User login
Quetiapine for the Treatment of Delirium
Delirium is an acute fluctuation in mental status that includes symptoms of inattention, disorganized thinking, and altered level of consciousness occurring over a short time period.[1] Prevalence of delirium ranges from 10% to 30% in the hospitalized medically ill and ranges from 40% to 60% in intensive care unit (ICU) patients who are not receiving mechanical ventilation.[2, 3] Patients experience delirium more often if they are of older age; have dementia, cancer, or acquired immune deficiency syndrome; have undergone surgery; are terminally ill; or have received multiple psychoactive medications, particularly benzodiazepines or opioids.[3, 4, 5, 6, 7] Delirium is associated with high rates of morbidity and mortality,[8] with patients more likely to develop complications such as pneumonia, decubitus ulcers, and long‐term cognitive deficits.[9, 10, 11, 12] These complications, in turn, lead to longer hospital stays and increased costs of care.[13, 14] Currently, there are no antipsychotics approved by the US Food and Drug Administration for the treatment of delirium.
Both the 2002 American Society of Critical Care Medicine[12] and the 2004 American Psychiatric Association guidelines[15] on delirium recommend haloperidol as the antipsychotic of choice due to its potent tranquilizing effect, lack of active metabolites, and limited anticholinergic and sedating side effects. However, when given intravenously or at high cumulative doses (generally >35 mg/day), haloperidol has been shown to cause QT interval prolongation potentially leading to torsades de pointes and sudden cardiac death.[15] Recent research in delirium treatment has focused on the second‐generation antipsychotics, and these studies have reported positive findings,[16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29] although no significant differences have been found compared to haloperidol.[16, 17, 18, 22, 23, 24] Systematic reviews have also failed to show a significant difference in efficacy or safety between second‐generation and first‐generation antipsychotics and unfortunately report a number of limitations including poor study designs, small sample sizes, lack of a placebo control group, exclusion of ICU patients, and weak primary outcomes.[8, 30, 31, 32] Attempting to correct for a number of these limitations, recent research with quetiapine has reported promising results in 2 controlled studies.[19, 20]
Quetiapine is a second‐generation antipsychotic with a very low affinity for dopamine receptors and a very high affinity for serotonin receptors.[33, 34] Additionally, quetiapine has a high affinity for histamine and 1‐adrenergic receptors, but a very low affinity for M1 muscarinic receptors.[34] This mechanism of action may allow quetiapine to effectively treat delirium and provide sedation without causing significant extrapyramidal side effects associated with potent dopamine receptor antagonism or precipitating delirium through muscarinic receptor antagonism. Quetiapine has a rapid absorption and a short half‐life (36 hours), giving it a quick onset of action and a fast elimination from the body.[35] Unlike haloperidol, it is only available in oral dosage forms, but can be crushed to administer via enteral tube. Common reported adverse effects include somnolence, hypotension, and dizziness. Although quetiapine has been shown to prolong QTc, Harrigan et al. reported a mean increase in QTc from baseline of 5.7 msec; there was no significant effect on this change in the presence of a metabolic inhibitor, ketoconazole.[36] The mean change in QTc from baseline with quetiapine was lower than the change reported with oral haloperidol, 5.7 msec compared to 7.1 msec, respectively. No statistical comparison was performed between these 2 drugs on this measure. Quetiapine does carry a black box warning for increased mortality in elderly patients with dementia‐related psychosis.[35] However, this risk has also been found with first‐generation antipsychotics.[37, 38, 39] A recent study found this risk of sudden cardiac death extended to adult users of both first‐ and second‐generation antipsychotics.[40] In contrast, Elie et al. found no increased risk of mortality in elderly patients with delirium receiving antipsychoticsover 90% were prescribed either haloperidol or risperidonein their nested case‐controlled study.[41] Quetiapine has demonstrated some benefit with limited side effects in studies utilizing antipsychotics for the treatment of delirium. The purpose of this review was to evaluate the role of quetiapine for the treatment of delirium.
LITERATURE REVIEW
We performed an English‐language literature search of MEDLINE and Embase databases to identify journal articles published between January 1960 and December 2012. Keywords included quetiapine, second‐generation antipsychotic, atypical antipsychotic, delirium, and agitation. The search was limited to English‐language articles and adult subjects (>18 years). Based on our review of abstracts, we included both controlled and noncontrolled trials as long as treatment of delirium was the primary focus. We eliminated case reports, foreign language articles, and poster presentations. We identified 8 trials[19, 20, 24, 25, 26, 27, 28, 29] that included 2 double‐blind, randomized, placebo‐controlled trials, which are described in the text below.[19, 20] Six other trials, 5 open‐label[25, 26, 27, 28, 29] and 1 retrospective cohort,[24] are described in Table 1.
| Study | Study Design | No. of Patients Included in Analyses | Patient Type | Treatment | Baseline Delirium Scores | Quetiapine Dose (mg/day) (MeanSD) | Efficacy Measures | Results | Side Effects |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Schwartz et al. (2000)[24] | RC | 22 | General hospital | Quetiapine or haloperidol flexible dose | DRS score 20.9 Q; 18.5 H | 211.4 Q; 3.4 H | >50% improvement in DRS scores | 10/11 in each treatment group had >50% improvement in DRS scores | EPS 2/11 H and 0/11 Q; mild‐to‐moderate sedation 0/11 H and 2/11 Q |
| Pae et al. (2004)[25] | OL | 22 | Neurosurgery and orthopedic surgery and oncology | Flexible dose quetiapine | DRS‐R‐98 21.83.2 and CGI‐s 4.90.8 | 127.172.2 | DRS‐R‐98 and CGI‐s score reduction | DRS‐R‐98 9.33.8 (P<0.0001) and CGI‐s 2.11.1 (P<0.0001) reduction from baseline; 19/22 (86.3%) and 17/22 (77.3%) showed >50% score reduction for DRS‐R‐98 and CGI‐s, respectively | EPS none; sedation requiring discontinuation 2; mild sedation 3; serious side effects none |
| Maneeton et al. (2007)[26] | OL | 17 | General hospital | Quetiapine 25100 mg/day in 1 or 2 divided doses | DRS 24.53.2 and CGI‐s 4.90.9 | 45.728.7 | 50% reduction in DRS scores | 15/17 (88.2%) had 50% reduction in DRS score; all DRS and CGI‐s scores on days 17 of the 7‐day treatment course were significantly lower than baseline scores | EPS tremor 2; hypotension 2; daytime sleepiness 13; nightmare 3; dry mouth 2; nausea 1 |
| Kim et al. (2003)[27] | OL | 12 (all male and age 64 years) | General hospital | Quetiapine 25 mg BID and increased by 25 mg every 2 days until patient maximally stabilized | DRS 18.256.05; MMSE 14.505.90; CGI‐s 3.000.43; CDT 3.25 2.77 | 93.7523.31 | Reduction in DRS, MMSE, CGI‐s, or CDT scores | All scores were statistically significantly improved from baseline; DRS at end of study 0.631.21 (P=0.03) | No dropouts due to side effects; EPS none; sedation 2; vivid dreams 1 |
| Sasaki et al. (2003)[28] | OL | 12 | General hospital | Quetiapine started at 25 or 50 mg/day and titrated to maximal clinical effect | DRS‐J 18.14.2 | 44.931.0 | Remission was DRS‐J score <12 (cutoff for delirium) and resolution of delirium symptoms | Remission occurred in all patients; mean DRS‐J score was reduced to 9.31.6 after remission | No statistically significant change from baseline in EPS; no excessive daytime sedation, anticholinergic effects, vital sign, or lab parameter changes |
| Lee et al. (2005)[29] | OL | 31 | Neurosurgery and orthopedic surgery, internal medicine, neurology and rehabilitation medicine | Flexible dose quetiapine or amisulpride | DRS‐R‐98 10.14.1 Q and 10.54.1 A | 11385.5 Q; 156.497.5 A | Reduction in DRS‐R‐98 score | DRS‐R‐98 scores significantly reduced from baseline in both groups with 3.52.6 Q (P=0.001) and 3.51.4 A (P=0.000); no difference between groups (P=0.842); 12 (80%) Q and 13 (81.3%) A had >50% reduction in DRS‐R‐98 score | No serious side effects; dropouts 5 Q and 4 A; oversedation 1 Q and 1 A; patient withdrawal 2 Q and 1 A; no statistical differences in total sleep time P=0.767 or quality of sleep P=0.984 |
Randomized Controlled Trials
Tahir et al. published a double‐blind, randomized, placebo‐controlled trial examining the efficacy and tolerability of quetiapine in the treatment of delirium.[19] Inclusion criteria were a Diagnostic and Statistical Manual of Mental Disorders‐IV diagnosis of delirium and a Delirium Rating Scale Revised 98 (DRS‐R‐98) total score of at least 15, indicating the presence of delirium. Subjects were excluded for major preexisting cognitive deficits, alcohol withdrawal, preexisting psychosis, substance dependence, inability to comply with the constraints of the trial, and concurrent medications that interact with quetiapine.
Forty‐two general medicine subjects were enrolled and randomized in the study with no difference in baseline characteristics. Patients received either placebo or quetiapine 25 mg once daily. Doses were titrated by 25 mg daily to a maximum daily dose of 175 mg in divided doses. The primary end point was DRS‐R‐98 total mean score assessed on days 1, 2, 3, 4, 7, and 10, with follow‐up assessment on day 30. Secondary outcome measures were Mini‐Mental State Examination (MMSE), the Brief Psychiatric Rating Scale, and the Clinical Global Improvement Scale. Tolerability was assessed using the Abnormal Involuntary Movements Scale and by clinical examination.
No differences in total mean DRS‐R‐98 score at individual time points reached statistical significance, but these scores improved more quickly in the quetiapine group than placebo. The secondary outcome of rate of delirium improvement on severity score did reach statistical significance; differences on mean severity scores were 0.8270.37 (P=0.026), suggesting that severity scores improved 82% more quickly than placebo. There were no significant differences between groups for any of the other secondary outcomes. Seven patients died within 30 days of entering the study (4 in the quetiapine group and 3 in the placebo group) due to serious medical conditions and not due to study medication as determined through clinical review. One patient withdrew from quetiapine due to sedation. Results of this study are limited by a small sample size, as it was underpowered to detect a statistically significant difference in the primary outcome. Other limitations include subjects who were older, with mean age of 84 years, which may have prevented titration of quetiapine, and subjects with minor cognitive deficits were included, which may have reduced the impact of treatment on the study outcomes. Additionally, strict criteria for exclusion kept those patients most likely to develop delirium from being included, limiting the studies external validity. Finally, the use of DRS‐R‐98 mean total and severity scores is subject to error as outliers in the mean could potentially skew the results.
Devlin et al. investigated the efficacy and safety of quetiapine for ICU delirium in critically ill patients in a double‐blind, randomized, placebo‐controlled trial.[20] Inclusion criteria were an Intensive Care Delirium Screening Checklist Score (ICDSC) >4, which indicates the presence of delirium, an order for as‐needed haloperidol, and tolerating enteral nutrition. Patients were excluded if they had a complicating neurologic condition, current treatment with dexmedetomidine or with medications that interact with quetiapine, baseline QTc interval >500, pregnancy, or poor prognosis. Thirty‐six critically ill subjects were randomized with no significant differences in baseline characteristics including exposure to fentanyl, haloperidol, and benzodiazepines, and in particular, midazolam. If the subject received at least 1 dose of as‐needed haloperidol in the previous 24 hours, then either placebo or quetiapine was given. Quetiapine was initiated at 50 mg every 12 hours and titrated up by 50 mg every 12 hours to a maximum dose of 200 mg every 12 hours. Patients could receive intravenous haloperidol, 1 to 10 mg up to every 2 hours as needed. The primary outcome was time to first resolution of delirium defined as time from administration of the first dose of study drug until an ICDSC <3 was first detected, indicating an absence of delirium. Secondary outcomes were total hours in delirium, total hours spent deeply sedated (Sedation‐Agitation Scale [SAS] <2) or agitated (SAS >5), episodes of subject‐initiated device removal, use of haloperidol therapy including total dose in milligrams, number of doses and number of days of therapy, the use of sedatives (converted to midazolam equivalents) and analgesics, duration of study‐drug administration, average daily and maximum study‐drug dose, length of mechanical ventilation, duration of both ICU and hospital stay, hospital mortality, and disposition of subjects after hospital discharge. Safety measures were total number of adverse and serious adverse events, episodes of somnolence, incidence of extrapyramidal symptoms, and episodes of QTc interval prolongation.
The time to first resolution of delirium was shorter with quetiapine compared to placebo (median [interquartile range]: 1.0 [0.53.0] vs 4.5 days [2.0‐7.0]; P=0.001). Resolution of delirium occurred at least once in all quetiapine patients and in 78% of placebo patients (P=0.05). Statistical significance with quetiapine was reached on the following secondary end points: time spent in delirium (36 [1287] vs 120 hours [60195], P=0.006); shorter duration of study drug (102 [84168] vs 186 hours [108228], P=0.04); lower daily study drug dose (110 [88191] vs 210 mg [116293], P=0.01); less upregulation of the study medication dose (200 [100313] vs 375 mg [25400], P=0.02); fewer hours of agitation (6 [038] vs 36 hours [1166], P=0.02); shorter duration of haloperidol therapy (3 [24] vs 4 days [38], P=0.05); and fewer days of fentanyl (0 [03] vs 4 days [19], P=0.03). There were no significant differences between groups for any other secondary outcomes. As‐needed haloperidol use in the quetiapine versus the placebo group was lower but not statistically significant (1.9 vs 4.3 mg per day; P=0.26). Two main limitations were a small sample size and strict exclusion criteria. Additionally, the primary end point of time to first resolution of delirium may be interpreted as a less rigorous measure, because there is no standard definition for delirium resolution, and delirium is a condition that waxes and wanes on its own.
A post hoc analysis by Devlin et al. was conducted on the above study to compare duration and time to first resolution of 10 delirium symptoms in 29 patients.[21] Symptoms included agitation, decreased level of consciousness, inattention, disorientation, hallucinations/delusions, hyperactivity, hypoactivity, inappropriate speech or mood, sleep/wake cycle disturbance, and symptom fluctuation. Only symptom fluctuation (P=0.009), time to first resolution of symptom fluctuation (P=0.004), time with inattention (47 vs 78 hours, P=0.025), and time with symptom fluctuation (47 vs 89 hours; P =0.04) reached statistical significance with quetiapine compared to placebo. However, quetiapine subjects had a longer time to resolution of agitation (3 vs 1 day, P=0.04) and hyperactivity (5 vs 1 day, P<0.04). The authors attribute these findings to the higher use of as‐needed haloperidol in the placebo group (1.9 vs 4.3 mg per day, P=0.26). These results may also be due to a limitation in the study design, which self‐selected for agitation delirium by requiring the use of as‐needed haloperidol as inclusion criteria, as symptoms of agitation would be more likely to receive as‐needed haloperidol doses. In addition, subjects were allowed to receive 1 to 10 mg of haloperidol up to every 2 hours as needed, but there is no discussion of controlling total daily haloperidol dose in each group in the study. Although 1.9 versus 4.3 mg per day is neither statistically nor likely clinically significant, haloperidol is a treatment for delirium, so the study design would be stronger if quetiapine could be directly compared to an equivalent daily dose of haloperidol or if both groups received the same daily dose of haloperidol. Study results are also limited by the nature of a post hoc analysis, missing documentation for individual delirium symptoms, symptoms of delirium not being well characterized and subjective, and delay in enrollment of subjects.
Noncontrolled Trials
Six additional trials are included in the table of this review including 5 open label trials and 1 retrospective cohort study.[24, 25, 26, 27, 28, 29] Schwartz and Masand[24] and Lee et al.[29] compared the efficacy of quetiapine to haloperidol and amisulpride, a second‐generation antipsychotic unavailable in the United States, respectively. In a retrospective cohort of 22 general hospital patients, Schwartz and Masand found quetiapine (average dose, 211.4 mg/day) was as efficacious as haloperidol (average dose, 3.4 mg/day) in improving delirium rating scale (DRS) scores by more than 50% in 10/11 subjects in each group. Lee et al. also found quetiapine (average dose, 113.0 mg/day) to be equally efficacious to amisulpride (156.4 mg/day) in statistically significantly improving DRS‐R‐98 scores in 31 neurosurgery, orthopedic surgery, internal medicine, neurology, and rehabilitation medicine patients. Four open‐label studies tested the efficacy of flexible doses of quetiapine in a total of 63 general hospital, neurosurgery, orthopedic surgery, and oncology patients.[25, 26, 27, 28] Pae et al.,[25] Maneeton et al.,[26] and Kim et al.[27] found that quetiapine statistically significantly reduced DRS‐R‐98, Clinical Global Impression Scale‐Severity, DRS, MMSE, and Clock Drawing Test scores from baseline. Sasaki et al.[28] found all 12 general hospital patients in their study reached remission with an average daily quetiapine dose of 44.9 mg/day. Although these studies have a limited level of evidence due to their small sample sizes, study designs, and heterogeneous subjects, they do still suggest that quetiapine may effectively treat delirium in various patient populations.
DISCUSSION
Although the role of quetiapine for the treatment of delirium continues to develop, the studies evaluated here suggest that quetiapine may be effective and safe for this usage. Resolution of delirium from baseline was shown in all studies. Quetiapine resolved symptoms of delirium more quickly than placebo and had equal efficacy to other antipsychotics, haloperidol and amisulpride. Quetiapine may be most useful for patients with symptom fluctuation as opposed to agitation and hyperactivity or in those patients who may not tolerate haloperidol well. Both randomized control and open‐label trials found a low incidence of adverse effects with quetiapine. There were fewer incidences of QT prolongation and extrapyramidal symptoms, but higher a rate of somnolence with quetiapine compared to other antipsychotics in these trials. However, none of these differences in adverse effects reached statistical significance.
Drawing definitive conclusions about the efficacy of quetiapine in the treatment of delirium is difficult due to multiple study limitations. First, the body of literature is small, with only 2 randomized controlled trials, both of which had limitations. Tahir et al.[19] was underpowered and found no statistically significant difference in the primary end point, DRS‐R‐98 total mean score. Devlin et al.[20] tested the primary end point of time to first resolution of delirium, which may be interpreted as a less rigorous measure because there is no standard definition for delirium resolution. Furthermore, both randomized controlled and observational trials tested the efficacy of quetiapine using different tests, making comparison between these trials difficult. All studies included in this review were carried out in small patient populations, with the largest trial having 42 subjects, and had highly restrictive exclusion criteria limiting the generalizability of the study population to the general hospital population. Although most of the patients included in these studies are general hospital populations, Devlin et al. was performed in critically ill patients, which creates the additional limitation of having heterogeneous study populations. Last, superiority of any 1 antipsychotic is not possible given that none of these studies have done a head‐to‐head comparison of quetiapine with another atypical antipsychotic.
Given the comparable efficacy and safety of antipsychotics, cost is a relevant factor in treatment decisions. Haloperidol is supplied as either a suspension for injection or a tablet. One vial of 5 mg/mL haloperidol is $8.32. Haloperidol 5 mg tablets are approximately $0.29 each, whereas 25 mg tablets of quetiapine are approximately $3.53 each. However, these prices are for consumers and will vary depending on institutional contracts.[42]
CONCLUSION
Quetiapine appears to be an effective and safe agent for the treatment of delirium in both general medicine and ICU patients. Superiority of quetiapine over other antipsychotics for hospital‐associated delirium has not been shown due to limitations in quality and quantity of data. Large, randomized, double‐blind, active control studies with longer study durations are needed to elucidate the efficacy and niche of quetiapine in the treatment of delirium.
Acknowledgment
Disclosure: Nothing to report.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: APA; 1994.
- American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156:1–20.
- , . Pharmacological and nonpharmacological management of delirium in critically ill patients. Neurotherapeutics. 2012;9:158–175.
- , . Delirium in cancer patients. Int Psychogeriatr. 1991;3:333–336.
- . Organic mental disorders caused by HIV: update on early‐diagnosis and treatment. Am J Psychiatry. 1990;147:696–710.
- . Post‐operative delirium. Int Psychogeriatr. 1991;3:325–332.
- , , . Delirium in terminally ill cancer patients. Am J Psychiatry. 1983;140:1048–1050.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Acute confusional states in the hospitalized elderly: incidence, risk factors and complications [abstract]. Clin Res. 1989;37:524A.
- , . Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41–46.
- , , . Duration of delirium shortened by the correction of electrolyte imbalance. Jpn J Psychiatry Neurol. 1988;42:81–88.
- , , , et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–141.
- , , , , , . Intensive care unit delirium is an independent predictor of longer hospital stay: a prospective analysis of 261 non‐ventilated patients. Crit Care. 2005;9:R375–R381.
- , , , et al. Costs associated with delirium in mechanically ventilated patients. Crit Care Med. 2004;32:955–962.
- Cook IA: Guideline Watch: Practice Guideline for the Treatment of Patients With Delirium. Arlington, VA:American Psychiatric Association, 2004. Available at: http://www.psych.org/psych_pract/treatg/pg/prac_guide.cfm. Accessed March 5, 2012.
- , . A double‐blind trial of risperdone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297–301.
- , , , . Olanzapine vs haloperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30:444–449.
- , , , et al. Feasibility, efficacy, and safety of antipsychotics for intensive care unit delirium: the MIND randomized, placebo‐controlled trial. Crit Care Med. 2010;38:428–437.
- , , , et al. A randomized controlled trial of quetiapine versus placebo in the treatment of delirium. Psychosom Res. 2010;69:485–490.
- , , , et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double‐blind, placebo‐controlled pilot study. Crit Care Med. 2010;38:419–427.
- , , , et al. Impact of quetiapine on resolution of individual delirium symptoms in critically ill patients with delirium: a post‐hoc analysis of a double‐blind, randomized, placebo‐controlled study. Critical Care. 2011;15:R215.
- , . Olanzapine in the treatment of delirium. Psychosomatics. 1998;39:422–430.
- , , . Comparative efficacy study of haloperidol, olanzapine and risperidone in delirium. J Psychosom Res. 2011;71:277–281.
- , . Treatment of delirium with quetiapine. J Clin Psychiatry. 2000;2:10–12.
- , , , , . A pilot trial of quetiapine for the treatment of patients with delirium. Hum Psychopharmacol Clin Exp. 2004;19:125–127.
- , , . An open‐label study of quetiapine for delirium. J Med Assoc Thai. 2007;10:2158–2163.
- , , , . Treatment of delirium in older adults with quetiapine. J Geriatr Psychiatry Neurol. 2003;16:29–31.
- , , , et al. A prospective, open‐label, flexible‐dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64:1316–1321.
- , , , et al. Amisulpride versus quetiapine for the treatment of delirium: a randomized, open prospective study. Int Clin Psychopharmacol. 2005;20:311–314.
- , , , et al. Pharmacological management of delirium in hospitalized adults‐a systematic evidence review. J Gen Intern Med. 2009;24:848–853.
- , , . Systematic review of antipsychotics for the treatment of hospital‐associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40:1966–1973.
- , , , . Atypical antipsychotics versus haloperidol for treatment of delirium in acutely ill patients. Pharmacotherapy. 2007;27:588–594.
- , , . Delirium: prevention, treatment, and outcome studies. J Geriatr Psychiatry Neurol. 1998;11:126–137.
- , . Seroquel: biochemical profile of a potential atypical antipsychotic. Psychopharmacology. 1993;112:285–292.
- Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; December 2011.
- , , , et al. A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol. 2004;24:62–69.
- , , , et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164:1568–1576.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , , . Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med. 2009;360:225–235.
- , , , , , . A retrospective, exploratory, secondary analysis of the association between antipsychotic use and mortality in elderly patients with delirium. Int Psychogeriatr. 2009;21(3):588–592.
- Walgreens Co. Price your drugs Available at: http://www.walgreens.com/pharmacy/psc/drugpricing/psc_drug_pricing.jsp. Accessed December 17, 2012.
Delirium is an acute fluctuation in mental status that includes symptoms of inattention, disorganized thinking, and altered level of consciousness occurring over a short time period.[1] Prevalence of delirium ranges from 10% to 30% in the hospitalized medically ill and ranges from 40% to 60% in intensive care unit (ICU) patients who are not receiving mechanical ventilation.[2, 3] Patients experience delirium more often if they are of older age; have dementia, cancer, or acquired immune deficiency syndrome; have undergone surgery; are terminally ill; or have received multiple psychoactive medications, particularly benzodiazepines or opioids.[3, 4, 5, 6, 7] Delirium is associated with high rates of morbidity and mortality,[8] with patients more likely to develop complications such as pneumonia, decubitus ulcers, and long‐term cognitive deficits.[9, 10, 11, 12] These complications, in turn, lead to longer hospital stays and increased costs of care.[13, 14] Currently, there are no antipsychotics approved by the US Food and Drug Administration for the treatment of delirium.
Both the 2002 American Society of Critical Care Medicine[12] and the 2004 American Psychiatric Association guidelines[15] on delirium recommend haloperidol as the antipsychotic of choice due to its potent tranquilizing effect, lack of active metabolites, and limited anticholinergic and sedating side effects. However, when given intravenously or at high cumulative doses (generally >35 mg/day), haloperidol has been shown to cause QT interval prolongation potentially leading to torsades de pointes and sudden cardiac death.[15] Recent research in delirium treatment has focused on the second‐generation antipsychotics, and these studies have reported positive findings,[16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29] although no significant differences have been found compared to haloperidol.[16, 17, 18, 22, 23, 24] Systematic reviews have also failed to show a significant difference in efficacy or safety between second‐generation and first‐generation antipsychotics and unfortunately report a number of limitations including poor study designs, small sample sizes, lack of a placebo control group, exclusion of ICU patients, and weak primary outcomes.[8, 30, 31, 32] Attempting to correct for a number of these limitations, recent research with quetiapine has reported promising results in 2 controlled studies.[19, 20]
Quetiapine is a second‐generation antipsychotic with a very low affinity for dopamine receptors and a very high affinity for serotonin receptors.[33, 34] Additionally, quetiapine has a high affinity for histamine and 1‐adrenergic receptors, but a very low affinity for M1 muscarinic receptors.[34] This mechanism of action may allow quetiapine to effectively treat delirium and provide sedation without causing significant extrapyramidal side effects associated with potent dopamine receptor antagonism or precipitating delirium through muscarinic receptor antagonism. Quetiapine has a rapid absorption and a short half‐life (36 hours), giving it a quick onset of action and a fast elimination from the body.[35] Unlike haloperidol, it is only available in oral dosage forms, but can be crushed to administer via enteral tube. Common reported adverse effects include somnolence, hypotension, and dizziness. Although quetiapine has been shown to prolong QTc, Harrigan et al. reported a mean increase in QTc from baseline of 5.7 msec; there was no significant effect on this change in the presence of a metabolic inhibitor, ketoconazole.[36] The mean change in QTc from baseline with quetiapine was lower than the change reported with oral haloperidol, 5.7 msec compared to 7.1 msec, respectively. No statistical comparison was performed between these 2 drugs on this measure. Quetiapine does carry a black box warning for increased mortality in elderly patients with dementia‐related psychosis.[35] However, this risk has also been found with first‐generation antipsychotics.[37, 38, 39] A recent study found this risk of sudden cardiac death extended to adult users of both first‐ and second‐generation antipsychotics.[40] In contrast, Elie et al. found no increased risk of mortality in elderly patients with delirium receiving antipsychoticsover 90% were prescribed either haloperidol or risperidonein their nested case‐controlled study.[41] Quetiapine has demonstrated some benefit with limited side effects in studies utilizing antipsychotics for the treatment of delirium. The purpose of this review was to evaluate the role of quetiapine for the treatment of delirium.
LITERATURE REVIEW
We performed an English‐language literature search of MEDLINE and Embase databases to identify journal articles published between January 1960 and December 2012. Keywords included quetiapine, second‐generation antipsychotic, atypical antipsychotic, delirium, and agitation. The search was limited to English‐language articles and adult subjects (>18 years). Based on our review of abstracts, we included both controlled and noncontrolled trials as long as treatment of delirium was the primary focus. We eliminated case reports, foreign language articles, and poster presentations. We identified 8 trials[19, 20, 24, 25, 26, 27, 28, 29] that included 2 double‐blind, randomized, placebo‐controlled trials, which are described in the text below.[19, 20] Six other trials, 5 open‐label[25, 26, 27, 28, 29] and 1 retrospective cohort,[24] are described in Table 1.
| Study | Study Design | No. of Patients Included in Analyses | Patient Type | Treatment | Baseline Delirium Scores | Quetiapine Dose (mg/day) (MeanSD) | Efficacy Measures | Results | Side Effects |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Schwartz et al. (2000)[24] | RC | 22 | General hospital | Quetiapine or haloperidol flexible dose | DRS score 20.9 Q; 18.5 H | 211.4 Q; 3.4 H | >50% improvement in DRS scores | 10/11 in each treatment group had >50% improvement in DRS scores | EPS 2/11 H and 0/11 Q; mild‐to‐moderate sedation 0/11 H and 2/11 Q |
| Pae et al. (2004)[25] | OL | 22 | Neurosurgery and orthopedic surgery and oncology | Flexible dose quetiapine | DRS‐R‐98 21.83.2 and CGI‐s 4.90.8 | 127.172.2 | DRS‐R‐98 and CGI‐s score reduction | DRS‐R‐98 9.33.8 (P<0.0001) and CGI‐s 2.11.1 (P<0.0001) reduction from baseline; 19/22 (86.3%) and 17/22 (77.3%) showed >50% score reduction for DRS‐R‐98 and CGI‐s, respectively | EPS none; sedation requiring discontinuation 2; mild sedation 3; serious side effects none |
| Maneeton et al. (2007)[26] | OL | 17 | General hospital | Quetiapine 25100 mg/day in 1 or 2 divided doses | DRS 24.53.2 and CGI‐s 4.90.9 | 45.728.7 | 50% reduction in DRS scores | 15/17 (88.2%) had 50% reduction in DRS score; all DRS and CGI‐s scores on days 17 of the 7‐day treatment course were significantly lower than baseline scores | EPS tremor 2; hypotension 2; daytime sleepiness 13; nightmare 3; dry mouth 2; nausea 1 |
| Kim et al. (2003)[27] | OL | 12 (all male and age 64 years) | General hospital | Quetiapine 25 mg BID and increased by 25 mg every 2 days until patient maximally stabilized | DRS 18.256.05; MMSE 14.505.90; CGI‐s 3.000.43; CDT 3.25 2.77 | 93.7523.31 | Reduction in DRS, MMSE, CGI‐s, or CDT scores | All scores were statistically significantly improved from baseline; DRS at end of study 0.631.21 (P=0.03) | No dropouts due to side effects; EPS none; sedation 2; vivid dreams 1 |
| Sasaki et al. (2003)[28] | OL | 12 | General hospital | Quetiapine started at 25 or 50 mg/day and titrated to maximal clinical effect | DRS‐J 18.14.2 | 44.931.0 | Remission was DRS‐J score <12 (cutoff for delirium) and resolution of delirium symptoms | Remission occurred in all patients; mean DRS‐J score was reduced to 9.31.6 after remission | No statistically significant change from baseline in EPS; no excessive daytime sedation, anticholinergic effects, vital sign, or lab parameter changes |
| Lee et al. (2005)[29] | OL | 31 | Neurosurgery and orthopedic surgery, internal medicine, neurology and rehabilitation medicine | Flexible dose quetiapine or amisulpride | DRS‐R‐98 10.14.1 Q and 10.54.1 A | 11385.5 Q; 156.497.5 A | Reduction in DRS‐R‐98 score | DRS‐R‐98 scores significantly reduced from baseline in both groups with 3.52.6 Q (P=0.001) and 3.51.4 A (P=0.000); no difference between groups (P=0.842); 12 (80%) Q and 13 (81.3%) A had >50% reduction in DRS‐R‐98 score | No serious side effects; dropouts 5 Q and 4 A; oversedation 1 Q and 1 A; patient withdrawal 2 Q and 1 A; no statistical differences in total sleep time P=0.767 or quality of sleep P=0.984 |
Randomized Controlled Trials
Tahir et al. published a double‐blind, randomized, placebo‐controlled trial examining the efficacy and tolerability of quetiapine in the treatment of delirium.[19] Inclusion criteria were a Diagnostic and Statistical Manual of Mental Disorders‐IV diagnosis of delirium and a Delirium Rating Scale Revised 98 (DRS‐R‐98) total score of at least 15, indicating the presence of delirium. Subjects were excluded for major preexisting cognitive deficits, alcohol withdrawal, preexisting psychosis, substance dependence, inability to comply with the constraints of the trial, and concurrent medications that interact with quetiapine.
Forty‐two general medicine subjects were enrolled and randomized in the study with no difference in baseline characteristics. Patients received either placebo or quetiapine 25 mg once daily. Doses were titrated by 25 mg daily to a maximum daily dose of 175 mg in divided doses. The primary end point was DRS‐R‐98 total mean score assessed on days 1, 2, 3, 4, 7, and 10, with follow‐up assessment on day 30. Secondary outcome measures were Mini‐Mental State Examination (MMSE), the Brief Psychiatric Rating Scale, and the Clinical Global Improvement Scale. Tolerability was assessed using the Abnormal Involuntary Movements Scale and by clinical examination.
No differences in total mean DRS‐R‐98 score at individual time points reached statistical significance, but these scores improved more quickly in the quetiapine group than placebo. The secondary outcome of rate of delirium improvement on severity score did reach statistical significance; differences on mean severity scores were 0.8270.37 (P=0.026), suggesting that severity scores improved 82% more quickly than placebo. There were no significant differences between groups for any of the other secondary outcomes. Seven patients died within 30 days of entering the study (4 in the quetiapine group and 3 in the placebo group) due to serious medical conditions and not due to study medication as determined through clinical review. One patient withdrew from quetiapine due to sedation. Results of this study are limited by a small sample size, as it was underpowered to detect a statistically significant difference in the primary outcome. Other limitations include subjects who were older, with mean age of 84 years, which may have prevented titration of quetiapine, and subjects with minor cognitive deficits were included, which may have reduced the impact of treatment on the study outcomes. Additionally, strict criteria for exclusion kept those patients most likely to develop delirium from being included, limiting the studies external validity. Finally, the use of DRS‐R‐98 mean total and severity scores is subject to error as outliers in the mean could potentially skew the results.
Devlin et al. investigated the efficacy and safety of quetiapine for ICU delirium in critically ill patients in a double‐blind, randomized, placebo‐controlled trial.[20] Inclusion criteria were an Intensive Care Delirium Screening Checklist Score (ICDSC) >4, which indicates the presence of delirium, an order for as‐needed haloperidol, and tolerating enteral nutrition. Patients were excluded if they had a complicating neurologic condition, current treatment with dexmedetomidine or with medications that interact with quetiapine, baseline QTc interval >500, pregnancy, or poor prognosis. Thirty‐six critically ill subjects were randomized with no significant differences in baseline characteristics including exposure to fentanyl, haloperidol, and benzodiazepines, and in particular, midazolam. If the subject received at least 1 dose of as‐needed haloperidol in the previous 24 hours, then either placebo or quetiapine was given. Quetiapine was initiated at 50 mg every 12 hours and titrated up by 50 mg every 12 hours to a maximum dose of 200 mg every 12 hours. Patients could receive intravenous haloperidol, 1 to 10 mg up to every 2 hours as needed. The primary outcome was time to first resolution of delirium defined as time from administration of the first dose of study drug until an ICDSC <3 was first detected, indicating an absence of delirium. Secondary outcomes were total hours in delirium, total hours spent deeply sedated (Sedation‐Agitation Scale [SAS] <2) or agitated (SAS >5), episodes of subject‐initiated device removal, use of haloperidol therapy including total dose in milligrams, number of doses and number of days of therapy, the use of sedatives (converted to midazolam equivalents) and analgesics, duration of study‐drug administration, average daily and maximum study‐drug dose, length of mechanical ventilation, duration of both ICU and hospital stay, hospital mortality, and disposition of subjects after hospital discharge. Safety measures were total number of adverse and serious adverse events, episodes of somnolence, incidence of extrapyramidal symptoms, and episodes of QTc interval prolongation.
The time to first resolution of delirium was shorter with quetiapine compared to placebo (median [interquartile range]: 1.0 [0.53.0] vs 4.5 days [2.0‐7.0]; P=0.001). Resolution of delirium occurred at least once in all quetiapine patients and in 78% of placebo patients (P=0.05). Statistical significance with quetiapine was reached on the following secondary end points: time spent in delirium (36 [1287] vs 120 hours [60195], P=0.006); shorter duration of study drug (102 [84168] vs 186 hours [108228], P=0.04); lower daily study drug dose (110 [88191] vs 210 mg [116293], P=0.01); less upregulation of the study medication dose (200 [100313] vs 375 mg [25400], P=0.02); fewer hours of agitation (6 [038] vs 36 hours [1166], P=0.02); shorter duration of haloperidol therapy (3 [24] vs 4 days [38], P=0.05); and fewer days of fentanyl (0 [03] vs 4 days [19], P=0.03). There were no significant differences between groups for any other secondary outcomes. As‐needed haloperidol use in the quetiapine versus the placebo group was lower but not statistically significant (1.9 vs 4.3 mg per day; P=0.26). Two main limitations were a small sample size and strict exclusion criteria. Additionally, the primary end point of time to first resolution of delirium may be interpreted as a less rigorous measure, because there is no standard definition for delirium resolution, and delirium is a condition that waxes and wanes on its own.
A post hoc analysis by Devlin et al. was conducted on the above study to compare duration and time to first resolution of 10 delirium symptoms in 29 patients.[21] Symptoms included agitation, decreased level of consciousness, inattention, disorientation, hallucinations/delusions, hyperactivity, hypoactivity, inappropriate speech or mood, sleep/wake cycle disturbance, and symptom fluctuation. Only symptom fluctuation (P=0.009), time to first resolution of symptom fluctuation (P=0.004), time with inattention (47 vs 78 hours, P=0.025), and time with symptom fluctuation (47 vs 89 hours; P =0.04) reached statistical significance with quetiapine compared to placebo. However, quetiapine subjects had a longer time to resolution of agitation (3 vs 1 day, P=0.04) and hyperactivity (5 vs 1 day, P<0.04). The authors attribute these findings to the higher use of as‐needed haloperidol in the placebo group (1.9 vs 4.3 mg per day, P=0.26). These results may also be due to a limitation in the study design, which self‐selected for agitation delirium by requiring the use of as‐needed haloperidol as inclusion criteria, as symptoms of agitation would be more likely to receive as‐needed haloperidol doses. In addition, subjects were allowed to receive 1 to 10 mg of haloperidol up to every 2 hours as needed, but there is no discussion of controlling total daily haloperidol dose in each group in the study. Although 1.9 versus 4.3 mg per day is neither statistically nor likely clinically significant, haloperidol is a treatment for delirium, so the study design would be stronger if quetiapine could be directly compared to an equivalent daily dose of haloperidol or if both groups received the same daily dose of haloperidol. Study results are also limited by the nature of a post hoc analysis, missing documentation for individual delirium symptoms, symptoms of delirium not being well characterized and subjective, and delay in enrollment of subjects.
Noncontrolled Trials
Six additional trials are included in the table of this review including 5 open label trials and 1 retrospective cohort study.[24, 25, 26, 27, 28, 29] Schwartz and Masand[24] and Lee et al.[29] compared the efficacy of quetiapine to haloperidol and amisulpride, a second‐generation antipsychotic unavailable in the United States, respectively. In a retrospective cohort of 22 general hospital patients, Schwartz and Masand found quetiapine (average dose, 211.4 mg/day) was as efficacious as haloperidol (average dose, 3.4 mg/day) in improving delirium rating scale (DRS) scores by more than 50% in 10/11 subjects in each group. Lee et al. also found quetiapine (average dose, 113.0 mg/day) to be equally efficacious to amisulpride (156.4 mg/day) in statistically significantly improving DRS‐R‐98 scores in 31 neurosurgery, orthopedic surgery, internal medicine, neurology, and rehabilitation medicine patients. Four open‐label studies tested the efficacy of flexible doses of quetiapine in a total of 63 general hospital, neurosurgery, orthopedic surgery, and oncology patients.[25, 26, 27, 28] Pae et al.,[25] Maneeton et al.,[26] and Kim et al.[27] found that quetiapine statistically significantly reduced DRS‐R‐98, Clinical Global Impression Scale‐Severity, DRS, MMSE, and Clock Drawing Test scores from baseline. Sasaki et al.[28] found all 12 general hospital patients in their study reached remission with an average daily quetiapine dose of 44.9 mg/day. Although these studies have a limited level of evidence due to their small sample sizes, study designs, and heterogeneous subjects, they do still suggest that quetiapine may effectively treat delirium in various patient populations.
DISCUSSION
Although the role of quetiapine for the treatment of delirium continues to develop, the studies evaluated here suggest that quetiapine may be effective and safe for this usage. Resolution of delirium from baseline was shown in all studies. Quetiapine resolved symptoms of delirium more quickly than placebo and had equal efficacy to other antipsychotics, haloperidol and amisulpride. Quetiapine may be most useful for patients with symptom fluctuation as opposed to agitation and hyperactivity or in those patients who may not tolerate haloperidol well. Both randomized control and open‐label trials found a low incidence of adverse effects with quetiapine. There were fewer incidences of QT prolongation and extrapyramidal symptoms, but higher a rate of somnolence with quetiapine compared to other antipsychotics in these trials. However, none of these differences in adverse effects reached statistical significance.
Drawing definitive conclusions about the efficacy of quetiapine in the treatment of delirium is difficult due to multiple study limitations. First, the body of literature is small, with only 2 randomized controlled trials, both of which had limitations. Tahir et al.[19] was underpowered and found no statistically significant difference in the primary end point, DRS‐R‐98 total mean score. Devlin et al.[20] tested the primary end point of time to first resolution of delirium, which may be interpreted as a less rigorous measure because there is no standard definition for delirium resolution. Furthermore, both randomized controlled and observational trials tested the efficacy of quetiapine using different tests, making comparison between these trials difficult. All studies included in this review were carried out in small patient populations, with the largest trial having 42 subjects, and had highly restrictive exclusion criteria limiting the generalizability of the study population to the general hospital population. Although most of the patients included in these studies are general hospital populations, Devlin et al. was performed in critically ill patients, which creates the additional limitation of having heterogeneous study populations. Last, superiority of any 1 antipsychotic is not possible given that none of these studies have done a head‐to‐head comparison of quetiapine with another atypical antipsychotic.
Given the comparable efficacy and safety of antipsychotics, cost is a relevant factor in treatment decisions. Haloperidol is supplied as either a suspension for injection or a tablet. One vial of 5 mg/mL haloperidol is $8.32. Haloperidol 5 mg tablets are approximately $0.29 each, whereas 25 mg tablets of quetiapine are approximately $3.53 each. However, these prices are for consumers and will vary depending on institutional contracts.[42]
CONCLUSION
Quetiapine appears to be an effective and safe agent for the treatment of delirium in both general medicine and ICU patients. Superiority of quetiapine over other antipsychotics for hospital‐associated delirium has not been shown due to limitations in quality and quantity of data. Large, randomized, double‐blind, active control studies with longer study durations are needed to elucidate the efficacy and niche of quetiapine in the treatment of delirium.
Acknowledgment
Disclosure: Nothing to report.
