User login
Better psychiatric documentation: From SOAP to PROMISE
Discuss this article at www.facebook.com/CurrentPsychiatry
Because documentation is an important part of medical practice,1 numerous tools have been developed to help physicians across all specialties, including the best-known acronym SOAP, which stands for Subjective, Objective, Assessment, and Plan. The SOAP note has been used in mental health settings,2 although this format may fall short for psychiatrists because objective tests are not diagnostic. Also, there’s no clear guidance to document specific information, such as behavioral risk assessment.
The acronym PROMISE—Problems, Resolved, Outcomes, Medications, Instructions, Safety, and Education—may be better suited for psychiatric documentation. The PROMISE note provides an easy-to-remember method to document specific information that might be overlooked in a less detailed format, such as normal findings, adherence and tolerability to medications, outcome ratings, and risk assessment.
Problems are described as ongoing symptoms, signs, and stressors. Resolved indicates improvement and normal findings. Outcome measures include patient or clinician rating scales. Medications documents the effectiveness and tolerability of current and past medications. Instructions are directives given; the rationale—cost-benefit analysis—can be documented in this section as well. Safety describes a behavioral risk assessment, including demographic, historical, clinical, and environmental risk and protective factors regarding suicidal or homicidal behavior. Education describes the verbal or written material shared with the patient.
Psychotherapists can use the same template. For them the M would stand for Methods of psychotherapy practiced in the session.
For an example of the PROMISE note used in practice, see the Table.
Table
Example of a patient’s PROMISE note
| Problems | Ongoing depressive symptoms: low mood, negative thinking, low interest level; patient has no insurance, pays out of pocket |
| Resolved | Mild improvement in motivation noted; sleeping and concentration both OK; continues to work full-time; spends time with parents |
| Outcomes | Clinical Global Impression-Severity Scale score: 4; PHQ-9 depression rating scale score: 12/27, indicating moderate depression (score 1 month ago was 15/27; 20% reduction) |
| Medications | Current treatment: citalopram, 20 mg/d, nortriptyline, 50 mg/d Prior medications: bupropion, citalopram, clomipramine, fluoxetine, MAOIs, sertraline, and venlafaxine. Patient’s adherence to medication is good Tolerability issues: sweating, constipation, dry mouth |
| Instructions | Increase both medications (20% improvement noted; recommend increase in nortriptyline; patient requests increase in citalopram). Ongoing moderate depression; initial side effects may subside |
| Safety | Identified risk or protective factors for suicidal, aggressive, or homicidal behavior: chronic depression without remission No current SI, HI, SIB, hopelessness, anxiety, agitation, insomnia, substance use, psychosis, or interpersonal aggression. No access to weapons. No history of suicide attempts. Good supports. Risk assessment: low |
| Education |
|
| HI: homicidal ideation; MAOIs: monoamine oxidase inhibitors; PHQ-9: 9-Question Patient Health Questionnaire; SI: suicidal ideation; SIB: self-injurious behavior | |
Disclosure
Dr. Bastiaens reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Because documentation is an important part of medical practice,1 numerous tools have been developed to help physicians across all specialties, including the best-known acronym SOAP, which stands for Subjective, Objective, Assessment, and Plan. The SOAP note has been used in mental health settings,2 although this format may fall short for psychiatrists because objective tests are not diagnostic. Also, there’s no clear guidance to document specific information, such as behavioral risk assessment.
The acronym PROMISE—Problems, Resolved, Outcomes, Medications, Instructions, Safety, and Education—may be better suited for psychiatric documentation. The PROMISE note provides an easy-to-remember method to document specific information that might be overlooked in a less detailed format, such as normal findings, adherence and tolerability to medications, outcome ratings, and risk assessment.
Problems are described as ongoing symptoms, signs, and stressors. Resolved indicates improvement and normal findings. Outcome measures include patient or clinician rating scales. Medications documents the effectiveness and tolerability of current and past medications. Instructions are directives given; the rationale—cost-benefit analysis—can be documented in this section as well. Safety describes a behavioral risk assessment, including demographic, historical, clinical, and environmental risk and protective factors regarding suicidal or homicidal behavior. Education describes the verbal or written material shared with the patient.
Psychotherapists can use the same template. For them the M would stand for Methods of psychotherapy practiced in the session.
For an example of the PROMISE note used in practice, see the Table.
Table
Example of a patient’s PROMISE note
| Problems | Ongoing depressive symptoms: low mood, negative thinking, low interest level; patient has no insurance, pays out of pocket |
| Resolved | Mild improvement in motivation noted; sleeping and concentration both OK; continues to work full-time; spends time with parents |
| Outcomes | Clinical Global Impression-Severity Scale score: 4; PHQ-9 depression rating scale score: 12/27, indicating moderate depression (score 1 month ago was 15/27; 20% reduction) |
| Medications | Current treatment: citalopram, 20 mg/d, nortriptyline, 50 mg/d Prior medications: bupropion, citalopram, clomipramine, fluoxetine, MAOIs, sertraline, and venlafaxine. Patient’s adherence to medication is good Tolerability issues: sweating, constipation, dry mouth |
| Instructions | Increase both medications (20% improvement noted; recommend increase in nortriptyline; patient requests increase in citalopram). Ongoing moderate depression; initial side effects may subside |
| Safety | Identified risk or protective factors for suicidal, aggressive, or homicidal behavior: chronic depression without remission No current SI, HI, SIB, hopelessness, anxiety, agitation, insomnia, substance use, psychosis, or interpersonal aggression. No access to weapons. No history of suicide attempts. Good supports. Risk assessment: low |
| Education |
|
| HI: homicidal ideation; MAOIs: monoamine oxidase inhibitors; PHQ-9: 9-Question Patient Health Questionnaire; SI: suicidal ideation; SIB: self-injurious behavior | |
Disclosure
Dr. Bastiaens reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Because documentation is an important part of medical practice,1 numerous tools have been developed to help physicians across all specialties, including the best-known acronym SOAP, which stands for Subjective, Objective, Assessment, and Plan. The SOAP note has been used in mental health settings,2 although this format may fall short for psychiatrists because objective tests are not diagnostic. Also, there’s no clear guidance to document specific information, such as behavioral risk assessment.
The acronym PROMISE—Problems, Resolved, Outcomes, Medications, Instructions, Safety, and Education—may be better suited for psychiatric documentation. The PROMISE note provides an easy-to-remember method to document specific information that might be overlooked in a less detailed format, such as normal findings, adherence and tolerability to medications, outcome ratings, and risk assessment.
Problems are described as ongoing symptoms, signs, and stressors. Resolved indicates improvement and normal findings. Outcome measures include patient or clinician rating scales. Medications documents the effectiveness and tolerability of current and past medications. Instructions are directives given; the rationale—cost-benefit analysis—can be documented in this section as well. Safety describes a behavioral risk assessment, including demographic, historical, clinical, and environmental risk and protective factors regarding suicidal or homicidal behavior. Education describes the verbal or written material shared with the patient.
Psychotherapists can use the same template. For them the M would stand for Methods of psychotherapy practiced in the session.
For an example of the PROMISE note used in practice, see the Table.
Table
Example of a patient’s PROMISE note
| Problems | Ongoing depressive symptoms: low mood, negative thinking, low interest level; patient has no insurance, pays out of pocket |
| Resolved | Mild improvement in motivation noted; sleeping and concentration both OK; continues to work full-time; spends time with parents |
| Outcomes | Clinical Global Impression-Severity Scale score: 4; PHQ-9 depression rating scale score: 12/27, indicating moderate depression (score 1 month ago was 15/27; 20% reduction) |
| Medications | Current treatment: citalopram, 20 mg/d, nortriptyline, 50 mg/d Prior medications: bupropion, citalopram, clomipramine, fluoxetine, MAOIs, sertraline, and venlafaxine. Patient’s adherence to medication is good Tolerability issues: sweating, constipation, dry mouth |
| Instructions | Increase both medications (20% improvement noted; recommend increase in nortriptyline; patient requests increase in citalopram). Ongoing moderate depression; initial side effects may subside |
| Safety | Identified risk or protective factors for suicidal, aggressive, or homicidal behavior: chronic depression without remission No current SI, HI, SIB, hopelessness, anxiety, agitation, insomnia, substance use, psychosis, or interpersonal aggression. No access to weapons. No history of suicide attempts. Good supports. Risk assessment: low |
| Education |
|
| HI: homicidal ideation; MAOIs: monoamine oxidase inhibitors; PHQ-9: 9-Question Patient Health Questionnaire; SI: suicidal ideation; SIB: self-injurious behavior | |
Disclosure
Dr. Bastiaens reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Psychiatric ‘holds’ for nonpsychiatric patients
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).

Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).
Metabolic disturbance and dementia: A modifiable link
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.

Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.
Can topiramate reduce nightmares in posttraumatic stress disorder?
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.
How to adapt cognitive-behavioral therapy for older adults
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT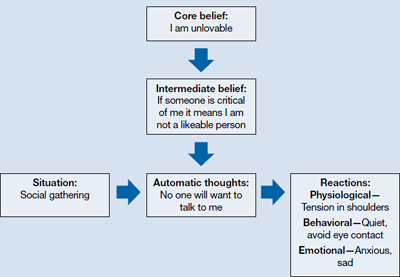
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
Autoimmune Hemolytic Anemia
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF:
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF:
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons. AIHA is mediated by antibodies, and in the majority of cases immunglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. This manual reviews the most common types of AIHA, with emphasis on diagnosis and treatment.
To read the full article in PDF:
Recent recommendations on steroid-induced osteoporosis: More targeted, but more complicated
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT

For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22

Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group

Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
KEY POINTS
- The risk of fracture should be assessed at the start of glucocorticoid therapy.
- Factors that affect the decision to prescribe osteoporosis drugs include the patient’s risk of fractures as assessed with FRAX (www.shef.ac.uk/FRAX), the dose of glucocorticoid, and the projected duration of treatment.
- Since FRAX treats glucocorticoid use simply as a yes-or-no question, it likely underestimates the fracture risk in current users and at high doses. The estimate of risk should be adjusted upward in these situations.
Cardiac tamponade: 12 pearls in diagnosis and management
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
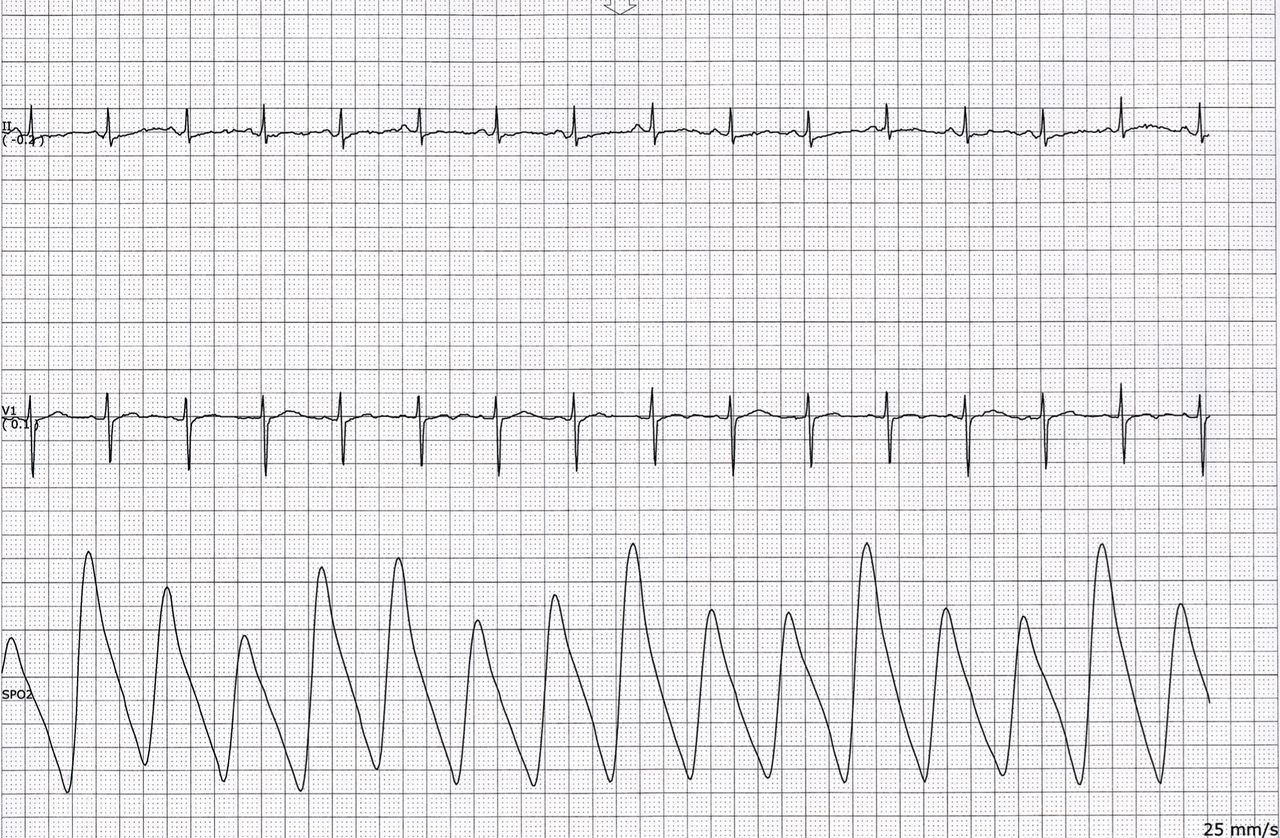
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
Cardiac tamponade is a life-threatening condition that can be palliated or cured, depending on its cause and on the timeliness of treatment. Making a timely diagnosis and providing the appropriate treatment can be gratifying for both patient and physician.
