce aggression, in patients with psychiatric illness.
Inflammation and psychiatric symptoms
An overactivated immune response has been empirically correlated to the development of psychiatric symptoms. Inducing systemic inflammation has adverse effects on cognition and behavior, whereas suppressing inflammation can dramatically improve sensorium and mood. Brain regions involved in arousal and alarm are particularly susceptible to inflammation. Subcortical areas, such as the basal ganglia, and cortical circuits, such as the amygdala and anterior insula, are affected by neuroinflammation. Several modifiable factors, including a diet rich in high glycemic food, improper sleep hygiene, tobacco use, a sedentary lifestyle, obesity, and excess psychosocial stressors, can contribute to systemic inflammation and the development of psychiatric symptoms. Oral diseases, such as tooth decay, periodontitis, and gingivitis, also contribute significantly to overall inflammation.
Anti-inflammatory agents
Using nonsteroidal anti-inflammatory drugs as augmentation to standard treatments has shown promise in several psychiatric illnesses. For example, low-dose aspirin, 81 mg/d, has demonstrated reliable results as an adjunctive treatment for depression.3 Research also has shown that the use of ibuprofen may reduce the chances of individuals seeking psychiatric care.3
Individuals who are at high risk for psychosis and schizophrenia have measurable increases in inflammatory microglial activity.4 The severity of psychotic symptoms corresponds to the magnitude of the immune response; this suggests that neuroinflammation is a risk factor for psychosis, and that anti-inflammatory treatments might help prevent or ameliorate psychosis.
In a double-blind, placebo-controlled study, 70 patients diagnosed with schizophrenia who were taking an antipsychotic were randomized to adjunctive aspirin, 1,000 mg/d, or placebo.5 Participants who received aspirin had significant improvement as measured by changes in Positive and Negative Syndrome Scale total score.5
Targeting C-reactive protein
Inflammation has long been associated with impulsive aggression. C-reactive protein (CRP) is a biomarker produced in the liver in response to inflammatory triggers. In a study of 213 inpatients with schizophrenia, researchers compared 57 patients with higher levels of CRP (>1 mg/dL) with 156 patients with normal levels (<1 mg/dL).2 Compared with patients with normal CRP levels, those with higher levels displayed increased aggressive behavior. Researchers found that the chance of being physically restrained during hospitalization was almost 2.5 times greater for patients with elevated CRP levels on admission compared with those with normal CRP levels.
Statins have long been used to reduce C-reactive peptides in patients with cardiovascular conditions. The use of simvastatin has been shown to significantly reduce negative symptoms in patients with schizophrenia.6
Continue to: Vitamin C also can effectively...
Vitamin C also can effectively lower CRP levels. In a 2-month study, 396 participants with elevated CRP levels received vitamin C, 1,000 mg/d, vitamin E, 800 IU/d, or placebo.7 Although vitamin E didn’t reduce CRP levels, vitamin C reduced CRP by 25.3% compared with placebo. Vitamin C is as effective as statins in controlling this biomarker.
Several nonpharmacologic measures also can help reduce the immune system’s activation of CRP, including increased physical activity, increased intake of low glycemic food and supplemental omega-3 fatty acids, improved dental hygiene, and enhanced sleep.
Using a relatively simple and inexpensive laboratory test for measuring CRP might help predict or stratify the risk of aggressive behavior among psychiatric inpatients. For psychiatric patients with elevated inflammatory markers, the interventions described here may be useful as adjunctive treatments to help reduce aggression and injury in an inpatient setting.
1. Coccaro EF, Lee R, Coussons-Read M. Elevated plasma inflammatory markers in individuals with intermittent explosive disorder and correlation with aggression in humans. JAMA Psychiatry. 2014;71(2):158-165.
2. Barzilay R, Lobel T, Krivoy A, et al. Elevated C-reactive protein levels in schizophrenia inpatients is associated with aggressive behavior. Eur Psychiatry. 2016;31:8-12.
3. Köhler O, Peterson L, Mors O, et al. Inflammation and depression: combined use of selective serotonin reuptake inhibitors and NSAIDs or paracetamol and psychiatric outcomes. Brain and Behavior. 2015;5(8):e00338. doi: 10.1002/brb3.338.
4. Bloomfield PS, Selvaraj S, Veronese M, et al. M icroglial activity in people at ultra high risk of psychosis and in schizophrenia; an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173(1):44-52.
5. Laan W, Grobbee DE, Selten JP, et al. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2010;71(5):520-527.
6. Tajik-Esmaeeli S, Moazen-Zadeh E, Abbasi N, et al. Simvastatin adjunct therapy for negative symptoms of schizophrenia: a randomized double-blind placebo-controlled trial. Int Clin Psychopharmacol. 2017;32(2):87-94.
7. Block G, Jensen CD, Dalvi TB, et al. Vitamin C treatment reduces elevated C-reactive protein. Free Radic Biol Med. 2009;46(1):70-77.
Several psychiatric disorders, including depression, schizophrenia, bipolar disorder, Alzheimer’s disease, traumatic brain injury, autism, and posttraumatic stress disorder, are associated with a dysregulated immune response and elevated levels of inflammatory biomarkers. Inflammation has long been associated with an increased risk of aggressive behavior.1,2 By taming immune system dysregulation, we might be able to more effectively reduce inflammation, and thus reduce aggression, in patients with psychiatric illness.
Inflammation and psychiatric symptoms
An overactivated immune response has been empirically correlated to the development of psychiatric symptoms. Inducing systemic inflammation has adverse effects on cognition and behavior, whereas suppressing inflammation can dramatically improve sensorium and mood. Brain regions involved in arousal and alarm are particularly susceptible to inflammation. Subcortical areas, such as the basal ganglia, and cortical circuits, such as the amygdala and anterior insula, are affected by neuroinflammation. Several modifiable factors, including a diet rich in high glycemic food, improper sleep hygiene, tobacco use, a sedentary lifestyle, obesity, and excess psychosocial stressors, can contribute to systemic inflammation and the development of psychiatric symptoms. Oral diseases, such as tooth decay, periodontitis, and gingivitis, also contribute significantly to overall inflammation.
Anti-inflammatory agents
Using nonsteroidal anti-inflammatory drugs as augmentation to standard treatments has shown promise in several psychiatric illnesses. For example, low-dose aspirin, 81 mg/d, has demonstrated reliable results as an adjunctive treatment for depression.3 Research also has shown that the use of ibuprofen may reduce the chances of individuals seeking psychiatric care.3
Individuals who are at high risk for psychosis and schizophrenia have measurable increases in inflammatory microglial activity.4 The severity of psychotic symptoms corresponds to the magnitude of the immune response; this suggests that neuroinflammation is a risk factor for psychosis, and that anti-inflammatory treatments might help prevent or ameliorate psychosis.
In a double-blind, placebo-controlled study, 70 patients diagnosed with schizophrenia who were taking an antipsychotic were randomized to adjunctive aspirin, 1,000 mg/d, or placebo.5 Participants who received aspirin had significant improvement as measured by changes in Positive and Negative Syndrome Scale total score.5
Targeting C-reactive protein
Inflammation has long been associated with impulsive aggression. C-reactive protein (CRP) is a biomarker produced in the liver in response to inflammatory triggers. In a study of 213 inpatients with schizophrenia, researchers compared 57 patients with higher levels of CRP (>1 mg/dL) with 156 patients with normal levels (<1 mg/dL).2 Compared with patients with normal CRP levels, those with higher levels displayed increased aggressive behavior. Researchers found that the chance of being physically restrained during hospitalization was almost 2.5 times greater for patients with elevated CRP levels on admission compared with those with normal CRP levels.
Statins have long been used to reduce C-reactive peptides in patients with cardiovascular conditions. The use of simvastatin has been shown to significantly reduce negative symptoms in patients with schizophrenia.6
Continue to: Vitamin C also can effectively...
Vitamin C also can effectively lower CRP levels. In a 2-month study, 396 participants with elevated CRP levels received vitamin C, 1,000 mg/d, vitamin E, 800 IU/d, or placebo.7 Although vitamin E didn’t reduce CRP levels, vitamin C reduced CRP by 25.3% compared with placebo. Vitamin C is as effective as statins in controlling this biomarker.
Several nonpharmacologic measures also can help reduce the immune system’s activation of CRP, including increased physical activity, increased intake of low glycemic food and supplemental omega-3 fatty acids, improved dental hygiene, and enhanced sleep.
Using a relatively simple and inexpensive laboratory test for measuring CRP might help predict or stratify the risk of aggressive behavior among psychiatric inpatients. For psychiatric patients with elevated inflammatory markers, the interventions described here may be useful as adjunctive treatments to help reduce aggression and injury in an inpatient setting.
Several psychiatric disorders, including depression, schizophrenia, bipolar disorder, Alzheimer’s disease, traumatic brain injury, autism, and posttraumatic stress disorder, are associated with a dysregulated immune response and elevated levels of inflammatory biomarkers. Inflammation has long been associated with an increased risk of aggressive behavior.1,2 By taming immune system dysregulation, we might be able to more effectively reduce inflammation, and thus reduce aggression, in patients with psychiatric illness.
Inflammation and psychiatric symptoms
An overactivated immune response has been empirically correlated to the development of psychiatric symptoms. Inducing systemic inflammation has adverse effects on cognition and behavior, whereas suppressing inflammation can dramatically improve sensorium and mood. Brain regions involved in arousal and alarm are particularly susceptible to inflammation. Subcortical areas, such as the basal ganglia, and cortical circuits, such as the amygdala and anterior insula, are affected by neuroinflammation. Several modifiable factors, including a diet rich in high glycemic food, improper sleep hygiene, tobacco use, a sedentary lifestyle, obesity, and excess psychosocial stressors, can contribute to systemic inflammation and the development of psychiatric symptoms. Oral diseases, such as tooth decay, periodontitis, and gingivitis, also contribute significantly to overall inflammation.
Anti-inflammatory agents
Using nonsteroidal anti-inflammatory drugs as augmentation to standard treatments has shown promise in several psychiatric illnesses. For example, low-dose aspirin, 81 mg/d, has demonstrated reliable results as an adjunctive treatment for depression.3 Research also has shown that the use of ibuprofen may reduce the chances of individuals seeking psychiatric care.3
Individuals who are at high risk for psychosis and schizophrenia have measurable increases in inflammatory microglial activity.4 The severity of psychotic symptoms corresponds to the magnitude of the immune response; this suggests that neuroinflammation is a risk factor for psychosis, and that anti-inflammatory treatments might help prevent or ameliorate psychosis.
In a double-blind, placebo-controlled study, 70 patients diagnosed with schizophrenia who were taking an antipsychotic were randomized to adjunctive aspirin, 1,000 mg/d, or placebo.5 Participants who received aspirin had significant improvement as measured by changes in Positive and Negative Syndrome Scale total score.5
Targeting C-reactive protein
Inflammation has long been associated with impulsive aggression. C-reactive protein (CRP) is a biomarker produced in the liver in response to inflammatory triggers. In a study of 213 inpatients with schizophrenia, researchers compared 57 patients with higher levels of CRP (>1 mg/dL) with 156 patients with normal levels (<1 mg/dL).2 Compared with patients with normal CRP levels, those with higher levels displayed increased aggressive behavior. Researchers found that the chance of being physically restrained during hospitalization was almost 2.5 times greater for patients with elevated CRP levels on admission compared with those with normal CRP levels.
Statins have long been used to reduce C-reactive peptides in patients with cardiovascular conditions. The use of simvastatin has been shown to significantly reduce negative symptoms in patients with schizophrenia.6
Continue to: Vitamin C also can effectively...
Vitamin C also can effectively lower CRP levels. In a 2-month study, 396 participants with elevated CRP levels received vitamin C, 1,000 mg/d, vitamin E, 800 IU/d, or placebo.7 Although vitamin E didn’t reduce CRP levels, vitamin C reduced CRP by 25.3% compared with placebo. Vitamin C is as effective as statins in controlling this biomarker.
Several nonpharmacologic measures also can help reduce the immune system’s activation of CRP, including increased physical activity, increased intake of low glycemic food and supplemental omega-3 fatty acids, improved dental hygiene, and enhanced sleep.
Using a relatively simple and inexpensive laboratory test for measuring CRP might help predict or stratify the risk of aggressive behavior among psychiatric inpatients. For psychiatric patients with elevated inflammatory markers, the interventions described here may be useful as adjunctive treatments to help reduce aggression and injury in an inpatient setting.
1. Coccaro EF, Lee R, Coussons-Read M. Elevated plasma inflammatory markers in individuals with intermittent explosive disorder and correlation with aggression in humans. JAMA Psychiatry. 2014;71(2):158-165.
2. Barzilay R, Lobel T, Krivoy A, et al. Elevated C-reactive protein levels in schizophrenia inpatients is associated with aggressive behavior. Eur Psychiatry. 2016;31:8-12.
3. Köhler O, Peterson L, Mors O, et al. Inflammation and depression: combined use of selective serotonin reuptake inhibitors and NSAIDs or paracetamol and psychiatric outcomes. Brain and Behavior. 2015;5(8):e00338. doi: 10.1002/brb3.338.
4. Bloomfield PS, Selvaraj S, Veronese M, et al. M icroglial activity in people at ultra high risk of psychosis and in schizophrenia; an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173(1):44-52.
5. Laan W, Grobbee DE, Selten JP, et al. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2010;71(5):520-527.
6. Tajik-Esmaeeli S, Moazen-Zadeh E, Abbasi N, et al. Simvastatin adjunct therapy for negative symptoms of schizophrenia: a randomized double-blind placebo-controlled trial. Int Clin Psychopharmacol. 2017;32(2):87-94.
7. Block G, Jensen CD, Dalvi TB, et al. Vitamin C treatment reduces elevated C-reactive protein. Free Radic Biol Med. 2009;46(1):70-77.
1. Coccaro EF, Lee R, Coussons-Read M. Elevated plasma inflammatory markers in individuals with intermittent explosive disorder and correlation with aggression in humans. JAMA Psychiatry. 2014;71(2):158-165.
2. Barzilay R, Lobel T, Krivoy A, et al. Elevated C-reactive protein levels in schizophrenia inpatients is associated with aggressive behavior. Eur Psychiatry. 2016;31:8-12.
3. Köhler O, Peterson L, Mors O, et al. Inflammation and depression: combined use of selective serotonin reuptake inhibitors and NSAIDs or paracetamol and psychiatric outcomes. Brain and Behavior. 2015;5(8):e00338. doi: 10.1002/brb3.338.
4. Bloomfield PS, Selvaraj S, Veronese M, et al. M icroglial activity in people at ultra high risk of psychosis and in schizophrenia; an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173(1):44-52.
5. Laan W, Grobbee DE, Selten JP, et al. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2010;71(5):520-527.
6. Tajik-Esmaeeli S, Moazen-Zadeh E, Abbasi N, et al. Simvastatin adjunct therapy for negative symptoms of schizophrenia: a randomized double-blind placebo-controlled trial. Int Clin Psychopharmacol. 2017;32(2):87-94.
7. Block G, Jensen CD, Dalvi TB, et al. Vitamin C treatment reduces elevated C-reactive protein. Free Radic Biol Med. 2009;46(1):70-77.
Differentiating serotonin syndrome and neuroleptic malignant syndrome
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).

It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.

Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.

Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.

Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.

Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.

Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.

Older-age bipolar disorder: A case series
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
")
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.

A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
")
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.

Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.

The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.

Antidepressants for chronic pain
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.

Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88

Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.

Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.

Monoclonal gammopathy of undetermined significance: A primary care guide


MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.

A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?

Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.

Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?

that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.

Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.

The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.
A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?
Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.
Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?
that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.
Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.
The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.
A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?
Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.
Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?
that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.
Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.
The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
KEY POINTS
- MGUS is the most common of the monoclonal gammopathies.
- The overall risk of MGUS progressing to myeloma and other lymphoproliferative disorders is 1% per year.
- Low-risk MGUS is defined by an immunoglobulin G monoclonal protein at a concentration less than 1.5 g/dL and a normal serum free light-chain ratio.
- Low-risk MGUS carries a much lower risk of progression than intermediate- and high-risk MGUS, may not require subspecialty referral, and can be followed by the outpatient provider.
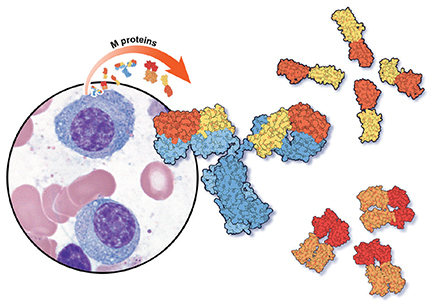
Hypertension guidelines: Treat patients, not numbers
When treating high blood pressure, how low should we try to go? Debate continues about optimal blood pressure goals after publication of guidelines from the American College of Cardiology and American Heart Association (ACC/AHA) in 2017 that set or permitted a treatment goal of less than 130 mm Hg, depending on the population.1
In this article, we summarize the evolution of hypertension guidelines and the evidence behind them.
HOW THE GOALS EVOLVED
JNC 7, 2003: 140/90 or 130/80
The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7),2 published in 2003, specified treatment goals of:
- < 140/90 mm Hg for most patients
- < 130/80 mm Hg for those with diabetes or chronic kidney disease.

JNC 7 provided much-needed clarity and uniformity to managing hypertension. Since then, various scientific groups have published their own guidelines (Table 1).1–9
ACC/AHA/CDC 2014: 140/90
In 2014, the ACC, AHA, and US Centers for Disease Control and Prevention (CDC) published an evidence-based algorithm for hypertension management.3 As in JNC 7, they suggested a blood pressure goal of less than 140/90 mm Hg, lifestyle modification, and polytherapy, eg, a thiazide diuretic for stage 1 hypertension (< 160/100 mm Hg) and combination therapy with a thiazide diuretic and an angiotensin-converting enzyme (ACE) inhibitor, angiotensin II receptor blocker (ARB), or calcium channel blocker for stage 2 hypertension (≥ 160/100 mm Hg).
JNC 8 2014: 140/90 or 150/90
Soon after, the much-anticipated report of the panel members appointed to the eighth JNC (JNC 8) was published.4 Previous JNC reports were written and published under the auspices of the National Heart, Lung, and Blood Institute, but while the JNC 8 report was being prepared, this government body announced it would no longer publish guidelines.
In contrast to JNC 7, the JNC 8 panel based its recommendations on a systematic review of randomized clinical trials. However, the process and methodology were controversial, especially as the panel excluded some important clinical trials from the analysis.
JNC 8 relaxed the targets in several subgroups, such as patients over age 60 and those with diabetes and chronic kidney disease, due to a lack of definitive evidence on the impact of blood pressure targets lower than 140/90 mm Hg in these groups. Thus, their goals were:
- < 140/90 mm Hg for patients under age 60
- < 150/90 mm Hg for patients age 60 and older.

Of note, a minority of the JNC 8 panel disagreed with the new targets and provided evidence for keeping the systolic blood pressure target below 140 mm Hg for patients 60 and older.5 Further, the JNC 8 report was not endorsed by several important societies, ie, the AHA, ACC, National Heart, Lung, and Blood Institute, and American Society of Hypertension (ASH). These issues compromised the acceptance and applicability of the guidelines.
ASH/ISH 2014: 140/90 or 150/90
Also in 2014, the ASH and the International Society of Hypertension released their own report.6 Their goals:
- < 140/90 mm Hg for most patients
- < 150/90 mm Hg for patients age 80 and older.
AHA/ACC/ASH 2015: Goals in subgroups
In 2015, the AHA, ACC, and ASH released a joint scientific statement outlining hypertension goals for specific patient populations7:
- < 150/90 mm Hg for those age 80 and older
- < 140/90 mm Hg for those with coronary artery disease
- < 130/80 mm Hg for those with comorbidities such as diabetes and cardiovascular disease.
ADA 2016: Goals for patients with diabetes
In 2016, the American Diabetes Association (ADA) set the following blood pressure goals for patients with diabetes8:
- < 140/90 mm Hg for adults with diabetes
- < 130/80 mm Hg for younger adults with diabetes and adults with a high risk of cardiovascular disease
- 120–160/80–105 mm Hg for pregnant patients with diabetes and preexisting hypertension who are treated with antihypertensive therapy.
ACP/AAFP 2017: Systolic 150 or 130
In 2017, the American College of Physicians (ACP) and the American Academy of Family Physicians (AAFP) recommended a relaxed systolic blood pressure target, ie, below 150 mm Hg, for adults over age 60, but a tighter goal of less than 140 mm Hg for the same age group if they have transient ischemic attack, stroke, or high cardiovascular risk.9
ACC/AHA 2017: 130/80
The 2017 ACC/AHA guidelines recommended a more aggressive goal of below 130/80 for all, including patients age 65 and older.1
This is a class I (strong) recommendation for patients with known cardiovascular disease or a 10-year risk of a cardiovascular event of 10% or higher, with a B-R level of evidence for the systolic goal (ie, moderate-quality, based on systematic review of randomized controlled trials) and a C-EO level of evidence for the diastolic goal (ie, based on expert opinion).
For patients who do not have cardiovascular disease and who are at lower risk of it, this is a class IIb (weak) recommendation, ie, it “may be reasonable,” with a B-NR level of evidence (moderate-quality, based on nonrandomized studies) for the systolic goal and C-EO (expert opinion) for the diastolic goal.
For many patients, this involves drug treatment. For those with known cardiovascular disease or a 10-year risk of an atherosclerotic cardiovascular disease event of 10% or higher, the ACC/AHA guidelines say that drug treatment “is recommended” if their average blood pressure is 130/80 mm Hg or higher (class I recommendation, based on strong evidence for the systolic threshold and expert option for the diastolic). For those without cardiovascular disease and at lower risk, drug treatment is recommended if their average blood pressure is 140/90 mm Hg or higher (also class I, but based on limited data).
EVERYONE AGREES ON LIFESTYLE
Although the guidelines differ in their blood pressure targets, they consistently recommend lifestyle modifications.
Lifestyle modifications, first described in JNC 7, included weight loss, sodium restriction, and the DASH diet, which is rich in fruits, vegetables, low-fat dairy products, whole grains, poultry, and fish, and low in red meat, sweets, cholesterol, and total and saturated fat.2
These recommendations were based on results from 3 large randomized controlled trials in patients with and without hypertension.10–12 In patients with no history of hypertension, interventions to promote weight loss and sodium restriction significantly reduced blood pressure and the incidence of hypertension (the latter by as much as 77%) compared with usual care.10,11
In patients with and without hypertension, lowering sodium intake in conjunction with the DASH diet was associated with substantially larger reductions in systolic blood pressure.12
The recommendation to lower sodium intake has not changed in the guideline revisions. Meanwhile, other modifications have been added, such as incorporating both aerobic and resistance exercise and moderating alcohol intake. These recommendations have a class I level of evidence (ie, strongest level) in the 2017 ACC/AHA guidelines.1
HYPERTENSION BEGINS AT 130/80
The definition of hypertension changed in the 2017 ACC/AHA guidelines1: previously set at 140/90 mm Hg or higher, it is now 130/80 mm Hg or higher for all age groups. Adults with systolic blood pressure of 130 to 139 mm Hg or diastolic blood pressure of 80 to 89 mm Hg are now classified as having stage 1 hypertension.
Under the new definition, the number of US adults who have hypertension expanded to 45.6% of the general population,13 up from 31.9% under the JNC 7 definition. Thus, overall, 103.3 million US adults now have hypertension, compared with 72.2 million under the JNC 7 criteria.
In addition, the new guidelines expanded the population of adults for whom antihypertensive drug treatment is recommended to 36.2% (81.9 million). However, this represents only a 1.9% absolute increase over the JNC 7 recommendations (34.3%) and a 5.1% absolute increase over the JNC 8 recommendations.14
SPRINT: INTENSIVE TREATMENT IS BENEFICIAL
The new ACC/AHA guidelines1 were based on evidence from several trials, including the Systolic Blood Pressure Intervention Trial (SPRINT).15
This multicenter trial investigated the effect of intensive blood pressure treatment on cardiovascular disease risk.16 The primary outcome was a composite of myocardial infarction, acute coronary syndrome, stroke, and heart failure.
The trial enrolled 9,361 participants at least 50 years of age with systolic blood pressure 130 mm Hg or higher and at least 1 additional risk factor for cardiovascular disease. It excluded anyone with a history of diabetes mellitus, stroke, symptomatic heart failure, or end-stage renal disease.
Two interventions were compared:
- Intensive treatment, with a systolic blood pressure goal of less than 120 mm Hg: the protocol called for polytherapy, even for participants who were 75 or older if their blood pressure was 140 mm Hg or higher
- Standard treatment, with a systolic blood pressure goal of less than 140 mm Hg: it used polytherapy for patients whose systolic blood pressure was 160 mm Hg or higher.
The trial was intended to last 5 years but was stopped early at a median of 3.26 years owing to a significantly lower rate of the primary composite outcome in the intensive-treatment group: 1.65% per year vs 2.19%, a 25% relative risk reduction (P < .001) or a 0.54% absolute risk reduction. We calculate the number needed to treat (NNT) for 1 year to prevent 1 event as 185, and over the 3.26 years of the trial, the investigators calculated the NNT as 61. Similarly, the rate of death from any cause was also lower with intensive treatment, 1.03% per year vs 1.40% per year, a 27% relative risk reduction (P = .003) or a 0.37% absolute risk reduction, NNT 270.
Using these findings, Bress et al16 estimated that implementing intensive blood pressure goals could prevent 107,500 deaths annually.
The downside is adverse effects. In SPRINT,15 the intensive-treatment group experienced significantly higher rates of serious adverse effects than the standard-treatment group, ie:
- Hypotension 2.4% vs 1.4%, P = .001
- Syncope 2.3% vs 1.7%, P = .05
- Electrolyte abnormalities 3.1% vs 2.3%, P = .02)
- Acute kidney injury or kidney failure 4.1% vs 2.5%, P < .001
- Any treatment-related adverse event 4.7% vs 2.5%, P = .001.
Thus, Bress et al16 estimated that fully implementing the intensive-treatment goals could cause an additional 56,100 episodes of hypotension per year, 34,400 cases of syncope, 43,400 serious electrolyte disorders, and 88,700 cases of acute kidney injury. All told, about 3 million Americans could suffer a serious adverse effect under the intensive-treatment goals.
SPRINT caveats and limitations
SPRINT15 was stopped early, after 3.26 years instead of the planned 5 years. The true risk-benefit ratio may have been different if the trial had been extended longer.
In addition, SPRINT used automated office blood pressure measurements in which patients were seated alone and a device (Model 907, Omron Healthcare) took 3 blood pressure measurements at 1-minute intervals after 5 minutes of quiet rest. This was designed to reduce elevated blood pressure readings in the presence of a healthcare professional in a medical setting (ie, “white coat” hypertension).
Many physicians are still taking blood pressure manually, which tends to give higher readings. Therefore, if they aim for a lower goal, they may risk overtreating the patient.
About 50% of patients did not achieve the target systolic blood pressure (< 120 mm Hg) despite receiving an average of 2.8 antihypertensive medications in the intensive-treatment group and 1.8 in the standard-treatment group. The use of antihypertensive medications, however, was not a controlled variable in the trial, and practitioners chose the appropriate drugs for their patients.
Diastolic pressure, which can be markedly lower in older hypertensive patients, was largely ignored, although lower diastolic pressure may have contributed to higher syncope rates in response to alpha blockers and calcium blockers.
Moreover, the trial excluded those with significant comorbidities and those younger than 50 (the mean age was 67.9), which limits the generalizability of the results.
JNC 8 VS SPRINT GOALS: WHAT'S THE EFFECT ON OUTCOMES?
JNC 84 recommended a relaxed target of less than 140/90 mm Hg for adults younger than 60, including those with chronic kidney disease or diabetes, and less than 150/90 mm Hg for adults 60 and older. The SPRINT findings upended those recommendations, showing that intensive treatment in adults age 75 or older significantly improved the composite cardiovascular disease outcome (2.59 vs 3.85 events per year; P < .001) and all-cause mortality (1.78 vs 2.63 events per year; P < .05) compared with standard treatment.17 Also, a subset review of SPRINT trial data found no difference in benefit based on chronic kidney disease status.18
A meta-analysis of 74 clinical trials (N = 306,273) offers a compromise between the SPRINT findings and the JNC 8 recommendations.19 It found that the beneficial effect of blood pressure treatment depended on the patient’s baseline systolic blood pressure. In those with a baseline systolic pressure of 160 mm Hg or higher, treatment reduced cardiovascular mortality by about 15% (relative risk [RR] 0.85; 95% confidence interval [CI] 0.77–0.95). In patients with systolic pressure below 140 mm Hg, treatment effects were neutral (RR 1.03, 95% CI 0.87–1.20) and not associated with any benefit as primary prevention, although data suggest it may reduce the risk of adverse outcomes in patients with coronary heart disease.
OTHER TRIALS THAT INFLUENCED THE GUIDELINES