Delirium is an acute fluctuation in mental status that includes symptoms of inattention, disorganized thinking, and altered level of consciousness occurring over a short time period.[1] Prevalence of delirium ranges from 10% to 30% in the hospitalized medically ill and ranges from 40% to 60% in intensive care unit (ICU) patients who are not receiving mechanical ventilation.[2, 3] Patients experience delirium more often if they are of older age; have dementia, cancer, or acquired immune deficiency syndrome; have undergone surgery; are terminally ill; or have received multiple psychoactive medications, particularly benzodiazepines or opioids.[3, 4, 5, 6, 7] Delirium is associated with high rates of morbidity and mortality,[8] with patients more likely to develop complications such as pneumonia, decubitus ulcers, and long‐term cognitive deficits.[9, 10, 11, 12] These complications, in turn, lead to longer hospital stays and increased costs of care.[13, 14] Currently, there are no antipsychotics approved by the US Food and Drug Administration for the treatment of delirium.
Both the 2002 American Society of Critical Care Medicine[12] and the 2004 American Psychiatric Association guidelines[15] on delirium recommend haloperidol as the antipsychotic of choice due to its potent tranquilizing effect, lack of active metabolites, and limited anticholinergic and sedating side effects. However, when given intravenously or at high cumulative doses (generally >35 mg/day), haloperidol has been shown to cause QT interval prolongation potentially leading to torsades de pointes and sudden cardiac death.[15] Recent research in delirium treatment has focused on the second‐generation antipsychotics, and these studies have reported positive findings,[16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29] although no significant differences have been found compared to haloperidol.[16, 17, 18, 22, 23, 24] Systematic reviews have also failed to show a significant difference in efficacy or safety between second‐generation and first‐generation antipsychotics and unfortunately report a number of limitations including poor study designs, small sample sizes, lack of a placebo control group, exclusion of ICU patients, and weak primary outcomes.[8, 30, 31, 32] Attempting to correct for a number of these limitations, recent research with quetiapine has reported promising results in 2 controlled studies.[19, 20]
Quetiapine is a second‐generation antipsychotic with a very low affinity for dopamine receptors and a very high affinity for serotonin receptors.[33, 34] Additionally, quetiapine has a high affinity for histamine and 1‐adrenergic receptors, but a very low affinity for M1 muscarinic receptors.[34] This mechanism of action may allow quetiapine to effectively treat delirium and provide sedation without causing significant extrapyramidal side effects associated with potent dopamine receptor antagonism or precipitating delirium through muscarinic receptor antagonism. Quetiapine has a rapid absorption and a short half‐life (36 hours), giving it a quick onset of action and a fast elimination from the body.[35] Unlike haloperidol, it is only available in oral dosage forms, but can be crushed to administer via enteral tube. Common reported adverse effects include somnolence, hypotension, and dizziness. Although quetiapine has been shown to prolong QTc, Harrigan et al. reported a mean increase in QTc from baseline of 5.7 msec; there was no significant effect on this change in the presence of a metabolic inhibitor, ketoconazole.[36] The mean change in QTc from baseline with quetiapine was lower than the change reported with oral haloperidol, 5.7 msec compared to 7.1 msec, respectively. No statistical comparison was performed between these 2 drugs on this measure. Quetiapine does carry a black box warning for increased mortality in elderly patients with dementia‐related psychosis.[35] However, this risk has also been found with first‐generation antipsychotics.[37, 38, 39] A recent study found this risk of sudden cardiac death extended to adult users of both first‐ and second‐generation antipsychotics.[40] In contrast, Elie et al. found no increased risk of mortality in elderly patients with delirium receiving antipsychoticsover 90% were prescribed either haloperidol or risperidonein their nested case‐controlled study.[41] Quetiapine has demonstrated some benefit with limited side effects in studies utilizing antipsychotics for the treatment of delirium. The purpose of this review was to evaluate the role of quetiapine for the treatment of delirium.
LITERATURE REVIEW
We performed an English‐language literature search of MEDLINE and Embase databases to identify journal articles published between January 1960 and December 2012. Keywords included quetiapine, second‐generation antipsychotic, atypical antipsychotic, delirium, and agitation. The search was limited to English‐language articles and adult subjects (>18 years). Based on our review of abstracts, we included both controlled and noncontrolled trials as long as treatment of delirium was the primary focus. We eliminated case reports, foreign language articles, and poster presentations. We identified 8 trials[19, 20, 24, 25, 26, 27, 28, 29] that included 2 double‐blind, randomized, placebo‐controlled trials, which are described in the text below.[19, 20] Six other trials, 5 open‐label[25, 26, 27, 28, 29] and 1 retrospective cohort,[24] are described in Table 1.
| Study | Study Design | No. of Patients Included in Analyses | Patient Type | Treatment | Baseline Delirium Scores | Quetiapine Dose (mg/day) (MeanSD) | Efficacy Measures | Results | Side Effects |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Schwartz et al. (2000)[24] | RC | 22 | General hospital | Quetiapine or haloperidol flexible dose | DRS score 20.9 Q; 18.5 H | 211.4 Q; 3.4 H | >50% improvement in DRS scores | 10/11 in each treatment group had >50% improvement in DRS scores | EPS 2/11 H and 0/11 Q; mild‐to‐moderate sedation 0/11 H and 2/11 Q |
| Pae et al. (2004)[25] | OL | 22 | Neurosurgery and orthopedic surgery and oncology | Flexible dose quetiapine | DRS‐R‐98 21.83.2 and CGI‐s 4.90.8 | 127.172.2 | DRS‐R‐98 and CGI‐s score reduction | DRS‐R‐98 9.33.8 (P<0.0001) and CGI‐s 2.11.1 (P<0.0001) reduction from baseline; 19/22 (86.3%) and 17/22 (77.3%) showed >50% score reduction for DRS‐R‐98 and CGI‐s, respectively | EPS none; sedation requiring discontinuation 2; mild sedation 3; serious side effects none |
| Maneeton et al. (2007)[26] | OL | 17 | General hospital | Quetiapine 25100 mg/day in 1 or 2 divided doses | DRS 24.53.2 and CGI‐s 4.90.9 | 45.728.7 | 50% reduction in DRS scores | 15/17 (88.2%) had 50% reduction in DRS score; all DRS and CGI‐s scores on days 17 of the 7‐day treatment course were significantly lower than baseline scores | EPS tremor 2; hypotension 2; daytime sleepiness 13; nightmare 3; dry mouth 2; nausea 1 |
| Kim et al. (2003)[27] | OL | 12 (all male and age 64 years) | General hospital | Quetiapine 25 mg BID and increased by 25 mg every 2 days until patient maximally stabilized | DRS 18.256.05; MMSE 14.505.90; CGI‐s 3.000.43; CDT 3.25 2.77 | 93.7523.31 | Reduction in DRS, MMSE, CGI‐s, or CDT scores | All scores were statistically significantly improved from baseline; DRS at end of study 0.631.21 (P=0.03) | No dropouts due to side effects; EPS none; sedation 2; vivid dreams 1 |
| Sasaki et al. (2003)[28] | OL | 12 | General hospital | Quetiapine started at 25 or 50 mg/day and titrated to maximal clinical effect | DRS‐J 18.14.2 | 44.931.0 | Remission was DRS‐J score <12 (cutoff for delirium) and resolution of delirium symptoms | Remission occurred in all patients; mean DRS‐J score was reduced to 9.31.6 after remission | No statistically significant change from baseline in EPS; no excessive daytime sedation, anticholinergic effects, vital sign, or lab parameter changes |
| Lee et al. (2005)[29] | OL | 31 | Neurosurgery and orthopedic surgery, internal medicine, neurology and rehabilitation medicine | Flexible dose quetiapine or amisulpride | DRS‐R‐98 10.14.1 Q and 10.54.1 A | 11385.5 Q; 156.497.5 A | Reduction in DRS‐R‐98 score | DRS‐R‐98 scores significantly reduced from baseline in both groups with 3.52.6 Q (P=0.001) and 3.51.4 A (P=0.000); no difference between groups (P=0.842); 12 (80%) Q and 13 (81.3%) A had >50% reduction in DRS‐R‐98 score | No serious side effects; dropouts 5 Q and 4 A; oversedation 1 Q and 1 A; patient withdrawal 2 Q and 1 A; no statistical differences in total sleep time P=0.767 or quality of sleep P=0.984 |
Randomized Controlled Trials
Tahir et al. published a double‐blind, randomized, placebo‐controlled trial examining the efficacy and tolerability of quetiapine in the treatment of delirium.[19] Inclusion criteria were a Diagnostic and Statistical Manual of Mental Disorders‐IV diagnosis of delirium and a Delirium Rating Scale Revised 98 (DRS‐R‐98) total score of at least 15, indicating the presence of delirium. Subjects were excluded for major preexisting cognitive deficits, alcohol withdrawal, preexisting psychosis, substance dependence, inability to comply with the constraints of the trial, and concurrent medications that interact with quetiapine.
Forty‐two general medicine subjects were enrolled and randomized in the study with no difference in baseline characteristics. Patients received either placebo or quetiapine 25 mg once daily. Doses were titrated by 25 mg daily to a maximum daily dose of 175 mg in divided doses. The primary end point was DRS‐R‐98 total mean score assessed on days 1, 2, 3, 4, 7, and 10, with follow‐up assessment on day 30. Secondary outcome measures were Mini‐Mental State Examination (MMSE), the Brief Psychiatric Rating Scale, and the Clinical Global Improvement Scale. Tolerability was assessed using the Abnormal Involuntary Movements Scale and by clinical examination.
No differences in total mean DRS‐R‐98 score at individual time points reached statistical significance, but these scores improved more quickly in the quetiapine group than placebo. The secondary outcome of rate of delirium improvement on severity score did reach statistical significance; differences on mean severity scores were 0.8270.37 (P=0.026), suggesting that severity scores improved 82% more quickly than placebo. There were no significant differences between groups for any of the other secondary outcomes. Seven patients died within 30 days of entering the study (4 in the quetiapine group and 3 in the placebo group) due to serious medical conditions and not due to study medication as determined through clinical review. One patient withdrew from quetiapine due to sedation. Results of this study are limited by a small sample size, as it was underpowered to detect a statistically significant difference in the primary outcome. Other limitations include subjects who were older, with mean age of 84 years, which may have prevented titration of quetiapine, and subjects with minor cognitive deficits were included, which may have reduced the impact of treatment on the study outcomes. Additionally, strict criteria for exclusion kept those patients most likely to develop delirium from being included, limiting the studies external validity. Finally, the use of DRS‐R‐98 mean total and severity scores is subject to error as outliers in the mean could potentially skew the results.
Devlin et al. investigated the efficacy and safety of quetiapine for ICU delirium in critically ill patients in a double‐blind, randomized, placebo‐controlled trial.[20] Inclusion criteria were an Intensive Care Delirium Screening Checklist Score (ICDSC) >4, which indicates the presence of delirium, an order for as‐needed haloperidol, and tolerating enteral nutrition. Patients were excluded if they had a complicating neurologic condition, current treatment with dexmedetomidine or with medications that interact with quetiapine, baseline QTc interval >500, pregnancy, or poor prognosis. Thirty‐six critically ill subjects were randomized with no significant differences in baseline characteristics including exposure to fentanyl, haloperidol, and benzodiazepines, and in particular, midazolam. If the subject received at least 1 dose of as‐needed haloperidol in the previous 24 hours, then either placebo or quetiapine was given. Quetiapine was initiated at 50 mg every 12 hours and titrated up by 50 mg every 12 hours to a maximum dose of 200 mg every 12 hours. Patients could receive intravenous haloperidol, 1 to 10 mg up to every 2 hours as needed. The primary outcome was time to first resolution of delirium defined as time from administration of the first dose of study drug until an ICDSC <3 was first detected, indicating an absence of delirium. Secondary outcomes were total hours in delirium, total hours spent deeply sedated (Sedation‐Agitation Scale [SAS] <2) or agitated (SAS >5), episodes of subject‐initiated device removal, use of haloperidol therapy including total dose in milligrams, number of doses and number of days of therapy, the use of sedatives (converted to midazolam equivalents) and analgesics, duration of study‐drug administration, average daily and maximum study‐drug dose, length of mechanical ventilation, duration of both ICU and hospital stay, hospital mortality, and disposition of subjects after hospital discharge. Safety measures were total number of adverse and serious adverse events, episodes of somnolence, incidence of extrapyramidal symptoms, and episodes of QTc interval prolongation.
The time to first resolution of delirium was shorter with quetiapine compared to placebo (median [interquartile range]: 1.0 [0.53.0] vs 4.5 days [2.0‐7.0]; P=0.001). Resolution of delirium occurred at least once in all quetiapine patients and in 78% of placebo patients (P=0.05). Statistical significance with quetiapine was reached on the following secondary end points: time spent in delirium (36 [1287] vs 120 hours [60195], P=0.006); shorter duration of study drug (102 [84168] vs 186 hours [108228], P=0.04); lower daily study drug dose (110 [88191] vs 210 mg [116293], P=0.01); less upregulation of the study medication dose (200 [100313] vs 375 mg [25400], P=0.02); fewer hours of agitation (6 [038] vs 36 hours [1166], P=0.02); shorter duration of haloperidol therapy (3 [24] vs 4 days [38], P=0.05); and fewer days of fentanyl (0 [03] vs 4 days [19], P=0.03). There were no significant differences between groups for any other secondary outcomes. As‐needed haloperidol use in the quetiapine versus the placebo group was lower but not statistically significant (1.9 vs 4.3 mg per day; P=0.26). Two main limitations were a small sample size and strict exclusion criteria. Additionally, the primary end point of time to first resolution of delirium may be interpreted as a less rigorous measure, because there is no standard definition for delirium resolution, and delirium is a condition that waxes and wanes on its own.
A post hoc analysis by Devlin et al. was conducted on the above study to compare duration and time to first resolution of 10 delirium symptoms in 29 patients.[21] Symptoms included agitation, decreased level of consciousness, inattention, disorientation, hallucinations/delusions, hyperactivity, hypoactivity, inappropriate speech or mood, sleep/wake cycle disturbance, and symptom fluctuation. Only symptom fluctuation (P=0.009), time to first resolution of symptom fluctuation (P=0.004), time with inattention (47 vs 78 hours, P=0.025), and time with symptom fluctuation (47 vs 89 hours; P =0.04) reached statistical significance with quetiapine compared to placebo. However, quetiapine subjects had a longer time to resolution of agitation (3 vs 1 day, P=0.04) and hyperactivity (5 vs 1 day, P<0.04). The authors attribute these findings to the higher use of as‐needed haloperidol in the placebo group (1.9 vs 4.3 mg per day, P=0.26). These results may also be due to a limitation in the study design, which self‐selected for agitation delirium by requiring the use of as‐needed haloperidol as inclusion criteria, as symptoms of agitation would be more likely to receive as‐needed haloperidol doses. In addition, subjects were allowed to receive 1 to 10 mg of haloperidol up to every 2 hours as needed, but there is no discussion of controlling total daily haloperidol dose in each group in the study. Although 1.9 versus 4.3 mg per day is neither statistically nor likely clinically significant, haloperidol is a treatment for delirium, so the study design would be stronger if quetiapine could be directly compared to an equivalent daily dose of haloperidol or if both groups received the same daily dose of haloperidol. Study results are also limited by the nature of a post hoc analysis, missing documentation for individual delirium symptoms, symptoms of delirium not being well characterized and subjective, and delay in enrollment of subjects.
Noncontrolled Trials
Six additional trials are included in the table of this review including 5 open label trials and 1 retrospective cohort study.[24, 25, 26, 27, 28, 29] Schwartz and Masand[24] and Lee et al.[29] compared the efficacy of quetiapine to haloperidol and amisulpride, a second‐generation antipsychotic unavailable in the United States, respectively. In a retrospective cohort of 22 general hospital patients, Schwartz and Masand found quetiapine (average dose, 211.4 mg/day) was as efficacious as haloperidol (average dose, 3.4 mg/day) in improving delirium rating scale (DRS) scores by more than 50% in 10/11 subjects in each group. Lee et al. also found quetiapine (average dose, 113.0 mg/day) to be equally efficacious to amisulpride (156.4 mg/day) in statistically significantly improving DRS‐R‐98 scores in 31 neurosurgery, orthopedic surgery, internal medicine, neurology, and rehabilitation medicine patients. Four open‐label studies tested the efficacy of flexible doses of quetiapine in a total of 63 general hospital, neurosurgery, orthopedic surgery, and oncology patients.[25, 26, 27, 28] Pae et al.,[25] Maneeton et al.,[26] and Kim et al.[27] found that quetiapine statistically significantly reduced DRS‐R‐98, Clinical Global Impression Scale‐Severity, DRS, MMSE, and Clock Drawing Test scores from baseline. Sasaki et al.[28] found all 12 general hospital patients in their study reached remission with an average daily quetiapine dose of 44.9 mg/day. Although these studies have a limited level of evidence due to their small sample sizes, study designs, and heterogeneous subjects, they do still suggest that quetiapine may effectively treat delirium in various patient populations.
DISCUSSION
Although the role of quetiapine for the treatment of delirium continues to develop, the studies evaluated here suggest that quetiapine may be effective and safe for this usage. Resolution of delirium from baseline was shown in all studies. Quetiapine resolved symptoms of delirium more quickly than placebo and had equal efficacy to other antipsychotics, haloperidol and amisulpride. Quetiapine may be most useful for patients with symptom fluctuation as opposed to agitation and hyperactivity or in those patients who may not tolerate haloperidol well. Both randomized control and open‐label trials found a low incidence of adverse effects with quetiapine. There were fewer incidences of QT prolongation and extrapyramidal symptoms, but higher a rate of somnolence with quetiapine compared to other antipsychotics in these trials. However, none of these differences in adverse effects reached statistical significance.
Drawing definitive conclusions about the efficacy of quetiapine in the treatment of delirium is difficult due to multiple study limitations. First, the body of literature is small, with only 2 randomized controlled trials, both of which had limitations. Tahir et al.[19] was underpowered and found no statistically significant difference in the primary end point, DRS‐R‐98 total mean score. Devlin et al.[20] tested the primary end point of time to first resolution of delirium, which may be interpreted as a less rigorous measure because there is no standard definition for delirium resolution. Furthermore, both randomized controlled and observational trials tested the efficacy of quetiapine using different tests, making comparison between these trials difficult. All studies included in this review were carried out in small patient populations, with the largest trial having 42 subjects, and had highly restrictive exclusion criteria limiting the generalizability of the study population to the general hospital population. Although most of the patients included in these studies are general hospital populations, Devlin et al. was performed in critically ill patients, which creates the additional limitation of having heterogeneous study populations. Last, superiority of any 1 antipsychotic is not possible given that none of these studies have done a head‐to‐head comparison of quetiapine with another atypical antipsychotic.
Given the comparable efficacy and safety of antipsychotics, cost is a relevant factor in treatment decisions. Haloperidol is supplied as either a suspension for injection or a tablet. One vial of 5 mg/mL haloperidol is $8.32. Haloperidol 5 mg tablets are approximately $0.29 each, whereas 25 mg tablets of quetiapine are approximately $3.53 each. However, these prices are for consumers and will vary depending on institutional contracts.[42]
CONCLUSION
Quetiapine appears to be an effective and safe agent for the treatment of delirium in both general medicine and ICU patients. Superiority of quetiapine over other antipsychotics for hospital‐associated delirium has not been shown due to limitations in quality and quantity of data. Large, randomized, double‐blind, active control studies with longer study durations are needed to elucidate the efficacy and niche of quetiapine in the treatment of delirium.
Acknowledgment
Disclosure: Nothing to report.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: APA; 1994.
- American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156:1–20.
- , . Pharmacological and nonpharmacological management of delirium in critically ill patients. Neurotherapeutics. 2012;9:158–175.
- , . Delirium in cancer patients. Int Psychogeriatr. 1991;3:333–336.
- . Organic mental disorders caused by HIV: update on early‐diagnosis and treatment. Am J Psychiatry. 1990;147:696–710.
- . Post‐operative delirium. Int Psychogeriatr. 1991;3:325–332.
- , , . Delirium in terminally ill cancer patients. Am J Psychiatry. 1983;140:1048–1050.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Acute confusional states in the hospitalized elderly: incidence, risk factors and complications [abstract]. Clin Res. 1989;37:524A.
- , . Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41–46.
- , , . Duration of delirium shortened by the correction of electrolyte imbalance. Jpn J Psychiatry Neurol. 1988;42:81–88.
- , , , et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–141.
- , , , , , . Intensive care unit delirium is an independent predictor of longer hospital stay: a prospective analysis of 261 non‐ventilated patients. Crit Care. 2005;9:R375–R381.
- , , , et al. Costs associated with delirium in mechanically ventilated patients. Crit Care Med. 2004;32:955–962.
- Cook IA: Guideline Watch: Practice Guideline for the Treatment of Patients With Delirium. Arlington, VA:American Psychiatric Association, 2004. Available at: http://www.psych.org/psych_pract/treatg/pg/prac_guide.cfm. Accessed March 5, 2012.
- , . A double‐blind trial of risperdone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297–301.
- , , , . Olanzapine vs haloperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30:444–449.
- , , , et al. Feasibility, efficacy, and safety of antipsychotics for intensive care unit delirium: the MIND randomized, placebo‐controlled trial. Crit Care Med. 2010;38:428–437.
- , , , et al. A randomized controlled trial of quetiapine versus placebo in the treatment of delirium. Psychosom Res. 2010;69:485–490.
- , , , et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double‐blind, placebo‐controlled pilot study. Crit Care Med. 2010;38:419–427.
- , , , et al. Impact of quetiapine on resolution of individual delirium symptoms in critically ill patients with delirium: a post‐hoc analysis of a double‐blind, randomized, placebo‐controlled study. Critical Care. 2011;15:R215.
- , . Olanzapine in the treatment of delirium. Psychosomatics. 1998;39:422–430.
- , , . Comparative efficacy study of haloperidol, olanzapine and risperidone in delirium. J Psychosom Res. 2011;71:277–281.
- , . Treatment of delirium with quetiapine. J Clin Psychiatry. 2000;2:10–12.
- , , , , . A pilot trial of quetiapine for the treatment of patients with delirium. Hum Psychopharmacol Clin Exp. 2004;19:125–127.
- , , . An open‐label study of quetiapine for delirium. J Med Assoc Thai. 2007;10:2158–2163.
- , , , . Treatment of delirium in older adults with quetiapine. J Geriatr Psychiatry Neurol. 2003;16:29–31.
- , , , et al. A prospective, open‐label, flexible‐dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64:1316–1321.
- , , , et al. Amisulpride versus quetiapine for the treatment of delirium: a randomized, open prospective study. Int Clin Psychopharmacol. 2005;20:311–314.
- , , , et al. Pharmacological management of delirium in hospitalized adults‐a systematic evidence review. J Gen Intern Med. 2009;24:848–853.
- , , . Systematic review of antipsychotics for the treatment of hospital‐associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40:1966–1973.
- , , , . Atypical antipsychotics versus haloperidol for treatment of delirium in acutely ill patients. Pharmacotherapy. 2007;27:588–594.
- , , . Delirium: prevention, treatment, and outcome studies. J Geriatr Psychiatry Neurol. 1998;11:126–137.
- , . Seroquel: biochemical profile of a potential atypical antipsychotic. Psychopharmacology. 1993;112:285–292.
- Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; December 2011.
- , , , et al. A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol. 2004;24:62–69.
- , , , et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164:1568–1576.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , , . Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med. 2009;360:225–235.
- , , , , , . A retrospective, exploratory, secondary analysis of the association between antipsychotic use and mortality in elderly patients with delirium. Int Psychogeriatr. 2009;21(3):588–592.
- Walgreens Co. Price your drugs Available at: http://www.walgreens.com/pharmacy/psc/drugpricing/psc_drug_pricing.jsp. Accessed December 17, 2012.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: APA; 1994.
- American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156:1–20.
- , . Pharmacological and nonpharmacological management of delirium in critically ill patients. Neurotherapeutics. 2012;9:158–175.
- , . Delirium in cancer patients. Int Psychogeriatr. 1991;3:333–336.
- . Organic mental disorders caused by HIV: update on early‐diagnosis and treatment. Am J Psychiatry. 1990;147:696–710.
- . Post‐operative delirium. Int Psychogeriatr. 1991;3:325–332.
- , , . Delirium in terminally ill cancer patients. Am J Psychiatry. 1983;140:1048–1050.
- , , . Antipsychotics in the treatment of delirium: a systematic review. J Clin Psychiatry. 2007;68:11–21.
- , , , et al. Acute confusional states in the hospitalized elderly: incidence, risk factors and complications [abstract]. Clin Res. 1989;37:524A.
- , . Prognosis of delirium in elderly hospital patients. CMAJ. 1993;149:41–46.
- , , . Duration of delirium shortened by the correction of electrolyte imbalance. Jpn J Psychiatry Neurol. 1988;42:81–88.
- , , , et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–141.
- , , , , , . Intensive care unit delirium is an independent predictor of longer hospital stay: a prospective analysis of 261 non‐ventilated patients. Crit Care. 2005;9:R375–R381.
- , , , et al. Costs associated with delirium in mechanically ventilated patients. Crit Care Med. 2004;32:955–962.
- Cook IA: Guideline Watch: Practice Guideline for the Treatment of Patients With Delirium. Arlington, VA:American Psychiatric Association, 2004. Available at: http://www.psych.org/psych_pract/treatg/pg/prac_guide.cfm. Accessed March 5, 2012.
- , . A double‐blind trial of risperdone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297–301.
- , , , . Olanzapine vs haloperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30:444–449.
- , , , et al. Feasibility, efficacy, and safety of antipsychotics for intensive care unit delirium: the MIND randomized, placebo‐controlled trial. Crit Care Med. 2010;38:428–437.
- , , , et al. A randomized controlled trial of quetiapine versus placebo in the treatment of delirium. Psychosom Res. 2010;69:485–490.
- , , , et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double‐blind, placebo‐controlled pilot study. Crit Care Med. 2010;38:419–427.
- , , , et al. Impact of quetiapine on resolution of individual delirium symptoms in critically ill patients with delirium: a post‐hoc analysis of a double‐blind, randomized, placebo‐controlled study. Critical Care. 2011;15:R215.
- , . Olanzapine in the treatment of delirium. Psychosomatics. 1998;39:422–430.
- , , . Comparative efficacy study of haloperidol, olanzapine and risperidone in delirium. J Psychosom Res. 2011;71:277–281.
- , . Treatment of delirium with quetiapine. J Clin Psychiatry. 2000;2:10–12.
- , , , , . A pilot trial of quetiapine for the treatment of patients with delirium. Hum Psychopharmacol Clin Exp. 2004;19:125–127.
- , , . An open‐label study of quetiapine for delirium. J Med Assoc Thai. 2007;10:2158–2163.
- , , , . Treatment of delirium in older adults with quetiapine. J Geriatr Psychiatry Neurol. 2003;16:29–31.
- , , , et al. A prospective, open‐label, flexible‐dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64:1316–1321.
- , , , et al. Amisulpride versus quetiapine for the treatment of delirium: a randomized, open prospective study. Int Clin Psychopharmacol. 2005;20:311–314.
- , , , et al. Pharmacological management of delirium in hospitalized adults‐a systematic evidence review. J Gen Intern Med. 2009;24:848–853.
- , , . Systematic review of antipsychotics for the treatment of hospital‐associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40:1966–1973.
- , , , . Atypical antipsychotics versus haloperidol for treatment of delirium in acutely ill patients. Pharmacotherapy. 2007;27:588–594.
- , , . Delirium: prevention, treatment, and outcome studies. J Geriatr Psychiatry Neurol. 1998;11:126–137.
- , . Seroquel: biochemical profile of a potential atypical antipsychotic. Psychopharmacology. 1993;112:285–292.
- Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; December 2011.
- , , , et al. A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol. 2004;24:62–69.
- , , , et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164:1568–1576.
- , , , et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775–786.
- , , , et al. Risk of death in elderly users of conventional vs. atypical antipsychotic medications. N Engl J Med. 2005;353:2335–2341.
- , , , , . Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med. 2009;360:225–235.
- , , , , , . A retrospective, exploratory, secondary analysis of the association between antipsychotic use and mortality in elderly patients with delirium. Int Psychogeriatr. 2009;21(3):588–592.
- Walgreens Co. Price your drugs Available at: http://www.walgreens.com/pharmacy/psc/drugpricing/psc_drug_pricing.jsp. Accessed December 17, 2012.
Initiating palliative care conversations: lessons from Jewish bioethics
What are the ethical responsibilities of the medical staff (doctors, nurses, social workers, and chaplains) regarding thepreservation of meaningful life for their patients who are approaching the end of life (EOL)? In particular, what is the staff’sethical responsibility to initiate a conversation with their patient regarding palliative care? By subjecting traditional Jewish teachings to an ethical analysis and then exploring the underlying universal principles, we will suggest a general ethical duty toinform patients of the different care options, especially in a manner that preserves hope. The principle that we can derive from Jewish bioethics teaches that the medical staff has a responsibility to help our patients live in a way that is consistent with how they understand their task or responsibility in life. For some patients, the best way to preserve a meaningful life in which they can fulfill their sense of purpose in the time that remains is to focus on palliation. For this reason, although palliative and supportive care are provided from the time of diagnosis, it is critical we make sure our patients realize that they have the opportunity to make a decision between either pursuing additional active treatments or choosing to focus primarily on palliative therapies to maximize quality of life. The Jewish tradition and our experience in spiritual care suggest the importance of helping patients preserve hope while, simultaneously, honestly acknowledging their situation. Staff members can play a vital role in helping patients make the most of this new period of their lives.
Click on the PDF icon at the top of this introduction to read the full article.
What are the ethical responsibilities of the medical staff (doctors, nurses, social workers, and chaplains) regarding thepreservation of meaningful life for their patients who are approaching the end of life (EOL)? In particular, what is the staff’sethical responsibility to initiate a conversation with their patient regarding palliative care? By subjecting traditional Jewish teachings to an ethical analysis and then exploring the underlying universal principles, we will suggest a general ethical duty toinform patients of the different care options, especially in a manner that preserves hope. The principle that we can derive from Jewish bioethics teaches that the medical staff has a responsibility to help our patients live in a way that is consistent with how they understand their task or responsibility in life. For some patients, the best way to preserve a meaningful life in which they can fulfill their sense of purpose in the time that remains is to focus on palliation. For this reason, although palliative and supportive care are provided from the time of diagnosis, it is critical we make sure our patients realize that they have the opportunity to make a decision between either pursuing additional active treatments or choosing to focus primarily on palliative therapies to maximize quality of life. The Jewish tradition and our experience in spiritual care suggest the importance of helping patients preserve hope while, simultaneously, honestly acknowledging their situation. Staff members can play a vital role in helping patients make the most of this new period of their lives.
Click on the PDF icon at the top of this introduction to read the full article.
What are the ethical responsibilities of the medical staff (doctors, nurses, social workers, and chaplains) regarding thepreservation of meaningful life for their patients who are approaching the end of life (EOL)? In particular, what is the staff’sethical responsibility to initiate a conversation with their patient regarding palliative care? By subjecting traditional Jewish teachings to an ethical analysis and then exploring the underlying universal principles, we will suggest a general ethical duty toinform patients of the different care options, especially in a manner that preserves hope. The principle that we can derive from Jewish bioethics teaches that the medical staff has a responsibility to help our patients live in a way that is consistent with how they understand their task or responsibility in life. For some patients, the best way to preserve a meaningful life in which they can fulfill their sense of purpose in the time that remains is to focus on palliation. For this reason, although palliative and supportive care are provided from the time of diagnosis, it is critical we make sure our patients realize that they have the opportunity to make a decision between either pursuing additional active treatments or choosing to focus primarily on palliative therapies to maximize quality of life. The Jewish tradition and our experience in spiritual care suggest the importance of helping patients preserve hope while, simultaneously, honestly acknowledging their situation. Staff members can play a vital role in helping patients make the most of this new period of their lives.
Click on the PDF icon at the top of this introduction to read the full article.

Septic shock: The initial moments and beyond
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).

The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.

Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).
The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.
Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
Considerably fewer patients who develop sepsis are dying of it now, thanks to a number of studies of how to reverse sepsis-induced tissue hypoxia.1 The greatest strides in improving outcomes have been attributed to better early management, which includes prompt recognition of sepsis, rapid initiation of antimicrobial therapy, elimination of the source of infection, and early goal-directed therapy. Thus, even though the incidence of severe sepsis and septic shock is increasing,2,3 the Surviving Sepsis Campaign has documented a significant decrease in unadjusted mortality rates (37% to 30.8%) associated with the bundled approach in the management of sepsis.4 (We will talk about this later in the article.)
This review will summarize the evidence for the early management of septic shock and will evaluate the various treatment decisions beyond the initial phases of resuscitation.
INFLAMMATION AND VASODILATION
Sepsis syndrome starts with an infection that leads to a proinflammatory state with a complex interaction between anti-inflammatory and proinflammatory mediators, enhanced coagulation, and impaired fibrinolysis.5,6
Sepsis induces vasodilation by way of inappropriate activation of vasodilatory mechanisms (increased synthesis of nitric oxide and vasopressin deficiency) and failure of vasoconstrictor mechanisms (activation of ATP-sensitive potassium channels in vascular smooth muscle).7 Thus, the hemodynamic abnormalities are multifactorial, and the resultant tissue hypoperfusion further contributes to the proinflammatory and procoagulant state, precipitating multiorgan dysfunction and, often, death.
DEFINITIONS
- Sepsis—infection together with systemic manifestation of inflammatory response
- Severe sepsis—sepsis plus induced organ dysfunction or evidence of tissue hypoperfusion
- Septic shock—sepsis-induced hypotension persisting despite adequate fluid resuscitation.
EARLY MANAGEMENT OF SEPTIC SHOCK
Early in the course of septic shock, the physician’s job is to:
- Recognize it promptly
- Begin empiric antibiotic therapy quickly
- Eliminate the source of infection, if applicable, eg, by removing an infected central venous catheter
- Give fluid resuscitation, titrated to specific goals
- Give vasopressor therapy to maintain blood pressure, organ perfusion, and oxygen delivery (Table 1).
The line between “early” and “late” is not clear. Traditionally, it has been drawn at 6 hours from presentation, and this cutoff was used in some of the studies we will discuss here.
Recognizing severe sepsis early in its course
The diagnosis of severe sepsis may be challenging, since up to 40% of patients may present with cryptic shock. These patients may not be hemodynamically compromised but may show evidence of tissue hypoxia, eg, an elevated serum lactate concentration or a low central venous oxygen saturation (Scvo2), or both.8 In view of this, much effort has gone into finding a biomarker that, in addition to clinical features, can help identify patients in an early stage of sepsis.
Procalcitonin levels rise in response to severe bacterial infection,9 and they correlate with sepsis-related organ failure scores and outcomes.10,11 Thus, the serum procalcitonin level may help in assessing the severity of sepsis, especially when combined with standard clinical and laboratory variables. However, controversy exists about the threshold to use in making decisions about antibiotic therapy and the value of this test in differentiating severe noninfectious inflammatory reactions from infectious causes of shock.12 Therefore, it is not widely used in clinical practice.
Serum lactate has been used for decades as a marker of tissue hypoperfusion. It is typically elevated in patients with severe sepsis and septic shock, and although the hyperlactatemia could be a result of global hypoperfusion, it can also be secondary to sepsis-induced mitochondrial dysfunction,13 impaired pyruvate dehydrogenase activity,14 increased aerobic glycolysis by catecholamine-stimulated sodium-potassium pump hyperactivity,15 and even impaired clearance.16
But whatever the mechanism, elevated lactate in severe sepsis and septic shock predicts a poor outcome and may help guide aggressive resuscitation. In fact, early lactate clearance (ie, normalization of an elevated value on repeat testing within the first 6 hours) is associated with better outcomes in patients with severe sepsis and septic shock.17,18
Panels of biomarkers. A literature search revealed over 3,000 papers on 178 different biomarkers in sepsis.19 Many of these biomarkers lack sufficient specificity and sensitivity for clinical use, and thus some investigators have suggested using a panel of them to enhance their predictive ability. Shapiro et al20 evaluated 971 patients admitted to the emergency department with suspected infection and discovered that a panel of three biomarkers (neutrophil gelatinase-associated lipocalin, protein C, and interleukin-1 receptor antagonist) was highly predictive of severe sepsis, septic shock, and death.
Starting empiric antibiotic therapy early
As soon as severe sepsis and septic shock are recognized, it is imperative that adequate empiric antibiotic treatment be started, along with infectious source control if applicable.21 The Surviving Sepsis Campaign guidelines recommend starting intravenous antibiotics as early as possible—within the first hour of recognition of severe sepsis with or without septic shock.22
Kumar et al,23 in a multicenter retrospective study of patients with septic shock, found that each hour of delay in giving appropriate antimicrobial agents in the first 6 hours from the onset of hypotension was associated with a 7.6% decrease in the in-hospital survival rate.
In a similar study,24 the same investigators analyzed data from 5,715 septic shock patients regarding the impact of starting the right antimicrobial therapy. Appropriate antimicrobial agents (ie, those having in vitro activity against the isolated pathogens) were given in 80.1% of cases, and the survival rate in those who received appropriate antibiotics was drastically higher than in those who received inappropriate ones (52.0% vs 10.3%, P < .0001).
In addition, two recent studies evaluated the importance of early empiric antibiotic therapy in conjunction with resuscitative protocols.25,26 In a preplanned analysis of early antimicrobial use in a study comparing lactate clearance and Scvo2 as goals of therapy, Puskarich et al26 found that fewer patients who received antibiotics before shock was recognized (according to formal criteria) died. Similarly, in a retrospective study in patients presenting to the emergency department and treated with early goal-directed therapy (defined below), Gaieski et al25 found that the mortality rate was drastically lower when antibiotics were started within 1 hour of either triage or initiation of early goal-directed therapy.
In short, it is imperative to promptly start the most appropriate broad-spectrum antibiotics to target the most likely pathogens based on site of infection, patient risk of multidrug-resistant pathogens, and local susceptibility patterns.
Goal-directed resuscitative therapy
As with antimicrobial therapy, resuscitative therapy should be started early and directed at defined goals.
Rivers et al27 conducted a randomized, controlled study in patients with severe sepsis or septic shock presenting to an emergency department of an urban teaching hospital. The patients were at high risk and had either persistent hypotension after a fluid challenge or serum lactate levels of 4 mmol/L or higher.
Two hundred sixty patients were randomized to receive either early goal-directed therapy in a protocol aimed at maximizing the intravascular volume and correcting global tissue hypoxia or standard therapy in the first 6 hours after presentation. The goals in the goal-directed therapy group were:
- Central venous pressure 8 to 12 mm Hg (achieved with aggressive fluid resuscitation with crystalloids)
- Mean arterial blood pressure greater than 65 mm Hg (maintained with vasoactive drugs, if necessary)
- Scvo2 above 70%. To achieve this third goal, packed red blood cells were infused to reach a target hematocrit of greater than 30%. For patients with a hematocrit higher than 30% but still with an Scvo2 less than 70%, inotropic agents were added and titrated to the Scvo2 goal of 70%.
Goal-directed therapy reduced the in-hospital mortality rate by 16% (the mortality rates were 30.5% in the goal-directed group and 46.5% in the standard therapy group, P = .009) and also reduced the 28- and 60-day mortality rates by similar proportions.27
Subsequent studies of a protocol for early recognition and treatment of sepsis have concluded that early aggressive fluid resuscitation decreases the ensuing need for vasopressor support.28 A resuscitation strategy based on early goal-directed therapy is a major component of the initial resuscitation bundle recommended by the Surviving Sepsis Campaign.22 (A “bundle” refers to the implementation of a core set of recommendations involving the simultaneous adaptation of a number of interventions.)
Areas of debate. However, concerns have been raised about the design of the study by Rivers et al and the mortality rate in the control group, which was higher than one would expect from the patients’ Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.29 In particular, the bundled approach they used precludes the ability to differentiate which interventions were responsible for the outcome benefits. Indeed, there were two major interventions in the early goal-directed therapy group: a protocol for achieving the goals described and the use of Scvo2 as a goal.
Aggressive fluid resuscitation is considered the most critical aspect of all the major interventions, and there is little argument on its value. The debate centers on central venous pressure as a preload marker, since after the publication of the early goal-directed therapy trial,27 several studies showed that central venous pressure may not be a valid measure to predict fluid responsiveness (discussed later in this paper).30,31
The choice of colloids or crystalloids for fluid resuscitation is another area of debate. Clinical evidence suggests that albumin is equivalent to normal saline in a heterogeneous intensive care unit population,32 but subgroup analyses suggest albumin may be superior in patients with septic shock.33 Studies are ongoing (NCT00707122, NCT01337934, and NCT00318942). The use of hydroxyethyl starch in severe sepsis is associated with higher rates of acute renal failure and need for renal replacement therapy than Ringer’s lactate,34 and is generally not recommended. This is further substantiated by two recent randomized controlled studies, which found that the use of hydroxyethyl starch for fluid resuscitation in severe sepsis, compared with crystalloids, did not reduce the mortality rate (and even increased it in one study), and was associated with more need for renal replacement therapy.35,36
The use of Scvo2 is yet another topic of debate, and other monitoring variables have been evaluated. A recent study assessed the noninferiority of incorporating venous lactate clearance into the early goal-directed therapy protocol vs Scvo2.37 Both groups had identical goals for central venous pressure and mean arterial pressure but differed in the use of lactate clearance (defined as at least a 10% decline) or Scvo2 (> 70%) as the goal for improving tissue hypoxia. There were no significant differences between groups in their in-hospital mortality rates (17% in the lactate clearance group vs 23% in the Scvo2 group; criteria for noninferiority met). This suggests that lactate may be an alternative to Scvo2 as a goal in early goal-directed therapy. However, a secondary analysis of the data revealed a lack of concordance in achieving lactate clearance and Scvo2 goals, which suggests that these parameters may be measuring distinct physiologic processes.38 Since the hemodynamic profiles of septic shock patients are complex, it may be prudent to use both of these markers of resuscitation until further studies are completed.
Given the debate, a number of prospective randomized trials are under way to evaluate resuscitative interventions. These include the Protocolized Care for Early Septic Shock trial (NCT00510835), the Australasian Resuscitation in Sepsis Evaluation trial (NCT00975793), and the Protocolised Management of Sepsis (ProMISe) trial in the United Kingdom (ISRCTN 36307479). These three trials will evaluate, collectively, close to 4,000 patients and will provide considerable insights into resuscitative interventions in septic shock.
Vasopressors: Which one to use?
If fluid therapy does not restore perfusion, vasopressors should be promptly initiated, as the longer that hypotension goes on, the lower the survival rate.39
But which vasopressor should be used? The early goal-directed therapy protocol used in the study by Rivers et al27 did not specify which vasopressor should be used to keep the mean arterial pressure above 65 mm Hg.
The Surviving Sepsis Campaign22 recommends norepinephrine as the first-choice vasopressor, with dopamine as an alternative only in selected patients, such as those with absolute or relative bradycardia.
The guidelines also recommend epinephrine to be added to or substituted for norepinephrine when an additional catecholamine is needed to maintain adequate blood pressure.22 Furthermore, vasopressin at a dose of 0.03 units/min can be added to norepinephrine with the intent of raising the blood pressure or decreasing the norepinephrine requirement. Higher doses of vasopressin should be reserved for salvage therapy.
Regarding phenylephrine, the guidelines recommend against its use except when norepinephrine use is associated with significant tachyarrhythmias, cardiac output is known to be higher, or as a salvage therapy.22
This is a topic of debate, with recent clinical studies offering further insight.