Cardiac tamponade occurs when fluid in the pericardial space reaches a pressure exceeding central venous pressure. This leads to jugular venous distention, visceral organ engorgement, edema, and elevated pulmonary venous pressure that causes dyspnea. Despite compensatory tachycardia, the decrease in cardiac filling leads to a fall in cardiac output and to arterial hypoperfusion of vital organs.
PEARL 1: SLOW ACCUMULATION LEADS TO EDEMA
The rate at which pericardial fluid accumulates influences the clinical presentation of cardiac tamponade, in particular whether or not there is edema. Whereas rapid accumulation is characterized more by hypotension than by edema, the slow accumulation of pericardial fluid affords the patient time to drink enough liquid to keep the central venous pressure higher than the rising pericardial pressure. Thus, edema and dyspnea are more prominent features of cardiac tamponade when there is a slow rise in pericardial pressure.
PEARL 2: EDEMA IS NOT ALWAYS TREATED WITH A DIURETIC
Edema is not always treated with a diuretic. In a patient who has a pericardial effusion that has developed slowly and who has been drinking enough fluid to keep the central venous pressure higher than the pericardial pressure, a diuretic can remove enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure and thus convert a benign pericardial effusion to potentially lethal cardiac tamponade.
One must understand the cause of edema or low urine output before treating it. This underscores the importance of the history and the physical examination. All of the following must be assessed:
- Symptoms and time course of the illness
- Concurrent medical illnesses
- Neck veins
- Blood pressure and its response to inspiration
- Heart sounds
- Heart rate and rhythm
- Abdominal organ engorgement
- Edema (or its absence).
PEARL 3: UNDERSTANDING THE CAUSE IS ESSENTIAL
Understanding the cause of cardiac tamponade is essential.
A trauma patient first encountered in the emergency department may have an underlying disease, but the focus is squarely on the effects of trauma or violent injury. In a patient with multiple trauma, hypotension and tachycardia that do not respond to intravenous volume replacement when there is an obvious rise in central venous pressure should be clues to cardiac tamponade.1
If the patient has recently undergone a cardiac procedure (for example, cardiac surgery, myocardial biopsy, coronary intervention, electrophysiologic study with intracardiac electrodes, transvenous pacemaker placement, pacemaker lead extraction, or radiofrequency ablation), knowing about the procedure narrows the differential diagnosis when hypotension, tachycardia, and jugular venous distention develop.
PEARL 4: CARDIAC OR AORTIC RUPTURE REQUIRES SURGERY
When the etiology of cardiac tamponade is cardiac or aortic rupture, the treatment is surgical.
Painful sudden causes of cardiac tamponade include hemopericardium due to rupture of the free wall after myocardial infarction, and spontaneous or posttraumatic dissection and rupture of the ascending aorta. Prompt diagnosis is necessary, but since these lesions will not close and heal spontaneously, the definitive treatment should be surgery. Moreover, needle removal of intrapericardial blood that has been opposing further bleeding is sure to permit bleeding to recur, often with lethal consequences.2
Causes of cardiac tamponade that have a less-acute onset are likely to be complications of medical problems. Medical illnesses known to be associated with cardiac tamponade include:
- Infectious disease (idiopathic or viral, associated with smallpox vaccination, mycobacterial, purulent bacterial, fungal)
- Metastatic cancer (lung, breast, esophagus, lymphoma, pancreas, liver, leukemia, stomach, melanoma)3
- Connective tissue disease (rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, scleroderma, Wegener granulomatosis, acute rheumatic fever)
- Endocrine disease (hypothyroidism)
- Drug side effects (procainamide, isoniazid, hydralazine, minoxidil, phenytoin, anticoagulants, methysergide)
- Inflammatory bowel disease (Crohn disease, ulcerative colitis)
- Congestive heart failure
- Uremia
- Radiation therapy
- Postmyocardial infarction syndrome (Dressler syndrome)
- Postpericardiotomy syndrome.
PEARL 5: REVIEW IMAGING BEFORE DIAGNOSING
What often brings a patient with cardiac tamponade to the attention of the physician is a finding on echocardiography, computed tomography, or magnetic resonance imaging of the chest.
Always review the imaging studies before making the diagnosis of cardiac tamponade. These tests must be reviewed to assess the anatomy and the size and location of the effusion. Particularly, one must look for atrial and right ventricular collapse and inferior vena caval plethora, which are echocardiographic signs of cardiac tamponade.4 Figures 1, 2, and 3 show imaging studies in a patient who presented with worsening cough 2 weeks after undergoing a cardiac procedure and who was found to have cardiac tamponade.
When the history and these imaging studies place cardiac tamponade high in the differential diagnosis as the cause of edema or dyspnea, it is time to reexamine the patient. The best first step is to measure pulsus paradoxus.
HOW PULSUS PARADOXUS OCCURS
To fully appreciate the subtleties of the next pearls, it is necessary to understand the pathophysiology of cardiac tamponade.
When pericardial fluid accumulation raises the pericardial pressure above the central venous pressure and pulmonary venous pressure (intravascular pressure), blood will not passively return to the right side of the heart from the venae cavae nor to the left side of the heart from the pulmonary veins unless it is influenced by the effects of respiration on intrathoracic pressure. During respiration, the right and left sides of the heart are alternately filled and deprived of their respective venous return.
During inspiration, as the intrathoracic pressure decreases, blood in the venae cavae empties into the right side of the heart, while blood in the pulmonary veins preferentially remains in the pulmonary veins, underfilling the left side of the heart. Since the right ventricle is more filled than the left ventricle during inspiration, the ventricular septum shifts from right to left, further opposing pulmonary venous return. As a result, during cardiac tamponade, the systemic blood pressure falls with inspiration.
During expiration the opposite occurs. Expiration decreases the intrathoracic volume, so the intrathoracic pressure rises. This tends to oppose vena caval return to the right side of the heart and to favor pulmonary venous return to the left side of the heart. The ventricular septum shifts from left to right, further accommodating left ventricular filling, raising stroke volume, and increasing blood pressure. This exaggerated alternate filling of the right and left sides of the heart during cardiac tamponade is what accounts for pulsus paradoxus, an inspiratory fall in systolic blood pressure of greater than 10 mm Hg.
If intravascular pressure is low (due to hemorrhage, dehydration, or diuretic therapy), the pressure in the pericardial space needed to oppose venous return is much less. In this low-pressure scenario, the results are low cardiac output and hypotension, which are treated by giving intravenous fluids to maintain intravascular volume.
PEARL 6: MEASURE PULSUS PARADOXUS
When cardiac tamponade is considered, one must always measure the pulsus paradoxus.
The term pulsus paradoxus was coined by Adolph Kussmaul in 1873, before physicians could even measure blood pressure. All they could do at that time was palpate the pulse and listen to the heart. Kussmaul described his observation as a conspicuous discrepancy between the cardiac action and the arterial pulse.
Although not described by Kussmaul, another explanation for this finding might be more suited to the use of the word “paradoxical.” When the pulse is palpated in a normal patient, with inspiration the pulse rate will increase via the Bainbridge reflex, and with expiration it will decrease. But in a patient with cardiac tamponade, there is a paradoxical inspiratory slowing of the pulse (because the decreased magnitude of the pulse at times makes it imperceptible) and an expiratory increase in pulse rate as the magnitude of the pulse again makes it palpable.
The magnitude of the fall in systolic blood pressure during inspiration has been used to estimate the level of hemodynamic impairment resulting from pericardial effusion.5 A rapidly accumulating pericardial effusion can have more hemodynamic impact than a much larger one that accumulates slowly. Thus, the intrapericardial pressure must be considered more than the volume of pericardial fluid.
When there is severe cardiac tamponade and overt pulsus paradoxus, simple palpation of a proximal arterial pulse can detect a marked inspiratory decrease or loss of the pulse, which returns with expiration. Tachycardia is almost always present, unless the cause is hypothyroidism.6
How to measure pulsus paradoxus with a manual sphygmomanometer
A stethoscope and manual sphygmomanometer are all that is needed to measure pulsus paradoxus. A noninvasive blood pressure monitor that averages multiple measurements cannot detect or quantify pulsus paradoxus.
The patient should be supine with the head elevated 30° to 45°, and the examiner should be seated comfortably at the patient’s side. The manometer should be on the opposite side of the patient in plain view of the examiner. Position the cuff on the arm above the elbow and place your stethoscope on the antecubital fossa. Then:
- Inflate the cuff 20 mm Hg above the highest systolic pressure previously auscultated.
- Slowly decrease the manometer pressure by 5 mm Hg and hold it there through two or three respiratory cycles while listening for the first Korotkoff (systolic) sound. Repeat this until you can hear the systolic sound (but only during expiration) and mentally note the pressure.
- Continue to decrease the manometer pressure by 5-mm Hg increments while listening. When the Korotkoff sounds no longer disappear with inspiration, mentally note this second value as well. The pulsus paradoxus is the difference between these values.
- When the Korotkoff sounds disappear as the manometer pressure is decreased, note this final value. This is the diastolic blood pressure.
PEARL 7: THE PLETHYSMOGRAM WAVE-FORM PARALLELS PULSUS PARADOXUS
Manual measurement of blood pressure and pulsus paradoxus can be difficult, especially in an obese patient or one with a fat-distorted arm on which the cuff does not maintain its position. In such patients, increased girth of the neck and abdomen also make it difficult to assess the jugular venous distention and visceral organ engorgement that characterize cardiac tamponade.
When the use of a sphygmomanometer is not possible, an arterial catheter can be inserted to demonstrate pulsus paradoxus. Simpler, however, is the novel use of another noninvasive instrument to detect and coarsely quantify pulsus paradoxus.7 The waveform on finger pulse oximetry can demonstrate pulsus paradoxus. The plethysmogram of the finger pulse oximeter can demonstrate the decrease in magnitude of the waveform with each inspiration (Figure 4).
Caution must be taken when interpreting this waveform, as with any measurement of pulsus paradoxus, to exclude a concomitant arrhythmia.
PEARL 8: PULSUS PARADOXUS WITHOUT CARDIAC TAMPONADE
Pulsus paradoxus can be present in the absence of cardiac tamponade. Once pulsus paradoxus of more than 10 mm Hg is measured, one must be sure the patient does not have a condition that can cause pulsus paradoxus without cardiac tamponade. Most of these are pulmonary conditions that necessitate an exaggerated inspiratory effort that can lower intrathoracic pressure sufficiently to oppose pulmonary venous return and cause a fall in systemic blood pressure:
- Chronic bronchitis
- Emphysema
- Mucus plug
- Pneumothorax
- Pulmonary embolism
- Stridor.
In these, there may be pulsus paradoxus, but not due to cardiac tamponade.
PEARL 9: CARDIAC TAMPONADE CAN BE PRESENT WITHOUT PULSUS PARADOXUS
Cardiac tamponade can be present without pulsus paradoxus. This occurs when certain conditions prevent inspiratory underfilling of the left ventricle relative to the filling of the right ventricle.8
How does this work? In cardiac tamponade, factors that drive the exaggerated fall in arterial pressure with inspiration (pulsus paradoxus) are the augmented right ventricular filling and the decreased left ventricular filling, both due to the lowering of the intrathoracic pressure. As the vena caval emptying is augmented, the right ventricular filling is increased, the ventricular septum shifts to the left, and pulmonary venous return to the heart is decreased.