SHEP and HYVET (the Systolic Hypertension in the Elderly Program20 and the Hypertension in the Very Elderly Trial)21 supported intensive blood pressure treatment for older patients by reporting a reduction in fatal and nonfatal stroke risks for those with a systolic blood pressure above 160 mm Hg.
FEVER (the Felodipine Event Reduction study)22 found that treatment with a calcium channel blocker in even a low dose can significantly decrease cardiovascular events, cardiovascular disease, and heart failure compared with no treatment.
JATOS and VALISH (the Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients23 and the Valsartan in Elderly Isolated Systolic Hypertension study)24 found that outcomes were similar with intensive vs standard treatment.
Ettehad et al25 performed a meta-analysis of 123 studies with more than 600,000 participants that provided strong evidence supporting blood pressure treatment goals below 130/90 mm Hg, in line with the SPRINT trial results.
BLOOD PRESSURE ISN’T EVERYTHING
Other trials remind us that although blood pressure is important, it is not the only factor affecting cardiovascular risk.
HOPE (the Heart Outcomes Prevention Evaluation)26 investigated the use of ramipril (an ACE inhibitor) in preventing myocardial infarction, stroke, or cardiovascular death in patients at high risk of cardiovascular events. The study included 9,297 participants over age 55 (mean age 66) with a baseline blood pressure 139/79 mm Hg. Follow-up was 4.5 years.
Ramipril was better than placebo, with significantly fewer patients experiencing adverse end points in the ramipril group compared with the placebo group:
- Myocardial infarction 9.9% vs 12.3%, RR 0.80, P < .001
- Cardiovascular death 6.1% vs 8.1%, RR 0.74, P < .001
- Stroke 3.4% vs 4.9%, RR = .68, P < .001
- The composite end point 14.0% vs 17.8%, RR 0.78, P < .001).
Results were even better in the subset of patients who had diabetes.27 However, the decrease in blood pressure attributable to antihypertensive therapy with ramipril was minimal (3–4 mm Hg systolic and 1–2 mm Hg diastolic). This slight change should not have been enough to produce significant differences in clinical outcomes, a major limitation of this trial. The investigators speculated that the positive results may be due to a class effect of ACE inhibitors.26
HOPE 328–30 explored the effect of blood pressure- and cholesterol-controlling drugs on the same primary end points but in patients at intermediate risk of major cardiovascular events. Investigators randomized the 12,705 patients to 4 treatment groups:
- Blood pressure control with candesartan (an ARB) plus hydrochlorothiazide (a thiazide diuretic)
- Cholesterol control with rosuvastatin (a statin)
- Blood pressure plus cholesterol control
- Placebo.
Therapy was started at a systolic blood pressure above 140 mm Hg.
Compared with placebo, the rate of composite events was significantly reduced in the rosuvastatin group (3.7% vs 4.8%, HR 0.76, P = .002)28 and the candesartan-hydrochlorothiazide-rosuvastatin group (3.6% vs 5.0%, HR 0.71; P = .005)29 but not in the candesartan-hydrochlorothiazide group (4.1% vs 4.4%; HR 0.93; P = .40).30
In addition, a subgroup analysis comparing active treatment vs placebo found a significant reduction in major cardiovascular events for treated patients whose baseline systolic blood pressure was in the upper third (> 143.5 mm Hg, mean 154.1 mm Hg), while treated patients in the lower middle and lower thirds had no significant reduction.30
These results suggest that intensive treatment to achieve a systolic blood pressure below 140 mm Hg in patients at intermediate risk may not be helpful. Nevertheless, there seems to be agreement that intensive treatment generally leads to a reduction in cardiovascular events. The results also show the benefit of lowering cholesterol.
Bundy et al31 performed a meta-analysis that provides support for intensive antihypertensive treatment. Reviewing 42 clinical trials in more than 144,000 patients, they found that treating to reach a target systolic blood pressure of 120 to 124 mm Hg can reduce cardiovascular events and all-cause mortality.
The trade-off is a minimal increase in the risk of adverse events. Also, the risk-benefit ratio of intensive treatment seems to vary in different patient subgroups.
WHAT ABOUT PATIENTS WITH COMORBIDITIES?
The debate over intensive vs standard treatment in blood pressure management extends beyond hypertension and includes important comorbidities such as diabetes, stroke, and renal disease. Patients with a history of stroke or end-stage renal disease have only a minimal mention in the AHA/ACC guidelines.
Diabetes
Emdin et al,32 in a meta-analysis of 40 trials that included more than 100,000 patients with diabetes, concluded that a 10-mm Hg lowering of systolic blood pressure significantly reduces the rates of all-cause mortality, cardiovascular disease, coronary heart disease, stroke, albuminuria, and retinopathy. Stratifying the results according to the systolic blood pressure achieved (≥ 130 or < 130 mm Hg), the relative risks of mortality, coronary heart disease, cardiovascular disease, heart failure, and albuminuria were actually lower in the higher stratum than in the lower.
ACCORD (the Action to Control Cardiovascular Risk in Diabetes)33 study provides contrary results. It examined intensive and standard blood pressure control targets in patients with type 2 diabetes at high risk of cardiovascular events, using primary outcome measures similar to those in SPRINT. It found no significant difference in fatal and nonfatal cardiovascular events between the intensive and standard blood pressure target arms.
Despite those results, the ACC/AHA guidelines still advocate for more intensive treatment (goal < 130/80 mm Hg) in all patients, including those with diabetes.1
The ADA position statement (September 2017) recommended a target below 140/90 mm Hg in patients with diabetes and hypertension.8 However, they also noted that lower systolic and diastolic blood pressure targets, such as below 130/80 mm Hg, may be appropriate for patients at high risk of cardiovascular disease “if they can be achieved without undue treatment burden.”8 Thus, it is not clear which blood pressure targets in patients with diabetes are the best.
Stroke
In patients with stroke, AHA/ACC guidelines1 recommend treatment if the blood pressure is 140/90 mm Hg or higher because antihypertensive therapy has been associated with a decrease in the recurrence of transient ischemic attack and stroke. The ideal target blood pressure is not known, but a goal of less than 130/80 mm Hg may be reasonable.
In the Secondary Prevention of Small Subcortical Strokes (SPS3) trial, a retrospective open-label trial, a target blood pressure below 130/80 mm Hg in patients with a history of lacunar stroke was associated with a lower risk of intracranial hemorrhage, but the difference was not statistically significant.34 For this reason, the ACC/AHA guidelines consider it reasonable to aim for a systolic blood pressure below 130 mm Hg in these patients.1
Renal disease
The ACC/AHA guidelines do not address how to manage hypertension in patients with end-stage renal disease, but for patients with chronic kidney disease they recommend a blood pressure target below 130/80 mm Hg.1 This recommendation is derived from the SPRINT trial,15 in which patients with stage 3 or 4 chronic kidney disease accounted for 28% of the study population. In that subgroup, intensive blood pressure control seemed to provide the same benefits for reduction in cardiovascular death and all-cause mortality.
TREAT PATIENTS, NOT NUMBERS
Blood pressure targets should be applied in the appropriate clinical context and on a patient-by-patient basis. In clinical practice, one size does not always fit all, as special cases exist.
For example, blood pressure can oscillate widely in patients with autonomic nerve disorders, making it difficult to strive for a specific target, especially an intensive one. Thus, it may be necessary to allow higher systolic blood pressure in these patients. Similarly, patients with diabetes or chronic kidney disease may be at higher risk of kidney injury with more intensive blood pressure management.
Treating numbers rather than patients may result in unbalanced patient care. The optimal approach to blood pressure management relies on a comprehensive risk factor assessment and shared decision-making with the patient before setting specific blood pressure targets.
OUR APPROACH
We aim for a blood pressure goal below 130/80 mm Hg for all patients with cardiovascular disease, according to the AHA/ACC guidelines. We aim for that same target in patients without cardiovascular disease but who have an elevated estimated cardiovascular risk (> 10%) over the next 10 years.
We recognize, however, that the benefits of aggressive blood pressure reduction may not be as clear in all patients, such as those with diabetes. We also recognize that some patient subgroups are at high risk of adverse events, including those with low diastolic pressure, chronic kidney disease, a history of falls, and older age. In those patients, we are extremely judicious when titrating antihypertensive medications. We often make smaller titrations, at longer intervals, and with more frequent laboratory testing and in-office follow-up.
Our process of managing hypertension through intensive blood pressure control to achieve lower systolic blood pressure targets requires a concerted effort among healthcare providers at all levels. It especially requires more involvement and investment from primary care providers to individualize treatment in their patients. This process has helped us to reach our treatment goals while limiting adverse effects of lower blood pressure targets.
MOVING FORWARD
Hypertension is a major risk factor for cardiovascular disease, and intensive blood pressure control has the potential to significantly reduce rates of morbidity and death associated with cardiovascular disease. Thus, a general consensus on the definition of hypertension and treatment goals is essential to reduce the risk of cardiovascular events in this large patient population.
Intensive blood pressure treatment has shown efficacy, but it has a small accompanying risk of adverse events, which varies in patient subgroups and affects the benefit-risk ratio of this therapy. For example, the cardiovascular benefit of intensive treatment is less clear in diabetic patients, and the risk of adverse events may be higher in older patients with chronic kidney disease.
Moving forward, more research is needed into the effects of intensive and standard treatment on patients of all ages, those with common comorbid conditions, and those with other important factors such as diastolic hypertension.
Finally, the various medical societies should collaborate on hypertension guideline development. This would require considerable planning and coordination but would ultimately be useful in creating a generalizable approach to hypertension management.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289(19):2560–2572. doi:10.1001/jama.289.19.2560
- Go AS, Bauman MA, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2014; 63(4):878–885. doi:10.1161/HYP.0000000000000003
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311(5):507–520. doi:10.1001/jama.2013.284427
- Wright JT Jr, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014; 160(7):499–503. doi:10.7326/M13-2981
- Weber MA, Schiffrin EL, White WB, et al. Notice of duplicate publication [duplicate publication of Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens 2014; 16(1):14–26. doi:10.1111/jch.12237] J Hypertens 2014; 32(1):3–15. doi:10.1097/HJH.0000000000000065
- Rosendorff C, Lackland DT, Allison M, et al. Treatment of hypertension in patients with coronary artery disease: a scientific statement from the American Heart Association, American College of Cardiology, and American Society of Hypertension. J Am Soc Hypertens 2015; 9(6):453–498. doi:10.1016/j.jash.2015.03.002
- de Boer IH, Bangalore S, Benetos A, et al. Diabetes and hypertension: a position statement by the American Diabetes Association. Diabetes Care 2017; 40(9):1273–1284. doi:10.2337/dci17-0026
- Qaseem A, Wilt TJ, Rich R, Humphrey LL, Frost J, Forciea MA. Pharmacologic treatment of hypertension in adults aged 60 years or older to higher versus lower blood pressure targets: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2017; 166(6):430–437. doi:10.7326/M16-1785
- The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in over-weight people with high normal blood pressure: the Trials of Hypertension Prevention, phase II. Arch Intern Med 1997; 157(6):657–667. pmid:9080920
- He J, Whelton PK, Appel LJ, Charleston J, Klag MJ. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension 2000; 35(2):544–549. pmid:10679495
- Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med 2001; 344(1):3–10. doi:10.1056/NEJM200101043440101
- Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for US adults: National Health Interview Survey, 2012. National Center for Health Statistics. Vital Health Stat 10; 2014(260):1–161. pmid:24819891
- Muntner P, Carey RM, Gidding S, et al. Potential US population impact of the 2017 ACC/AHA high blood pressure guideline. J Am Coll Cardiol 2018; 71(2):109–118. doi:10.1016/j.jacc.2017.10.073
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Bress AP, Kramer H, Khatib R, et al. Potential deaths averted and serious adverse events incurred from adoption of the SPRINT (Systolic Blood Pressure Intervention Trial) intensive blood pressure regimen in the United States: Projections from NHANES (National Health and Nutrition Examination Survey). Circulation 2017; 135(17):1617–1628. doi:10.1161/CIRCULATIONAHA.116.025322
- Williamson JD, Supiano MA, Applegate WB, et al. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315(24):2673–2682. doi:10.1001/jama.2016.7050
- Beddhu S, Rocco MV, Toto R, et al. Effects of intensive systolic blood pressure control on kidney and cardiovascular outcomes in persons without kidney disease: a secondary analysis of a randomized trial. Ann Intern Med 2017; 167(6):375–383. doi:10.7326/M16-2966
- Brunström M, Carlberg B. Association of blood pressure lowering with mortality and cardiovascular disease across blood pressure levels: a systematic review and meta-analysis. JAMA Intern Med 2018; 178(1):28–36. doi:10.1001/jamainternmed.2017.6015
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265(24):3255–3264. pmid:2046107
- Bulpitt CJ, Beckett NS, Cooke J, et al. Results of the pilot study for the Hypertension in the Very Elderly Trial. J Hypertens 2003; 21(12):2409–2417. doi:10.1097/01.hjh.0000084782.15238.a2
- Liu L, Zhang Y, Liu G, et al. The Felodipine Event Reduction (FEVER) study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23(12):2157–2172. pmid:16269957
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31(12):2115–2127. doi:10.1291/hypres.31.2115
- Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56(2):196–202. doi:10.1161/HYPERTENSIONAHA.109.146035
- Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet 2016; 387(10022):957–967. doi:10.1016/S0140-6736(15)01225-8
- Sleight P. The HOPE study (Heart Outcomes Prevention Evaluation). J Renin Angiotensin Aldosterone Syst 2000; 1(1):18–20. doi:10.3317/jraas.2000.002
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000; 355(9200):253–259. pmid:10675071
- Yusuf S, Bosch J, Dagenais G, et al. Cholesterol lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2021–2031. doi:10.1056/NEJMoa1600176
- Yusuf S, Lonn E, Pais P, et al. Blood-pressure and cholesterol lowering in persons without cardiovascular disease. N Engl J Med 2016; 374(21):2032–2043. doi:10.1056/NEJMoa1600177
- Lonn EM, Bosch J, López-Jaramillo P, et al. Blood-pressure lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2009–2020. doi:10.1056/NEJMoa1600175
- Bundy JD, Li C, Stuchlik P, et al. Systolic blood pressure reduction and risk of cardiovascular disease and mortality: a systematic review and network meta-analysis. JAMA Cardiol 2017; 2(7):775–781. doi:10.1001/jamacardio.2017.1421
- Emdin CA, Rahimi K, Neal B, Callender T, Perkovic V, Patel A. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA 2015; 313(6):603–615. doi:10.1001/jama.2014.18574
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362(17):1575–1585. doi:10.1056/NEJMoa1001286
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382(9891):507–515. doi:10.1016/S0140-6736(13)60852-1
When treating high blood pressure, how low should we try to go? Debate continues about optimal blood pressure goals after publication of guidelines from the American College of Cardiology and American Heart Association (ACC/AHA) in 2017 that set or permitted a treatment goal of less than 130 mm Hg, depending on the population.1
In this article, we summarize the evolution of hypertension guidelines and the evidence behind them.
HOW THE GOALS EVOLVED
JNC 7, 2003: 140/90 or 130/80
The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7),2 published in 2003, specified treatment goals of:
- < 140/90 mm Hg for most patients
- < 130/80 mm Hg for those with diabetes or chronic kidney disease.
JNC 7 provided much-needed clarity and uniformity to managing hypertension. Since then, various scientific groups have published their own guidelines (Table 1).1–9
ACC/AHA/CDC 2014: 140/90
In 2014, the ACC, AHA, and US Centers for Disease Control and Prevention (CDC) published an evidence-based algorithm for hypertension management.3 As in JNC 7, they suggested a blood pressure goal of less than 140/90 mm Hg, lifestyle modification, and polytherapy, eg, a thiazide diuretic for stage 1 hypertension (< 160/100 mm Hg) and combination therapy with a thiazide diuretic and an angiotensin-converting enzyme (ACE) inhibitor, angiotensin II receptor blocker (ARB), or calcium channel blocker for stage 2 hypertension (≥ 160/100 mm Hg).
JNC 8 2014: 140/90 or 150/90
Soon after, the much-anticipated report of the panel members appointed to the eighth JNC (JNC 8) was published.4 Previous JNC reports were written and published under the auspices of the National Heart, Lung, and Blood Institute, but while the JNC 8 report was being prepared, this government body announced it would no longer publish guidelines.
In contrast to JNC 7, the JNC 8 panel based its recommendations on a systematic review of randomized clinical trials. However, the process and methodology were controversial, especially as the panel excluded some important clinical trials from the analysis.
JNC 8 relaxed the targets in several subgroups, such as patients over age 60 and those with diabetes and chronic kidney disease, due to a lack of definitive evidence on the impact of blood pressure targets lower than 140/90 mm Hg in these groups. Thus, their goals were:
- < 140/90 mm Hg for patients under age 60
- < 150/90 mm Hg for patients age 60 and older.
Of note, a minority of the JNC 8 panel disagreed with the new targets and provided evidence for keeping the systolic blood pressure target below 140 mm Hg for patients 60 and older.5 Further, the JNC 8 report was not endorsed by several important societies, ie, the AHA, ACC, National Heart, Lung, and Blood Institute, and American Society of Hypertension (ASH). These issues compromised the acceptance and applicability of the guidelines.
ASH/ISH 2014: 140/90 or 150/90
Also in 2014, the ASH and the International Society of Hypertension released their own report.6 Their goals:
- < 140/90 mm Hg for most patients
- < 150/90 mm Hg for patients age 80 and older.
AHA/ACC/ASH 2015: Goals in subgroups
In 2015, the AHA, ACC, and ASH released a joint scientific statement outlining hypertension goals for specific patient populations7:
- < 150/90 mm Hg for those age 80 and older
- < 140/90 mm Hg for those with coronary artery disease
- < 130/80 mm Hg for those with comorbidities such as diabetes and cardiovascular disease.
ADA 2016: Goals for patients with diabetes
In 2016, the American Diabetes Association (ADA) set the following blood pressure goals for patients with diabetes8:
- < 140/90 mm Hg for adults with diabetes
- < 130/80 mm Hg for younger adults with diabetes and adults with a high risk of cardiovascular disease
- 120–160/80–105 mm Hg for pregnant patients with diabetes and preexisting hypertension who are treated with antihypertensive therapy.
ACP/AAFP 2017: Systolic 150 or 130
In 2017, the American College of Physicians (ACP) and the American Academy of Family Physicians (AAFP) recommended a relaxed systolic blood pressure target, ie, below 150 mm Hg, for adults over age 60, but a tighter goal of less than 140 mm Hg for the same age group if they have transient ischemic attack, stroke, or high cardiovascular risk.9
ACC/AHA 2017: 130/80
The 2017 ACC/AHA guidelines recommended a more aggressive goal of below 130/80 for all, including patients age 65 and older.1
This is a class I (strong) recommendation for patients with known cardiovascular disease or a 10-year risk of a cardiovascular event of 10% or higher, with a B-R level of evidence for the systolic goal (ie, moderate-quality, based on systematic review of randomized controlled trials) and a C-EO level of evidence for the diastolic goal (ie, based on expert opinion).
For patients who do not have cardiovascular disease and who are at lower risk of it, this is a class IIb (weak) recommendation, ie, it “may be reasonable,” with a B-NR level of evidence (moderate-quality, based on nonrandomized studies) for the systolic goal and C-EO (expert opinion) for the diastolic goal.
For many patients, this involves drug treatment. For those with known cardiovascular disease or a 10-year risk of an atherosclerotic cardiovascular disease event of 10% or higher, the ACC/AHA guidelines say that drug treatment “is recommended” if their average blood pressure is 130/80 mm Hg or higher (class I recommendation, based on strong evidence for the systolic threshold and expert option for the diastolic). For those without cardiovascular disease and at lower risk, drug treatment is recommended if their average blood pressure is 140/90 mm Hg or higher (also class I, but based on limited data).
EVERYONE AGREES ON LIFESTYLE
Although the guidelines differ in their blood pressure targets, they consistently recommend lifestyle modifications.
Lifestyle modifications, first described in JNC 7, included weight loss, sodium restriction, and the DASH diet, which is rich in fruits, vegetables, low-fat dairy products, whole grains, poultry, and fish, and low in red meat, sweets, cholesterol, and total and saturated fat.2
These recommendations were based on results from 3 large randomized controlled trials in patients with and without hypertension.10–12 In patients with no history of hypertension, interventions to promote weight loss and sodium restriction significantly reduced blood pressure and the incidence of hypertension (the latter by as much as 77%) compared with usual care.10,11
In patients with and without hypertension, lowering sodium intake in conjunction with the DASH diet was associated with substantially larger reductions in systolic blood pressure.12
The recommendation to lower sodium intake has not changed in the guideline revisions. Meanwhile, other modifications have been added, such as incorporating both aerobic and resistance exercise and moderating alcohol intake. These recommendations have a class I level of evidence (ie, strongest level) in the 2017 ACC/AHA guidelines.1
HYPERTENSION BEGINS AT 130/80
The definition of hypertension changed in the 2017 ACC/AHA guidelines1: previously set at 140/90 mm Hg or higher, it is now 130/80 mm Hg or higher for all age groups. Adults with systolic blood pressure of 130 to 139 mm Hg or diastolic blood pressure of 80 to 89 mm Hg are now classified as having stage 1 hypertension.
Under the new definition, the number of US adults who have hypertension expanded to 45.6% of the general population,13 up from 31.9% under the JNC 7 definition. Thus, overall, 103.3 million US adults now have hypertension, compared with 72.2 million under the JNC 7 criteria.
In addition, the new guidelines expanded the population of adults for whom antihypertensive drug treatment is recommended to 36.2% (81.9 million). However, this represents only a 1.9% absolute increase over the JNC 7 recommendations (34.3%) and a 5.1% absolute increase over the JNC 8 recommendations.14
SPRINT: INTENSIVE TREATMENT IS BENEFICIAL
The new ACC/AHA guidelines1 were based on evidence from several trials, including the Systolic Blood Pressure Intervention Trial (SPRINT).15
This multicenter trial investigated the effect of intensive blood pressure treatment on cardiovascular disease risk.16 The primary outcome was a composite of myocardial infarction, acute coronary syndrome, stroke, and heart failure.
The trial enrolled 9,361 participants at least 50 years of age with systolic blood pressure 130 mm Hg or higher and at least 1 additional risk factor for cardiovascular disease. It excluded anyone with a history of diabetes mellitus, stroke, symptomatic heart failure, or end-stage renal disease.
Two interventions were compared:
- Intensive treatment, with a systolic blood pressure goal of less than 120 mm Hg: the protocol called for polytherapy, even for participants who were 75 or older if their blood pressure was 140 mm Hg or higher
- Standard treatment, with a systolic blood pressure goal of less than 140 mm Hg: it used polytherapy for patients whose systolic blood pressure was 160 mm Hg or higher.
The trial was intended to last 5 years but was stopped early at a median of 3.26 years owing to a significantly lower rate of the primary composite outcome in the intensive-treatment group: 1.65% per year vs 2.19%, a 25% relative risk reduction (P < .001) or a 0.54% absolute risk reduction. We calculate the number needed to treat (NNT) for 1 year to prevent 1 event as 185, and over the 3.26 years of the trial, the investigators calculated the NNT as 61. Similarly, the rate of death from any cause was also lower with intensive treatment, 1.03% per year vs 1.40% per year, a 27% relative risk reduction (P = .003) or a 0.37% absolute risk reduction, NNT 270.
Using these findings, Bress et al16 estimated that implementing intensive blood pressure goals could prevent 107,500 deaths annually.
The downside is adverse effects. In SPRINT,15 the intensive-treatment group experienced significantly higher rates of serious adverse effects than the standard-treatment group, ie:
- Hypotension 2.4% vs 1.4%, P = .001
- Syncope 2.3% vs 1.7%, P = .05
- Electrolyte abnormalities 3.1% vs 2.3%, P = .02)
- Acute kidney injury or kidney failure 4.1% vs 2.5%, P < .001
- Any treatment-related adverse event 4.7% vs 2.5%, P = .001.
Thus, Bress et al16 estimated that fully implementing the intensive-treatment goals could cause an additional 56,100 episodes of hypotension per year, 34,400 cases of syncope, 43,400 serious electrolyte disorders, and 88,700 cases of acute kidney injury. All told, about 3 million Americans could suffer a serious adverse effect under the intensive-treatment goals.
SPRINT caveats and limitations
SPRINT15 was stopped early, after 3.26 years instead of the planned 5 years. The true risk-benefit ratio may have been different if the trial had been extended longer.
In addition, SPRINT used automated office blood pressure measurements in which patients were seated alone and a device (Model 907, Omron Healthcare) took 3 blood pressure measurements at 1-minute intervals after 5 minutes of quiet rest. This was designed to reduce elevated blood pressure readings in the presence of a healthcare professional in a medical setting (ie, “white coat” hypertension).
Many physicians are still taking blood pressure manually, which tends to give higher readings. Therefore, if they aim for a lower goal, they may risk overtreating the patient.
About 50% of patients did not achieve the target systolic blood pressure (< 120 mm Hg) despite receiving an average of 2.8 antihypertensive medications in the intensive-treatment group and 1.8 in the standard-treatment group. The use of antihypertensive medications, however, was not a controlled variable in the trial, and practitioners chose the appropriate drugs for their patients.
Diastolic pressure, which can be markedly lower in older hypertensive patients, was largely ignored, although lower diastolic pressure may have contributed to higher syncope rates in response to alpha blockers and calcium blockers.
Moreover, the trial excluded those with significant comorbidities and those younger than 50 (the mean age was 67.9), which limits the generalizability of the results.
JNC 8 VS SPRINT GOALS: WHAT'S THE EFFECT ON OUTCOMES?
JNC 84 recommended a relaxed target of less than 140/90 mm Hg for adults younger than 60, including those with chronic kidney disease or diabetes, and less than 150/90 mm Hg for adults 60 and older. The SPRINT findings upended those recommendations, showing that intensive treatment in adults age 75 or older significantly improved the composite cardiovascular disease outcome (2.59 vs 3.85 events per year; P < .001) and all-cause mortality (1.78 vs 2.63 events per year; P < .05) compared with standard treatment.17 Also, a subset review of SPRINT trial data found no difference in benefit based on chronic kidney disease status.18
A meta-analysis of 74 clinical trials (N = 306,273) offers a compromise between the SPRINT findings and the JNC 8 recommendations.19 It found that the beneficial effect of blood pressure treatment depended on the patient’s baseline systolic blood pressure. In those with a baseline systolic pressure of 160 mm Hg or higher, treatment reduced cardiovascular mortality by about 15% (relative risk [RR] 0.85; 95% confidence interval [CI] 0.77–0.95). In patients with systolic pressure below 140 mm Hg, treatment effects were neutral (RR 1.03, 95% CI 0.87–1.20) and not associated with any benefit as primary prevention, although data suggest it may reduce the risk of adverse outcomes in patients with coronary heart disease.
OTHER TRIALS THAT INFLUENCED THE GUIDELINES
SHEP and HYVET (the Systolic Hypertension in the Elderly Program20 and the Hypertension in the Very Elderly Trial)21 supported intensive blood pressure treatment for older patients by reporting a reduction in fatal and nonfatal stroke risks for those with a systolic blood pressure above 160 mm Hg.
FEVER (the Felodipine Event Reduction study)22 found that treatment with a calcium channel blocker in even a low dose can significantly decrease cardiovascular events, cardiovascular disease, and heart failure compared with no treatment.
JATOS and VALISH (the Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients23 and the Valsartan in Elderly Isolated Systolic Hypertension study)24 found that outcomes were similar with intensive vs standard treatment.
Ettehad et al25 performed a meta-analysis of 123 studies with more than 600,000 participants that provided strong evidence supporting blood pressure treatment goals below 130/90 mm Hg, in line with the SPRINT trial results.
BLOOD PRESSURE ISN’T EVERYTHING
Other trials remind us that although blood pressure is important, it is not the only factor affecting cardiovascular risk.
HOPE (the Heart Outcomes Prevention Evaluation)26 investigated the use of ramipril (an ACE inhibitor) in preventing myocardial infarction, stroke, or cardiovascular death in patients at high risk of cardiovascular events. The study included 9,297 participants over age 55 (mean age 66) with a baseline blood pressure 139/79 mm Hg. Follow-up was 4.5 years.
Ramipril was better than placebo, with significantly fewer patients experiencing adverse end points in the ramipril group compared with the placebo group:
- Myocardial infarction 9.9% vs 12.3%, RR 0.80, P < .001
- Cardiovascular death 6.1% vs 8.1%, RR 0.74, P < .001
- Stroke 3.4% vs 4.9%, RR = .68, P < .001
- The composite end point 14.0% vs 17.8%, RR 0.78, P < .001).
Results were even better in the subset of patients who had diabetes.27 However, the decrease in blood pressure attributable to antihypertensive therapy with ramipril was minimal (3–4 mm Hg systolic and 1–2 mm Hg diastolic). This slight change should not have been enough to produce significant differences in clinical outcomes, a major limitation of this trial. The investigators speculated that the positive results may be due to a class effect of ACE inhibitors.26
HOPE 328–30 explored the effect of blood pressure- and cholesterol-controlling drugs on the same primary end points but in patients at intermediate risk of major cardiovascular events. Investigators randomized the 12,705 patients to 4 treatment groups:
- Blood pressure control with candesartan (an ARB) plus hydrochlorothiazide (a thiazide diuretic)
- Cholesterol control with rosuvastatin (a statin)
- Blood pressure plus cholesterol control
- Placebo.
Therapy was started at a systolic blood pressure above 140 mm Hg.
Compared with placebo, the rate of composite events was significantly reduced in the rosuvastatin group (3.7% vs 4.8%, HR 0.76, P = .002)28 and the candesartan-hydrochlorothiazide-rosuvastatin group (3.6% vs 5.0%, HR 0.71; P = .005)29 but not in the candesartan-hydrochlorothiazide group (4.1% vs 4.4%; HR 0.93; P = .40).30
In addition, a subgroup analysis comparing active treatment vs placebo found a significant reduction in major cardiovascular events for treated patients whose baseline systolic blood pressure was in the upper third (> 143.5 mm Hg, mean 154.1 mm Hg), while treated patients in the lower middle and lower thirds had no significant reduction.30
These results suggest that intensive treatment to achieve a systolic blood pressure below 140 mm Hg in patients at intermediate risk may not be helpful. Nevertheless, there seems to be agreement that intensive treatment generally leads to a reduction in cardiovascular events. The results also show the benefit of lowering cholesterol.
Bundy et al31 performed a meta-analysis that provides support for intensive antihypertensive treatment. Reviewing 42 clinical trials in more than 144,000 patients, they found that treating to reach a target systolic blood pressure of 120 to 124 mm Hg can reduce cardiovascular events and all-cause mortality.
The trade-off is a minimal increase in the risk of adverse events. Also, the risk-benefit ratio of intensive treatment seems to vary in different patient subgroups.
WHAT ABOUT PATIENTS WITH COMORBIDITIES?
The debate over intensive vs standard treatment in blood pressure management extends beyond hypertension and includes important comorbidities such as diabetes, stroke, and renal disease. Patients with a history of stroke or end-stage renal disease have only a minimal mention in the AHA/ACC guidelines.
Diabetes
Emdin et al,32 in a meta-analysis of 40 trials that included more than 100,000 patients with diabetes, concluded that a 10-mm Hg lowering of systolic blood pressure significantly reduces the rates of all-cause mortality, cardiovascular disease, coronary heart disease, stroke, albuminuria, and retinopathy. Stratifying the results according to the systolic blood pressure achieved (≥ 130 or < 130 mm Hg), the relative risks of mortality, coronary heart disease, cardiovascular disease, heart failure, and albuminuria were actually lower in the higher stratum than in the lower.
ACCORD (the Action to Control Cardiovascular Risk in Diabetes)33 study provides contrary results. It examined intensive and standard blood pressure control targets in patients with type 2 diabetes at high risk of cardiovascular events, using primary outcome measures similar to those in SPRINT. It found no significant difference in fatal and nonfatal cardiovascular events between the intensive and standard blood pressure target arms.
Despite those results, the ACC/AHA guidelines still advocate for more intensive treatment (goal < 130/80 mm Hg) in all patients, including those with diabetes.1
The ADA position statement (September 2017) recommended a target below 140/90 mm Hg in patients with diabetes and hypertension.8 However, they also noted that lower systolic and diastolic blood pressure targets, such as below 130/80 mm Hg, may be appropriate for patients at high risk of cardiovascular disease “if they can be achieved without undue treatment burden.”8 Thus, it is not clear which blood pressure targets in patients with diabetes are the best.
Stroke
In patients with stroke, AHA/ACC guidelines1 recommend treatment if the blood pressure is 140/90 mm Hg or higher because antihypertensive therapy has been associated with a decrease in the recurrence of transient ischemic attack and stroke. The ideal target blood pressure is not known, but a goal of less than 130/80 mm Hg may be reasonable.
In the Secondary Prevention of Small Subcortical Strokes (SPS3) trial, a retrospective open-label trial, a target blood pressure below 130/80 mm Hg in patients with a history of lacunar stroke was associated with a lower risk of intracranial hemorrhage, but the difference was not statistically significant.34 For this reason, the ACC/AHA guidelines consider it reasonable to aim for a systolic blood pressure below 130 mm Hg in these patients.1
Renal disease
The ACC/AHA guidelines do not address how to manage hypertension in patients with end-stage renal disease, but for patients with chronic kidney disease they recommend a blood pressure target below 130/80 mm Hg.1 This recommendation is derived from the SPRINT trial,15 in which patients with stage 3 or 4 chronic kidney disease accounted for 28% of the study population. In that subgroup, intensive blood pressure control seemed to provide the same benefits for reduction in cardiovascular death and all-cause mortality.
TREAT PATIENTS, NOT NUMBERS
Blood pressure targets should be applied in the appropriate clinical context and on a patient-by-patient basis. In clinical practice, one size does not always fit all, as special cases exist.
For example, blood pressure can oscillate widely in patients with autonomic nerve disorders, making it difficult to strive for a specific target, especially an intensive one. Thus, it may be necessary to allow higher systolic blood pressure in these patients. Similarly, patients with diabetes or chronic kidney disease may be at higher risk of kidney injury with more intensive blood pressure management.
Treating numbers rather than patients may result in unbalanced patient care. The optimal approach to blood pressure management relies on a comprehensive risk factor assessment and shared decision-making with the patient before setting specific blood pressure targets.
OUR APPROACH
We aim for a blood pressure goal below 130/80 mm Hg for all patients with cardiovascular disease, according to the AHA/ACC guidelines. We aim for that same target in patients without cardiovascular disease but who have an elevated estimated cardiovascular risk (> 10%) over the next 10 years.