De Backer et al40 compared the effects of dopamine vs norepinephrine for the treatment of shock in 1,679 patients, 62% of whom had septic shock. Overall, there was a trend towards better outcomes with norepinephrine, but no significant difference in mortality rates at 28 days (52.5% with dopamine vs 48.5% with norepinephrine, P = .10). Importantly, fewer patients who were randomized to norepinephrine developed arrhythmias (12.4% vs 24.1%, P < .001), and the norepinephrine group required fewer days of study drug (11.0 vs 12.5, P = .01) and open-label vasopressors (12.6 vs 14.2, P = .007). Of note, patients with cardiogenic shock randomized to norepinephrine had a significantly lower mortality rate than those randomized to dopamine. Although no significant difference in outcome was found between the two vasopressors in the subgroup of patients with septic shock, the overall improvements in secondary surrogate markers suggest that norepinephrine should be the first-line agent.
Norepinephrine has also been compared with “secondary” vasopressors. Annane et al,41 in a prospective multicenter randomized controlled study, evaluated the effect of norepinephrine plus dobutamine vs epinephrine alone in managing septic shock. There was no significant difference in the primary outcome measure of 28-day mortality (34% with norepinephrine plus dobutamine vs 40% with epinephrine alone, P = .31). However, the study was powered to evaluate for an absolute risk reduction of 20% in the mortality rate, which would be a big reduction. A smaller reduction in the mortality rate, which would not have been statistically significant in this study, might still be considered clinically significant. Furthermore, the group randomized to norepinephrine plus dobutamine had more vasopressor-free days (20 days vs 22 days, P = .05) and less acidosis on days 1 to 4 than the group randomized to epinephrine.
Norepinephrine was also compared with phenylephrine as a first-line vasopressor in a randomized controlled trial in 32 patients with septic shock. No difference was found in cardiopulmonary performance, global oxygen transport, or regional hemodynamics between phenylephrine and norepinephrine.42
While encouraging, these preliminary data need to be verified in a larger randomized controlled trial with concrete outcome measures before being clinically adapted. Taken together, the above studies suggest that norepinephrine should be the initial vasopressor of choice for patients with septic shock.
CONTINUED MANAGEMENT OF SEPTIC SHOCK
How to manage septic shock after the initial stages is much less defined.
Uncertainty persists about the importance of achieving the early goals of resuscitation in patients who did not reach them in the initial 6 hours of treatment. Although there are data suggesting that extending the goals beyond the initial 6 hours may be beneficial, clinicians should use caution when interpreting these results in light of the observational design of the studies.43,44 For the purpose of this discussion, “continued management” of septic shock will mean after the first 6 hours and after all the early goals are met.
The clinical decisions necessary after the initial stages of resuscitation include:
- Whether further fluid resuscitation is needed
- Assessment for further and additional hemodynamic therapies
- Consideration of adjunctive therapies
- Reevaluation of antibiotic choices (Table 2).
Is more fluid needed? How can we tell?
There is considerable debate about the ideal method for assessing fluid responsiveness. In fact, one of the criticisms of the early goal-directed therapy study27 was that it used central venous pressure as a marker of fluid responsiveness.
Several studies have shown that central venous pressure or pulmonary artery occlusion pressure may not be valid measures of fluid responsiveness.45 In fact, in a retrospective study of 150 volume challenges, the area under the receiver-operating-characteristics curve of central venous pressure as a marker of fluid responsiveness was only 0.58. (Recall that the closer the area under the curve is to 1.0, the better the test; a value of 0.50 is the same as chance.) The area under the curve for pulmonary artery occlusion pressure was 0.63.46
In contrast, several dynamic indices have been proposed to better guide fluid resuscitation in mechanically ventilated patients.31 These are based on changes in stroke volume, aortic blood flow, or arterial pulse pressure in response to the ventilator cycle or passive leg-raising. A detailed review of these markers can be found elsewhere,31 but taken together, they have a sensitivity and specificity of over 90% for predicting fluid responsiveness. Clinicians may consider using dynamic markers of fluid responsiveness to determine when to give additional fluids, particularly after the first 6 hours of shock, in which data supporting the use of central venous pressure are lacking.
Optimal use of fluids is particularly important, since some studies suggest that “overresuscitation” has negative consequences. In a multicenter observational study of 1,177 patients with sepsis, after adjusting for a number of comorbidities and baseline severity of illness, the cumulative fluid balance in the first 72 hours after the onset of sepsis was independently associated with a worse mortality rate.47
Furthermore, in a retrospective analysis of a randomized controlled trial of vasopressin in conjunction with norepinephrine for septic shock, patients in the highest quartile of fluid balance (more fluid in than out) at 12 hours and 4 days after presentation had significantly higher mortality rates than those in the lowest two quartiles.48 The worse outcome with a positive fluid balance might be explained by worsening oxygenation and prolonged mechanical ventilation, as demonstrated by the Fluid and Catheter Treatment Trial in patients with acute lung injury or acute respiratory distress syndrome (ALI/ARDS).49 Indeed, when fluid balance in patients with septic shockinduced ALI/ARDS was evaluated, patients with both adequate initial fluid resuscitation and conservative late fluid management had a lower mortality rate than those with either one alone.50
In view of these findings, especially beyond the initial hours of resuscitation, clinicians should remember that further unnecessary fluid administration may have detrimental effects. Therefore, given the superior predictive abilities of dynamic markers of fluid responsiveness, these should be used to determine the need for further fluid boluses.
In cases in which patients are no longer fluid-responsive and need increasing levels of hemodynamic support, clinicians still have a number of options. These include increasing the current vasopressor dose or starting an additional therapy such as an alternative catecholamine vasopressor, vasopressin, inotropic therapy, or an adjunctive therapy such as a corticosteroid. The intervention could also be a combination of the above choices.
Adding catecholamines
The optimal time point or vasopressor dose at which to consider initiating additional therapies is unknown. However, the Vasopressin and Septic Shock Trial (VASST) provides some insight.51
This study compared two strategies: escalating doses of norepinephrine vs adding vasopressin to norepinephrine. Overall, adding vasopressin showed no benefit in terms of a lower mortality rate. However, in the subgroup of patients with norepinephrine requirements of 5 to 14 μg/min at study enrollment (ie, a low dose, reflecting less-severe sepsis) vasopressin was associated with a lower 28-day mortality rate (26.5% vs 35.7%, P = .05) and 90-day mortality rate (35.8% vs 46.1%, P = .04). Benefit was also noted in patients with other markers of lower disease severity such as low lactate levels or having received a single vasopressor at baseline.51
Although subgroup analyses should not generally be used to guide treatment decisions, a prospective trial may never be done to evaluate adding vasopressin to catecholamines earlier vs later. Thus, clinicians who choose to use vasopressin may consider starting this therapy when catecholamine doses are relatively low or before profound hyperlactatemia from prolonged tissue hypoxia has developed.
There is less evidence to guide clinicians who are considering adding a different catecholamine. The theoretical concerns of splanchnic ischemia and cardiac arrhythmia associated with higher doses of catecholamines are usually the impetus to limit a single catecholamine to a “maximum” dose. However, studies that have evaluated combination catecholamine therapies have generally studied combinations of vasopressors with inotropes and lacked standardization in their protocols, thus making them difficult to interpret.52–54 One could also argue that additional catecholamine therapies, which all function similarly, may have additive effects and cause even more adverse effects. As such, adding another vasopressor should be reserved for patients experiencing noticeable adverse effects (such as tachycardia) on first-line therapy.
Inotropic support
Left ventricular function should be assessed in all patients who continue to be hypotensive despite adequate fluid resuscitation and vasopressor therapy. In a study of patients with septic shock in whom echocardiography was performed daily for the first 3 days of hemodynamic support, new-onset left ventricular hypokinesia was found in 26 (39%) of 67 patients on presentation and in an additional 14 patients (21%) after at least 24 hours of norepinephrine.55 Adding inotropic support with dobutamine or epinephrine led to decreases in vasopressor dose and enhanced left ventricular ejection fraction.
In short, left ventricular hypokinesia is common in septic shock, may occur at presentation or after a period of vasopressor support, and is usually correctable with the addition of inotropic support.
Corticosteroids
Beyond hemodynamic support with fluids and catecholamines or vasopressin (or both), clinicians should also consider adjunctive corticosteroid therapy. However, for many years the issue has been controversial for patients with severe sepsis and septic shock.
Annane et al56 conducted a large, multicenter, randomized, double-blind, placebocontrolled trial to assess the effect of low doses of corticosteroids in patients with refractory septic shock. Overall, the 28-day mortality rate was 61% in the treatment group and 55% in the placebo group, which was not statistically significant (adjusted odds ratio 0.65, 95% confidence interval 0.39–1.07, P value .09). However, when separated by response to cosyntropin stimulation, those with a change in cortisol of 9 ug/dL or less (nonresponders) randomized to receive corticosteroids had significantly higher survival rates in the short term (28 days) and the long term (1 year). The positive results of this study led to the adoption of low-dose hydrocortisone as standard practice in most patients with septic shock.57
But then, to evaluate the effects of corticosteroids in a broader intensive-care population with septic shock, another trial was designed: the Corticosteroid Therapy of Septic Shock (CORTICUS) trial.58 Surprisingly, this multicenter, randomized, double-blind, placebo-controlled trial found no significant difference in survival between the group that received hydrocortisone and the placebo group, regardless of response to a cosyntropin stimulation test.
Taking into account the above studies and other randomized controlled trials, the 2012 Surviving Sepsis Campaign guidelines and the International Task Force for the Diagnosis and Management of Corticosteroid Insufficiency in Critically Ill Adult Patients recommend intravenous hydrocortisone therapy in adults with septic shock whose blood pressure responds poorly to fluid resuscitation and vasopressor therapy. These consensus statements do not recommend the cosyntropin stimulation test to identify patients with septic shock who should receive corticosteroids.22,59 The guidelines, however, do not explicitly define poor response to initial therapy.
Of note, in the Annane study, which found a lower mortality rate with corticosteroids, the patients were severely ill, with a mean baseline norepinephrine dose of 1.1 μg/kg/min. In contrast, in the CORTICUS study (which found no benefit of hydrocortisone), patients had lower baseline vasopressor doses, with a mean norepinephrine dose of 0.5 μg/kg/min.
While corticosteroids are associated with a higher rate of shock reversal 7 days after initiation, 59 this has not translated into a consistent reduction in the death rate. If a clinician is considering adding corticosteroids to decrease the risk of death, it would seem prudent to add this therapy in patients receiving norepinephrine in doses above 0.5 μg/kg/min.
The ideal sequence and combination of the above therapies including fluids, catecholamine vasopressors, vasopressin, inotropes, and vasopressors have not been elucidated. However, some preliminary evidence suggests an advantage with the combination of vasopressin and corticosteroids. In a subgroup analysis of the VASST study, in patients who received corticosteroids, the combination of vasopressin plus norepinephrine was associated with a lower 28-day mortality rate than with norepinephrine alone (35.9% vs 44.7%, P = .03).60 These findings have been replicated in other studies,61,62 prompting suggestions for a study of vasopressin with and without corticosteroids in patients on norepinephrine to elucidate the role of each therapy individually and in combination.
Tight glycemic control
As with corticosteroids, the pendulum for tight glycemic control in critically ill patients has swung widely in recent years. Enthusiasm was high at first after the publication of a study by van den Berghe et al, which described a 3.4% absolute reduction in mortality with intensive insulin therapy to maintain blood glucose at or below 110 mg/dL.63 However, the significant benefits found in this study were never replicated.
In fact, recent evidence suggests that tight glycemic control is associated with no benefit and a higher risk of hypoglycemia.34,64 In the largest randomized controlled trial of this topic, with more than 6,000 patients, intensive insulin therapy with a target blood glucose level of 81 to 108 mg/dL was associated with a significantly higher mortality rate (odds ratio 1.14, 95% confidence interval 1.02–1.28, P = .02) than with a target glucose level of less than 180 mg/dL.65 Furthermore, in a recent follow-up analysis,66 moderate hypoglycemia (serum glucose 41–70 mg/dL) and severe hypoglycemia (serum glucose < 41 mg/dL) were associated with a higher rate of death in a dose-response relationship.66
Taking this information together, clinicians should be aware that there is no additional benefit in lowering blood glucose below the range of 140 to 180 mg/dL, and that doing so may be harmful.
Drotecogin alfa
Drotecogin alfa (Xigris) was another adjunctive therapy that has fallen from favor. It was approved for the treatment of severe sepsis in light of promising findings in initial studies.67
However, on October 25, 2011, drotecogin alfa was voluntarily withdrawn from the market by the manufacturer after another study found no beneficial effect on the mortality rates at 28 days or at 90 days.68 Furthermore, no difference could be found regarding any predetermined primary or secondary outcome measures.
Continued antibiotic therapy
The decision whether to continue initial empiric antimicrobial coverage, broaden it, or de-escalate must be faced for all patients with septic shock, and is ultimately clinical.
The serum procalcitonin level has been proposed to guide antibiotic discontinuation in several clinical settings, although there are still questions about the safety of such an approach. The largest randomized trial published to date reported that a procalcitoninguided strategy to treat suspected bacterial infections in nonsurgical patients could reduce antibiotic exposure with no apparent adverse outcomes.69 On the other hand, other data discourage the use of procalcitonin-guided antimicrobial escalation, as this approach did not improve survival and worsened organ function and length of stay in the intensive care unit.70
The Surviving Sepsis Campaign guidelines recommend combination antibiotic therapy for no longer than 3 to 5 days and limiting the duration of antibiotics in most cases to 7 to 10 days.22
TRIALS ARE ONGOING
The understanding of the pathophysiology and treatment of sepsis has greatly advanced over the last decade. Adoption of evidence-based protocols for managing patients with septic shock has improved outcomes. Nevertheless, many multicenter trials are being conducted worldwide to look into some of the most controversial therapies, and their results will guide therapy in the future.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
- Kumar G, Kumar N, Taneja A, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 2011; 140:1223–1231.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 2003; 168:165–172.
- Levy MM, Dellinger RP, Townsend SR, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010; 36:222–231.
- Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intensive Care Med 2004; 30:1032–1040.
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–150.
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med 2001; 345:588–595.
- Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically ill in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med 1996; 14:218–225.
- Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993; 34:515–518.
- Muller B, Becker KL, Schachinger H, et al. Calcitonin precursors are reliable markers of sepsis in a medical intensive care unit. Crit Care Med 2000; 28:977–983.
- Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (London, England) 1999; 3:45–50.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007; 7:210–217.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Levraut J, Ciebiera JP, Chave S, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than over-production. Am J Respir Crit Care Med 1998; 157:1021–1026.
- Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Pierrakos C, Vincent JL. Sepsis biomarkers: a review. Crit Care 2010; 14:R15.
- Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96–104.
- Marshall JC, al Naqbi A. Principles of source control in the management of sepsis. Crit Care Clin 2009; 25:753–768,viii–ix.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34:1589–1596.
- Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest 2009; 136:1237–1248.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med 2011; 39:2066–2071.
- Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- Micek ST, Roubinian N, Heuring T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006; 34:2707–2713.
- Schmidt GA. Counterpoint: adherence to early goal-directed therapy: does it really matter? No. Both risks and benefits require further study. Chest 2010; 138:480–483; discussion 483–484.
- Jain RK, Antonio BL, Bowton DL, Houle TT, MacGregor DA. Variability in central venous pressure measurements and the potential impact on fluid management. Shock 2009; 33:253–257.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- Perner A, Haase N, Guttormsen AB, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Myburgh JA, Finfer S, Bellomo R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Puskarich MA, Trzciak S, Shapiro NI, Kline JA, Jones AE. Concordance and prognostic value of central venous oxygen saturation and lactate clearance in emergency department patients with septic shock. Acad Emerg Med 2011; 19:S159–S160.
- Dunser MW, Takala J, Ulmer H, et al. Arterial blood pressure during early sepsis and outcome. Intensive Care Med 2009; 35:1225–1233.
- De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Morelli A, Ertmer C, Rehberg S, et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care (London, England) 2008; 12:R143.
- Coba V, Whitmill M, Mooney R, et al. Resuscitation bundle compliance in severe sepsis and septic shock: improves survival, is better late than never. J Intensive Care Med 2011 Jan 10[Epub ahead of print].
- Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Ortiz F, Llorca J, Delgado-Rodriguez M. Late compliance with the sepsis resuscitation bundle: impact on mortality. Shock 2011; 36:542–547.
- Marik PE, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest 2008; 134:172–178.
- Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med 2007; 35:64–68.
- Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Murphy CV, Schramm GE, Doherty JA, et al. The importance of fluid management in acute lung injury secondary to septic shock. Chest 2009; 136:102–109.
- Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Vincent JL, Roman A, Kahn RJ. Dobutamine administration in septic shock: addition to a standard protocol. Crit Care Med 1990; 18:689–693.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med 1993; 19:151–154.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008; 36:1701–1706.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002; 288:862–871.
- Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858–873.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36:1937–1949.
- Russell JA, Walley KR, Gordon AC, et al. Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 2009; 37:811–818.
- Bauer SR, Lam SW, Cha SS, Oyen LJ. Effect of corticosteroids on arginine vasopressin-containing vasopressor therapy for septic shock: a case control study. J Crit Care 2008; 23:500–506.
- Torgersen C, Luckner G, Schroder DC, et al. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: association with mortality. Intensive Care Med 2011; 37:1432–1437.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- Preiser JC, Devos P, Ruiz-Santana S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med 2009; 35:1738–1748.
- Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Finfer S, Liu B, Chittock DR, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med 2012; 367:1108–1118.
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709.
- Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064.
- Bouadma L, Luyt CE, Tubach F, et al. Use of procalcitonin to reduce patients’ exposure to antibiotics in intensive care units (PRORATA trial): a multicentre randomised controlled trial. Lancet 2009; 375:463–474.
- Jensen JU, Hein L, Lundgren B, et al. Procalcitonin-guided interventions against infections to increase early appropriate antibiotics and improve survival in the intensive care unit: a randomized trial. Crit Care Med 2011; 39:2048–2058.
KEY POINTS
- Managing septic shock in the first 6 hours involves prompt recognition, empiric antibiotic therapy, elimination of the source of infection (if applicable), fluid resuscitation titrated to specific goals, and vasopressor therapy.
- A number of biomarkers have been proposed to help recognize septic shock early in its course.
- A delay in starting appropriate antibiotic treatment is associated with higher risk of death.
- The ideal measure of the adequacy of fluid resuscitation remains a topic of study and debate.
- Preliminary studies suggest that norepinephrine should be the initial vasopressor.
- Management after the first 6 hours is less well defined. Decisions in this period include whether to give further fluid resuscitation, further and additional hemodynamic therapies, adjunctive therapies, and antibiotics.
Frailty in older adults: Implications for end-of-life care
As people get older, they have more things wrong with them. And the more things they have wrong with them, the more likely they are to die. But everyone accumulates deficits at a different rate, and not all people of the same age have the same short-term risk of dying. This variable susceptibility to death and other adverse outcomes in older people of the same age is called frailty.1
Frailty poses special challenges to how we organize and deliver health care. These challenges are sometimes seen most starkly when people are most frail, especially as they approach the end of life.
In this paper, we will review how frailty is conceptualized and defined, consider how frailty affects the care of people at the end of their lives, and suggest practices that can make end-of-life care better for frail older adults.
DEFINING FRAILTY
As with all complex systems, when frail people become acutely unwell their highest-order functions fail first. Thus, cognitive impairment, functional decline, impaired mobility, and social withdrawal are hallmark presentations of the further accumulation of deficits in vulnerable seniors.
Delirium and falls are important clues that a person’s resilience is becoming compromised and that the person is at risk of further insults in a downward spiral or acceleration of things going wrong.1,2 Frailty is associated with poor health outcomes, from disability to institutionalization and death.3
This idea of frailty as vulnerability arising from dysregulation of multiple physiologic systems is reasonably non-controversial. Even so, there are competing views on how to systematically quantify those who are at an increased risk of adverse sequelae.
Quantifying frailty is particularly important if it can tell us if a patient is at high risk of further decline and death. As frailty advances, it is appropriate to shift the focus of care to palliation, with the goal of optimizing quality of life and easing symptoms.4 Identifying someone as frail can aid decision-making in the setting of critical illness, where the system commonly defaults to an “always do everything” mode without considering the ramifications of such an approach. Furthermore, without a routine means of measuring frailty, it is often left to critical care units or rapid-response medical teams to initiate a discussion about whether an aggressive course of care is appropriate or desired.5,6
Frailty as a syndrome
Fried et al7 defined frailty as a syndrome arising from the “physiologic triad” of sarcopenia and immune and neuroendocrine dysregulation. Patients are considered frail if they have three or more of the following five criteria:
- Reduced activity
- Slowing of mobility
- Weight loss
- Diminished handgrip strength
- Exhaustion.
Someone who has only one or two of these items is said to be “pre-frail”; someone with none is said to be “robust.”
The frailty index
An alternative viewpoint is that frailty is a state arising from the accumulation of deficits, which can be counted in a frailty index.
The frailty index is based on the concept that frailty is a consequence of interacting physical, psychological, and social factors. As deficits accumulate, people become increasingly vulnerable to adverse outcomes.
The frailty index is calculated as the number of deficits the patient has, divided by the number of deficits considered. For example, in a frailty index based on a comprehensive geriatric assessment, an individual with impairments in 4 of 10 domains and with 10 of 24 possible comorbidities would have 14 of 34 possible deficits, for a frailty index of 0.41.8
A criticism of the frailty index is that it includes functional dependence as a deficit. The criticism stems from the view that frailty should be seen as occurring prior to disability. According to this view, including dependence in instrumental and basic activities of daily living as a deficit confuses disability with frailty.
Proponents of the frailty index counter that frailty is not “all or none” and needs to be graded. The frailty index can distinguish between people with and without disability by means of the number of deficits that they have, which is most important. For example, a person disabled by a paraplegic injury would have a lower frailty index score and therefore would be considered less frail than a person with advanced cancer affecting multiple body systems. (This is assuming the person who has suffered the injury resulting in paraplegia doesn’t have a concomitant condition such as renal failure or heart disease. In the absence of other health insults, such patients are less at risk of further morbidity or death than the patient with advanced cancer until they get another health insult or insults added to their frailty.)
In any case, functional capacity is fundamental in medical decision-making and when estimating prognoses. An example is the use of the Eastern Cooperative Oncology Group’s functional status measure.9,10
Sum of physical and psychological stressors
Consensus is growing for the concept that frail people are made more vulnerable by the combination of both physical and psychological stressors. This is particularly important to bear in mind for patients who may appear physically robust but whose total health burden makes them vulnerable to further insults.
For example, think of a relatively young overweight patient with hypertension, diabetes, dyslipidemia, and ischemic white matter changes (which can manifest as low mood and even mild vascular cognitive impairment). In such a patient, an acute illness could result in cognitive and functional decline that can be permanent.
Balance of assets and deficits
About 20 years ago, we used the metaphor of a balance beam to describe how frailty comes about in older adults. In this view, there is an interplay of physiological and functional health determinants. Assets such as health, resources, and caregivers are balanced against deficits such as illnesses, dependency on others, and support burden.8
For the most part, later concepts of frailty have focused on the individual, with social factors construed separately as social vulnerability.11
Tools for assessing frailty in people who are not yet disabled
Several tools exist to clinically assess frailty in people who are not yet disabled.
The FRAIL scale.12 The Geriatric Advisory Panel of the International Academy of Nutrition and Aging formulated a scale for measuring frailty as a “pre-disability state.” The FRAIL scale consists of five easily remembered items:
- Fatigue
- Resistance (inability to climb one flight of stairs)
- Ambulation (inability to walk one block)
- Illnesses (more than five)
- Loss of weight (> 5%).
Like the “reduced activity” criterion of the frailty syndrome mentioned above (in practical terms, described as the inability to do heavy household chores),13 the FRAIL scale seems to blur the distinction between disability (here, the inability to climb stairs or to walk a block) and “pre-disability,” to an uncertain end. It also seems to blend the notion of a state and a syndrome; these points will need to be clarified in due course.
The Tilburg Frailty Indicator14 was constructed around the multidimensional viewpoint of frailty, beyond disease or disability state, to identify frail community-dwelling older individuals. The first part of this two-part questionnaire consists of 10 questions on frailty determinants and medical comorbidities, while the second part contains physical, psychological, and social variables strongly associated with frailty, as well as information about disability in walking and balance. Interestingly, although it includes both social and physical factors, it does not include cognition.
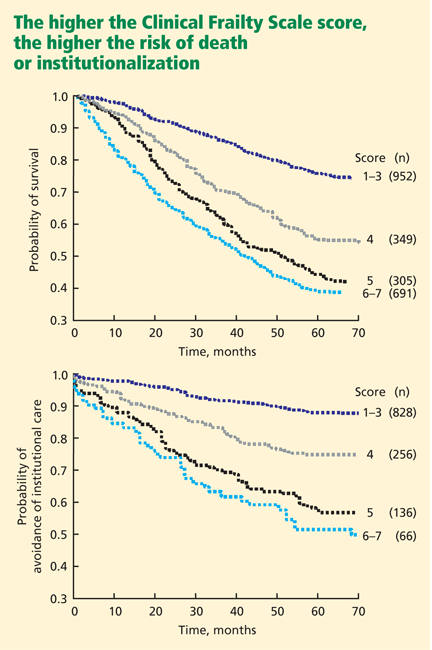
The Clinical Frailty Scale was developed as a practical approach to assess frailty using physical and functional indicators of health and illness burden. The descriptors for this 7-point scale guide clinicians in quantifying the degree of frailty present. It ranges from 1 (very fit) to 7 (severely frail).7 The higher the score, the higher the risks of death or institutionalization. Even mild frailty is associated with a 50% 5-year mortality rate in community-dwelling older adults (Figure 1).8
The Edmonton Frail Scale,15 like the Clinical Frailty Scale, was developed to be practical and usable at the bedside. It is based on the following domains: cognition, general health status, functional independence, social support, medication use, nutrition, mood, continence, and functional performance.
In a community-based sample, the Edmonton Frail Scale compared favorably with the clinical assessment of geriatric specialists who completed a comprehensive evaluation (Pearson’s correlation coefficient 0.64, P < .001).15
FRAILTY AS A PROGNOSTIC INDICATOR
Using frailty scales to aid in prognostication can be useful to clinicians. Survival prognostication is inherently challenging in individuals with multiple comorbidities and variable trajectories of decline, but it remains a vital clinical skill for all clinicians. Framing these difficult discussions in the context of degree of frailty provides a unifying concept, beyond a single-system construct, for care providers, patients, and their loved ones.
Patients nearing the end of their lives need this kind of clarity and support. Regardless of their diagnoses, patients typically want to know when they are at high risk of dying, as do their families and caregivers. People in general look for such information so that they can align medical decision-making congruently with predicted prognosis.16,17 They also use it to plan for the final chapter of their life and their death.
The frailty index is strongly correlated with risk of death
The frailty index is strongly correlated with the risk of death, with a correlation coefficient greater than 0.95. As such, an individual’s frailty index score is considered an estimate of biologic age, which has greater correlation with associated morbidity and death than does chronological age.18,19 In the general population, more than 99% of people have a frailty index value of less than 0.7. As people approach this value, the chance of survival is greatly diminished; indeed, one report suggested that of those who have a frailty index value of more than 0.5 (based on a comprehensive geriatric assessment), 100% are dead by about 20 months later.20,21
In short, there is a limit to which deficits can be added before the system fails. In this sense, the frailty index is akin to the concept of physiologic reserve. Reserve is finite, and as a system loses redundancy it can no longer survive new stresses.
What does this information mean for individual patients?
Even so, prognostication for individual patients remains probabilistic. Any patient has a chance to improve, stabilize, worsen, or die. However, a patient can reach an upper limit of frailty. At that point, instead of accumulating another deficit, death is much more likely. Similarly, although improvement can happen, the chance of improvement is low, and the improvement is typically modest.
Framing survival possibilities in terms of the number of things that people have wrong with them and the chance of death or of change (and the extent of change) makes sense to physicians, patients, and families. Being able to do so offers a much greater opportunity for realistic discussions of the likely outcomes of medical care than the foreseeable scenario of a junior doctor asking a senior citizen, “If your heart stops, do you want us to save your life?”
Understanding prognosis in the face of not just disease but also frailty can also help us focus not on disease but on health consequences of illness. Can the person think? Walk? Care for herself or himself? Interact with others? These questions need to be considered when end-of-life decisions are being discussed.22
Since making predictions about survival is most challenging when multiple comorbidities are present, using the concept of accumulating deficits to better define the slope of decline can be very helpful when discussing “the road ahead” with patients and their families. Visually mapping out the slope of decline and how it is accelerating as conditions progress and deficits accumulate can aid in medical decision-making. Looking individually at the deficits themselves and associated markers of progression can also help with prognostic discussions.
For example, a patient with chronic obstructive pulmonary disease could very well be unaware of the progression and ultimately terminal prognosis of this disease. The slope of clinical decline can be initially shallow, with saw-tooth fluctuations from acute exacerbations that seemingly “resolve to baseline” when antibiotic and steroid courses are completed. Talking with these patients and their families about heralding markers, such as more hospitalizations and cognitive decline with acute exacerbations, can clarify the steepening slope of decline and the way comorbidities interact.
FRAILTY AND END-OF-LIFE CARE
Frailty is progressive, and as it worsens, integrating a palliative approach is appropriate, with a focus on optimizing quality of life and relieving symptoms.4 This principle holds true regardless of the care setting, from acute care hospitals to hospice facilities and long-term care residences.
The principles of end-of-life care are applicable to frail individuals with progressive conditions from the time of diagnosis throughout the course of decline. As the population ages, more people suffer and die from progressive chronic conditions such as cerebrovascular disease, respiratory disease, and dementia.23 An interdisciplinary team approach can ensure all components of palliation are effectively delivered, such as easing symptoms, providing psychosocial and spiritual support, and improving quality of life.24
Pain management
Pain is widely underassessed and undertreated in older patients. Its management at the end of life is particularly challenging if the patient’s language is compromised, as in dementia.23,25,26
A recent cross-sectional analysis of self-reported pain in a longitudinal study of community-dwelling older adults showed an independent association between moderate or higher pain and frailty. The authors propose that persistent pain goes beyond physical discomfort in that it may contribute to homeostenosis (progressive diminishment of homeostatic reserve) and directly worsen frailty.27
Examine the medication list
In palliative care, medical interventions focus on optimizing quality of life.
This especially includes reexamining long medication lists that increase the chance of adverse drug effects.28 Many patients are on disease-modifying medications that may or may not help control symptoms—and might well exacerbate them. For example, betablockers for ischemic heart disease and angiotensin-converting enzyme inhibitors for diabetic nephropathy both can cause hypotension-induced lassitude or even falls due to orthostasis.
A sensible approach is to keep the drugs that may still contribute to quality of life, while discontinuing other drugs that may be causing side effects or that are unlikely to provide meaningful benefit in terms of prevention in patients who have limited life expectancy.29 Discontinuing ineffective, poorly tolerated, and duplicated medications also makes it easier to introduce new medications to manage symptoms—there will be fewer drug interactions, and fewer pills to take, an increasingly important issue in the setting of gastrointestinal symptoms such as dysphagia and gastroparesis or compliance issues as frequently encountered when cognitive impairment is present.29,30
In managing symptoms, start low and go slow—but get there
In managing symptoms in frail elderly patients we use the same classes of medications as in younger patients. The trick is to use appropriate doses.
The concept of “start low and go slow” is key, but so is “get there”—ie, reach the therapeutic goal. The principal drugs for symptom control, such as opioids for pain and dyspnea and anxiolytics for anxiety and restlessness, are associated with a higher rate of and more severe adverse effects in frail older adults.
Even so, most frail older adults appear to be undertreated in this regard, particularly if they are cognitively impaired.23 This fact, coupled with the reality that behavioral symptoms associated with advanced dementia can represent unmet care needs including undertreated pain, highlights the critical need to control symptoms optimally in frail seniors.
This is particularly relevant for those who can no longer verbally articulate their symptoms. Nonverbal pain scales and vigilant assessment of behavioral signs of pain are paramount skills for clinicians providing palliative care to patients with cognitive impairment. Caregivers and loved ones should be included in the assessment process.26,31
Adjunctive therapies for pain control
Maximizing adjunctive therapies can optimize pain control in this drug-sensitive population. Heat and cold packs to affected areas, acupuncture, massage therapy, and structured exercise regimens are some options that can improve quality of life. Cognitive behavioral therapy may offer coping strategies, provided the patient can participate in this process from a cognitive perspective.26,27 Topical preparations are often well tolerated and may include medicinal ingredients that are helpful without systemic effects, such as anti-inflammatory drugs or analgesics.
Optimal use of nonopioid drugs may help reduce the need for narcotics, particularly in the presence of musculoskeletal pain. An example is acetaminophen in regular doses—we would recommend no more than 3 g per day. Acetaminophen is preferred for older adults rather than nonsteroidal anti-inflammatory drugs, given the potential gastric and cardiovascular side effects of the latter medications.
Antidepressants and anticonvulsants such as gabapentin can also be considered as adjuncts for pain control, particularly in the setting of neuropathic pain, with careful monitoring for tolerance.25
When opioids are used, vigilance for constipation is essential.
Establishing goals of care
Goals of care need to be established incrementally along the course of clinical decline and as early as possible so that palliative support can be promptly implemented as symptoms worsen.32
End-of-life care can still include treatments with curative intent, depending on the overall prognosis and the state of the underlying terminal illness. On the other hand, frail older adults who are subjected to invasive treatments that are unlikely to provide cure, such as Whipple surgery, need special intervention postoperatively if they are not to suffer complications such as persisting delirium and functional decline.33
In this regard, geriatric palliative care is frequently about not “crossing a threshold.” Patients may still be receiving active management and be hospitalized for acute exacerbations of progressing chronic conditions, such as chronic obstructive pulmonary disease and heart failure, while palliative principles are introduced and increasingly become the focus of care.
To align goals of care with frailty burden, it is crucial to quantify frailty and to review the patient’s comorbidities. Particularly when dementia is present, lack of communication between the patient’s doctors or between the doctor and the family about disease burden can lead to inappropriately aggressive care.
Many family members and even clinicians do not recognize that advanced dementia is terminal.34,35 In light of this, a palliative approach to care may not even be considered as an appropriate plan when hallmark complications associated with progressing cognitive decline occur, such as aspiration pneumonia or dehydration. Education about dementia and other conditions with progressive organ failure should be done as soon as possible after diagnosis and readdressed at intervals throughout the patient’s clinical decline.
Earlier discussions also ensure that patients themselves can be involved in decision-making more often before cognitive impairment advances to the point where proxy discussions take over.16
BETTER PALLIATIVE CARE FOR ALL
Palliative care, developed initially to provide holistic and timely symptom-based care for patients with noncurable cancer, should also be available and offered to patients with nonmalignant, life-limiting diseases.23,36,37 Meeting this standard of geriatric care is not easy, given the burden of frailty in this population. Needed are multimodal palliative efforts across the spectrum of settings, from the home to the hospital and nursing home.23
To do this, we need to embrace the complexity posed by each person’s presentation and view care through the frailty lens. This will give us a common language in which to engage in a conversation with the same goal in mind: optimizing quality of life.
Furthermore, quantifying frailty can help minimize interventions that are futile or burdensome, that are not expected to ease symptoms, and that can worsen cognition and function. At the end of a patient’s life, we do not want to add to his or her frailty burden but rather minimize the morbidity associated with it.
The concept of frailty assessment is therefore essential for the timely delivery of holistic palliative care in geriatric patients who have progressive and ultimately terminal conditions.
- Rockwood K, Mitnitski A. Frailty defined by deficit accumulation and geriatric medicine defined by frailty. Clin Geriatr Med 2011; 27:17–26.
- Michel JP, Bonin-Guillame S, Gold G, Herrmann F. Cognition and frailty: possible interrelations. In:Carey JR, Robine JM, Michel JP, Christen Y, editors. Longevity and Frailty. Berlin Heidelberg: Springer; 2005:119–124. Research and Perspectives in Longevity.
- Espinoza S, Walston JD. Frailty in older adults: insights and interventions. Cleve Clin J Med 2005; 72:1105–1112.
- Boockvar KS, Meier DE. Palliative care for frail older adults: “there are things I can’t do anymore that I wish I could . . . “. JAMA 2006; 296:2245–2253.
- Abellan van Kan G, Rolland Y, Houles M, Gillette-Guyonnet S, Soto M, Vellas B. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–286.
- McDermid RC, Bagshaw SM. ICU and critical care outreach for the elderly. Best Pract Res Clin Anaesthesiol 2011; 25:439–449.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Rockwood K, Fox RA, Stolee P, Robertson D, Beattie BL. Frailty in elderly people: an evolving concept. CMAJ 1994; 150:489–495.
- Puts MT, Monette J, Girre V, et al. Are frailty markers useful for predicting treatment toxicity and mortality in older newly diagnosed cancer patients? Results from a prospective pilot study. Crit Rev Oncol Hematol 2011; 78:138–149.
- Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982; 5:649–655.
- Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS One 2008; 3:e2232.
- Abellan van Kan G, Rolland YM, Morley JE, Vellas B. Frailty: toward a clinical definition. J Am Med Dir Assoc 2008; 9:71–72.
- Eckel SP, Bandeen-Roche K, Chaves PH, Fried LP, Louis TA. Surrogate screening models for the low physical activity criterion of frailty. Aging Clin Exp Res 2011; 23:209–216.
- Gobbens RJ, van Assen MA, Luijkx KG, Wijnen-Sponselee MT, Schols JM. The Tilburg Frailty Indicator: psychometric properties. J Am Med Dir Assoc 2010; 11:344–355.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- McDermid RC, Stelfox HT, Bagshaw SM. Frailty in the critically ill: a novel concept. Crit Care 2011; 15:301.
- Kulminski AM, Ukraintseva SV, Kulminskaya IV, Arbeev KG, Land K, Yashin AI. Cumulative deficits better characterize susceptibility to death in elderly people than phenotypic frailty: lessons from the Cardiovascular Health Study. J Am Geriatr Soc 2008; 56:898–903.
- Rockwood K, Mitnitski A. Frailty in relation to the accumulation of deficits. J Gerontol A Biol Sci Med Sci 2007; 62:722–727.
- Rockwood K, Mitnitski A. Limits to deficit accumulation in elderly people. Mech Ageing Dev 2006; 127:494–496.
- Rockwood K, Rockwood MR, Mitnitski A. Physiological redundancy in older adults in relation to the change with age in the slope of a frailty index. J Am Geriatr Soc 2010; 58:318–323.
- Kulminski A, Yashin A, Ukraintseva S, et al. Accumulation of health disorders as a systemic measure of aging: findings from the NLTCS data. Mech Ageing Dev 2006; 127:840–848.
- Davies E, Higginson IJ; WHO Europe. Better Palliative Care for Older People. Milan, Italy: Tipolitografia Trabella Sr; 2004.
- Raudonis BM, Daniel K. Frailty: an indication for palliative care. Geriatr Nurs 2010; 31:379–384.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1436.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6):S205–S224.
- Shega JW, Dale W, Andrew M, Paice J, Rockwood K, Weiner DK. Persistent pain and frailty: a case for homeostenosis. J Am Geriatr Soc 2012; 60:113–117.
- Hanlon JT, Perera S, Sevick MA, Rodriguez KL, Jaffe EJ. Pain and its treatment in older nursing home hospice/palliative care residents. J Am Med Dir Assoc 2010; 11:579–583.
- Currow DC, Stevenson JP, Abernethy AP, Plummer J, Shelby-James TM. Prescribing in palliative care as death approaches. J Am Geriatr Soc 2007; 55:590–595.
- Holmes HM, Hayley DC, Alexander GC, Sachs GA. Reconsidering medication appropriateness for patients late in life. Arch Intern Med 2006; 166:605–609.
- Kapo J, Morrison LJ, Liao S. Palliative care for the older adult. J Palliat Med 2007; 10:185–209.
- Gaertner J, Wolf J, Frechen S, et al. Recommending early integration of palliative care—does it work? Support Care Cancer 2012; 20:507–513.
- Chen CC, Lin MT, Tien YW, Yen CJ, Huang GH, Inouye SK. Modified hospital elder life program: effects on abdominal surgery patients. J Am Coll Surg 2011; 213:245–252.
- Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med 2009; 361:1529–1538.
- Arcand M, Monette J, Monette M, et al. Educating nursing home staff about the progression of dementia and the comfort care option: impact on family satisfaction with end-of-life care. J Am Med Dir Assoc 2009; 10:50–55.
- Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med 2004; 164:321–326.
- Birch D, Draper J. A critical literature review exploring the challenges of delivering effective palliative care to older people with dementia. J Clin Nurs 2008; 17:1144–1163.
As people get older, they have more things wrong with them. And the more things they have wrong with them, the more likely they are to die. But everyone accumulates deficits at a different rate, and not all people of the same age have the same short-term risk of dying. This variable susceptibility to death and other adverse outcomes in older people of the same age is called frailty.1
Frailty poses special challenges to how we organize and deliver health care. These challenges are sometimes seen most starkly when people are most frail, especially as they approach the end of life.
In this paper, we will review how frailty is conceptualized and defined, consider how frailty affects the care of people at the end of their lives, and suggest practices that can make end-of-life care better for frail older adults.
DEFINING FRAILTY
As with all complex systems, when frail people become acutely unwell their highest-order functions fail first. Thus, cognitive impairment, functional decline, impaired mobility, and social withdrawal are hallmark presentations of the further accumulation of deficits in vulnerable seniors.
Delirium and falls are important clues that a person’s resilience is becoming compromised and that the person is at risk of further insults in a downward spiral or acceleration of things going wrong.1,2 Frailty is associated with poor health outcomes, from disability to institutionalization and death.3
This idea of frailty as vulnerability arising from dysregulation of multiple physiologic systems is reasonably non-controversial. Even so, there are competing views on how to systematically quantify those who are at an increased risk of adverse sequelae.
Quantifying frailty is particularly important if it can tell us if a patient is at high risk of further decline and death. As frailty advances, it is appropriate to shift the focus of care to palliation, with the goal of optimizing quality of life and easing symptoms.4 Identifying someone as frail can aid decision-making in the setting of critical illness, where the system commonly defaults to an “always do everything” mode without considering the ramifications of such an approach. Furthermore, without a routine means of measuring frailty, it is often left to critical care units or rapid-response medical teams to initiate a discussion about whether an aggressive course of care is appropriate or desired.5,6
Frailty as a syndrome
Fried et al7 defined frailty as a syndrome arising from the “physiologic triad” of sarcopenia and immune and neuroendocrine dysregulation. Patients are considered frail if they have three or more of the following five criteria:
- Reduced activity
- Slowing of mobility
- Weight loss
- Diminished handgrip strength
- Exhaustion.
Someone who has only one or two of these items is said to be “pre-frail”; someone with none is said to be “robust.”
The frailty index
An alternative viewpoint is that frailty is a state arising from the accumulation of deficits, which can be counted in a frailty index.
The frailty index is based on the concept that frailty is a consequence of interacting physical, psychological, and social factors. As deficits accumulate, people become increasingly vulnerable to adverse outcomes.