Factors that can oppose pulsus paradoxus:
- Positive pressure ventilation prevents pulsus paradoxus by preventing the fall in intrathoracic pressure.
- Severe aortic regurgitation does not permit underfilling of the left ventricle during inspiration.
- An atrial septal defect will always equalize the right and left atrial pressures, preventing differential right ventricular and left ventricular filling with inspiration.
- Severe left ventricular hypertrophy does not permit the inspiratory shift of the ventricular septum from right to left that would otherwise lead to decreased left ventricular filling.
- Severe left ventricular dysfunction, with its low stroke volume and severe elevation of left ventricular end-diastolic pressure, never permits underfilling of the left ventricle, despite cardiac tamponade and an inspiratory decrease in intrathoracic pressure.
- Intravascular volume depletion due to hemorrhage, hemodialysis, or mistaken use of diuretics to treat edema can cause marked hypotension, making pulsus paradoxus impossible to detect.
Knowledge of underlying medical conditions, the likelihood of their causing cardiac tamponade, and the appearance of the echocardiogram prompt the physician to look further when the presence or absence of pulsus paradoxus does not fit with the working diagnosis.
The echocardiogram can give hints to the etiology of a pericardial effusion, such as clotted blood after trauma or a cardiac-perforating procedure, tumor studding of the epicardium,9 or fibrin strands indicating chronicity or an inflammatory process.10 Diastolic collapse of the right ventricle, more than collapse of the right atrium or left atrium, speaks for the severity of cardiac tamponade. With hemodynamically significant pericardial effusion and cardiac tamponade, the inferior vena cava is distended and does not decrease in size with inspiration unless there is severe intravascular volume depletion, at which time the inferior vena cava is underfilled throughout the respiratory cycle.
PEARL 10: PLAN HOW TO DRAIN
The size and location of the pericardial effusion and the patient’s hemodynamics must be integrated when deciding how to relieve cardiac tamponade. When cardiac tamponade is indeed severe and the patient and physician agree that it must be drained, the options are percutaneous needle aspiration (pericardiocentesis) and surgical pericardiostomy (creation of a pericardial window). Here again, as assessed by echocardiography, the access to the pericardial fluid should influence the choice.
Pericardiocentesis can be safely done if certain criteria are met. The patient must be able to lie still in the supine position, perhaps with the head of the bed elevated 30 degrees. Anticoagulation must be reversed or allowed time to resolve if drainage is not an emergency.
Pericardiocentesis can be risky or unsuccessful if there is not enough pericardial fluid to permit respiratory cardiac motion without perforating the heart with the needle; if the effusion is loculated (confined to a pocket) posteriorly; or if it is too far from the skin to permit precise control and placement of a spinal needle into the pericardial space. In cases of cardiac tamponade in which the anatomy indicates surgical pericardiostomy but severe hypotension prevents the induction of anesthesia and positive-pressure ventilation—which can result in profound, irreversible hypotension—percutaneous needle drainage (pericardiocentesis) should be performed in the operating room to relieve the tamponade before the induction of anesthesia and the surgical drainage.11
To reiterate, a suspected cardiac or aortic rupture that causes cardiac tamponade is usually large and not apt to self-seal. In such cases, the halt in the accumulation of pericardial blood is due to hypotension and not due to spontaneous resolution. Open surgical drainage is required from the outset because an initial success of pericardiocentesis yields to the recurrence of cardiac tamponade.
PEARL 11: ANTICIPATE WHAT THE FLUID SHOULD LOOK LIKE
Before performing pericardiocentesis, anticipate the appearance of the pericardial fluid on the basis of the presumed etiology, ie:
- Sanguinous—trauma, heart surgery, cardiac perforation from a procedure, anticoagulation, uremia, or malignancy
- Serous—congestive heart failure, acute radiation therapy
- Purulent—infections (natural or postoperative)
- Turbid (like gold paint)—mycobacterial infection, rheumatoid arthritis, myxedema
- Chylous—pericardium fistulized to the thoracic duct by a natural or postsurgical cause.
Sanguinous pericardial effusion encountered during a pericardiocentesis, if not anticipated, can be daunting and can cause the operator to question if it is the result of inadvertent needle placement in a cardiac chamber. If the needle is indeed in the heart, blood often surges out under pressure in pulses, which strongly suggests that the needle is not in the pericardial space and should be removed; but if confirmation of the location is needed before removing the needle, it can be done by injecting 2 mL of agitated sterile saline through the pericardiocentesis needle during echocardiographic imaging.12
Before inserting the needle, the ideal access location and needle angle must be determined by the operator with echocardiographic transducer in hand. The distance from skin to a point just through the parietal pericardium can also be measured at this time.
Once the needle is in the pericardial fluid (and you are confident of its placement), removal of 50 to 100 mL of the fluid with a large syringe can be enough to afford the patient easier breathing, higher blood pressure, and lower pulsus paradoxus—and even the physician will breathe easier. The same syringe can be filled and emptied multiple times. Less traumatic and more complete removal of pericardial fluid requires insertion of a multihole pigtail catheter over a J-tipped guidewire that is introduced through the needle.
PEARL 12: DRAIN SLOWLY TO AVOID PULMONARY EDEMA
Pulmonary edema is an uncommon complication of pericardiocentesis that might be avoidable. Heralded by sudden coughing and pink, frothy sputum, it can rapidly deteriorate into respiratory failure. The mechanism has been attributed to a sudden increase in right ventricular stroke volume and resultant left ventricular filling after the excess pericardial fluid has been removed, before the systemic arteries, which constrict to keep the systemic blood pressure up during cardiac tamponade, have had time to relax.13
To avoid this complication, if the volume of pericardial fluid responsible for cardiac tamponade is large, it should be removed slowly,14 stopping for a several-minute rest after each 250 mL. Catheter removal of pericardial fluid by gravity drainage over 24 hours has been suggested.15 A drawback to this approach is catheter clotting or sludging before all the fluid has been removed. It is helpful to keep the drainage catheter close to the patient’s body temperature to make the fluid less viscous. Output should be monitored hourly.
When the pericardial fluid has been completely drained, one must decide how long to leave the catheter in. One reason to remove the catheter at this time is that it causes pleuritic pain; another is to avoid introducing infection. A reason to leave the catheter in is to observe the effect of medical treatment on the hourly pericardial fluid output. Nonsteroidal anti-inflammatory drugs are the drugs of first choice when treating pericardial inflammation and suppressing production of pericardial fluid.16 In most cases the catheter should not be left in place for more than 3 days.
Laboratory analysis of the pericardial fluid should shed light on its suspected cause. Analysis usually involves chemistry testing, microscopic inspection of blood cell smears, cytology, microbiologic stains and cultures, and immunologic tests. Results often take days. Meyers and colleagues17 expound on this subject.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
- Schiavone WA, Ghumrawi BK, Catalano DR, et al. The use of echocardiography in the emergency management of nonpenetrating traumatic cardiac rupture. Ann Emerg Med 1991; 20:1248–1250.
- Manuchehry A, Fontana GP, Gurudevan S, Marchevsky AM, Siegel RJ. Missed diagnosis of limited ascending aortic dissection by multiple imaging modalities leading to fatal cardiac tamponade and aortic rupture. Echocardiography 2011; 28:E187–E190.
- Lam KY, Dickens P, Chan AC. Tumors of the heart. A 20-year experience with a review of 12,485 consecutive autopsies. Arch Pathol Lab Med 1993; 117:1027–1031.
- Tsang TS, Oh JK, Seward JB, Tajik AJ. Diagnostic value of echocardiography in cardiac tamponade. Herz 2000; 25:734–740.
- Curtiss EI, Reddy PS, Uretsky BF, Cecchetti AA. Pulsus paradoxus: definition and relation to the severity of cardiac tamponade. Am Heart J 1988; 115:391–398.
- Wang JL, Hsieh MJ, Lee CH, et al. Hypothyroid cardiac tamponade: clinical features, electrocardiography, pericardial fluid and management. Am J Med Sci 2010; 340:276–281.
- Tamburro RF, Ring JC, Womback K. Detection of pulsus paradoxus associated with large pericardial effusions in pediatric patients by analysis of the pulse-oximetry waveform. Pediatrics 2002; 109:673–677.
- Spodick DH. Pulsus paradoxus. In:Spodick DH, editor. The Pericardium: A Comprehensive Textbook. New York, NY: Marcel Dekker; 1997:191–199.
- Burke A, Jeudy J, Virmani R. Cardiac tumors. In:Topol EJ, editor. Textbook of Cardiovascular Medicine. 3rd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007:710–720.
- Roberts WC. Pericardial heart disease: Its morphologic features and its causes. Proc (Bayl Univ Med Cent) 2005; 18:38–55.
- Stoelting RK, Miller RD, editors. Basics of Anesthesia. 4th ed. New York, NY: Churchill Livingstone; 2000:264–265.
- Ainsworth CD, Salehian O. Echo-guided pericardiocentesis: let the bubbles show the way. Circulation 2011; 123:e210–e211.
- Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases executive summary; The Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2004; 25:587–610.
- Vandyke WH, Cure J, Chakko CS, Gheorghiade M. Pulmonary edema after pericardiocentesis for cardiac tamponade. N Engl J Med 1983; 309:595–596.
- Bernal JM, Pradhan J, Li T, Tchokonte R, Afonso L. Acute pulmonary edema following pericardiocentesis for cardiac tamponade. Can J Cardiol 2007; 23:1155–1156.
- Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 2011; 3:135–143.
- Meyers DG, Meyers RE, Prendergast TW. The usefulness of diagnostic tests on pericardial fluid. Chest 1997; 111:1213–1221.
KEY POINTS
- Slow accumulation of pericardial fluid can result in edema, whereas rapid accumulation leads to hypotension.
- Diuretics can worsen tamponade by removing enough volume from the circulation to lower the central venous pressure below the intrapericardial pressure.
- Try to determine why cardiac tamponade has occurred. Cardiac or aortic rupture requires surgery. If the gross appearance of the pericardial fluid does not match the presumed etiology, reconsider your diagnosis.
- Always review imaging studies before making the diagnosis of cardiac tamponade.
- When cardiac tamponade is considered, pulsus paradoxus must be measured, and if present, integrated with other physical findings and the echocardiogram. However, pulsus paradoxus can be present in the absence of cardiac tamponade, and vice versa.
- Consider the size and location of the pericardial effusion and the patient’s hemodynamic status when deciding between surgery and needle aspiration.
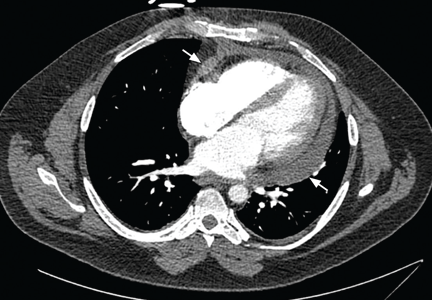
Fecal microbiota transplantation for recurrent C difficile infection: Ready for prime time?
If you had a serious disease, would you agree to an alternative treatment that was cheap, safe, and effective—but seemed disgusting? Would you recommend it to patients?
Such a disease is recurrent Clostridium difficile infection, and such a treatment is fecal microbiota transplantation—instillation of blenderized feces from a healthy donor (ideally, the patient’s spouse or “significant other”) into the patient’s colon to restore a healthy population of bacteria.1,2 The rationale behind this procedure is simple: antibiotics and other factors disrupt the normal balance of the colonic flora, allowing C difficile to proliferate, but the imbalance can be corrected by reintroducing the normal flora.1
In this article, we will review how recurrent C difficile infection occurs and the importance of the gut microbiota in resisting colonization with this pathogen. We will also describe the protocol used for fecal microbiota transplantation.