We recognize, however, that the benefits of aggressive blood pressure reduction may not be as clear in all patients, such as those with diabetes. We also recognize that some patient subgroups are at high risk of adverse events, including those with low diastolic pressure, chronic kidney disease, a history of falls, and older age. In those patients, we are extremely judicious when titrating antihypertensive medications. We often make smaller titrations, at longer intervals, and with more frequent laboratory testing and in-office follow-up.
Our process of managing hypertension through intensive blood pressure control to achieve lower systolic blood pressure targets requires a concerted effort among healthcare providers at all levels. It especially requires more involvement and investment from primary care providers to individualize treatment in their patients. This process has helped us to reach our treatment goals while limiting adverse effects of lower blood pressure targets.
MOVING FORWARD
Hypertension is a major risk factor for cardiovascular disease, and intensive blood pressure control has the potential to significantly reduce rates of morbidity and death associated with cardiovascular disease. Thus, a general consensus on the definition of hypertension and treatment goals is essential to reduce the risk of cardiovascular events in this large patient population.
Intensive blood pressure treatment has shown efficacy, but it has a small accompanying risk of adverse events, which varies in patient subgroups and affects the benefit-risk ratio of this therapy. For example, the cardiovascular benefit of intensive treatment is less clear in diabetic patients, and the risk of adverse events may be higher in older patients with chronic kidney disease.
Moving forward, more research is needed into the effects of intensive and standard treatment on patients of all ages, those with common comorbid conditions, and those with other important factors such as diastolic hypertension.
Finally, the various medical societies should collaborate on hypertension guideline development. This would require considerable planning and coordination but would ultimately be useful in creating a generalizable approach to hypertension management.
When treating high blood pressure, how low should we try to go? Debate continues about optimal blood pressure goals after publication of guidelines from the American College of Cardiology and American Heart Association (ACC/AHA) in 2017 that set or permitted a treatment goal of less than 130 mm Hg, depending on the population.1
In this article, we summarize the evolution of hypertension guidelines and the evidence behind them.
HOW THE GOALS EVOLVED
JNC 7, 2003: 140/90 or 130/80
The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7),2 published in 2003, specified treatment goals of:
- < 140/90 mm Hg for most patients
- < 130/80 mm Hg for those with diabetes or chronic kidney disease.
JNC 7 provided much-needed clarity and uniformity to managing hypertension. Since then, various scientific groups have published their own guidelines (Table 1).1–9
ACC/AHA/CDC 2014: 140/90
In 2014, the ACC, AHA, and US Centers for Disease Control and Prevention (CDC) published an evidence-based algorithm for hypertension management.3 As in JNC 7, they suggested a blood pressure goal of less than 140/90 mm Hg, lifestyle modification, and polytherapy, eg, a thiazide diuretic for stage 1 hypertension (< 160/100 mm Hg) and combination therapy with a thiazide diuretic and an angiotensin-converting enzyme (ACE) inhibitor, angiotensin II receptor blocker (ARB), or calcium channel blocker for stage 2 hypertension (≥ 160/100 mm Hg).
JNC 8 2014: 140/90 or 150/90
Soon after, the much-anticipated report of the panel members appointed to the eighth JNC (JNC 8) was published.4 Previous JNC reports were written and published under the auspices of the National Heart, Lung, and Blood Institute, but while the JNC 8 report was being prepared, this government body announced it would no longer publish guidelines.
In contrast to JNC 7, the JNC 8 panel based its recommendations on a systematic review of randomized clinical trials. However, the process and methodology were controversial, especially as the panel excluded some important clinical trials from the analysis.
JNC 8 relaxed the targets in several subgroups, such as patients over age 60 and those with diabetes and chronic kidney disease, due to a lack of definitive evidence on the impact of blood pressure targets lower than 140/90 mm Hg in these groups. Thus, their goals were:
- < 140/90 mm Hg for patients under age 60
- < 150/90 mm Hg for patients age 60 and older.
Of note, a minority of the JNC 8 panel disagreed with the new targets and provided evidence for keeping the systolic blood pressure target below 140 mm Hg for patients 60 and older.5 Further, the JNC 8 report was not endorsed by several important societies, ie, the AHA, ACC, National Heart, Lung, and Blood Institute, and American Society of Hypertension (ASH). These issues compromised the acceptance and applicability of the guidelines.
ASH/ISH 2014: 140/90 or 150/90
Also in 2014, the ASH and the International Society of Hypertension released their own report.6 Their goals:
- < 140/90 mm Hg for most patients
- < 150/90 mm Hg for patients age 80 and older.
AHA/ACC/ASH 2015: Goals in subgroups
In 2015, the AHA, ACC, and ASH released a joint scientific statement outlining hypertension goals for specific patient populations7:
- < 150/90 mm Hg for those age 80 and older
- < 140/90 mm Hg for those with coronary artery disease
- < 130/80 mm Hg for those with comorbidities such as diabetes and cardiovascular disease.
ADA 2016: Goals for patients with diabetes
In 2016, the American Diabetes Association (ADA) set the following blood pressure goals for patients with diabetes8:
- < 140/90 mm Hg for adults with diabetes
- < 130/80 mm Hg for younger adults with diabetes and adults with a high risk of cardiovascular disease
- 120–160/80–105 mm Hg for pregnant patients with diabetes and preexisting hypertension who are treated with antihypertensive therapy.
ACP/AAFP 2017: Systolic 150 or 130
In 2017, the American College of Physicians (ACP) and the American Academy of Family Physicians (AAFP) recommended a relaxed systolic blood pressure target, ie, below 150 mm Hg, for adults over age 60, but a tighter goal of less than 140 mm Hg for the same age group if they have transient ischemic attack, stroke, or high cardiovascular risk.9
ACC/AHA 2017: 130/80
The 2017 ACC/AHA guidelines recommended a more aggressive goal of below 130/80 for all, including patients age 65 and older.1
This is a class I (strong) recommendation for patients with known cardiovascular disease or a 10-year risk of a cardiovascular event of 10% or higher, with a B-R level of evidence for the systolic goal (ie, moderate-quality, based on systematic review of randomized controlled trials) and a C-EO level of evidence for the diastolic goal (ie, based on expert opinion).
For patients who do not have cardiovascular disease and who are at lower risk of it, this is a class IIb (weak) recommendation, ie, it “may be reasonable,” with a B-NR level of evidence (moderate-quality, based on nonrandomized studies) for the systolic goal and C-EO (expert opinion) for the diastolic goal.
For many patients, this involves drug treatment. For those with known cardiovascular disease or a 10-year risk of an atherosclerotic cardiovascular disease event of 10% or higher, the ACC/AHA guidelines say that drug treatment “is recommended” if their average blood pressure is 130/80 mm Hg or higher (class I recommendation, based on strong evidence for the systolic threshold and expert option for the diastolic). For those without cardiovascular disease and at lower risk, drug treatment is recommended if their average blood pressure is 140/90 mm Hg or higher (also class I, but based on limited data).
EVERYONE AGREES ON LIFESTYLE
Although the guidelines differ in their blood pressure targets, they consistently recommend lifestyle modifications.
Lifestyle modifications, first described in JNC 7, included weight loss, sodium restriction, and the DASH diet, which is rich in fruits, vegetables, low-fat dairy products, whole grains, poultry, and fish, and low in red meat, sweets, cholesterol, and total and saturated fat.2
These recommendations were based on results from 3 large randomized controlled trials in patients with and without hypertension.10–12 In patients with no history of hypertension, interventions to promote weight loss and sodium restriction significantly reduced blood pressure and the incidence of hypertension (the latter by as much as 77%) compared with usual care.10,11
In patients with and without hypertension, lowering sodium intake in conjunction with the DASH diet was associated with substantially larger reductions in systolic blood pressure.12
The recommendation to lower sodium intake has not changed in the guideline revisions. Meanwhile, other modifications have been added, such as incorporating both aerobic and resistance exercise and moderating alcohol intake. These recommendations have a class I level of evidence (ie, strongest level) in the 2017 ACC/AHA guidelines.1
HYPERTENSION BEGINS AT 130/80
The definition of hypertension changed in the 2017 ACC/AHA guidelines1: previously set at 140/90 mm Hg or higher, it is now 130/80 mm Hg or higher for all age groups. Adults with systolic blood pressure of 130 to 139 mm Hg or diastolic blood pressure of 80 to 89 mm Hg are now classified as having stage 1 hypertension.
Under the new definition, the number of US adults who have hypertension expanded to 45.6% of the general population,13 up from 31.9% under the JNC 7 definition. Thus, overall, 103.3 million US adults now have hypertension, compared with 72.2 million under the JNC 7 criteria.
In addition, the new guidelines expanded the population of adults for whom antihypertensive drug treatment is recommended to 36.2% (81.9 million). However, this represents only a 1.9% absolute increase over the JNC 7 recommendations (34.3%) and a 5.1% absolute increase over the JNC 8 recommendations.14
SPRINT: INTENSIVE TREATMENT IS BENEFICIAL
The new ACC/AHA guidelines1 were based on evidence from several trials, including the Systolic Blood Pressure Intervention Trial (SPRINT).15
This multicenter trial investigated the effect of intensive blood pressure treatment on cardiovascular disease risk.16 The primary outcome was a composite of myocardial infarction, acute coronary syndrome, stroke, and heart failure.
The trial enrolled 9,361 participants at least 50 years of age with systolic blood pressure 130 mm Hg or higher and at least 1 additional risk factor for cardiovascular disease. It excluded anyone with a history of diabetes mellitus, stroke, symptomatic heart failure, or end-stage renal disease.
Two interventions were compared:
- Intensive treatment, with a systolic blood pressure goal of less than 120 mm Hg: the protocol called for polytherapy, even for participants who were 75 or older if their blood pressure was 140 mm Hg or higher
- Standard treatment, with a systolic blood pressure goal of less than 140 mm Hg: it used polytherapy for patients whose systolic blood pressure was 160 mm Hg or higher.
The trial was intended to last 5 years but was stopped early at a median of 3.26 years owing to a significantly lower rate of the primary composite outcome in the intensive-treatment group: 1.65% per year vs 2.19%, a 25% relative risk reduction (P < .001) or a 0.54% absolute risk reduction. We calculate the number needed to treat (NNT) for 1 year to prevent 1 event as 185, and over the 3.26 years of the trial, the investigators calculated the NNT as 61. Similarly, the rate of death from any cause was also lower with intensive treatment, 1.03% per year vs 1.40% per year, a 27% relative risk reduction (P = .003) or a 0.37% absolute risk reduction, NNT 270.
Using these findings, Bress et al16 estimated that implementing intensive blood pressure goals could prevent 107,500 deaths annually.
The downside is adverse effects. In SPRINT,15 the intensive-treatment group experienced significantly higher rates of serious adverse effects than the standard-treatment group, ie:
- Hypotension 2.4% vs 1.4%, P = .001
- Syncope 2.3% vs 1.7%, P = .05
- Electrolyte abnormalities 3.1% vs 2.3%, P = .02)
- Acute kidney injury or kidney failure 4.1% vs 2.5%, P < .001
- Any treatment-related adverse event 4.7% vs 2.5%, P = .001.
Thus, Bress et al16 estimated that fully implementing the intensive-treatment goals could cause an additional 56,100 episodes of hypotension per year, 34,400 cases of syncope, 43,400 serious electrolyte disorders, and 88,700 cases of acute kidney injury. All told, about 3 million Americans could suffer a serious adverse effect under the intensive-treatment goals.
SPRINT caveats and limitations
SPRINT15 was stopped early, after 3.26 years instead of the planned 5 years. The true risk-benefit ratio may have been different if the trial had been extended longer.
In addition, SPRINT used automated office blood pressure measurements in which patients were seated alone and a device (Model 907, Omron Healthcare) took 3 blood pressure measurements at 1-minute intervals after 5 minutes of quiet rest. This was designed to reduce elevated blood pressure readings in the presence of a healthcare professional in a medical setting (ie, “white coat” hypertension).
Many physicians are still taking blood pressure manually, which tends to give higher readings. Therefore, if they aim for a lower goal, they may risk overtreating the patient.
About 50% of patients did not achieve the target systolic blood pressure (< 120 mm Hg) despite receiving an average of 2.8 antihypertensive medications in the intensive-treatment group and 1.8 in the standard-treatment group. The use of antihypertensive medications, however, was not a controlled variable in the trial, and practitioners chose the appropriate drugs for their patients.
Diastolic pressure, which can be markedly lower in older hypertensive patients, was largely ignored, although lower diastolic pressure may have contributed to higher syncope rates in response to alpha blockers and calcium blockers.
Moreover, the trial excluded those with significant comorbidities and those younger than 50 (the mean age was 67.9), which limits the generalizability of the results.
JNC 8 VS SPRINT GOALS: WHAT'S THE EFFECT ON OUTCOMES?
JNC 84 recommended a relaxed target of less than 140/90 mm Hg for adults younger than 60, including those with chronic kidney disease or diabetes, and less than 150/90 mm Hg for adults 60 and older. The SPRINT findings upended those recommendations, showing that intensive treatment in adults age 75 or older significantly improved the composite cardiovascular disease outcome (2.59 vs 3.85 events per year; P < .001) and all-cause mortality (1.78 vs 2.63 events per year; P < .05) compared with standard treatment.17 Also, a subset review of SPRINT trial data found no difference in benefit based on chronic kidney disease status.18
A meta-analysis of 74 clinical trials (N = 306,273) offers a compromise between the SPRINT findings and the JNC 8 recommendations.19 It found that the beneficial effect of blood pressure treatment depended on the patient’s baseline systolic blood pressure. In those with a baseline systolic pressure of 160 mm Hg or higher, treatment reduced cardiovascular mortality by about 15% (relative risk [RR] 0.85; 95% confidence interval [CI] 0.77–0.95). In patients with systolic pressure below 140 mm Hg, treatment effects were neutral (RR 1.03, 95% CI 0.87–1.20) and not associated with any benefit as primary prevention, although data suggest it may reduce the risk of adverse outcomes in patients with coronary heart disease.
OTHER TRIALS THAT INFLUENCED THE GUIDELINES
SHEP and HYVET (the Systolic Hypertension in the Elderly Program20 and the Hypertension in the Very Elderly Trial)21 supported intensive blood pressure treatment for older patients by reporting a reduction in fatal and nonfatal stroke risks for those with a systolic blood pressure above 160 mm Hg.
FEVER (the Felodipine Event Reduction study)22 found that treatment with a calcium channel blocker in even a low dose can significantly decrease cardiovascular events, cardiovascular disease, and heart failure compared with no treatment.
JATOS and VALISH (the Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients23 and the Valsartan in Elderly Isolated Systolic Hypertension study)24 found that outcomes were similar with intensive vs standard treatment.
Ettehad et al25 performed a meta-analysis of 123 studies with more than 600,000 participants that provided strong evidence supporting blood pressure treatment goals below 130/90 mm Hg, in line with the SPRINT trial results.
BLOOD PRESSURE ISN’T EVERYTHING
Other trials remind us that although blood pressure is important, it is not the only factor affecting cardiovascular risk.
HOPE (the Heart Outcomes Prevention Evaluation)26 investigated the use of ramipril (an ACE inhibitor) in preventing myocardial infarction, stroke, or cardiovascular death in patients at high risk of cardiovascular events. The study included 9,297 participants over age 55 (mean age 66) with a baseline blood pressure 139/79 mm Hg. Follow-up was 4.5 years.
Ramipril was better than placebo, with significantly fewer patients experiencing adverse end points in the ramipril group compared with the placebo group:
- Myocardial infarction 9.9% vs 12.3%, RR 0.80, P < .001
- Cardiovascular death 6.1% vs 8.1%, RR 0.74, P < .001
- Stroke 3.4% vs 4.9%, RR = .68, P < .001
- The composite end point 14.0% vs 17.8%, RR 0.78, P < .001).
Results were even better in the subset of patients who had diabetes.27 However, the decrease in blood pressure attributable to antihypertensive therapy with ramipril was minimal (3–4 mm Hg systolic and 1–2 mm Hg diastolic). This slight change should not have been enough to produce significant differences in clinical outcomes, a major limitation of this trial. The investigators speculated that the positive results may be due to a class effect of ACE inhibitors.26
HOPE 328–30 explored the effect of blood pressure- and cholesterol-controlling drugs on the same primary end points but in patients at intermediate risk of major cardiovascular events. Investigators randomized the 12,705 patients to 4 treatment groups:
- Blood pressure control with candesartan (an ARB) plus hydrochlorothiazide (a thiazide diuretic)
- Cholesterol control with rosuvastatin (a statin)
- Blood pressure plus cholesterol control
- Placebo.
Therapy was started at a systolic blood pressure above 140 mm Hg.
Compared with placebo, the rate of composite events was significantly reduced in the rosuvastatin group (3.7% vs 4.8%, HR 0.76, P = .002)28 and the candesartan-hydrochlorothiazide-rosuvastatin group (3.6% vs 5.0%, HR 0.71; P = .005)29 but not in the candesartan-hydrochlorothiazide group (4.1% vs 4.4%; HR 0.93; P = .40).30
In addition, a subgroup analysis comparing active treatment vs placebo found a significant reduction in major cardiovascular events for treated patients whose baseline systolic blood pressure was in the upper third (> 143.5 mm Hg, mean 154.1 mm Hg), while treated patients in the lower middle and lower thirds had no significant reduction.30
These results suggest that intensive treatment to achieve a systolic blood pressure below 140 mm Hg in patients at intermediate risk may not be helpful. Nevertheless, there seems to be agreement that intensive treatment generally leads to a reduction in cardiovascular events. The results also show the benefit of lowering cholesterol.
Bundy et al31 performed a meta-analysis that provides support for intensive antihypertensive treatment. Reviewing 42 clinical trials in more than 144,000 patients, they found that treating to reach a target systolic blood pressure of 120 to 124 mm Hg can reduce cardiovascular events and all-cause mortality.
The trade-off is a minimal increase in the risk of adverse events. Also, the risk-benefit ratio of intensive treatment seems to vary in different patient subgroups.
WHAT ABOUT PATIENTS WITH COMORBIDITIES?
The debate over intensive vs standard treatment in blood pressure management extends beyond hypertension and includes important comorbidities such as diabetes, stroke, and renal disease. Patients with a history of stroke or end-stage renal disease have only a minimal mention in the AHA/ACC guidelines.
Diabetes
Emdin et al,32 in a meta-analysis of 40 trials that included more than 100,000 patients with diabetes, concluded that a 10-mm Hg lowering of systolic blood pressure significantly reduces the rates of all-cause mortality, cardiovascular disease, coronary heart disease, stroke, albuminuria, and retinopathy. Stratifying the results according to the systolic blood pressure achieved (≥ 130 or < 130 mm Hg), the relative risks of mortality, coronary heart disease, cardiovascular disease, heart failure, and albuminuria were actually lower in the higher stratum than in the lower.
ACCORD (the Action to Control Cardiovascular Risk in Diabetes)33 study provides contrary results. It examined intensive and standard blood pressure control targets in patients with type 2 diabetes at high risk of cardiovascular events, using primary outcome measures similar to those in SPRINT. It found no significant difference in fatal and nonfatal cardiovascular events between the intensive and standard blood pressure target arms.
Despite those results, the ACC/AHA guidelines still advocate for more intensive treatment (goal < 130/80 mm Hg) in all patients, including those with diabetes.1
The ADA position statement (September 2017) recommended a target below 140/90 mm Hg in patients with diabetes and hypertension.8 However, they also noted that lower systolic and diastolic blood pressure targets, such as below 130/80 mm Hg, may be appropriate for patients at high risk of cardiovascular disease “if they can be achieved without undue treatment burden.”8 Thus, it is not clear which blood pressure targets in patients with diabetes are the best.
Stroke
In patients with stroke, AHA/ACC guidelines1 recommend treatment if the blood pressure is 140/90 mm Hg or higher because antihypertensive therapy has been associated with a decrease in the recurrence of transient ischemic attack and stroke. The ideal target blood pressure is not known, but a goal of less than 130/80 mm Hg may be reasonable.
In the Secondary Prevention of Small Subcortical Strokes (SPS3) trial, a retrospective open-label trial, a target blood pressure below 130/80 mm Hg in patients with a history of lacunar stroke was associated with a lower risk of intracranial hemorrhage, but the difference was not statistically significant.34 For this reason, the ACC/AHA guidelines consider it reasonable to aim for a systolic blood pressure below 130 mm Hg in these patients.1
Renal disease
The ACC/AHA guidelines do not address how to manage hypertension in patients with end-stage renal disease, but for patients with chronic kidney disease they recommend a blood pressure target below 130/80 mm Hg.1 This recommendation is derived from the SPRINT trial,15 in which patients with stage 3 or 4 chronic kidney disease accounted for 28% of the study population. In that subgroup, intensive blood pressure control seemed to provide the same benefits for reduction in cardiovascular death and all-cause mortality.
TREAT PATIENTS, NOT NUMBERS
Blood pressure targets should be applied in the appropriate clinical context and on a patient-by-patient basis. In clinical practice, one size does not always fit all, as special cases exist.
For example, blood pressure can oscillate widely in patients with autonomic nerve disorders, making it difficult to strive for a specific target, especially an intensive one. Thus, it may be necessary to allow higher systolic blood pressure in these patients. Similarly, patients with diabetes or chronic kidney disease may be at higher risk of kidney injury with more intensive blood pressure management.
Treating numbers rather than patients may result in unbalanced patient care. The optimal approach to blood pressure management relies on a comprehensive risk factor assessment and shared decision-making with the patient before setting specific blood pressure targets.
OUR APPROACH
We aim for a blood pressure goal below 130/80 mm Hg for all patients with cardiovascular disease, according to the AHA/ACC guidelines. We aim for that same target in patients without cardiovascular disease but who have an elevated estimated cardiovascular risk (> 10%) over the next 10 years.
We recognize, however, that the benefits of aggressive blood pressure reduction may not be as clear in all patients, such as those with diabetes. We also recognize that some patient subgroups are at high risk of adverse events, including those with low diastolic pressure, chronic kidney disease, a history of falls, and older age. In those patients, we are extremely judicious when titrating antihypertensive medications. We often make smaller titrations, at longer intervals, and with more frequent laboratory testing and in-office follow-up.
Our process of managing hypertension through intensive blood pressure control to achieve lower systolic blood pressure targets requires a concerted effort among healthcare providers at all levels. It especially requires more involvement and investment from primary care providers to individualize treatment in their patients. This process has helped us to reach our treatment goals while limiting adverse effects of lower blood pressure targets.
MOVING FORWARD
Hypertension is a major risk factor for cardiovascular disease, and intensive blood pressure control has the potential to significantly reduce rates of morbidity and death associated with cardiovascular disease. Thus, a general consensus on the definition of hypertension and treatment goals is essential to reduce the risk of cardiovascular events in this large patient population.
Intensive blood pressure treatment has shown efficacy, but it has a small accompanying risk of adverse events, which varies in patient subgroups and affects the benefit-risk ratio of this therapy. For example, the cardiovascular benefit of intensive treatment is less clear in diabetic patients, and the risk of adverse events may be higher in older patients with chronic kidney disease.
Moving forward, more research is needed into the effects of intensive and standard treatment on patients of all ages, those with common comorbid conditions, and those with other important factors such as diastolic hypertension.
Finally, the various medical societies should collaborate on hypertension guideline development. This would require considerable planning and coordination but would ultimately be useful in creating a generalizable approach to hypertension management.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289(19):2560–2572. doi:10.1001/jama.289.19.2560
- Go AS, Bauman MA, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2014; 63(4):878–885. doi:10.1161/HYP.0000000000000003
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311(5):507–520. doi:10.1001/jama.2013.284427
- Wright JT Jr, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014; 160(7):499–503. doi:10.7326/M13-2981
- Weber MA, Schiffrin EL, White WB, et al. Notice of duplicate publication [duplicate publication of Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens 2014; 16(1):14–26. doi:10.1111/jch.12237] J Hypertens 2014; 32(1):3–15. doi:10.1097/HJH.0000000000000065
- Rosendorff C, Lackland DT, Allison M, et al. Treatment of hypertension in patients with coronary artery disease: a scientific statement from the American Heart Association, American College of Cardiology, and American Society of Hypertension. J Am Soc Hypertens 2015; 9(6):453–498. doi:10.1016/j.jash.2015.03.002
- de Boer IH, Bangalore S, Benetos A, et al. Diabetes and hypertension: a position statement by the American Diabetes Association. Diabetes Care 2017; 40(9):1273–1284. doi:10.2337/dci17-0026
- Qaseem A, Wilt TJ, Rich R, Humphrey LL, Frost J, Forciea MA. Pharmacologic treatment of hypertension in adults aged 60 years or older to higher versus lower blood pressure targets: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2017; 166(6):430–437. doi:10.7326/M16-1785
- The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in over-weight people with high normal blood pressure: the Trials of Hypertension Prevention, phase II. Arch Intern Med 1997; 157(6):657–667. pmid:9080920
- He J, Whelton PK, Appel LJ, Charleston J, Klag MJ. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension 2000; 35(2):544–549. pmid:10679495
- Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med 2001; 344(1):3–10. doi:10.1056/NEJM200101043440101
- Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for US adults: National Health Interview Survey, 2012. National Center for Health Statistics. Vital Health Stat 10; 2014(260):1–161. pmid:24819891
- Muntner P, Carey RM, Gidding S, et al. Potential US population impact of the 2017 ACC/AHA high blood pressure guideline. J Am Coll Cardiol 2018; 71(2):109–118. doi:10.1016/j.jacc.2017.10.073
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Bress AP, Kramer H, Khatib R, et al. Potential deaths averted and serious adverse events incurred from adoption of the SPRINT (Systolic Blood Pressure Intervention Trial) intensive blood pressure regimen in the United States: Projections from NHANES (National Health and Nutrition Examination Survey). Circulation 2017; 135(17):1617–1628. doi:10.1161/CIRCULATIONAHA.116.025322
- Williamson JD, Supiano MA, Applegate WB, et al. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315(24):2673–2682. doi:10.1001/jama.2016.7050
- Beddhu S, Rocco MV, Toto R, et al. Effects of intensive systolic blood pressure control on kidney and cardiovascular outcomes in persons without kidney disease: a secondary analysis of a randomized trial. Ann Intern Med 2017; 167(6):375–383. doi:10.7326/M16-2966
- Brunström M, Carlberg B. Association of blood pressure lowering with mortality and cardiovascular disease across blood pressure levels: a systematic review and meta-analysis. JAMA Intern Med 2018; 178(1):28–36. doi:10.1001/jamainternmed.2017.6015
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265(24):3255–3264. pmid:2046107
- Bulpitt CJ, Beckett NS, Cooke J, et al. Results of the pilot study for the Hypertension in the Very Elderly Trial. J Hypertens 2003; 21(12):2409–2417. doi:10.1097/01.hjh.0000084782.15238.a2
- Liu L, Zhang Y, Liu G, et al. The Felodipine Event Reduction (FEVER) study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23(12):2157–2172. pmid:16269957
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31(12):2115–2127. doi:10.1291/hypres.31.2115
- Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56(2):196–202. doi:10.1161/HYPERTENSIONAHA.109.146035
- Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet 2016; 387(10022):957–967. doi:10.1016/S0140-6736(15)01225-8
- Sleight P. The HOPE study (Heart Outcomes Prevention Evaluation). J Renin Angiotensin Aldosterone Syst 2000; 1(1):18–20. doi:10.3317/jraas.2000.002
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000; 355(9200):253–259. pmid:10675071
- Yusuf S, Bosch J, Dagenais G, et al. Cholesterol lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2021–2031. doi:10.1056/NEJMoa1600176
- Yusuf S, Lonn E, Pais P, et al. Blood-pressure and cholesterol lowering in persons without cardiovascular disease. N Engl J Med 2016; 374(21):2032–2043. doi:10.1056/NEJMoa1600177
- Lonn EM, Bosch J, López-Jaramillo P, et al. Blood-pressure lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2009–2020. doi:10.1056/NEJMoa1600175
- Bundy JD, Li C, Stuchlik P, et al. Systolic blood pressure reduction and risk of cardiovascular disease and mortality: a systematic review and network meta-analysis. JAMA Cardiol 2017; 2(7):775–781. doi:10.1001/jamacardio.2017.1421
- Emdin CA, Rahimi K, Neal B, Callender T, Perkovic V, Patel A. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA 2015; 313(6):603–615. doi:10.1001/jama.2014.18574
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362(17):1575–1585. doi:10.1056/NEJMoa1001286
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382(9891):507–515. doi:10.1016/S0140-6736(13)60852-1
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289(19):2560–2572. doi:10.1001/jama.289.19.2560
- Go AS, Bauman MA, King SM, et al. An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 2014; 63(4):878–885. doi:10.1161/HYP.0000000000000003
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311(5):507–520. doi:10.1001/jama.2013.284427
- Wright JT Jr, Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence supporting a systolic blood pressure goal of less than 150 mm Hg in patients aged 60 years or older: the minority view. Ann Intern Med 2014; 160(7):499–503. doi:10.7326/M13-2981
- Weber MA, Schiffrin EL, White WB, et al. Notice of duplicate publication [duplicate publication of Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens 2014; 16(1):14–26. doi:10.1111/jch.12237] J Hypertens 2014; 32(1):3–15. doi:10.1097/HJH.0000000000000065
- Rosendorff C, Lackland DT, Allison M, et al. Treatment of hypertension in patients with coronary artery disease: a scientific statement from the American Heart Association, American College of Cardiology, and American Society of Hypertension. J Am Soc Hypertens 2015; 9(6):453–498. doi:10.1016/j.jash.2015.03.002
- de Boer IH, Bangalore S, Benetos A, et al. Diabetes and hypertension: a position statement by the American Diabetes Association. Diabetes Care 2017; 40(9):1273–1284. doi:10.2337/dci17-0026
- Qaseem A, Wilt TJ, Rich R, Humphrey LL, Frost J, Forciea MA. Pharmacologic treatment of hypertension in adults aged 60 years or older to higher versus lower blood pressure targets: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2017; 166(6):430–437. doi:10.7326/M16-1785
- The Trials of Hypertension Prevention Collaborative Research Group. Effects of weight loss and sodium reduction intervention on blood pressure and hypertension incidence in over-weight people with high normal blood pressure: the Trials of Hypertension Prevention, phase II. Arch Intern Med 1997; 157(6):657–667. pmid:9080920
- He J, Whelton PK, Appel LJ, Charleston J, Klag MJ. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension 2000; 35(2):544–549. pmid:10679495
- Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med 2001; 344(1):3–10. doi:10.1056/NEJM200101043440101
- Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for US adults: National Health Interview Survey, 2012. National Center for Health Statistics. Vital Health Stat 10; 2014(260):1–161. pmid:24819891
- Muntner P, Carey RM, Gidding S, et al. Potential US population impact of the 2017 ACC/AHA high blood pressure guideline. J Am Coll Cardiol 2018; 71(2):109–118. doi:10.1016/j.jacc.2017.10.073
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Bress AP, Kramer H, Khatib R, et al. Potential deaths averted and serious adverse events incurred from adoption of the SPRINT (Systolic Blood Pressure Intervention Trial) intensive blood pressure regimen in the United States: Projections from NHANES (National Health and Nutrition Examination Survey). Circulation 2017; 135(17):1617–1628. doi:10.1161/CIRCULATIONAHA.116.025322
- Williamson JD, Supiano MA, Applegate WB, et al. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315(24):2673–2682. doi:10.1001/jama.2016.7050
- Beddhu S, Rocco MV, Toto R, et al. Effects of intensive systolic blood pressure control on kidney and cardiovascular outcomes in persons without kidney disease: a secondary analysis of a randomized trial. Ann Intern Med 2017; 167(6):375–383. doi:10.7326/M16-2966
- Brunström M, Carlberg B. Association of blood pressure lowering with mortality and cardiovascular disease across blood pressure levels: a systematic review and meta-analysis. JAMA Intern Med 2018; 178(1):28–36. doi:10.1001/jamainternmed.2017.6015
- Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265(24):3255–3264. pmid:2046107
- Bulpitt CJ, Beckett NS, Cooke J, et al. Results of the pilot study for the Hypertension in the Very Elderly Trial. J Hypertens 2003; 21(12):2409–2417. doi:10.1097/01.hjh.0000084782.15238.a2
- Liu L, Zhang Y, Liu G, et al. The Felodipine Event Reduction (FEVER) study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23(12):2157–2172. pmid:16269957
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31(12):2115–2127. doi:10.1291/hypres.31.2115
- Ogihara T, Saruta T, Rakugi H, et al. Target blood pressure for treatment of isolated systolic hypertension in the elderly: valsartan in elderly isolated systolic hypertension study. Hypertension 2010; 56(2):196–202. doi:10.1161/HYPERTENSIONAHA.109.146035
- Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet 2016; 387(10022):957–967. doi:10.1016/S0140-6736(15)01225-8
- Sleight P. The HOPE study (Heart Outcomes Prevention Evaluation). J Renin Angiotensin Aldosterone Syst 2000; 1(1):18–20. doi:10.3317/jraas.2000.002
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000; 355(9200):253–259. pmid:10675071
- Yusuf S, Bosch J, Dagenais G, et al. Cholesterol lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2021–2031. doi:10.1056/NEJMoa1600176
- Yusuf S, Lonn E, Pais P, et al. Blood-pressure and cholesterol lowering in persons without cardiovascular disease. N Engl J Med 2016; 374(21):2032–2043. doi:10.1056/NEJMoa1600177
- Lonn EM, Bosch J, López-Jaramillo P, et al. Blood-pressure lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med 2016; 374(21):2009–2020. doi:10.1056/NEJMoa1600175
- Bundy JD, Li C, Stuchlik P, et al. Systolic blood pressure reduction and risk of cardiovascular disease and mortality: a systematic review and network meta-analysis. JAMA Cardiol 2017; 2(7):775–781. doi:10.1001/jamacardio.2017.1421
- Emdin CA, Rahimi K, Neal B, Callender T, Perkovic V, Patel A. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA 2015; 313(6):603–615. doi:10.1001/jama.2014.18574
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362(17):1575–1585. doi:10.1056/NEJMoa1001286
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382(9891):507–515. doi:10.1016/S0140-6736(13)60852-1
KEY POINTS
- The 2017 ACC/AHA guidelines lowered the definition of hypertension to 130/80 mm Hg or higher, thereby in-creasing the number of US adults with hypertension from 31.9% to 45.6%.
- For patients with known cardiovascular disease or a 10-year risk of an atherosclerotic cardiovascular disease event of 10% or higher, drug treatment “is recommended” if the average blood pressure is 130/80 mm Hg or higher. For those without cardiovascular disease and at lower risk, drug treatment is recommended if the aver-age blood pressure is 140/90 mm Hg or higher.
- A treatment goal of less than 130/80 mm Hg “is recommended” for patients with hypertension and known car-diovascular disease or a 10-year risk of an atherosclerotic cardiovascular disease event of 10% or higher, and “may be reasonable” for those without additional markers of increased cardiovascular risk.
- Intensive blood pressure control has the potential to significantly reduce rates of morbidity and death associated with cardiovascular disease, at the price of causing more adverse effects.