The frailty index is calculated as the number of deficits the patient has, divided by the number of deficits considered. For example, in a frailty index based on a comprehensive geriatric assessment, an individual with impairments in 4 of 10 domains and with 10 of 24 possible comorbidities would have 14 of 34 possible deficits, for a frailty index of 0.41.8
A criticism of the frailty index is that it includes functional dependence as a deficit. The criticism stems from the view that frailty should be seen as occurring prior to disability. According to this view, including dependence in instrumental and basic activities of daily living as a deficit confuses disability with frailty.
Proponents of the frailty index counter that frailty is not “all or none” and needs to be graded. The frailty index can distinguish between people with and without disability by means of the number of deficits that they have, which is most important. For example, a person disabled by a paraplegic injury would have a lower frailty index score and therefore would be considered less frail than a person with advanced cancer affecting multiple body systems. (This is assuming the person who has suffered the injury resulting in paraplegia doesn’t have a concomitant condition such as renal failure or heart disease. In the absence of other health insults, such patients are less at risk of further morbidity or death than the patient with advanced cancer until they get another health insult or insults added to their frailty.)
In any case, functional capacity is fundamental in medical decision-making and when estimating prognoses. An example is the use of the Eastern Cooperative Oncology Group’s functional status measure.9,10
Sum of physical and psychological stressors
Consensus is growing for the concept that frail people are made more vulnerable by the combination of both physical and psychological stressors. This is particularly important to bear in mind for patients who may appear physically robust but whose total health burden makes them vulnerable to further insults.
For example, think of a relatively young overweight patient with hypertension, diabetes, dyslipidemia, and ischemic white matter changes (which can manifest as low mood and even mild vascular cognitive impairment). In such a patient, an acute illness could result in cognitive and functional decline that can be permanent.
Balance of assets and deficits
About 20 years ago, we used the metaphor of a balance beam to describe how frailty comes about in older adults. In this view, there is an interplay of physiological and functional health determinants. Assets such as health, resources, and caregivers are balanced against deficits such as illnesses, dependency on others, and support burden.8
For the most part, later concepts of frailty have focused on the individual, with social factors construed separately as social vulnerability.11
Tools for assessing frailty in people who are not yet disabled
Several tools exist to clinically assess frailty in people who are not yet disabled.
The FRAIL scale.12 The Geriatric Advisory Panel of the International Academy of Nutrition and Aging formulated a scale for measuring frailty as a “pre-disability state.” The FRAIL scale consists of five easily remembered items:
- Fatigue
- Resistance (inability to climb one flight of stairs)
- Ambulation (inability to walk one block)
- Illnesses (more than five)
- Loss of weight (> 5%).
Like the “reduced activity” criterion of the frailty syndrome mentioned above (in practical terms, described as the inability to do heavy household chores),13 the FRAIL scale seems to blur the distinction between disability (here, the inability to climb stairs or to walk a block) and “pre-disability,” to an uncertain end. It also seems to blend the notion of a state and a syndrome; these points will need to be clarified in due course.
The Tilburg Frailty Indicator14 was constructed around the multidimensional viewpoint of frailty, beyond disease or disability state, to identify frail community-dwelling older individuals. The first part of this two-part questionnaire consists of 10 questions on frailty determinants and medical comorbidities, while the second part contains physical, psychological, and social variables strongly associated with frailty, as well as information about disability in walking and balance. Interestingly, although it includes both social and physical factors, it does not include cognition.
The Clinical Frailty Scale was developed as a practical approach to assess frailty using physical and functional indicators of health and illness burden. The descriptors for this 7-point scale guide clinicians in quantifying the degree of frailty present. It ranges from 1 (very fit) to 7 (severely frail).7 The higher the score, the higher the risks of death or institutionalization. Even mild frailty is associated with a 50% 5-year mortality rate in community-dwelling older adults (Figure 1).8
The Edmonton Frail Scale,15 like the Clinical Frailty Scale, was developed to be practical and usable at the bedside. It is based on the following domains: cognition, general health status, functional independence, social support, medication use, nutrition, mood, continence, and functional performance.
In a community-based sample, the Edmonton Frail Scale compared favorably with the clinical assessment of geriatric specialists who completed a comprehensive evaluation (Pearson’s correlation coefficient 0.64, P < .001).15
FRAILTY AS A PROGNOSTIC INDICATOR
Using frailty scales to aid in prognostication can be useful to clinicians. Survival prognostication is inherently challenging in individuals with multiple comorbidities and variable trajectories of decline, but it remains a vital clinical skill for all clinicians. Framing these difficult discussions in the context of degree of frailty provides a unifying concept, beyond a single-system construct, for care providers, patients, and their loved ones.
Patients nearing the end of their lives need this kind of clarity and support. Regardless of their diagnoses, patients typically want to know when they are at high risk of dying, as do their families and caregivers. People in general look for such information so that they can align medical decision-making congruently with predicted prognosis.16,17 They also use it to plan for the final chapter of their life and their death.
The frailty index is strongly correlated with risk of death
The frailty index is strongly correlated with the risk of death, with a correlation coefficient greater than 0.95. As such, an individual’s frailty index score is considered an estimate of biologic age, which has greater correlation with associated morbidity and death than does chronological age.18,19 In the general population, more than 99% of people have a frailty index value of less than 0.7. As people approach this value, the chance of survival is greatly diminished; indeed, one report suggested that of those who have a frailty index value of more than 0.5 (based on a comprehensive geriatric assessment), 100% are dead by about 20 months later.20,21
In short, there is a limit to which deficits can be added before the system fails. In this sense, the frailty index is akin to the concept of physiologic reserve. Reserve is finite, and as a system loses redundancy it can no longer survive new stresses.
What does this information mean for individual patients?
Even so, prognostication for individual patients remains probabilistic. Any patient has a chance to improve, stabilize, worsen, or die. However, a patient can reach an upper limit of frailty. At that point, instead of accumulating another deficit, death is much more likely. Similarly, although improvement can happen, the chance of improvement is low, and the improvement is typically modest.
Framing survival possibilities in terms of the number of things that people have wrong with them and the chance of death or of change (and the extent of change) makes sense to physicians, patients, and families. Being able to do so offers a much greater opportunity for realistic discussions of the likely outcomes of medical care than the foreseeable scenario of a junior doctor asking a senior citizen, “If your heart stops, do you want us to save your life?”
Understanding prognosis in the face of not just disease but also frailty can also help us focus not on disease but on health consequences of illness. Can the person think? Walk? Care for herself or himself? Interact with others? These questions need to be considered when end-of-life decisions are being discussed.22
Since making predictions about survival is most challenging when multiple comorbidities are present, using the concept of accumulating deficits to better define the slope of decline can be very helpful when discussing “the road ahead” with patients and their families. Visually mapping out the slope of decline and how it is accelerating as conditions progress and deficits accumulate can aid in medical decision-making. Looking individually at the deficits themselves and associated markers of progression can also help with prognostic discussions.
For example, a patient with chronic obstructive pulmonary disease could very well be unaware of the progression and ultimately terminal prognosis of this disease. The slope of clinical decline can be initially shallow, with saw-tooth fluctuations from acute exacerbations that seemingly “resolve to baseline” when antibiotic and steroid courses are completed. Talking with these patients and their families about heralding markers, such as more hospitalizations and cognitive decline with acute exacerbations, can clarify the steepening slope of decline and the way comorbidities interact.
FRAILTY AND END-OF-LIFE CARE
Frailty is progressive, and as it worsens, integrating a palliative approach is appropriate, with a focus on optimizing quality of life and relieving symptoms.4 This principle holds true regardless of the care setting, from acute care hospitals to hospice facilities and long-term care residences.
The principles of end-of-life care are applicable to frail individuals with progressive conditions from the time of diagnosis throughout the course of decline. As the population ages, more people suffer and die from progressive chronic conditions such as cerebrovascular disease, respiratory disease, and dementia.23 An interdisciplinary team approach can ensure all components of palliation are effectively delivered, such as easing symptoms, providing psychosocial and spiritual support, and improving quality of life.24
Pain management
Pain is widely underassessed and undertreated in older patients. Its management at the end of life is particularly challenging if the patient’s language is compromised, as in dementia.23,25,26
A recent cross-sectional analysis of self-reported pain in a longitudinal study of community-dwelling older adults showed an independent association between moderate or higher pain and frailty. The authors propose that persistent pain goes beyond physical discomfort in that it may contribute to homeostenosis (progressive diminishment of homeostatic reserve) and directly worsen frailty.27
Examine the medication list
In palliative care, medical interventions focus on optimizing quality of life.
This especially includes reexamining long medication lists that increase the chance of adverse drug effects.28 Many patients are on disease-modifying medications that may or may not help control symptoms—and might well exacerbate them. For example, betablockers for ischemic heart disease and angiotensin-converting enzyme inhibitors for diabetic nephropathy both can cause hypotension-induced lassitude or even falls due to orthostasis.
A sensible approach is to keep the drugs that may still contribute to quality of life, while discontinuing other drugs that may be causing side effects or that are unlikely to provide meaningful benefit in terms of prevention in patients who have limited life expectancy.29 Discontinuing ineffective, poorly tolerated, and duplicated medications also makes it easier to introduce new medications to manage symptoms—there will be fewer drug interactions, and fewer pills to take, an increasingly important issue in the setting of gastrointestinal symptoms such as dysphagia and gastroparesis or compliance issues as frequently encountered when cognitive impairment is present.29,30
In managing symptoms, start low and go slow—but get there
In managing symptoms in frail elderly patients we use the same classes of medications as in younger patients. The trick is to use appropriate doses.
The concept of “start low and go slow” is key, but so is “get there”—ie, reach the therapeutic goal. The principal drugs for symptom control, such as opioids for pain and dyspnea and anxiolytics for anxiety and restlessness, are associated with a higher rate of and more severe adverse effects in frail older adults.
Even so, most frail older adults appear to be undertreated in this regard, particularly if they are cognitively impaired.23 This fact, coupled with the reality that behavioral symptoms associated with advanced dementia can represent unmet care needs including undertreated pain, highlights the critical need to control symptoms optimally in frail seniors.
This is particularly relevant for those who can no longer verbally articulate their symptoms. Nonverbal pain scales and vigilant assessment of behavioral signs of pain are paramount skills for clinicians providing palliative care to patients with cognitive impairment. Caregivers and loved ones should be included in the assessment process.26,31
Adjunctive therapies for pain control
Maximizing adjunctive therapies can optimize pain control in this drug-sensitive population. Heat and cold packs to affected areas, acupuncture, massage therapy, and structured exercise regimens are some options that can improve quality of life. Cognitive behavioral therapy may offer coping strategies, provided the patient can participate in this process from a cognitive perspective.26,27 Topical preparations are often well tolerated and may include medicinal ingredients that are helpful without systemic effects, such as anti-inflammatory drugs or analgesics.
Optimal use of nonopioid drugs may help reduce the need for narcotics, particularly in the presence of musculoskeletal pain. An example is acetaminophen in regular doses—we would recommend no more than 3 g per day. Acetaminophen is preferred for older adults rather than nonsteroidal anti-inflammatory drugs, given the potential gastric and cardiovascular side effects of the latter medications.
Antidepressants and anticonvulsants such as gabapentin can also be considered as adjuncts for pain control, particularly in the setting of neuropathic pain, with careful monitoring for tolerance.25
When opioids are used, vigilance for constipation is essential.
Establishing goals of care
Goals of care need to be established incrementally along the course of clinical decline and as early as possible so that palliative support can be promptly implemented as symptoms worsen.32
End-of-life care can still include treatments with curative intent, depending on the overall prognosis and the state of the underlying terminal illness. On the other hand, frail older adults who are subjected to invasive treatments that are unlikely to provide cure, such as Whipple surgery, need special intervention postoperatively if they are not to suffer complications such as persisting delirium and functional decline.33
In this regard, geriatric palliative care is frequently about not “crossing a threshold.” Patients may still be receiving active management and be hospitalized for acute exacerbations of progressing chronic conditions, such as chronic obstructive pulmonary disease and heart failure, while palliative principles are introduced and increasingly become the focus of care.
To align goals of care with frailty burden, it is crucial to quantify frailty and to review the patient’s comorbidities. Particularly when dementia is present, lack of communication between the patient’s doctors or between the doctor and the family about disease burden can lead to inappropriately aggressive care.
Many family members and even clinicians do not recognize that advanced dementia is terminal.34,35 In light of this, a palliative approach to care may not even be considered as an appropriate plan when hallmark complications associated with progressing cognitive decline occur, such as aspiration pneumonia or dehydration. Education about dementia and other conditions with progressive organ failure should be done as soon as possible after diagnosis and readdressed at intervals throughout the patient’s clinical decline.
Earlier discussions also ensure that patients themselves can be involved in decision-making more often before cognitive impairment advances to the point where proxy discussions take over.16
BETTER PALLIATIVE CARE FOR ALL
Palliative care, developed initially to provide holistic and timely symptom-based care for patients with noncurable cancer, should also be available and offered to patients with nonmalignant, life-limiting diseases.23,36,37 Meeting this standard of geriatric care is not easy, given the burden of frailty in this population. Needed are multimodal palliative efforts across the spectrum of settings, from the home to the hospital and nursing home.23
To do this, we need to embrace the complexity posed by each person’s presentation and view care through the frailty lens. This will give us a common language in which to engage in a conversation with the same goal in mind: optimizing quality of life.
Furthermore, quantifying frailty can help minimize interventions that are futile or burdensome, that are not expected to ease symptoms, and that can worsen cognition and function. At the end of a patient’s life, we do not want to add to his or her frailty burden but rather minimize the morbidity associated with it.
The concept of frailty assessment is therefore essential for the timely delivery of holistic palliative care in geriatric patients who have progressive and ultimately terminal conditions.
As people get older, they have more things wrong with them. And the more things they have wrong with them, the more likely they are to die. But everyone accumulates deficits at a different rate, and not all people of the same age have the same short-term risk of dying. This variable susceptibility to death and other adverse outcomes in older people of the same age is called frailty.1
Frailty poses special challenges to how we organize and deliver health care. These challenges are sometimes seen most starkly when people are most frail, especially as they approach the end of life.
In this paper, we will review how frailty is conceptualized and defined, consider how frailty affects the care of people at the end of their lives, and suggest practices that can make end-of-life care better for frail older adults.
DEFINING FRAILTY
As with all complex systems, when frail people become acutely unwell their highest-order functions fail first. Thus, cognitive impairment, functional decline, impaired mobility, and social withdrawal are hallmark presentations of the further accumulation of deficits in vulnerable seniors.
Delirium and falls are important clues that a person’s resilience is becoming compromised and that the person is at risk of further insults in a downward spiral or acceleration of things going wrong.1,2 Frailty is associated with poor health outcomes, from disability to institutionalization and death.3
This idea of frailty as vulnerability arising from dysregulation of multiple physiologic systems is reasonably non-controversial. Even so, there are competing views on how to systematically quantify those who are at an increased risk of adverse sequelae.
Quantifying frailty is particularly important if it can tell us if a patient is at high risk of further decline and death. As frailty advances, it is appropriate to shift the focus of care to palliation, with the goal of optimizing quality of life and easing symptoms.4 Identifying someone as frail can aid decision-making in the setting of critical illness, where the system commonly defaults to an “always do everything” mode without considering the ramifications of such an approach. Furthermore, without a routine means of measuring frailty, it is often left to critical care units or rapid-response medical teams to initiate a discussion about whether an aggressive course of care is appropriate or desired.5,6
Frailty as a syndrome
Fried et al7 defined frailty as a syndrome arising from the “physiologic triad” of sarcopenia and immune and neuroendocrine dysregulation. Patients are considered frail if they have three or more of the following five criteria:
- Reduced activity
- Slowing of mobility
- Weight loss
- Diminished handgrip strength
- Exhaustion.
Someone who has only one or two of these items is said to be “pre-frail”; someone with none is said to be “robust.”
The frailty index
An alternative viewpoint is that frailty is a state arising from the accumulation of deficits, which can be counted in a frailty index.
The frailty index is based on the concept that frailty is a consequence of interacting physical, psychological, and social factors. As deficits accumulate, people become increasingly vulnerable to adverse outcomes.
The frailty index is calculated as the number of deficits the patient has, divided by the number of deficits considered. For example, in a frailty index based on a comprehensive geriatric assessment, an individual with impairments in 4 of 10 domains and with 10 of 24 possible comorbidities would have 14 of 34 possible deficits, for a frailty index of 0.41.8
A criticism of the frailty index is that it includes functional dependence as a deficit. The criticism stems from the view that frailty should be seen as occurring prior to disability. According to this view, including dependence in instrumental and basic activities of daily living as a deficit confuses disability with frailty.
Proponents of the frailty index counter that frailty is not “all or none” and needs to be graded. The frailty index can distinguish between people with and without disability by means of the number of deficits that they have, which is most important. For example, a person disabled by a paraplegic injury would have a lower frailty index score and therefore would be considered less frail than a person with advanced cancer affecting multiple body systems. (This is assuming the person who has suffered the injury resulting in paraplegia doesn’t have a concomitant condition such as renal failure or heart disease. In the absence of other health insults, such patients are less at risk of further morbidity or death than the patient with advanced cancer until they get another health insult or insults added to their frailty.)
In any case, functional capacity is fundamental in medical decision-making and when estimating prognoses. An example is the use of the Eastern Cooperative Oncology Group’s functional status measure.9,10
Sum of physical and psychological stressors
Consensus is growing for the concept that frail people are made more vulnerable by the combination of both physical and psychological stressors. This is particularly important to bear in mind for patients who may appear physically robust but whose total health burden makes them vulnerable to further insults.
For example, think of a relatively young overweight patient with hypertension, diabetes, dyslipidemia, and ischemic white matter changes (which can manifest as low mood and even mild vascular cognitive impairment). In such a patient, an acute illness could result in cognitive and functional decline that can be permanent.
Balance of assets and deficits
About 20 years ago, we used the metaphor of a balance beam to describe how frailty comes about in older adults. In this view, there is an interplay of physiological and functional health determinants. Assets such as health, resources, and caregivers are balanced against deficits such as illnesses, dependency on others, and support burden.8
For the most part, later concepts of frailty have focused on the individual, with social factors construed separately as social vulnerability.11
Tools for assessing frailty in people who are not yet disabled
Several tools exist to clinically assess frailty in people who are not yet disabled.
The FRAIL scale.12 The Geriatric Advisory Panel of the International Academy of Nutrition and Aging formulated a scale for measuring frailty as a “pre-disability state.” The FRAIL scale consists of five easily remembered items:
- Fatigue
- Resistance (inability to climb one flight of stairs)
- Ambulation (inability to walk one block)
- Illnesses (more than five)
- Loss of weight (> 5%).
Like the “reduced activity” criterion of the frailty syndrome mentioned above (in practical terms, described as the inability to do heavy household chores),13 the FRAIL scale seems to blur the distinction between disability (here, the inability to climb stairs or to walk a block) and “pre-disability,” to an uncertain end. It also seems to blend the notion of a state and a syndrome; these points will need to be clarified in due course.
The Tilburg Frailty Indicator14 was constructed around the multidimensional viewpoint of frailty, beyond disease or disability state, to identify frail community-dwelling older individuals. The first part of this two-part questionnaire consists of 10 questions on frailty determinants and medical comorbidities, while the second part contains physical, psychological, and social variables strongly associated with frailty, as well as information about disability in walking and balance. Interestingly, although it includes both social and physical factors, it does not include cognition.
The Clinical Frailty Scale was developed as a practical approach to assess frailty using physical and functional indicators of health and illness burden. The descriptors for this 7-point scale guide clinicians in quantifying the degree of frailty present. It ranges from 1 (very fit) to 7 (severely frail).7 The higher the score, the higher the risks of death or institutionalization. Even mild frailty is associated with a 50% 5-year mortality rate in community-dwelling older adults (Figure 1).8
The Edmonton Frail Scale,15 like the Clinical Frailty Scale, was developed to be practical and usable at the bedside. It is based on the following domains: cognition, general health status, functional independence, social support, medication use, nutrition, mood, continence, and functional performance.
In a community-based sample, the Edmonton Frail Scale compared favorably with the clinical assessment of geriatric specialists who completed a comprehensive evaluation (Pearson’s correlation coefficient 0.64, P < .001).15
FRAILTY AS A PROGNOSTIC INDICATOR
Using frailty scales to aid in prognostication can be useful to clinicians. Survival prognostication is inherently challenging in individuals with multiple comorbidities and variable trajectories of decline, but it remains a vital clinical skill for all clinicians. Framing these difficult discussions in the context of degree of frailty provides a unifying concept, beyond a single-system construct, for care providers, patients, and their loved ones.
Patients nearing the end of their lives need this kind of clarity and support. Regardless of their diagnoses, patients typically want to know when they are at high risk of dying, as do their families and caregivers. People in general look for such information so that they can align medical decision-making congruently with predicted prognosis.16,17 They also use it to plan for the final chapter of their life and their death.
The frailty index is strongly correlated with risk of death
The frailty index is strongly correlated with the risk of death, with a correlation coefficient greater than 0.95. As such, an individual’s frailty index score is considered an estimate of biologic age, which has greater correlation with associated morbidity and death than does chronological age.18,19 In the general population, more than 99% of people have a frailty index value of less than 0.7. As people approach this value, the chance of survival is greatly diminished; indeed, one report suggested that of those who have a frailty index value of more than 0.5 (based on a comprehensive geriatric assessment), 100% are dead by about 20 months later.20,21
In short, there is a limit to which deficits can be added before the system fails. In this sense, the frailty index is akin to the concept of physiologic reserve. Reserve is finite, and as a system loses redundancy it can no longer survive new stresses.
What does this information mean for individual patients?
Even so, prognostication for individual patients remains probabilistic. Any patient has a chance to improve, stabilize, worsen, or die. However, a patient can reach an upper limit of frailty. At that point, instead of accumulating another deficit, death is much more likely. Similarly, although improvement can happen, the chance of improvement is low, and the improvement is typically modest.
Framing survival possibilities in terms of the number of things that people have wrong with them and the chance of death or of change (and the extent of change) makes sense to physicians, patients, and families. Being able to do so offers a much greater opportunity for realistic discussions of the likely outcomes of medical care than the foreseeable scenario of a junior doctor asking a senior citizen, “If your heart stops, do you want us to save your life?”
Understanding prognosis in the face of not just disease but also frailty can also help us focus not on disease but on health consequences of illness. Can the person think? Walk? Care for herself or himself? Interact with others? These questions need to be considered when end-of-life decisions are being discussed.22
Since making predictions about survival is most challenging when multiple comorbidities are present, using the concept of accumulating deficits to better define the slope of decline can be very helpful when discussing “the road ahead” with patients and their families. Visually mapping out the slope of decline and how it is accelerating as conditions progress and deficits accumulate can aid in medical decision-making. Looking individually at the deficits themselves and associated markers of progression can also help with prognostic discussions.
For example, a patient with chronic obstructive pulmonary disease could very well be unaware of the progression and ultimately terminal prognosis of this disease. The slope of clinical decline can be initially shallow, with saw-tooth fluctuations from acute exacerbations that seemingly “resolve to baseline” when antibiotic and steroid courses are completed. Talking with these patients and their families about heralding markers, such as more hospitalizations and cognitive decline with acute exacerbations, can clarify the steepening slope of decline and the way comorbidities interact.
FRAILTY AND END-OF-LIFE CARE
Frailty is progressive, and as it worsens, integrating a palliative approach is appropriate, with a focus on optimizing quality of life and relieving symptoms.4 This principle holds true regardless of the care setting, from acute care hospitals to hospice facilities and long-term care residences.
The principles of end-of-life care are applicable to frail individuals with progressive conditions from the time of diagnosis throughout the course of decline. As the population ages, more people suffer and die from progressive chronic conditions such as cerebrovascular disease, respiratory disease, and dementia.23 An interdisciplinary team approach can ensure all components of palliation are effectively delivered, such as easing symptoms, providing psychosocial and spiritual support, and improving quality of life.24
Pain management
Pain is widely underassessed and undertreated in older patients. Its management at the end of life is particularly challenging if the patient’s language is compromised, as in dementia.23,25,26
A recent cross-sectional analysis of self-reported pain in a longitudinal study of community-dwelling older adults showed an independent association between moderate or higher pain and frailty. The authors propose that persistent pain goes beyond physical discomfort in that it may contribute to homeostenosis (progressive diminishment of homeostatic reserve) and directly worsen frailty.27
Examine the medication list
In palliative care, medical interventions focus on optimizing quality of life.
This especially includes reexamining long medication lists that increase the chance of adverse drug effects.28 Many patients are on disease-modifying medications that may or may not help control symptoms—and might well exacerbate them. For example, betablockers for ischemic heart disease and angiotensin-converting enzyme inhibitors for diabetic nephropathy both can cause hypotension-induced lassitude or even falls due to orthostasis.
A sensible approach is to keep the drugs that may still contribute to quality of life, while discontinuing other drugs that may be causing side effects or that are unlikely to provide meaningful benefit in terms of prevention in patients who have limited life expectancy.29 Discontinuing ineffective, poorly tolerated, and duplicated medications also makes it easier to introduce new medications to manage symptoms—there will be fewer drug interactions, and fewer pills to take, an increasingly important issue in the setting of gastrointestinal symptoms such as dysphagia and gastroparesis or compliance issues as frequently encountered when cognitive impairment is present.29,30
In managing symptoms, start low and go slow—but get there
In managing symptoms in frail elderly patients we use the same classes of medications as in younger patients. The trick is to use appropriate doses.
The concept of “start low and go slow” is key, but so is “get there”—ie, reach the therapeutic goal. The principal drugs for symptom control, such as opioids for pain and dyspnea and anxiolytics for anxiety and restlessness, are associated with a higher rate of and more severe adverse effects in frail older adults.
Even so, most frail older adults appear to be undertreated in this regard, particularly if they are cognitively impaired.23 This fact, coupled with the reality that behavioral symptoms associated with advanced dementia can represent unmet care needs including undertreated pain, highlights the critical need to control symptoms optimally in frail seniors.
This is particularly relevant for those who can no longer verbally articulate their symptoms. Nonverbal pain scales and vigilant assessment of behavioral signs of pain are paramount skills for clinicians providing palliative care to patients with cognitive impairment. Caregivers and loved ones should be included in the assessment process.26,31
Adjunctive therapies for pain control
Maximizing adjunctive therapies can optimize pain control in this drug-sensitive population. Heat and cold packs to affected areas, acupuncture, massage therapy, and structured exercise regimens are some options that can improve quality of life. Cognitive behavioral therapy may offer coping strategies, provided the patient can participate in this process from a cognitive perspective.26,27 Topical preparations are often well tolerated and may include medicinal ingredients that are helpful without systemic effects, such as anti-inflammatory drugs or analgesics.
Optimal use of nonopioid drugs may help reduce the need for narcotics, particularly in the presence of musculoskeletal pain. An example is acetaminophen in regular doses—we would recommend no more than 3 g per day. Acetaminophen is preferred for older adults rather than nonsteroidal anti-inflammatory drugs, given the potential gastric and cardiovascular side effects of the latter medications.
Antidepressants and anticonvulsants such as gabapentin can also be considered as adjuncts for pain control, particularly in the setting of neuropathic pain, with careful monitoring for tolerance.25
When opioids are used, vigilance for constipation is essential.
Establishing goals of care
Goals of care need to be established incrementally along the course of clinical decline and as early as possible so that palliative support can be promptly implemented as symptoms worsen.32
End-of-life care can still include treatments with curative intent, depending on the overall prognosis and the state of the underlying terminal illness. On the other hand, frail older adults who are subjected to invasive treatments that are unlikely to provide cure, such as Whipple surgery, need special intervention postoperatively if they are not to suffer complications such as persisting delirium and functional decline.33
In this regard, geriatric palliative care is frequently about not “crossing a threshold.” Patients may still be receiving active management and be hospitalized for acute exacerbations of progressing chronic conditions, such as chronic obstructive pulmonary disease and heart failure, while palliative principles are introduced and increasingly become the focus of care.
To align goals of care with frailty burden, it is crucial to quantify frailty and to review the patient’s comorbidities. Particularly when dementia is present, lack of communication between the patient’s doctors or between the doctor and the family about disease burden can lead to inappropriately aggressive care.
Many family members and even clinicians do not recognize that advanced dementia is terminal.34,35 In light of this, a palliative approach to care may not even be considered as an appropriate plan when hallmark complications associated with progressing cognitive decline occur, such as aspiration pneumonia or dehydration. Education about dementia and other conditions with progressive organ failure should be done as soon as possible after diagnosis and readdressed at intervals throughout the patient’s clinical decline.
Earlier discussions also ensure that patients themselves can be involved in decision-making more often before cognitive impairment advances to the point where proxy discussions take over.16
BETTER PALLIATIVE CARE FOR ALL
Palliative care, developed initially to provide holistic and timely symptom-based care for patients with noncurable cancer, should also be available and offered to patients with nonmalignant, life-limiting diseases.23,36,37 Meeting this standard of geriatric care is not easy, given the burden of frailty in this population. Needed are multimodal palliative efforts across the spectrum of settings, from the home to the hospital and nursing home.23
To do this, we need to embrace the complexity posed by each person’s presentation and view care through the frailty lens. This will give us a common language in which to engage in a conversation with the same goal in mind: optimizing quality of life.
Furthermore, quantifying frailty can help minimize interventions that are futile or burdensome, that are not expected to ease symptoms, and that can worsen cognition and function. At the end of a patient’s life, we do not want to add to his or her frailty burden but rather minimize the morbidity associated with it.
The concept of frailty assessment is therefore essential for the timely delivery of holistic palliative care in geriatric patients who have progressive and ultimately terminal conditions.
- Rockwood K, Mitnitski A. Frailty defined by deficit accumulation and geriatric medicine defined by frailty. Clin Geriatr Med 2011; 27:17–26.
- Michel JP, Bonin-Guillame S, Gold G, Herrmann F. Cognition and frailty: possible interrelations. In:Carey JR, Robine JM, Michel JP, Christen Y, editors. Longevity and Frailty. Berlin Heidelberg: Springer; 2005:119–124. Research and Perspectives in Longevity.
- Espinoza S, Walston JD. Frailty in older adults: insights and interventions. Cleve Clin J Med 2005; 72:1105–1112.
- Boockvar KS, Meier DE. Palliative care for frail older adults: “there are things I can’t do anymore that I wish I could . . . “. JAMA 2006; 296:2245–2253.
- Abellan van Kan G, Rolland Y, Houles M, Gillette-Guyonnet S, Soto M, Vellas B. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–286.
- McDermid RC, Bagshaw SM. ICU and critical care outreach for the elderly. Best Pract Res Clin Anaesthesiol 2011; 25:439–449.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Rockwood K, Fox RA, Stolee P, Robertson D, Beattie BL. Frailty in elderly people: an evolving concept. CMAJ 1994; 150:489–495.
- Puts MT, Monette J, Girre V, et al. Are frailty markers useful for predicting treatment toxicity and mortality in older newly diagnosed cancer patients? Results from a prospective pilot study. Crit Rev Oncol Hematol 2011; 78:138–149.
- Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982; 5:649–655.
- Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS One 2008; 3:e2232.
- Abellan van Kan G, Rolland YM, Morley JE, Vellas B. Frailty: toward a clinical definition. J Am Med Dir Assoc 2008; 9:71–72.
- Eckel SP, Bandeen-Roche K, Chaves PH, Fried LP, Louis TA. Surrogate screening models for the low physical activity criterion of frailty. Aging Clin Exp Res 2011; 23:209–216.
- Gobbens RJ, van Assen MA, Luijkx KG, Wijnen-Sponselee MT, Schols JM. The Tilburg Frailty Indicator: psychometric properties. J Am Med Dir Assoc 2010; 11:344–355.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- McDermid RC, Stelfox HT, Bagshaw SM. Frailty in the critically ill: a novel concept. Crit Care 2011; 15:301.
- Kulminski AM, Ukraintseva SV, Kulminskaya IV, Arbeev KG, Land K, Yashin AI. Cumulative deficits better characterize susceptibility to death in elderly people than phenotypic frailty: lessons from the Cardiovascular Health Study. J Am Geriatr Soc 2008; 56:898–903.
- Rockwood K, Mitnitski A. Frailty in relation to the accumulation of deficits. J Gerontol A Biol Sci Med Sci 2007; 62:722–727.
- Rockwood K, Mitnitski A. Limits to deficit accumulation in elderly people. Mech Ageing Dev 2006; 127:494–496.
- Rockwood K, Rockwood MR, Mitnitski A. Physiological redundancy in older adults in relation to the change with age in the slope of a frailty index. J Am Geriatr Soc 2010; 58:318–323.
- Kulminski A, Yashin A, Ukraintseva S, et al. Accumulation of health disorders as a systemic measure of aging: findings from the NLTCS data. Mech Ageing Dev 2006; 127:840–848.
- Davies E, Higginson IJ; WHO Europe. Better Palliative Care for Older People. Milan, Italy: Tipolitografia Trabella Sr; 2004.
- Raudonis BM, Daniel K. Frailty: an indication for palliative care. Geriatr Nurs 2010; 31:379–384.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1436.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6):S205–S224.
- Shega JW, Dale W, Andrew M, Paice J, Rockwood K, Weiner DK. Persistent pain and frailty: a case for homeostenosis. J Am Geriatr Soc 2012; 60:113–117.
- Hanlon JT, Perera S, Sevick MA, Rodriguez KL, Jaffe EJ. Pain and its treatment in older nursing home hospice/palliative care residents. J Am Med Dir Assoc 2010; 11:579–583.
- Currow DC, Stevenson JP, Abernethy AP, Plummer J, Shelby-James TM. Prescribing in palliative care as death approaches. J Am Geriatr Soc 2007; 55:590–595.
- Holmes HM, Hayley DC, Alexander GC, Sachs GA. Reconsidering medication appropriateness for patients late in life. Arch Intern Med 2006; 166:605–609.
- Kapo J, Morrison LJ, Liao S. Palliative care for the older adult. J Palliat Med 2007; 10:185–209.
- Gaertner J, Wolf J, Frechen S, et al. Recommending early integration of palliative care—does it work? Support Care Cancer 2012; 20:507–513.
- Chen CC, Lin MT, Tien YW, Yen CJ, Huang GH, Inouye SK. Modified hospital elder life program: effects on abdominal surgery patients. J Am Coll Surg 2011; 213:245–252.
- Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med 2009; 361:1529–1538.
- Arcand M, Monette J, Monette M, et al. Educating nursing home staff about the progression of dementia and the comfort care option: impact on family satisfaction with end-of-life care. J Am Med Dir Assoc 2009; 10:50–55.
- Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med 2004; 164:321–326.
- Birch D, Draper J. A critical literature review exploring the challenges of delivering effective palliative care to older people with dementia. J Clin Nurs 2008; 17:1144–1163.
- Rockwood K, Mitnitski A. Frailty defined by deficit accumulation and geriatric medicine defined by frailty. Clin Geriatr Med 2011; 27:17–26.
- Michel JP, Bonin-Guillame S, Gold G, Herrmann F. Cognition and frailty: possible interrelations. In:Carey JR, Robine JM, Michel JP, Christen Y, editors. Longevity and Frailty. Berlin Heidelberg: Springer; 2005:119–124. Research and Perspectives in Longevity.
- Espinoza S, Walston JD. Frailty in older adults: insights and interventions. Cleve Clin J Med 2005; 72:1105–1112.
- Boockvar KS, Meier DE. Palliative care for frail older adults: “there are things I can’t do anymore that I wish I could . . . “. JAMA 2006; 296:2245–2253.
- Abellan van Kan G, Rolland Y, Houles M, Gillette-Guyonnet S, Soto M, Vellas B. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–286.
- McDermid RC, Bagshaw SM. ICU and critical care outreach for the elderly. Best Pract Res Clin Anaesthesiol 2011; 25:439–449.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Rockwood K, Fox RA, Stolee P, Robertson D, Beattie BL. Frailty in elderly people: an evolving concept. CMAJ 1994; 150:489–495.
- Puts MT, Monette J, Girre V, et al. Are frailty markers useful for predicting treatment toxicity and mortality in older newly diagnosed cancer patients? Results from a prospective pilot study. Crit Rev Oncol Hematol 2011; 78:138–149.
- Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982; 5:649–655.
- Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS One 2008; 3:e2232.
- Abellan van Kan G, Rolland YM, Morley JE, Vellas B. Frailty: toward a clinical definition. J Am Med Dir Assoc 2008; 9:71–72.
- Eckel SP, Bandeen-Roche K, Chaves PH, Fried LP, Louis TA. Surrogate screening models for the low physical activity criterion of frailty. Aging Clin Exp Res 2011; 23:209–216.
- Gobbens RJ, van Assen MA, Luijkx KG, Wijnen-Sponselee MT, Schols JM. The Tilburg Frailty Indicator: psychometric properties. J Am Med Dir Assoc 2010; 11:344–355.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- McDermid RC, Stelfox HT, Bagshaw SM. Frailty in the critically ill: a novel concept. Crit Care 2011; 15:301.
- Kulminski AM, Ukraintseva SV, Kulminskaya IV, Arbeev KG, Land K, Yashin AI. Cumulative deficits better characterize susceptibility to death in elderly people than phenotypic frailty: lessons from the Cardiovascular Health Study. J Am Geriatr Soc 2008; 56:898–903.
- Rockwood K, Mitnitski A. Frailty in relation to the accumulation of deficits. J Gerontol A Biol Sci Med Sci 2007; 62:722–727.
- Rockwood K, Mitnitski A. Limits to deficit accumulation in elderly people. Mech Ageing Dev 2006; 127:494–496.
- Rockwood K, Rockwood MR, Mitnitski A. Physiological redundancy in older adults in relation to the change with age in the slope of a frailty index. J Am Geriatr Soc 2010; 58:318–323.
- Kulminski A, Yashin A, Ukraintseva S, et al. Accumulation of health disorders as a systemic measure of aging: findings from the NLTCS data. Mech Ageing Dev 2006; 127:840–848.
- Davies E, Higginson IJ; WHO Europe. Better Palliative Care for Older People. Milan, Italy: Tipolitografia Trabella Sr; 2004.
- Raudonis BM, Daniel K. Frailty: an indication for palliative care. Geriatr Nurs 2010; 31:379–384.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1436.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6):S205–S224.
- Shega JW, Dale W, Andrew M, Paice J, Rockwood K, Weiner DK. Persistent pain and frailty: a case for homeostenosis. J Am Geriatr Soc 2012; 60:113–117.
- Hanlon JT, Perera S, Sevick MA, Rodriguez KL, Jaffe EJ. Pain and its treatment in older nursing home hospice/palliative care residents. J Am Med Dir Assoc 2010; 11:579–583.
- Currow DC, Stevenson JP, Abernethy AP, Plummer J, Shelby-James TM. Prescribing in palliative care as death approaches. J Am Geriatr Soc 2007; 55:590–595.
- Holmes HM, Hayley DC, Alexander GC, Sachs GA. Reconsidering medication appropriateness for patients late in life. Arch Intern Med 2006; 166:605–609.
- Kapo J, Morrison LJ, Liao S. Palliative care for the older adult. J Palliat Med 2007; 10:185–209.
- Gaertner J, Wolf J, Frechen S, et al. Recommending early integration of palliative care—does it work? Support Care Cancer 2012; 20:507–513.
- Chen CC, Lin MT, Tien YW, Yen CJ, Huang GH, Inouye SK. Modified hospital elder life program: effects on abdominal surgery patients. J Am Coll Surg 2011; 213:245–252.
- Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med 2009; 361:1529–1538.
- Arcand M, Monette J, Monette M, et al. Educating nursing home staff about the progression of dementia and the comfort care option: impact on family satisfaction with end-of-life care. J Am Med Dir Assoc 2009; 10:50–55.
- Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med 2004; 164:321–326.
- Birch D, Draper J. A critical literature review exploring the challenges of delivering effective palliative care to older people with dementia. J Clin Nurs 2008; 17:1144–1163.
KEY POINTS
- Frail older adults are more susceptible to delirium, functional decline, impaired mobility, falls, social withdrawal, and death.
- Evaluating the health care needs of people who are frail requires assessment of their cognition, function, mobility, balance, and social circumstances, in addition to understanding their medical problems.
- When people are so frail that they cannot withstand interventions that can cause significant injury, such as surgery or chemotherapy, then appropriate end-of-life care should focus on maintaining their highest-order functions.
- End-of-life care can include curative treatments of some episodes if they threaten cognition, mobility, or function or cause pain and suffering, even in the context of an overall palliative care plan.
Cervical cancer screening: What’s new and what’s coming?
Advances in our understanding of the pathogenesis of cervical cancer, new tests for human papillomavirus (HPV), and the development of HPV vaccines in the last decade are transforming the way we screen for cervical cancer.
As a result, screening guidelines are evolving rapidly, requiring clinicians to keep up-to-date with the evidence and rationales supporting the latest guidelines to properly convey best practices to patients.1–3
For example, we must understand why it is safe to extend the screening interval in women at low risk (as recommended in the new guidelines), and we need to be familiar with the options for women who test positive for HPV. Patients and providers may often find such new recommendations frustrating, and patients may feel that they are being denied something necessary by insurers rather than being treated according to scientific evidence.
This article will review the newest screening guidelines and the evidence supporting these recommendations for primary care providers. We will also review the potential role of novel biomarkers, newer HPV tests, and possible future strategies for cervical cancer screening.
WHAT’S NEW IN THE LATEST SCREENING GUIDELINES
Over the years, various organizations have issued separate screening guidelines, sometimes agreeing with each other, sometimes disagreeing.4 Now, for the first time, several of these organizations have developed guidelines collaboratively, and we have consensus in the screening recommendations.
Shortly after the American Congress of Obstetricians and Gynecologists (ACOG) issued its screening guidelines in December 2009,1 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), and American Society for Clinical Pathology (ASCP) convened an expert panel to review the available evidence and develop a new joint screening guideline. Concurrently, the US Preventive Services Task Force (USPSTF) commissioned a targeted systematic review of the latest evidence.
