User login
Managing aneurysmal subarachnoid hemorrhage: It takes a team
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
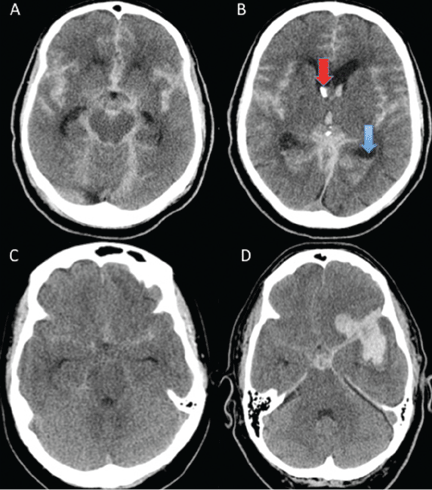
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.


The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
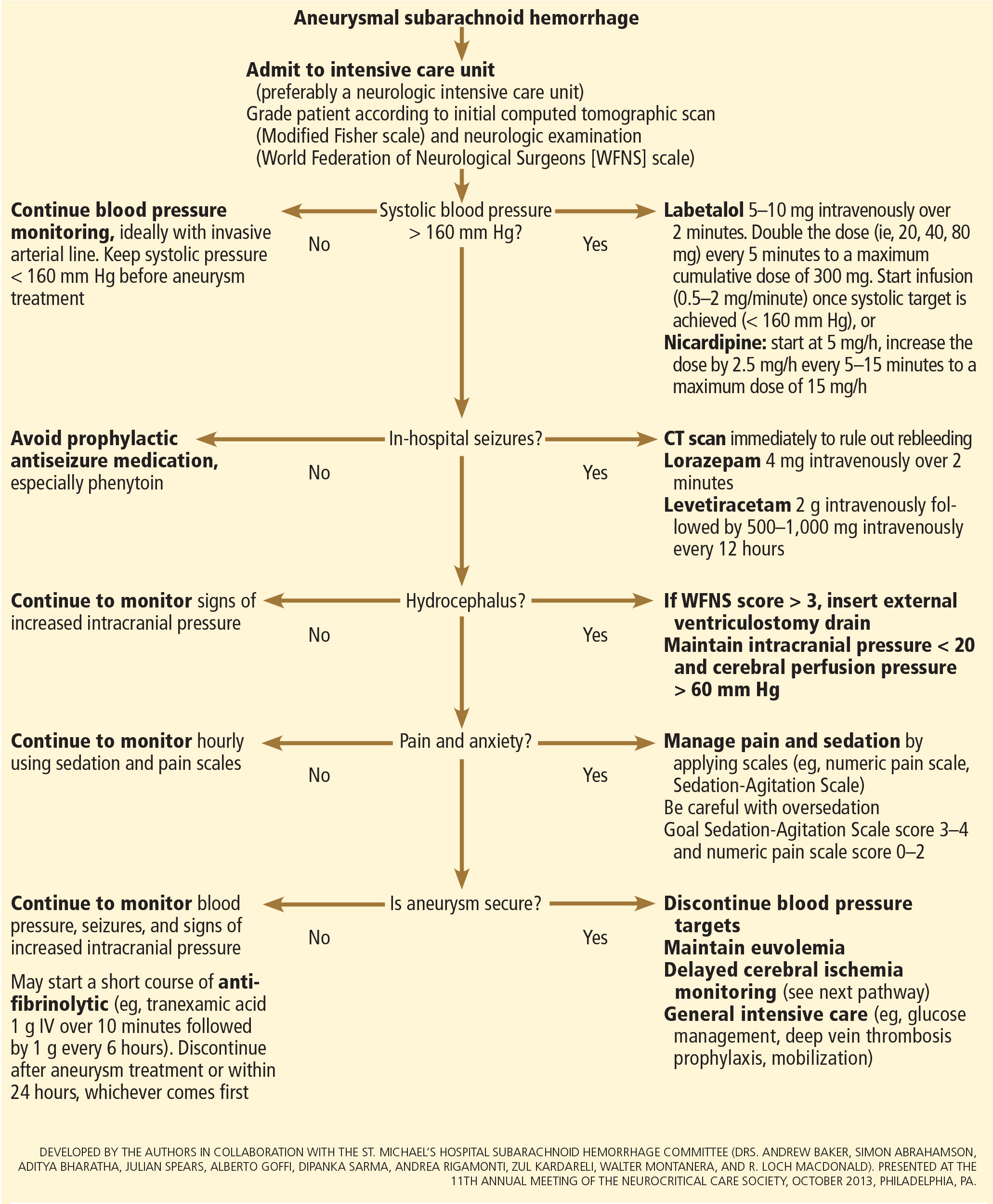
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
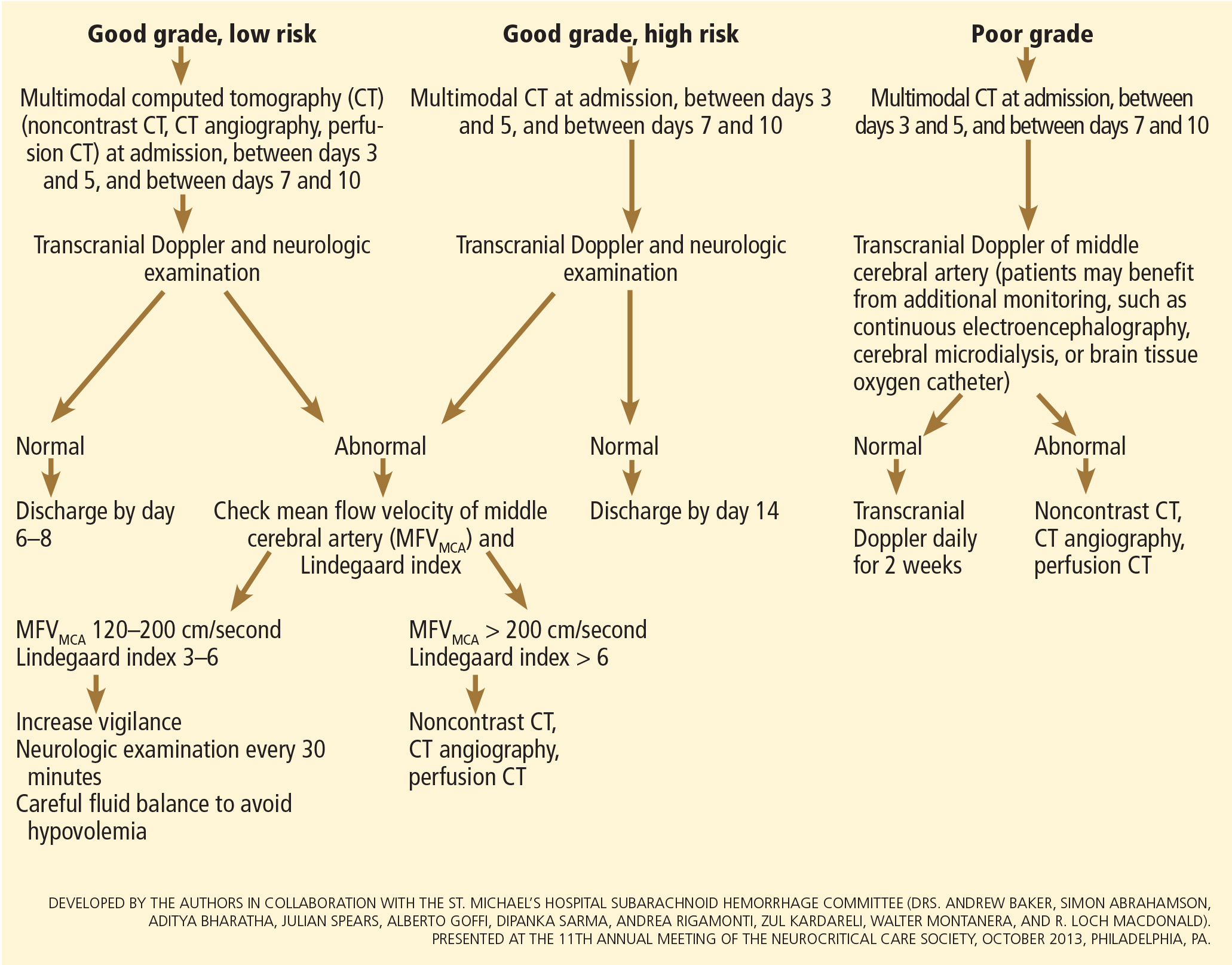
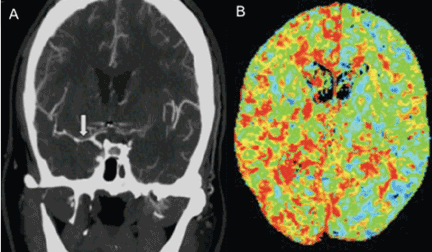
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