Common benign breast concerns for the primary care physician
Breast concerns account for approximately 3% of all female visits to a primary care practice.1 The most common symptoms are breast lumps and breast pain.

Because breast cancer is the most common malignancy in women in the United States, affecting nearly 1 in 8 women in their lifetime, women with breast problems often fear the worst. However, only about 3.5% of women reporting a concern have cancer; most problems are benign (Table 1).1
Here, we present an evidence-based review of common breast problems in primary care practice and discuss how to evaluate and manage them.
GENERAL APPROACH
The evaluation of a breast concern requires a systematic approach, beginning with a history that documents the onset, severity, and frequency of symptoms. If the concern is a lump or mass, ask whether it becomes more tender or increases in size at any point during the menstrual cycle.
Focus the physical examination on the cervical, supraclavicular, infraclavicular, and axillary lymph nodes and on the breast itself. Assess breast symmetry, note any skin changes such as dimpling, and check the nipples for discharge and inversion. Palpate the breasts for masses.
PALPABLE BREAST MASS: IMAGING NEEDED
If a mass is present, it is more likely to be malignant if any of the following is true:
- Firm to hard texture or indistinct margins
- Attached to the underlying deep fascia or skin
- Associated nipple inversion or skin dimpling.2
Breast masses are more likely benign if they have discrete, well-defined margins, are mobile with a soft to rubbery consistency, and change with the menstrual cycle. However, clinical features are unreliable indicators of cause, and thus additional investigation with breast imaging is warranted.
Mammography remains the diagnostic test of choice for all women age 30 or older who have a palpable breast mass. It is less effective in younger women because they are more likely to have extremely dense fibroglandular tissue that will limit its sensitivity to imaging.
Order diagnostic mammography, which includes additional views focused on the area of concern, rather than screening mammography, which includes only standard craniocaudal and mediolateral oblique views. A skin marker should be applied over the palpable lump to aid imaging. Because a breast that contains a mass may be denser than the opposite breast or may show asymmetry, both breasts should be imaged. The sensitivity of diagnostic mammography varies from 85% to 90%, so a negative mammogram does not rule out malignancy.2,3
Targeted ultrasonography of the palpable mass helps identify solid masses such as fibroadenomas or malignant tumors, classifies the margins (lobulated, smooth, or irregular), and assesses vascularity. Ultrasonography is particularly useful for characterizing cystic lesions (eg, simple, septated, or clustered cysts) and cysts with internal echoes. It can also identify lipomas or sebaceous cysts.
If the findings on both mammography and ultrasonography are benign, the likelihood of cancer is very low, with an estimated negative predictive value of 97% to 100%.2,3 Additionally, the likelihood of nonmalignant findings on biopsy after benign imaging is approximately 99%.3
Although radiologic imaging can define palpable masses, it is intended as a clinical aid. Suspicious findings on clinical examination should never be ignored even if findings on imaging are reassuring, as studies have documented that about 5% of breast cancers may be detected on clinical breast examination alone.4
Other imaging tests such as magnetic resonance imaging may be considered occasionally if clinical suspicion remains high after negative mammography and ultrasonography, but they cannot confirm a diagnosis of malignancy. In that case, refer the patient to a surgeon for consideration of excisional biopsy.
Patients with an indeterminate lesion can return in 3 to 12 weeks for a follow-up examination and repeat imaging, which helps assess interval clinical stability. The latter option is especially helpful for patients with masses that are of low suspicion or for patients who prefer to avoid invasive tissue biopsy.
Patients with clinical and radiologic findings that suggest a benign cause can return for short-term follow-up in 6 months or in 12 months for their regular mammogram.
BREAST PAIN: RARELY MALIGNANT
More than 50% of women experience breast pain at some point in their life.5 Of these, 35% report that the pain adversely affects their sleep, and 41% note that the pain detrimentally affects their sexual quality of life. Up to 66% of breast pain correlates directly with the patient’s menstrual cycle.5 Breast pain is rarely associated with malignancy.
Regardless of its severity and the low likelihood of malignancy, breast pain can be a significant source of distress for the patient, primarily because of concerns about underlying malignancy. If the patient has a focal area of pain on examination, order mammography in combination with targeted ultrasonography. The sensitivity and negative predictive value of benign findings on combination mammography and ultrasonography in this setting are as high as 100%. The incidence of underlying cancer in patients with focal breast pain and no palpable mass is approximately 1.2%.6
The long-term prognosis in women with diffuse, often bilateral breast pain (in the absence of additional clinical findings) is excellent. In one study, the incidence of a breast cancer diagnosis was 1.8% after a median of 51 months of follow-up.7 Therefore, patients presenting with diffuse pain, no palpable abnormalities, and benign imaging can be safely reassured. Magnetic resonance imaging is rarely indicated in patients with breast pain unless other clinical findings, such as a mass or skin changes, are noted and the results of mammography and ultrasonography are negative.
Treating breast pain
Treating breast pain remains a challenge. The first step is to reassure the patient about her prognosis and help her make appropriate lifestyle modifications.
A well-fitting bra. Suggest getting a professional bra fitting. Wearing a well-fitted bra that offers lift, support, and compression and reduces excess motion can help improve benign breast pain. A bra fitting is especially important for women with large breasts because it can be difficult for these women to get an accurate size. Wearing a lightly fitted bra at night may also provide comfort if there is nighttime pain with breast tissue movement.
Reducing daily caffeine intake is often advised for pain management, but strong evidence of its efficacy is lacking.
Anti-inflammatory drugs can be beneficial if used short-term, especially if costochondritis is suspected.
Danazol improves pain in more than 70% of patients with cyclical symptoms and in up to 48% of those with noncyclical symptoms.
Bromocriptine is effective in up to 54% of those with cyclical symptoms and in up to 33% of those with noncyclical symptoms.8 However, the US Food and Drug Administration (FDA) withdrew approval for this indication because of adverse effects.
Tamoxifen, in contrast, provides relief in 94% of those with cyclical symptoms and in 56% of those with noncyclical symptoms.9
Adverse effects, however, limit the use of danazol, bromocriptine, and tamoxifen, and they should be prescribed only for short-term use (3 to 6 months) and only in women with chronic debilitating pain.
A few small studies have evaluated alternative options.
Toremifene is a triphenylethylene derivative similar to tamoxifen that is also used in the adjuvant treatment of postmenopausal breast cancer (but with fewer adverse effects). It has been documented to have a significant effect on premenstrual breast pain, with a 64% reduction in breast pain scores compared with a 26% reduction with placebo.10 However, the FDA has not approved it for this indication, and it can be cost-prohibitive.
Over-the-counter medications that may provide relief for cyclic breast pain include vitamin E or B6, products containing oil of Vitex agnus castus (chaste tree or chasteberry), and flaxseed.11,12
Acupuncture has been evaluated in patients with noncyclic breast pain and was found to reduce pain by 56% to 67% in one study,13 although it did not affect quality of life.
NIPPLE DISCHARGE
From 5% to 7% of women seek medical attention for nipple discharge.14,15 Breast cancer is found in 5% to 15% of women who undergo surgery for nipple discharge.16,17
Review the patient’s current medications and inquire about health conditions such as thyroid dysfunction or visual field changes that suggest a pituitary mass (which can lead to nipple discharge by causing hormonal dysregulation or hyperprolactinemia).
Palpate the breasts for an underlying mass, look for lesions on the nipple, and assess the color of the fluid. Also note whether there is discharge from one or both breasts, whether it is spontaneous or expressive, and whether it occurs from a single or multiple ducts. Nipple lesions may require further testing with punch biopsy.
Nonlactational nipple discharge is classified as physiologic or pathologic. Physiologic nipple discharge is typically bilateral, involving multiple ducts, and is often clear or straw-colored but may also be green, gray, or brown.
White, opaque fluid is often related to galactorrhea as a result of hyperprolactinemia, hypothyroidism, or medications such as antipsychotic drugs (eg, haloperidol and fluphenazine) and gastrointestinal motility agents such as metoclopramide. Discharge also commonly results from benign underlying ductal abnormalities such as intraductal papilloma, periductal mastitis, and duct ectasia.
Pathologic nipple discharge is often unilateral and persistent, occurring spontaneously from a solitary duct, and may be bloody or serous.
For women with pathologic nipple discharge who are 30 or older, diagnostic imaging with mammography and subareolar ultrasonography is recommended. If the patient is younger than 30, ultrasonography of the subareolar region alone can be used. Targeted ultrasonography of any palpable area is also advised.
Cytologic assessment of the fluid is not recommended because it can often lead to a false-positive finding of atypical cells. Imaging studies such as ductography, duct lavage, ductoscopy, and magnetic resonance imaging are also generally unnecessary; instead, a persistent clinical concern should prompt a surgical referral for consideration of duct excision.
When a patient has pathologic nipple discharge with a negative physical examination and breast imaging, studies have shown that the risk of cancer is 3% or less.18
Patients with spontaneous bloody or serous single-duct discharge with negative results on mammography and ultrasonography should be reassured that they have a low risk of underlying cancer. If the patient prefers, one approachto management is follow-up mammography and ultrasonography at 6 months and clinical examination for up to 2 years or until the discharge resolves on its own.
On the other hand, if the discharge is distressing to the patient, subareolar duct excision can be performed with both a diagnostic and therapeutic purpose.
NIPPLE-AREOLAR RASH: CONSIDER PAGET DISEASE
A rash on the nipple or areolar region warrants careful evaluation because it may be the first sign of Paget disease of the breast.
In the clinical breast examination, assess the extent of the rash and the presence of any underlying breast mass or nipple discharge. Dermatitis often starts on the areola and resolves quickly with topical therapy. However, Paget disease tends to start directly on the nipple itself, is unresponsive or only partially responsive to topical therapy, and progresses gradually, leading to erosions and ultimately effacement of the nipple itself.
If the clinical examination suggests mild dermatitis and the results of breast imaging are negative, treat the patient with a topical medication because benign conditions such as dermatitis and eczema are common. However, continued follow-up is mandatory until the rash completely resolves: Paget disease sometimes initially improves with topical therapy due to its inflammatory nature.
If you suspect Paget disease or the rash does not fully resolve after 2 to 3 weeks of topical therapy, refer the patient to a dermatologist for full-thickness punch biopsy to establish the diagnosis.
Paget disease of the breast may or may not be associated with underlying ductal carcinoma in situ or invasive breast cancer.19 The absence of clinical or imaging abnormalities in a patient with Paget disease does not rule out underlying malignancy.20
DENSE BREASTS


Increased breast density has been shown to be a risk factor for breast cancer and may be prognostically useful when combined with the Tyrer-Cuzick model or the Gail model of breast cancer risk.24
Additionally, increased density can mask cancers on mammography, significantly reducing its sensitivity. In women with heterogeneously or extremely dense breasts, the sensitivity of mammography for detecting cancer is only 25% to 50%.21 Due to this low sensitivity, supplemental imaging is helpful, particularly in women already at risk of breast cancer based on family history.
Supplemental screening
Digital mammography with tomosynthesis was approved by the FDA in 2011 for use in combination with standard digital mammography for breast cancer screening. Compared with traditional 2-dimensional mammography alone, adding 3-D tomosynthesis decreases the recall rate and increases the cancer detection rate.25
Tomosynthesis tends to perform better in women with heterogeneously dense breasts (BI-RADS category C). There is no significant improvement in cancer detection in women with extremely dense breasts (BI-RADS category D).26
Depending on the methodology, radiation exposure can be either higher or lower than with traditional mammography. However, in all forms, the very small amount of radiation is considered safe.
Whole breast ultrasonography. When whole breast ultrasonography is used to supplement mammography, the recall rate is higher than when mammography is used alone (14% vs 7%–11%).22 It also increases the cancer detection rate by 4.4 additional cancers per 1,000 examinations. However, the false-positive rate with whole breast ultrasonography is higher; the positive predictive value of combined mammography and ultrasonography is 11.2% vs 22.6% for mammography alone.22 Therefore, we do not generally recommend whole breast ultrasonography as a supplement to mammography in women with dense breast tissue unless other studies are not an option.
Molecular breast imaging is not widely available because it requires special equipment, injection of a radiopharamceutical (technetium Tc 99m sestamibi), and a radiologist who specializes in breast imaging to interpret the results. When it is available, however, it increases the cancer detection rate by 8.8 in 1,000 examinations; the positive predictive value is similar to that of screening mammography alone.21 It is particularly useful in patients with dense breasts who do not qualify for screening magnetic resonance imaging (lifetime risk of < 20% to 25%).
Technetium sestamibi is associated with a minimal amount of radiation exposure (2.4 mSv vs 1.2 mSV with standard mammography). However, this exposure is much less than background radiation exposure and is considered safe.21
IF THE PATIENT HAS AN ABNORMAL SCREENING MAMMOGRAM

Screening mammography can disclose abnormalities such as calcifications, masses, asymmetry, or architectural distortion.27 Abnormalities are reported using standardized BI-RADS categories designated with the numbers 0 through 6 (Table 3).23
A report of BI-RADS category 0 (incomplete), 4 (suspicious), or 5 (highly suspicious) requires additional workup.
Category 1 (negative) requires no further follow-up, and the patient should resume age-appropriate screening.
For patients with category 2 (benign) findings, routine screening is recommended, whereas patients with category 3 (probably benign) are advised to come back in 6 months for follow-up imaging.
Diagnostic mammography includes additional assessments for focal symptoms or areas of abnormality noted on screening imaging or clinical examination. These may include spot magnification views of areas of asymmetry, mass, architectural distortion, or calcifications. Ultrasonography of focal breast abnormalities can help determine if there is an underlying cyst or solid mass.
MANAGEMENT OF BENIGN FINDINGS ON BREAST BIOPSY