C DIFFICILE INFECTION OFTEN RECURS
C difficile is the most common cause of hospital-acquired diarrhea and an important cause of morbidity and death in hospitalized patients.3,4 The cost of this infection is estimated to be more than $1.1 billion per year and its incidence is rising, partly because of the emergence of more-virulent strains that make treatment of recurrent infection more difficult.5,6
C difficile infection is characterized by diarrhea associated with findings suggestive of pseudomembranous colitis or, in fulminant cases, ileus or megacolon.7 Recurrent C difficile infection is defined as the return of symptoms within 8 weeks after successful treatment.7
C difficile produces two types of toxins. Toxin A is an enterotoxin, causing increased intestinal permeability and fluid secretion, while toxin B is a cytotoxin, causing intense colonic inflammation. People who have a poor host immune response to these toxins tend to develop more diarrhea and colonic inflammation.8
A more virulent strain of C difficile has emerged. Known as BI/NAP1/027, this strain is resistant to quinolones, and it also produces a binary toxin that has a partial gene deletion that allows for increased production of toxins A and B in vitro.9,10 More cases of severe and recurrent C difficile infection have been associated with the increasing number of people infected with this hypervirulent strain.9,10
C difficile infection recurs in about 20% to 30% of cases after antibiotic treatment for it, usually within 30 days, and the risk of a subsequent episode doubles after two or more occurrences.10,11 Metronidazole (Flagyl) and vancomycin are the primary treatments; alternative treatments include fidaxomicin (Dificid), 10 rifaximin (Xifaxan),12 nitazoxanide,13 and tolevamer (a novel polymer that binds C difficile toxins).14

Table 1 summarizes the treatment regimen for C difficile infection in adults, based on clinical practice guidelines from the US Centers for Disease Control and Prevention (CDC).7
THE NORMAL GUT MICROBIOTA KEEPS PATHOGENS OUT
Immediately after birth, the sterile human gut becomes colonized by a diverse community of microorganisms.15 This gut microbiota performs various functions, such as synthesizing vitamin K and vitamin B complex, helping digest food, maintaining the mucosal integrity of the gut, and priming the mucosal immune response to maintain homeostasis of commensal microbiota.16
However, the most important role of the gut microbiota is “colonization resistance” or preventing exogenous or potentially pathogenic organisms from establishing a colony within the gut.17 It involves competition for nutrients and occupation of binding sites on the gut epithelium by indigenous flora.16 Other factors such as the mucosal barrier, salivation, swallowing, gastric acidity, desquamation of mucosal membrane cells, intestinal motility, and secretion of antibodies also play major roles in colonization resistance.17
ANTIBIOTICS DISRUPT THE GUT FLORA
Physical or chemical injuries (the latter by antimicrobial or antineoplastic agents, eg) may disrupt the gut microbiota. In this situation, opportunistic pathogens such as C difficile colonize the gut mucosa, stimulate an immune reaction, and release toxins that cause diarrhea and inflammation.18C difficile will try to compete for nutrients and adhesion sites until it dominates the intestinal tract.
When C difficile spores are ingested, they replicate in the gut and eventually release toxins. Antibiotic therapy may eliminate C difficile bacteria but not the spores; hence, C difficile infection can recur after the antibiotic is discontinued unless the indigenous bacteria can restrain C difficile from spreading.19
HOW DOES FECAL MICROBIOTA TRANSPLANTATION WORK?
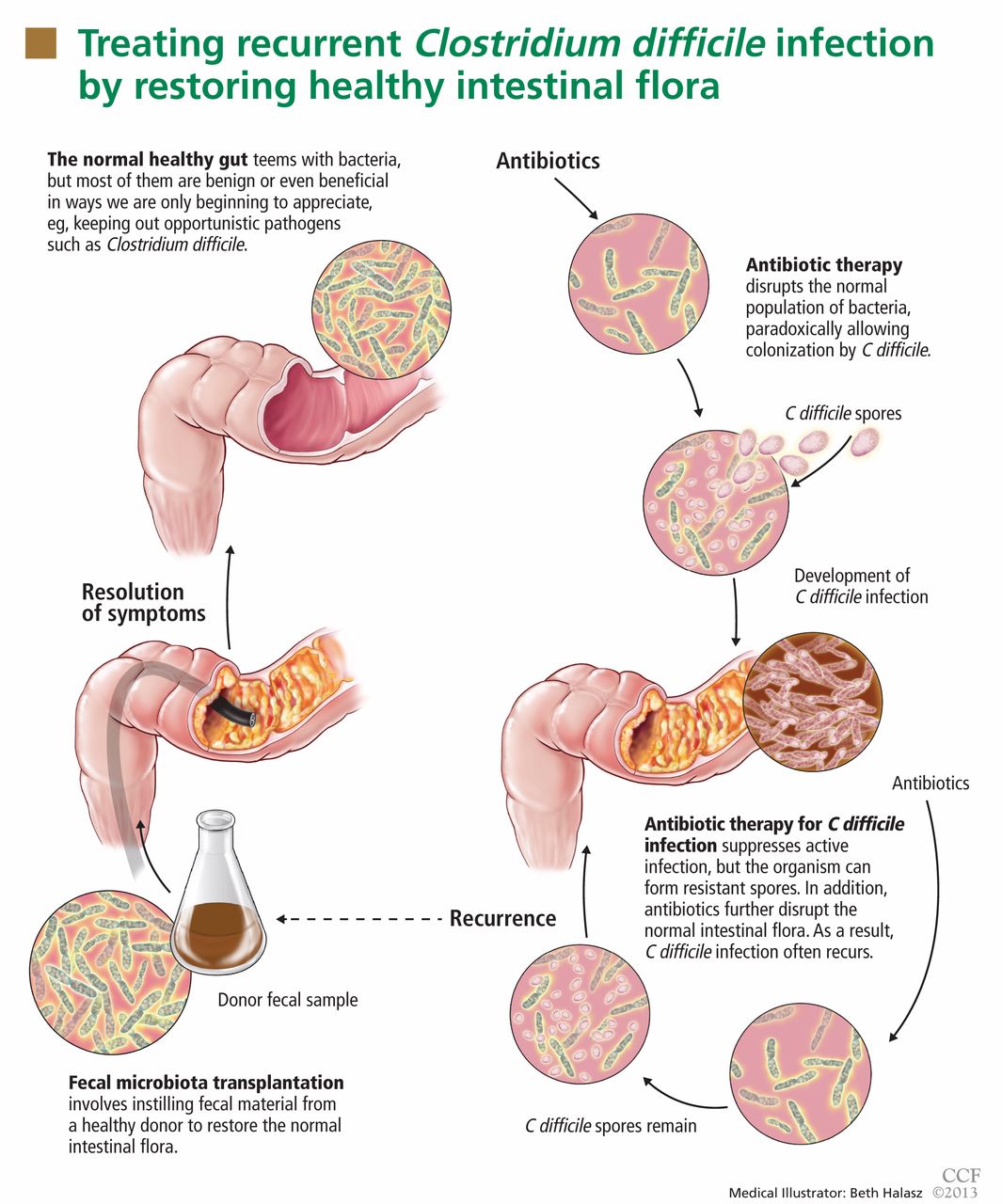
Fecal microbiota transplantation involves instilling processed stool that contains essential intestinal bacteria (eg, Bacteroides species) from a healthy screened donor into the diseased gastrointestinal tract of a suitable recipient (Figure 1).1
The aim of this procedure is to reestablish the normal composition of the gut flora, restore balance in metabolism, and stimulate both the acquired and the humoral immune responses in the intestinal mucosa after disruption of the normal flora.20–23 One study showed that patients who have recurrent C difficile infections have fewer protective microorganisms (ie, Firmicutes and Bacteriodetes) in their gut, but after fecal microbiota transplantation their microbiota was found to be similar to that of the donor, and their symptoms promptly resolved.18
STUDIES UP TO NOW
The principle of transplanting donor stool to treat various gastrointestinal diseases has been practiced in veterinary medicine for decades in a process known as transfaunation.24 Fecal microbiota transplantation was first performed in humans in the late 1950s in patients with fulminant pseudomembranous colitis that did not respond to standard antibiotic therapy for C difficile infection.25 Since then, a number of case reports and case series have described instillation of donor stool via nasogastric tube,26 via colonoscope,27–31 and via enema.32 Regardless of the protocols used, disease resolution has been shown in 92% of cases and few adverse effects have been reported, even though transmission of infectious pathogens is theoretically possible.33
A recent multicenter long-term follow-up study34 showed that diarrhea resolved within 90 days after fecal microbiota transplantation in 70 (91%) of 77 patients, while resolution of C difficile infection after a further course of antibiotics with or without repeating fecal microbiota transplantation was seen in 76 (98%) of 77 patients.34 Some patients were reported to have improvement of preexisting allergies, and a few patients developed peripheral neuropathy and autoimmune diseases such as Sjögren syndrome, idiopathic thrombocytopenic purpura, and rheumatoid arthritis.33
As the important role of the gut microbiota in resisting colonization by C difficile is becoming more recognized, scientists are beginning to understand and explore the additional potential benefits of fecal microbiota transplantation on other microbiotarelated dysfunctions.2 The Human Microbiome Project is focusing on characterizing and understanding the role of the microbial components of the human genetic and metabolic landscape in relation to human health and disease.35 Earlier observational studies showed fecal microbiota transplantation to be beneficial in inflammatory bowel disease, 36,37 irritable bowel syndrome,38,39 multiple sclerosis,40 rheumatologic40 and autoimmune diseases,41 and metabolic syndrome,42 likely owing to the role of the microbiota in immunity and energy metabolism. Although these reports may provide insight into the unexplored possibilities of fecal microbiota transplantation, further clinical investigations with randomized controlled trials are still necessary.
THE CURRENT PROTOCOL FOR FECAL MICROBIOTA TRANSPLANTATION
As yet, there is no standardized protocol for fecal microbiota transplantation, since no completed randomized trial supporting its efficacy and safety has been published. However, a group of experts in infectious disease and gastroenterology have published a formal standard practice guideline,19 as summarized below.
Primary indications for fecal microbiota transplantation
- Recurrent C difficile infection—at least three episodes of mild to moderate C difficile infection and failure of a 6- to 8-week taper with vancomycin with or without an alternative antibiotic such as rifaximin or nitazoxanide, or at least two episodes of severe C difficile infection resulting in hospitalization and associated with significant morbidity
- Mild to moderate C difficile infection not responding to standard therapy for at least 1 week
- Severe or fulminant C difficile colitis that has not responded to standard therapy after 48 hours.
Who is a likely donor?
The gut microbiota is continuously replenished with bacteria from the environment in which we live, and we constantly acquire organisms from people who live in that same environment. Hence, the preferred donor is someone who has intimate physical contact with the recipient.33,43,44 The preferred stool donor (in order of preference) is a spouse or significant partner, a family household member, or any other healthy donor.26,36
Who should not be a donor?
It is the responsibility of the physician performing the fecal microbiota transplantation to make sure that the possibility of transmitting disease to the recipient is minimized. Extensive history-taking and physical examination must never be omitted, since not all diseases or conditions can be detected by laboratory screening alone, especially if testing was done during the early stage or window period of a given disease.19 Nevertheless, the donor’s blood and stool should be screened for transmissible diseases such as human immunodeficiency virus (HIV), hepatitis, syphilis, enteric bacteria, parasites, and C difficile.
![]()
The recipient has the option to be tested for transmissible diseases such as HIV and hepatitis in order to avoid future questions about transmission after fecal microbiota transplantation. A positive screening test must always be verified with confirmatory testing.19
Table 2 summarizes the exclusion criteria and screening tests performed for donors according to the practice guidelines for fecal microbiota transplantation formulated by Bakken et al.19
Preprocedure instructions and stool preparation
![]()
The physician should orient both the donor and recipient regarding “do’s and don’ts” before fecal microbiota transplantation. Table 3 summarizes the preprocedure instructions and steps for stool preparation.
Route of administration
The route of administration may vary depending on the clinical situation. Upper-gastrointestinal administration is performed via nasogastric or nasojejunal tube or gastroscopy. Lower-gastrointestinal administration is performed via colonoscopy (the route of choice) or retention enema.
The upper-gastrointestinal route (nasogastric tube, jejunal catheter, or gastroscope). The nasogastric or nasojejunal tube or gastroscope is inserted into the upper-gastrointestinal tract, and positioning is confirmed by radiography. From 25 to 50 mL of stool suspension is drawn up in a syringe and instilled into the tubing followed by flushing with 25 mL of normal saline.26 Immediately after instillation, the tube is removed and the patient is allowed to go home and continue with his or her usual diet.
This approach is easier to perform, costs less, and poses lower risk of intestinal perforation than the colonoscopic approach. Disadvantages include the possibility that stool suspension may not reach distal areas of the colon, especially in patients with ileus and small-bowel obstruction. There is also a higher risk of bacterial overgrowth in elderly patients who have lower gastric acid levels.33
The lower-gastrointestinal route (colonoscopy, retention enema). Colonoscopy is currently considered the first-line approach for fecal microbiota transplantation.45 After giving informed consent, the patient undergoes standard colonoscopy under sedation. An initial colonoscopic examination is performed, and biopsy specimans are obtained if necessary. Approximately 20 mL of stool suspension is drawn up in a syringe and injected via the biopsy channel of the colonoscope every 5 to 10 cm as the scope is withdrawn, for a total volume of 250 to 500 mL.19,27 The patient should be advised to refrain from defecating for 30 to 45 minutes after fecal microbiota transplantation.46
This approach allows direct visualization of the entire colon, allowing instillation of stool suspension in certain areas where C difficile may predominate or hide (eg, in diverticuli).27,47 One disadvantage to this route of administration is the risk of colon perforation, especially if the patient has toxic colitis.