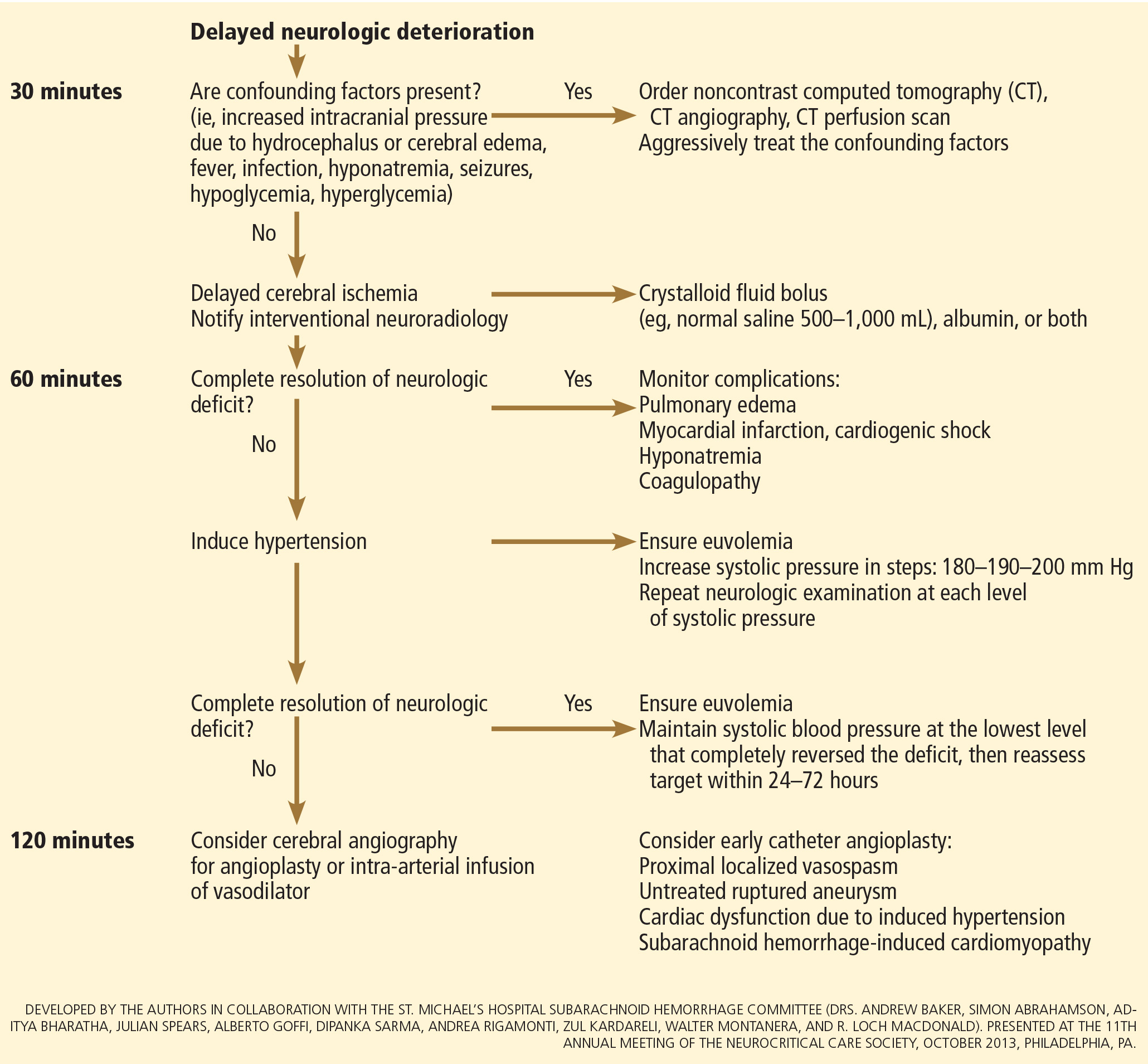
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
KEY POINTS
- The key symptom is the abrupt onset of severe headache, commonly described as “the worst headache of my life.
- Computed tomography without contrast should be done promptly when this condition is suspected.
- Outcomes are improved when patients are managed in a high-volume center with a specialized neurointensive care unit and access to an interdisciplinary team.
- Early aneurysm repair by surgical clipping or endovascular coiling is considered the standard of care and is the best strategy to reduce the risk of rebleeding.
- Medical and neurologic complications are extremely common and include hydrocephalus, increased intracranial pressure, seizures, delayed cerebral ischemia, hyponatremia, hypovolemia, and cardiac and pulmonary abnormalities.
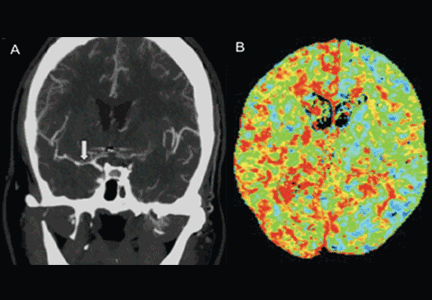
Is triglyceride therapy worth the effort?