Benign breast disease is diagnosed when a patient with a palpable or radiographic abnormality undergoes breast biopsy with benign findings.28,29 It can be largely grouped into 3 categories: nonproliferative, proliferative without atypia, and proliferative with atypia (Table 4).28,29
If core-needle biopsy study results are benign, the next step is to establish radiologic-pathologic and clinical-pathologic concordance. If the findings on clinical examination or imaging are not consistent with those on pathologic study, excisional biopsy should be performed, as imaging-directed biopsy may not have adequately sampled the lesion.30
Nonproliferative lesions account for about 65% of findings on core-needle biopsy and include simple cysts, fibroadenomas, columnar cell changes, apocrine metaplasia, and mild ductal hyperplasia of the usual type. These lesions do not significantly increase the risk of breast cancer; the relative risk is 1.2 to 1.4.28,29 Additionally, the risk of “upstaging” after excisional biopsy—ie, to a higher-risk lesion or to malignancy—is minimal. Therefore, no additional action is necessary when these findings alone are noted on core-needle biopsy.
Proliferative lesions without atypia account for about 30% of biopsy results and include usual ductal hyperplasia, sclerosing adenosis, columnar hyperplasia, papilloma, and radial scar. Generally, there is a slightly increased risk of subsequent breast cancer, with a relative risk of 1.7 to 2.1.28 Usual ductal hyperplasia and columnar hyperplasia have little risk of upstaging with excision, and therefore, surgical consultation is not recommended.
Previously, surgical excision was recommended for any intraductal papilloma due to risk of upgrade in pathologic diagnosis at the time of excision. However, more recent data suggest that the upgrade rate is about 2.2% for a solitary papilloma that is less than 1 cm in diameter and without associated mass lesion (either clinically or radiographically), is concordant with radiographic findings, and has no associated atypical cells on biopsy.31 In this case, observation and short-interval clinical follow-up are reasonable. If there are multiple papillomas, the patient has symptoms such as persistent bloody nipple discharge, or any of the above criteria are not met, surgical excision is recommended.28
Similarly, radial scars and complex sclerosing lesions are increasingly likely to be associated with malignancy based on size. Upstaging ranges from 0% to 12%. It is again important when evaluating radial scars that there is pathologic concordance and that there were no associated high-risk lesions on pathology. If this is the case, it is reasonable to clinically monitor patients with small radial scars, particularly in those who do not have an elevated risk of developing breast cancer.30
For all patients who have undergone biopsy and whose pathology study results are benign, a thorough risk evaluation should be performed, including calculation of their lifetime risk of breast cancer. This can be done with the National Cancer Institute Breast Cancer Risk Assessment Tool, the International Breast Cancer Intervention Study (IBIS) risk calculator, or other model using family history as a basis for calculations. Patients found to have a lifetime risk of breast cancer of greater than 20% to 25% should be offered annual screening with magnetic resonance imaging in addition to mammography.
ATYPICAL HYPERPLASIA: INCREASED RISK
When biopsy study shows atypical ductal hyperplasia or atypical lobular hyperplasia, there is an increased risk of breast cancer.28,32 The absolute overall risk of developing breast cancer in 25 years is 30%, and that risk is further stratified based on the number of foci of atypia noted in the specimen.29
When core-needle biopsy study reveals atypical ductal hyperplasia in the tissue, there is a 15% to 30% risk of finding breast cancer with surgical excision.28 Surgical excision is therefore recommended for atypical ductal hyperplasia noted on core-needle biopsy.28
In contrast, when atypical lobular hyperplasia alone is noted, the risk of upstagingto malignancy varies widely—from 0% to 67%—although recent studies have noted risks of 1% to 3%.33,34 Thus, the decision for surgical excision is more variable. Generally, if the atypical lobular hyperplasia is noted incidentally, is not associated with a higher grade lesion, and is concordant with imaging, it is reasonable to closely monitor with serial imaging and physical examination. Excision is unnecessary.35
Patients found to have atypical hyperplasia on breast biopsy should receive counseling about risk-reducing medications. Selective estrogen receptor modulators such as tamoxifen and raloxifene have been shown to reduce the risk of breast cancer by as much as 86% in patients with atypical hyperplasia.36 Similarly, aromatase inhibitors such as exemestane and anastrozole reduce breast cancer risk by approximately 65%.37
- Eberl MM, Phillips RL Jr, Lamberts H, Okkes I, Mahoney MC. Characterizing breast symptoms in family practice. Ann Fam Med 2008; 6(6):528–533. doi:10.1370/afm.905
- Harvey JA, Mahoney MC, Newell MS, et al. ACR appropriateness criteria palpable breast masses. J Am Coll Radiol 2013; 10(10):742–749.e3. doi:10.1016/j.jacr.2013.06.013
- Ha R, Kim H, Mango V, Wynn R, Comstock C. Ultrasonographic features and clinical implications of benign palpable breast lesions in young women. Ultrasonography 2015; 34(1):66–70. doi:10.14366/usg.14043
- Provencher L, Hogue JC, Desbiens C, et al. Is clinical breast examination important for breast cancer detection? Curr Oncol 2016; 23(4):e332–e339. doi:10.3747/co.23.2881
- Scurr J, Hedger W, Morris P, Brown N. The prevalence, severity, and impact of breast pain in the general population. Breast J 2014; 20(5):508–513. doi:10.1111/tbj.12305
- Leddy R, Irshad A, Zerwas E, et al. Role of breast ultrasound and mammography in evaluating patients presenting with focal breast pain in the absence of a palpable lump. Breast J 2013; 19(6):582–589. doi:10.1111/tbj.12178
- Noroozian M, Stein LF, Gaetke-Udager K, Helvie MA. Long-term clinical outcomes in women with breast pain in the absence of additional clinical findings: mammography remains indicated. Breast Cancer Res Treat 2015; 149(2):417–424. doi:10.1007/s10549-014-3257-3
- Gateley CA, Miers M, Mansel RE, Hughes LE. Drug treatments for mastalgia: 17 years experience in the Cardiff Mastalgia Clinic. J R Soc Med 1992; 85(1):12–15. pmid:1548647
- Fentiman IS, Caleffi M, Hamed H, Chaudary MA. Dosage and duration of tamoxifen treatment for mastalgia: a controlled trial. Br J Surg 1988; 75(9):845–846. pmid:3052691
- Oksa S, Luukkaala T, Mäenpää J. Toremifene for premenstrual mastalgia: a randomised, placebo-controlled crossover study. BJOG 2006; 113(6):713–718. doi:10.1111/j.1471-0528.2006.00943.x
- Mirghafourvand M, Mohammad-Alizadeh-Charandabi S, Ahmadpour P, Javadzadeh Y. Effects of Vitex agnus and flaxseed on cyclic mastalgia: a randomized controlled trial. Complement Ther Med 2016; 24:90–95. doi:10.1016/j.ctim.2015.12.009
- Shobeiri F, Oshvandi K, Nazari M. Clinical effectiveness of vitamin E and vitamin B6 for improving pain severity in cyclic mastalgia. Iran J Nurs Midwifery Res 2015; 20(6):723–727. doi:10.4103/1735-9066.170003
- Thicke LA, Hazelton JK, Bauer BA, et al. Acupuncture for treatment of noncyclic breast pain: a pilot study. Am J Chin Med 2011; 39(6):1117–1129. doi:10.1142/S0192415X11009445
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353(3):275–285. doi:10.1056/NEJMra035692
- Gülay H, Bora S, Kìlìçturgay S, Hamaloglu E, Göksel HA. Management of nipple discharge. J Am Coll Surg 1994; 178(5):471–474. pmid:8167884
- Murad TM, Contesso G, Mouriesse H. Nipple discharge from the breast. Ann Surg 1982; 195(3):259–264. pmid:6277258
- Sakorafas GH. Nipple discharge: current diagnostic and therapeutic approaches. Cancer Treat Rev 2001; 27(5):275–282. doi:10.1053/ctrv.2001.0234
- Ashfaq A, Senior D, Pockaj BA, et al. Validation study of a modern treatment algorithm for nipple discharge. Am J Surg 2014; 208(2):222–227. doi:10.1016/j.amjsurg.2013.12.035
- Chen CY, Sun LM, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the US. Cancer 2006; 107(7):1448–1458. doi:10.1002/cncr.22137
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE 3rd. Paget's disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187(2):171–177. pmid:9704964
- Hruska CB. Molecular breast imaging for screening in dense breasts: state of the art and future directions. AJR Am J Roentgenol 2017; 208(2):275–283. doi:10.2214/AJR.16.17131
- Melnikow J, Fenton JJ, Whitlock EP, et al. Supplemental screening for breast cancer in women with dense breasts: a systematic review for the US Preventive Services Task Force. Ann Intern Med 2016; 164(4):268–278. doi:10.7326/M15-1789
- American College of Radiology. Breast imaging reporting and data system (BI-RADS). Reston, VA: American College of Radiology; 2013.
- Brentnall AR, Harkness EF, Astley SM, et al. Mammographic density adds accuracy to both the Tyrer-Cuzick and Gail breast cancer risk models in a prospective UK screening cohort. Breast Cancer Res 2015; 17(1):147. doi:10.1186/s13058-015-0653-5
- Friedewald SM, Rafferty EA, Rose SL, et al. Breast cancer screening using tomosynthesis in combination with digital mammography. JAMA 2014; 311(24):2499–2507. doi:10.1001/jama.2014.6095
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA 2016; 315(16):1784–1786. doi:10.1001/jama.2016.1708
- Venkatesan A, Chu P, Kerlikowske K, Sickles EA, Smith-Bindman R. Positive predictive value of specific mammographic findings according to reader and patient variables. Radiology 2009; 250(3):648–657. doi:10.1148/radiol.2503080541
- Hartmann LC, Sellers TA, Frost MH, et al. Benign breast disease and the risk of breast cancer. N Engl J Med 2005; 353(3):229–237. doi:10.1056/NEJMoa044383
- Hartmann LC, Degnim AC, Santen RJ, DuPont WD, Ghosh K. Atypical hyperplasia of the breast—risk assessment and management options. N Engl J Med 2015; 372(1):78–89. doi:10.1056/NEJMsr1407164
- Neal L, Sandhu NP, Hieken TJ, et al. Diagnosis and management of benign, atypical, and indeterminate breast lesions detected on core needle biopsy. Mayo Clin Proc 2014; 89(4):536–547. doi:10.1016/j.mayocp.2014.02.004
- Nakhlis F, Ahmadiyeh N, Lester S, Raza S, Lotfi P, Golshan M. Papilloma on core biopsy: excision vs observation. Ann Surg Oncol 2015; 22(5):1479–1482. doi:10.1245/s10434-014-4091-x
- Degnim AC, Dupont WE, Radisky DC, et al. Extent of atypical hyperplasia stratifies breast cancer risk in 2 independent cohorts of women. Cancer 2016; 122(19):2971-2978. doi:10.1002/cncr.30153
- Sen LQ, Berg WA, Hooley RJ, Carter GJ, Desouki MM, Sumkin JH. Core breast biopsies showing lobular carcinoma in situ should be excised and surveillance is reasonable for atypical lobular hyperplasia. AJR Am J Roentgenol 2016; 207(5):1132–1145. doi:10.2214/AJR.15.15425
- Nakhlis F, Gilmore L, Gelman R, et al. Incidence of adjacent synchronous invasive carcinoma and/or ductal carcinoma in situ in patient with lobular neoplasia on core biopsy: results from a prospective multi-institutional registry (TBCRC 020). Ann Surg Oncol 2016; 23(3):722–728. doi:10.1245/s10434-015-4922-4
- Racz JM, Carter JM, Degnim AC. Lobular neoplasia and atypical ductal hyperplasia on core biopsy: current surgical management recommendations. Ann Surg Oncol 2017; 24(10):2848–2854. doi:10.1245/s10434-017-5978-0
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388. doi:10.1093/jnci/dji372
- Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011; 364(25):2381–2391. doi:10.1056/NEJMoa1103507
Breast concerns account for approximately 3% of all female visits to a primary care practice.1 The most common symptoms are breast lumps and breast pain.
Because breast cancer is the most common malignancy in women in the United States, affecting nearly 1 in 8 women in their lifetime, women with breast problems often fear the worst. However, only about 3.5% of women reporting a concern have cancer; most problems are benign (Table 1).1
Here, we present an evidence-based review of common breast problems in primary care practice and discuss how to evaluate and manage them.
GENERAL APPROACH
The evaluation of a breast concern requires a systematic approach, beginning with a history that documents the onset, severity, and frequency of symptoms. If the concern is a lump or mass, ask whether it becomes more tender or increases in size at any point during the menstrual cycle.
Focus the physical examination on the cervical, supraclavicular, infraclavicular, and axillary lymph nodes and on the breast itself. Assess breast symmetry, note any skin changes such as dimpling, and check the nipples for discharge and inversion. Palpate the breasts for masses.
PALPABLE BREAST MASS: IMAGING NEEDED
If a mass is present, it is more likely to be malignant if any of the following is true:
- Firm to hard texture or indistinct margins
- Attached to the underlying deep fascia or skin
- Associated nipple inversion or skin dimpling.2
Breast masses are more likely benign if they have discrete, well-defined margins, are mobile with a soft to rubbery consistency, and change with the menstrual cycle. However, clinical features are unreliable indicators of cause, and thus additional investigation with breast imaging is warranted.
Mammography remains the diagnostic test of choice for all women age 30 or older who have a palpable breast mass. It is less effective in younger women because they are more likely to have extremely dense fibroglandular tissue that will limit its sensitivity to imaging.
Order diagnostic mammography, which includes additional views focused on the area of concern, rather than screening mammography, which includes only standard craniocaudal and mediolateral oblique views. A skin marker should be applied over the palpable lump to aid imaging. Because a breast that contains a mass may be denser than the opposite breast or may show asymmetry, both breasts should be imaged. The sensitivity of diagnostic mammography varies from 85% to 90%, so a negative mammogram does not rule out malignancy.2,3
Targeted ultrasonography of the palpable mass helps identify solid masses such as fibroadenomas or malignant tumors, classifies the margins (lobulated, smooth, or irregular), and assesses vascularity. Ultrasonography is particularly useful for characterizing cystic lesions (eg, simple, septated, or clustered cysts) and cysts with internal echoes. It can also identify lipomas or sebaceous cysts.
If the findings on both mammography and ultrasonography are benign, the likelihood of cancer is very low, with an estimated negative predictive value of 97% to 100%.2,3 Additionally, the likelihood of nonmalignant findings on biopsy after benign imaging is approximately 99%.3
Although radiologic imaging can define palpable masses, it is intended as a clinical aid. Suspicious findings on clinical examination should never be ignored even if findings on imaging are reassuring, as studies have documented that about 5% of breast cancers may be detected on clinical breast examination alone.4
Other imaging tests such as magnetic resonance imaging may be considered occasionally if clinical suspicion remains high after negative mammography and ultrasonography, but they cannot confirm a diagnosis of malignancy. In that case, refer the patient to a surgeon for consideration of excisional biopsy.
Patients with an indeterminate lesion can return in 3 to 12 weeks for a follow-up examination and repeat imaging, which helps assess interval clinical stability. The latter option is especially helpful for patients with masses that are of low suspicion or for patients who prefer to avoid invasive tissue biopsy.
Patients with clinical and radiologic findings that suggest a benign cause can return for short-term follow-up in 6 months or in 12 months for their regular mammogram.
BREAST PAIN: RARELY MALIGNANT
More than 50% of women experience breast pain at some point in their life.5 Of these, 35% report that the pain adversely affects their sleep, and 41% note that the pain detrimentally affects their sexual quality of life. Up to 66% of breast pain correlates directly with the patient’s menstrual cycle.5 Breast pain is rarely associated with malignancy.
Regardless of its severity and the low likelihood of malignancy, breast pain can be a significant source of distress for the patient, primarily because of concerns about underlying malignancy. If the patient has a focal area of pain on examination, order mammography in combination with targeted ultrasonography. The sensitivity and negative predictive value of benign findings on combination mammography and ultrasonography in this setting are as high as 100%. The incidence of underlying cancer in patients with focal breast pain and no palpable mass is approximately 1.2%.6
The long-term prognosis in women with diffuse, often bilateral breast pain (in the absence of additional clinical findings) is excellent. In one study, the incidence of a breast cancer diagnosis was 1.8% after a median of 51 months of follow-up.7 Therefore, patients presenting with diffuse pain, no palpable abnormalities, and benign imaging can be safely reassured. Magnetic resonance imaging is rarely indicated in patients with breast pain unless other clinical findings, such as a mass or skin changes, are noted and the results of mammography and ultrasonography are negative.
Treating breast pain
Treating breast pain remains a challenge. The first step is to reassure the patient about her prognosis and help her make appropriate lifestyle modifications.
A well-fitting bra. Suggest getting a professional bra fitting. Wearing a well-fitted bra that offers lift, support, and compression and reduces excess motion can help improve benign breast pain. A bra fitting is especially important for women with large breasts because it can be difficult for these women to get an accurate size. Wearing a lightly fitted bra at night may also provide comfort if there is nighttime pain with breast tissue movement.
Reducing daily caffeine intake is often advised for pain management, but strong evidence of its efficacy is lacking.
Anti-inflammatory drugs can be beneficial if used short-term, especially if costochondritis is suspected.
Danazol improves pain in more than 70% of patients with cyclical symptoms and in up to 48% of those with noncyclical symptoms.
Bromocriptine is effective in up to 54% of those with cyclical symptoms and in up to 33% of those with noncyclical symptoms.8 However, the US Food and Drug Administration (FDA) withdrew approval for this indication because of adverse effects.
Tamoxifen, in contrast, provides relief in 94% of those with cyclical symptoms and in 56% of those with noncyclical symptoms.9
Adverse effects, however, limit the use of danazol, bromocriptine, and tamoxifen, and they should be prescribed only for short-term use (3 to 6 months) and only in women with chronic debilitating pain.
A few small studies have evaluated alternative options.
Toremifene is a triphenylethylene derivative similar to tamoxifen that is also used in the adjuvant treatment of postmenopausal breast cancer (but with fewer adverse effects). It has been documented to have a significant effect on premenstrual breast pain, with a 64% reduction in breast pain scores compared with a 26% reduction with placebo.10 However, the FDA has not approved it for this indication, and it can be cost-prohibitive.
Over-the-counter medications that may provide relief for cyclic breast pain include vitamin E or B6, products containing oil of Vitex agnus castus (chaste tree or chasteberry), and flaxseed.11,12
Acupuncture has been evaluated in patients with noncyclic breast pain and was found to reduce pain by 56% to 67% in one study,13 although it did not affect quality of life.
NIPPLE DISCHARGE
From 5% to 7% of women seek medical attention for nipple discharge.14,15 Breast cancer is found in 5% to 15% of women who undergo surgery for nipple discharge.16,17
Review the patient’s current medications and inquire about health conditions such as thyroid dysfunction or visual field changes that suggest a pituitary mass (which can lead to nipple discharge by causing hormonal dysregulation or hyperprolactinemia).
Palpate the breasts for an underlying mass, look for lesions on the nipple, and assess the color of the fluid. Also note whether there is discharge from one or both breasts, whether it is spontaneous or expressive, and whether it occurs from a single or multiple ducts. Nipple lesions may require further testing with punch biopsy.
Nonlactational nipple discharge is classified as physiologic or pathologic. Physiologic nipple discharge is typically bilateral, involving multiple ducts, and is often clear or straw-colored but may also be green, gray, or brown.
White, opaque fluid is often related to galactorrhea as a result of hyperprolactinemia, hypothyroidism, or medications such as antipsychotic drugs (eg, haloperidol and fluphenazine) and gastrointestinal motility agents such as metoclopramide. Discharge also commonly results from benign underlying ductal abnormalities such as intraductal papilloma, periductal mastitis, and duct ectasia.
Pathologic nipple discharge is often unilateral and persistent, occurring spontaneously from a solitary duct, and may be bloody or serous.
For women with pathologic nipple discharge who are 30 or older, diagnostic imaging with mammography and subareolar ultrasonography is recommended. If the patient is younger than 30, ultrasonography of the subareolar region alone can be used. Targeted ultrasonography of any palpable area is also advised.
Cytologic assessment of the fluid is not recommended because it can often lead to a false-positive finding of atypical cells. Imaging studies such as ductography, duct lavage, ductoscopy, and magnetic resonance imaging are also generally unnecessary; instead, a persistent clinical concern should prompt a surgical referral for consideration of duct excision.
When a patient has pathologic nipple discharge with a negative physical examination and breast imaging, studies have shown that the risk of cancer is 3% or less.18
Patients with spontaneous bloody or serous single-duct discharge with negative results on mammography and ultrasonography should be reassured that they have a low risk of underlying cancer. If the patient prefers, one approachto management is follow-up mammography and ultrasonography at 6 months and clinical examination for up to 2 years or until the discharge resolves on its own.
On the other hand, if the discharge is distressing to the patient, subareolar duct excision can be performed with both a diagnostic and therapeutic purpose.
NIPPLE-AREOLAR RASH: CONSIDER PAGET DISEASE
A rash on the nipple or areolar region warrants careful evaluation because it may be the first sign of Paget disease of the breast.
In the clinical breast examination, assess the extent of the rash and the presence of any underlying breast mass or nipple discharge. Dermatitis often starts on the areola and resolves quickly with topical therapy. However, Paget disease tends to start directly on the nipple itself, is unresponsive or only partially responsive to topical therapy, and progresses gradually, leading to erosions and ultimately effacement of the nipple itself.
If the clinical examination suggests mild dermatitis and the results of breast imaging are negative, treat the patient with a topical medication because benign conditions such as dermatitis and eczema are common. However, continued follow-up is mandatory until the rash completely resolves: Paget disease sometimes initially improves with topical therapy due to its inflammatory nature.
If you suspect Paget disease or the rash does not fully resolve after 2 to 3 weeks of topical therapy, refer the patient to a dermatologist for full-thickness punch biopsy to establish the diagnosis.
Paget disease of the breast may or may not be associated with underlying ductal carcinoma in situ or invasive breast cancer.19 The absence of clinical or imaging abnormalities in a patient with Paget disease does not rule out underlying malignancy.20
DENSE BREASTS
Increased breast density has been shown to be a risk factor for breast cancer and may be prognostically useful when combined with the Tyrer-Cuzick model or the Gail model of breast cancer risk.24
Additionally, increased density can mask cancers on mammography, significantly reducing its sensitivity. In women with heterogeneously or extremely dense breasts, the sensitivity of mammography for detecting cancer is only 25% to 50%.21 Due to this low sensitivity, supplemental imaging is helpful, particularly in women already at risk of breast cancer based on family history.
Supplemental screening
Digital mammography with tomosynthesis was approved by the FDA in 2011 for use in combination with standard digital mammography for breast cancer screening. Compared with traditional 2-dimensional mammography alone, adding 3-D tomosynthesis decreases the recall rate and increases the cancer detection rate.25
Tomosynthesis tends to perform better in women with heterogeneously dense breasts (BI-RADS category C). There is no significant improvement in cancer detection in women with extremely dense breasts (BI-RADS category D).26
Depending on the methodology, radiation exposure can be either higher or lower than with traditional mammography. However, in all forms, the very small amount of radiation is considered safe.
Whole breast ultrasonography. When whole breast ultrasonography is used to supplement mammography, the recall rate is higher than when mammography is used alone (14% vs 7%–11%).22 It also increases the cancer detection rate by 4.4 additional cancers per 1,000 examinations. However, the false-positive rate with whole breast ultrasonography is higher; the positive predictive value of combined mammography and ultrasonography is 11.2% vs 22.6% for mammography alone.22 Therefore, we do not generally recommend whole breast ultrasonography as a supplement to mammography in women with dense breast tissue unless other studies are not an option.
Molecular breast imaging is not widely available because it requires special equipment, injection of a radiopharamceutical (technetium Tc 99m sestamibi), and a radiologist who specializes in breast imaging to interpret the results. When it is available, however, it increases the cancer detection rate by 8.8 in 1,000 examinations; the positive predictive value is similar to that of screening mammography alone.21 It is particularly useful in patients with dense breasts who do not qualify for screening magnetic resonance imaging (lifetime risk of < 20% to 25%).
Technetium sestamibi is associated with a minimal amount of radiation exposure (2.4 mSv vs 1.2 mSV with standard mammography). However, this exposure is much less than background radiation exposure and is considered safe.21
IF THE PATIENT HAS AN ABNORMAL SCREENING MAMMOGRAM
Screening mammography can disclose abnormalities such as calcifications, masses, asymmetry, or architectural distortion.27 Abnormalities are reported using standardized BI-RADS categories designated with the numbers 0 through 6 (Table 3).23
A report of BI-RADS category 0 (incomplete), 4 (suspicious), or 5 (highly suspicious) requires additional workup.
Category 1 (negative) requires no further follow-up, and the patient should resume age-appropriate screening.
For patients with category 2 (benign) findings, routine screening is recommended, whereas patients with category 3 (probably benign) are advised to come back in 6 months for follow-up imaging.
Diagnostic mammography includes additional assessments for focal symptoms or areas of abnormality noted on screening imaging or clinical examination. These may include spot magnification views of areas of asymmetry, mass, architectural distortion, or calcifications. Ultrasonography of focal breast abnormalities can help determine if there is an underlying cyst or solid mass.
MANAGEMENT OF BENIGN FINDINGS ON BREAST BIOPSY
Benign breast disease is diagnosed when a patient with a palpable or radiographic abnormality undergoes breast biopsy with benign findings.28,29 It can be largely grouped into 3 categories: nonproliferative, proliferative without atypia, and proliferative with atypia (Table 4).28,29
If core-needle biopsy study results are benign, the next step is to establish radiologic-pathologic and clinical-pathologic concordance. If the findings on clinical examination or imaging are not consistent with those on pathologic study, excisional biopsy should be performed, as imaging-directed biopsy may not have adequately sampled the lesion.30
Nonproliferative lesions account for about 65% of findings on core-needle biopsy and include simple cysts, fibroadenomas, columnar cell changes, apocrine metaplasia, and mild ductal hyperplasia of the usual type. These lesions do not significantly increase the risk of breast cancer; the relative risk is 1.2 to 1.4.28,29 Additionally, the risk of “upstaging” after excisional biopsy—ie, to a higher-risk lesion or to malignancy—is minimal. Therefore, no additional action is necessary when these findings alone are noted on core-needle biopsy.
Proliferative lesions without atypia account for about 30% of biopsy results and include usual ductal hyperplasia, sclerosing adenosis, columnar hyperplasia, papilloma, and radial scar. Generally, there is a slightly increased risk of subsequent breast cancer, with a relative risk of 1.7 to 2.1.28 Usual ductal hyperplasia and columnar hyperplasia have little risk of upstaging with excision, and therefore, surgical consultation is not recommended.
Previously, surgical excision was recommended for any intraductal papilloma due to risk of upgrade in pathologic diagnosis at the time of excision. However, more recent data suggest that the upgrade rate is about 2.2% for a solitary papilloma that is less than 1 cm in diameter and without associated mass lesion (either clinically or radiographically), is concordant with radiographic findings, and has no associated atypical cells on biopsy.31 In this case, observation and short-interval clinical follow-up are reasonable. If there are multiple papillomas, the patient has symptoms such as persistent bloody nipple discharge, or any of the above criteria are not met, surgical excision is recommended.28
Similarly, radial scars and complex sclerosing lesions are increasingly likely to be associated with malignancy based on size. Upstaging ranges from 0% to 12%. It is again important when evaluating radial scars that there is pathologic concordance and that there were no associated high-risk lesions on pathology. If this is the case, it is reasonable to clinically monitor patients with small radial scars, particularly in those who do not have an elevated risk of developing breast cancer.30
For all patients who have undergone biopsy and whose pathology study results are benign, a thorough risk evaluation should be performed, including calculation of their lifetime risk of breast cancer. This can be done with the National Cancer Institute Breast Cancer Risk Assessment Tool, the International Breast Cancer Intervention Study (IBIS) risk calculator, or other model using family history as a basis for calculations. Patients found to have a lifetime risk of breast cancer of greater than 20% to 25% should be offered annual screening with magnetic resonance imaging in addition to mammography.
ATYPICAL HYPERPLASIA: INCREASED RISK
When biopsy study shows atypical ductal hyperplasia or atypical lobular hyperplasia, there is an increased risk of breast cancer.28,32 The absolute overall risk of developing breast cancer in 25 years is 30%, and that risk is further stratified based on the number of foci of atypia noted in the specimen.29
When core-needle biopsy study reveals atypical ductal hyperplasia in the tissue, there is a 15% to 30% risk of finding breast cancer with surgical excision.28 Surgical excision is therefore recommended for atypical ductal hyperplasia noted on core-needle biopsy.28
In contrast, when atypical lobular hyperplasia alone is noted, the risk of upstagingto malignancy varies widely—from 0% to 67%—although recent studies have noted risks of 1% to 3%.33,34 Thus, the decision for surgical excision is more variable. Generally, if the atypical lobular hyperplasia is noted incidentally, is not associated with a higher grade lesion, and is concordant with imaging, it is reasonable to closely monitor with serial imaging and physical examination. Excision is unnecessary.35
Patients found to have atypical hyperplasia on breast biopsy should receive counseling about risk-reducing medications. Selective estrogen receptor modulators such as tamoxifen and raloxifene have been shown to reduce the risk of breast cancer by as much as 86% in patients with atypical hyperplasia.36 Similarly, aromatase inhibitors such as exemestane and anastrozole reduce breast cancer risk by approximately 65%.37
Breast concerns account for approximately 3% of all female visits to a primary care practice.1 The most common symptoms are breast lumps and breast pain.
Because breast cancer is the most common malignancy in women in the United States, affecting nearly 1 in 8 women in their lifetime, women with breast problems often fear the worst. However, only about 3.5% of women reporting a concern have cancer; most problems are benign (Table 1).1
Here, we present an evidence-based review of common breast problems in primary care practice and discuss how to evaluate and manage them.
GENERAL APPROACH
The evaluation of a breast concern requires a systematic approach, beginning with a history that documents the onset, severity, and frequency of symptoms. If the concern is a lump or mass, ask whether it becomes more tender or increases in size at any point during the menstrual cycle.
Focus the physical examination on the cervical, supraclavicular, infraclavicular, and axillary lymph nodes and on the breast itself. Assess breast symmetry, note any skin changes such as dimpling, and check the nipples for discharge and inversion. Palpate the breasts for masses.
PALPABLE BREAST MASS: IMAGING NEEDED
If a mass is present, it is more likely to be malignant if any of the following is true:
- Firm to hard texture or indistinct margins
- Attached to the underlying deep fascia or skin
- Associated nipple inversion or skin dimpling.2
Breast masses are more likely benign if they have discrete, well-defined margins, are mobile with a soft to rubbery consistency, and change with the menstrual cycle. However, clinical features are unreliable indicators of cause, and thus additional investigation with breast imaging is warranted.
Mammography remains the diagnostic test of choice for all women age 30 or older who have a palpable breast mass. It is less effective in younger women because they are more likely to have extremely dense fibroglandular tissue that will limit its sensitivity to imaging.
Order diagnostic mammography, which includes additional views focused on the area of concern, rather than screening mammography, which includes only standard craniocaudal and mediolateral oblique views. A skin marker should be applied over the palpable lump to aid imaging. Because a breast that contains a mass may be denser than the opposite breast or may show asymmetry, both breasts should be imaged. The sensitivity of diagnostic mammography varies from 85% to 90%, so a negative mammogram does not rule out malignancy.2,3
Targeted ultrasonography of the palpable mass helps identify solid masses such as fibroadenomas or malignant tumors, classifies the margins (lobulated, smooth, or irregular), and assesses vascularity. Ultrasonography is particularly useful for characterizing cystic lesions (eg, simple, septated, or clustered cysts) and cysts with internal echoes. It can also identify lipomas or sebaceous cysts.
If the findings on both mammography and ultrasonography are benign, the likelihood of cancer is very low, with an estimated negative predictive value of 97% to 100%.2,3 Additionally, the likelihood of nonmalignant findings on biopsy after benign imaging is approximately 99%.3
Although radiologic imaging can define palpable masses, it is intended as a clinical aid. Suspicious findings on clinical examination should never be ignored even if findings on imaging are reassuring, as studies have documented that about 5% of breast cancers may be detected on clinical breast examination alone.4
Other imaging tests such as magnetic resonance imaging may be considered occasionally if clinical suspicion remains high after negative mammography and ultrasonography, but they cannot confirm a diagnosis of malignancy. In that case, refer the patient to a surgeon for consideration of excisional biopsy.
Patients with an indeterminate lesion can return in 3 to 12 weeks for a follow-up examination and repeat imaging, which helps assess interval clinical stability. The latter option is especially helpful for patients with masses that are of low suspicion or for patients who prefer to avoid invasive tissue biopsy.
Patients with clinical and radiologic findings that suggest a benign cause can return for short-term follow-up in 6 months or in 12 months for their regular mammogram.
BREAST PAIN: RARELY MALIGNANT
More than 50% of women experience breast pain at some point in their life.5 Of these, 35% report that the pain adversely affects their sleep, and 41% note that the pain detrimentally affects their sexual quality of life. Up to 66% of breast pain correlates directly with the patient’s menstrual cycle.5 Breast pain is rarely associated with malignancy.
Regardless of its severity and the low likelihood of malignancy, breast pain can be a significant source of distress for the patient, primarily because of concerns about underlying malignancy. If the patient has a focal area of pain on examination, order mammography in combination with targeted ultrasonography. The sensitivity and negative predictive value of benign findings on combination mammography and ultrasonography in this setting are as high as 100%. The incidence of underlying cancer in patients with focal breast pain and no palpable mass is approximately 1.2%.6