Instillation via retention enema may be done at home with a standard enema kit.32 Disadvantages include the need for multiple instillations over 3 to 5 days,36 back-leakage of stool suspension causing discomfort to patients, and stool suspension reaching only to the splenic flexure.48
MEASUREMENT OF OUTCOME
Fecal microbiota transplantation is considered successful if symptoms resolve and there is no relapse within 8 weeks. Testing for C difficile in asymptomatic patients is not recommended since patients can be colonized with C difficile without necessarily developing disease.19 There is currently no consensus on treatment recommendations for patients who do not respond to fecal microbiota transplantation, although some reports showed resolution of diarrhea after a repeat 2-week standard course of oral vancomycin26 or repeated instillation of feces collected from new donors.49
IS IT READY FOR PRIME TIME?
Fecal microbiota transplantation has been used primarily as an alternative treatment for recurrent C difficile infection, although other indications for its use are currently being identified and studied. This procedure is now being done in several specialized centers in the United States and abroad, and although the protocol may vary by institution, the clinical outcomes have been consistently promising.
The Fecal Therapy to Eliminate Associated Long-standing Diarrhea (FECAL) trial, currently underway, is the first randomized trial to assess the efficacy of fecal microbiota transplantation for treatment of recurrent C difficile infection.50 Clinical trials such as this one should satisfy our doubts about the efficacy of fecal microbiota transplantation and hopefully pave the way for its application in the near future.
An increasing number of patients are learning to overcome the “yuck factor” associated with fecal microbiota transplantation once they understand its safety and benefits.51 Moreover, the Human Microbiome Project is attempting to identify specific organisms in stool that may specifically treat C difficile infection, hence eliminating the need for whole-stool transplantation in the near future. Although fecal microbiota transplantation is still in its infancy, its low cost, safety, and effectiveness in treating recurrent C difficile infection will likely lead to the procedure becoming widely adopted in mainstream clinical practice.
Editor’s note: On January 16, 2013, after this article was completed, a randomized controlled trial of fecal microbiota transplantation was published in the New England Journal of Medicine. That trial, “Duodenal infusion of donor feces for recurrent Clostridium difficile,” found: “The infusion of donor feces was significantly more effective for the treatment of recurrent C difficile infection than the use of vancomycin.” The study is available online at http://www.nejm.org/doi/full/10.1056/NEJMoa1205037 (subscription required).
- Brandt L, Reddy S. Fecal microbiota transplantation for recurrent Clostridium difficile infection. J Clin Gastroenterol 2011; 45(suppl):S159–S167.
- Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol 2011; 9:88–96.
- Lipp MJ, Nero DC, Callahan MA. The impact of hospital-acquired Clostridium difficile. J Gastroenterol Hepatol 2012; 27:1733–1737.
- Kyne L, Sougioultzis S, McFarland LV, Kelly CP. Underlying disease severity as a major risk factor for nosocomial Clostridium difficile diarrhea. Infect Control Hosp Epidemiol 2002; 23:653–659.
- Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis 2002; 34:346–353.
- Gorbach SL. Antibiotics and Clostridium difficile. N Engl J Med 1999; 341:1690–1691.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Beales IL. Intravenous immunoglobulin for recurrent Clostridium difficile diarrhoea. Gut 2002; 51:456.
- O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 2009; 136:1913–1924.
- Louie TJ, Miller MA, Mullane KM, et al; OPT-80-003 Clinical Study Group. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 2011; 364:422–431.
- Kelly CP, LaMont JT. Clostridium difficile—more difficult than ever. N Engl J Med 2008; 359:1932–1940.
- Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin Infect Dis 2007; 44:846–848.
- Musher DM, Logan N, Hamill RJ, et al Nitazoxanide for the treatment of Clostridium difficile colitis. Clin Infect Dis 2006; 43:421–427.
- Louie TJ, Peppe J, Watt CK, et al. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis 2006; 43:411–420.
- Reid G, Younes JA, Van der Mei HC, Gloor GB, Knight R, Busscher JH. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Microbiol 2011; 9:27–38.
- Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996; 4:430–435.
- Vollaard EJ, Clasener HA. Colonization resistance. Antimicrob Agents Chemother 1994; 38:409–414.
- Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 2010; 44:354–360.
- Bakken JS, Borody T, Brandt LJ, et al; Fecal Microbiota Transplantation Workgroup. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol 2011; 9:1044–1049.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Neish AS, Gewirtz AT, Rao AS, et al. Non-pathogenic bacteria may block epithelial responses: Attenuation of IKB ubiquitination as a novel, physiologic mode of antiinflammation. Gastroenterology 2000; 118:A3754.
- Helwig U, Rizzello F, Cifone G, et al. Elevated IL-10 levels in pouch-tissue after probiotic therapy. Immunol Lett. 1999; 69:159.
- Rager KD, George LW, House JK, DePeters EJ. Evaluation of rumen transfaunation after surgical correction of left-sided displacement of the abomasum in cows. J Am Vet Med Assoc 2004; 225:915–920.
- Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 1958; 44:854–859.
- Aas J, Gessert CE, Bakken JS. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin Infect Dis 2003; 36:580–585.
- Yoon SS, Brandt LJ. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J Clin Gastroenterol 2010; 44:562–566.
- Mattila E, Uusitalo-Seppälä R, Wuorela M, et al. Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterology 2012; 142:490–496.
- Garborg K, Waagsbø B, Stallemo A, Matre J, Sundøy A. Results of faecal donor instillation therapy for recurrent Clostridium difficile-associated diarrhoea. Scand J Infect Dis 2010; 42:857–861.
- Mellow MH, Kanatzar A. Colonoscopic fecal bacteriotherapy in the treatment of recurrent Clostridium difficile infection–results and follow-up. J Okla State Med Assoc 2011; 104:89–91.
- Rohlke F, Surawicz CM, Stollman N. Fecal flora reconstitution for recurrent Clostridium difficile infection: results and methodology. J Clin Gastroenterol 2010; 44:567–570.
- Silverman MS, Davis I, Pillai DR. Success of self-administered home fecal transplantation for chronic Clostridium difficile infection. Clin Gastroenterol Hepatol 2010; 8:471–473.
- Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 2011; 53:994–1002.
- Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol 2012; 107:1079–1087.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- Borody TJ, Warren EF, Leis S, Surace R, Ashman O. Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol 2003; 37:42–47.
- Borody TJ, Torres M, Campbell J, et al. Reversal of inflammatory bowel disease (IBD) with recurrent fecal microbiota transplants (FMT). Am J Gastroenterol 2011; 106:S352.
- Andrews P, Borody TJ, Shortis NP, Thompson S. Bacteriotherapy for chronic constipation—long term follow-up. (abstract). Gastroenterology 1995; 108:A563.
- Borody TJ. Bacteriotherapy for chronic fatigue syndrome: a long-term follow up study. Presented at the 1995 Chronic Fatigue Syndrome National Consensus Conference.
- Borody TJ, Leis S, Campbell J, et al. Fecal microbiota transplantation (FMT) in multiple sclerosis (MS) (abstract). Am J Gastroenterol 2011; 106:S352.
- Borody TJ, Campbell J, Torres M, et al. Reversal of idiopathic thrombocytopenic purpura (ITP) with fecal microbiota transplantation (FMT) (abstract). Am J Gastroenterol 2011; 106:S352.
- Vrieze AF, Holleman MJ, Serlie MT, Ackermans GM, Dallinga-Thie GM, Groen AK. Metabolic effects of transplanting gut microbiota from lean donors to subjects with metabolic syndrome (abstract). Diabetologia 2010; 53:S44.
- Bakken JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 2009; 15:285–289.
- Bjørneklett A. [To repair an ecosystem] (In Norwegian). Tidsskr Nor Laegeforen 1998; 118:1026.
- Brandt LJ, Borody TJ, Campbell J. Endoscopic fecal microbiota transplantation: “first-line” treatment for severe Clostridium difficile infection? J Clin Gastroenterol 2011; 45:655–657.
- Kelly CR, de Leon L, Jasutkar N. Fecal microbiota transplantation for relapsing Clostridium difficile infection in 26 patients: methodology and results. J Clin Gastroenterol 2012; 46:145–149.
- Thanjan AJ, Southern W, Anand N, et al. Is Clostridium difficile infection (CDI) more difficult to eradicate in patients with diverticulosis? (abstract) Am J Gastroenterol 2008; 103:S195.
- Persky SE, Brandt LJ. Treatment of recurrent Clostridium difficile-associated diarrhea by administration of donated stool directly through a colonoscope. Am J Gastroenterol 2000; 95:3283–3285.
- Nieuwdorp M, van Nood E, Speelman P, et al. [Treatment of recurrent Clostridium difficile-associated diarrhoea with a suspension of donor faeces] (In Dutch). Ned Tijdschr Geneeskd 2008; 152:1927–1932.
- van Nood E, Speelman P, Kuijper EJ, Keller JJ. Struggling with recurrent Clostridium difficile infections: is donor faeces the solution? Euro Surveill 2009; 14. doi:pii:19316.
- Kahn SA, Gorawara-Bhat R, Rubin DT. Fecal bacteriotherapy for ulcerative colitis: patients are ready, are we? Inflamm Bowel Dis 2012; 18:676–684.
If you had a serious disease, would you agree to an alternative treatment that was cheap, safe, and effective—but seemed disgusting? Would you recommend it to patients?
Such a disease is recurrent Clostridium difficile infection, and such a treatment is fecal microbiota transplantation—instillation of blenderized feces from a healthy donor (ideally, the patient’s spouse or “significant other”) into the patient’s colon to restore a healthy population of bacteria.1,2 The rationale behind this procedure is simple: antibiotics and other factors disrupt the normal balance of the colonic flora, allowing C difficile to proliferate, but the imbalance can be corrected by reintroducing the normal flora.1
In this article, we will review how recurrent C difficile infection occurs and the importance of the gut microbiota in resisting colonization with this pathogen. We will also describe the protocol used for fecal microbiota transplantation.
C DIFFICILE INFECTION OFTEN RECURS
C difficile is the most common cause of hospital-acquired diarrhea and an important cause of morbidity and death in hospitalized patients.3,4 The cost of this infection is estimated to be more than $1.1 billion per year and its incidence is rising, partly because of the emergence of more-virulent strains that make treatment of recurrent infection more difficult.5,6
C difficile infection is characterized by diarrhea associated with findings suggestive of pseudomembranous colitis or, in fulminant cases, ileus or megacolon.7 Recurrent C difficile infection is defined as the return of symptoms within 8 weeks after successful treatment.7
C difficile produces two types of toxins. Toxin A is an enterotoxin, causing increased intestinal permeability and fluid secretion, while toxin B is a cytotoxin, causing intense colonic inflammation. People who have a poor host immune response to these toxins tend to develop more diarrhea and colonic inflammation.8
A more virulent strain of C difficile has emerged. Known as BI/NAP1/027, this strain is resistant to quinolones, and it also produces a binary toxin that has a partial gene deletion that allows for increased production of toxins A and B in vitro.9,10 More cases of severe and recurrent C difficile infection have been associated with the increasing number of people infected with this hypervirulent strain.9,10
C difficile infection recurs in about 20% to 30% of cases after antibiotic treatment for it, usually within 30 days, and the risk of a subsequent episode doubles after two or more occurrences.10,11 Metronidazole (Flagyl) and vancomycin are the primary treatments; alternative treatments include fidaxomicin (Dificid), 10 rifaximin (Xifaxan),12 nitazoxanide,13 and tolevamer (a novel polymer that binds C difficile toxins).14
Table 1 summarizes the treatment regimen for C difficile infection in adults, based on clinical practice guidelines from the US Centers for Disease Control and Prevention (CDC).7
THE NORMAL GUT MICROBIOTA KEEPS PATHOGENS OUT
Immediately after birth, the sterile human gut becomes colonized by a diverse community of microorganisms.15 This gut microbiota performs various functions, such as synthesizing vitamin K and vitamin B complex, helping digest food, maintaining the mucosal integrity of the gut, and priming the mucosal immune response to maintain homeostasis of commensal microbiota.16
However, the most important role of the gut microbiota is “colonization resistance” or preventing exogenous or potentially pathogenic organisms from establishing a colony within the gut.17 It involves competition for nutrients and occupation of binding sites on the gut epithelium by indigenous flora.16 Other factors such as the mucosal barrier, salivation, swallowing, gastric acidity, desquamation of mucosal membrane cells, intestinal motility, and secretion of antibodies also play major roles in colonization resistance.17
ANTIBIOTICS DISRUPT THE GUT FLORA
Physical or chemical injuries (the latter by antimicrobial or antineoplastic agents, eg) may disrupt the gut microbiota. In this situation, opportunistic pathogens such as C difficile colonize the gut mucosa, stimulate an immune reaction, and release toxins that cause diarrhea and inflammation.18C difficile will try to compete for nutrients and adhesion sites until it dominates the intestinal tract.