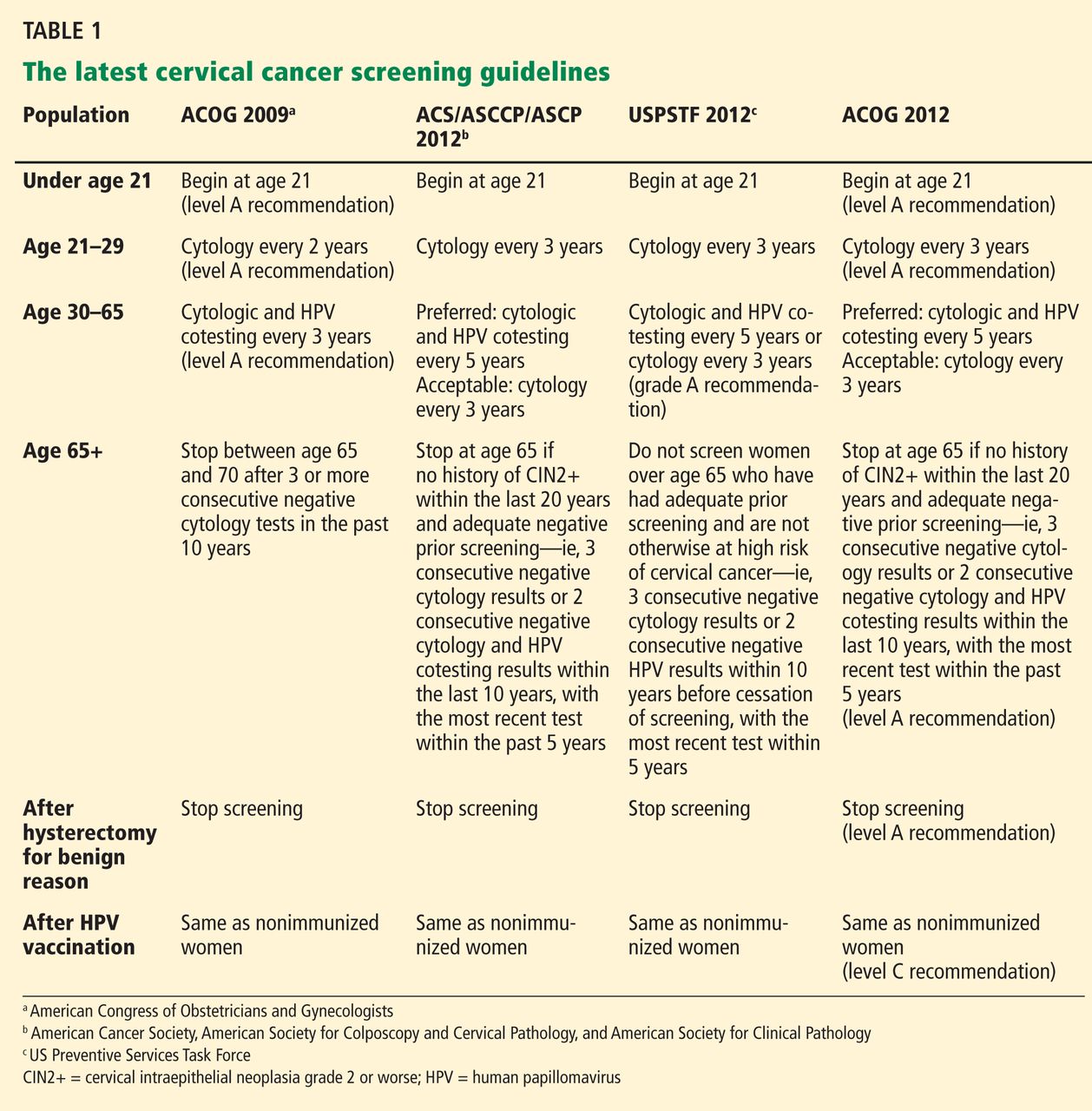
Both the ACS/ASCCP/ASCP group2 and the USPSTF3 released their new guidelines on March 14, 2012. In November 2012, ACOG issued its latest recommendation on cervical cancer screening.4 The following discussion highlights the consensus recommendations from these organizations (Table 1).
These guidelines apply to the general population only. They do not apply to women at high risk who may require more intensive screening, such as those who have a history of cervical cancer, are immunocompromised (eg, positive for human immunodeficiency virus [HIV]), or were exposed in utero to diethylstilbestrol.
Start screening at age 21
According to the new guidelines, women younger than 21 years should not be screened, regardless of the age at which they start having sex.1–3 This is a change from the 2002 and 2003 ACS recommendations, which said screening should begin 3 years after the onset of vaginal intercourse.5,6
Evidence. The rationale for the recommendation not to screen before age 21 stems from two pieces of evidence:
- Invasive cervical cancer is rare in this age group.7
- Screening can cause harm. For example, unnecessary treatment of preinvasive lesions can lead to long-term complications such as cervical stenosis, preterm delivery, and preterm premature rupture of membranes.8,9
Additionally, one study found that screening before age 21 has little or no impact on the incidence of invasive cervical cancer.10
Longer screening intervals
The 2012 ACS/ASCCP/ASCP guidelines2 and the latest ACOG guidelines4 lengthen the interval between cytology (Papanicolaou) testing to every 3 years in women age 21 to 29. Previous recommendations from these groups were to screen every 2 years, and the USPSTF first recommended the 3-year interval in 2003.11
For women age 30 to 65, the ACS/ASCCP/ASCP, ACOG, and the USPSTF now recommend screening every 5 years if the patient’s results on combined cytology and HPV testing are negative. However, cytologic testing alone every 3 years is also acceptable.2–4
Evidence. The evidence supporting a 3-year screening interval in women age 21 to 29 is primarily from modeling studies—no randomized clinical trial has been done. These studies found no significant difference in outcomes with a 2-year vs a 3-year screening interval.12,13 In particular, the predicted lifetime risk of cervical cancer in women screened every 3 years was 5 to 8 new cases of cancer per 1,000 women, compared with 4 to 6 cases per 1,000 women screened every 2 years.14
Similarly, screening women younger than age 30 at 2-year or 3-year intervals carried the same predicted lifetime risk of death from cervical cancer of 0.05 per 1,000 women, yet women screened every 2 years underwent 40% more colposcopies than those screened every 3 years.2 Therefore, screening every 3 years offers the best balance of benefits and risks in this age group.
Adding HPV testing to cytologic testing increases the sensitivity of screening—thus the recommendation to lengthen the screening interval to every 5 years in women age 30 to 65 who are at low risk and who have negative results on both tests. (Previously, the interval was 3 years.)
Specifically, adding HPV testing improves the sensitivity of screening for cervical intraepithelial neoplasia grade 3 (CIN3), so that, in subsequent rounds of screening, fewer cases of CIN3 or worse (CIN3+) or cancer are detected.15–17 The longer diagnostic lead time with combined testing is associated with a lower risk of CIN3+ or cancer following a double-negative test result than screening with cytology alone at shorter intervals. Combined testing at 5-year intervals is associated with a similar or lower cancer risk than cytology-alone screening at 3-year intervals.9
Moreover, modeling studies have shown that combined testing of women age 30 and older at 5-year intervals leads to fewer colposcopies and a similar or lower cancer risk than with cytology screening at 3-year intervals.18,19
A stronger endorsement for HPV testing
Combined cytologic and HPV testing has received its strongest endorsement to date from the ACS/ASCCP/ASCP, ACOG, and USPSTF in their latest guidelines.2–4
In 2003, ACOG gave HPV and cytology combined testing an “optional” recommendation for women over age 30; in 2009, it upgraded its recommendation to the highest level of recommendation.1 At that time, the USPSTF did not recommend for or against HPV testing, while the ACS did recommend HPV testing (with cytology testing alone every 2 to 3 years as an alternative screening strategy).5
Now, the ACS/ASCCP/ASCP and ACOG recommend HPV and cytology combined testing as the preferred strategy for screening women age 30 or over.2,4 Similarly, the USPSTF gives combined testing for women age 30 to 65 a grade A (its highest) recommendation.3 (In 2003, it had given it a grade I—insufficient evidence to assess the balance of benefit and harm.)
Evidence. Several recent studies provide compelling evidence that HPV testing has high sensitivity and excellent negative predictive value, supporting the stronger endorsement of HPV testing and longer screening intervals.
The Joint European Cohort study,20 in 24,295 women, conclusively showed that the 6-year risk of CIN3+ following a negative HPV test was significantly lower than that following a negative cytology result alone (0.27% vs 0.97%).
Katki et al,21 in another retrospective study, analyzed data from 330,000 women age 30 and older who underwent combined HPV and cytology testing. Looking at the tests separately, they found the risk of CIN3+ was comparable in the 3 years following a negative cytology test by itself and in the 5 years following negative combined HPV and cytology testing. In fact, combined testing at 5- or 6-year intervals offered better protection than cytology alone at 3-year intervals.
Furthermore, combined testing is also more sensitive for detecting cervical adenocarcinoma.22 (Most cancers of the cervix are squamous cell carcinomas, but approximately 10% are adenocarcinomas.)
Stop screening sooner
In 2002, the ACS recommended ending screening at age 70,11 and in 2009 ACOG said to stop at age 65 to 70.1 Now, the ACS/ASCCP/ASCP group2 and ACOG4 recommend stopping screening sooner—at age 65—provided that:
- The patient has had adequate negative screening until then. (Adequate negative prior screening is defined as three consecutive negative cytology results or two consecutive negative combined HPV and cytologic testing results within the 10 years before ceasing screening, with the most recent test performed within the last 5 years.)
- The patient has no history of CIN2+ within the last 20 years.
- The patient is not at high risk of cervical cancer, eg, no history of a high-grade precancerous cervical lesion or cervical cancer, in utero exposure to diethylstilbestrol, or immunosuppression (eg, HIV infection).
The USPSTF had already adopted this position.
Evidence. In women over age 65 who have had good screening, cervical cancer is rare and CIN2+ is uncommon.2,23,24 Kulasingam et al,9 in a modeling study performed for the USPSTF, calculated that continuing to screen until age 90 prevents only 1.6 cancer cases and 0.5 cancer deaths and extends life expectancy by only 1 year per 1,000 women.
Other studies also suggest that newly acquired high-risk HPV infection in women age 65 or older is associated with a very low absolute risk of HPV persistence and CIN3+ progression.25,26
In addition, cervical cancer takes a median of 20 to 25 years to develop after infection with high-risk HPV.2 Also, continuing to screen this older population will detect only a very small number of new cases of CIN2+ and may lead to harm from overtreatment.
Finally, postmenopausal women often have smaller and less accessible cervical transformation zones that may require more interventions to obtain adequate samples and to treat.
Stop screening after hysterectomy
The ACS/ASCCP/ASCP group, ACOG, and the USPSTF reaffirmed their recommendation against screening in women who have had a hysterectomy with removal of the cervix for a reason other than cancer and who have had no history of CIN2+ or cervical cancer.2–4
Evidence. Several lines of evidence suggest stopping screening after a woman has a hysterectomy. The incidence of vaginal cancer is extremely low,27 and the positive predictive value of cytologic testing of the vaginal cuff for vaginal cancer was zero in one study.28 Also, a large cross-sectional study of 5,330 screening cytology tests in women who had a hysterectomy found only one case of dysplasia and no cancer.29
Continue to screen after HPV vaccination
For the first time since HPV vaccines were introduced in 2006, the ACS/ASCCP/ASCP, ACOG, and the USPSTF have had to consider what to do for vaccinated women. All of their new guidelines say to keep screening them.
Evidence. The currently available HPV vaccines protect against cervical cancer,30 but only against cervical cancer caused by HPV types 16 and 18. Other oncogenic types of HPV exist, and the current vaccines do not protect against them.
Furthermore, many women are vaccinated who are already infected. In addition, as of 2010, only about 32% of eligible girls and women in the United States had received all three recommended doses of the vaccine.31 And modeling studies predict that the impact of the HPV vaccine will not be apparent for at least another decade.32
HPV 16/18 genotyping
The ACS/ASCCP/ASCP and ACOG now recommend HPV 16/18 genotyping as a triage option in women who have positive results on HPV testing but negative cytology results, and immediate referral for colposcopy if the genotyping test is positive.2 The alternative option in this situation is to repeat combined HPV and cytologic testing in 12 months.2,33
Evidence. The standard tests for HPV can detect DNA from about a dozen of the oncogenic types of HPV depending on the test, but they do not tell you which one the patient has. This information may be relevant, since not all “high-risk” HPV types are equally bad. HPV 16 and HPV 18 are the worst of all, together accounting for more than 70% of cases of cervical cancer.
Large cohort studies34,35 have shown that the risk of CIN3 reaches 10% over 1 to 4 years in women who test positive for HPV 16, and over 2 to 5 years if they test positive for HPV 18. This clinically relevant short-term risk supports immediate referral for colposcopy.
In March 2009, the US Food and Drug Administration (FDA) approved a test for HPV 16 and HPV 18—Cervista HPV 16/18 (Hologic, Bedford, MA).36
More recently, researchers from the Addressing the Need for Advanced HPV Diagnostics (ATHENA) trial,37 in 47,208 women, reported that they found CIN2+ in 11.4% of women who tested positive for either HPV 16 or HPV 18, and CIN3+ in 9.8%. Of those who were positive for HPV 16, 13.6% had CIN2+ and 11.7% had CIN3+.
WHAT’S COMING?
As we gain knowledge of the molecular oncogenesis of cervical cancer, we appreciate more the complex relation between HPV oncoproteins and cervical dysplasia. Recent studies demonstrated the clinical utility of detecting novel markers in women who have positive HPV results.38,39
At present, however, there is insufficient evidence to integrate these strategies into our standard of care for cervical cancer screening.
Novel biomarkers: p16 and Ki-67
Although HPV testing is sensitive, it has poor specificity and positive predictive value.40,41 In a primary screening setting, women with normal cytology results who test positive for high-risk HPV may carry a risk of only 3% to 7% for high-grade CIN.42,43
HPV 16/18 genotyping can be useful in this situation (see above). However, not everyone who carries HPV 16 or 18 goes on to develop CIN or cancer.44

A novel biomarker, p16, has been shown to be overexpressed in cervical dysplasia and associated with high-risk HPV oncogenic transformation. Another novel marker, Ki-67, can be regarded as a surrogate marker of deregulated cell proliferation (Figure 1).38
A recent study reported that a combined test for both of these markers (dual-stained cytology) had a sensitivity of 91.9% for detecting CIN2+ and 96.4% for CIN3+. This test was also highly specific: 82.1% for CIN2+ and 76.9% for CIN3+.38
An Italian randomized trial reported that p16 immunostaining improved the specificity of HPV testing in detecting CIN2+.45
In addition, the European Equivocal or Mildly Abnormal Papanicolaou Cytology Study46 found that the dual-stained cytology test had excellent sensitivity for CIN2+ in women with atypical squamous cells of undetermined significance (ASCUS) or low-grade squamous intraepithelial lesion (LSIL) cytology results (92.2% for ASCUS, 94.2% for LSIL). The specificity for CIN2+ in ASCUS and LSIL was 80.6% and 68%, respectively.
A US study also showed that the sensitivity and specificity to detect CIN3+ by using p16/Ki-67 were 97.2% and 60%, respectively, in women age 30 and older.47
If confirmed in more studies, p16/Ki-67 dual staining could help us in deciding which women who have positive HPV but negative cytology results should be referred for colposcopy.
HPV oncogene E6/E7 mRNA testing
In October 2011, the FDA approved the clinical use of a new-generation HPV test, the Aptima HPV assay (Hologic Gen-Probe, San Diego, CA), which detects mRNA for the proteins E6 and E7 from high-risk HPV.39
HPV E6/E7 mRNA expression has been found in virtually all HPV-positive cancer cases and demonstrates a stronger correlation with cervical disease than detection of HPV DNA.48 High-risk HPV E6 and E7 proteins immortalize and malignantly transform infected cells by inhibiting two host cellular anticancer proteins, p53 and retinoblastoma protein (pRB).44,49
The recent FDA approval was based on data from the CLEAR (Clinical Evaluation of Aptima HPV RNA) trial.39 In this trial, in more than 11,000 women, the test was as sensitive for detecting CIN2+ as the HPV DNA-based test, and it was more specific. This advantage was statistically significant. The higher specificity may reduce the number of unnecessary colposcopies and allow for more effective management.50,51
A promising future screening strategy: HPV testing first, then cytology
HPV testing is more sensitive than cytology, while cytology is more specific. Thus, it would be logical to test for HPV first, and then to perform cytologic testing in patients who have positive results on HPV testing.
In the past 5 years, several large randomized clinical trials within national screening programs in Italy, England, Sweden, and the Netherlands examined the value of a primary HPV-based screening strategy.15–17,52 These studies confirmed the superior sensitivity of HPV testing for detection of CIN2+.
A large Canadian randomized trial53 compared HPV testing and cytologic testing as screening tests in women age 30 to 69. HPV DNA testing was 94.6% sensitive in detecting CIN2 or CIN3, compared with 55.4% for cytology. The specificity of HPV testing was nearly as high as that of cytology, 94.1% vs 96.8%. Furthermore, HPV testing followed (in those positive for HPV) by cytology resulted in a lower referral rate for colposcopy than did either test alone (1.1% vs 2.9% with cytology alone or 6.1% with HPV testing alone).
More randomized trial data are needed to evaluate the validity of this promising new approach in varied populations. The HPV FOCAL trial is comparing HPV-then-cytology testing vs cytology-then (in women with ASCUS)-HPV testing.54 In addition, the aforementioned novel biomarkers for HPV oncogenic activity may eventually play a greater role in primary screening.
With the latest evidence-based screening guidelines, we can implement a more sensitive and effective screening strategy for better prevention and early detection of cervical cancer. Newer cutting-edge molecular technologies appear promising; however, their cost-effectiveness needs to be further evaluated.
A MORAL AND ETHICAL RESPONSIBILITY
Our unscreened and underscreened populations carry a higher burden of cervical cancer and of death from cervical cancer. Identifying and reaching out to these women is our moral and ethical responsibility and yet poses the biggest challenge in screening. Arguably, this could have the most significant impact on rates of death from cervical cancer.
Innovative measures in overcoming healthcare barriers and in making testing cheaper will help to close the gap between well-screened and underscreened populations in the United States and globally. Examples would be a low-cost, point-of-care screening test for the general population, and a government-subsidized global vaccination program. It is entirely conceivable that women will no longer die from cervical cancer in the near future, thanks to global effective screening and preventive efforts through widespread HPV vaccination.
- ACOG Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin no. 109: cervical cytology screening. Obstet Gynecol 2009; 114:1409–1420.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VAUS Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin 2010; 60:99–119.
- US Preventive Services Task Force. Screening for cervical cancer. Recommendations and rationale. AHRQ Publication No. 03-515A. Rockville, MD: Agency for Healthcare Research and Quality, 2003.
- Castle PE, Carreon JD. Practice improvement in cervical screening and management: symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:238–340.
- Moscicki AB, Cox JT. Practice improvement in cervical screening and management (PICSM): symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:73–80.
- Kulasingam SL, Havrilesky L, Ghebre R, Myers ER. Screening for cervical cancer: a decision analysis for the US Preventive Services Task Force. AHRQ Publication No. 11-05157-EF-1. Rockville, MD: Agency for Healthcare Research and Quality, 2011.
- Sasieni P, Castanon A, Cuzick J. Effectiveness of cervical screening with age: population based case-control study of prospectively recorded data. BMJ 2009; 339:b2968.
- Saslow D, Runowicz CD, Solomon D, et al; American Cancer Society. American Cancer Society guideline for the early detection of cervical neoplasia and cancer. CA Cancer J Clin 2002; 52:342–362.
- Sasieni PD, Cuzick J, Lynch-Farmery E. Estimating the efficacy of screening by auditing smear histories of women with and without cervical cancer. The National Co-ordinating Network for Cervical Screening Working Group. Br J Cancer 1996; 73:1001–1005.
- Sasieni P, Adams J, Cuzick J. Benefit of cervical screening at different ages: evidence from the UK audit of screening histories. Br J Cancer 2003; 89:88–93.
- Goldie SJ, Kim JJ, Wright TC. Cost-effectiveness of human papillomavirus DNA testing for cervical cancer screening in women aged 30 years or more. Obstet Gynecol 2004; 103:619–631.
- Naucler P, Ryd W, Törnberg S, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med 2007; 357:1589–1597.
- Bulkmans NW, Berkhof J, Rozendaal L, et al. Human papillomavirus DNA testing for the detection of cervical intraepithelial neoplasia grade 3 and cancer: 5-year follow-up of a randomised controlled implementation trial. Lancet 2007; 370:1764–1772.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomised controlled trial. Lancet Oncol 2010; 11:249–257.
- Vijayaraghavan A, Efrusy MB, Mayrand MH, Santas CC, Goggin P. Cost-effectiveness of high-risk human papillomavirus testing for cervical cancer screening in Québec, Canada. Can J Public Health 2010; 101:220–225.
- Koliopoulos G, Arbyn M, Martin-Hirsch P, Kyrgiou M, Prendiville W, Paraskevaidis E. Diagnostic accuracy of human papillomavirus testing in primary cervical screening: a systematic review and metaanalysis of non-randomized studies. Gynecol Oncol 2007; 104:232–246.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Anttila A, Kotaniemi-Talonen L, Leinonen M, et al. Rate of cervical cancer, severe intraepithelial neoplasia, and adenocarcinoma in situ in primary HPV DNA screening with cytology triage: randomised study within organised screening programme. BMJ 2010; 340:c1804.
- Castle PE, Schiffman M, Wheeler CM, Solomon D. Evidence for frequent regression of cervical intraepithelial neoplasia-grade 2. Obstet Gynecol 2009; 113:18–25.
- Copeland G, Datta SD, Spivak G, Garvin AD, Cote ML. Total burden and incidence of in situ and invasive cervical carcinoma in Michigan, 1985–2003. Cancer 2008; 113(suppl 10):2946–2954.
- Chen HC, Schiffman M, Lin CY, et al; CBCSP-HPV Study Group. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J Natl Cancer Inst 2011; 103:1387–1396.
- Rodríguez AC, Schiffman M, Herrero R, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst 2010; 102:315–324.
- Wu X, Matanoski G, Chen VW, et al. Descriptive epidemiology of vaginal cancer incidence and survival by race, ethnicity, and age in the United States. Cancer 2008; 113(suppl 10):2873–2882.
- Pearce KF, Haefner HK, Sarwar SF, Nolan TE. Cytopathological findings on vaginal Papanicolaou smears after hysterectomy for benign gynecologic disease. N Engl J Med 1996; 335:1559–1562.
- Fox J, Remington P, Layde P, Klein G. The effect of hysterectomy on the risk of an abnormal screening Papanicolaou test result. Am J Obstet Gynecol 1999; 180:1104–1109.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
- Wright TC, Massad LS, Dunton CJ, Spitzer M, Wilkinson EJ, Solomon D; 2006 ASCCP-Sponsored Consensus Conference. 2006 consensus guidelines for the management of women with abnormal cervical screening tests. J Low Genit Tract Dis 2007; 11:201–222.
- Kjær SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- US Food and Drug Administration (FDA). FDA approved first DNA test for two types of human papillomavirus: agency also approved second DNA test for wider range of HPV types. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2009/ucm149544.htm. Accessed February 5, 2013.
- Wright TC, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing THE Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Petry KU, Schmidt D, Scherbring S, et al. Triaging Pap cytology negative, HPV positive cervical cancer screening results with p16/Ki-67 dual-stained cytology. Gynecol Oncol 2011; 121:505–509.
- Clad A, Reuschenbach M, Weinschenk J, Grote R, Rahmsdorf J, Freudenberg N. Performance of the Aptima high-risk human papillomavirus mRNA assay in a referral population in comparison with Hybrid Capture 2 and cytology. J Clin Microbiol 2011; 49:1071–1076.
- Cárdenas-Turanzas M, Nogueras-Gonzalez GM, Scheurer ME, et al. The performance of human papillomavirus high-risk DNA testing in the screening and diagnostic settings. Cancer Epidemiol Biomarkers Prev 2008; 17:2865–2871.
- Kulasingam SL, Hughes JP, Kiviat NB, et al. Evaluation of human papillomavirus testing in primary screening for cervical abnormalities: comparison of sensitivity, specificity, and frequency of referral. JAMA 2002; 288:1749–1757.
- Petry KU, Menton S, Menton M, et al. Inclusion of HPV testing in routine cervical cancer screening for women above 29 years in Germany: results for 8466 patients. Br J Cancer 2003; 88:1570–1577.
- Castle PE, Fetterman B, Poitras N, Lorey T, Shaber R, Kinney W. Fiveyear experience of human papillomavirus DNA and Papanicolaou test cotesting. Obstet Gynecol 2009; 113:595–600.
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006; 110:525–541.
- Carozzi F, Confortini M, Dalla Palma P, et al; New Technologies for Cervival Cancer Screening (NTCC) Working Group. Use of p16-INK4A overexpression to increase the specificity of human papillomavirus testing: a nested substudy of the NTCC randomised controlled trial. Lancet Oncol 2008; 9:937–945.
- Schmidt D, Bergeron C, Denton KJ, Ridder R; European CINtec Cytology Study Group. p16/ki-67 dual-stain cytology in the triage of ASCUS and LSIL papanicolaou cytology: results from the European equivocal or mildly abnormal Papanicolaou cytology study. Cancer Cytopathol 2011; 119:158–166.
- Wentzensen N, Schwartz L, Zuna RE, et al. Performance of p16/Ki-67 immunostaining to detect cervical cancer precursors in a colposcopy referral population. Clin Cancer Res 2012; 18:4154–4162.
- Nakagawa S, Yoshikawa H, Yasugi T, et al. Ubiquitous presence of E6 and E7 transcripts in human papillomavirus-positive cervical carcinomas regardless of its type. J Med Virol 2000; 62:251–258.
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10:431–442.
- Cuschieri K, Wentzensen N. Human papillomavirus mRNA and p16 detection as biomarkers for the improved diagnosis of cervical neoplasia. Cancer Epidemiol Biomarkers Prev 2008; 17:2536–2545.
- Dockter J, Schroder A, Hill C, Guzenski L, Monsonego J, Giachetti C. Clinical performance of the APTIMA HPV Assay for the detection of high-risk HPV and high-grade cervical lesions. J Clin Virol 2009; 45(suppl 1):S55–S61.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomised controlled trial. Lancet Oncol 2009; 10:672–682.
- Mayrand MH, Duarte-Franco E, Rodrigues I, et al; Canadian Cervical Cancer Screening Trial Study Group. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N Engl J Med 2007; 357:1579–1588.
- Ogilvie GS, van Niekerk DJ, Krajden M, et al. A randomized controlled trial of human papillomavirus (HPV) testing for cervical cancer screening: trial design and preliminary results (HPV FOCAL Trial). BMC Cancer 2010; 10:111.
Advances in our understanding of the pathogenesis of cervical cancer, new tests for human papillomavirus (HPV), and the development of HPV vaccines in the last decade are transforming the way we screen for cervical cancer.
As a result, screening guidelines are evolving rapidly, requiring clinicians to keep up-to-date with the evidence and rationales supporting the latest guidelines to properly convey best practices to patients.1–3
For example, we must understand why it is safe to extend the screening interval in women at low risk (as recommended in the new guidelines), and we need to be familiar with the options for women who test positive for HPV. Patients and providers may often find such new recommendations frustrating, and patients may feel that they are being denied something necessary by insurers rather than being treated according to scientific evidence.
This article will review the newest screening guidelines and the evidence supporting these recommendations for primary care providers. We will also review the potential role of novel biomarkers, newer HPV tests, and possible future strategies for cervical cancer screening.
WHAT’S NEW IN THE LATEST SCREENING GUIDELINES
Over the years, various organizations have issued separate screening guidelines, sometimes agreeing with each other, sometimes disagreeing.4 Now, for the first time, several of these organizations have developed guidelines collaboratively, and we have consensus in the screening recommendations.
Shortly after the American Congress of Obstetricians and Gynecologists (ACOG) issued its screening guidelines in December 2009,1 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), and American Society for Clinical Pathology (ASCP) convened an expert panel to review the available evidence and develop a new joint screening guideline. Concurrently, the US Preventive Services Task Force (USPSTF) commissioned a targeted systematic review of the latest evidence.
Both the ACS/ASCCP/ASCP group2 and the USPSTF3 released their new guidelines on March 14, 2012. In November 2012, ACOG issued its latest recommendation on cervical cancer screening.4 The following discussion highlights the consensus recommendations from these organizations (Table 1).
These guidelines apply to the general population only. They do not apply to women at high risk who may require more intensive screening, such as those who have a history of cervical cancer, are immunocompromised (eg, positive for human immunodeficiency virus [HIV]), or were exposed in utero to diethylstilbestrol.
Start screening at age 21
According to the new guidelines, women younger than 21 years should not be screened, regardless of the age at which they start having sex.1–3 This is a change from the 2002 and 2003 ACS recommendations, which said screening should begin 3 years after the onset of vaginal intercourse.5,6
Evidence. The rationale for the recommendation not to screen before age 21 stems from two pieces of evidence:
- Invasive cervical cancer is rare in this age group.7
- Screening can cause harm. For example, unnecessary treatment of preinvasive lesions can lead to long-term complications such as cervical stenosis, preterm delivery, and preterm premature rupture of membranes.8,9
Additionally, one study found that screening before age 21 has little or no impact on the incidence of invasive cervical cancer.10
Longer screening intervals
The 2012 ACS/ASCCP/ASCP guidelines2 and the latest ACOG guidelines4 lengthen the interval between cytology (Papanicolaou) testing to every 3 years in women age 21 to 29. Previous recommendations from these groups were to screen every 2 years, and the USPSTF first recommended the 3-year interval in 2003.11
For women age 30 to 65, the ACS/ASCCP/ASCP, ACOG, and the USPSTF now recommend screening every 5 years if the patient’s results on combined cytology and HPV testing are negative. However, cytologic testing alone every 3 years is also acceptable.2–4
Evidence. The evidence supporting a 3-year screening interval in women age 21 to 29 is primarily from modeling studies—no randomized clinical trial has been done. These studies found no significant difference in outcomes with a 2-year vs a 3-year screening interval.12,13 In particular, the predicted lifetime risk of cervical cancer in women screened every 3 years was 5 to 8 new cases of cancer per 1,000 women, compared with 4 to 6 cases per 1,000 women screened every 2 years.14
Similarly, screening women younger than age 30 at 2-year or 3-year intervals carried the same predicted lifetime risk of death from cervical cancer of 0.05 per 1,000 women, yet women screened every 2 years underwent 40% more colposcopies than those screened every 3 years.2 Therefore, screening every 3 years offers the best balance of benefits and risks in this age group.
Adding HPV testing to cytologic testing increases the sensitivity of screening—thus the recommendation to lengthen the screening interval to every 5 years in women age 30 to 65 who are at low risk and who have negative results on both tests. (Previously, the interval was 3 years.)
Specifically, adding HPV testing improves the sensitivity of screening for cervical intraepithelial neoplasia grade 3 (CIN3), so that, in subsequent rounds of screening, fewer cases of CIN3 or worse (CIN3+) or cancer are detected.15–17 The longer diagnostic lead time with combined testing is associated with a lower risk of CIN3+ or cancer following a double-negative test result than screening with cytology alone at shorter intervals. Combined testing at 5-year intervals is associated with a similar or lower cancer risk than cytology-alone screening at 3-year intervals.9
Moreover, modeling studies have shown that combined testing of women age 30 and older at 5-year intervals leads to fewer colposcopies and a similar or lower cancer risk than with cytology screening at 3-year intervals.18,19
A stronger endorsement for HPV testing
Combined cytologic and HPV testing has received its strongest endorsement to date from the ACS/ASCCP/ASCP, ACOG, and USPSTF in their latest guidelines.2–4
In 2003, ACOG gave HPV and cytology combined testing an “optional” recommendation for women over age 30; in 2009, it upgraded its recommendation to the highest level of recommendation.1 At that time, the USPSTF did not recommend for or against HPV testing, while the ACS did recommend HPV testing (with cytology testing alone every 2 to 3 years as an alternative screening strategy).5
Now, the ACS/ASCCP/ASCP and ACOG recommend HPV and cytology combined testing as the preferred strategy for screening women age 30 or over.2,4 Similarly, the USPSTF gives combined testing for women age 30 to 65 a grade A (its highest) recommendation.3 (In 2003, it had given it a grade I—insufficient evidence to assess the balance of benefit and harm.)
Evidence. Several recent studies provide compelling evidence that HPV testing has high sensitivity and excellent negative predictive value, supporting the stronger endorsement of HPV testing and longer screening intervals.
The Joint European Cohort study,20 in 24,295 women, conclusively showed that the 6-year risk of CIN3+ following a negative HPV test was significantly lower than that following a negative cytology result alone (0.27% vs 0.97%).
Katki et al,21 in another retrospective study, analyzed data from 330,000 women age 30 and older who underwent combined HPV and cytology testing. Looking at the tests separately, they found the risk of CIN3+ was comparable in the 3 years following a negative cytology test by itself and in the 5 years following negative combined HPV and cytology testing. In fact, combined testing at 5- or 6-year intervals offered better protection than cytology alone at 3-year intervals.
Furthermore, combined testing is also more sensitive for detecting cervical adenocarcinoma.22 (Most cancers of the cervix are squamous cell carcinomas, but approximately 10% are adenocarcinomas.)
Stop screening sooner
In 2002, the ACS recommended ending screening at age 70,11 and in 2009 ACOG said to stop at age 65 to 70.1 Now, the ACS/ASCCP/ASCP group2 and ACOG4 recommend stopping screening sooner—at age 65—provided that:
- The patient has had adequate negative screening until then. (Adequate negative prior screening is defined as three consecutive negative cytology results or two consecutive negative combined HPV and cytologic testing results within the 10 years before ceasing screening, with the most recent test performed within the last 5 years.)
- The patient has no history of CIN2+ within the last 20 years.
- The patient is not at high risk of cervical cancer, eg, no history of a high-grade precancerous cervical lesion or cervical cancer, in utero exposure to diethylstilbestrol, or immunosuppression (eg, HIV infection).
The USPSTF had already adopted this position.
Evidence. In women over age 65 who have had good screening, cervical cancer is rare and CIN2+ is uncommon.2,23,24 Kulasingam et al,9 in a modeling study performed for the USPSTF, calculated that continuing to screen until age 90 prevents only 1.6 cancer cases and 0.5 cancer deaths and extends life expectancy by only 1 year per 1,000 women.
Other studies also suggest that newly acquired high-risk HPV infection in women age 65 or older is associated with a very low absolute risk of HPV persistence and CIN3+ progression.25,26
In addition, cervical cancer takes a median of 20 to 25 years to develop after infection with high-risk HPV.2 Also, continuing to screen this older population will detect only a very small number of new cases of CIN2+ and may lead to harm from overtreatment.
Finally, postmenopausal women often have smaller and less accessible cervical transformation zones that may require more interventions to obtain adequate samples and to treat.
Stop screening after hysterectomy
The ACS/ASCCP/ASCP group, ACOG, and the USPSTF reaffirmed their recommendation against screening in women who have had a hysterectomy with removal of the cervix for a reason other than cancer and who have had no history of CIN2+ or cervical cancer.2–4
Evidence. Several lines of evidence suggest stopping screening after a woman has a hysterectomy. The incidence of vaginal cancer is extremely low,27 and the positive predictive value of cytologic testing of the vaginal cuff for vaginal cancer was zero in one study.28 Also, a large cross-sectional study of 5,330 screening cytology tests in women who had a hysterectomy found only one case of dysplasia and no cancer.29
Continue to screen after HPV vaccination
For the first time since HPV vaccines were introduced in 2006, the ACS/ASCCP/ASCP, ACOG, and the USPSTF have had to consider what to do for vaccinated women. All of their new guidelines say to keep screening them.
Evidence. The currently available HPV vaccines protect against cervical cancer,30 but only against cervical cancer caused by HPV types 16 and 18. Other oncogenic types of HPV exist, and the current vaccines do not protect against them.
Furthermore, many women are vaccinated who are already infected. In addition, as of 2010, only about 32% of eligible girls and women in the United States had received all three recommended doses of the vaccine.31 And modeling studies predict that the impact of the HPV vaccine will not be apparent for at least another decade.32
HPV 16/18 genotyping
The ACS/ASCCP/ASCP and ACOG now recommend HPV 16/18 genotyping as a triage option in women who have positive results on HPV testing but negative cytology results, and immediate referral for colposcopy if the genotyping test is positive.2 The alternative option in this situation is to repeat combined HPV and cytologic testing in 12 months.2,33
Evidence. The standard tests for HPV can detect DNA from about a dozen of the oncogenic types of HPV depending on the test, but they do not tell you which one the patient has. This information may be relevant, since not all “high-risk” HPV types are equally bad. HPV 16 and HPV 18 are the worst of all, together accounting for more than 70% of cases of cervical cancer.
Large cohort studies34,35 have shown that the risk of CIN3 reaches 10% over 1 to 4 years in women who test positive for HPV 16, and over 2 to 5 years if they test positive for HPV 18. This clinically relevant short-term risk supports immediate referral for colposcopy.
In March 2009, the US Food and Drug Administration (FDA) approved a test for HPV 16 and HPV 18—Cervista HPV 16/18 (Hologic, Bedford, MA).36
More recently, researchers from the Addressing the Need for Advanced HPV Diagnostics (ATHENA) trial,37 in 47,208 women, reported that they found CIN2+ in 11.4% of women who tested positive for either HPV 16 or HPV 18, and CIN3+ in 9.8%. Of those who were positive for HPV 16, 13.6% had CIN2+ and 11.7% had CIN3+.
WHAT’S COMING?
As we gain knowledge of the molecular oncogenesis of cervical cancer, we appreciate more the complex relation between HPV oncoproteins and cervical dysplasia. Recent studies demonstrated the clinical utility of detecting novel markers in women who have positive HPV results.38,39
At present, however, there is insufficient evidence to integrate these strategies into our standard of care for cervical cancer screening.
Novel biomarkers: p16 and Ki-67
Although HPV testing is sensitive, it has poor specificity and positive predictive value.40,41 In a primary screening setting, women with normal cytology results who test positive for high-risk HPV may carry a risk of only 3% to 7% for high-grade CIN.42,43
HPV 16/18 genotyping can be useful in this situation (see above). However, not everyone who carries HPV 16 or 18 goes on to develop CIN or cancer.44
A novel biomarker, p16, has been shown to be overexpressed in cervical dysplasia and associated with high-risk HPV oncogenic transformation. Another novel marker, Ki-67, can be regarded as a surrogate marker of deregulated cell proliferation (Figure 1).38
A recent study reported that a combined test for both of these markers (dual-stained cytology) had a sensitivity of 91.9% for detecting CIN2+ and 96.4% for CIN3+. This test was also highly specific: 82.1% for CIN2+ and 76.9% for CIN3+.38
An Italian randomized trial reported that p16 immunostaining improved the specificity of HPV testing in detecting CIN2+.45
In addition, the European Equivocal or Mildly Abnormal Papanicolaou Cytology Study46 found that the dual-stained cytology test had excellent sensitivity for CIN2+ in women with atypical squamous cells of undetermined significance (ASCUS) or low-grade squamous intraepithelial lesion (LSIL) cytology results (92.2% for ASCUS, 94.2% for LSIL). The specificity for CIN2+ in ASCUS and LSIL was 80.6% and 68%, respectively.
A US study also showed that the sensitivity and specificity to detect CIN3+ by using p16/Ki-67 were 97.2% and 60%, respectively, in women age 30 and older.47
If confirmed in more studies, p16/Ki-67 dual staining could help us in deciding which women who have positive HPV but negative cytology results should be referred for colposcopy.
HPV oncogene E6/E7 mRNA testing
In October 2011, the FDA approved the clinical use of a new-generation HPV test, the Aptima HPV assay (Hologic Gen-Probe, San Diego, CA), which detects mRNA for the proteins E6 and E7 from high-risk HPV.39
HPV E6/E7 mRNA expression has been found in virtually all HPV-positive cancer cases and demonstrates a stronger correlation with cervical disease than detection of HPV DNA.48 High-risk HPV E6 and E7 proteins immortalize and malignantly transform infected cells by inhibiting two host cellular anticancer proteins, p53 and retinoblastoma protein (pRB).44,49
The recent FDA approval was based on data from the CLEAR (Clinical Evaluation of Aptima HPV RNA) trial.39 In this trial, in more than 11,000 women, the test was as sensitive for detecting CIN2+ as the HPV DNA-based test, and it was more specific. This advantage was statistically significant. The higher specificity may reduce the number of unnecessary colposcopies and allow for more effective management.50,51
A promising future screening strategy: HPV testing first, then cytology
HPV testing is more sensitive than cytology, while cytology is more specific. Thus, it would be logical to test for HPV first, and then to perform cytologic testing in patients who have positive results on HPV testing.
In the past 5 years, several large randomized clinical trials within national screening programs in Italy, England, Sweden, and the Netherlands examined the value of a primary HPV-based screening strategy.15–17,52 These studies confirmed the superior sensitivity of HPV testing for detection of CIN2+.
A large Canadian randomized trial53 compared HPV testing and cytologic testing as screening tests in women age 30 to 69. HPV DNA testing was 94.6% sensitive in detecting CIN2 or CIN3, compared with 55.4% for cytology. The specificity of HPV testing was nearly as high as that of cytology, 94.1% vs 96.8%. Furthermore, HPV testing followed (in those positive for HPV) by cytology resulted in a lower referral rate for colposcopy than did either test alone (1.1% vs 2.9% with cytology alone or 6.1% with HPV testing alone).
More randomized trial data are needed to evaluate the validity of this promising new approach in varied populations. The HPV FOCAL trial is comparing HPV-then-cytology testing vs cytology-then (in women with ASCUS)-HPV testing.54 In addition, the aforementioned novel biomarkers for HPV oncogenic activity may eventually play a greater role in primary screening.
With the latest evidence-based screening guidelines, we can implement a more sensitive and effective screening strategy for better prevention and early detection of cervical cancer. Newer cutting-edge molecular technologies appear promising; however, their cost-effectiveness needs to be further evaluated.
A MORAL AND ETHICAL RESPONSIBILITY
Our unscreened and underscreened populations carry a higher burden of cervical cancer and of death from cervical cancer. Identifying and reaching out to these women is our moral and ethical responsibility and yet poses the biggest challenge in screening. Arguably, this could have the most significant impact on rates of death from cervical cancer.
Innovative measures in overcoming healthcare barriers and in making testing cheaper will help to close the gap between well-screened and underscreened populations in the United States and globally. Examples would be a low-cost, point-of-care screening test for the general population, and a government-subsidized global vaccination program. It is entirely conceivable that women will no longer die from cervical cancer in the near future, thanks to global effective screening and preventive efforts through widespread HPV vaccination.
Advances in our understanding of the pathogenesis of cervical cancer, new tests for human papillomavirus (HPV), and the development of HPV vaccines in the last decade are transforming the way we screen for cervical cancer.
As a result, screening guidelines are evolving rapidly, requiring clinicians to keep up-to-date with the evidence and rationales supporting the latest guidelines to properly convey best practices to patients.1–3
For example, we must understand why it is safe to extend the screening interval in women at low risk (as recommended in the new guidelines), and we need to be familiar with the options for women who test positive for HPV. Patients and providers may often find such new recommendations frustrating, and patients may feel that they are being denied something necessary by insurers rather than being treated according to scientific evidence.
This article will review the newest screening guidelines and the evidence supporting these recommendations for primary care providers. We will also review the potential role of novel biomarkers, newer HPV tests, and possible future strategies for cervical cancer screening.
WHAT’S NEW IN THE LATEST SCREENING GUIDELINES
Over the years, various organizations have issued separate screening guidelines, sometimes agreeing with each other, sometimes disagreeing.4 Now, for the first time, several of these organizations have developed guidelines collaboratively, and we have consensus in the screening recommendations.
Shortly after the American Congress of Obstetricians and Gynecologists (ACOG) issued its screening guidelines in December 2009,1 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), and American Society for Clinical Pathology (ASCP) convened an expert panel to review the available evidence and develop a new joint screening guideline. Concurrently, the US Preventive Services Task Force (USPSTF) commissioned a targeted systematic review of the latest evidence.
Both the ACS/ASCCP/ASCP group2 and the USPSTF3 released their new guidelines on March 14, 2012. In November 2012, ACOG issued its latest recommendation on cervical cancer screening.4 The following discussion highlights the consensus recommendations from these organizations (Table 1).
These guidelines apply to the general population only. They do not apply to women at high risk who may require more intensive screening, such as those who have a history of cervical cancer, are immunocompromised (eg, positive for human immunodeficiency virus [HIV]), or were exposed in utero to diethylstilbestrol.