The long-term prognosis in women with diffuse, often bilateral breast pain (in the absence of additional clinical findings) is excellent. In one study, the incidence of a breast cancer diagnosis was 1.8% after a median of 51 months of follow-up.7 Therefore, patients presenting with diffuse pain, no palpable abnormalities, and benign imaging can be safely reassured. Magnetic resonance imaging is rarely indicated in patients with breast pain unless other clinical findings, such as a mass or skin changes, are noted and the results of mammography and ultrasonography are negative.
Treating breast pain
Treating breast pain remains a challenge. The first step is to reassure the patient about her prognosis and help her make appropriate lifestyle modifications.
A well-fitting bra. Suggest getting a professional bra fitting. Wearing a well-fitted bra that offers lift, support, and compression and reduces excess motion can help improve benign breast pain. A bra fitting is especially important for women with large breasts because it can be difficult for these women to get an accurate size. Wearing a lightly fitted bra at night may also provide comfort if there is nighttime pain with breast tissue movement.
Reducing daily caffeine intake is often advised for pain management, but strong evidence of its efficacy is lacking.
Anti-inflammatory drugs can be beneficial if used short-term, especially if costochondritis is suspected.
Danazol improves pain in more than 70% of patients with cyclical symptoms and in up to 48% of those with noncyclical symptoms.
Bromocriptine is effective in up to 54% of those with cyclical symptoms and in up to 33% of those with noncyclical symptoms.8 However, the US Food and Drug Administration (FDA) withdrew approval for this indication because of adverse effects.
Tamoxifen, in contrast, provides relief in 94% of those with cyclical symptoms and in 56% of those with noncyclical symptoms.9
Adverse effects, however, limit the use of danazol, bromocriptine, and tamoxifen, and they should be prescribed only for short-term use (3 to 6 months) and only in women with chronic debilitating pain.
A few small studies have evaluated alternative options.
Toremifene is a triphenylethylene derivative similar to tamoxifen that is also used in the adjuvant treatment of postmenopausal breast cancer (but with fewer adverse effects). It has been documented to have a significant effect on premenstrual breast pain, with a 64% reduction in breast pain scores compared with a 26% reduction with placebo.10 However, the FDA has not approved it for this indication, and it can be cost-prohibitive.
Over-the-counter medications that may provide relief for cyclic breast pain include vitamin E or B6, products containing oil of Vitex agnus castus (chaste tree or chasteberry), and flaxseed.11,12
Acupuncture has been evaluated in patients with noncyclic breast pain and was found to reduce pain by 56% to 67% in one study,13 although it did not affect quality of life.
NIPPLE DISCHARGE
From 5% to 7% of women seek medical attention for nipple discharge.14,15 Breast cancer is found in 5% to 15% of women who undergo surgery for nipple discharge.16,17
Review the patient’s current medications and inquire about health conditions such as thyroid dysfunction or visual field changes that suggest a pituitary mass (which can lead to nipple discharge by causing hormonal dysregulation or hyperprolactinemia).
Palpate the breasts for an underlying mass, look for lesions on the nipple, and assess the color of the fluid. Also note whether there is discharge from one or both breasts, whether it is spontaneous or expressive, and whether it occurs from a single or multiple ducts. Nipple lesions may require further testing with punch biopsy.
Nonlactational nipple discharge is classified as physiologic or pathologic. Physiologic nipple discharge is typically bilateral, involving multiple ducts, and is often clear or straw-colored but may also be green, gray, or brown.
White, opaque fluid is often related to galactorrhea as a result of hyperprolactinemia, hypothyroidism, or medications such as antipsychotic drugs (eg, haloperidol and fluphenazine) and gastrointestinal motility agents such as metoclopramide. Discharge also commonly results from benign underlying ductal abnormalities such as intraductal papilloma, periductal mastitis, and duct ectasia.
Pathologic nipple discharge is often unilateral and persistent, occurring spontaneously from a solitary duct, and may be bloody or serous.
For women with pathologic nipple discharge who are 30 or older, diagnostic imaging with mammography and subareolar ultrasonography is recommended. If the patient is younger than 30, ultrasonography of the subareolar region alone can be used. Targeted ultrasonography of any palpable area is also advised.
Cytologic assessment of the fluid is not recommended because it can often lead to a false-positive finding of atypical cells. Imaging studies such as ductography, duct lavage, ductoscopy, and magnetic resonance imaging are also generally unnecessary; instead, a persistent clinical concern should prompt a surgical referral for consideration of duct excision.
When a patient has pathologic nipple discharge with a negative physical examination and breast imaging, studies have shown that the risk of cancer is 3% or less.18
Patients with spontaneous bloody or serous single-duct discharge with negative results on mammography and ultrasonography should be reassured that they have a low risk of underlying cancer. If the patient prefers, one approachto management is follow-up mammography and ultrasonography at 6 months and clinical examination for up to 2 years or until the discharge resolves on its own.
On the other hand, if the discharge is distressing to the patient, subareolar duct excision can be performed with both a diagnostic and therapeutic purpose.
NIPPLE-AREOLAR RASH: CONSIDER PAGET DISEASE
A rash on the nipple or areolar region warrants careful evaluation because it may be the first sign of Paget disease of the breast.
In the clinical breast examination, assess the extent of the rash and the presence of any underlying breast mass or nipple discharge. Dermatitis often starts on the areola and resolves quickly with topical therapy. However, Paget disease tends to start directly on the nipple itself, is unresponsive or only partially responsive to topical therapy, and progresses gradually, leading to erosions and ultimately effacement of the nipple itself.
If the clinical examination suggests mild dermatitis and the results of breast imaging are negative, treat the patient with a topical medication because benign conditions such as dermatitis and eczema are common. However, continued follow-up is mandatory until the rash completely resolves: Paget disease sometimes initially improves with topical therapy due to its inflammatory nature.
If you suspect Paget disease or the rash does not fully resolve after 2 to 3 weeks of topical therapy, refer the patient to a dermatologist for full-thickness punch biopsy to establish the diagnosis.
Paget disease of the breast may or may not be associated with underlying ductal carcinoma in situ or invasive breast cancer.19 The absence of clinical or imaging abnormalities in a patient with Paget disease does not rule out underlying malignancy.20
DENSE BREASTS
Increased breast density has been shown to be a risk factor for breast cancer and may be prognostically useful when combined with the Tyrer-Cuzick model or the Gail model of breast cancer risk.24
Additionally, increased density can mask cancers on mammography, significantly reducing its sensitivity. In women with heterogeneously or extremely dense breasts, the sensitivity of mammography for detecting cancer is only 25% to 50%.21 Due to this low sensitivity, supplemental imaging is helpful, particularly in women already at risk of breast cancer based on family history.
Supplemental screening
Digital mammography with tomosynthesis was approved by the FDA in 2011 for use in combination with standard digital mammography for breast cancer screening. Compared with traditional 2-dimensional mammography alone, adding 3-D tomosynthesis decreases the recall rate and increases the cancer detection rate.25
Tomosynthesis tends to perform better in women with heterogeneously dense breasts (BI-RADS category C). There is no significant improvement in cancer detection in women with extremely dense breasts (BI-RADS category D).26
Depending on the methodology, radiation exposure can be either higher or lower than with traditional mammography. However, in all forms, the very small amount of radiation is considered safe.
Whole breast ultrasonography. When whole breast ultrasonography is used to supplement mammography, the recall rate is higher than when mammography is used alone (14% vs 7%–11%).22 It also increases the cancer detection rate by 4.4 additional cancers per 1,000 examinations. However, the false-positive rate with whole breast ultrasonography is higher; the positive predictive value of combined mammography and ultrasonography is 11.2% vs 22.6% for mammography alone.22 Therefore, we do not generally recommend whole breast ultrasonography as a supplement to mammography in women with dense breast tissue unless other studies are not an option.
Molecular breast imaging is not widely available because it requires special equipment, injection of a radiopharamceutical (technetium Tc 99m sestamibi), and a radiologist who specializes in breast imaging to interpret the results. When it is available, however, it increases the cancer detection rate by 8.8 in 1,000 examinations; the positive predictive value is similar to that of screening mammography alone.21 It is particularly useful in patients with dense breasts who do not qualify for screening magnetic resonance imaging (lifetime risk of < 20% to 25%).
Technetium sestamibi is associated with a minimal amount of radiation exposure (2.4 mSv vs 1.2 mSV with standard mammography). However, this exposure is much less than background radiation exposure and is considered safe.21
IF THE PATIENT HAS AN ABNORMAL SCREENING MAMMOGRAM
Screening mammography can disclose abnormalities such as calcifications, masses, asymmetry, or architectural distortion.27 Abnormalities are reported using standardized BI-RADS categories designated with the numbers 0 through 6 (Table 3).23
A report of BI-RADS category 0 (incomplete), 4 (suspicious), or 5 (highly suspicious) requires additional workup.
Category 1 (negative) requires no further follow-up, and the patient should resume age-appropriate screening.
For patients with category 2 (benign) findings, routine screening is recommended, whereas patients with category 3 (probably benign) are advised to come back in 6 months for follow-up imaging.
Diagnostic mammography includes additional assessments for focal symptoms or areas of abnormality noted on screening imaging or clinical examination. These may include spot magnification views of areas of asymmetry, mass, architectural distortion, or calcifications. Ultrasonography of focal breast abnormalities can help determine if there is an underlying cyst or solid mass.
MANAGEMENT OF BENIGN FINDINGS ON BREAST BIOPSY
Benign breast disease is diagnosed when a patient with a palpable or radiographic abnormality undergoes breast biopsy with benign findings.28,29 It can be largely grouped into 3 categories: nonproliferative, proliferative without atypia, and proliferative with atypia (Table 4).28,29
If core-needle biopsy study results are benign, the next step is to establish radiologic-pathologic and clinical-pathologic concordance. If the findings on clinical examination or imaging are not consistent with those on pathologic study, excisional biopsy should be performed, as imaging-directed biopsy may not have adequately sampled the lesion.30
Nonproliferative lesions account for about 65% of findings on core-needle biopsy and include simple cysts, fibroadenomas, columnar cell changes, apocrine metaplasia, and mild ductal hyperplasia of the usual type. These lesions do not significantly increase the risk of breast cancer; the relative risk is 1.2 to 1.4.28,29 Additionally, the risk of “upstaging” after excisional biopsy—ie, to a higher-risk lesion or to malignancy—is minimal. Therefore, no additional action is necessary when these findings alone are noted on core-needle biopsy.
Proliferative lesions without atypia account for about 30% of biopsy results and include usual ductal hyperplasia, sclerosing adenosis, columnar hyperplasia, papilloma, and radial scar. Generally, there is a slightly increased risk of subsequent breast cancer, with a relative risk of 1.7 to 2.1.28 Usual ductal hyperplasia and columnar hyperplasia have little risk of upstaging with excision, and therefore, surgical consultation is not recommended.
Previously, surgical excision was recommended for any intraductal papilloma due to risk of upgrade in pathologic diagnosis at the time of excision. However, more recent data suggest that the upgrade rate is about 2.2% for a solitary papilloma that is less than 1 cm in diameter and without associated mass lesion (either clinically or radiographically), is concordant with radiographic findings, and has no associated atypical cells on biopsy.31 In this case, observation and short-interval clinical follow-up are reasonable. If there are multiple papillomas, the patient has symptoms such as persistent bloody nipple discharge, or any of the above criteria are not met, surgical excision is recommended.28
Similarly, radial scars and complex sclerosing lesions are increasingly likely to be associated with malignancy based on size. Upstaging ranges from 0% to 12%. It is again important when evaluating radial scars that there is pathologic concordance and that there were no associated high-risk lesions on pathology. If this is the case, it is reasonable to clinically monitor patients with small radial scars, particularly in those who do not have an elevated risk of developing breast cancer.30
For all patients who have undergone biopsy and whose pathology study results are benign, a thorough risk evaluation should be performed, including calculation of their lifetime risk of breast cancer. This can be done with the National Cancer Institute Breast Cancer Risk Assessment Tool, the International Breast Cancer Intervention Study (IBIS) risk calculator, or other model using family history as a basis for calculations. Patients found to have a lifetime risk of breast cancer of greater than 20% to 25% should be offered annual screening with magnetic resonance imaging in addition to mammography.
ATYPICAL HYPERPLASIA: INCREASED RISK
When biopsy study shows atypical ductal hyperplasia or atypical lobular hyperplasia, there is an increased risk of breast cancer.28,32 The absolute overall risk of developing breast cancer in 25 years is 30%, and that risk is further stratified based on the number of foci of atypia noted in the specimen.29
When core-needle biopsy study reveals atypical ductal hyperplasia in the tissue, there is a 15% to 30% risk of finding breast cancer with surgical excision.28 Surgical excision is therefore recommended for atypical ductal hyperplasia noted on core-needle biopsy.28
In contrast, when atypical lobular hyperplasia alone is noted, the risk of upstagingto malignancy varies widely—from 0% to 67%—although recent studies have noted risks of 1% to 3%.33,34 Thus, the decision for surgical excision is more variable. Generally, if the atypical lobular hyperplasia is noted incidentally, is not associated with a higher grade lesion, and is concordant with imaging, it is reasonable to closely monitor with serial imaging and physical examination. Excision is unnecessary.35
Patients found to have atypical hyperplasia on breast biopsy should receive counseling about risk-reducing medications. Selective estrogen receptor modulators such as tamoxifen and raloxifene have been shown to reduce the risk of breast cancer by as much as 86% in patients with atypical hyperplasia.36 Similarly, aromatase inhibitors such as exemestane and anastrozole reduce breast cancer risk by approximately 65%.37
- Eberl MM, Phillips RL Jr, Lamberts H, Okkes I, Mahoney MC. Characterizing breast symptoms in family practice. Ann Fam Med 2008; 6(6):528–533. doi:10.1370/afm.905
- Harvey JA, Mahoney MC, Newell MS, et al. ACR appropriateness criteria palpable breast masses. J Am Coll Radiol 2013; 10(10):742–749.e3. doi:10.1016/j.jacr.2013.06.013
- Ha R, Kim H, Mango V, Wynn R, Comstock C. Ultrasonographic features and clinical implications of benign palpable breast lesions in young women. Ultrasonography 2015; 34(1):66–70. doi:10.14366/usg.14043
- Provencher L, Hogue JC, Desbiens C, et al. Is clinical breast examination important for breast cancer detection? Curr Oncol 2016; 23(4):e332–e339. doi:10.3747/co.23.2881
- Scurr J, Hedger W, Morris P, Brown N. The prevalence, severity, and impact of breast pain in the general population. Breast J 2014; 20(5):508–513. doi:10.1111/tbj.12305
- Leddy R, Irshad A, Zerwas E, et al. Role of breast ultrasound and mammography in evaluating patients presenting with focal breast pain in the absence of a palpable lump. Breast J 2013; 19(6):582–589. doi:10.1111/tbj.12178
- Noroozian M, Stein LF, Gaetke-Udager K, Helvie MA. Long-term clinical outcomes in women with breast pain in the absence of additional clinical findings: mammography remains indicated. Breast Cancer Res Treat 2015; 149(2):417–424. doi:10.1007/s10549-014-3257-3
- Gateley CA, Miers M, Mansel RE, Hughes LE. Drug treatments for mastalgia: 17 years experience in the Cardiff Mastalgia Clinic. J R Soc Med 1992; 85(1):12–15. pmid:1548647
- Fentiman IS, Caleffi M, Hamed H, Chaudary MA. Dosage and duration of tamoxifen treatment for mastalgia: a controlled trial. Br J Surg 1988; 75(9):845–846. pmid:3052691
- Oksa S, Luukkaala T, Mäenpää J. Toremifene for premenstrual mastalgia: a randomised, placebo-controlled crossover study. BJOG 2006; 113(6):713–718. doi:10.1111/j.1471-0528.2006.00943.x
- Mirghafourvand M, Mohammad-Alizadeh-Charandabi S, Ahmadpour P, Javadzadeh Y. Effects of Vitex agnus and flaxseed on cyclic mastalgia: a randomized controlled trial. Complement Ther Med 2016; 24:90–95. doi:10.1016/j.ctim.2015.12.009
- Shobeiri F, Oshvandi K, Nazari M. Clinical effectiveness of vitamin E and vitamin B6 for improving pain severity in cyclic mastalgia. Iran J Nurs Midwifery Res 2015; 20(6):723–727. doi:10.4103/1735-9066.170003
- Thicke LA, Hazelton JK, Bauer BA, et al. Acupuncture for treatment of noncyclic breast pain: a pilot study. Am J Chin Med 2011; 39(6):1117–1129. doi:10.1142/S0192415X11009445
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353(3):275–285. doi:10.1056/NEJMra035692
- Gülay H, Bora S, Kìlìçturgay S, Hamaloglu E, Göksel HA. Management of nipple discharge. J Am Coll Surg 1994; 178(5):471–474. pmid:8167884
- Murad TM, Contesso G, Mouriesse H. Nipple discharge from the breast. Ann Surg 1982; 195(3):259–264. pmid:6277258
- Sakorafas GH. Nipple discharge: current diagnostic and therapeutic approaches. Cancer Treat Rev 2001; 27(5):275–282. doi:10.1053/ctrv.2001.0234
- Ashfaq A, Senior D, Pockaj BA, et al. Validation study of a modern treatment algorithm for nipple discharge. Am J Surg 2014; 208(2):222–227. doi:10.1016/j.amjsurg.2013.12.035
- Chen CY, Sun LM, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the US. Cancer 2006; 107(7):1448–1458. doi:10.1002/cncr.22137
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE 3rd. Paget's disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187(2):171–177. pmid:9704964
- Hruska CB. Molecular breast imaging for screening in dense breasts: state of the art and future directions. AJR Am J Roentgenol 2017; 208(2):275–283. doi:10.2214/AJR.16.17131
- Melnikow J, Fenton JJ, Whitlock EP, et al. Supplemental screening for breast cancer in women with dense breasts: a systematic review for the US Preventive Services Task Force. Ann Intern Med 2016; 164(4):268–278. doi:10.7326/M15-1789
- American College of Radiology. Breast imaging reporting and data system (BI-RADS). Reston, VA: American College of Radiology; 2013.
- Brentnall AR, Harkness EF, Astley SM, et al. Mammographic density adds accuracy to both the Tyrer-Cuzick and Gail breast cancer risk models in a prospective UK screening cohort. Breast Cancer Res 2015; 17(1):147. doi:10.1186/s13058-015-0653-5
- Friedewald SM, Rafferty EA, Rose SL, et al. Breast cancer screening using tomosynthesis in combination with digital mammography. JAMA 2014; 311(24):2499–2507. doi:10.1001/jama.2014.6095
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA 2016; 315(16):1784–1786. doi:10.1001/jama.2016.1708
- Venkatesan A, Chu P, Kerlikowske K, Sickles EA, Smith-Bindman R. Positive predictive value of specific mammographic findings according to reader and patient variables. Radiology 2009; 250(3):648–657. doi:10.1148/radiol.2503080541
- Hartmann LC, Sellers TA, Frost MH, et al. Benign breast disease and the risk of breast cancer. N Engl J Med 2005; 353(3):229–237. doi:10.1056/NEJMoa044383
- Hartmann LC, Degnim AC, Santen RJ, DuPont WD, Ghosh K. Atypical hyperplasia of the breast—risk assessment and management options. N Engl J Med 2015; 372(1):78–89. doi:10.1056/NEJMsr1407164
- Neal L, Sandhu NP, Hieken TJ, et al. Diagnosis and management of benign, atypical, and indeterminate breast lesions detected on core needle biopsy. Mayo Clin Proc 2014; 89(4):536–547. doi:10.1016/j.mayocp.2014.02.004
- Nakhlis F, Ahmadiyeh N, Lester S, Raza S, Lotfi P, Golshan M. Papilloma on core biopsy: excision vs observation. Ann Surg Oncol 2015; 22(5):1479–1482. doi:10.1245/s10434-014-4091-x
- Degnim AC, Dupont WE, Radisky DC, et al. Extent of atypical hyperplasia stratifies breast cancer risk in 2 independent cohorts of women. Cancer 2016; 122(19):2971-2978. doi:10.1002/cncr.30153
- Sen LQ, Berg WA, Hooley RJ, Carter GJ, Desouki MM, Sumkin JH. Core breast biopsies showing lobular carcinoma in situ should be excised and surveillance is reasonable for atypical lobular hyperplasia. AJR Am J Roentgenol 2016; 207(5):1132–1145. doi:10.2214/AJR.15.15425
- Nakhlis F, Gilmore L, Gelman R, et al. Incidence of adjacent synchronous invasive carcinoma and/or ductal carcinoma in situ in patient with lobular neoplasia on core biopsy: results from a prospective multi-institutional registry (TBCRC 020). Ann Surg Oncol 2016; 23(3):722–728. doi:10.1245/s10434-015-4922-4
- Racz JM, Carter JM, Degnim AC. Lobular neoplasia and atypical ductal hyperplasia on core biopsy: current surgical management recommendations. Ann Surg Oncol 2017; 24(10):2848–2854. doi:10.1245/s10434-017-5978-0
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388. doi:10.1093/jnci/dji372
- Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011; 364(25):2381–2391. doi:10.1056/NEJMoa1103507
- Eberl MM, Phillips RL Jr, Lamberts H, Okkes I, Mahoney MC. Characterizing breast symptoms in family practice. Ann Fam Med 2008; 6(6):528–533. doi:10.1370/afm.905
- Harvey JA, Mahoney MC, Newell MS, et al. ACR appropriateness criteria palpable breast masses. J Am Coll Radiol 2013; 10(10):742–749.e3. doi:10.1016/j.jacr.2013.06.013
- Ha R, Kim H, Mango V, Wynn R, Comstock C. Ultrasonographic features and clinical implications of benign palpable breast lesions in young women. Ultrasonography 2015; 34(1):66–70. doi:10.14366/usg.14043
- Provencher L, Hogue JC, Desbiens C, et al. Is clinical breast examination important for breast cancer detection? Curr Oncol 2016; 23(4):e332–e339. doi:10.3747/co.23.2881
- Scurr J, Hedger W, Morris P, Brown N. The prevalence, severity, and impact of breast pain in the general population. Breast J 2014; 20(5):508–513. doi:10.1111/tbj.12305
- Leddy R, Irshad A, Zerwas E, et al. Role of breast ultrasound and mammography in evaluating patients presenting with focal breast pain in the absence of a palpable lump. Breast J 2013; 19(6):582–589. doi:10.1111/tbj.12178
- Noroozian M, Stein LF, Gaetke-Udager K, Helvie MA. Long-term clinical outcomes in women with breast pain in the absence of additional clinical findings: mammography remains indicated. Breast Cancer Res Treat 2015; 149(2):417–424. doi:10.1007/s10549-014-3257-3
- Gateley CA, Miers M, Mansel RE, Hughes LE. Drug treatments for mastalgia: 17 years experience in the Cardiff Mastalgia Clinic. J R Soc Med 1992; 85(1):12–15. pmid:1548647
- Fentiman IS, Caleffi M, Hamed H, Chaudary MA. Dosage and duration of tamoxifen treatment for mastalgia: a controlled trial. Br J Surg 1988; 75(9):845–846. pmid:3052691
- Oksa S, Luukkaala T, Mäenpää J. Toremifene for premenstrual mastalgia: a randomised, placebo-controlled crossover study. BJOG 2006; 113(6):713–718. doi:10.1111/j.1471-0528.2006.00943.x
- Mirghafourvand M, Mohammad-Alizadeh-Charandabi S, Ahmadpour P, Javadzadeh Y. Effects of Vitex agnus and flaxseed on cyclic mastalgia: a randomized controlled trial. Complement Ther Med 2016; 24:90–95. doi:10.1016/j.ctim.2015.12.009
- Shobeiri F, Oshvandi K, Nazari M. Clinical effectiveness of vitamin E and vitamin B6 for improving pain severity in cyclic mastalgia. Iran J Nurs Midwifery Res 2015; 20(6):723–727. doi:10.4103/1735-9066.170003
- Thicke LA, Hazelton JK, Bauer BA, et al. Acupuncture for treatment of noncyclic breast pain: a pilot study. Am J Chin Med 2011; 39(6):1117–1129. doi:10.1142/S0192415X11009445
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353(3):275–285. doi:10.1056/NEJMra035692
- Gülay H, Bora S, Kìlìçturgay S, Hamaloglu E, Göksel HA. Management of nipple discharge. J Am Coll Surg 1994; 178(5):471–474. pmid:8167884
- Murad TM, Contesso G, Mouriesse H. Nipple discharge from the breast. Ann Surg 1982; 195(3):259–264. pmid:6277258
- Sakorafas GH. Nipple discharge: current diagnostic and therapeutic approaches. Cancer Treat Rev 2001; 27(5):275–282. doi:10.1053/ctrv.2001.0234
- Ashfaq A, Senior D, Pockaj BA, et al. Validation study of a modern treatment algorithm for nipple discharge. Am J Surg 2014; 208(2):222–227. doi:10.1016/j.amjsurg.2013.12.035
- Chen CY, Sun LM, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the US. Cancer 2006; 107(7):1448–1458. doi:10.1002/cncr.22137
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE 3rd. Paget's disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187(2):171–177. pmid:9704964
- Hruska CB. Molecular breast imaging for screening in dense breasts: state of the art and future directions. AJR Am J Roentgenol 2017; 208(2):275–283. doi:10.2214/AJR.16.17131
- Melnikow J, Fenton JJ, Whitlock EP, et al. Supplemental screening for breast cancer in women with dense breasts: a systematic review for the US Preventive Services Task Force. Ann Intern Med 2016; 164(4):268–278. doi:10.7326/M15-1789
- American College of Radiology. Breast imaging reporting and data system (BI-RADS). Reston, VA: American College of Radiology; 2013.
- Brentnall AR, Harkness EF, Astley SM, et al. Mammographic density adds accuracy to both the Tyrer-Cuzick and Gail breast cancer risk models in a prospective UK screening cohort. Breast Cancer Res 2015; 17(1):147. doi:10.1186/s13058-015-0653-5
- Friedewald SM, Rafferty EA, Rose SL, et al. Breast cancer screening using tomosynthesis in combination with digital mammography. JAMA 2014; 311(24):2499–2507. doi:10.1001/jama.2014.6095
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA 2016; 315(16):1784–1786. doi:10.1001/jama.2016.1708
- Venkatesan A, Chu P, Kerlikowske K, Sickles EA, Smith-Bindman R. Positive predictive value of specific mammographic findings according to reader and patient variables. Radiology 2009; 250(3):648–657. doi:10.1148/radiol.2503080541
- Hartmann LC, Sellers TA, Frost MH, et al. Benign breast disease and the risk of breast cancer. N Engl J Med 2005; 353(3):229–237. doi:10.1056/NEJMoa044383
- Hartmann LC, Degnim AC, Santen RJ, DuPont WD, Ghosh K. Atypical hyperplasia of the breast—risk assessment and management options. N Engl J Med 2015; 372(1):78–89. doi:10.1056/NEJMsr1407164
- Neal L, Sandhu NP, Hieken TJ, et al. Diagnosis and management of benign, atypical, and indeterminate breast lesions detected on core needle biopsy. Mayo Clin Proc 2014; 89(4):536–547. doi:10.1016/j.mayocp.2014.02.004
- Nakhlis F, Ahmadiyeh N, Lester S, Raza S, Lotfi P, Golshan M. Papilloma on core biopsy: excision vs observation. Ann Surg Oncol 2015; 22(5):1479–1482. doi:10.1245/s10434-014-4091-x
- Degnim AC, Dupont WE, Radisky DC, et al. Extent of atypical hyperplasia stratifies breast cancer risk in 2 independent cohorts of women. Cancer 2016; 122(19):2971-2978. doi:10.1002/cncr.30153
- Sen LQ, Berg WA, Hooley RJ, Carter GJ, Desouki MM, Sumkin JH. Core breast biopsies showing lobular carcinoma in situ should be excised and surveillance is reasonable for atypical lobular hyperplasia. AJR Am J Roentgenol 2016; 207(5):1132–1145. doi:10.2214/AJR.15.15425
- Nakhlis F, Gilmore L, Gelman R, et al. Incidence of adjacent synchronous invasive carcinoma and/or ductal carcinoma in situ in patient with lobular neoplasia on core biopsy: results from a prospective multi-institutional registry (TBCRC 020). Ann Surg Oncol 2016; 23(3):722–728. doi:10.1245/s10434-015-4922-4
- Racz JM, Carter JM, Degnim AC. Lobular neoplasia and atypical ductal hyperplasia on core biopsy: current surgical management recommendations. Ann Surg Oncol 2017; 24(10):2848–2854. doi:10.1245/s10434-017-5978-0
- Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371–1388. doi:10.1093/jnci/dji372
- Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011; 364(25):2381–2391. doi:10.1056/NEJMoa1103507
KEY POINTS
- The two most common breast symptoms are lumps and pain.
- Most breast problems are not caused by cancer.
- Evaluation of any breast problem begins with a focused history followed by a breast examination and, when necessary, imaging.
- If the results of the breast examination and imaging suggest a benign cause, no further follow-up is necessary.
- If there is discordance between imaging and breast examination results, or if there is a high clinical suspicion of cancer, then consider serial follow-up examinations at short intervals, referral to a breast surgeon for excision, or both.
Who needs to carry an epinephrine autoinjector?
Anaphylaxis is potentially fatal but can be prevented if the trigger is identified and avoided, and death can be avoided if episodes are treated promptly.
A consensus definition of anaphylaxis has been difficult to achieve, with slight variations among international guidelines. The World Allergy Organization classifies anaphylaxis as immunologic, nonimmunologic, or idiopathic.1 The National Institute of Allergy and Infectious Diseases and the Food Allergy and Anaphylaxis Network highlight clinical symptoms and criteria.2 The International Consensus on Food Allergy describes reactions as being immunoglobulin E (IgE)-mediated, cell-mediated, or a combination of the 2 mechanisms.3
Despite the subtle differences in these definitions, all 3 international organizations have a common recommendation for anaphylaxis: once it is diagnosed, epinephrine is the treatment of choice.
EPINEPHRINE IS THE TREATMENT OF CHOICE FOR ANAPHYLAXIS
Anaphylaxis commonly results from exposure to foods, medications, and Hymenoptera venom.4 Avoiding triggers is key in preventing anaphylaxis but is not always possible.
Although epinephrine is the cornerstone of the emergency treatment of anaphylaxis, many patients instead receive antihistamines and corticosteroids as initial therapy. Some take these medications on their own, and some receive them in emergency departments and outpatient clinics.5
Diphenhydramine, a histamine 1 receptor antagonist, is often used as a first-line medication. But diphenhydramine has a slow onset of action, taking 80 minutes after an oral dose to suppress a histamine-induced cutaneous flare by 50%, and taking 52 minutes with intramuscular administration.6 Corticosteroids also have a slow onset of action. These drugs cannot prevent death in anaphylaxis, a condition in which the median time to respiratory or cardiac arrest is 30 minutes after ingestion of food, 15 minutes after envenomation, and 5 minutes after iatrogenic reactions.7
Combination therapy with diphenhydramine and a histamine 2 receptor antagonist (eg, cimetidine, famotidine) is also commonly used,8 but this combination offers no advantage in terms of onset of action, and a Cochrane review could find no definitive evidence for or against the use of histamine 2 receptor antagonists.9
Because of their slow onset of action, all of these should be second-line therapies, given after epinephrine. Epinephrine is the first line of treatment because it has a maximal pharmacokinetic effect (time to maximal peak serum level) within 10 minutes of intramuscular injection into the thigh.10,11
In addition, epinephrine acts on numerous receptors to antagonize the multiple pathologic effects of the mediators released during an anaphylactic episode. In contrast, antihistamines block only 1 mediator, while mediators other than histamine can be responsible for severe events and deaths.12,13
It is crucial that epinephrine be given immediately, as delay has been associated with fatalities.14 In addition, guidelines recommend repeating epinephrine dosing after 5 to 15 minutes if the response to the first dose is suboptimal.1,2 From 16% to 36% of patients may need a second dose.15–18 Therefore, many physicians recommend that patients at risk of anaphylaxis keep not 1 but 2 epinephrine autoinjectors on hand at all times, and so say the US guidelines for the management of anaphylaxis.19
WHO SHOULD CARRY AN EPINEPHRINE AUTOINJECTOR?
All published guidelines recommend epinephrine as the drug of choice for anaphylaxis. And an epinephrine autoinjector is indicated for anyone who has experienced an anaphylactic event or is at risk of one, and these patients should carry it with them at all times. Such individuals include those with food allergy or Hymenoptera hypersensitivity.
Food allergy
The foods that most often cause anaphylaxis are peanuts, tree nuts, fish, shellfish, milk, and eggs, but any food can cause a reaction.
The prevalence of food allergy has increased over time, and treatments are limited. Some food desensitization protocols look promising but are still in the research stages. The best treatment at this time is to avoid the offending food, but there are accidental exposures.
Hymenoptera hypersensitivity
Patients who have had anaphylaxis after being stung by insects such as bees, wasps, yellow-faced hornets, white-faced hornets, yellow jackets, and fire ants should be evaluated by an allergist. Skin testing and serum IgE testing helps properly diagnose Hymenoptera hypersensitivity.
Once the diagnosis is confirmed, venom immunotherapy should be considered. Some patients choose only to carry an epinephrine autoinjector and to avoid these insects as much as possible. However, most patients also choose to receive venom immunotherapy, because 80% to 90% of those who receive this treatment for 3 to 5 years do not have a systemic reaction if they are stung again.20
Regardless of whether they choose to undergo immunotherapy, sensitive patients should always carry an epinephrine autoinjector. This is also the case after treatment ends, since the therapy is not 100% effective.
PATIENTS FOR WHOM THE NEED MAY BE LESS CLEAR
In other patients who may be at increased risk, the mandate for an epinephrine autoinjector is less clear, and the decision to carry one is determined on an individual basis. Such individuals are those receiving allergen immunotherapy, with large local reactions to insect stings, with oral allergy syndrome, with mastocytosis, and with drug allergy. In these cases, the benefit vs the burden of carrying an autoinjector should be discussed with the patient.
Patients on allergen immunotherapy
National guidelines recommend that all patients who receive allergen immunotherapy be monitored in the clinic under a physician’s supervision for 30 minutes after the injection. Fortunately, life-threatening reactions occurring after 30 minutes are rare. But delayed systemic reactions can occur and may account for up to 50% of such events.21
Therefore, many physicians consider it prudent for patients on immunotherapy to carry an epinephrine autoinjector, but there is no consensus. A survey22 found that 13.5% of allergists did not prescribe the autoinjector for patients on immunotherapy, while 33.3% prescribed it for all their patients on immunotherapy, and the rest prescribed based on risk.
Since there are no national guidelines on epinephrine autoinjectors for patients on immunotherapy, the decision should be based on the patient’s risks and comorbidities and informed by discussion between the individual patient and his or her allergist.
Patients with large local reactions to insect stings
From 5% to 10% of patients who have large local reactions to insect stings are at risk of systemic reactions.20
Patients with oral allergy syndrome
Oral allergy syndrome, also known as pollen-food allergy, causes itching and mild swelling of the mouth, lips, and throat after eating fresh fruits and vegetables. The prevalence ranges from 2% to 10% of patients with allergies.23
A survey of allergists found that 20% of patients with oral allergy syndrome had experienced systemic symptoms.24 The survey also showed that the decision to prescribe an epinephrine autoinjector to these patients was highly variable. Only about 30% of allergists recommend epinephrine autoinjectors to patients with oral allergy syndrome, while most believe that the decision should be based on the individual’s symptoms and risk.
More research is needed in the area of food allergy. Because data are limited, there are no national guidelines on whether these patients should carry an epinephrine autoinjector. We agree with the Joint Task Force on Practice Parameters14 recommendation that the decision be made on an individual basis following discussion between the patient and physician.
Patients with mastocytosis
Patients with mastocytosis and a history of anaphylaxis are at increased risk for systemic reactions to Hymenoptera venom.
Patients with medication allergy
Once medication allergy has been diagnosed, avoidance is usually effective, obviating the need for an epinephrine autoinjector, although the physician has the option of prescribing one.
CAUTIONS, NOT CONTRAINDICATIONS
Physicians may be reluctant to prescribe an epinephrine autoinjector because of the risk of an adverse reaction in patients with hypertension, coronary artery disease, or arrhythmias, and in elderly patients taking multiple drugs, especially drugs that can interact with epinephrine. Nevertheless, there is no absolute contraindication to the use of epinephrine in anaphylaxis.