When C difficile spores are ingested, they replicate in the gut and eventually release toxins. Antibiotic therapy may eliminate C difficile bacteria but not the spores; hence, C difficile infection can recur after the antibiotic is discontinued unless the indigenous bacteria can restrain C difficile from spreading.19
HOW DOES FECAL MICROBIOTA TRANSPLANTATION WORK?
Fecal microbiota transplantation involves instilling processed stool that contains essential intestinal bacteria (eg, Bacteroides species) from a healthy screened donor into the diseased gastrointestinal tract of a suitable recipient (Figure 1).1
The aim of this procedure is to reestablish the normal composition of the gut flora, restore balance in metabolism, and stimulate both the acquired and the humoral immune responses in the intestinal mucosa after disruption of the normal flora.20–23 One study showed that patients who have recurrent C difficile infections have fewer protective microorganisms (ie, Firmicutes and Bacteriodetes) in their gut, but after fecal microbiota transplantation their microbiota was found to be similar to that of the donor, and their symptoms promptly resolved.18
STUDIES UP TO NOW
The principle of transplanting donor stool to treat various gastrointestinal diseases has been practiced in veterinary medicine for decades in a process known as transfaunation.24 Fecal microbiota transplantation was first performed in humans in the late 1950s in patients with fulminant pseudomembranous colitis that did not respond to standard antibiotic therapy for C difficile infection.25 Since then, a number of case reports and case series have described instillation of donor stool via nasogastric tube,26 via colonoscope,27–31 and via enema.32 Regardless of the protocols used, disease resolution has been shown in 92% of cases and few adverse effects have been reported, even though transmission of infectious pathogens is theoretically possible.33
A recent multicenter long-term follow-up study34 showed that diarrhea resolved within 90 days after fecal microbiota transplantation in 70 (91%) of 77 patients, while resolution of C difficile infection after a further course of antibiotics with or without repeating fecal microbiota transplantation was seen in 76 (98%) of 77 patients.34 Some patients were reported to have improvement of preexisting allergies, and a few patients developed peripheral neuropathy and autoimmune diseases such as Sjögren syndrome, idiopathic thrombocytopenic purpura, and rheumatoid arthritis.33
As the important role of the gut microbiota in resisting colonization by C difficile is becoming more recognized, scientists are beginning to understand and explore the additional potential benefits of fecal microbiota transplantation on other microbiotarelated dysfunctions.2 The Human Microbiome Project is focusing on characterizing and understanding the role of the microbial components of the human genetic and metabolic landscape in relation to human health and disease.35 Earlier observational studies showed fecal microbiota transplantation to be beneficial in inflammatory bowel disease, 36,37 irritable bowel syndrome,38,39 multiple sclerosis,40 rheumatologic40 and autoimmune diseases,41 and metabolic syndrome,42 likely owing to the role of the microbiota in immunity and energy metabolism. Although these reports may provide insight into the unexplored possibilities of fecal microbiota transplantation, further clinical investigations with randomized controlled trials are still necessary.
THE CURRENT PROTOCOL FOR FECAL MICROBIOTA TRANSPLANTATION
As yet, there is no standardized protocol for fecal microbiota transplantation, since no completed randomized trial supporting its efficacy and safety has been published. However, a group of experts in infectious disease and gastroenterology have published a formal standard practice guideline,19 as summarized below.
Primary indications for fecal microbiota transplantation
- Recurrent C difficile infection—at least three episodes of mild to moderate C difficile infection and failure of a 6- to 8-week taper with vancomycin with or without an alternative antibiotic such as rifaximin or nitazoxanide, or at least two episodes of severe C difficile infection resulting in hospitalization and associated with significant morbidity
- Mild to moderate C difficile infection not responding to standard therapy for at least 1 week
- Severe or fulminant C difficile colitis that has not responded to standard therapy after 48 hours.
Who is a likely donor?
The gut microbiota is continuously replenished with bacteria from the environment in which we live, and we constantly acquire organisms from people who live in that same environment. Hence, the preferred donor is someone who has intimate physical contact with the recipient.33,43,44 The preferred stool donor (in order of preference) is a spouse or significant partner, a family household member, or any other healthy donor.26,36
Who should not be a donor?
It is the responsibility of the physician performing the fecal microbiota transplantation to make sure that the possibility of transmitting disease to the recipient is minimized. Extensive history-taking and physical examination must never be omitted, since not all diseases or conditions can be detected by laboratory screening alone, especially if testing was done during the early stage or window period of a given disease.19 Nevertheless, the donor’s blood and stool should be screened for transmissible diseases such as human immunodeficiency virus (HIV), hepatitis, syphilis, enteric bacteria, parasites, and C difficile.
![]()
The recipient has the option to be tested for transmissible diseases such as HIV and hepatitis in order to avoid future questions about transmission after fecal microbiota transplantation. A positive screening test must always be verified with confirmatory testing.19
Table 2 summarizes the exclusion criteria and screening tests performed for donors according to the practice guidelines for fecal microbiota transplantation formulated by Bakken et al.19
Preprocedure instructions and stool preparation
![]()
The physician should orient both the donor and recipient regarding “do’s and don’ts” before fecal microbiota transplantation. Table 3 summarizes the preprocedure instructions and steps for stool preparation.
Route of administration
The route of administration may vary depending on the clinical situation. Upper-gastrointestinal administration is performed via nasogastric or nasojejunal tube or gastroscopy. Lower-gastrointestinal administration is performed via colonoscopy (the route of choice) or retention enema.
The upper-gastrointestinal route (nasogastric tube, jejunal catheter, or gastroscope). The nasogastric or nasojejunal tube or gastroscope is inserted into the upper-gastrointestinal tract, and positioning is confirmed by radiography. From 25 to 50 mL of stool suspension is drawn up in a syringe and instilled into the tubing followed by flushing with 25 mL of normal saline.26 Immediately after instillation, the tube is removed and the patient is allowed to go home and continue with his or her usual diet.
This approach is easier to perform, costs less, and poses lower risk of intestinal perforation than the colonoscopic approach. Disadvantages include the possibility that stool suspension may not reach distal areas of the colon, especially in patients with ileus and small-bowel obstruction. There is also a higher risk of bacterial overgrowth in elderly patients who have lower gastric acid levels.33
The lower-gastrointestinal route (colonoscopy, retention enema). Colonoscopy is currently considered the first-line approach for fecal microbiota transplantation.45 After giving informed consent, the patient undergoes standard colonoscopy under sedation. An initial colonoscopic examination is performed, and biopsy specimans are obtained if necessary. Approximately 20 mL of stool suspension is drawn up in a syringe and injected via the biopsy channel of the colonoscope every 5 to 10 cm as the scope is withdrawn, for a total volume of 250 to 500 mL.19,27 The patient should be advised to refrain from defecating for 30 to 45 minutes after fecal microbiota transplantation.46
This approach allows direct visualization of the entire colon, allowing instillation of stool suspension in certain areas where C difficile may predominate or hide (eg, in diverticuli).27,47 One disadvantage to this route of administration is the risk of colon perforation, especially if the patient has toxic colitis.
Instillation via retention enema may be done at home with a standard enema kit.32 Disadvantages include the need for multiple instillations over 3 to 5 days,36 back-leakage of stool suspension causing discomfort to patients, and stool suspension reaching only to the splenic flexure.48
MEASUREMENT OF OUTCOME
Fecal microbiota transplantation is considered successful if symptoms resolve and there is no relapse within 8 weeks. Testing for C difficile in asymptomatic patients is not recommended since patients can be colonized with C difficile without necessarily developing disease.19 There is currently no consensus on treatment recommendations for patients who do not respond to fecal microbiota transplantation, although some reports showed resolution of diarrhea after a repeat 2-week standard course of oral vancomycin26 or repeated instillation of feces collected from new donors.49
IS IT READY FOR PRIME TIME?
Fecal microbiota transplantation has been used primarily as an alternative treatment for recurrent C difficile infection, although other indications for its use are currently being identified and studied. This procedure is now being done in several specialized centers in the United States and abroad, and although the protocol may vary by institution, the clinical outcomes have been consistently promising.
The Fecal Therapy to Eliminate Associated Long-standing Diarrhea (FECAL) trial, currently underway, is the first randomized trial to assess the efficacy of fecal microbiota transplantation for treatment of recurrent C difficile infection.50 Clinical trials such as this one should satisfy our doubts about the efficacy of fecal microbiota transplantation and hopefully pave the way for its application in the near future.
An increasing number of patients are learning to overcome the “yuck factor” associated with fecal microbiota transplantation once they understand its safety and benefits.51 Moreover, the Human Microbiome Project is attempting to identify specific organisms in stool that may specifically treat C difficile infection, hence eliminating the need for whole-stool transplantation in the near future. Although fecal microbiota transplantation is still in its infancy, its low cost, safety, and effectiveness in treating recurrent C difficile infection will likely lead to the procedure becoming widely adopted in mainstream clinical practice.
Editor’s note: On January 16, 2013, after this article was completed, a randomized controlled trial of fecal microbiota transplantation was published in the New England Journal of Medicine. That trial, “Duodenal infusion of donor feces for recurrent Clostridium difficile,” found: “The infusion of donor feces was significantly more effective for the treatment of recurrent C difficile infection than the use of vancomycin.” The study is available online at http://www.nejm.org/doi/full/10.1056/NEJMoa1205037 (subscription required).
If you had a serious disease, would you agree to an alternative treatment that was cheap, safe, and effective—but seemed disgusting? Would you recommend it to patients?
Such a disease is recurrent Clostridium difficile infection, and such a treatment is fecal microbiota transplantation—instillation of blenderized feces from a healthy donor (ideally, the patient’s spouse or “significant other”) into the patient’s colon to restore a healthy population of bacteria.1,2 The rationale behind this procedure is simple: antibiotics and other factors disrupt the normal balance of the colonic flora, allowing C difficile to proliferate, but the imbalance can be corrected by reintroducing the normal flora.1
In this article, we will review how recurrent C difficile infection occurs and the importance of the gut microbiota in resisting colonization with this pathogen. We will also describe the protocol used for fecal microbiota transplantation.
C DIFFICILE INFECTION OFTEN RECURS
C difficile is the most common cause of hospital-acquired diarrhea and an important cause of morbidity and death in hospitalized patients.3,4 The cost of this infection is estimated to be more than $1.1 billion per year and its incidence is rising, partly because of the emergence of more-virulent strains that make treatment of recurrent infection more difficult.5,6
C difficile infection is characterized by diarrhea associated with findings suggestive of pseudomembranous colitis or, in fulminant cases, ileus or megacolon.7 Recurrent C difficile infection is defined as the return of symptoms within 8 weeks after successful treatment.7
C difficile produces two types of toxins. Toxin A is an enterotoxin, causing increased intestinal permeability and fluid secretion, while toxin B is a cytotoxin, causing intense colonic inflammation. People who have a poor host immune response to these toxins tend to develop more diarrhea and colonic inflammation.8
A more virulent strain of C difficile has emerged. Known as BI/NAP1/027, this strain is resistant to quinolones, and it also produces a binary toxin that has a partial gene deletion that allows for increased production of toxins A and B in vitro.9,10 More cases of severe and recurrent C difficile infection have been associated with the increasing number of people infected with this hypervirulent strain.9,10
C difficile infection recurs in about 20% to 30% of cases after antibiotic treatment for it, usually within 30 days, and the risk of a subsequent episode doubles after two or more occurrences.10,11 Metronidazole (Flagyl) and vancomycin are the primary treatments; alternative treatments include fidaxomicin (Dificid), 10 rifaximin (Xifaxan),12 nitazoxanide,13 and tolevamer (a novel polymer that binds C difficile toxins).14
Table 1 summarizes the treatment regimen for C difficile infection in adults, based on clinical practice guidelines from the US Centers for Disease Control and Prevention (CDC).7
THE NORMAL GUT MICROBIOTA KEEPS PATHOGENS OUT
Immediately after birth, the sterile human gut becomes colonized by a diverse community of microorganisms.15 This gut microbiota performs various functions, such as synthesizing vitamin K and vitamin B complex, helping digest food, maintaining the mucosal integrity of the gut, and priming the mucosal immune response to maintain homeostasis of commensal microbiota.16
However, the most important role of the gut microbiota is “colonization resistance” or preventing exogenous or potentially pathogenic organisms from establishing a colony within the gut.17 It involves competition for nutrients and occupation of binding sites on the gut epithelium by indigenous flora.16 Other factors such as the mucosal barrier, salivation, swallowing, gastric acidity, desquamation of mucosal membrane cells, intestinal motility, and secretion of antibodies also play major roles in colonization resistance.17
ANTIBIOTICS DISRUPT THE GUT FLORA
Physical or chemical injuries (the latter by antimicrobial or antineoplastic agents, eg) may disrupt the gut microbiota. In this situation, opportunistic pathogens such as C difficile colonize the gut mucosa, stimulate an immune reaction, and release toxins that cause diarrhea and inflammation.18C difficile will try to compete for nutrients and adhesion sites until it dominates the intestinal tract.