Start screening at age 21
According to the new guidelines, women younger than 21 years should not be screened, regardless of the age at which they start having sex.1–3 This is a change from the 2002 and 2003 ACS recommendations, which said screening should begin 3 years after the onset of vaginal intercourse.5,6
Evidence. The rationale for the recommendation not to screen before age 21 stems from two pieces of evidence:
- Invasive cervical cancer is rare in this age group.7
- Screening can cause harm. For example, unnecessary treatment of preinvasive lesions can lead to long-term complications such as cervical stenosis, preterm delivery, and preterm premature rupture of membranes.8,9
Additionally, one study found that screening before age 21 has little or no impact on the incidence of invasive cervical cancer.10
Longer screening intervals
The 2012 ACS/ASCCP/ASCP guidelines2 and the latest ACOG guidelines4 lengthen the interval between cytology (Papanicolaou) testing to every 3 years in women age 21 to 29. Previous recommendations from these groups were to screen every 2 years, and the USPSTF first recommended the 3-year interval in 2003.11
For women age 30 to 65, the ACS/ASCCP/ASCP, ACOG, and the USPSTF now recommend screening every 5 years if the patient’s results on combined cytology and HPV testing are negative. However, cytologic testing alone every 3 years is also acceptable.2–4
Evidence. The evidence supporting a 3-year screening interval in women age 21 to 29 is primarily from modeling studies—no randomized clinical trial has been done. These studies found no significant difference in outcomes with a 2-year vs a 3-year screening interval.12,13 In particular, the predicted lifetime risk of cervical cancer in women screened every 3 years was 5 to 8 new cases of cancer per 1,000 women, compared with 4 to 6 cases per 1,000 women screened every 2 years.14
Similarly, screening women younger than age 30 at 2-year or 3-year intervals carried the same predicted lifetime risk of death from cervical cancer of 0.05 per 1,000 women, yet women screened every 2 years underwent 40% more colposcopies than those screened every 3 years.2 Therefore, screening every 3 years offers the best balance of benefits and risks in this age group.
Adding HPV testing to cytologic testing increases the sensitivity of screening—thus the recommendation to lengthen the screening interval to every 5 years in women age 30 to 65 who are at low risk and who have negative results on both tests. (Previously, the interval was 3 years.)
Specifically, adding HPV testing improves the sensitivity of screening for cervical intraepithelial neoplasia grade 3 (CIN3), so that, in subsequent rounds of screening, fewer cases of CIN3 or worse (CIN3+) or cancer are detected.15–17 The longer diagnostic lead time with combined testing is associated with a lower risk of CIN3+ or cancer following a double-negative test result than screening with cytology alone at shorter intervals. Combined testing at 5-year intervals is associated with a similar or lower cancer risk than cytology-alone screening at 3-year intervals.9
Moreover, modeling studies have shown that combined testing of women age 30 and older at 5-year intervals leads to fewer colposcopies and a similar or lower cancer risk than with cytology screening at 3-year intervals.18,19
A stronger endorsement for HPV testing
Combined cytologic and HPV testing has received its strongest endorsement to date from the ACS/ASCCP/ASCP, ACOG, and USPSTF in their latest guidelines.2–4
In 2003, ACOG gave HPV and cytology combined testing an “optional” recommendation for women over age 30; in 2009, it upgraded its recommendation to the highest level of recommendation.1 At that time, the USPSTF did not recommend for or against HPV testing, while the ACS did recommend HPV testing (with cytology testing alone every 2 to 3 years as an alternative screening strategy).5
Now, the ACS/ASCCP/ASCP and ACOG recommend HPV and cytology combined testing as the preferred strategy for screening women age 30 or over.2,4 Similarly, the USPSTF gives combined testing for women age 30 to 65 a grade A (its highest) recommendation.3 (In 2003, it had given it a grade I—insufficient evidence to assess the balance of benefit and harm.)
Evidence. Several recent studies provide compelling evidence that HPV testing has high sensitivity and excellent negative predictive value, supporting the stronger endorsement of HPV testing and longer screening intervals.
The Joint European Cohort study,20 in 24,295 women, conclusively showed that the 6-year risk of CIN3+ following a negative HPV test was significantly lower than that following a negative cytology result alone (0.27% vs 0.97%).
Katki et al,21 in another retrospective study, analyzed data from 330,000 women age 30 and older who underwent combined HPV and cytology testing. Looking at the tests separately, they found the risk of CIN3+ was comparable in the 3 years following a negative cytology test by itself and in the 5 years following negative combined HPV and cytology testing. In fact, combined testing at 5- or 6-year intervals offered better protection than cytology alone at 3-year intervals.
Furthermore, combined testing is also more sensitive for detecting cervical adenocarcinoma.22 (Most cancers of the cervix are squamous cell carcinomas, but approximately 10% are adenocarcinomas.)
Stop screening sooner
In 2002, the ACS recommended ending screening at age 70,11 and in 2009 ACOG said to stop at age 65 to 70.1 Now, the ACS/ASCCP/ASCP group2 and ACOG4 recommend stopping screening sooner—at age 65—provided that:
- The patient has had adequate negative screening until then. (Adequate negative prior screening is defined as three consecutive negative cytology results or two consecutive negative combined HPV and cytologic testing results within the 10 years before ceasing screening, with the most recent test performed within the last 5 years.)
- The patient has no history of CIN2+ within the last 20 years.
- The patient is not at high risk of cervical cancer, eg, no history of a high-grade precancerous cervical lesion or cervical cancer, in utero exposure to diethylstilbestrol, or immunosuppression (eg, HIV infection).
The USPSTF had already adopted this position.
Evidence. In women over age 65 who have had good screening, cervical cancer is rare and CIN2+ is uncommon.2,23,24 Kulasingam et al,9 in a modeling study performed for the USPSTF, calculated that continuing to screen until age 90 prevents only 1.6 cancer cases and 0.5 cancer deaths and extends life expectancy by only 1 year per 1,000 women.
Other studies also suggest that newly acquired high-risk HPV infection in women age 65 or older is associated with a very low absolute risk of HPV persistence and CIN3+ progression.25,26
In addition, cervical cancer takes a median of 20 to 25 years to develop after infection with high-risk HPV.2 Also, continuing to screen this older population will detect only a very small number of new cases of CIN2+ and may lead to harm from overtreatment.
Finally, postmenopausal women often have smaller and less accessible cervical transformation zones that may require more interventions to obtain adequate samples and to treat.
Stop screening after hysterectomy
The ACS/ASCCP/ASCP group, ACOG, and the USPSTF reaffirmed their recommendation against screening in women who have had a hysterectomy with removal of the cervix for a reason other than cancer and who have had no history of CIN2+ or cervical cancer.2–4
Evidence. Several lines of evidence suggest stopping screening after a woman has a hysterectomy. The incidence of vaginal cancer is extremely low,27 and the positive predictive value of cytologic testing of the vaginal cuff for vaginal cancer was zero in one study.28 Also, a large cross-sectional study of 5,330 screening cytology tests in women who had a hysterectomy found only one case of dysplasia and no cancer.29
Continue to screen after HPV vaccination
For the first time since HPV vaccines were introduced in 2006, the ACS/ASCCP/ASCP, ACOG, and the USPSTF have had to consider what to do for vaccinated women. All of their new guidelines say to keep screening them.
Evidence. The currently available HPV vaccines protect against cervical cancer,30 but only against cervical cancer caused by HPV types 16 and 18. Other oncogenic types of HPV exist, and the current vaccines do not protect against them.
Furthermore, many women are vaccinated who are already infected. In addition, as of 2010, only about 32% of eligible girls and women in the United States had received all three recommended doses of the vaccine.31 And modeling studies predict that the impact of the HPV vaccine will not be apparent for at least another decade.32
HPV 16/18 genotyping
The ACS/ASCCP/ASCP and ACOG now recommend HPV 16/18 genotyping as a triage option in women who have positive results on HPV testing but negative cytology results, and immediate referral for colposcopy if the genotyping test is positive.2 The alternative option in this situation is to repeat combined HPV and cytologic testing in 12 months.2,33
Evidence. The standard tests for HPV can detect DNA from about a dozen of the oncogenic types of HPV depending on the test, but they do not tell you which one the patient has. This information may be relevant, since not all “high-risk” HPV types are equally bad. HPV 16 and HPV 18 are the worst of all, together accounting for more than 70% of cases of cervical cancer.
Large cohort studies34,35 have shown that the risk of CIN3 reaches 10% over 1 to 4 years in women who test positive for HPV 16, and over 2 to 5 years if they test positive for HPV 18. This clinically relevant short-term risk supports immediate referral for colposcopy.
In March 2009, the US Food and Drug Administration (FDA) approved a test for HPV 16 and HPV 18—Cervista HPV 16/18 (Hologic, Bedford, MA).36
More recently, researchers from the Addressing the Need for Advanced HPV Diagnostics (ATHENA) trial,37 in 47,208 women, reported that they found CIN2+ in 11.4% of women who tested positive for either HPV 16 or HPV 18, and CIN3+ in 9.8%. Of those who were positive for HPV 16, 13.6% had CIN2+ and 11.7% had CIN3+.
WHAT’S COMING?
As we gain knowledge of the molecular oncogenesis of cervical cancer, we appreciate more the complex relation between HPV oncoproteins and cervical dysplasia. Recent studies demonstrated the clinical utility of detecting novel markers in women who have positive HPV results.38,39
At present, however, there is insufficient evidence to integrate these strategies into our standard of care for cervical cancer screening.
Novel biomarkers: p16 and Ki-67
Although HPV testing is sensitive, it has poor specificity and positive predictive value.40,41 In a primary screening setting, women with normal cytology results who test positive for high-risk HPV may carry a risk of only 3% to 7% for high-grade CIN.42,43
HPV 16/18 genotyping can be useful in this situation (see above). However, not everyone who carries HPV 16 or 18 goes on to develop CIN or cancer.44
A novel biomarker, p16, has been shown to be overexpressed in cervical dysplasia and associated with high-risk HPV oncogenic transformation. Another novel marker, Ki-67, can be regarded as a surrogate marker of deregulated cell proliferation (Figure 1).38
A recent study reported that a combined test for both of these markers (dual-stained cytology) had a sensitivity of 91.9% for detecting CIN2+ and 96.4% for CIN3+. This test was also highly specific: 82.1% for CIN2+ and 76.9% for CIN3+.38
An Italian randomized trial reported that p16 immunostaining improved the specificity of HPV testing in detecting CIN2+.45
In addition, the European Equivocal or Mildly Abnormal Papanicolaou Cytology Study46 found that the dual-stained cytology test had excellent sensitivity for CIN2+ in women with atypical squamous cells of undetermined significance (ASCUS) or low-grade squamous intraepithelial lesion (LSIL) cytology results (92.2% for ASCUS, 94.2% for LSIL). The specificity for CIN2+ in ASCUS and LSIL was 80.6% and 68%, respectively.
A US study also showed that the sensitivity and specificity to detect CIN3+ by using p16/Ki-67 were 97.2% and 60%, respectively, in women age 30 and older.47
If confirmed in more studies, p16/Ki-67 dual staining could help us in deciding which women who have positive HPV but negative cytology results should be referred for colposcopy.
HPV oncogene E6/E7 mRNA testing
In October 2011, the FDA approved the clinical use of a new-generation HPV test, the Aptima HPV assay (Hologic Gen-Probe, San Diego, CA), which detects mRNA for the proteins E6 and E7 from high-risk HPV.39
HPV E6/E7 mRNA expression has been found in virtually all HPV-positive cancer cases and demonstrates a stronger correlation with cervical disease than detection of HPV DNA.48 High-risk HPV E6 and E7 proteins immortalize and malignantly transform infected cells by inhibiting two host cellular anticancer proteins, p53 and retinoblastoma protein (pRB).44,49
The recent FDA approval was based on data from the CLEAR (Clinical Evaluation of Aptima HPV RNA) trial.39 In this trial, in more than 11,000 women, the test was as sensitive for detecting CIN2+ as the HPV DNA-based test, and it was more specific. This advantage was statistically significant. The higher specificity may reduce the number of unnecessary colposcopies and allow for more effective management.50,51
A promising future screening strategy: HPV testing first, then cytology
HPV testing is more sensitive than cytology, while cytology is more specific. Thus, it would be logical to test for HPV first, and then to perform cytologic testing in patients who have positive results on HPV testing.
In the past 5 years, several large randomized clinical trials within national screening programs in Italy, England, Sweden, and the Netherlands examined the value of a primary HPV-based screening strategy.15–17,52 These studies confirmed the superior sensitivity of HPV testing for detection of CIN2+.
A large Canadian randomized trial53 compared HPV testing and cytologic testing as screening tests in women age 30 to 69. HPV DNA testing was 94.6% sensitive in detecting CIN2 or CIN3, compared with 55.4% for cytology. The specificity of HPV testing was nearly as high as that of cytology, 94.1% vs 96.8%. Furthermore, HPV testing followed (in those positive for HPV) by cytology resulted in a lower referral rate for colposcopy than did either test alone (1.1% vs 2.9% with cytology alone or 6.1% with HPV testing alone).
More randomized trial data are needed to evaluate the validity of this promising new approach in varied populations. The HPV FOCAL trial is comparing HPV-then-cytology testing vs cytology-then (in women with ASCUS)-HPV testing.54 In addition, the aforementioned novel biomarkers for HPV oncogenic activity may eventually play a greater role in primary screening.
With the latest evidence-based screening guidelines, we can implement a more sensitive and effective screening strategy for better prevention and early detection of cervical cancer. Newer cutting-edge molecular technologies appear promising; however, their cost-effectiveness needs to be further evaluated.
A MORAL AND ETHICAL RESPONSIBILITY
Our unscreened and underscreened populations carry a higher burden of cervical cancer and of death from cervical cancer. Identifying and reaching out to these women is our moral and ethical responsibility and yet poses the biggest challenge in screening. Arguably, this could have the most significant impact on rates of death from cervical cancer.
Innovative measures in overcoming healthcare barriers and in making testing cheaper will help to close the gap between well-screened and underscreened populations in the United States and globally. Examples would be a low-cost, point-of-care screening test for the general population, and a government-subsidized global vaccination program. It is entirely conceivable that women will no longer die from cervical cancer in the near future, thanks to global effective screening and preventive efforts through widespread HPV vaccination.
- ACOG Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin no. 109: cervical cytology screening. Obstet Gynecol 2009; 114:1409–1420.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VAUS Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin 2010; 60:99–119.
- US Preventive Services Task Force. Screening for cervical cancer. Recommendations and rationale. AHRQ Publication No. 03-515A. Rockville, MD: Agency for Healthcare Research and Quality, 2003.
- Castle PE, Carreon JD. Practice improvement in cervical screening and management: symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:238–340.
- Moscicki AB, Cox JT. Practice improvement in cervical screening and management (PICSM): symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:73–80.
- Kulasingam SL, Havrilesky L, Ghebre R, Myers ER. Screening for cervical cancer: a decision analysis for the US Preventive Services Task Force. AHRQ Publication No. 11-05157-EF-1. Rockville, MD: Agency for Healthcare Research and Quality, 2011.
- Sasieni P, Castanon A, Cuzick J. Effectiveness of cervical screening with age: population based case-control study of prospectively recorded data. BMJ 2009; 339:b2968.
- Saslow D, Runowicz CD, Solomon D, et al; American Cancer Society. American Cancer Society guideline for the early detection of cervical neoplasia and cancer. CA Cancer J Clin 2002; 52:342–362.
- Sasieni PD, Cuzick J, Lynch-Farmery E. Estimating the efficacy of screening by auditing smear histories of women with and without cervical cancer. The National Co-ordinating Network for Cervical Screening Working Group. Br J Cancer 1996; 73:1001–1005.
- Sasieni P, Adams J, Cuzick J. Benefit of cervical screening at different ages: evidence from the UK audit of screening histories. Br J Cancer 2003; 89:88–93.
- Goldie SJ, Kim JJ, Wright TC. Cost-effectiveness of human papillomavirus DNA testing for cervical cancer screening in women aged 30 years or more. Obstet Gynecol 2004; 103:619–631.
- Naucler P, Ryd W, Törnberg S, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med 2007; 357:1589–1597.
- Bulkmans NW, Berkhof J, Rozendaal L, et al. Human papillomavirus DNA testing for the detection of cervical intraepithelial neoplasia grade 3 and cancer: 5-year follow-up of a randomised controlled implementation trial. Lancet 2007; 370:1764–1772.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomised controlled trial. Lancet Oncol 2010; 11:249–257.
- Vijayaraghavan A, Efrusy MB, Mayrand MH, Santas CC, Goggin P. Cost-effectiveness of high-risk human papillomavirus testing for cervical cancer screening in Québec, Canada. Can J Public Health 2010; 101:220–225.
- Koliopoulos G, Arbyn M, Martin-Hirsch P, Kyrgiou M, Prendiville W, Paraskevaidis E. Diagnostic accuracy of human papillomavirus testing in primary cervical screening: a systematic review and metaanalysis of non-randomized studies. Gynecol Oncol 2007; 104:232–246.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Anttila A, Kotaniemi-Talonen L, Leinonen M, et al. Rate of cervical cancer, severe intraepithelial neoplasia, and adenocarcinoma in situ in primary HPV DNA screening with cytology triage: randomised study within organised screening programme. BMJ 2010; 340:c1804.
- Castle PE, Schiffman M, Wheeler CM, Solomon D. Evidence for frequent regression of cervical intraepithelial neoplasia-grade 2. Obstet Gynecol 2009; 113:18–25.
- Copeland G, Datta SD, Spivak G, Garvin AD, Cote ML. Total burden and incidence of in situ and invasive cervical carcinoma in Michigan, 1985–2003. Cancer 2008; 113(suppl 10):2946–2954.
- Chen HC, Schiffman M, Lin CY, et al; CBCSP-HPV Study Group. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J Natl Cancer Inst 2011; 103:1387–1396.
- Rodríguez AC, Schiffman M, Herrero R, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst 2010; 102:315–324.
- Wu X, Matanoski G, Chen VW, et al. Descriptive epidemiology of vaginal cancer incidence and survival by race, ethnicity, and age in the United States. Cancer 2008; 113(suppl 10):2873–2882.
- Pearce KF, Haefner HK, Sarwar SF, Nolan TE. Cytopathological findings on vaginal Papanicolaou smears after hysterectomy for benign gynecologic disease. N Engl J Med 1996; 335:1559–1562.
- Fox J, Remington P, Layde P, Klein G. The effect of hysterectomy on the risk of an abnormal screening Papanicolaou test result. Am J Obstet Gynecol 1999; 180:1104–1109.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
- Wright TC, Massad LS, Dunton CJ, Spitzer M, Wilkinson EJ, Solomon D; 2006 ASCCP-Sponsored Consensus Conference. 2006 consensus guidelines for the management of women with abnormal cervical screening tests. J Low Genit Tract Dis 2007; 11:201–222.
- Kjær SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- US Food and Drug Administration (FDA). FDA approved first DNA test for two types of human papillomavirus: agency also approved second DNA test for wider range of HPV types. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2009/ucm149544.htm. Accessed February 5, 2013.
- Wright TC, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing THE Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Petry KU, Schmidt D, Scherbring S, et al. Triaging Pap cytology negative, HPV positive cervical cancer screening results with p16/Ki-67 dual-stained cytology. Gynecol Oncol 2011; 121:505–509.
- Clad A, Reuschenbach M, Weinschenk J, Grote R, Rahmsdorf J, Freudenberg N. Performance of the Aptima high-risk human papillomavirus mRNA assay in a referral population in comparison with Hybrid Capture 2 and cytology. J Clin Microbiol 2011; 49:1071–1076.
- Cárdenas-Turanzas M, Nogueras-Gonzalez GM, Scheurer ME, et al. The performance of human papillomavirus high-risk DNA testing in the screening and diagnostic settings. Cancer Epidemiol Biomarkers Prev 2008; 17:2865–2871.
- Kulasingam SL, Hughes JP, Kiviat NB, et al. Evaluation of human papillomavirus testing in primary screening for cervical abnormalities: comparison of sensitivity, specificity, and frequency of referral. JAMA 2002; 288:1749–1757.
- Petry KU, Menton S, Menton M, et al. Inclusion of HPV testing in routine cervical cancer screening for women above 29 years in Germany: results for 8466 patients. Br J Cancer 2003; 88:1570–1577.
- Castle PE, Fetterman B, Poitras N, Lorey T, Shaber R, Kinney W. Fiveyear experience of human papillomavirus DNA and Papanicolaou test cotesting. Obstet Gynecol 2009; 113:595–600.
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006; 110:525–541.
- Carozzi F, Confortini M, Dalla Palma P, et al; New Technologies for Cervival Cancer Screening (NTCC) Working Group. Use of p16-INK4A overexpression to increase the specificity of human papillomavirus testing: a nested substudy of the NTCC randomised controlled trial. Lancet Oncol 2008; 9:937–945.
- Schmidt D, Bergeron C, Denton KJ, Ridder R; European CINtec Cytology Study Group. p16/ki-67 dual-stain cytology in the triage of ASCUS and LSIL papanicolaou cytology: results from the European equivocal or mildly abnormal Papanicolaou cytology study. Cancer Cytopathol 2011; 119:158–166.
- Wentzensen N, Schwartz L, Zuna RE, et al. Performance of p16/Ki-67 immunostaining to detect cervical cancer precursors in a colposcopy referral population. Clin Cancer Res 2012; 18:4154–4162.
- Nakagawa S, Yoshikawa H, Yasugi T, et al. Ubiquitous presence of E6 and E7 transcripts in human papillomavirus-positive cervical carcinomas regardless of its type. J Med Virol 2000; 62:251–258.
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10:431–442.
- Cuschieri K, Wentzensen N. Human papillomavirus mRNA and p16 detection as biomarkers for the improved diagnosis of cervical neoplasia. Cancer Epidemiol Biomarkers Prev 2008; 17:2536–2545.
- Dockter J, Schroder A, Hill C, Guzenski L, Monsonego J, Giachetti C. Clinical performance of the APTIMA HPV Assay for the detection of high-risk HPV and high-grade cervical lesions. J Clin Virol 2009; 45(suppl 1):S55–S61.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomised controlled trial. Lancet Oncol 2009; 10:672–682.
- Mayrand MH, Duarte-Franco E, Rodrigues I, et al; Canadian Cervical Cancer Screening Trial Study Group. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N Engl J Med 2007; 357:1579–1588.
- Ogilvie GS, van Niekerk DJ, Krajden M, et al. A randomized controlled trial of human papillomavirus (HPV) testing for cervical cancer screening: trial design and preliminary results (HPV FOCAL Trial). BMC Cancer 2010; 10:111.
- ACOG Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin no. 109: cervical cytology screening. Obstet Gynecol 2009; 114:1409–1420.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VAUS Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin 2010; 60:99–119.
- US Preventive Services Task Force. Screening for cervical cancer. Recommendations and rationale. AHRQ Publication No. 03-515A. Rockville, MD: Agency for Healthcare Research and Quality, 2003.
- Castle PE, Carreon JD. Practice improvement in cervical screening and management: symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:238–340.
- Moscicki AB, Cox JT. Practice improvement in cervical screening and management (PICSM): symposium on management of cervical abnormalities in adolescents and young women. J Low Genit Tract Dis 2010; 14:73–80.
- Kulasingam SL, Havrilesky L, Ghebre R, Myers ER. Screening for cervical cancer: a decision analysis for the US Preventive Services Task Force. AHRQ Publication No. 11-05157-EF-1. Rockville, MD: Agency for Healthcare Research and Quality, 2011.
- Sasieni P, Castanon A, Cuzick J. Effectiveness of cervical screening with age: population based case-control study of prospectively recorded data. BMJ 2009; 339:b2968.
- Saslow D, Runowicz CD, Solomon D, et al; American Cancer Society. American Cancer Society guideline for the early detection of cervical neoplasia and cancer. CA Cancer J Clin 2002; 52:342–362.
- Sasieni PD, Cuzick J, Lynch-Farmery E. Estimating the efficacy of screening by auditing smear histories of women with and without cervical cancer. The National Co-ordinating Network for Cervical Screening Working Group. Br J Cancer 1996; 73:1001–1005.
- Sasieni P, Adams J, Cuzick J. Benefit of cervical screening at different ages: evidence from the UK audit of screening histories. Br J Cancer 2003; 89:88–93.
- Goldie SJ, Kim JJ, Wright TC. Cost-effectiveness of human papillomavirus DNA testing for cervical cancer screening in women aged 30 years or more. Obstet Gynecol 2004; 103:619–631.
- Naucler P, Ryd W, Törnberg S, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med 2007; 357:1589–1597.
- Bulkmans NW, Berkhof J, Rozendaal L, et al. Human papillomavirus DNA testing for the detection of cervical intraepithelial neoplasia grade 3 and cancer: 5-year follow-up of a randomised controlled implementation trial. Lancet 2007; 370:1764–1772.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomised controlled trial. Lancet Oncol 2010; 11:249–257.
- Vijayaraghavan A, Efrusy MB, Mayrand MH, Santas CC, Goggin P. Cost-effectiveness of high-risk human papillomavirus testing for cervical cancer screening in Québec, Canada. Can J Public Health 2010; 101:220–225.
- Koliopoulos G, Arbyn M, Martin-Hirsch P, Kyrgiou M, Prendiville W, Paraskevaidis E. Diagnostic accuracy of human papillomavirus testing in primary cervical screening: a systematic review and metaanalysis of non-randomized studies. Gynecol Oncol 2007; 104:232–246.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Anttila A, Kotaniemi-Talonen L, Leinonen M, et al. Rate of cervical cancer, severe intraepithelial neoplasia, and adenocarcinoma in situ in primary HPV DNA screening with cytology triage: randomised study within organised screening programme. BMJ 2010; 340:c1804.
- Castle PE, Schiffman M, Wheeler CM, Solomon D. Evidence for frequent regression of cervical intraepithelial neoplasia-grade 2. Obstet Gynecol 2009; 113:18–25.
- Copeland G, Datta SD, Spivak G, Garvin AD, Cote ML. Total burden and incidence of in situ and invasive cervical carcinoma in Michigan, 1985–2003. Cancer 2008; 113(suppl 10):2946–2954.
- Chen HC, Schiffman M, Lin CY, et al; CBCSP-HPV Study Group. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J Natl Cancer Inst 2011; 103:1387–1396.
- Rodríguez AC, Schiffman M, Herrero R, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst 2010; 102:315–324.
- Wu X, Matanoski G, Chen VW, et al. Descriptive epidemiology of vaginal cancer incidence and survival by race, ethnicity, and age in the United States. Cancer 2008; 113(suppl 10):2873–2882.
- Pearce KF, Haefner HK, Sarwar SF, Nolan TE. Cytopathological findings on vaginal Papanicolaou smears after hysterectomy for benign gynecologic disease. N Engl J Med 1996; 335:1559–1562.
- Fox J, Remington P, Layde P, Klein G. The effect of hysterectomy on the risk of an abnormal screening Papanicolaou test result. Am J Obstet Gynecol 1999; 180:1104–1109.
- FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
- Centers for Disease Control and Prevention (CDC). National and state vaccination coverage among adolescents aged 13 through 17 years—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:1117–1123.
- Cuzick J, Castañón A, Sasieni P. Predicted impact of vaccination against human papillomavirus 16/18 on cancer incidence and cervical abnormalities in women aged 20–29 in the UK. Br J Cancer 2010; 102:933–939.
- Wright TC, Massad LS, Dunton CJ, Spitzer M, Wilkinson EJ, Solomon D; 2006 ASCCP-Sponsored Consensus Conference. 2006 consensus guidelines for the management of women with abnormal cervical screening tests. J Low Genit Tract Dis 2007; 11:201–222.
- Kjær SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- US Food and Drug Administration (FDA). FDA approved first DNA test for two types of human papillomavirus: agency also approved second DNA test for wider range of HPV types. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2009/ucm149544.htm. Accessed February 5, 2013.
- Wright TC, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing THE Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Petry KU, Schmidt D, Scherbring S, et al. Triaging Pap cytology negative, HPV positive cervical cancer screening results with p16/Ki-67 dual-stained cytology. Gynecol Oncol 2011; 121:505–509.
- Clad A, Reuschenbach M, Weinschenk J, Grote R, Rahmsdorf J, Freudenberg N. Performance of the Aptima high-risk human papillomavirus mRNA assay in a referral population in comparison with Hybrid Capture 2 and cytology. J Clin Microbiol 2011; 49:1071–1076.
- Cárdenas-Turanzas M, Nogueras-Gonzalez GM, Scheurer ME, et al. The performance of human papillomavirus high-risk DNA testing in the screening and diagnostic settings. Cancer Epidemiol Biomarkers Prev 2008; 17:2865–2871.
- Kulasingam SL, Hughes JP, Kiviat NB, et al. Evaluation of human papillomavirus testing in primary screening for cervical abnormalities: comparison of sensitivity, specificity, and frequency of referral. JAMA 2002; 288:1749–1757.
- Petry KU, Menton S, Menton M, et al. Inclusion of HPV testing in routine cervical cancer screening for women above 29 years in Germany: results for 8466 patients. Br J Cancer 2003; 88:1570–1577.
- Castle PE, Fetterman B, Poitras N, Lorey T, Shaber R, Kinney W. Fiveyear experience of human papillomavirus DNA and Papanicolaou test cotesting. Obstet Gynecol 2009; 113:595–600.
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006; 110:525–541.
- Carozzi F, Confortini M, Dalla Palma P, et al; New Technologies for Cervival Cancer Screening (NTCC) Working Group. Use of p16-INK4A overexpression to increase the specificity of human papillomavirus testing: a nested substudy of the NTCC randomised controlled trial. Lancet Oncol 2008; 9:937–945.
- Schmidt D, Bergeron C, Denton KJ, Ridder R; European CINtec Cytology Study Group. p16/ki-67 dual-stain cytology in the triage of ASCUS and LSIL papanicolaou cytology: results from the European equivocal or mildly abnormal Papanicolaou cytology study. Cancer Cytopathol 2011; 119:158–166.
- Wentzensen N, Schwartz L, Zuna RE, et al. Performance of p16/Ki-67 immunostaining to detect cervical cancer precursors in a colposcopy referral population. Clin Cancer Res 2012; 18:4154–4162.
- Nakagawa S, Yoshikawa H, Yasugi T, et al. Ubiquitous presence of E6 and E7 transcripts in human papillomavirus-positive cervical carcinomas regardless of its type. J Med Virol 2000; 62:251–258.
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ 2003; 10:431–442.
- Cuschieri K, Wentzensen N. Human papillomavirus mRNA and p16 detection as biomarkers for the improved diagnosis of cervical neoplasia. Cancer Epidemiol Biomarkers Prev 2008; 17:2536–2545.
- Dockter J, Schroder A, Hill C, Guzenski L, Monsonego J, Giachetti C. Clinical performance of the APTIMA HPV Assay for the detection of high-risk HPV and high-grade cervical lesions. J Clin Virol 2009; 45(suppl 1):S55–S61.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomised controlled trial. Lancet Oncol 2009; 10:672–682.
- Mayrand MH, Duarte-Franco E, Rodrigues I, et al; Canadian Cervical Cancer Screening Trial Study Group. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N Engl J Med 2007; 357:1579–1588.
- Ogilvie GS, van Niekerk DJ, Krajden M, et al. A randomized controlled trial of human papillomavirus (HPV) testing for cervical cancer screening: trial design and preliminary results (HPV FOCAL Trial). BMC Cancer 2010; 10:111.
KEY POINTS
- The new guidelines still recommend starting screening with cytologic (Papanicolaou) testing at age 21, but now recommend repeating the test less often, ie, every 3 years rather than every 2 years for women age 21 to 29.
- Women age 30 and older who are screened by combined cytologic and HPV testing should be rescreened every 5 years if both tests are negative (instead of every 3 years, as previously recommended). Alternatively, they can be screened by cytology alone every 3 years.
- We can stop screening women at age 65 if they have had adequate screening until then and no history of cervical intraepithelial neoplasia grade 2 or worse (CIN2+) in the past 20 years. Once screening is discontinued, it should not resume, even if the patient has a new sexual partner.
- Screening should not change after HPV vaccination.
- When women have negative cytology but positive HPV results, tests for the HPV 16 and 18 genotypes can help to identify those at higher risk of developing CIN2+.
EHRs and medicolegal risk: How they help, when they could hurt
Survey: Many physicians plan to leave or scale down practice
Janelle Yates (February 2012)
Is private ObGyn practice on its way out?
Lucia DiVenere, MA (October 2011)
The medical record has evolved considerably since it originated in ancient Greece as a narrative of cure.1 For one thing, it’s now electronic. For another, it’s no longer a medical record but a health record. According to the US Department of Health and Human Services, the distinction is not a trivial one. A medical record is used by clinicians mostly for diagnosis and treatment, whereas the health record focuses on the total wellbeing of the patient.2 The medical record is used primarily within a practice. The electronic health record (EHR) reaches across borders to other offices, institutions, and clinicians.
Use of the EHR has been stimulated by the Health Information Technology for Economic and Clinical Health Act,3 which offers grants and incentives for “meaningful use” of electronic records.4 After 2014, medical practices that do not use EHRs will face a financial penalty that amounts to 2% of 2013 clinical revenue.
EHRs have been hailed as a panacea and derided as anathema. Whatever your perspective, there is no denying that they dramatically increase the immediate and easy availability of information and, therefore, influence decision-making in regard to medical care, cost-effectiveness, and patient safety. EHRs have the potential to improve communication, broaden access to information, and help guide clinical decision-making through the use of best-practice algorithms. When used properly—which means taking advantage of the EHR’s full potential and adapting to the way information is organized and analyzed—the EHR can reduce adverse events and help defend the appropriateness of the care provided. This lowers your medicolegal risk. When used improperly or haphazardly, they may increase that risk. In this article, we elaborate on both.
EHRs have many benefits
Improved communication. EHRs facilitate communication between healthcare providers. A primary care physician can access a consultant’s report practically as it is written. Providers also can carry on a dialogue electronically, planning together for care that will best serve the patient, with less redundancy and time.
The EHR also facilitates communication between physician and patient, allowing the physician to see the patient’s recent history and plan her management while speaking to her on the phone. Issues can be addressed with greater accuracy and expediency, leading to reduced anxiety for the patient and increased compliance.
Seamless integration. Information can be entered into the EHR and integrated into the full record more seamlessly than it is with written records. And data can be entered once and used many times.
Enhanced decision-making. Decision-making depends on careful analysis of a clinical scenario. Protocols, templates, and order sets embedded in the EHR can reduce medical errors by identifying scenarios for the physician to review.5,6
The EHR can also highlight adverse drug-drug interactions and help avoid potential allergic reactions. Murphy and colleagues reported a reduction of medical errors by utilizing a pharmacy-driven EHR component—a reduction from 90% to 47% on the surgical unit and from 57% to 33% on the medicine unit.7
Improved documentation. The EHR can enhance documentation by offering specific and detailed templates for informed consent, making it more comprehensive than a handwritten notation of the risks and benefits.
Decipherability is another strength of the EHR. Because physicians are notorious for poor handwriting skills, some hospitals now require a writing sample as part of their privileging process. The EHR avoids this issue entirely.8 Typos and grammatical errors are minimized by spellchecking and grammar-correcting programs written into the EHR.
Quality assurance. Timely evaluation of approaches to clinical care is available to physicians as well as hospitals that use EHRs.9 An individual physician can perform personal quality-assurance audits. And hospital management can gather cumulative statistics more quickly and easily.5,6,10,11
Patient data can be accessed independent of medical department, with lab tests, imaging studies, and pathology reports readily available for review. And accessibility is available regardless of geographic location.
EHRs are not perfect, and neither are their users. EHRs present the potential for problems related to absent or erroneous data entry, patient privacy issues, misunderstanding and misuse of software, and development of metadata.
With initial use, EHRs can create documentation gaps with the transition from paper to electronic records. In addition, inadequate provider training can create new error pathways, and a failure to use EHRs consistently can lead to loss of data and communication errors. These gaps and errors can increase medicolegal risk, as can the more extensive documentation often seen with early use, which creates more discoverable data. The temptation to cut and paste risks repeating earlier errors and omitting new information.
Another area of risk involves communication with the patient via email. A failure to reply could result in claims of negligence, and information overload could obscure pertinent pieces of information. And a departure from clinical decision support could be used by the patient to defend allegations of negligence.
With widespread use of EHRs, improved access to data could change the “duty” owed to the patient. In addition, clinical decision support embedded within the software could become the de facto “standard of care.”
The learning curve can be steep
The learning curve for EHRs may be steep and, at times, discouraging. One reason is that data are organized differently than in the conventional paper record, where information is read and analyzed in a progressive and stepwise manner, as in an analog or vertical system. The EHR is a digital format, so finding information requires digital (horizontal) inquiry. Information is, therefore, utilized in both horizontal and vertical formats in everyday situations. If data are entered incorrectly, all subsequent decisions could be flawed. And if the EHR suggests a plan, and that plan is not performed by the provider, the risk of liability could increase.
Inadvertent violation of the Health Insurance Portability and Accountability Act (HIPAA) with an EHR could increase medicolegal risk. For example, HIPAA allows for patients to make corrections to inaccurate information in their personal documents, but access by the patient could require the physician to review all records viewed by the patient after visit notes have been entered. This could drive up the cost of practice and reduce face-to-face time between physician and patient. Patients are not necessarily the best judges of which information is most important in their medical records.
Internet access raises concerns about the privacy of sensitive issues and misuse of information. Making a patient’s protected health information accessible electronically leaves physicians and hospitals at risk for a government fine or lawsuit. In several instances, the US Department of Health and Human Services (HHS) has levied fines against small practices and government agencies.
In one case, HHS fined Phoenix Cardiac Surgery in Phoenix, Arizona, $100,000 for posting surgery and appointment schedules on an Internet-based calendar that was accessible to the public.12 In another, HHS fined the Massachusetts Eye and Ear Infirmary in Boston $1.5 million after it reported the loss of an encrypted personal laptop containing the protected health information of patients and research subjects.13 The Alaska Department of Health and Social Services (DHSS) agreed to pay HHS $1.7 million after it reported the loss of a USB drive—possibly containing protected health information—from the vehicle of a DHSS employee.14
In traditional physician practices that employ handwritten records, the potential for compromise of patient information is limited. An organization may lose a few patient charts in the office and recover from the loss without incident. With the EHR, the loss poses a significant threat. The cases mentioned above were attributed to negligence or ignorance. The consequences could be worse if the compromise of EHR data is determined to be intentional. On September 4, 2010, hackers may have exposed the personal information of approximately 9,493 patients at Southwest Seattle Orthopaedics and Sports Medicine in Burien, Washington. Even with the best encryption technology, any electronic system remains vulnerable to external attack.
Metadata reveal how original data are used
Another concern regarding EHRs involves metadata—”data about data content.”15 Metadata is structured information that describes, locates, explains, or manages information. Metadata relevant to the EHR includes the data and time it was reviewed by the provider and whether it was manipulated in any way. Clearly, there is a potential for use and misuse by third-party reviewers.
Specialty-specific EHRs are recommended
Many ObGyns have found that most EHR systems are inadequate to the task of recording and analyzing information relevant to their specialty. Obstetric care is episodic and frequent. Data are added into the flow that must be considered at each visit, such as gestational age, fetal growth, labs (and normative values), prenatal diagnostic studies, and so on, representing both vertical and horizontal processing.16
The legal discovery process poses challenges that have not yet been resolved
The legal discovery process grants all parties to a lawsuit equal access to information. Under ideal circumstances, the EHR can provide comprehensive data more quickly than traditional records can. The problem is determining what constitutes relevant data and which party has the burden or benefit of making that decision. Uncontrolled access has the potential to violate privacy and privilege requirements.
Rules regarding discovery are still being debated in regard to their applicability to digital discovery.17 Even before a lawsuit is filed, the potential for “data mining” by third parties could lead to allegations of malpractice.
How to use EHRs responsibly without increasing risk
Good communication between patient and provider is paramount in the provision of quality medical care. Adherence to evidence-based standards with thorough documentation always serves the best interests of both patients and providers. The EHR can facilitate this process.
Our recommendations for appropriate use of your EHR include:
- Spend time learning the ins and outs of your particular EHR, and make sure your staff does the same. This will help reduce the likelihood that errors will be introduced into the record and ensure consistent use.
- Use individual sign-ons for anyone involved in data entry. This step facilitates the identification of users responsible for inaccurate use or errors, so that the situation can be addressed efficiently.
- Do not let third parties enter or manipulate data. This could jeopardize patient privacy, as well as the integrity of the record itself.
- Track all data entry on a regular basis. The frequency of tracking should be a function of routine as well as clinical circumstance. All new data from the previous interval should be reviewed at the time of the subsequent visit in order to direct care and ensure proper data entry.
Because of the considerable risk of liability claims in ObGyn practice, it is critical that the medical record accurately and precisely reflects the circumstances of each case. The EHR can be an effective and useful tool to document what occurred (and when) in a clinical scenario.18 As with all medical records, completeness and accuracy are the first and best defense against allegations of medical malpractice.
1. The Casebooks Project. History of Medical Record-keeping. http://www.magicandmedicine.hps.cam.ac.uk/on-astrological-medicine/further-reading/history-of-medical-record-keeping/. Accessed February 26 2013.
2. US Department of Health and Human Services. EMR vs EHR—What is the difference? Health IT Buzz. http://www.healthit.gov/buzz-blog/electronic-health-and-medical-records/emr-vs-ehr-difference/. Accessed February 20, 2013.
3. Health Information Technology for Economic and Clinical Health Act of 2009. HITECH Act. Pub L No 111-5 Div A tit XIII Div B tit IV Feb 17 2009, 123 stat 226, 467. Codified in scattered sections of 42 USCA.
4. Mangalmurti S, Murtagh L, Mello M. Medical malpractice liability in the age of electronic health records. N Engl J Med. 2010;363(21):2060-2067.
5. Reid P, Compton D, Grossman J, et al. Building a Better Delivery System: A New Engineering/Healthcare Partnership. Committee on Engineering and the Health Care System, Institute of Medicine and the National Academy of Engineering. Washington, DC: National Academies Press; 2005.
6. Grossman J. Disruptive innovation in healthcare: challenges for engineering. The Bridge. 2008;38:10-16.
7. Murphy E, Oxencis C, Klauck J, et al. Medication reconciliation at an academic medical center; implementation of a comprehensive program from admission to discharge. Am J Health-System Pharmacy. 2009;66(23):2126-2131.
8. Schuler R. The smart grid: a bridge between emerging technologies society and the environment. The Bridge. 2010;40:42-49.
9. Haberman S, Feldman J, Merhi Z, et al. Effect of clinical decision support on documentation compliance in an electronic medical record. Obstet Gynecol. 2009;114(2 Pt 1):311-317.
10. Hasley S. Decision support and patient safety: the time has come. Am J Obstet Gynecol. 2011;204(6):461-465.
11. Lagrew D, Stutman H, Sicaeros L. Voluntary physician adoption of an inpatient electronic medical record by obstetrician-gynecologists. Am J Obstet Gynecol. 2008;198(6):690.e1-e6.
12. Dolan PL. $100,000 HIPAA fine designed to send message to small physician practices. American Medical News. 2012. http://www.ama-assn.org/amednews/2012/04/30/bisd0502.htm. Accessed February 26, 2013.
13. US Department of Health and Human Services. Massachusetts provider settles HIPAA case for $1.5 million [news release]. September 17 2012. http://www.hhs.gov/news/press/2012pres/09/20120917a.html. Accessed February 26, 2013.