In patients with atherosclerosis and cardiovascular disease
Epinephrine increases vasoconstriction, heart rate, and cardiac force of contraction. These effects are beneficial during anaphylaxis, but in rare cases patients have experienced myocardial infarction and acute coronary syndrome after receiving intravenous epinephrine.25 These incidents have naturally prompted reluctance to prescribe it in susceptible patients with coronary disease during anaphylaxis.
Yet epinephrine may not be solely to blame for these adverse responses. Mast cells are abundant in the heart, and their release of mediators can also result in adverse cardiac manifestations, including myocardial infarction.26
Conversely, some drugs used to treat cardiovascular disease can worsen anaphylaxis.
Beta-blockers can cause bronchospasm and decrease cardiac contractility. They can also blunt the pharmacologic effects of epinephrine. There is concern that epinephrine may produce dangerous elevations of blood pressure in patients taking beta-blockers by unopposed alpha-adrenergic stimulation and reflex vagotonic effects.27 And there is evidence that beta-blockers may increase the risk and severity of reactions. One study reported that patients taking beta-blockers are more than 8 times more likely to be hospitalized due to anaphylactoid reaction with bronchospasm.28
Beta-blockers and, to a lesser extent, angiotensin-converting enzyme inhibitors have been shown to increase the risk of anaphylaxis in the emergency department.29,30 However, some investigators have not found beta-blockers to be a risk factor. A study evaluating anaphylactoid reactions from contrast media found no statistically significant higher risk in patients taking beta-blockers.31 Similarly, a study of 3,178 patients on beta-blockers receiving venom immunotherapy or allergen immunotherapy found no increase in the frequency of systemic reactions.32 Nevertheless, overall, more studies support the hypothesis that beta-blockers may be an additional risk factor in anaphylaxis.33
Thus, clinicians treating patients with cardiovascular disease and anaphylaxis face a dilemma. Although there is concern in this population, epinephrine should not be withheld in patients with cardiovascular disease who are experiencing an anaphylactic event.33 If epinephrine is not administered, the patient could die.
Elderly patients on multiple medications
Older patients are also at risk of anaphylaxis. But clinicians are reluctant to treat older patients with epinephrine because of concerns about adverse effects.
Epinephrine dispensing rates vary substantially in different age groups: 1.44% for patients under age 17, 0.9% for those ages 17 to 64, and 0.32% for those age 65 or older.34 A Canadian study of 492 patients with anaphylaxis in the emergency department showed that those over age 50 received epinephrine less often than younger patients (36.1% vs 60.5%).35 Cardiovascular complications were more frequent in the older group, occurring in 4 (9.1%) of the 44 older patients who received epinephrine compared with 1 (0.4%) of the 225 younger patients who received it. On the other hand, the rate of adverse effects from subcutaneous epinephrine was no different in older asthma patients compared with younger patients.36
Many older patients take multiple medications, raising concern about adverse effects. Commonly prescribed medications in the elderly can affect the actions of epinephrine. Monoamine oxidase inhibitors retard the catabolism of epinephrine. Tricyclic antidepressants may decrease the reuptake of catecholamines by neurons and thus interfere with the degradation of epinephrine. Digoxin has a narrow therapeutic window and can potentially increase the risk of arrhythmias when given with epinephrine.
Although the clinician must be cautious in treating older patients who have comorbidities, these are not sufficient to withhold prescribing an epinephrine autoinjector to elderly patients at risk of anaphylaxis.
INJECTOR OPTIONS
Epinephrine autoinjectors come preloaded for prompt delivery of the drug. They are intended primarily for use by patients themselves in unsupervised settings in suspected anaphylaxis. Simplicity of use and safety must be considered in such a setting so that patients can use the device correctly and are not incorrectly dosed.
Several models are commercially available, with different ergonomic designs and sizes. EpiPen, the first one marketed in the United States, was introduced in 1987. One device (Auvi-Q) contains an audio chip that gives step-by-step instructions at the time of use. It is hoped that this device will reduce errors in usage during this stressful time for patients and caregivers.
In the United States, epinephrine autoinjectors contain either 0.15 or 0.30 mg of the drug, but some clinicians believe this may not be enough. The UK Resuscitation Council recommends 0.50 mg for patients over age 12,37 and an epinephrine autoinjector with that dose is available in Europe.
Subcutaneous vs intramuscular delivery
The package insert for some epinephrine autoinjectors says the injector can be used to treat anaphylaxis by both subcutaneous and intramuscular administration. However, the routes are not equivalent.
The goal in anaphylaxis is to quickly achieve high tissue and plasma epinephrine concentrations, and studies have found that injection into the vastus lateralis muscle, but not the deltoid muscle, results in faster time to peak plasma concentration: 8 minutes for injection in the vastus lateralis muscle and 34 minutes for subcutaneous delivery.10,11 In addition, injection in the vastus lateralis muscle results in a higher peak plasma concentration than the subcutaneous or deltoid route. Based on these data, intramuscular injection into the vastus lateralis muscle in the thigh appears to be the preferred route of administration of epinephrine.
Obese patients may need a longer needle
Research on the original autoinjector was conducted by the US military, which wanted a rapidly effective and easy-to-use antidote for battlefield exposure to poison gas. The resulting device had 2 separate spring-loaded syringes, 1 containing pralidoxime chloride and the other atropine sulfate. To enable its use through the thick fabric of a chemical warfare suit, the needles were 2.2 cm long.
The first commercial autoinjector to contain epinephrine was made by Survival Technology (Bethesda, MD) in the mid-1970s. The manufacturer considered a 2.2-cm needle to be too long, and the first commercially available epinephrine autoinjector, EpiPen, had a 1.43-cm needle for adult use.
Since then, needle lengths have ranged from 1.17 to 2.5 cm to accommodate different skin-to-muscle depths, with shorter needles for children and longer needles for obese adults.38
However, the prevalence of obesity is high and continues to rise.39 Obesity raises concern that the needles in epinephrine autoinjectors may be too short for the preferred intramuscular delivery, resulting in subcutaneous deposition.
A study that used computed tomography of the thigh found that 1 (2%) of 50 men and 21 (42%) of 50 women studied had a subcutaneous tissue depth greater than 1.43 cm, the needle length in EpiPen. These were not anaphylaxis patients, but the findings suggest that many patients—especially women—may be getting subcutaneous instead of intramuscular delivery with this device.40
Another study that used ultrasonography showed that the 1.43-cm EpiPen needle was too short for 36 (31%) of 116 adults.41 Women were 6.4 times more likely than men to encounter this problem. Other risk factors include higher body mass index, short height, and thicker thighs.
Emerade, an injector with a 2.5-cm needle, is available in some European countries. A longer needle may be helpful in some cases. but we do not yet have enough data to determine the optimal needle length.
Conversely, some children may need shorter needles and may in fact be at risk of having the needle penetrate bone.42 The US Food and Drug Administration recently approved a shorter needle for an epinephrine autoinjector (Auvi-Q) to be used in children weighing 7.5 kg to 15 kg.
BARRIERS TO USING EPINEPHRINE AUTOINJECTORS
Many patients do not use their epinephrine autoinjector in times of anaphylaxis or do not have one with them. Common reasons cited by respondents in a survey43 of 1,385 patients included the following:
They took an oral antihistamine instead (38%).
They never received a prescription for an epinephrine autoinjector (28%).
They thought their symptoms were mild and would resolve with time (13%).
They were afraid (6%). There are reports of accidental injection, typically into fingers, hands, and thumbs. Fortunately, most accidental injections do not require a hand surgeon evaluation or surgery.44 Conservative therapy and monitoring of the injection site are sufficient in most cases.
They could not afford an epinephrine autoinjector (1%).43 Mylan Pharmaceuticals infamously increased the price of its EpiPen to more than $600 for a package of 2 pens. Generic devices are available in the United States but are still too expensive for some patients and are cumbersome to carry.
However, even expensive epinephrine autoinjectors may be cost-effective. Epidemiologic studies have found that patients who did not use an epinephrine autoinjector incurred a higher burden of cost due to emergency department visits and inpatient hospitalizations.45
As a do-it-yourself option, some resourceful patients are obtaining autoinjectors intended for insulin injection, replacing the needle, and filling the injector with epinephrine, at a cost of about $30. (The manufacturer does not endorse this off-label use of their device—www.owenmumford.com/us/patients/if-you-need-to-inject.) Least costly of all is to prescribe multidose vials of epinephrine and regular syringes and teach patients and their caregivers how to draw up the proper dose and give themselves an injection—in essence going back to what was done before 1987.
It was past its expiration date (2%).43 Failure to refill the prescription is common. A California Kaiser Permanente study46 showed that only 46% of patients refilled their epinephrine autoinjector prescription at least once, and the refill rate decreased over time: 43% at 1 to 2 year follow-up, 35% at 3 to 4 years, and 30% at 5 years or longer. Based on these data, it is imperative to educate patients regarding the importance of replacing the epinephrine autoinjector when the old one expires.
NEED FOR PATIENT EDUCATION
Even though prompt treatment with epinephrine decreases fatalities, it continues to be underused in the community. In addition, it is often prescribed without adequate training in its use and appropriate emphasis on the need to keep the device on hand at all times and to replace it in a timely manner if it is used or has expired. Physicians need to educate patients on how to avoid triggers and how to recognize symptoms of anaphylaxis whenever they prescribe an epinephrine autoinjector.
- Simons FE, Ardusso LR, Bilò MB, et al. International consensus on (ICON) anaphylaxis. World Allergy Organ J 2014; 7(1):9. doi:10.1186/1939-4551-7-9
- NIAID-Sponsored Expert Panel; Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol 2010; 126(6 suppl):S1–S58. doi:10.1016/j.jaci.2010.10.007
- Burks AW, Tang M, Sicherer S, et al. ICON: food allergy. J Allergy Clin Immunol 2012; 129(4):906–920. doi:10.1016/j.jaci.2012.02.001
- Lieberman P, Carmago CA Jr, Bohlke K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma, and Immunology. Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol 2006; 97(5):596–602. doi:10.1016/S1081-1206(10)61086-1
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis—a statement of the World Allergy Organization. World Allergy Organ J 2008; 1(suppl 7):S18–S26. doi:10.1097/WOX.0b013e31817c9338
- Jones DH, Romero FA, Casale TB. Time-dependent inhibition of histamine-induced cutaneous responses by oral and intramuscular diphenhydramine and oral fexofenadine. Ann Allergy Asthma Immunol 2008; 100(5):452–456. doi:10.1016/S1081-1206(10)60470-X
- Pumphrey RS. Lessons for management of anaphylaxis from a study of fatal reactions. Clin Exp Allerg 2000; 30(8):1144–1150. pmid:10931122
- Runge JW, Martinez JC, Caravati EM, Williamson SG, Hartsell SC. Histamine antagonists in the treatment of acute allergic reactions. Ann Emerg Med 1992; 21:237–242. pmid:1536481
- Sheikh A, Simons FE, Barbour V, Worth A. Adrenaline auto-injectors for the treatment of anaphylaxis with and without cardiovascular collapse in the community. Cochrane Database Syst Rev 2012; (8):CD008935. doi:10.1002/14651858.CD008935.pub2
- Simons FE, Gu X, Simons KJ. Epinephrine absorption in adults: intramuscular versus subcutaneous injection. J Allergy Clin Immunol 2001; 108(5):871–873. doi:10.1067/mai.2001.119409
- Simons FE, Roberts JR, Gu X, Simons KJ. Epinephrine absorption in children with a history of anaphylaxis. J Allergy Clin Immunol 1998; 101(1 pt 1):33–37. doi:10.1016/S0091-6749(98)70190-3
- Vadas P. The platelet-activating factor pathway in food allergy and anaphylaxis. Ann Allergy Asthma Immunol 2016; 117(5):455–457. doi:10.1016/j.anai.2016.05.003
- Stone SF, Brown SG. Mediators released during human anaphylaxis. Curr Allergy Asthma Rep 2012; 12(1):33–41. doi:10.1007/s11882-011-0231-6
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126(3):477–480.e1–e42. doi:10.1016/j.jaci.2010.06.022
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis. A statement of the World Allergy Organization. Allergy 2008; 63(8):1061–1070. doi:10.1111/j.1398-9995.2008.01733.x
- Oren E, Banderji A, Clark S, Camargo CA Jr. Food-induced anaphylaxis and repeated epinephrine treatments. Ann Allergy Asthma Immunol 2007; 99(5):429–432. doi:10.1016/S1081-1206(10)60568-6
- Uguz A, Lack G, Pumphrey R, et al. Allergic reactions in the community: a questionnaire survey of members of the anaphylaxis campaign. Clin Exp Allergy 2005; 35(6):746–750. doi:10.1111/j.1365-2222.2005.02257.x
- Kelso JM. A second dose of epinephrine for anaphylaxis: how often needed and how to carry. J Allergy Clin Immunol 2006; 117(2):464–465. doi:10.1016/j.jaci.2005.11.015
- Lieberman P, Nicklas RA, Randolph C, et al. Anaphylaxis—a practice parameter update 2015. Ann Allergy Asthma Immunol 2015; 115(5):341–384. doi:10.1016/j.anai.2015.07.019
- Golden BK, Demain J, Freeman T, et al. Stinging insect hypersensitivity: a practice parameter update 2016. Ann Allergy Asthma Immunol 2017; 118(1):28–54. doi:10.1016/j.anai.2016.10.031
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55. doi:10.1016/j.jaci.2010.09.034
- Gupta P, Gerrish PK, Silverman B, Schneider A. Current practices among allergists on writing self-injectable epinephrine prescriptions for immunotherapy patients. J Allergy Clin Immunol 2012; 129(2):571–572.e1-e2. doi:10.1016/j.jaci.2011.09.033
- Ortolani C, Pastorello EA, Farioli L, et al. IgE-mediated allergy from vegetable allergens. Ann Allergy 1993; 71:470–476. pmid: 8250353
- Ma S, Shcherer SH, Nowak-Wegrzyn A. A survey on the management of pollen food allergy syndrome in allergy practices. J Allergy Clin Immunol 2003;112:784–788. doi:10.1016/S0091-6749(03)02008-6
- Shaver KJ, Adams C, Weiss SJ. Acute myocardial infarction after administration of low dose intravenous epinephrine for anaphylaxis. CJEM 2006; 8(4):289–294. pmid:17324313
- Triggiani M, Patella V, Staiano RI, Granata F, Marone G. Allergy and the cardiovascular system. Clin Exp Immunol 2008; 153(suppl 1):7–11. doi:10.1111/j.1365-2249.2008.03714.x
- Gilman AG, Rail TW, Nies AS, Taylor P, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York, NY: Pergamon Press; 1990.
- Lang DM, Alpern MB, Visintainer PF, Smith ST. Increased risk for anaphylactoid reaction from contrast media in patients on beta-adrenergic blockers or with asthma. Ann Intern Med 1991; 115(14):270–276. pmid:1677239
- Nassiri M, Babina M, Dölle S, Edenharter G, Ruëff F, Worm M. Ramipril and metoprolol intake aggravate human and murine anaphylaxis: evidence for direct mast cell priming. J Allergy Clin Immunol 2015; 135(2):491–499. doi:10.1016/j.jaci.2014.09.004
- Lee S, Hess EP, Nestler DM, et al. Antihypertensive medication use is associated with increased organ system involvement and hospitalization in emergency department patients with anaphylaxis. J Allergy Clin Immunol 2013; 131(4):1103–1108. doi:10.1016/j.jaci.2013.01.011
- Greenberger PA, Meyers SN, Kramer BL, Kramer BL. Effects of beta-adrenergic and calcium antagonists on the development of anaphylactoid reactions from radiographic contrast media during cardiac angiography. J Allergy Clin Immunol 1987; 80(5):698–702. pmid:2890682
- Hepner MJ, Ownby DR, Anderson JA, Rowe MS, Sears-Ewald D, Brown EB. Risk of systemic reactions in patients taking beta-blocker drugs receiving allergen immunotherapy injections. J Allergy Clin Immunol 1990; 86(3 pt 1):407–411. pmid:1976666
- Lieberman P, Simons FE. Anaphylaxis and cardiovascular disease: therapeutic dilemmas. Clin Exp Allergy 2015; 45(8):1288–1295. doi:10.1111/cea.12520
- Simons FE, Peterson S, Black CD. Epinephrine dispensing patterns for an out-of-hospital population: a novel approach to studying the epidemiology of anaphylaxis. J Allergy Clin Immunol 2002; 110(4):647–651. pmid:12373275
- Kawano T, Scheuermeyer FX, Stenstrom R, Rowe BH, Grafstein E, Grunau B. Epinephrine use in older patients with anaphylaxis: clinical outcomes and cardiovascular complications. Resuscitation 2017; 112:53–58. doi:10.1016/j.resuscitation.2016.12.020
- Cydulka R, Davison R, Grammer L, Parker M, Mathews J 4th. The use of epinephrine in the treatment of older adult asthmatics. Ann Emerg Med 1988; 17(4):322–326. pmid:3354935
- Soar J, Pumphrey R, Cant A, et al; Working Group of the Resuscitation Council (UK). Emergency treatment of anaphylactic reactions—guidelines for healthcare providers. Resuscitation 2008; 77(2):157–169. doi:10.1016/j.resuscitation.2008.02.001
- Dreborg S, Wen X, Kim L, et al. Do epinephrine auto-injectors have an unsuitable needle length in children and adolescents at risk for anaphylaxis from food allergy? Allergy Asthma Clin Immunol 2016; 12:11. doi:10.1186/s13223-016-0110-8
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014; 311(8):806–814. doi:10.1001/jama.2014.732
- Song TT, Nelson MR, Chang JH, Engler RJ, Chowdhury BA. Adequacy of the epinephrine autoinjector needle length in delivering epinephrine to the intramuscular tissues. Ann Allergy Asthma Immunol 2005; 94(5):539–542. doi:10.1016/S1081-1206(10)61130-1
- Bhalla MC, Gable BD, Frey JA, Reichenbach MR, Wilber ST. Predictors of epinephrine autoinjector needle length inadequacy. Am J Emerg Med 2013; 31(12):1671–1676. doi:10.1016/j.ajem.2013.09.001
- Kim H, Dinakar C, McInnis P, et al. Inadequacy of current pediatric epinephrine autoinjector needle length for use in infants and toddlers. Ann Allergy Asthma Immunol 2017; 118(6):719–725.e1. doi:10.1016/j.anai.2017.03.017
- Simons FE, Clark S, Camargo CA Jr. Anaphylaxis in the community: learning from the survivors. J Allergy Clin Immunol 2009; 124(2):301–306. doi:10.1016/j.jaci.2009.03.050
- Muck AE, Bebarta VS, Borys DJ, Morgan DL. Six years of epinephrine digital injections: absence of significant local or systemic effects. Ann Emerg Med 2010; 56(3):270–274. doi:10.1016/j.annemergmed.2010.02.019
- Fleming JT, Clark S, Camargo CA Jr, Rudders SA. Early treatment of food-induced anaphylaxis with epinephrine is associated with a lower risk of hospitalization. J Allergy Clin Immunol Pract 2015; 3(1):57–62. doi:10.1016/j.jaip.2014.07.004
- Kaplan MS, Jung SY, Chiang ML. Epinephrine autoinjector refill history in an HMO. Curr Allergy Asthma Rep 2011; 11(1):65–70. doi:10.1007/s11882-010-0155-6
Anaphylaxis is potentially fatal but can be prevented if the trigger is identified and avoided, and death can be avoided if episodes are treated promptly.
A consensus definition of anaphylaxis has been difficult to achieve, with slight variations among international guidelines. The World Allergy Organization classifies anaphylaxis as immunologic, nonimmunologic, or idiopathic.1 The National Institute of Allergy and Infectious Diseases and the Food Allergy and Anaphylaxis Network highlight clinical symptoms and criteria.2 The International Consensus on Food Allergy describes reactions as being immunoglobulin E (IgE)-mediated, cell-mediated, or a combination of the 2 mechanisms.3
Despite the subtle differences in these definitions, all 3 international organizations have a common recommendation for anaphylaxis: once it is diagnosed, epinephrine is the treatment of choice.
EPINEPHRINE IS THE TREATMENT OF CHOICE FOR ANAPHYLAXIS
Anaphylaxis commonly results from exposure to foods, medications, and Hymenoptera venom.4 Avoiding triggers is key in preventing anaphylaxis but is not always possible.
Although epinephrine is the cornerstone of the emergency treatment of anaphylaxis, many patients instead receive antihistamines and corticosteroids as initial therapy. Some take these medications on their own, and some receive them in emergency departments and outpatient clinics.5
Diphenhydramine, a histamine 1 receptor antagonist, is often used as a first-line medication. But diphenhydramine has a slow onset of action, taking 80 minutes after an oral dose to suppress a histamine-induced cutaneous flare by 50%, and taking 52 minutes with intramuscular administration.6 Corticosteroids also have a slow onset of action. These drugs cannot prevent death in anaphylaxis, a condition in which the median time to respiratory or cardiac arrest is 30 minutes after ingestion of food, 15 minutes after envenomation, and 5 minutes after iatrogenic reactions.7
Combination therapy with diphenhydramine and a histamine 2 receptor antagonist (eg, cimetidine, famotidine) is also commonly used,8 but this combination offers no advantage in terms of onset of action, and a Cochrane review could find no definitive evidence for or against the use of histamine 2 receptor antagonists.9
Because of their slow onset of action, all of these should be second-line therapies, given after epinephrine. Epinephrine is the first line of treatment because it has a maximal pharmacokinetic effect (time to maximal peak serum level) within 10 minutes of intramuscular injection into the thigh.10,11
In addition, epinephrine acts on numerous receptors to antagonize the multiple pathologic effects of the mediators released during an anaphylactic episode. In contrast, antihistamines block only 1 mediator, while mediators other than histamine can be responsible for severe events and deaths.12,13
It is crucial that epinephrine be given immediately, as delay has been associated with fatalities.14 In addition, guidelines recommend repeating epinephrine dosing after 5 to 15 minutes if the response to the first dose is suboptimal.1,2 From 16% to 36% of patients may need a second dose.15–18 Therefore, many physicians recommend that patients at risk of anaphylaxis keep not 1 but 2 epinephrine autoinjectors on hand at all times, and so say the US guidelines for the management of anaphylaxis.19
WHO SHOULD CARRY AN EPINEPHRINE AUTOINJECTOR?
All published guidelines recommend epinephrine as the drug of choice for anaphylaxis. And an epinephrine autoinjector is indicated for anyone who has experienced an anaphylactic event or is at risk of one, and these patients should carry it with them at all times. Such individuals include those with food allergy or Hymenoptera hypersensitivity.
Food allergy
The foods that most often cause anaphylaxis are peanuts, tree nuts, fish, shellfish, milk, and eggs, but any food can cause a reaction.
The prevalence of food allergy has increased over time, and treatments are limited. Some food desensitization protocols look promising but are still in the research stages. The best treatment at this time is to avoid the offending food, but there are accidental exposures.
Hymenoptera hypersensitivity
Patients who have had anaphylaxis after being stung by insects such as bees, wasps, yellow-faced hornets, white-faced hornets, yellow jackets, and fire ants should be evaluated by an allergist. Skin testing and serum IgE testing helps properly diagnose Hymenoptera hypersensitivity.
Once the diagnosis is confirmed, venom immunotherapy should be considered. Some patients choose only to carry an epinephrine autoinjector and to avoid these insects as much as possible. However, most patients also choose to receive venom immunotherapy, because 80% to 90% of those who receive this treatment for 3 to 5 years do not have a systemic reaction if they are stung again.20
Regardless of whether they choose to undergo immunotherapy, sensitive patients should always carry an epinephrine autoinjector. This is also the case after treatment ends, since the therapy is not 100% effective.
PATIENTS FOR WHOM THE NEED MAY BE LESS CLEAR
In other patients who may be at increased risk, the mandate for an epinephrine autoinjector is less clear, and the decision to carry one is determined on an individual basis. Such individuals are those receiving allergen immunotherapy, with large local reactions to insect stings, with oral allergy syndrome, with mastocytosis, and with drug allergy. In these cases, the benefit vs the burden of carrying an autoinjector should be discussed with the patient.
Patients on allergen immunotherapy
National guidelines recommend that all patients who receive allergen immunotherapy be monitored in the clinic under a physician’s supervision for 30 minutes after the injection. Fortunately, life-threatening reactions occurring after 30 minutes are rare. But delayed systemic reactions can occur and may account for up to 50% of such events.21
Therefore, many physicians consider it prudent for patients on immunotherapy to carry an epinephrine autoinjector, but there is no consensus. A survey22 found that 13.5% of allergists did not prescribe the autoinjector for patients on immunotherapy, while 33.3% prescribed it for all their patients on immunotherapy, and the rest prescribed based on risk.
Since there are no national guidelines on epinephrine autoinjectors for patients on immunotherapy, the decision should be based on the patient’s risks and comorbidities and informed by discussion between the individual patient and his or her allergist.
Patients with large local reactions to insect stings
From 5% to 10% of patients who have large local reactions to insect stings are at risk of systemic reactions.20
Patients with oral allergy syndrome
Oral allergy syndrome, also known as pollen-food allergy, causes itching and mild swelling of the mouth, lips, and throat after eating fresh fruits and vegetables. The prevalence ranges from 2% to 10% of patients with allergies.23
A survey of allergists found that 20% of patients with oral allergy syndrome had experienced systemic symptoms.24 The survey also showed that the decision to prescribe an epinephrine autoinjector to these patients was highly variable. Only about 30% of allergists recommend epinephrine autoinjectors to patients with oral allergy syndrome, while most believe that the decision should be based on the individual’s symptoms and risk.
More research is needed in the area of food allergy. Because data are limited, there are no national guidelines on whether these patients should carry an epinephrine autoinjector. We agree with the Joint Task Force on Practice Parameters14 recommendation that the decision be made on an individual basis following discussion between the patient and physician.
Patients with mastocytosis
Patients with mastocytosis and a history of anaphylaxis are at increased risk for systemic reactions to Hymenoptera venom.
Patients with medication allergy
Once medication allergy has been diagnosed, avoidance is usually effective, obviating the need for an epinephrine autoinjector, although the physician has the option of prescribing one.
CAUTIONS, NOT CONTRAINDICATIONS
Physicians may be reluctant to prescribe an epinephrine autoinjector because of the risk of an adverse reaction in patients with hypertension, coronary artery disease, or arrhythmias, and in elderly patients taking multiple drugs, especially drugs that can interact with epinephrine. Nevertheless, there is no absolute contraindication to the use of epinephrine in anaphylaxis.
In patients with atherosclerosis and cardiovascular disease
Epinephrine increases vasoconstriction, heart rate, and cardiac force of contraction. These effects are beneficial during anaphylaxis, but in rare cases patients have experienced myocardial infarction and acute coronary syndrome after receiving intravenous epinephrine.25 These incidents have naturally prompted reluctance to prescribe it in susceptible patients with coronary disease during anaphylaxis.
Yet epinephrine may not be solely to blame for these adverse responses. Mast cells are abundant in the heart, and their release of mediators can also result in adverse cardiac manifestations, including myocardial infarction.26
Conversely, some drugs used to treat cardiovascular disease can worsen anaphylaxis.
Beta-blockers can cause bronchospasm and decrease cardiac contractility. They can also blunt the pharmacologic effects of epinephrine. There is concern that epinephrine may produce dangerous elevations of blood pressure in patients taking beta-blockers by unopposed alpha-adrenergic stimulation and reflex vagotonic effects.27 And there is evidence that beta-blockers may increase the risk and severity of reactions. One study reported that patients taking beta-blockers are more than 8 times more likely to be hospitalized due to anaphylactoid reaction with bronchospasm.28
Beta-blockers and, to a lesser extent, angiotensin-converting enzyme inhibitors have been shown to increase the risk of anaphylaxis in the emergency department.29,30 However, some investigators have not found beta-blockers to be a risk factor. A study evaluating anaphylactoid reactions from contrast media found no statistically significant higher risk in patients taking beta-blockers.31 Similarly, a study of 3,178 patients on beta-blockers receiving venom immunotherapy or allergen immunotherapy found no increase in the frequency of systemic reactions.32 Nevertheless, overall, more studies support the hypothesis that beta-blockers may be an additional risk factor in anaphylaxis.33
Thus, clinicians treating patients with cardiovascular disease and anaphylaxis face a dilemma. Although there is concern in this population, epinephrine should not be withheld in patients with cardiovascular disease who are experiencing an anaphylactic event.33 If epinephrine is not administered, the patient could die.
Elderly patients on multiple medications
Older patients are also at risk of anaphylaxis. But clinicians are reluctant to treat older patients with epinephrine because of concerns about adverse effects.
Epinephrine dispensing rates vary substantially in different age groups: 1.44% for patients under age 17, 0.9% for those ages 17 to 64, and 0.32% for those age 65 or older.34 A Canadian study of 492 patients with anaphylaxis in the emergency department showed that those over age 50 received epinephrine less often than younger patients (36.1% vs 60.5%).35 Cardiovascular complications were more frequent in the older group, occurring in 4 (9.1%) of the 44 older patients who received epinephrine compared with 1 (0.4%) of the 225 younger patients who received it. On the other hand, the rate of adverse effects from subcutaneous epinephrine was no different in older asthma patients compared with younger patients.36
Many older patients take multiple medications, raising concern about adverse effects. Commonly prescribed medications in the elderly can affect the actions of epinephrine. Monoamine oxidase inhibitors retard the catabolism of epinephrine. Tricyclic antidepressants may decrease the reuptake of catecholamines by neurons and thus interfere with the degradation of epinephrine. Digoxin has a narrow therapeutic window and can potentially increase the risk of arrhythmias when given with epinephrine.
Although the clinician must be cautious in treating older patients who have comorbidities, these are not sufficient to withhold prescribing an epinephrine autoinjector to elderly patients at risk of anaphylaxis.
INJECTOR OPTIONS
Epinephrine autoinjectors come preloaded for prompt delivery of the drug. They are intended primarily for use by patients themselves in unsupervised settings in suspected anaphylaxis. Simplicity of use and safety must be considered in such a setting so that patients can use the device correctly and are not incorrectly dosed.
Several models are commercially available, with different ergonomic designs and sizes. EpiPen, the first one marketed in the United States, was introduced in 1987. One device (Auvi-Q) contains an audio chip that gives step-by-step instructions at the time of use. It is hoped that this device will reduce errors in usage during this stressful time for patients and caregivers.
In the United States, epinephrine autoinjectors contain either 0.15 or 0.30 mg of the drug, but some clinicians believe this may not be enough. The UK Resuscitation Council recommends 0.50 mg for patients over age 12,37 and an epinephrine autoinjector with that dose is available in Europe.
Subcutaneous vs intramuscular delivery
The package insert for some epinephrine autoinjectors says the injector can be used to treat anaphylaxis by both subcutaneous and intramuscular administration. However, the routes are not equivalent.
The goal in anaphylaxis is to quickly achieve high tissue and plasma epinephrine concentrations, and studies have found that injection into the vastus lateralis muscle, but not the deltoid muscle, results in faster time to peak plasma concentration: 8 minutes for injection in the vastus lateralis muscle and 34 minutes for subcutaneous delivery.10,11 In addition, injection in the vastus lateralis muscle results in a higher peak plasma concentration than the subcutaneous or deltoid route. Based on these data, intramuscular injection into the vastus lateralis muscle in the thigh appears to be the preferred route of administration of epinephrine.
Obese patients may need a longer needle
Research on the original autoinjector was conducted by the US military, which wanted a rapidly effective and easy-to-use antidote for battlefield exposure to poison gas. The resulting device had 2 separate spring-loaded syringes, 1 containing pralidoxime chloride and the other atropine sulfate. To enable its use through the thick fabric of a chemical warfare suit, the needles were 2.2 cm long.
The first commercial autoinjector to contain epinephrine was made by Survival Technology (Bethesda, MD) in the mid-1970s. The manufacturer considered a 2.2-cm needle to be too long, and the first commercially available epinephrine autoinjector, EpiPen, had a 1.43-cm needle for adult use.
Since then, needle lengths have ranged from 1.17 to 2.5 cm to accommodate different skin-to-muscle depths, with shorter needles for children and longer needles for obese adults.38
However, the prevalence of obesity is high and continues to rise.39 Obesity raises concern that the needles in epinephrine autoinjectors may be too short for the preferred intramuscular delivery, resulting in subcutaneous deposition.
A study that used computed tomography of the thigh found that 1 (2%) of 50 men and 21 (42%) of 50 women studied had a subcutaneous tissue depth greater than 1.43 cm, the needle length in EpiPen. These were not anaphylaxis patients, but the findings suggest that many patients—especially women—may be getting subcutaneous instead of intramuscular delivery with this device.40
Another study that used ultrasonography showed that the 1.43-cm EpiPen needle was too short for 36 (31%) of 116 adults.41 Women were 6.4 times more likely than men to encounter this problem. Other risk factors include higher body mass index, short height, and thicker thighs.
Emerade, an injector with a 2.5-cm needle, is available in some European countries. A longer needle may be helpful in some cases. but we do not yet have enough data to determine the optimal needle length.
Conversely, some children may need shorter needles and may in fact be at risk of having the needle penetrate bone.42 The US Food and Drug Administration recently approved a shorter needle for an epinephrine autoinjector (Auvi-Q) to be used in children weighing 7.5 kg to 15 kg.
BARRIERS TO USING EPINEPHRINE AUTOINJECTORS
Many patients do not use their epinephrine autoinjector in times of anaphylaxis or do not have one with them. Common reasons cited by respondents in a survey43 of 1,385 patients included the following:
They took an oral antihistamine instead (38%).
They never received a prescription for an epinephrine autoinjector (28%).
They thought their symptoms were mild and would resolve with time (13%).
They were afraid (6%). There are reports of accidental injection, typically into fingers, hands, and thumbs. Fortunately, most accidental injections do not require a hand surgeon evaluation or surgery.44 Conservative therapy and monitoring of the injection site are sufficient in most cases.
They could not afford an epinephrine autoinjector (1%).43 Mylan Pharmaceuticals infamously increased the price of its EpiPen to more than $600 for a package of 2 pens. Generic devices are available in the United States but are still too expensive for some patients and are cumbersome to carry.
However, even expensive epinephrine autoinjectors may be cost-effective. Epidemiologic studies have found that patients who did not use an epinephrine autoinjector incurred a higher burden of cost due to emergency department visits and inpatient hospitalizations.45
As a do-it-yourself option, some resourceful patients are obtaining autoinjectors intended for insulin injection, replacing the needle, and filling the injector with epinephrine, at a cost of about $30. (The manufacturer does not endorse this off-label use of their device—www.owenmumford.com/us/patients/if-you-need-to-inject.) Least costly of all is to prescribe multidose vials of epinephrine and regular syringes and teach patients and their caregivers how to draw up the proper dose and give themselves an injection—in essence going back to what was done before 1987.
It was past its expiration date (2%).43 Failure to refill the prescription is common. A California Kaiser Permanente study46 showed that only 46% of patients refilled their epinephrine autoinjector prescription at least once, and the refill rate decreased over time: 43% at 1 to 2 year follow-up, 35% at 3 to 4 years, and 30% at 5 years or longer. Based on these data, it is imperative to educate patients regarding the importance of replacing the epinephrine autoinjector when the old one expires.