When C difficile spores are ingested, they replicate in the gut and eventually release toxins. Antibiotic therapy may eliminate C difficile bacteria but not the spores; hence, C difficile infection can recur after the antibiotic is discontinued unless the indigenous bacteria can restrain C difficile from spreading.19
HOW DOES FECAL MICROBIOTA TRANSPLANTATION WORK?
Fecal microbiota transplantation involves instilling processed stool that contains essential intestinal bacteria (eg, Bacteroides species) from a healthy screened donor into the diseased gastrointestinal tract of a suitable recipient (Figure 1).1
The aim of this procedure is to reestablish the normal composition of the gut flora, restore balance in metabolism, and stimulate both the acquired and the humoral immune responses in the intestinal mucosa after disruption of the normal flora.20–23 One study showed that patients who have recurrent C difficile infections have fewer protective microorganisms (ie, Firmicutes and Bacteriodetes) in their gut, but after fecal microbiota transplantation their microbiota was found to be similar to that of the donor, and their symptoms promptly resolved.18
STUDIES UP TO NOW
The principle of transplanting donor stool to treat various gastrointestinal diseases has been practiced in veterinary medicine for decades in a process known as transfaunation.24 Fecal microbiota transplantation was first performed in humans in the late 1950s in patients with fulminant pseudomembranous colitis that did not respond to standard antibiotic therapy for C difficile infection.25 Since then, a number of case reports and case series have described instillation of donor stool via nasogastric tube,26 via colonoscope,27–31 and via enema.32 Regardless of the protocols used, disease resolution has been shown in 92% of cases and few adverse effects have been reported, even though transmission of infectious pathogens is theoretically possible.33
A recent multicenter long-term follow-up study34 showed that diarrhea resolved within 90 days after fecal microbiota transplantation in 70 (91%) of 77 patients, while resolution of C difficile infection after a further course of antibiotics with or without repeating fecal microbiota transplantation was seen in 76 (98%) of 77 patients.34 Some patients were reported to have improvement of preexisting allergies, and a few patients developed peripheral neuropathy and autoimmune diseases such as Sjögren syndrome, idiopathic thrombocytopenic purpura, and rheumatoid arthritis.33
As the important role of the gut microbiota in resisting colonization by C difficile is becoming more recognized, scientists are beginning to understand and explore the additional potential benefits of fecal microbiota transplantation on other microbiotarelated dysfunctions.2 The Human Microbiome Project is focusing on characterizing and understanding the role of the microbial components of the human genetic and metabolic landscape in relation to human health and disease.35 Earlier observational studies showed fecal microbiota transplantation to be beneficial in inflammatory bowel disease, 36,37 irritable bowel syndrome,38,39 multiple sclerosis,40 rheumatologic40 and autoimmune diseases,41 and metabolic syndrome,42 likely owing to the role of the microbiota in immunity and energy metabolism. Although these reports may provide insight into the unexplored possibilities of fecal microbiota transplantation, further clinical investigations with randomized controlled trials are still necessary.
THE CURRENT PROTOCOL FOR FECAL MICROBIOTA TRANSPLANTATION
As yet, there is no standardized protocol for fecal microbiota transplantation, since no completed randomized trial supporting its efficacy and safety has been published. However, a group of experts in infectious disease and gastroenterology have published a formal standard practice guideline,19 as summarized below.
Primary indications for fecal microbiota transplantation
- Recurrent C difficile infection—at least three episodes of mild to moderate C difficile infection and failure of a 6- to 8-week taper with vancomycin with or without an alternative antibiotic such as rifaximin or nitazoxanide, or at least two episodes of severe C difficile infection resulting in hospitalization and associated with significant morbidity
- Mild to moderate C difficile infection not responding to standard therapy for at least 1 week
- Severe or fulminant C difficile colitis that has not responded to standard therapy after 48 hours.
Who is a likely donor?
The gut microbiota is continuously replenished with bacteria from the environment in which we live, and we constantly acquire organisms from people who live in that same environment. Hence, the preferred donor is someone who has intimate physical contact with the recipient.33,43,44 The preferred stool donor (in order of preference) is a spouse or significant partner, a family household member, or any other healthy donor.26,36
Who should not be a donor?
It is the responsibility of the physician performing the fecal microbiota transplantation to make sure that the possibility of transmitting disease to the recipient is minimized. Extensive history-taking and physical examination must never be omitted, since not all diseases or conditions can be detected by laboratory screening alone, especially if testing was done during the early stage or window period of a given disease.19 Nevertheless, the donor’s blood and stool should be screened for transmissible diseases such as human immunodeficiency virus (HIV), hepatitis, syphilis, enteric bacteria, parasites, and C difficile.
![]()
The recipient has the option to be tested for transmissible diseases such as HIV and hepatitis in order to avoid future questions about transmission after fecal microbiota transplantation. A positive screening test must always be verified with confirmatory testing.19
Table 2 summarizes the exclusion criteria and screening tests performed for donors according to the practice guidelines for fecal microbiota transplantation formulated by Bakken et al.19
Preprocedure instructions and stool preparation
![]()
The physician should orient both the donor and recipient regarding “do’s and don’ts” before fecal microbiota transplantation. Table 3 summarizes the preprocedure instructions and steps for stool preparation.
Route of administration
The route of administration may vary depending on the clinical situation. Upper-gastrointestinal administration is performed via nasogastric or nasojejunal tube or gastroscopy. Lower-gastrointestinal administration is performed via colonoscopy (the route of choice) or retention enema.
The upper-gastrointestinal route (nasogastric tube, jejunal catheter, or gastroscope). The nasogastric or nasojejunal tube or gastroscope is inserted into the upper-gastrointestinal tract, and positioning is confirmed by radiography. From 25 to 50 mL of stool suspension is drawn up in a syringe and instilled into the tubing followed by flushing with 25 mL of normal saline.26 Immediately after instillation, the tube is removed and the patient is allowed to go home and continue with his or her usual diet.
This approach is easier to perform, costs less, and poses lower risk of intestinal perforation than the colonoscopic approach. Disadvantages include the possibility that stool suspension may not reach distal areas of the colon, especially in patients with ileus and small-bowel obstruction. There is also a higher risk of bacterial overgrowth in elderly patients who have lower gastric acid levels.33
The lower-gastrointestinal route (colonoscopy, retention enema). Colonoscopy is currently considered the first-line approach for fecal microbiota transplantation.45 After giving informed consent, the patient undergoes standard colonoscopy under sedation. An initial colonoscopic examination is performed, and biopsy specimans are obtained if necessary. Approximately 20 mL of stool suspension is drawn up in a syringe and injected via the biopsy channel of the colonoscope every 5 to 10 cm as the scope is withdrawn, for a total volume of 250 to 500 mL.19,27 The patient should be advised to refrain from defecating for 30 to 45 minutes after fecal microbiota transplantation.46
This approach allows direct visualization of the entire colon, allowing instillation of stool suspension in certain areas where C difficile may predominate or hide (eg, in diverticuli).27,47 One disadvantage to this route of administration is the risk of colon perforation, especially if the patient has toxic colitis.
Instillation via retention enema may be done at home with a standard enema kit.32 Disadvantages include the need for multiple instillations over 3 to 5 days,36 back-leakage of stool suspension causing discomfort to patients, and stool suspension reaching only to the splenic flexure.48
MEASUREMENT OF OUTCOME
Fecal microbiota transplantation is considered successful if symptoms resolve and there is no relapse within 8 weeks. Testing for C difficile in asymptomatic patients is not recommended since patients can be colonized with C difficile without necessarily developing disease.19 There is currently no consensus on treatment recommendations for patients who do not respond to fecal microbiota transplantation, although some reports showed resolution of diarrhea after a repeat 2-week standard course of oral vancomycin26 or repeated instillation of feces collected from new donors.49
IS IT READY FOR PRIME TIME?
Fecal microbiota transplantation has been used primarily as an alternative treatment for recurrent C difficile infection, although other indications for its use are currently being identified and studied. This procedure is now being done in several specialized centers in the United States and abroad, and although the protocol may vary by institution, the clinical outcomes have been consistently promising.
The Fecal Therapy to Eliminate Associated Long-standing Diarrhea (FECAL) trial, currently underway, is the first randomized trial to assess the efficacy of fecal microbiota transplantation for treatment of recurrent C difficile infection.50 Clinical trials such as this one should satisfy our doubts about the efficacy of fecal microbiota transplantation and hopefully pave the way for its application in the near future.
An increasing number of patients are learning to overcome the “yuck factor” associated with fecal microbiota transplantation once they understand its safety and benefits.51 Moreover, the Human Microbiome Project is attempting to identify specific organisms in stool that may specifically treat C difficile infection, hence eliminating the need for whole-stool transplantation in the near future. Although fecal microbiota transplantation is still in its infancy, its low cost, safety, and effectiveness in treating recurrent C difficile infection will likely lead to the procedure becoming widely adopted in mainstream clinical practice.
Editor’s note: On January 16, 2013, after this article was completed, a randomized controlled trial of fecal microbiota transplantation was published in the New England Journal of Medicine. That trial, “Duodenal infusion of donor feces for recurrent Clostridium difficile,” found: “The infusion of donor feces was significantly more effective for the treatment of recurrent C difficile infection than the use of vancomycin.” The study is available online at http://www.nejm.org/doi/full/10.1056/NEJMoa1205037 (subscription required).
- Brandt L, Reddy S. Fecal microbiota transplantation for recurrent Clostridium difficile infection. J Clin Gastroenterol 2011; 45(suppl):S159–S167.
- Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol 2011; 9:88–96.
- Lipp MJ, Nero DC, Callahan MA. The impact of hospital-acquired Clostridium difficile. J Gastroenterol Hepatol 2012; 27:1733–1737.
- Kyne L, Sougioultzis S, McFarland LV, Kelly CP. Underlying disease severity as a major risk factor for nosocomial Clostridium difficile diarrhea. Infect Control Hosp Epidemiol 2002; 23:653–659.
- Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis 2002; 34:346–353.
- Gorbach SL. Antibiotics and Clostridium difficile. N Engl J Med 1999; 341:1690–1691.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Beales IL. Intravenous immunoglobulin for recurrent Clostridium difficile diarrhoea. Gut 2002; 51:456.
- O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 2009; 136:1913–1924.
- Louie TJ, Miller MA, Mullane KM, et al; OPT-80-003 Clinical Study Group. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 2011; 364:422–431.
- Kelly CP, LaMont JT. Clostridium difficile—more difficult than ever. N Engl J Med 2008; 359:1932–1940.
- Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin Infect Dis 2007; 44:846–848.
- Musher DM, Logan N, Hamill RJ, et al Nitazoxanide for the treatment of Clostridium difficile colitis. Clin Infect Dis 2006; 43:421–427.
- Louie TJ, Peppe J, Watt CK, et al. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis 2006; 43:411–420.
- Reid G, Younes JA, Van der Mei HC, Gloor GB, Knight R, Busscher JH. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Microbiol 2011; 9:27–38.
- Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996; 4:430–435.
- Vollaard EJ, Clasener HA. Colonization resistance. Antimicrob Agents Chemother 1994; 38:409–414.
- Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 2010; 44:354–360.
- Bakken JS, Borody T, Brandt LJ, et al; Fecal Microbiota Transplantation Workgroup. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol 2011; 9:1044–1049.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Neish AS, Gewirtz AT, Rao AS, et al. Non-pathogenic bacteria may block epithelial responses: Attenuation of IKB ubiquitination as a novel, physiologic mode of antiinflammation. Gastroenterology 2000; 118:A3754.