14. US Department of Health and Human Services. Alaska settles HIPAA security case for $1,700,000 [news release]. June 26, 2012. http://www.hhs.gov/news/press/2012pres/06/20120626a.html. Accessed February 26, 2013.
15. National Information Standards Organization. Understanding Metadata. Bethesda MD: NISO Press; 2004. http://www.niso.org/publications/press/UnderstandingMetadata.pdf. Accessed February 26, 2013.
16. McCoy M, Diamond A, Strunk A. Special requirements of electronic medical record systems in obstetrics and gynecology. Obstet Gynecol. 2010;116(1):140-143.
17. The Berkman Center for Internet and Society at Harvard Law School. The Federal Rules of Civil Procedure: The Impact of Digital Discovery. http://cyber.law.harvard.edu/digitaldiscovery/digdisc_library_4.html. Accessed February 26 2013.
18. Quinn M, Kats A, Kleinman K, et al. The relationship between electronic health records and malpractice claims. Arch Intern Med. 2012;172(15):1187-1188.
Survey: Many physicians plan to leave or scale down practice
Janelle Yates (February 2012)
Is private ObGyn practice on its way out?
Lucia DiVenere, MA (October 2011)
The medical record has evolved considerably since it originated in ancient Greece as a narrative of cure.1 For one thing, it’s now electronic. For another, it’s no longer a medical record but a health record. According to the US Department of Health and Human Services, the distinction is not a trivial one. A medical record is used by clinicians mostly for diagnosis and treatment, whereas the health record focuses on the total wellbeing of the patient.2 The medical record is used primarily within a practice. The electronic health record (EHR) reaches across borders to other offices, institutions, and clinicians.
Use of the EHR has been stimulated by the Health Information Technology for Economic and Clinical Health Act,3 which offers grants and incentives for “meaningful use” of electronic records.4 After 2014, medical practices that do not use EHRs will face a financial penalty that amounts to 2% of 2013 clinical revenue.
EHRs have been hailed as a panacea and derided as anathema. Whatever your perspective, there is no denying that they dramatically increase the immediate and easy availability of information and, therefore, influence decision-making in regard to medical care, cost-effectiveness, and patient safety. EHRs have the potential to improve communication, broaden access to information, and help guide clinical decision-making through the use of best-practice algorithms. When used properly—which means taking advantage of the EHR’s full potential and adapting to the way information is organized and analyzed—the EHR can reduce adverse events and help defend the appropriateness of the care provided. This lowers your medicolegal risk. When used improperly or haphazardly, they may increase that risk. In this article, we elaborate on both.
EHRs have many benefits
Improved communication. EHRs facilitate communication between healthcare providers. A primary care physician can access a consultant’s report practically as it is written. Providers also can carry on a dialogue electronically, planning together for care that will best serve the patient, with less redundancy and time.
The EHR also facilitates communication between physician and patient, allowing the physician to see the patient’s recent history and plan her management while speaking to her on the phone. Issues can be addressed with greater accuracy and expediency, leading to reduced anxiety for the patient and increased compliance.
Seamless integration. Information can be entered into the EHR and integrated into the full record more seamlessly than it is with written records. And data can be entered once and used many times.
Enhanced decision-making. Decision-making depends on careful analysis of a clinical scenario. Protocols, templates, and order sets embedded in the EHR can reduce medical errors by identifying scenarios for the physician to review.5,6
The EHR can also highlight adverse drug-drug interactions and help avoid potential allergic reactions. Murphy and colleagues reported a reduction of medical errors by utilizing a pharmacy-driven EHR component—a reduction from 90% to 47% on the surgical unit and from 57% to 33% on the medicine unit.7
Improved documentation. The EHR can enhance documentation by offering specific and detailed templates for informed consent, making it more comprehensive than a handwritten notation of the risks and benefits.
Decipherability is another strength of the EHR. Because physicians are notorious for poor handwriting skills, some hospitals now require a writing sample as part of their privileging process. The EHR avoids this issue entirely.8 Typos and grammatical errors are minimized by spellchecking and grammar-correcting programs written into the EHR.
Quality assurance. Timely evaluation of approaches to clinical care is available to physicians as well as hospitals that use EHRs.9 An individual physician can perform personal quality-assurance audits. And hospital management can gather cumulative statistics more quickly and easily.5,6,10,11
Patient data can be accessed independent of medical department, with lab tests, imaging studies, and pathology reports readily available for review. And accessibility is available regardless of geographic location.
EHRs are not perfect, and neither are their users. EHRs present the potential for problems related to absent or erroneous data entry, patient privacy issues, misunderstanding and misuse of software, and development of metadata.
With initial use, EHRs can create documentation gaps with the transition from paper to electronic records. In addition, inadequate provider training can create new error pathways, and a failure to use EHRs consistently can lead to loss of data and communication errors. These gaps and errors can increase medicolegal risk, as can the more extensive documentation often seen with early use, which creates more discoverable data. The temptation to cut and paste risks repeating earlier errors and omitting new information.
Another area of risk involves communication with the patient via email. A failure to reply could result in claims of negligence, and information overload could obscure pertinent pieces of information. And a departure from clinical decision support could be used by the patient to defend allegations of negligence.
With widespread use of EHRs, improved access to data could change the “duty” owed to the patient. In addition, clinical decision support embedded within the software could become the de facto “standard of care.”
The learning curve can be steep
The learning curve for EHRs may be steep and, at times, discouraging. One reason is that data are organized differently than in the conventional paper record, where information is read and analyzed in a progressive and stepwise manner, as in an analog or vertical system. The EHR is a digital format, so finding information requires digital (horizontal) inquiry. Information is, therefore, utilized in both horizontal and vertical formats in everyday situations. If data are entered incorrectly, all subsequent decisions could be flawed. And if the EHR suggests a plan, and that plan is not performed by the provider, the risk of liability could increase.
Inadvertent violation of the Health Insurance Portability and Accountability Act (HIPAA) with an EHR could increase medicolegal risk. For example, HIPAA allows for patients to make corrections to inaccurate information in their personal documents, but access by the patient could require the physician to review all records viewed by the patient after visit notes have been entered. This could drive up the cost of practice and reduce face-to-face time between physician and patient. Patients are not necessarily the best judges of which information is most important in their medical records.
Internet access raises concerns about the privacy of sensitive issues and misuse of information. Making a patient’s protected health information accessible electronically leaves physicians and hospitals at risk for a government fine or lawsuit. In several instances, the US Department of Health and Human Services (HHS) has levied fines against small practices and government agencies.
In one case, HHS fined Phoenix Cardiac Surgery in Phoenix, Arizona, $100,000 for posting surgery and appointment schedules on an Internet-based calendar that was accessible to the public.12 In another, HHS fined the Massachusetts Eye and Ear Infirmary in Boston $1.5 million after it reported the loss of an encrypted personal laptop containing the protected health information of patients and research subjects.13 The Alaska Department of Health and Social Services (DHSS) agreed to pay HHS $1.7 million after it reported the loss of a USB drive—possibly containing protected health information—from the vehicle of a DHSS employee.14
In traditional physician practices that employ handwritten records, the potential for compromise of patient information is limited. An organization may lose a few patient charts in the office and recover from the loss without incident. With the EHR, the loss poses a significant threat. The cases mentioned above were attributed to negligence or ignorance. The consequences could be worse if the compromise of EHR data is determined to be intentional. On September 4, 2010, hackers may have exposed the personal information of approximately 9,493 patients at Southwest Seattle Orthopaedics and Sports Medicine in Burien, Washington. Even with the best encryption technology, any electronic system remains vulnerable to external attack.
Metadata reveal how original data are used
Another concern regarding EHRs involves metadata—”data about data content.”15 Metadata is structured information that describes, locates, explains, or manages information. Metadata relevant to the EHR includes the data and time it was reviewed by the provider and whether it was manipulated in any way. Clearly, there is a potential for use and misuse by third-party reviewers.
Specialty-specific EHRs are recommended
Many ObGyns have found that most EHR systems are inadequate to the task of recording and analyzing information relevant to their specialty. Obstetric care is episodic and frequent. Data are added into the flow that must be considered at each visit, such as gestational age, fetal growth, labs (and normative values), prenatal diagnostic studies, and so on, representing both vertical and horizontal processing.16
The legal discovery process poses challenges that have not yet been resolved
The legal discovery process grants all parties to a lawsuit equal access to information. Under ideal circumstances, the EHR can provide comprehensive data more quickly than traditional records can. The problem is determining what constitutes relevant data and which party has the burden or benefit of making that decision. Uncontrolled access has the potential to violate privacy and privilege requirements.
Rules regarding discovery are still being debated in regard to their applicability to digital discovery.17 Even before a lawsuit is filed, the potential for “data mining” by third parties could lead to allegations of malpractice.
How to use EHRs responsibly without increasing risk
Good communication between patient and provider is paramount in the provision of quality medical care. Adherence to evidence-based standards with thorough documentation always serves the best interests of both patients and providers. The EHR can facilitate this process.
Our recommendations for appropriate use of your EHR include:
- Spend time learning the ins and outs of your particular EHR, and make sure your staff does the same. This will help reduce the likelihood that errors will be introduced into the record and ensure consistent use.
- Use individual sign-ons for anyone involved in data entry. This step facilitates the identification of users responsible for inaccurate use or errors, so that the situation can be addressed efficiently.
- Do not let third parties enter or manipulate data. This could jeopardize patient privacy, as well as the integrity of the record itself.
- Track all data entry on a regular basis. The frequency of tracking should be a function of routine as well as clinical circumstance. All new data from the previous interval should be reviewed at the time of the subsequent visit in order to direct care and ensure proper data entry.
Because of the considerable risk of liability claims in ObGyn practice, it is critical that the medical record accurately and precisely reflects the circumstances of each case. The EHR can be an effective and useful tool to document what occurred (and when) in a clinical scenario.18 As with all medical records, completeness and accuracy are the first and best defense against allegations of medical malpractice.
Survey: Many physicians plan to leave or scale down practice
Janelle Yates (February 2012)
Is private ObGyn practice on its way out?
Lucia DiVenere, MA (October 2011)
The medical record has evolved considerably since it originated in ancient Greece as a narrative of cure.1 For one thing, it’s now electronic. For another, it’s no longer a medical record but a health record. According to the US Department of Health and Human Services, the distinction is not a trivial one. A medical record is used by clinicians mostly for diagnosis and treatment, whereas the health record focuses on the total wellbeing of the patient.2 The medical record is used primarily within a practice. The electronic health record (EHR) reaches across borders to other offices, institutions, and clinicians.
Use of the EHR has been stimulated by the Health Information Technology for Economic and Clinical Health Act,3 which offers grants and incentives for “meaningful use” of electronic records.4 After 2014, medical practices that do not use EHRs will face a financial penalty that amounts to 2% of 2013 clinical revenue.
EHRs have been hailed as a panacea and derided as anathema. Whatever your perspective, there is no denying that they dramatically increase the immediate and easy availability of information and, therefore, influence decision-making in regard to medical care, cost-effectiveness, and patient safety. EHRs have the potential to improve communication, broaden access to information, and help guide clinical decision-making through the use of best-practice algorithms. When used properly—which means taking advantage of the EHR’s full potential and adapting to the way information is organized and analyzed—the EHR can reduce adverse events and help defend the appropriateness of the care provided. This lowers your medicolegal risk. When used improperly or haphazardly, they may increase that risk. In this article, we elaborate on both.
EHRs have many benefits
Improved communication. EHRs facilitate communication between healthcare providers. A primary care physician can access a consultant’s report practically as it is written. Providers also can carry on a dialogue electronically, planning together for care that will best serve the patient, with less redundancy and time.
The EHR also facilitates communication between physician and patient, allowing the physician to see the patient’s recent history and plan her management while speaking to her on the phone. Issues can be addressed with greater accuracy and expediency, leading to reduced anxiety for the patient and increased compliance.
Seamless integration. Information can be entered into the EHR and integrated into the full record more seamlessly than it is with written records. And data can be entered once and used many times.
Enhanced decision-making. Decision-making depends on careful analysis of a clinical scenario. Protocols, templates, and order sets embedded in the EHR can reduce medical errors by identifying scenarios for the physician to review.5,6
The EHR can also highlight adverse drug-drug interactions and help avoid potential allergic reactions. Murphy and colleagues reported a reduction of medical errors by utilizing a pharmacy-driven EHR component—a reduction from 90% to 47% on the surgical unit and from 57% to 33% on the medicine unit.7
Improved documentation. The EHR can enhance documentation by offering specific and detailed templates for informed consent, making it more comprehensive than a handwritten notation of the risks and benefits.
Decipherability is another strength of the EHR. Because physicians are notorious for poor handwriting skills, some hospitals now require a writing sample as part of their privileging process. The EHR avoids this issue entirely.8 Typos and grammatical errors are minimized by spellchecking and grammar-correcting programs written into the EHR.
Quality assurance. Timely evaluation of approaches to clinical care is available to physicians as well as hospitals that use EHRs.9 An individual physician can perform personal quality-assurance audits. And hospital management can gather cumulative statistics more quickly and easily.5,6,10,11
Patient data can be accessed independent of medical department, with lab tests, imaging studies, and pathology reports readily available for review. And accessibility is available regardless of geographic location.
EHRs are not perfect, and neither are their users. EHRs present the potential for problems related to absent or erroneous data entry, patient privacy issues, misunderstanding and misuse of software, and development of metadata.
With initial use, EHRs can create documentation gaps with the transition from paper to electronic records. In addition, inadequate provider training can create new error pathways, and a failure to use EHRs consistently can lead to loss of data and communication errors. These gaps and errors can increase medicolegal risk, as can the more extensive documentation often seen with early use, which creates more discoverable data. The temptation to cut and paste risks repeating earlier errors and omitting new information.
Another area of risk involves communication with the patient via email. A failure to reply could result in claims of negligence, and information overload could obscure pertinent pieces of information. And a departure from clinical decision support could be used by the patient to defend allegations of negligence.
With widespread use of EHRs, improved access to data could change the “duty” owed to the patient. In addition, clinical decision support embedded within the software could become the de facto “standard of care.”
The learning curve can be steep
The learning curve for EHRs may be steep and, at times, discouraging. One reason is that data are organized differently than in the conventional paper record, where information is read and analyzed in a progressive and stepwise manner, as in an analog or vertical system. The EHR is a digital format, so finding information requires digital (horizontal) inquiry. Information is, therefore, utilized in both horizontal and vertical formats in everyday situations. If data are entered incorrectly, all subsequent decisions could be flawed. And if the EHR suggests a plan, and that plan is not performed by the provider, the risk of liability could increase.
Inadvertent violation of the Health Insurance Portability and Accountability Act (HIPAA) with an EHR could increase medicolegal risk. For example, HIPAA allows for patients to make corrections to inaccurate information in their personal documents, but access by the patient could require the physician to review all records viewed by the patient after visit notes have been entered. This could drive up the cost of practice and reduce face-to-face time between physician and patient. Patients are not necessarily the best judges of which information is most important in their medical records.
Internet access raises concerns about the privacy of sensitive issues and misuse of information. Making a patient’s protected health information accessible electronically leaves physicians and hospitals at risk for a government fine or lawsuit. In several instances, the US Department of Health and Human Services (HHS) has levied fines against small practices and government agencies.
In one case, HHS fined Phoenix Cardiac Surgery in Phoenix, Arizona, $100,000 for posting surgery and appointment schedules on an Internet-based calendar that was accessible to the public.12 In another, HHS fined the Massachusetts Eye and Ear Infirmary in Boston $1.5 million after it reported the loss of an encrypted personal laptop containing the protected health information of patients and research subjects.13 The Alaska Department of Health and Social Services (DHSS) agreed to pay HHS $1.7 million after it reported the loss of a USB drive—possibly containing protected health information—from the vehicle of a DHSS employee.14
In traditional physician practices that employ handwritten records, the potential for compromise of patient information is limited. An organization may lose a few patient charts in the office and recover from the loss without incident. With the EHR, the loss poses a significant threat. The cases mentioned above were attributed to negligence or ignorance. The consequences could be worse if the compromise of EHR data is determined to be intentional. On September 4, 2010, hackers may have exposed the personal information of approximately 9,493 patients at Southwest Seattle Orthopaedics and Sports Medicine in Burien, Washington. Even with the best encryption technology, any electronic system remains vulnerable to external attack.
Metadata reveal how original data are used
Another concern regarding EHRs involves metadata—”data about data content.”15 Metadata is structured information that describes, locates, explains, or manages information. Metadata relevant to the EHR includes the data and time it was reviewed by the provider and whether it was manipulated in any way. Clearly, there is a potential for use and misuse by third-party reviewers.
Specialty-specific EHRs are recommended
Many ObGyns have found that most EHR systems are inadequate to the task of recording and analyzing information relevant to their specialty. Obstetric care is episodic and frequent. Data are added into the flow that must be considered at each visit, such as gestational age, fetal growth, labs (and normative values), prenatal diagnostic studies, and so on, representing both vertical and horizontal processing.16
The legal discovery process poses challenges that have not yet been resolved
The legal discovery process grants all parties to a lawsuit equal access to information. Under ideal circumstances, the EHR can provide comprehensive data more quickly than traditional records can. The problem is determining what constitutes relevant data and which party has the burden or benefit of making that decision. Uncontrolled access has the potential to violate privacy and privilege requirements.
Rules regarding discovery are still being debated in regard to their applicability to digital discovery.17 Even before a lawsuit is filed, the potential for “data mining” by third parties could lead to allegations of malpractice.
How to use EHRs responsibly without increasing risk
Good communication between patient and provider is paramount in the provision of quality medical care. Adherence to evidence-based standards with thorough documentation always serves the best interests of both patients and providers. The EHR can facilitate this process.
Our recommendations for appropriate use of your EHR include:
- Spend time learning the ins and outs of your particular EHR, and make sure your staff does the same. This will help reduce the likelihood that errors will be introduced into the record and ensure consistent use.
- Use individual sign-ons for anyone involved in data entry. This step facilitates the identification of users responsible for inaccurate use or errors, so that the situation can be addressed efficiently.
- Do not let third parties enter or manipulate data. This could jeopardize patient privacy, as well as the integrity of the record itself.
- Track all data entry on a regular basis. The frequency of tracking should be a function of routine as well as clinical circumstance. All new data from the previous interval should be reviewed at the time of the subsequent visit in order to direct care and ensure proper data entry.
Because of the considerable risk of liability claims in ObGyn practice, it is critical that the medical record accurately and precisely reflects the circumstances of each case. The EHR can be an effective and useful tool to document what occurred (and when) in a clinical scenario.18 As with all medical records, completeness and accuracy are the first and best defense against allegations of medical malpractice.
1. The Casebooks Project. History of Medical Record-keeping. http://www.magicandmedicine.hps.cam.ac.uk/on-astrological-medicine/further-reading/history-of-medical-record-keeping/. Accessed February 26 2013.
2. US Department of Health and Human Services. EMR vs EHR—What is the difference? Health IT Buzz. http://www.healthit.gov/buzz-blog/electronic-health-and-medical-records/emr-vs-ehr-difference/. Accessed February 20, 2013.
3. Health Information Technology for Economic and Clinical Health Act of 2009. HITECH Act. Pub L No 111-5 Div A tit XIII Div B tit IV Feb 17 2009, 123 stat 226, 467. Codified in scattered sections of 42 USCA.
4. Mangalmurti S, Murtagh L, Mello M. Medical malpractice liability in the age of electronic health records. N Engl J Med. 2010;363(21):2060-2067.
5. Reid P, Compton D, Grossman J, et al. Building a Better Delivery System: A New Engineering/Healthcare Partnership. Committee on Engineering and the Health Care System, Institute of Medicine and the National Academy of Engineering. Washington, DC: National Academies Press; 2005.
6. Grossman J. Disruptive innovation in healthcare: challenges for engineering. The Bridge. 2008;38:10-16.
7. Murphy E, Oxencis C, Klauck J, et al. Medication reconciliation at an academic medical center; implementation of a comprehensive program from admission to discharge. Am J Health-System Pharmacy. 2009;66(23):2126-2131.
8. Schuler R. The smart grid: a bridge between emerging technologies society and the environment. The Bridge. 2010;40:42-49.
9. Haberman S, Feldman J, Merhi Z, et al. Effect of clinical decision support on documentation compliance in an electronic medical record. Obstet Gynecol. 2009;114(2 Pt 1):311-317.
10. Hasley S. Decision support and patient safety: the time has come. Am J Obstet Gynecol. 2011;204(6):461-465.
11. Lagrew D, Stutman H, Sicaeros L. Voluntary physician adoption of an inpatient electronic medical record by obstetrician-gynecologists. Am J Obstet Gynecol. 2008;198(6):690.e1-e6.
12. Dolan PL. $100,000 HIPAA fine designed to send message to small physician practices. American Medical News. 2012. http://www.ama-assn.org/amednews/2012/04/30/bisd0502.htm. Accessed February 26, 2013.
13. US Department of Health and Human Services. Massachusetts provider settles HIPAA case for $1.5 million [news release]. September 17 2012. http://www.hhs.gov/news/press/2012pres/09/20120917a.html. Accessed February 26, 2013.
14. US Department of Health and Human Services. Alaska settles HIPAA security case for $1,700,000 [news release]. June 26, 2012. http://www.hhs.gov/news/press/2012pres/06/20120626a.html. Accessed February 26, 2013.
15. National Information Standards Organization. Understanding Metadata. Bethesda MD: NISO Press; 2004. http://www.niso.org/publications/press/UnderstandingMetadata.pdf. Accessed February 26, 2013.
16. McCoy M, Diamond A, Strunk A. Special requirements of electronic medical record systems in obstetrics and gynecology. Obstet Gynecol. 2010;116(1):140-143.
17. The Berkman Center for Internet and Society at Harvard Law School. The Federal Rules of Civil Procedure: The Impact of Digital Discovery. http://cyber.law.harvard.edu/digitaldiscovery/digdisc_library_4.html. Accessed February 26 2013.
18. Quinn M, Kats A, Kleinman K, et al. The relationship between electronic health records and malpractice claims. Arch Intern Med. 2012;172(15):1187-1188.
1. The Casebooks Project. History of Medical Record-keeping. http://www.magicandmedicine.hps.cam.ac.uk/on-astrological-medicine/further-reading/history-of-medical-record-keeping/. Accessed February 26 2013.
2. US Department of Health and Human Services. EMR vs EHR—What is the difference? Health IT Buzz. http://www.healthit.gov/buzz-blog/electronic-health-and-medical-records/emr-vs-ehr-difference/. Accessed February 20, 2013.
3. Health Information Technology for Economic and Clinical Health Act of 2009. HITECH Act. Pub L No 111-5 Div A tit XIII Div B tit IV Feb 17 2009, 123 stat 226, 467. Codified in scattered sections of 42 USCA.
4. Mangalmurti S, Murtagh L, Mello M. Medical malpractice liability in the age of electronic health records. N Engl J Med. 2010;363(21):2060-2067.
5. Reid P, Compton D, Grossman J, et al. Building a Better Delivery System: A New Engineering/Healthcare Partnership. Committee on Engineering and the Health Care System, Institute of Medicine and the National Academy of Engineering. Washington, DC: National Academies Press; 2005.
6. Grossman J. Disruptive innovation in healthcare: challenges for engineering. The Bridge. 2008;38:10-16.
7. Murphy E, Oxencis C, Klauck J, et al. Medication reconciliation at an academic medical center; implementation of a comprehensive program from admission to discharge. Am J Health-System Pharmacy. 2009;66(23):2126-2131.
8. Schuler R. The smart grid: a bridge between emerging technologies society and the environment. The Bridge. 2010;40:42-49.
9. Haberman S, Feldman J, Merhi Z, et al. Effect of clinical decision support on documentation compliance in an electronic medical record. Obstet Gynecol. 2009;114(2 Pt 1):311-317.
10. Hasley S. Decision support and patient safety: the time has come. Am J Obstet Gynecol. 2011;204(6):461-465.
11. Lagrew D, Stutman H, Sicaeros L. Voluntary physician adoption of an inpatient electronic medical record by obstetrician-gynecologists. Am J Obstet Gynecol. 2008;198(6):690.e1-e6.
12. Dolan PL. $100,000 HIPAA fine designed to send message to small physician practices. American Medical News. 2012. http://www.ama-assn.org/amednews/2012/04/30/bisd0502.htm. Accessed February 26, 2013.
13. US Department of Health and Human Services. Massachusetts provider settles HIPAA case for $1.5 million [news release]. September 17 2012. http://www.hhs.gov/news/press/2012pres/09/20120917a.html. Accessed February 26, 2013.
14. US Department of Health and Human Services. Alaska settles HIPAA security case for $1,700,000 [news release]. June 26, 2012. http://www.hhs.gov/news/press/2012pres/06/20120626a.html. Accessed February 26, 2013.
15. National Information Standards Organization. Understanding Metadata. Bethesda MD: NISO Press; 2004. http://www.niso.org/publications/press/UnderstandingMetadata.pdf. Accessed February 26, 2013.
16. McCoy M, Diamond A, Strunk A. Special requirements of electronic medical record systems in obstetrics and gynecology. Obstet Gynecol. 2010;116(1):140-143.
17. The Berkman Center for Internet and Society at Harvard Law School. The Federal Rules of Civil Procedure: The Impact of Digital Discovery. http://cyber.law.harvard.edu/digitaldiscovery/digdisc_library_4.html. Accessed February 26 2013.
18. Quinn M, Kats A, Kleinman K, et al. The relationship between electronic health records and malpractice claims. Arch Intern Med. 2012;172(15):1187-1188.
Obese mother gains another 60 lb before delivery … and more

AN OBESE WOMAN with a family history of diabetes had previously given birth to a large baby. Even though she expressed her concern that this fetus would also be macrosomic, the ObGyn planned for spontaneous vaginal delivery. At 39 weeks’ gestation, after gaining 60 lb, she went to the hospital requesting induction of labor; the ObGyn reluctantly agreed. Labor was lengthy, forceps-assisted delivery was performed, and a shoulder dystocia was encountered. The baby was born with respiratory distress, a brachial plexus injury, bruises on his right cheek and both ears, and multiple rib fractures. After transfer to a children’s hospital, surgical exploration revealed avulsion of the C6 root nerve from the spinal cord and damage to C5, C7, and C8 nerve roots. Several surgical repairs and physical therapy have led to some improvement, but the child is permanently injured. His right arm is shorter than the left, his right hand is smaller, and he has less strength and range of motion in the right arm. He also has excessive tearing in the right eye and his right eyelid droops.
PARENTS’ CLAIM The ObGyn failed to recognize the risk of delivering a macrosomic baby and did not consider cesarean delivery. The brachial plexus injury was due to downward traction applied during delivery.
PHYSICIAN’S DEFENSE There was no negligence. The brachial plexus injury was not caused by downward traction.
VERDICT A $4.1 million Indiana verdict was returned, but was reduced to the state cap of $1.25 million.
Failure to follow-up on mass: $1.97M verdict
AFTER STAGE II OVARIAN CANCER was found in 1999, a woman underwent surgery and chemotherapy, and was told she was cancer-free. She had regular visits between 2000 and 2008 with another surgical oncologist after her first surgeon moved. In 2004, the oncologist documented finding a round fullness during a pelvic exam. A CT scan confirmed a mass in the pelvic cul-de-sac.
In August 2008, the patient was treated for deep venous thrombosis in her leg. The attending physician saw the pelvic mass on imaging, and a biopsy indicated a recurrence of ovarian cancer. After chemotherapy, the patient underwent surgery, but the tumor was unresectable. In early 2011, testing revealed metastasis to the spine, sternum, pelvic bone, arm, and lung.
PATIENT’S CLAIM The surgeon did not properly investigate the mass resulting in a delayed diagnosis of cancer recurrence. The patient alleged that the surgical oncologist repeatedly stated that the mass had not changed and was most likely fluid; it was nothing to worry about. Radiology reports indicated a suspicion of cancer.
DEFENDANTS’ DEFENSE The oncologist repeatedly told the patient that the mass should be biopsied, but the patient refused because she was dealing with other medical issues. The radiologist argued that reports to the oncologist included everything needed to diagnose the cancer.
VERDICT A Pennsylvania jury found the surgical oncologist fully at fault and returned a $1,971,455 verdict.
Incomplete tubal ligation
BEFORE DELIVERY OF HER THIRD CHILD, a 26-year-old woman requested sterilization using tubal ligation. After delivery, the ObGyn performed a bilateral tubal ligation. The pathologist’s report indicated that the ligation was incomplete: the left fallopian tube had not been fully removed. The ObGyn failed to note the report’s results in the patient’s record, nor did he advise the patient. Two years later, the patient delivered a fourth child.
PATIENT’S CLAIM The patient alleged wrongful birth against both the ObGyn and pathologist. The ObGyn was negligent for not reacting to the pathologist’s report of incomplete tubal ligation, and for not informing the patient. The pathologist should have verbally confirmed receipt of the report with the ObGyn.
PHYSICIANS’ DEFENSE The ObGyn settled before trial. The pathologist claimed he had properly interpreted the specimen and reported the results.
VERDICT A Louisiana jury found the ObGyn fully at fault and assessed additional damages of $56,252 to the $100,000 settlement.
A WOMAN SUFFERED FROM PELVIC PAIN caused by adhesions following two cesarean deliveries and a hysterectomy. In January 2003, her ObGyn performed laparotomy to reduce adhesions from prior surgeries and place Gore-Tex mesh to prevent future adhesions. In October 2010, the patient reported epigastric pain, and went to a different surgeon (her insurance changed). A CT scan identified a foreign body encapsulated in scar tissue in the patient’s lower abdomen/pelvis. The surgeon removed the foreign body.
PATIENT’S CLAIM The ObGyn and hospital were negligent in conducting the 2003 procedure; the foreign object was a retained surgical sponge.
DEFENDANTS’ DEFENSE The foreign body removed in 2010 was the Gore-Tex mesh placed in 2003. The mesh became encapsulated in scar tissue due to the patient’s propensity to develop adhesions, and then moved within the patient’s body. Surgical sponges have embedded radiopaque tracers; CT scans in 2003 and 2010 did not detect any radiopaque tracers.
VERDICT A California defense verdict was returned.
Massive bleed during sacrocolpopexy
AFTER A 72-YEAR-OLD WOMAN developed pelvic organ prolapse, her urologist performed an abdominal sacrocolpopexy. As the urologist attempted to gain access to the sacral prominence, a tear in the median sacral vein expanded to involve the inferior vena cava and left iliac vein. Massive bleeding occurred and multiple units of blood were transfused. A general surgeon successfully repaired the vascular injuries. The patient was hospitalized for 16 days, received home healthcare, and fully recovered.
PATIENT’S CLAIM The urologist was negligent in overaggressive manipulation of the median sacral vein, causing it to avulse.
PHYSICIAN’S DEFENSE Bleeds of this type are a known complication of the procedure.
VERDICT A Michigan defense verdict was returned.
Was it hypoxia or autism?
AFTER SEVERAL HOURS IN LABOR, a fetal heart-rate monitor indicated decreasing fetal heart rate that led to terminal bradycardia. The ObGyn was called and performed an emergency cesarean delivery. The child was diagnosed with brain damage at 2 years of age.
PARENTS’ CLAIM A cesarean delivery should have been planned because of the fetal weight (8 lb 11 oz). A hypoxic event occurred during labor. Ultrasonography would have shown that the fetus was inverted and that the baby’s face was covered by one of its hands. Delivery was not properly managed, and fetal distress was not reported to the ObGyn in a timely manner.
DEFENDANTS’ DEFENSE The infant’s weight was not sufficient to warrant a cesarean delivery. The infant did not suffer hypoxia. The child’s abnormalities only emerged in the second year of life. An MRI at that time did not indicate brain damage. The child’s development with subsequent regression suggests autism.
VERDICT A New York defense verdict was returned.
Should mammography have been diagnostic?
A 46-YEAR-OLD WOMAN with a family history of breast cancer had regular annual screenings. In December 2006, the patient reported pain, hardness, and burning in her left breast to her gynecologist. A radiologist interpreted the mammography as normal. In May 2007, the patient found a lump in her left breast. Testing indicated she had stage IV breast cancer. She died 2 months after the trial concluded.
PATIENT’S CLAIM The 2006 mammogram was performed as a screening mammography, but should have been diagnostic, considering her family history and reported symptoms. The radiologist improperly interpreted the films.
DEFENDANTS’ DEFENSE The hospital staff testified that the patient did not report pain, hardness, and burning in her left breast when she presented for the 2006 mammography. The radiologist claimed his screening and interpretation were appropriate.
VERDICT The Louisiana court granted the patient’s motion for judgment, and awarded $558,000 in medical costs and $1.3 million in noneconomic damages, totalling $1.808 million. This was reduced to the $500,000 statutory cap.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
AN OBESE WOMAN with a family history of diabetes had previously given birth to a large baby. Even though she expressed her concern that this fetus would also be macrosomic, the ObGyn planned for spontaneous vaginal delivery. At 39 weeks’ gestation, after gaining 60 lb, she went to the hospital requesting induction of labor; the ObGyn reluctantly agreed. Labor was lengthy, forceps-assisted delivery was performed, and a shoulder dystocia was encountered. The baby was born with respiratory distress, a brachial plexus injury, bruises on his right cheek and both ears, and multiple rib fractures. After transfer to a children’s hospital, surgical exploration revealed avulsion of the C6 root nerve from the spinal cord and damage to C5, C7, and C8 nerve roots. Several surgical repairs and physical therapy have led to some improvement, but the child is permanently injured. His right arm is shorter than the left, his right hand is smaller, and he has less strength and range of motion in the right arm. He also has excessive tearing in the right eye and his right eyelid droops.
PARENTS’ CLAIM The ObGyn failed to recognize the risk of delivering a macrosomic baby and did not consider cesarean delivery. The brachial plexus injury was due to downward traction applied during delivery.
PHYSICIAN’S DEFENSE There was no negligence. The brachial plexus injury was not caused by downward traction.
VERDICT A $4.1 million Indiana verdict was returned, but was reduced to the state cap of $1.25 million.
Failure to follow-up on mass: $1.97M verdict
AFTER STAGE II OVARIAN CANCER was found in 1999, a woman underwent surgery and chemotherapy, and was told she was cancer-free. She had regular visits between 2000 and 2008 with another surgical oncologist after her first surgeon moved. In 2004, the oncologist documented finding a round fullness during a pelvic exam. A CT scan confirmed a mass in the pelvic cul-de-sac.
In August 2008, the patient was treated for deep venous thrombosis in her leg. The attending physician saw the pelvic mass on imaging, and a biopsy indicated a recurrence of ovarian cancer. After chemotherapy, the patient underwent surgery, but the tumor was unresectable. In early 2011, testing revealed metastasis to the spine, sternum, pelvic bone, arm, and lung.
PATIENT’S CLAIM The surgeon did not properly investigate the mass resulting in a delayed diagnosis of cancer recurrence. The patient alleged that the surgical oncologist repeatedly stated that the mass had not changed and was most likely fluid; it was nothing to worry about. Radiology reports indicated a suspicion of cancer.
DEFENDANTS’ DEFENSE The oncologist repeatedly told the patient that the mass should be biopsied, but the patient refused because she was dealing with other medical issues. The radiologist argued that reports to the oncologist included everything needed to diagnose the cancer.
VERDICT A Pennsylvania jury found the surgical oncologist fully at fault and returned a $1,971,455 verdict.
Incomplete tubal ligation
BEFORE DELIVERY OF HER THIRD CHILD, a 26-year-old woman requested sterilization using tubal ligation. After delivery, the ObGyn performed a bilateral tubal ligation. The pathologist’s report indicated that the ligation was incomplete: the left fallopian tube had not been fully removed. The ObGyn failed to note the report’s results in the patient’s record, nor did he advise the patient. Two years later, the patient delivered a fourth child.
PATIENT’S CLAIM The patient alleged wrongful birth against both the ObGyn and pathologist. The ObGyn was negligent for not reacting to the pathologist’s report of incomplete tubal ligation, and for not informing the patient. The pathologist should have verbally confirmed receipt of the report with the ObGyn.
PHYSICIANS’ DEFENSE The ObGyn settled before trial. The pathologist claimed he had properly interpreted the specimen and reported the results.
VERDICT A Louisiana jury found the ObGyn fully at fault and assessed additional damages of $56,252 to the $100,000 settlement.
A WOMAN SUFFERED FROM PELVIC PAIN caused by adhesions following two cesarean deliveries and a hysterectomy. In January 2003, her ObGyn performed laparotomy to reduce adhesions from prior surgeries and place Gore-Tex mesh to prevent future adhesions. In October 2010, the patient reported epigastric pain, and went to a different surgeon (her insurance changed). A CT scan identified a foreign body encapsulated in scar tissue in the patient’s lower abdomen/pelvis. The surgeon removed the foreign body.
PATIENT’S CLAIM The ObGyn and hospital were negligent in conducting the 2003 procedure; the foreign object was a retained surgical sponge.
DEFENDANTS’ DEFENSE The foreign body removed in 2010 was the Gore-Tex mesh placed in 2003. The mesh became encapsulated in scar tissue due to the patient’s propensity to develop adhesions, and then moved within the patient’s body. Surgical sponges have embedded radiopaque tracers; CT scans in 2003 and 2010 did not detect any radiopaque tracers.
VERDICT A California defense verdict was returned.
Massive bleed during sacrocolpopexy
AFTER A 72-YEAR-OLD WOMAN developed pelvic organ prolapse, her urologist performed an abdominal sacrocolpopexy. As the urologist attempted to gain access to the sacral prominence, a tear in the median sacral vein expanded to involve the inferior vena cava and left iliac vein. Massive bleeding occurred and multiple units of blood were transfused. A general surgeon successfully repaired the vascular injuries. The patient was hospitalized for 16 days, received home healthcare, and fully recovered.
PATIENT’S CLAIM The urologist was negligent in overaggressive manipulation of the median sacral vein, causing it to avulse.
PHYSICIAN’S DEFENSE Bleeds of this type are a known complication of the procedure.
VERDICT A Michigan defense verdict was returned.
Was it hypoxia or autism?
AFTER SEVERAL HOURS IN LABOR, a fetal heart-rate monitor indicated decreasing fetal heart rate that led to terminal bradycardia. The ObGyn was called and performed an emergency cesarean delivery. The child was diagnosed with brain damage at 2 years of age.
PARENTS’ CLAIM A cesarean delivery should have been planned because of the fetal weight (8 lb 11 oz). A hypoxic event occurred during labor. Ultrasonography would have shown that the fetus was inverted and that the baby’s face was covered by one of its hands. Delivery was not properly managed, and fetal distress was not reported to the ObGyn in a timely manner.
DEFENDANTS’ DEFENSE The infant’s weight was not sufficient to warrant a cesarean delivery. The infant did not suffer hypoxia. The child’s abnormalities only emerged in the second year of life. An MRI at that time did not indicate brain damage. The child’s development with subsequent regression suggests autism.
VERDICT A New York defense verdict was returned.
Should mammography have been diagnostic?
A 46-YEAR-OLD WOMAN with a family history of breast cancer had regular annual screenings. In December 2006, the patient reported pain, hardness, and burning in her left breast to her gynecologist. A radiologist interpreted the mammography as normal. In May 2007, the patient found a lump in her left breast. Testing indicated she had stage IV breast cancer. She died 2 months after the trial concluded.
PATIENT’S CLAIM The 2006 mammogram was performed as a screening mammography, but should have been diagnostic, considering her family history and reported symptoms. The radiologist improperly interpreted the films.
DEFENDANTS’ DEFENSE The hospital staff testified that the patient did not report pain, hardness, and burning in her left breast when she presented for the 2006 mammography. The radiologist claimed his screening and interpretation were appropriate.
VERDICT The Louisiana court granted the patient’s motion for judgment, and awarded $558,000 in medical costs and $1.3 million in noneconomic damages, totalling $1.808 million. This was reduced to the $500,000 statutory cap.
AN OBESE WOMAN with a family history of diabetes had previously given birth to a large baby. Even though she expressed her concern that this fetus would also be macrosomic, the ObGyn planned for spontaneous vaginal delivery. At 39 weeks’ gestation, after gaining 60 lb, she went to the hospital requesting induction of labor; the ObGyn reluctantly agreed. Labor was lengthy, forceps-assisted delivery was performed, and a shoulder dystocia was encountered. The baby was born with respiratory distress, a brachial plexus injury, bruises on his right cheek and both ears, and multiple rib fractures. After transfer to a children’s hospital, surgical exploration revealed avulsion of the C6 root nerve from the spinal cord and damage to C5, C7, and C8 nerve roots. Several surgical repairs and physical therapy have led to some improvement, but the child is permanently injured. His right arm is shorter than the left, his right hand is smaller, and he has less strength and range of motion in the right arm. He also has excessive tearing in the right eye and his right eyelid droops.
PARENTS’ CLAIM The ObGyn failed to recognize the risk of delivering a macrosomic baby and did not consider cesarean delivery. The brachial plexus injury was due to downward traction applied during delivery.
PHYSICIAN’S DEFENSE There was no negligence. The brachial plexus injury was not caused by downward traction.
VERDICT A $4.1 million Indiana verdict was returned, but was reduced to the state cap of $1.25 million.
Failure to follow-up on mass: $1.97M verdict
AFTER STAGE II OVARIAN CANCER was found in 1999, a woman underwent surgery and chemotherapy, and was told she was cancer-free. She had regular visits between 2000 and 2008 with another surgical oncologist after her first surgeon moved. In 2004, the oncologist documented finding a round fullness during a pelvic exam. A CT scan confirmed a mass in the pelvic cul-de-sac.
In August 2008, the patient was treated for deep venous thrombosis in her leg. The attending physician saw the pelvic mass on imaging, and a biopsy indicated a recurrence of ovarian cancer. After chemotherapy, the patient underwent surgery, but the tumor was unresectable. In early 2011, testing revealed metastasis to the spine, sternum, pelvic bone, arm, and lung.
PATIENT’S CLAIM The surgeon did not properly investigate the mass resulting in a delayed diagnosis of cancer recurrence. The patient alleged that the surgical oncologist repeatedly stated that the mass had not changed and was most likely fluid; it was nothing to worry about. Radiology reports indicated a suspicion of cancer.
DEFENDANTS’ DEFENSE The oncologist repeatedly told the patient that the mass should be biopsied, but the patient refused because she was dealing with other medical issues. The radiologist argued that reports to the oncologist included everything needed to diagnose the cancer.
VERDICT A Pennsylvania jury found the surgical oncologist fully at fault and returned a $1,971,455 verdict.
Incomplete tubal ligation
BEFORE DELIVERY OF HER THIRD CHILD, a 26-year-old woman requested sterilization using tubal ligation. After delivery, the ObGyn performed a bilateral tubal ligation. The pathologist’s report indicated that the ligation was incomplete: the left fallopian tube had not been fully removed. The ObGyn failed to note the report’s results in the patient’s record, nor did he advise the patient. Two years later, the patient delivered a fourth child.
PATIENT’S CLAIM The patient alleged wrongful birth against both the ObGyn and pathologist. The ObGyn was negligent for not reacting to the pathologist’s report of incomplete tubal ligation, and for not informing the patient. The pathologist should have verbally confirmed receipt of the report with the ObGyn.
PHYSICIANS’ DEFENSE The ObGyn settled before trial. The pathologist claimed he had properly interpreted the specimen and reported the results.
VERDICT A Louisiana jury found the ObGyn fully at fault and assessed additional damages of $56,252 to the $100,000 settlement.