NEED FOR PATIENT EDUCATION
Even though prompt treatment with epinephrine decreases fatalities, it continues to be underused in the community. In addition, it is often prescribed without adequate training in its use and appropriate emphasis on the need to keep the device on hand at all times and to replace it in a timely manner if it is used or has expired. Physicians need to educate patients on how to avoid triggers and how to recognize symptoms of anaphylaxis whenever they prescribe an epinephrine autoinjector.
Anaphylaxis is potentially fatal but can be prevented if the trigger is identified and avoided, and death can be avoided if episodes are treated promptly.
A consensus definition of anaphylaxis has been difficult to achieve, with slight variations among international guidelines. The World Allergy Organization classifies anaphylaxis as immunologic, nonimmunologic, or idiopathic.1 The National Institute of Allergy and Infectious Diseases and the Food Allergy and Anaphylaxis Network highlight clinical symptoms and criteria.2 The International Consensus on Food Allergy describes reactions as being immunoglobulin E (IgE)-mediated, cell-mediated, or a combination of the 2 mechanisms.3
Despite the subtle differences in these definitions, all 3 international organizations have a common recommendation for anaphylaxis: once it is diagnosed, epinephrine is the treatment of choice.
EPINEPHRINE IS THE TREATMENT OF CHOICE FOR ANAPHYLAXIS
Anaphylaxis commonly results from exposure to foods, medications, and Hymenoptera venom.4 Avoiding triggers is key in preventing anaphylaxis but is not always possible.
Although epinephrine is the cornerstone of the emergency treatment of anaphylaxis, many patients instead receive antihistamines and corticosteroids as initial therapy. Some take these medications on their own, and some receive them in emergency departments and outpatient clinics.5
Diphenhydramine, a histamine 1 receptor antagonist, is often used as a first-line medication. But diphenhydramine has a slow onset of action, taking 80 minutes after an oral dose to suppress a histamine-induced cutaneous flare by 50%, and taking 52 minutes with intramuscular administration.6 Corticosteroids also have a slow onset of action. These drugs cannot prevent death in anaphylaxis, a condition in which the median time to respiratory or cardiac arrest is 30 minutes after ingestion of food, 15 minutes after envenomation, and 5 minutes after iatrogenic reactions.7
Combination therapy with diphenhydramine and a histamine 2 receptor antagonist (eg, cimetidine, famotidine) is also commonly used,8 but this combination offers no advantage in terms of onset of action, and a Cochrane review could find no definitive evidence for or against the use of histamine 2 receptor antagonists.9
Because of their slow onset of action, all of these should be second-line therapies, given after epinephrine. Epinephrine is the first line of treatment because it has a maximal pharmacokinetic effect (time to maximal peak serum level) within 10 minutes of intramuscular injection into the thigh.10,11
In addition, epinephrine acts on numerous receptors to antagonize the multiple pathologic effects of the mediators released during an anaphylactic episode. In contrast, antihistamines block only 1 mediator, while mediators other than histamine can be responsible for severe events and deaths.12,13
It is crucial that epinephrine be given immediately, as delay has been associated with fatalities.14 In addition, guidelines recommend repeating epinephrine dosing after 5 to 15 minutes if the response to the first dose is suboptimal.1,2 From 16% to 36% of patients may need a second dose.15–18 Therefore, many physicians recommend that patients at risk of anaphylaxis keep not 1 but 2 epinephrine autoinjectors on hand at all times, and so say the US guidelines for the management of anaphylaxis.19
WHO SHOULD CARRY AN EPINEPHRINE AUTOINJECTOR?
All published guidelines recommend epinephrine as the drug of choice for anaphylaxis. And an epinephrine autoinjector is indicated for anyone who has experienced an anaphylactic event or is at risk of one, and these patients should carry it with them at all times. Such individuals include those with food allergy or Hymenoptera hypersensitivity.
Food allergy
The foods that most often cause anaphylaxis are peanuts, tree nuts, fish, shellfish, milk, and eggs, but any food can cause a reaction.
The prevalence of food allergy has increased over time, and treatments are limited. Some food desensitization protocols look promising but are still in the research stages. The best treatment at this time is to avoid the offending food, but there are accidental exposures.
Hymenoptera hypersensitivity
Patients who have had anaphylaxis after being stung by insects such as bees, wasps, yellow-faced hornets, white-faced hornets, yellow jackets, and fire ants should be evaluated by an allergist. Skin testing and serum IgE testing helps properly diagnose Hymenoptera hypersensitivity.
Once the diagnosis is confirmed, venom immunotherapy should be considered. Some patients choose only to carry an epinephrine autoinjector and to avoid these insects as much as possible. However, most patients also choose to receive venom immunotherapy, because 80% to 90% of those who receive this treatment for 3 to 5 years do not have a systemic reaction if they are stung again.20
Regardless of whether they choose to undergo immunotherapy, sensitive patients should always carry an epinephrine autoinjector. This is also the case after treatment ends, since the therapy is not 100% effective.
PATIENTS FOR WHOM THE NEED MAY BE LESS CLEAR
In other patients who may be at increased risk, the mandate for an epinephrine autoinjector is less clear, and the decision to carry one is determined on an individual basis. Such individuals are those receiving allergen immunotherapy, with large local reactions to insect stings, with oral allergy syndrome, with mastocytosis, and with drug allergy. In these cases, the benefit vs the burden of carrying an autoinjector should be discussed with the patient.
Patients on allergen immunotherapy
National guidelines recommend that all patients who receive allergen immunotherapy be monitored in the clinic under a physician’s supervision for 30 minutes after the injection. Fortunately, life-threatening reactions occurring after 30 minutes are rare. But delayed systemic reactions can occur and may account for up to 50% of such events.21
Therefore, many physicians consider it prudent for patients on immunotherapy to carry an epinephrine autoinjector, but there is no consensus. A survey22 found that 13.5% of allergists did not prescribe the autoinjector for patients on immunotherapy, while 33.3% prescribed it for all their patients on immunotherapy, and the rest prescribed based on risk.
Since there are no national guidelines on epinephrine autoinjectors for patients on immunotherapy, the decision should be based on the patient’s risks and comorbidities and informed by discussion between the individual patient and his or her allergist.
Patients with large local reactions to insect stings
From 5% to 10% of patients who have large local reactions to insect stings are at risk of systemic reactions.20
Patients with oral allergy syndrome
Oral allergy syndrome, also known as pollen-food allergy, causes itching and mild swelling of the mouth, lips, and throat after eating fresh fruits and vegetables. The prevalence ranges from 2% to 10% of patients with allergies.23
A survey of allergists found that 20% of patients with oral allergy syndrome had experienced systemic symptoms.24 The survey also showed that the decision to prescribe an epinephrine autoinjector to these patients was highly variable. Only about 30% of allergists recommend epinephrine autoinjectors to patients with oral allergy syndrome, while most believe that the decision should be based on the individual’s symptoms and risk.
More research is needed in the area of food allergy. Because data are limited, there are no national guidelines on whether these patients should carry an epinephrine autoinjector. We agree with the Joint Task Force on Practice Parameters14 recommendation that the decision be made on an individual basis following discussion between the patient and physician.
Patients with mastocytosis
Patients with mastocytosis and a history of anaphylaxis are at increased risk for systemic reactions to Hymenoptera venom.
Patients with medication allergy
Once medication allergy has been diagnosed, avoidance is usually effective, obviating the need for an epinephrine autoinjector, although the physician has the option of prescribing one.
CAUTIONS, NOT CONTRAINDICATIONS
Physicians may be reluctant to prescribe an epinephrine autoinjector because of the risk of an adverse reaction in patients with hypertension, coronary artery disease, or arrhythmias, and in elderly patients taking multiple drugs, especially drugs that can interact with epinephrine. Nevertheless, there is no absolute contraindication to the use of epinephrine in anaphylaxis.
In patients with atherosclerosis and cardiovascular disease
Epinephrine increases vasoconstriction, heart rate, and cardiac force of contraction. These effects are beneficial during anaphylaxis, but in rare cases patients have experienced myocardial infarction and acute coronary syndrome after receiving intravenous epinephrine.25 These incidents have naturally prompted reluctance to prescribe it in susceptible patients with coronary disease during anaphylaxis.
Yet epinephrine may not be solely to blame for these adverse responses. Mast cells are abundant in the heart, and their release of mediators can also result in adverse cardiac manifestations, including myocardial infarction.26
Conversely, some drugs used to treat cardiovascular disease can worsen anaphylaxis.
Beta-blockers can cause bronchospasm and decrease cardiac contractility. They can also blunt the pharmacologic effects of epinephrine. There is concern that epinephrine may produce dangerous elevations of blood pressure in patients taking beta-blockers by unopposed alpha-adrenergic stimulation and reflex vagotonic effects.27 And there is evidence that beta-blockers may increase the risk and severity of reactions. One study reported that patients taking beta-blockers are more than 8 times more likely to be hospitalized due to anaphylactoid reaction with bronchospasm.28
Beta-blockers and, to a lesser extent, angiotensin-converting enzyme inhibitors have been shown to increase the risk of anaphylaxis in the emergency department.29,30 However, some investigators have not found beta-blockers to be a risk factor. A study evaluating anaphylactoid reactions from contrast media found no statistically significant higher risk in patients taking beta-blockers.31 Similarly, a study of 3,178 patients on beta-blockers receiving venom immunotherapy or allergen immunotherapy found no increase in the frequency of systemic reactions.32 Nevertheless, overall, more studies support the hypothesis that beta-blockers may be an additional risk factor in anaphylaxis.33
Thus, clinicians treating patients with cardiovascular disease and anaphylaxis face a dilemma. Although there is concern in this population, epinephrine should not be withheld in patients with cardiovascular disease who are experiencing an anaphylactic event.33 If epinephrine is not administered, the patient could die.
Elderly patients on multiple medications
Older patients are also at risk of anaphylaxis. But clinicians are reluctant to treat older patients with epinephrine because of concerns about adverse effects.
Epinephrine dispensing rates vary substantially in different age groups: 1.44% for patients under age 17, 0.9% for those ages 17 to 64, and 0.32% for those age 65 or older.34 A Canadian study of 492 patients with anaphylaxis in the emergency department showed that those over age 50 received epinephrine less often than younger patients (36.1% vs 60.5%).35 Cardiovascular complications were more frequent in the older group, occurring in 4 (9.1%) of the 44 older patients who received epinephrine compared with 1 (0.4%) of the 225 younger patients who received it. On the other hand, the rate of adverse effects from subcutaneous epinephrine was no different in older asthma patients compared with younger patients.36
Many older patients take multiple medications, raising concern about adverse effects. Commonly prescribed medications in the elderly can affect the actions of epinephrine. Monoamine oxidase inhibitors retard the catabolism of epinephrine. Tricyclic antidepressants may decrease the reuptake of catecholamines by neurons and thus interfere with the degradation of epinephrine. Digoxin has a narrow therapeutic window and can potentially increase the risk of arrhythmias when given with epinephrine.
Although the clinician must be cautious in treating older patients who have comorbidities, these are not sufficient to withhold prescribing an epinephrine autoinjector to elderly patients at risk of anaphylaxis.
INJECTOR OPTIONS
Epinephrine autoinjectors come preloaded for prompt delivery of the drug. They are intended primarily for use by patients themselves in unsupervised settings in suspected anaphylaxis. Simplicity of use and safety must be considered in such a setting so that patients can use the device correctly and are not incorrectly dosed.
Several models are commercially available, with different ergonomic designs and sizes. EpiPen, the first one marketed in the United States, was introduced in 1987. One device (Auvi-Q) contains an audio chip that gives step-by-step instructions at the time of use. It is hoped that this device will reduce errors in usage during this stressful time for patients and caregivers.
In the United States, epinephrine autoinjectors contain either 0.15 or 0.30 mg of the drug, but some clinicians believe this may not be enough. The UK Resuscitation Council recommends 0.50 mg for patients over age 12,37 and an epinephrine autoinjector with that dose is available in Europe.
Subcutaneous vs intramuscular delivery
The package insert for some epinephrine autoinjectors says the injector can be used to treat anaphylaxis by both subcutaneous and intramuscular administration. However, the routes are not equivalent.
The goal in anaphylaxis is to quickly achieve high tissue and plasma epinephrine concentrations, and studies have found that injection into the vastus lateralis muscle, but not the deltoid muscle, results in faster time to peak plasma concentration: 8 minutes for injection in the vastus lateralis muscle and 34 minutes for subcutaneous delivery.10,11 In addition, injection in the vastus lateralis muscle results in a higher peak plasma concentration than the subcutaneous or deltoid route. Based on these data, intramuscular injection into the vastus lateralis muscle in the thigh appears to be the preferred route of administration of epinephrine.
Obese patients may need a longer needle
Research on the original autoinjector was conducted by the US military, which wanted a rapidly effective and easy-to-use antidote for battlefield exposure to poison gas. The resulting device had 2 separate spring-loaded syringes, 1 containing pralidoxime chloride and the other atropine sulfate. To enable its use through the thick fabric of a chemical warfare suit, the needles were 2.2 cm long.
The first commercial autoinjector to contain epinephrine was made by Survival Technology (Bethesda, MD) in the mid-1970s. The manufacturer considered a 2.2-cm needle to be too long, and the first commercially available epinephrine autoinjector, EpiPen, had a 1.43-cm needle for adult use.
Since then, needle lengths have ranged from 1.17 to 2.5 cm to accommodate different skin-to-muscle depths, with shorter needles for children and longer needles for obese adults.38
However, the prevalence of obesity is high and continues to rise.39 Obesity raises concern that the needles in epinephrine autoinjectors may be too short for the preferred intramuscular delivery, resulting in subcutaneous deposition.
A study that used computed tomography of the thigh found that 1 (2%) of 50 men and 21 (42%) of 50 women studied had a subcutaneous tissue depth greater than 1.43 cm, the needle length in EpiPen. These were not anaphylaxis patients, but the findings suggest that many patients—especially women—may be getting subcutaneous instead of intramuscular delivery with this device.40
Another study that used ultrasonography showed that the 1.43-cm EpiPen needle was too short for 36 (31%) of 116 adults.41 Women were 6.4 times more likely than men to encounter this problem. Other risk factors include higher body mass index, short height, and thicker thighs.
Emerade, an injector with a 2.5-cm needle, is available in some European countries. A longer needle may be helpful in some cases. but we do not yet have enough data to determine the optimal needle length.
Conversely, some children may need shorter needles and may in fact be at risk of having the needle penetrate bone.42 The US Food and Drug Administration recently approved a shorter needle for an epinephrine autoinjector (Auvi-Q) to be used in children weighing 7.5 kg to 15 kg.
BARRIERS TO USING EPINEPHRINE AUTOINJECTORS
Many patients do not use their epinephrine autoinjector in times of anaphylaxis or do not have one with them. Common reasons cited by respondents in a survey43 of 1,385 patients included the following:
They took an oral antihistamine instead (38%).
They never received a prescription for an epinephrine autoinjector (28%).
They thought their symptoms were mild and would resolve with time (13%).
They were afraid (6%). There are reports of accidental injection, typically into fingers, hands, and thumbs. Fortunately, most accidental injections do not require a hand surgeon evaluation or surgery.44 Conservative therapy and monitoring of the injection site are sufficient in most cases.
They could not afford an epinephrine autoinjector (1%).43 Mylan Pharmaceuticals infamously increased the price of its EpiPen to more than $600 for a package of 2 pens. Generic devices are available in the United States but are still too expensive for some patients and are cumbersome to carry.
However, even expensive epinephrine autoinjectors may be cost-effective. Epidemiologic studies have found that patients who did not use an epinephrine autoinjector incurred a higher burden of cost due to emergency department visits and inpatient hospitalizations.45
As a do-it-yourself option, some resourceful patients are obtaining autoinjectors intended for insulin injection, replacing the needle, and filling the injector with epinephrine, at a cost of about $30. (The manufacturer does not endorse this off-label use of their device—www.owenmumford.com/us/patients/if-you-need-to-inject.) Least costly of all is to prescribe multidose vials of epinephrine and regular syringes and teach patients and their caregivers how to draw up the proper dose and give themselves an injection—in essence going back to what was done before 1987.
It was past its expiration date (2%).43 Failure to refill the prescription is common. A California Kaiser Permanente study46 showed that only 46% of patients refilled their epinephrine autoinjector prescription at least once, and the refill rate decreased over time: 43% at 1 to 2 year follow-up, 35% at 3 to 4 years, and 30% at 5 years or longer. Based on these data, it is imperative to educate patients regarding the importance of replacing the epinephrine autoinjector when the old one expires.
NEED FOR PATIENT EDUCATION
Even though prompt treatment with epinephrine decreases fatalities, it continues to be underused in the community. In addition, it is often prescribed without adequate training in its use and appropriate emphasis on the need to keep the device on hand at all times and to replace it in a timely manner if it is used or has expired. Physicians need to educate patients on how to avoid triggers and how to recognize symptoms of anaphylaxis whenever they prescribe an epinephrine autoinjector.
- Simons FE, Ardusso LR, Bilò MB, et al. International consensus on (ICON) anaphylaxis. World Allergy Organ J 2014; 7(1):9. doi:10.1186/1939-4551-7-9
- NIAID-Sponsored Expert Panel; Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol 2010; 126(6 suppl):S1–S58. doi:10.1016/j.jaci.2010.10.007
- Burks AW, Tang M, Sicherer S, et al. ICON: food allergy. J Allergy Clin Immunol 2012; 129(4):906–920. doi:10.1016/j.jaci.2012.02.001
- Lieberman P, Carmago CA Jr, Bohlke K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma, and Immunology. Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol 2006; 97(5):596–602. doi:10.1016/S1081-1206(10)61086-1
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis—a statement of the World Allergy Organization. World Allergy Organ J 2008; 1(suppl 7):S18–S26. doi:10.1097/WOX.0b013e31817c9338
- Jones DH, Romero FA, Casale TB. Time-dependent inhibition of histamine-induced cutaneous responses by oral and intramuscular diphenhydramine and oral fexofenadine. Ann Allergy Asthma Immunol 2008; 100(5):452–456. doi:10.1016/S1081-1206(10)60470-X
- Pumphrey RS. Lessons for management of anaphylaxis from a study of fatal reactions. Clin Exp Allerg 2000; 30(8):1144–1150. pmid:10931122
- Runge JW, Martinez JC, Caravati EM, Williamson SG, Hartsell SC. Histamine antagonists in the treatment of acute allergic reactions. Ann Emerg Med 1992; 21:237–242. pmid:1536481
- Sheikh A, Simons FE, Barbour V, Worth A. Adrenaline auto-injectors for the treatment of anaphylaxis with and without cardiovascular collapse in the community. Cochrane Database Syst Rev 2012; (8):CD008935. doi:10.1002/14651858.CD008935.pub2
- Simons FE, Gu X, Simons KJ. Epinephrine absorption in adults: intramuscular versus subcutaneous injection. J Allergy Clin Immunol 2001; 108(5):871–873. doi:10.1067/mai.2001.119409
- Simons FE, Roberts JR, Gu X, Simons KJ. Epinephrine absorption in children with a history of anaphylaxis. J Allergy Clin Immunol 1998; 101(1 pt 1):33–37. doi:10.1016/S0091-6749(98)70190-3
- Vadas P. The platelet-activating factor pathway in food allergy and anaphylaxis. Ann Allergy Asthma Immunol 2016; 117(5):455–457. doi:10.1016/j.anai.2016.05.003
- Stone SF, Brown SG. Mediators released during human anaphylaxis. Curr Allergy Asthma Rep 2012; 12(1):33–41. doi:10.1007/s11882-011-0231-6
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126(3):477–480.e1–e42. doi:10.1016/j.jaci.2010.06.022
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis. A statement of the World Allergy Organization. Allergy 2008; 63(8):1061–1070. doi:10.1111/j.1398-9995.2008.01733.x
- Oren E, Banderji A, Clark S, Camargo CA Jr. Food-induced anaphylaxis and repeated epinephrine treatments. Ann Allergy Asthma Immunol 2007; 99(5):429–432. doi:10.1016/S1081-1206(10)60568-6
- Uguz A, Lack G, Pumphrey R, et al. Allergic reactions in the community: a questionnaire survey of members of the anaphylaxis campaign. Clin Exp Allergy 2005; 35(6):746–750. doi:10.1111/j.1365-2222.2005.02257.x
- Kelso JM. A second dose of epinephrine for anaphylaxis: how often needed and how to carry. J Allergy Clin Immunol 2006; 117(2):464–465. doi:10.1016/j.jaci.2005.11.015
- Lieberman P, Nicklas RA, Randolph C, et al. Anaphylaxis—a practice parameter update 2015. Ann Allergy Asthma Immunol 2015; 115(5):341–384. doi:10.1016/j.anai.2015.07.019
- Golden BK, Demain J, Freeman T, et al. Stinging insect hypersensitivity: a practice parameter update 2016. Ann Allergy Asthma Immunol 2017; 118(1):28–54. doi:10.1016/j.anai.2016.10.031
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55. doi:10.1016/j.jaci.2010.09.034
- Gupta P, Gerrish PK, Silverman B, Schneider A. Current practices among allergists on writing self-injectable epinephrine prescriptions for immunotherapy patients. J Allergy Clin Immunol 2012; 129(2):571–572.e1-e2. doi:10.1016/j.jaci.2011.09.033
- Ortolani C, Pastorello EA, Farioli L, et al. IgE-mediated allergy from vegetable allergens. Ann Allergy 1993; 71:470–476. pmid: 8250353
- Ma S, Shcherer SH, Nowak-Wegrzyn A. A survey on the management of pollen food allergy syndrome in allergy practices. J Allergy Clin Immunol 2003;112:784–788. doi:10.1016/S0091-6749(03)02008-6
- Shaver KJ, Adams C, Weiss SJ. Acute myocardial infarction after administration of low dose intravenous epinephrine for anaphylaxis. CJEM 2006; 8(4):289–294. pmid:17324313
- Triggiani M, Patella V, Staiano RI, Granata F, Marone G. Allergy and the cardiovascular system. Clin Exp Immunol 2008; 153(suppl 1):7–11. doi:10.1111/j.1365-2249.2008.03714.x
- Gilman AG, Rail TW, Nies AS, Taylor P, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York, NY: Pergamon Press; 1990.
- Lang DM, Alpern MB, Visintainer PF, Smith ST. Increased risk for anaphylactoid reaction from contrast media in patients on beta-adrenergic blockers or with asthma. Ann Intern Med 1991; 115(14):270–276. pmid:1677239
- Nassiri M, Babina M, Dölle S, Edenharter G, Ruëff F, Worm M. Ramipril and metoprolol intake aggravate human and murine anaphylaxis: evidence for direct mast cell priming. J Allergy Clin Immunol 2015; 135(2):491–499. doi:10.1016/j.jaci.2014.09.004
- Lee S, Hess EP, Nestler DM, et al. Antihypertensive medication use is associated with increased organ system involvement and hospitalization in emergency department patients with anaphylaxis. J Allergy Clin Immunol 2013; 131(4):1103–1108. doi:10.1016/j.jaci.2013.01.011
- Greenberger PA, Meyers SN, Kramer BL, Kramer BL. Effects of beta-adrenergic and calcium antagonists on the development of anaphylactoid reactions from radiographic contrast media during cardiac angiography. J Allergy Clin Immunol 1987; 80(5):698–702. pmid:2890682
- Hepner MJ, Ownby DR, Anderson JA, Rowe MS, Sears-Ewald D, Brown EB. Risk of systemic reactions in patients taking beta-blocker drugs receiving allergen immunotherapy injections. J Allergy Clin Immunol 1990; 86(3 pt 1):407–411. pmid:1976666
- Lieberman P, Simons FE. Anaphylaxis and cardiovascular disease: therapeutic dilemmas. Clin Exp Allergy 2015; 45(8):1288–1295. doi:10.1111/cea.12520
- Simons FE, Peterson S, Black CD. Epinephrine dispensing patterns for an out-of-hospital population: a novel approach to studying the epidemiology of anaphylaxis. J Allergy Clin Immunol 2002; 110(4):647–651. pmid:12373275
- Kawano T, Scheuermeyer FX, Stenstrom R, Rowe BH, Grafstein E, Grunau B. Epinephrine use in older patients with anaphylaxis: clinical outcomes and cardiovascular complications. Resuscitation 2017; 112:53–58. doi:10.1016/j.resuscitation.2016.12.020
- Cydulka R, Davison R, Grammer L, Parker M, Mathews J 4th. The use of epinephrine in the treatment of older adult asthmatics. Ann Emerg Med 1988; 17(4):322–326. pmid:3354935
- Soar J, Pumphrey R, Cant A, et al; Working Group of the Resuscitation Council (UK). Emergency treatment of anaphylactic reactions—guidelines for healthcare providers. Resuscitation 2008; 77(2):157–169. doi:10.1016/j.resuscitation.2008.02.001
- Dreborg S, Wen X, Kim L, et al. Do epinephrine auto-injectors have an unsuitable needle length in children and adolescents at risk for anaphylaxis from food allergy? Allergy Asthma Clin Immunol 2016; 12:11. doi:10.1186/s13223-016-0110-8
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014; 311(8):806–814. doi:10.1001/jama.2014.732
- Song TT, Nelson MR, Chang JH, Engler RJ, Chowdhury BA. Adequacy of the epinephrine autoinjector needle length in delivering epinephrine to the intramuscular tissues. Ann Allergy Asthma Immunol 2005; 94(5):539–542. doi:10.1016/S1081-1206(10)61130-1
- Bhalla MC, Gable BD, Frey JA, Reichenbach MR, Wilber ST. Predictors of epinephrine autoinjector needle length inadequacy. Am J Emerg Med 2013; 31(12):1671–1676. doi:10.1016/j.ajem.2013.09.001
- Kim H, Dinakar C, McInnis P, et al. Inadequacy of current pediatric epinephrine autoinjector needle length for use in infants and toddlers. Ann Allergy Asthma Immunol 2017; 118(6):719–725.e1. doi:10.1016/j.anai.2017.03.017
- Simons FE, Clark S, Camargo CA Jr. Anaphylaxis in the community: learning from the survivors. J Allergy Clin Immunol 2009; 124(2):301–306. doi:10.1016/j.jaci.2009.03.050
- Muck AE, Bebarta VS, Borys DJ, Morgan DL. Six years of epinephrine digital injections: absence of significant local or systemic effects. Ann Emerg Med 2010; 56(3):270–274. doi:10.1016/j.annemergmed.2010.02.019
- Fleming JT, Clark S, Camargo CA Jr, Rudders SA. Early treatment of food-induced anaphylaxis with epinephrine is associated with a lower risk of hospitalization. J Allergy Clin Immunol Pract 2015; 3(1):57–62. doi:10.1016/j.jaip.2014.07.004
- Kaplan MS, Jung SY, Chiang ML. Epinephrine autoinjector refill history in an HMO. Curr Allergy Asthma Rep 2011; 11(1):65–70. doi:10.1007/s11882-010-0155-6
- Simons FE, Ardusso LR, Bilò MB, et al. International consensus on (ICON) anaphylaxis. World Allergy Organ J 2014; 7(1):9. doi:10.1186/1939-4551-7-9
- NIAID-Sponsored Expert Panel; Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol 2010; 126(6 suppl):S1–S58. doi:10.1016/j.jaci.2010.10.007
- Burks AW, Tang M, Sicherer S, et al. ICON: food allergy. J Allergy Clin Immunol 2012; 129(4):906–920. doi:10.1016/j.jaci.2012.02.001
- Lieberman P, Carmago CA Jr, Bohlke K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma, and Immunology. Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol 2006; 97(5):596–602. doi:10.1016/S1081-1206(10)61086-1
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis—a statement of the World Allergy Organization. World Allergy Organ J 2008; 1(suppl 7):S18–S26. doi:10.1097/WOX.0b013e31817c9338
- Jones DH, Romero FA, Casale TB. Time-dependent inhibition of histamine-induced cutaneous responses by oral and intramuscular diphenhydramine and oral fexofenadine. Ann Allergy Asthma Immunol 2008; 100(5):452–456. doi:10.1016/S1081-1206(10)60470-X
- Pumphrey RS. Lessons for management of anaphylaxis from a study of fatal reactions. Clin Exp Allerg 2000; 30(8):1144–1150. pmid:10931122
- Runge JW, Martinez JC, Caravati EM, Williamson SG, Hartsell SC. Histamine antagonists in the treatment of acute allergic reactions. Ann Emerg Med 1992; 21:237–242. pmid:1536481
- Sheikh A, Simons FE, Barbour V, Worth A. Adrenaline auto-injectors for the treatment of anaphylaxis with and without cardiovascular collapse in the community. Cochrane Database Syst Rev 2012; (8):CD008935. doi:10.1002/14651858.CD008935.pub2
- Simons FE, Gu X, Simons KJ. Epinephrine absorption in adults: intramuscular versus subcutaneous injection. J Allergy Clin Immunol 2001; 108(5):871–873. doi:10.1067/mai.2001.119409
- Simons FE, Roberts JR, Gu X, Simons KJ. Epinephrine absorption in children with a history of anaphylaxis. J Allergy Clin Immunol 1998; 101(1 pt 1):33–37. doi:10.1016/S0091-6749(98)70190-3
- Vadas P. The platelet-activating factor pathway in food allergy and anaphylaxis. Ann Allergy Asthma Immunol 2016; 117(5):455–457. doi:10.1016/j.anai.2016.05.003
- Stone SF, Brown SG. Mediators released during human anaphylaxis. Curr Allergy Asthma Rep 2012; 12(1):33–41. doi:10.1007/s11882-011-0231-6
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126(3):477–480.e1–e42. doi:10.1016/j.jaci.2010.06.022
- Kemp SF, Lockey RF, Simons FE; World Allergy Organization ad hoc Committee on Epinephrine in Anaphylaxis. Epinephrine: the drug of choice for anaphylaxis. A statement of the World Allergy Organization. Allergy 2008; 63(8):1061–1070. doi:10.1111/j.1398-9995.2008.01733.x
- Oren E, Banderji A, Clark S, Camargo CA Jr. Food-induced anaphylaxis and repeated epinephrine treatments. Ann Allergy Asthma Immunol 2007; 99(5):429–432. doi:10.1016/S1081-1206(10)60568-6
- Uguz A, Lack G, Pumphrey R, et al. Allergic reactions in the community: a questionnaire survey of members of the anaphylaxis campaign. Clin Exp Allergy 2005; 35(6):746–750. doi:10.1111/j.1365-2222.2005.02257.x
- Kelso JM. A second dose of epinephrine for anaphylaxis: how often needed and how to carry. J Allergy Clin Immunol 2006; 117(2):464–465. doi:10.1016/j.jaci.2005.11.015
- Lieberman P, Nicklas RA, Randolph C, et al. Anaphylaxis—a practice parameter update 2015. Ann Allergy Asthma Immunol 2015; 115(5):341–384. doi:10.1016/j.anai.2015.07.019
- Golden BK, Demain J, Freeman T, et al. Stinging insect hypersensitivity: a practice parameter update 2016. Ann Allergy Asthma Immunol 2017; 118(1):28–54. doi:10.1016/j.anai.2016.10.031
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55. doi:10.1016/j.jaci.2010.09.034
- Gupta P, Gerrish PK, Silverman B, Schneider A. Current practices among allergists on writing self-injectable epinephrine prescriptions for immunotherapy patients. J Allergy Clin Immunol 2012; 129(2):571–572.e1-e2. doi:10.1016/j.jaci.2011.09.033
- Ortolani C, Pastorello EA, Farioli L, et al. IgE-mediated allergy from vegetable allergens. Ann Allergy 1993; 71:470–476. pmid: 8250353
- Ma S, Shcherer SH, Nowak-Wegrzyn A. A survey on the management of pollen food allergy syndrome in allergy practices. J Allergy Clin Immunol 2003;112:784–788. doi:10.1016/S0091-6749(03)02008-6
- Shaver KJ, Adams C, Weiss SJ. Acute myocardial infarction after administration of low dose intravenous epinephrine for anaphylaxis. CJEM 2006; 8(4):289–294. pmid:17324313
- Triggiani M, Patella V, Staiano RI, Granata F, Marone G. Allergy and the cardiovascular system. Clin Exp Immunol 2008; 153(suppl 1):7–11. doi:10.1111/j.1365-2249.2008.03714.x
- Gilman AG, Rail TW, Nies AS, Taylor P, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York, NY: Pergamon Press; 1990.
- Lang DM, Alpern MB, Visintainer PF, Smith ST. Increased risk for anaphylactoid reaction from contrast media in patients on beta-adrenergic blockers or with asthma. Ann Intern Med 1991; 115(14):270–276. pmid:1677239
- Nassiri M, Babina M, Dölle S, Edenharter G, Ruëff F, Worm M. Ramipril and metoprolol intake aggravate human and murine anaphylaxis: evidence for direct mast cell priming. J Allergy Clin Immunol 2015; 135(2):491–499. doi:10.1016/j.jaci.2014.09.004
- Lee S, Hess EP, Nestler DM, et al. Antihypertensive medication use is associated with increased organ system involvement and hospitalization in emergency department patients with anaphylaxis. J Allergy Clin Immunol 2013; 131(4):1103–1108. doi:10.1016/j.jaci.2013.01.011
- Greenberger PA, Meyers SN, Kramer BL, Kramer BL. Effects of beta-adrenergic and calcium antagonists on the development of anaphylactoid reactions from radiographic contrast media during cardiac angiography. J Allergy Clin Immunol 1987; 80(5):698–702. pmid:2890682
- Hepner MJ, Ownby DR, Anderson JA, Rowe MS, Sears-Ewald D, Brown EB. Risk of systemic reactions in patients taking beta-blocker drugs receiving allergen immunotherapy injections. J Allergy Clin Immunol 1990; 86(3 pt 1):407–411. pmid:1976666
- Lieberman P, Simons FE. Anaphylaxis and cardiovascular disease: therapeutic dilemmas. Clin Exp Allergy 2015; 45(8):1288–1295. doi:10.1111/cea.12520
- Simons FE, Peterson S, Black CD. Epinephrine dispensing patterns for an out-of-hospital population: a novel approach to studying the epidemiology of anaphylaxis. J Allergy Clin Immunol 2002; 110(4):647–651. pmid:12373275
- Kawano T, Scheuermeyer FX, Stenstrom R, Rowe BH, Grafstein E, Grunau B. Epinephrine use in older patients with anaphylaxis: clinical outcomes and cardiovascular complications. Resuscitation 2017; 112:53–58. doi:10.1016/j.resuscitation.2016.12.020
- Cydulka R, Davison R, Grammer L, Parker M, Mathews J 4th. The use of epinephrine in the treatment of older adult asthmatics. Ann Emerg Med 1988; 17(4):322–326. pmid:3354935
- Soar J, Pumphrey R, Cant A, et al; Working Group of the Resuscitation Council (UK). Emergency treatment of anaphylactic reactions—guidelines for healthcare providers. Resuscitation 2008; 77(2):157–169. doi:10.1016/j.resuscitation.2008.02.001
- Dreborg S, Wen X, Kim L, et al. Do epinephrine auto-injectors have an unsuitable needle length in children and adolescents at risk for anaphylaxis from food allergy? Allergy Asthma Clin Immunol 2016; 12:11. doi:10.1186/s13223-016-0110-8
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014; 311(8):806–814. doi:10.1001/jama.2014.732
- Song TT, Nelson MR, Chang JH, Engler RJ, Chowdhury BA. Adequacy of the epinephrine autoinjector needle length in delivering epinephrine to the intramuscular tissues. Ann Allergy Asthma Immunol 2005; 94(5):539–542. doi:10.1016/S1081-1206(10)61130-1
- Bhalla MC, Gable BD, Frey JA, Reichenbach MR, Wilber ST. Predictors of epinephrine autoinjector needle length inadequacy. Am J Emerg Med 2013; 31(12):1671–1676. doi:10.1016/j.ajem.2013.09.001
- Kim H, Dinakar C, McInnis P, et al. Inadequacy of current pediatric epinephrine autoinjector needle length for use in infants and toddlers. Ann Allergy Asthma Immunol 2017; 118(6):719–725.e1. doi:10.1016/j.anai.2017.03.017
- Simons FE, Clark S, Camargo CA Jr. Anaphylaxis in the community: learning from the survivors. J Allergy Clin Immunol 2009; 124(2):301–306. doi:10.1016/j.jaci.2009.03.050
- Muck AE, Bebarta VS, Borys DJ, Morgan DL. Six years of epinephrine digital injections: absence of significant local or systemic effects. Ann Emerg Med 2010; 56(3):270–274. doi:10.1016/j.annemergmed.2010.02.019
- Fleming JT, Clark S, Camargo CA Jr, Rudders SA. Early treatment of food-induced anaphylaxis with epinephrine is associated with a lower risk of hospitalization. J Allergy Clin Immunol Pract 2015; 3(1):57–62. doi:10.1016/j.jaip.2014.07.004
- Kaplan MS, Jung SY, Chiang ML. Epinephrine autoinjector refill history in an HMO. Curr Allergy Asthma Rep 2011; 11(1):65–70. doi:10.1007/s11882-010-0155-6
KEY POINTS
- Based on current data, there is no absolute contraindication to epinephrine for anaphylaxis. And failure to give epinephrine promptly has resulted in deaths.
- Clinicians concerned about adverse effects of epinephrine may be reluctant to give it during anaphylaxis.
- Education about anaphylaxis and its prompt treatment with epinephrine is critical for patients and their caregivers.

Injectable extended-release naltrexone for opioid dependence: 3 studies
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).

1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).
1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).
1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