- Helwig U, Rizzello F, Cifone G, et al. Elevated IL-10 levels in pouch-tissue after probiotic therapy. Immunol Lett. 1999; 69:159.
- Rager KD, George LW, House JK, DePeters EJ. Evaluation of rumen transfaunation after surgical correction of left-sided displacement of the abomasum in cows. J Am Vet Med Assoc 2004; 225:915–920.
- Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 1958; 44:854–859.
- Aas J, Gessert CE, Bakken JS. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin Infect Dis 2003; 36:580–585.
- Yoon SS, Brandt LJ. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J Clin Gastroenterol 2010; 44:562–566.
- Mattila E, Uusitalo-Seppälä R, Wuorela M, et al. Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterology 2012; 142:490–496.
- Garborg K, Waagsbø B, Stallemo A, Matre J, Sundøy A. Results of faecal donor instillation therapy for recurrent Clostridium difficile-associated diarrhoea. Scand J Infect Dis 2010; 42:857–861.
- Mellow MH, Kanatzar A. Colonoscopic fecal bacteriotherapy in the treatment of recurrent Clostridium difficile infection–results and follow-up. J Okla State Med Assoc 2011; 104:89–91.
- Rohlke F, Surawicz CM, Stollman N. Fecal flora reconstitution for recurrent Clostridium difficile infection: results and methodology. J Clin Gastroenterol 2010; 44:567–570.
- Silverman MS, Davis I, Pillai DR. Success of self-administered home fecal transplantation for chronic Clostridium difficile infection. Clin Gastroenterol Hepatol 2010; 8:471–473.
- Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 2011; 53:994–1002.
- Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol 2012; 107:1079–1087.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- Borody TJ, Warren EF, Leis S, Surace R, Ashman O. Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol 2003; 37:42–47.
- Borody TJ, Torres M, Campbell J, et al. Reversal of inflammatory bowel disease (IBD) with recurrent fecal microbiota transplants (FMT). Am J Gastroenterol 2011; 106:S352.
- Andrews P, Borody TJ, Shortis NP, Thompson S. Bacteriotherapy for chronic constipation—long term follow-up. (abstract). Gastroenterology 1995; 108:A563.
- Borody TJ. Bacteriotherapy for chronic fatigue syndrome: a long-term follow up study. Presented at the 1995 Chronic Fatigue Syndrome National Consensus Conference.
- Borody TJ, Leis S, Campbell J, et al. Fecal microbiota transplantation (FMT) in multiple sclerosis (MS) (abstract). Am J Gastroenterol 2011; 106:S352.
- Borody TJ, Campbell J, Torres M, et al. Reversal of idiopathic thrombocytopenic purpura (ITP) with fecal microbiota transplantation (FMT) (abstract). Am J Gastroenterol 2011; 106:S352.
- Vrieze AF, Holleman MJ, Serlie MT, Ackermans GM, Dallinga-Thie GM, Groen AK. Metabolic effects of transplanting gut microbiota from lean donors to subjects with metabolic syndrome (abstract). Diabetologia 2010; 53:S44.
- Bakken JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 2009; 15:285–289.
- Bjørneklett A. [To repair an ecosystem] (In Norwegian). Tidsskr Nor Laegeforen 1998; 118:1026.
- Brandt LJ, Borody TJ, Campbell J. Endoscopic fecal microbiota transplantation: “first-line” treatment for severe Clostridium difficile infection? J Clin Gastroenterol 2011; 45:655–657.
- Kelly CR, de Leon L, Jasutkar N. Fecal microbiota transplantation for relapsing Clostridium difficile infection in 26 patients: methodology and results. J Clin Gastroenterol 2012; 46:145–149.
- Thanjan AJ, Southern W, Anand N, et al. Is Clostridium difficile infection (CDI) more difficult to eradicate in patients with diverticulosis? (abstract) Am J Gastroenterol 2008; 103:S195.
- Persky SE, Brandt LJ. Treatment of recurrent Clostridium difficile-associated diarrhea by administration of donated stool directly through a colonoscope. Am J Gastroenterol 2000; 95:3283–3285.
- Nieuwdorp M, van Nood E, Speelman P, et al. [Treatment of recurrent Clostridium difficile-associated diarrhoea with a suspension of donor faeces] (In Dutch). Ned Tijdschr Geneeskd 2008; 152:1927–1932.
- van Nood E, Speelman P, Kuijper EJ, Keller JJ. Struggling with recurrent Clostridium difficile infections: is donor faeces the solution? Euro Surveill 2009; 14. doi:pii:19316.
- Kahn SA, Gorawara-Bhat R, Rubin DT. Fecal bacteriotherapy for ulcerative colitis: patients are ready, are we? Inflamm Bowel Dis 2012; 18:676–684.
- Brandt L, Reddy S. Fecal microbiota transplantation for recurrent Clostridium difficile infection. J Clin Gastroenterol 2011; 45(suppl):S159–S167.
- Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol 2011; 9:88–96.
- Lipp MJ, Nero DC, Callahan MA. The impact of hospital-acquired Clostridium difficile. J Gastroenterol Hepatol 2012; 27:1733–1737.
- Kyne L, Sougioultzis S, McFarland LV, Kelly CP. Underlying disease severity as a major risk factor for nosocomial Clostridium difficile diarrhea. Infect Control Hosp Epidemiol 2002; 23:653–659.
- Kyne L, Hamel MB, Polavaram R, Kelly CP. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis 2002; 34:346–353.
- Gorbach SL. Antibiotics and Clostridium difficile. N Engl J Med 1999; 341:1690–1691.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Beales IL. Intravenous immunoglobulin for recurrent Clostridium difficile diarrhoea. Gut 2002; 51:456.
- O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 2009; 136:1913–1924.
- Louie TJ, Miller MA, Mullane KM, et al; OPT-80-003 Clinical Study Group. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 2011; 364:422–431.
- Kelly CP, LaMont JT. Clostridium difficile—more difficult than ever. N Engl J Med 2008; 359:1932–1940.
- Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin Infect Dis 2007; 44:846–848.
- Musher DM, Logan N, Hamill RJ, et al Nitazoxanide for the treatment of Clostridium difficile colitis. Clin Infect Dis 2006; 43:421–427.
- Louie TJ, Peppe J, Watt CK, et al. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis 2006; 43:411–420.
- Reid G, Younes JA, Van der Mei HC, Gloor GB, Knight R, Busscher JH. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Microbiol 2011; 9:27–38.
- Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996; 4:430–435.
- Vollaard EJ, Clasener HA. Colonization resistance. Antimicrob Agents Chemother 1994; 38:409–414.
- Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 2010; 44:354–360.
- Bakken JS, Borody T, Brandt LJ, et al; Fecal Microbiota Transplantation Workgroup. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol 2011; 9:1044–1049.
- Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 2007; 45:302–307.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271:1913–1918.
- Neish AS, Gewirtz AT, Rao AS, et al. Non-pathogenic bacteria may block epithelial responses: Attenuation of IKB ubiquitination as a novel, physiologic mode of antiinflammation. Gastroenterology 2000; 118:A3754.
- Helwig U, Rizzello F, Cifone G, et al. Elevated IL-10 levels in pouch-tissue after probiotic therapy. Immunol Lett. 1999; 69:159.
- Rager KD, George LW, House JK, DePeters EJ. Evaluation of rumen transfaunation after surgical correction of left-sided displacement of the abomasum in cows. J Am Vet Med Assoc 2004; 225:915–920.
- Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 1958; 44:854–859.
- Aas J, Gessert CE, Bakken JS. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin Infect Dis 2003; 36:580–585.
- Yoon SS, Brandt LJ. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J Clin Gastroenterol 2010; 44:562–566.
- Mattila E, Uusitalo-Seppälä R, Wuorela M, et al. Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterology 2012; 142:490–496.
- Garborg K, Waagsbø B, Stallemo A, Matre J, Sundøy A. Results of faecal donor instillation therapy for recurrent Clostridium difficile-associated diarrhoea. Scand J Infect Dis 2010; 42:857–861.
- Mellow MH, Kanatzar A. Colonoscopic fecal bacteriotherapy in the treatment of recurrent Clostridium difficile infection–results and follow-up. J Okla State Med Assoc 2011; 104:89–91.
- Rohlke F, Surawicz CM, Stollman N. Fecal flora reconstitution for recurrent Clostridium difficile infection: results and methodology. J Clin Gastroenterol 2010; 44:567–570.
- Silverman MS, Davis I, Pillai DR. Success of self-administered home fecal transplantation for chronic Clostridium difficile infection. Clin Gastroenterol Hepatol 2010; 8:471–473.
- Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 2011; 53:994–1002.
- Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol 2012; 107:1079–1087.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- Borody TJ, Warren EF, Leis S, Surace R, Ashman O. Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol 2003; 37:42–47.
- Borody TJ, Torres M, Campbell J, et al. Reversal of inflammatory bowel disease (IBD) with recurrent fecal microbiota transplants (FMT). Am J Gastroenterol 2011; 106:S352.
- Andrews P, Borody TJ, Shortis NP, Thompson S. Bacteriotherapy for chronic constipation—long term follow-up. (abstract). Gastroenterology 1995; 108:A563.
- Borody TJ. Bacteriotherapy for chronic fatigue syndrome: a long-term follow up study. Presented at the 1995 Chronic Fatigue Syndrome National Consensus Conference.
- Borody TJ, Leis S, Campbell J, et al. Fecal microbiota transplantation (FMT) in multiple sclerosis (MS) (abstract). Am J Gastroenterol 2011; 106:S352.
- Borody TJ, Campbell J, Torres M, et al. Reversal of idiopathic thrombocytopenic purpura (ITP) with fecal microbiota transplantation (FMT) (abstract). Am J Gastroenterol 2011; 106:S352.
- Vrieze AF, Holleman MJ, Serlie MT, Ackermans GM, Dallinga-Thie GM, Groen AK. Metabolic effects of transplanting gut microbiota from lean donors to subjects with metabolic syndrome (abstract). Diabetologia 2010; 53:S44.
- Bakken JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 2009; 15:285–289.
- Bjørneklett A. [To repair an ecosystem] (In Norwegian). Tidsskr Nor Laegeforen 1998; 118:1026.
- Brandt LJ, Borody TJ, Campbell J. Endoscopic fecal microbiota transplantation: “first-line” treatment for severe Clostridium difficile infection? J Clin Gastroenterol 2011; 45:655–657.
- Kelly CR, de Leon L, Jasutkar N. Fecal microbiota transplantation for relapsing Clostridium difficile infection in 26 patients: methodology and results. J Clin Gastroenterol 2012; 46:145–149.
- Thanjan AJ, Southern W, Anand N, et al. Is Clostridium difficile infection (CDI) more difficult to eradicate in patients with diverticulosis? (abstract) Am J Gastroenterol 2008; 103:S195.
- Persky SE, Brandt LJ. Treatment of recurrent Clostridium difficile-associated diarrhea by administration of donated stool directly through a colonoscope. Am J Gastroenterol 2000; 95:3283–3285.
- Nieuwdorp M, van Nood E, Speelman P, et al. [Treatment of recurrent Clostridium difficile-associated diarrhoea with a suspension of donor faeces] (In Dutch). Ned Tijdschr Geneeskd 2008; 152:1927–1932.
- van Nood E, Speelman P, Kuijper EJ, Keller JJ. Struggling with recurrent Clostridium difficile infections: is donor faeces the solution? Euro Surveill 2009; 14. doi:pii:19316.
- Kahn SA, Gorawara-Bhat R, Rubin DT. Fecal bacteriotherapy for ulcerative colitis: patients are ready, are we? Inflamm Bowel Dis 2012; 18:676–684.
KEY POINTS
- Fecal microbiota transplantation involves instilling gut microbiota from a healthy donor into the diseased gut of a patient who has recurrent or recalcitrant episodes of diarrhea despite antibiotic treatment for C difficile infection. The instillation can be done via nasogastric tube, endoscope, or enema.
- Donor screening is necessary to prevent transmission of communicable diseases to the recipient.
- Recently published studies indicate that this procedure is effective for treating recurrent C difficile infection. Randomized clinical trials to assess its efficacy and safety are underway.
- The field of microbiota therapy is rapidly progressing. More physicians are learning to embrace the concept of fecal microbiota transplantation, and patients are beginning to overcome the “yuck factor” and accept its benefits.
Inhaled loxapine for agitation
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.