A WOMAN SUFFERED FROM PELVIC PAIN caused by adhesions following two cesarean deliveries and a hysterectomy. In January 2003, her ObGyn performed laparotomy to reduce adhesions from prior surgeries and place Gore-Tex mesh to prevent future adhesions. In October 2010, the patient reported epigastric pain, and went to a different surgeon (her insurance changed). A CT scan identified a foreign body encapsulated in scar tissue in the patient’s lower abdomen/pelvis. The surgeon removed the foreign body.
PATIENT’S CLAIM The ObGyn and hospital were negligent in conducting the 2003 procedure; the foreign object was a retained surgical sponge.
DEFENDANTS’ DEFENSE The foreign body removed in 2010 was the Gore-Tex mesh placed in 2003. The mesh became encapsulated in scar tissue due to the patient’s propensity to develop adhesions, and then moved within the patient’s body. Surgical sponges have embedded radiopaque tracers; CT scans in 2003 and 2010 did not detect any radiopaque tracers.
VERDICT A California defense verdict was returned.
Massive bleed during sacrocolpopexy
AFTER A 72-YEAR-OLD WOMAN developed pelvic organ prolapse, her urologist performed an abdominal sacrocolpopexy. As the urologist attempted to gain access to the sacral prominence, a tear in the median sacral vein expanded to involve the inferior vena cava and left iliac vein. Massive bleeding occurred and multiple units of blood were transfused. A general surgeon successfully repaired the vascular injuries. The patient was hospitalized for 16 days, received home healthcare, and fully recovered.
PATIENT’S CLAIM The urologist was negligent in overaggressive manipulation of the median sacral vein, causing it to avulse.
PHYSICIAN’S DEFENSE Bleeds of this type are a known complication of the procedure.
VERDICT A Michigan defense verdict was returned.
Was it hypoxia or autism?
AFTER SEVERAL HOURS IN LABOR, a fetal heart-rate monitor indicated decreasing fetal heart rate that led to terminal bradycardia. The ObGyn was called and performed an emergency cesarean delivery. The child was diagnosed with brain damage at 2 years of age.
PARENTS’ CLAIM A cesarean delivery should have been planned because of the fetal weight (8 lb 11 oz). A hypoxic event occurred during labor. Ultrasonography would have shown that the fetus was inverted and that the baby’s face was covered by one of its hands. Delivery was not properly managed, and fetal distress was not reported to the ObGyn in a timely manner.
DEFENDANTS’ DEFENSE The infant’s weight was not sufficient to warrant a cesarean delivery. The infant did not suffer hypoxia. The child’s abnormalities only emerged in the second year of life. An MRI at that time did not indicate brain damage. The child’s development with subsequent regression suggests autism.
VERDICT A New York defense verdict was returned.
Should mammography have been diagnostic?
A 46-YEAR-OLD WOMAN with a family history of breast cancer had regular annual screenings. In December 2006, the patient reported pain, hardness, and burning in her left breast to her gynecologist. A radiologist interpreted the mammography as normal. In May 2007, the patient found a lump in her left breast. Testing indicated she had stage IV breast cancer. She died 2 months after the trial concluded.
PATIENT’S CLAIM The 2006 mammogram was performed as a screening mammography, but should have been diagnostic, considering her family history and reported symptoms. The radiologist improperly interpreted the films.
DEFENDANTS’ DEFENSE The hospital staff testified that the patient did not report pain, hardness, and burning in her left breast when she presented for the 2006 mammography. The radiologist claimed his screening and interpretation were appropriate.
VERDICT The Louisiana court granted the patient’s motion for judgment, and awarded $558,000 in medical costs and $1.3 million in noneconomic damages, totalling $1.808 million. This was reduced to the $500,000 statutory cap.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Four pillars of a successful practice: 1. Keep your current patients happy
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
The medical landscape has changed. No one is quite certain what the future will hold. One thing we do know: 20 million more Americans, half of them women, will enter the health-care system in the very near future, as the Affordable Care Act continues to unfold. This flood of new patients will affect nearly every ObGyn practice in the country because we lack an adequate increase in the number of physicians to care for them. In the meantime, what can you do to ensure the continued success of your practice? This series focuses on four key areas. I call them the “four pillars”:
- Keep existing patients happy
- Attract new patients
- Nurture relationships with your medical colleagues and other health-care providers, such as physician assistants and nurse practitioners
- Maintain the morale of your staff.
No pillar is more important than the others; all four are necessary to guarantee success.
It is more cost-effective to keep an existing customer than to attract
a new one
In this article, I explain why it is vital to ensure that every patient has a positive experience of your practice, from the moment they make their first telephone call for an appointment, through their wait in the reception area, to the moment they are seen by the staff or the physician, and beyond—when they tell others about their stellar experience.
It’s nice to get new patients, but it is more important to keep the ones you already have. In most professions and businesses, the cost of keeping an established customer is one-fifth the cost of acquiring a new one. Medical practices are no exception. If you are not doing a good job with the patients you already have, spending thousands of dollars on a marketing plan to bring in new patients is pointless. The patients you have right now are the backbone of your practice.
Give your practice a checkup
Look at your practice from your patients’ perspective
Today, it is critical to know the needs and expectations of your patients and referring physicians. The best way to do this is to ask your patients what they think, which also will reveal your practice’s strengths and weaknesses. This information can be obtained easily by surveying patients about various aspects of your practice. (Even practices that are full or closed need to evaluate their services periodically and listen to their patients. Changes always occur, and the cup may not “runneth over” forever.)
Tom Peters, the nationally renowned author of In Search of Excellence, has described two keys to success in business:
- Find out what the customer (patient) wants and give him or her more of it
- Find out what the customer (patient) does not want and be sure to avoid it.
Techniques to gather the patient’s perspective
There are five effective techniques for determining how patients perceive your practice and for evaluating your performance and reputation:
- Conduct personal interviews
- Conduct patient surveys
- Create a focus group
- Use a suggestion box
- Commission evaluation by a mystery shopper.
Although I have used all five techniques, my favorite and most cost-effective involves a survey card that is given to every patient on every office visit (FIGURE 1). The card is given to the patient when she checks in, and she can complete it in the reception area or exam room and return it to the receptionist before leaving the practice. The cards are reviewed by a nurse, who addresses positive and negative comments. Most negative comments are addressed with a phone call. If necessary, I respond to the patient’s complaint.
FIGURE 1: Patient survey card
There is another benefit to the survey card. The flip side of the card prompts the patient to write down the three questions she would like you to address on her current visit to the office (FIGURE 2). Conducting the appointment according to these concerns can help keep patients from initiating a last-minute discussion, often after you have closed the chart or electronic medical record, that you don’t have time to address adequately. Since my office has implemented use of this survey card, we seldom get follow-up phone calls from patients about issues they forgot to ask about. The survey card demonstrates that we are listening to the patient and want to be certain that all her questions have been answered at the time of her office visit.

FIGURE 2: Focus of the appointment
Develop an on-time practice philosophy
You know the adage: Timeliness is next to godliness. (Actually, it’s cleanliness that’s next to godliness, but timeliness is vital, too.) The most common complaint patients have about the health-care experience is “waiting for the doctor.” Spending time in the reception area probably accounts for more patient dissatisfaction than any other aspect of medical care. In one recent survey, nearly one in four patients (24%) claimed to have waited 30 minutes or longer. With so many more women entering the marketplace, this statistic is only going to get worse.
In order to gain an accurate picture of what is happening in regard to the schedule in your practice, I suggest conducting a “time and motion” study. For a period of 3 to 5 days, place a sheet on each patient’s record or superbill and log in the following:
- time of her appointment
- time she arrived
- time she left the office
- how much time she spent with her physician.
You will be amazed to discover that patients are waiting 1 or 2 hours or longer to see the physician, and that the physician is spending only 5 minutes with the patient. Ask any patient on a survey if she feels she is getting bang for her buck, and she will answer, “No!”
By conducting a time and motion study, you will discover that there are predictable periods when backlogs occur. Often, these delays are the result of “working a patient in” to the schedule. Unscheduled patients who call to report vaginal bleeding, pelvic pain, urinary tract infection, or another problem are often told to come in without an appointment, but they inevitably displace women already scheduled and delay their visit. This problem affects almost every ObGyn.
One way to avoid this scenario is to create “sacred” time slots. These are 15-minute intervals at the end of the morning or afternoon in which unscheduled patients can be worked into the round of visits. Instead of telling the patient to “just come in,” I tell her to report at a specific time. These time slots cannot be filled with routine appointments. Nor can they be filled prior to 9 am each day. This leaves two or three slots open for patients who must be seen immediately.
Few ObGyns can change health-care policy. But all of us can be more sensitive to our patients’ time and make an effort to see them as soon as possible, thereby eliminating one of patients’ most common complaints: the long wait to see their doctor.
Make the patient’s experience memorable
All of us can provide a diagnosis and treatment strategy for most women’s medical problems. But how many of us can make the experience memorable for the woman? Often, it is a few little things that can be easily and inexpensively performed that make a big difference.
Go with cloth, not paper. There’s a sharp contrast between a paper sheet, a paper gown, and a soft robe. You don’t step out of the shower in a fine hotel and put on a paper robe. If you are offering five-star service, you need to offer five-star amenities. If you want to attract special patients, treat your patients special. It doesn’t cost that much to add a few dozen robes to your office supplies, laundering them after each use and placing them on hangers or in a plastic bag that each patient can use during her visit. I can assure you that this single idea will set you apart from most other ObGyns in your community.
Stirrups are cold! Here’s another idea: Use pads to cover the metal of the stirrups for the pelvic exam. Those stirrups are cold steel, and no woman who is already naked and concerned about her dignity wants to place her feet on those chilly structures. You can have lamb’s wool pads created by a seamstress for a few dollars—or if you prefer to go low-tech, you can use potholders to cover the stirrups.
Warm the speculum. My wife shared with me how uncomfortable it is to have a metal speculum inserted and how much she appreciated her gynecologist warming the instrument with tap water before its insertion. I have found that this saves on the use of lubricant jelly, too, because the water serves as a lubricant!
Keep the temperature in mind. Most medical offices are kept at 70° to 72° F to keep the doctor and staff comfortable. However, when a woman puts on her gown or robe, she often becomes cold and uncomfortable. On days when it is cold outside or the office is cold, use an inexpensive heater to make the room comfortable for the patient.
Talk to your patient “eyeball to eyeball.” You make big points with your patient if you speak to her when she is fully dressed and when your eyes are at the same level as hers. A woman lying on her back in a gown or robe does not hear or recall what her doctor is telling her. However, if the doctor and nurse leave the room and allow the patient to get dressed, and if the doctor sits with the patient without barriers between them, she is far more likely to listen and recall what has been discussed.
Pick up the telephone. I am often asked for my “best” idea to keep patients happy. My numero uno suggestion is to take a few minutes to call the patient at home. Which patients should you call? Women undergoing outpatient studies or procedures, those recently discharged from the hospital, and those who require a little more hand holding and attention. You can be sure that every patient who undergoes a procedure or is discharged from the hospital has questions about the findings, any precautions, medications, and follow-up. A call from a nurse or doctor does a lot to allay her apprehension—and it often keeps the patient from calling the office with her questions and concerns.
My nurse identifies key patients and contacts them at the end of the workday. She is usually able to answer all the questions but may identify two or three that require my attention. She tells the patient what time I will call so she can keep the phone line free.
Calling patients usually takes no more than 5 to 10 minutes a day and provides me with great satisfaction. Patients are usually shocked—and happy—that their physician is calling them at home. The advantages of this strategy include:
- fewer calls from your patients
- more efficient use of your time
- deep appreciation by the patient.
One patient I called at home wrote me a note that I think is worth mentioning: “This is the first time a member of your profession has taken the time to call me at home and check on my condition. Undoubtedly, it will foster a better relationship between you and me.”
Results of an informal poll indicate the answer is mostly “Yes”
As outlined in the article by Neil Baum, MD, the need to keep existing patients happy—and to determine how they’re feeling about your practice—seems as though it should occupy a berth rather high on your list of priorities. To gauge how widespread the practice of measuring patient satisfaction is among ObGyns, we polled our Virtual Board of Editors (VBE) on the subject. Because these physicians range from private practitioners to academic professionals and hospital employees, we find them to be one bellwether of wider practice patterns.
When we asked, 65% of our VBE members reported that they regularly measure the satisfaction of their patients. Among the reasons given for this tactic were corporate policy, but the vast majority of respondents indicated that they “need to know what patients like and don’t like” in order to “improve our services.” As one physician noted, “all practices can improve in some respects.” Regular inquiries about patient satisfaction provide a method and rationale for improvement.
Another respondent observed that the information gained from assessments of patient satisfaction is useful during insurance contracting. Another said, simply, “It’s the right thing to do.”
When asked exactly how they measure patient satisfaction, almost 60% of respondents who regularly assess this component of practice said they use surveys to do so, compared with 14% who use interviews, 17% who make a suggestion box available, and 11% who employ a “mystery shopper” (The percentages add up to more than 100% because some VBE members employ more than one approach.) None of the VBE members reported convening a focus group.
When asked to rate the importance of patient-satisfaction assessments, just over half of all respondents characterized it as “very important.” Only one physician reported that the practice of measuring patient satisfaction is “not important.”
“It is critical—especially for doctors with younger practices who are trying to build a reputation or practice—to get feedback to improve care and increase their patient load,” noted one respondent. Another reported: “We do a detailed satisfaction survey after every surgery on every patient. We actively seek feedback and use our Web site and social media to find ways to improve.”
“We are in a large city (over 500,000 population), so there is competition, and patients have choices,” wrote another VBE member. “In smaller communities, patients may have fewer options and have to accept the few available providers. My basic method of achieving patient satisfaction is outdated: I spend a lot of time per patient, see few patients per day, and try to help with whatever issues they have (not just breast and pelvic). I make less money practicing this way—I accepted a long time ago that really caring for patients means spending more time and being paid less for it.”
Another VBE member said, “I promise what I’ll do for patients, and I keep that promise! I use the telephone as a tool. Patients are very impressed and thankful when I talk to them about their problems and test results.”
—Janelle Yates, Senior Editor
The bottom line
Word of mouth was the time-honored method of attracting new patients for thousands of years. That method still works today. Ensuring that your patients have an outstanding experience during their visit is one of the smartest strategies to market and promote your practice.
We want to hear from you! Tell us what you think.
CLICK HERE to access 10 recent articles on managing your ObGyn practice.
| Dr. Baum describes his number one strategy to retain patients |

Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
| Dr. Baum describes his number one strategy to retain patients |
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
| Dr. Baum describes his number one strategy to retain patients |
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
The medical landscape has changed. No one is quite certain what the future will hold. One thing we do know: 20 million more Americans, half of them women, will enter the health-care system in the very near future, as the Affordable Care Act continues to unfold. This flood of new patients will affect nearly every ObGyn practice in the country because we lack an adequate increase in the number of physicians to care for them. In the meantime, what can you do to ensure the continued success of your practice? This series focuses on four key areas. I call them the “four pillars”:
- Keep existing patients happy
- Attract new patients
- Nurture relationships with your medical colleagues and other health-care providers, such as physician assistants and nurse practitioners
- Maintain the morale of your staff.
No pillar is more important than the others; all four are necessary to guarantee success.
It is more cost-effective to keep an existing customer than to attract
a new one
In this article, I explain why it is vital to ensure that every patient has a positive experience of your practice, from the moment they make their first telephone call for an appointment, through their wait in the reception area, to the moment they are seen by the staff or the physician, and beyond—when they tell others about their stellar experience.
It’s nice to get new patients, but it is more important to keep the ones you already have. In most professions and businesses, the cost of keeping an established customer is one-fifth the cost of acquiring a new one. Medical practices are no exception. If you are not doing a good job with the patients you already have, spending thousands of dollars on a marketing plan to bring in new patients is pointless. The patients you have right now are the backbone of your practice.
Give your practice a checkup
Look at your practice from your patients’ perspective
Today, it is critical to know the needs and expectations of your patients and referring physicians. The best way to do this is to ask your patients what they think, which also will reveal your practice’s strengths and weaknesses. This information can be obtained easily by surveying patients about various aspects of your practice. (Even practices that are full or closed need to evaluate their services periodically and listen to their patients. Changes always occur, and the cup may not “runneth over” forever.)
Tom Peters, the nationally renowned author of In Search of Excellence, has described two keys to success in business:
- Find out what the customer (patient) wants and give him or her more of it
- Find out what the customer (patient) does not want and be sure to avoid it.
Techniques to gather the patient’s perspective
There are five effective techniques for determining how patients perceive your practice and for evaluating your performance and reputation:
- Conduct personal interviews
- Conduct patient surveys
- Create a focus group
- Use a suggestion box
- Commission evaluation by a mystery shopper.
Although I have used all five techniques, my favorite and most cost-effective involves a survey card that is given to every patient on every office visit (FIGURE 1). The card is given to the patient when she checks in, and she can complete it in the reception area or exam room and return it to the receptionist before leaving the practice. The cards are reviewed by a nurse, who addresses positive and negative comments. Most negative comments are addressed with a phone call. If necessary, I respond to the patient’s complaint.
FIGURE 1: Patient survey card
There is another benefit to the survey card. The flip side of the card prompts the patient to write down the three questions she would like you to address on her current visit to the office (FIGURE 2). Conducting the appointment according to these concerns can help keep patients from initiating a last-minute discussion, often after you have closed the chart or electronic medical record, that you don’t have time to address adequately. Since my office has implemented use of this survey card, we seldom get follow-up phone calls from patients about issues they forgot to ask about. The survey card demonstrates that we are listening to the patient and want to be certain that all her questions have been answered at the time of her office visit.
FIGURE 2: Focus of the appointment
Develop an on-time practice philosophy
You know the adage: Timeliness is next to godliness. (Actually, it’s cleanliness that’s next to godliness, but timeliness is vital, too.) The most common complaint patients have about the health-care experience is “waiting for the doctor.” Spending time in the reception area probably accounts for more patient dissatisfaction than any other aspect of medical care. In one recent survey, nearly one in four patients (24%) claimed to have waited 30 minutes or longer. With so many more women entering the marketplace, this statistic is only going to get worse.
In order to gain an accurate picture of what is happening in regard to the schedule in your practice, I suggest conducting a “time and motion” study. For a period of 3 to 5 days, place a sheet on each patient’s record or superbill and log in the following:
- time of her appointment
- time she arrived
- time she left the office
- how much time she spent with her physician.
You will be amazed to discover that patients are waiting 1 or 2 hours or longer to see the physician, and that the physician is spending only 5 minutes with the patient. Ask any patient on a survey if she feels she is getting bang for her buck, and she will answer, “No!”
By conducting a time and motion study, you will discover that there are predictable periods when backlogs occur. Often, these delays are the result of “working a patient in” to the schedule. Unscheduled patients who call to report vaginal bleeding, pelvic pain, urinary tract infection, or another problem are often told to come in without an appointment, but they inevitably displace women already scheduled and delay their visit. This problem affects almost every ObGyn.
One way to avoid this scenario is to create “sacred” time slots. These are 15-minute intervals at the end of the morning or afternoon in which unscheduled patients can be worked into the round of visits. Instead of telling the patient to “just come in,” I tell her to report at a specific time. These time slots cannot be filled with routine appointments. Nor can they be filled prior to 9 am each day. This leaves two or three slots open for patients who must be seen immediately.
Few ObGyns can change health-care policy. But all of us can be more sensitive to our patients’ time and make an effort to see them as soon as possible, thereby eliminating one of patients’ most common complaints: the long wait to see their doctor.
Make the patient’s experience memorable
All of us can provide a diagnosis and treatment strategy for most women’s medical problems. But how many of us can make the experience memorable for the woman? Often, it is a few little things that can be easily and inexpensively performed that make a big difference.
Go with cloth, not paper. There’s a sharp contrast between a paper sheet, a paper gown, and a soft robe. You don’t step out of the shower in a fine hotel and put on a paper robe. If you are offering five-star service, you need to offer five-star amenities. If you want to attract special patients, treat your patients special. It doesn’t cost that much to add a few dozen robes to your office supplies, laundering them after each use and placing them on hangers or in a plastic bag that each patient can use during her visit. I can assure you that this single idea will set you apart from most other ObGyns in your community.
Stirrups are cold! Here’s another idea: Use pads to cover the metal of the stirrups for the pelvic exam. Those stirrups are cold steel, and no woman who is already naked and concerned about her dignity wants to place her feet on those chilly structures. You can have lamb’s wool pads created by a seamstress for a few dollars—or if you prefer to go low-tech, you can use potholders to cover the stirrups.
Warm the speculum. My wife shared with me how uncomfortable it is to have a metal speculum inserted and how much she appreciated her gynecologist warming the instrument with tap water before its insertion. I have found that this saves on the use of lubricant jelly, too, because the water serves as a lubricant!
Keep the temperature in mind. Most medical offices are kept at 70° to 72° F to keep the doctor and staff comfortable. However, when a woman puts on her gown or robe, she often becomes cold and uncomfortable. On days when it is cold outside or the office is cold, use an inexpensive heater to make the room comfortable for the patient.
Talk to your patient “eyeball to eyeball.” You make big points with your patient if you speak to her when she is fully dressed and when your eyes are at the same level as hers. A woman lying on her back in a gown or robe does not hear or recall what her doctor is telling her. However, if the doctor and nurse leave the room and allow the patient to get dressed, and if the doctor sits with the patient without barriers between them, she is far more likely to listen and recall what has been discussed.
Pick up the telephone. I am often asked for my “best” idea to keep patients happy. My numero uno suggestion is to take a few minutes to call the patient at home. Which patients should you call? Women undergoing outpatient studies or procedures, those recently discharged from the hospital, and those who require a little more hand holding and attention. You can be sure that every patient who undergoes a procedure or is discharged from the hospital has questions about the findings, any precautions, medications, and follow-up. A call from a nurse or doctor does a lot to allay her apprehension—and it often keeps the patient from calling the office with her questions and concerns.
My nurse identifies key patients and contacts them at the end of the workday. She is usually able to answer all the questions but may identify two or three that require my attention. She tells the patient what time I will call so she can keep the phone line free.
Calling patients usually takes no more than 5 to 10 minutes a day and provides me with great satisfaction. Patients are usually shocked—and happy—that their physician is calling them at home. The advantages of this strategy include:
- fewer calls from your patients
- more efficient use of your time
- deep appreciation by the patient.
One patient I called at home wrote me a note that I think is worth mentioning: “This is the first time a member of your profession has taken the time to call me at home and check on my condition. Undoubtedly, it will foster a better relationship between you and me.”
Results of an informal poll indicate the answer is mostly “Yes”
As outlined in the article by Neil Baum, MD, the need to keep existing patients happy—and to determine how they’re feeling about your practice—seems as though it should occupy a berth rather high on your list of priorities. To gauge how widespread the practice of measuring patient satisfaction is among ObGyns, we polled our Virtual Board of Editors (VBE) on the subject. Because these physicians range from private practitioners to academic professionals and hospital employees, we find them to be one bellwether of wider practice patterns.
When we asked, 65% of our VBE members reported that they regularly measure the satisfaction of their patients. Among the reasons given for this tactic were corporate policy, but the vast majority of respondents indicated that they “need to know what patients like and don’t like” in order to “improve our services.” As one physician noted, “all practices can improve in some respects.” Regular inquiries about patient satisfaction provide a method and rationale for improvement.
Another respondent observed that the information gained from assessments of patient satisfaction is useful during insurance contracting. Another said, simply, “It’s the right thing to do.”
When asked exactly how they measure patient satisfaction, almost 60% of respondents who regularly assess this component of practice said they use surveys to do so, compared with 14% who use interviews, 17% who make a suggestion box available, and 11% who employ a “mystery shopper” (The percentages add up to more than 100% because some VBE members employ more than one approach.) None of the VBE members reported convening a focus group.
When asked to rate the importance of patient-satisfaction assessments, just over half of all respondents characterized it as “very important.” Only one physician reported that the practice of measuring patient satisfaction is “not important.”
“It is critical—especially for doctors with younger practices who are trying to build a reputation or practice—to get feedback to improve care and increase their patient load,” noted one respondent. Another reported: “We do a detailed satisfaction survey after every surgery on every patient. We actively seek feedback and use our Web site and social media to find ways to improve.”
“We are in a large city (over 500,000 population), so there is competition, and patients have choices,” wrote another VBE member. “In smaller communities, patients may have fewer options and have to accept the few available providers. My basic method of achieving patient satisfaction is outdated: I spend a lot of time per patient, see few patients per day, and try to help with whatever issues they have (not just breast and pelvic). I make less money practicing this way—I accepted a long time ago that really caring for patients means spending more time and being paid less for it.”
Another VBE member said, “I promise what I’ll do for patients, and I keep that promise! I use the telephone as a tool. Patients are very impressed and thankful when I talk to them about their problems and test results.”
—Janelle Yates, Senior Editor
The bottom line
Word of mouth was the time-honored method of attracting new patients for thousands of years. That method still works today. Ensuring that your patients have an outstanding experience during their visit is one of the smartest strategies to market and promote your practice.
We want to hear from you! Tell us what you think.
CLICK HERE to access 10 recent articles on managing your ObGyn practice.
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 3: Obtain and maintain physician referrals (June 2013)
Pillar 4: Motivate your staff (August 2013)
The medical landscape has changed. No one is quite certain what the future will hold. One thing we do know: 20 million more Americans, half of them women, will enter the health-care system in the very near future, as the Affordable Care Act continues to unfold. This flood of new patients will affect nearly every ObGyn practice in the country because we lack an adequate increase in the number of physicians to care for them. In the meantime, what can you do to ensure the continued success of your practice? This series focuses on four key areas. I call them the “four pillars”:
- Keep existing patients happy
- Attract new patients
- Nurture relationships with your medical colleagues and other health-care providers, such as physician assistants and nurse practitioners
- Maintain the morale of your staff.
No pillar is more important than the others; all four are necessary to guarantee success.
It is more cost-effective to keep an existing customer than to attract
a new one
In this article, I explain why it is vital to ensure that every patient has a positive experience of your practice, from the moment they make their first telephone call for an appointment, through their wait in the reception area, to the moment they are seen by the staff or the physician, and beyond—when they tell others about their stellar experience.
It’s nice to get new patients, but it is more important to keep the ones you already have. In most professions and businesses, the cost of keeping an established customer is one-fifth the cost of acquiring a new one. Medical practices are no exception. If you are not doing a good job with the patients you already have, spending thousands of dollars on a marketing plan to bring in new patients is pointless. The patients you have right now are the backbone of your practice.
Give your practice a checkup
Look at your practice from your patients’ perspective
Today, it is critical to know the needs and expectations of your patients and referring physicians. The best way to do this is to ask your patients what they think, which also will reveal your practice’s strengths and weaknesses. This information can be obtained easily by surveying patients about various aspects of your practice. (Even practices that are full or closed need to evaluate their services periodically and listen to their patients. Changes always occur, and the cup may not “runneth over” forever.)
Tom Peters, the nationally renowned author of In Search of Excellence, has described two keys to success in business:
- Find out what the customer (patient) wants and give him or her more of it
- Find out what the customer (patient) does not want and be sure to avoid it.
Techniques to gather the patient’s perspective
There are five effective techniques for determining how patients perceive your practice and for evaluating your performance and reputation:
- Conduct personal interviews
- Conduct patient surveys
- Create a focus group
- Use a suggestion box
- Commission evaluation by a mystery shopper.
Although I have used all five techniques, my favorite and most cost-effective involves a survey card that is given to every patient on every office visit (FIGURE 1). The card is given to the patient when she checks in, and she can complete it in the reception area or exam room and return it to the receptionist before leaving the practice. The cards are reviewed by a nurse, who addresses positive and negative comments. Most negative comments are addressed with a phone call. If necessary, I respond to the patient’s complaint.
FIGURE 1: Patient survey card
There is another benefit to the survey card. The flip side of the card prompts the patient to write down the three questions she would like you to address on her current visit to the office (FIGURE 2). Conducting the appointment according to these concerns can help keep patients from initiating a last-minute discussion, often after you have closed the chart or electronic medical record, that you don’t have time to address adequately. Since my office has implemented use of this survey card, we seldom get follow-up phone calls from patients about issues they forgot to ask about. The survey card demonstrates that we are listening to the patient and want to be certain that all her questions have been answered at the time of her office visit.
FIGURE 2: Focus of the appointment
Develop an on-time practice philosophy
You know the adage: Timeliness is next to godliness. (Actually, it’s cleanliness that’s next to godliness, but timeliness is vital, too.) The most common complaint patients have about the health-care experience is “waiting for the doctor.” Spending time in the reception area probably accounts for more patient dissatisfaction than any other aspect of medical care. In one recent survey, nearly one in four patients (24%) claimed to have waited 30 minutes or longer. With so many more women entering the marketplace, this statistic is only going to get worse.
In order to gain an accurate picture of what is happening in regard to the schedule in your practice, I suggest conducting a “time and motion” study. For a period of 3 to 5 days, place a sheet on each patient’s record or superbill and log in the following:
- time of her appointment
- time she arrived
- time she left the office
- how much time she spent with her physician.
You will be amazed to discover that patients are waiting 1 or 2 hours or longer to see the physician, and that the physician is spending only 5 minutes with the patient. Ask any patient on a survey if she feels she is getting bang for her buck, and she will answer, “No!”
By conducting a time and motion study, you will discover that there are predictable periods when backlogs occur. Often, these delays are the result of “working a patient in” to the schedule. Unscheduled patients who call to report vaginal bleeding, pelvic pain, urinary tract infection, or another problem are often told to come in without an appointment, but they inevitably displace women already scheduled and delay their visit. This problem affects almost every ObGyn.
One way to avoid this scenario is to create “sacred” time slots. These are 15-minute intervals at the end of the morning or afternoon in which unscheduled patients can be worked into the round of visits. Instead of telling the patient to “just come in,” I tell her to report at a specific time. These time slots cannot be filled with routine appointments. Nor can they be filled prior to 9 am each day. This leaves two or three slots open for patients who must be seen immediately.
Few ObGyns can change health-care policy. But all of us can be more sensitive to our patients’ time and make an effort to see them as soon as possible, thereby eliminating one of patients’ most common complaints: the long wait to see their doctor.
Make the patient’s experience memorable
All of us can provide a diagnosis and treatment strategy for most women’s medical problems. But how many of us can make the experience memorable for the woman? Often, it is a few little things that can be easily and inexpensively performed that make a big difference.
Go with cloth, not paper. There’s a sharp contrast between a paper sheet, a paper gown, and a soft robe. You don’t step out of the shower in a fine hotel and put on a paper robe. If you are offering five-star service, you need to offer five-star amenities. If you want to attract special patients, treat your patients special. It doesn’t cost that much to add a few dozen robes to your office supplies, laundering them after each use and placing them on hangers or in a plastic bag that each patient can use during her visit. I can assure you that this single idea will set you apart from most other ObGyns in your community.
Stirrups are cold! Here’s another idea: Use pads to cover the metal of the stirrups for the pelvic exam. Those stirrups are cold steel, and no woman who is already naked and concerned about her dignity wants to place her feet on those chilly structures. You can have lamb’s wool pads created by a seamstress for a few dollars—or if you prefer to go low-tech, you can use potholders to cover the stirrups.
Warm the speculum. My wife shared with me how uncomfortable it is to have a metal speculum inserted and how much she appreciated her gynecologist warming the instrument with tap water before its insertion. I have found that this saves on the use of lubricant jelly, too, because the water serves as a lubricant!
Keep the temperature in mind. Most medical offices are kept at 70° to 72° F to keep the doctor and staff comfortable. However, when a woman puts on her gown or robe, she often becomes cold and uncomfortable. On days when it is cold outside or the office is cold, use an inexpensive heater to make the room comfortable for the patient.
Talk to your patient “eyeball to eyeball.” You make big points with your patient if you speak to her when she is fully dressed and when your eyes are at the same level as hers. A woman lying on her back in a gown or robe does not hear or recall what her doctor is telling her. However, if the doctor and nurse leave the room and allow the patient to get dressed, and if the doctor sits with the patient without barriers between them, she is far more likely to listen and recall what has been discussed.
Pick up the telephone. I am often asked for my “best” idea to keep patients happy. My numero uno suggestion is to take a few minutes to call the patient at home. Which patients should you call? Women undergoing outpatient studies or procedures, those recently discharged from the hospital, and those who require a little more hand holding and attention. You can be sure that every patient who undergoes a procedure or is discharged from the hospital has questions about the findings, any precautions, medications, and follow-up. A call from a nurse or doctor does a lot to allay her apprehension—and it often keeps the patient from calling the office with her questions and concerns.
My nurse identifies key patients and contacts them at the end of the workday. She is usually able to answer all the questions but may identify two or three that require my attention. She tells the patient what time I will call so she can keep the phone line free.
Calling patients usually takes no more than 5 to 10 minutes a day and provides me with great satisfaction. Patients are usually shocked—and happy—that their physician is calling them at home. The advantages of this strategy include:
- fewer calls from your patients
- more efficient use of your time
- deep appreciation by the patient.
One patient I called at home wrote me a note that I think is worth mentioning: “This is the first time a member of your profession has taken the time to call me at home and check on my condition. Undoubtedly, it will foster a better relationship between you and me.”
Results of an informal poll indicate the answer is mostly “Yes”
As outlined in the article by Neil Baum, MD, the need to keep existing patients happy—and to determine how they’re feeling about your practice—seems as though it should occupy a berth rather high on your list of priorities. To gauge how widespread the practice of measuring patient satisfaction is among ObGyns, we polled our Virtual Board of Editors (VBE) on the subject. Because these physicians range from private practitioners to academic professionals and hospital employees, we find them to be one bellwether of wider practice patterns.
When we asked, 65% of our VBE members reported that they regularly measure the satisfaction of their patients. Among the reasons given for this tactic were corporate policy, but the vast majority of respondents indicated that they “need to know what patients like and don’t like” in order to “improve our services.” As one physician noted, “all practices can improve in some respects.” Regular inquiries about patient satisfaction provide a method and rationale for improvement.
Another respondent observed that the information gained from assessments of patient satisfaction is useful during insurance contracting. Another said, simply, “It’s the right thing to do.”
When asked exactly how they measure patient satisfaction, almost 60% of respondents who regularly assess this component of practice said they use surveys to do so, compared with 14% who use interviews, 17% who make a suggestion box available, and 11% who employ a “mystery shopper” (The percentages add up to more than 100% because some VBE members employ more than one approach.) None of the VBE members reported convening a focus group.
When asked to rate the importance of patient-satisfaction assessments, just over half of all respondents characterized it as “very important.” Only one physician reported that the practice of measuring patient satisfaction is “not important.”
“It is critical—especially for doctors with younger practices who are trying to build a reputation or practice—to get feedback to improve care and increase their patient load,” noted one respondent. Another reported: “We do a detailed satisfaction survey after every surgery on every patient. We actively seek feedback and use our Web site and social media to find ways to improve.”
“We are in a large city (over 500,000 population), so there is competition, and patients have choices,” wrote another VBE member. “In smaller communities, patients may have fewer options and have to accept the few available providers. My basic method of achieving patient satisfaction is outdated: I spend a lot of time per patient, see few patients per day, and try to help with whatever issues they have (not just breast and pelvic). I make less money practicing this way—I accepted a long time ago that really caring for patients means spending more time and being paid less for it.”
Another VBE member said, “I promise what I’ll do for patients, and I keep that promise! I use the telephone as a tool. Patients are very impressed and thankful when I talk to them about their problems and test results.”
—Janelle Yates, Senior Editor
The bottom line
Word of mouth was the time-honored method of attracting new patients for thousands of years. That method still works today. Ensuring that your patients have an outstanding experience during their visit is one of the smartest strategies to market and promote your practice.
We want to hear from you! Tell us what you think.
CLICK HERE to access 10 recent articles on managing your ObGyn practice.
Genetics of schizophrenia: What do we know?
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
Emergency brain imaging: CT or MRI?
Discuss this article at www.facebook.com/CurrentPsychiatry
Together with a clinical assessment, neuroimaging increases diagnostic accuracy of detecting neuropathology. Direct patient benefit from scanning is best documented in those with overt, new clinical signs and symptoms of neurologic or psychiatric disease.1,2
Computerized tomography (CT) and magnetic resonance imaging (MRI) are the most common head scanning techniques used in emergency medicine.3 CT is quicker and cheaper, has less movement artifact, and is excellent at delineating acute hemorrhage, calcification, and bony anatomy.3,4 Unfortunately, CT exposes patients to radiation and poorly visualizes white matter or posterior fossa pathology.4
MRI is outstanding for well-defined tissue contrast in multiplanar views and excellent for identifying demyelination or metastatic processes,5 but may be contraindicated for patients with implanted metallic objects such as pacemakers, certain vascular clips or stents, and certain orthopedic devices.3-5 Some patients cannot tolerate the narrow space surrounding them during an MRI.4,5
Safety concerns with CT during pregnancy are well established, but are less clear with MRI. The opposite is true of contrast enhancement; gadolinium with MRI is better tolerated than CT procedures, for which contrast risks include allergy and renal dysfunction. When scanning for a hemorrhage, select a CT scan for patients in whom you suspect bleeding developed within the past 3 days; MRI may be better at screening for older bleeds.
For a list of indications for which a patient should undergo a CT or MRI, see the Table.1-5
Table
Indications for CT or MRI
| New or first-onset psychiatric illness |
| Recent head trauma |
| Recent or advancing cognitive dysfunction |
| New or worsening instances of syncope, vertigo, loss of consciousness, etc. |
| New, worsening, or altered pattern headaches |
| New signs of brain pathology, eg, seizure, paresis, or brain-related visual alteration |
| New neurologic examination abnormalities |
| Concerns about intracranial infection, inflammation, metastases, or increased pressure |
| Change in mental status in persons age >50 |
| Prescreening patients who are candidates for electroconvulsive therapy |
| Source: References 1-5 |
Disclosure
Dr. Lippmann reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pary R, Lippmann S. Clinical review of head CT scans in psychiatric patients. VA Practitioner. 1986;3:48-53.
2. Capote HA. Neuroimaging in psychiatry. Neurol Clin. 2009;27(1):237-249.
3. Malhi GS, Lagopoulos J. Making sense of neuroimaging in psychiatry. Acta Psychiatr Scand. 2008;117(2):100-117.
4. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172.
5. Broderick DF. Neuroimaging in neuropsychiatry. Psychiatr Clin North Am. 2005;28(3):549-566,64.
Discuss this article at www.facebook.com/CurrentPsychiatry
Together with a clinical assessment, neuroimaging increases diagnostic accuracy of detecting neuropathology. Direct patient benefit from scanning is best documented in those with overt, new clinical signs and symptoms of neurologic or psychiatric disease.1,2
Computerized tomography (CT) and magnetic resonance imaging (MRI) are the most common head scanning techniques used in emergency medicine.3 CT is quicker and cheaper, has less movement artifact, and is excellent at delineating acute hemorrhage, calcification, and bony anatomy.3,4 Unfortunately, CT exposes patients to radiation and poorly visualizes white matter or posterior fossa pathology.4
MRI is outstanding for well-defined tissue contrast in multiplanar views and excellent for identifying demyelination or metastatic processes,5 but may be contraindicated for patients with implanted metallic objects such as pacemakers, certain vascular clips or stents, and certain orthopedic devices.3-5 Some patients cannot tolerate the narrow space surrounding them during an MRI.4,5
Safety concerns with CT during pregnancy are well established, but are less clear with MRI. The opposite is true of contrast enhancement; gadolinium with MRI is better tolerated than CT procedures, for which contrast risks include allergy and renal dysfunction. When scanning for a hemorrhage, select a CT scan for patients in whom you suspect bleeding developed within the past 3 days; MRI may be better at screening for older bleeds.
For a list of indications for which a patient should undergo a CT or MRI, see the Table.1-5
Table
Indications for CT or MRI
| New or first-onset psychiatric illness |
| Recent head trauma |
| Recent or advancing cognitive dysfunction |
| New or worsening instances of syncope, vertigo, loss of consciousness, etc. |
| New, worsening, or altered pattern headaches |
| New signs of brain pathology, eg, seizure, paresis, or brain-related visual alteration |
| New neurologic examination abnormalities |
| Concerns about intracranial infection, inflammation, metastases, or increased pressure |
| Change in mental status in persons age >50 |
| Prescreening patients who are candidates for electroconvulsive therapy |
| Source: References 1-5 |
Disclosure
Dr. Lippmann reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Together with a clinical assessment, neuroimaging increases diagnostic accuracy of detecting neuropathology. Direct patient benefit from scanning is best documented in those with overt, new clinical signs and symptoms of neurologic or psychiatric disease.1,2
Computerized tomography (CT) and magnetic resonance imaging (MRI) are the most common head scanning techniques used in emergency medicine.3 CT is quicker and cheaper, has less movement artifact, and is excellent at delineating acute hemorrhage, calcification, and bony anatomy.3,4 Unfortunately, CT exposes patients to radiation and poorly visualizes white matter or posterior fossa pathology.4
MRI is outstanding for well-defined tissue contrast in multiplanar views and excellent for identifying demyelination or metastatic processes,5 but may be contraindicated for patients with implanted metallic objects such as pacemakers, certain vascular clips or stents, and certain orthopedic devices.3-5 Some patients cannot tolerate the narrow space surrounding them during an MRI.4,5
Safety concerns with CT during pregnancy are well established, but are less clear with MRI. The opposite is true of contrast enhancement; gadolinium with MRI is better tolerated than CT procedures, for which contrast risks include allergy and renal dysfunction. When scanning for a hemorrhage, select a CT scan for patients in whom you suspect bleeding developed within the past 3 days; MRI may be better at screening for older bleeds.
For a list of indications for which a patient should undergo a CT or MRI, see the Table.1-5
Table
Indications for CT or MRI
| New or first-onset psychiatric illness |
| Recent head trauma |
| Recent or advancing cognitive dysfunction |
| New or worsening instances of syncope, vertigo, loss of consciousness, etc. |
| New, worsening, or altered pattern headaches |
| New signs of brain pathology, eg, seizure, paresis, or brain-related visual alteration |
| New neurologic examination abnormalities |
| Concerns about intracranial infection, inflammation, metastases, or increased pressure |
| Change in mental status in persons age >50 |
| Prescreening patients who are candidates for electroconvulsive therapy |
| Source: References 1-5 |
Disclosure
Dr. Lippmann reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pary R, Lippmann S. Clinical review of head CT scans in psychiatric patients. VA Practitioner. 1986;3:48-53.
2. Capote HA. Neuroimaging in psychiatry. Neurol Clin. 2009;27(1):237-249.
3. Malhi GS, Lagopoulos J. Making sense of neuroimaging in psychiatry. Acta Psychiatr Scand. 2008;117(2):100-117.
4. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172.
5. Broderick DF. Neuroimaging in neuropsychiatry. Psychiatr Clin North Am. 2005;28(3):549-566,64.
1. Pary R, Lippmann S. Clinical review of head CT scans in psychiatric patients. VA Practitioner. 1986;3:48-53.
2. Capote HA. Neuroimaging in psychiatry. Neurol Clin. 2009;27(1):237-249.
3. Malhi GS, Lagopoulos J. Making sense of neuroimaging in psychiatry. Acta Psychiatr Scand. 2008;117(2):100-117.
4. Small GW, Bookheimer SY, Thompson PM, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7(2):161-172.
5. Broderick DF. Neuroimaging in neuropsychiatry. Psychiatr Clin North Am. 2005;28(3):549-566,64.