Triglyceride levels do matter. Not only are they a marker of risk of cardiovascular disease, they may be mechanistically linked to it. Although we still lack evidence that specifically lowering elevated triglyceride levels reduces the risk of cardiovascular disease, the reason is that controlled trials have not yet been done. Such studies are under way to determine whether fish-oil-derived omega-3 preparations added to statin therapy can reduce coronary heart disease risk in high-risk patients with hypertriglyceridemia.
POSSIBLY MECHANISTICALLY LINKED TO CORONARY HEART DISEASE
Hypertriglyceridemia does not cause atherosclerosis directly, but there is evidence that it is mechanistically linked to it.
While lipolysis of triglyceride-rich lipoproteins, chylomicrons, and very-low-density lipoprotein cholesterol serves as a mammalian source of energy, the cholesterol-enriched byproducts are atherogenic.1 The higher the triglyceride level, the greater the likelihood of accumulation of atherogenic remnant particles.2 A high-triglyceride state, defined as a fasting level greater than 200 mg/dL, is associated with several atherogenic factors:
- Higher levels of apolipoprotein C3-containing particles, which promote inflammation and insulin resistance3
- A higher concentration of atherogenic low-density lipoprotein cholesterol (LDL-C) particles4
- Dysfunctional high-density lipoprotein cholesterol (HDL-C) particles.5
In general, the risk of death from cardiovascular disease is 25% higher with triglyceride levels above 200 mg/dL than with levels below 150 mg/dL.6 Hypertriglyceridemic phenotypes, most notably dysbetalipoproteinemia and mixed hyperlipidemia, may be particularly atherogenic in the presence of other risk factors for cardiovascular disease.7
LOW LEVELS ARE BEST
In US adults, the mean age-adjusted triglyceride level is 128 mg/dL in men and 110 mg/dL in women.1 Currently, a “desirable” fasting level is less than 150 mg/dL, with borderline-high levels between 150 and 199 mg/dL. In its 2011 scientific statement on triglycerides and cardiovascular disease,1 the American Heart Association defined a fasting triglyceride level of less than 100 mg/dL as “optimal” in order to define a metric of metabolic health.
Supporting the concept that low levels are best, a study of 1,962 middle-aged Norwegian men found that the risk of incident diabetes over a 7-year period was 2.6 times lower in the lowest triglyceride tertile (mean 69 mg/dL) compared with levels in higher tertiles (mean 177 mg/dL).8
Moreover, in contrast to the atherogenic phenotype of combined or mixed hyperlipidemia, levels below 100 mg/dL seem to pose a low risk of cardiovascular disease as seen in studies of hunter-gatherer populations.9 Recent genetic studies have extended these findings: specifically, mutations in the gene encoding apolipoprotein C3 (APOC3) have been associated with fasting triglyceride levels less than 100 mg/dL, reduced coronary calcification,10 and decreased risk of cardiovascular disease.11,12
The Baltimore Coronary Observational Long-term Study observed a 50% lower rate of recurrent coronary heart disease events in patients whose baseline triglyceride level was less than 100 mg/dL compared with higher levels.13 In the Copenhagen Heart Study,14 levels in the lowest quartile (< 89 mg/dL) were associated with a 41% lower risk of all-cause mortality compared with the highest quartile (> 265 mg/dL).

The Framingham Offspring Study15 recently reported that an isolated low level of HDL-C—ie, below the median of less than 42 mg/dL in men and less than 54 mg/dL in women—was associated with a very low risk of incident coronary heart disease when accompanied by a triglyceride level below 100 mg/dL and an LDL-C level below 100 mg/dL. However, at higher levels of triglyceride and LDL-C, the risk of myocardial infarction or death from cardiovascular disease was more than twice as high after adjustment for other covariates.15 This raises the possibility that “isolated” low HDL-C itself is not an atherogenic lipoprotein phenotype, but rather requires other triggers (eg, an increase in triglyceride-rich and LDL-C particles) to drive the process.
WHERE DO TRIGLYCERIDES FIT IN THE NEW CHOLESTEROL GUIDELINES?
The 2013 joint guidelines of the American College of Cardiology and the American Heart Association on the treatment of blood cholesterol16 provide evidenced-based recommendations from randomized clinical trials. While they recommend measuring the fasting triglyceride level if nonfasting levels exceed 500 mg/dL, there are no recommendations for triglyceride-lowering therapies unless fasting levels exceed 500 mg/dL.
This in no way implies that lowering triglyceride levels may not be beneficial when levels are below 500 mg/dL; rather, it stems from a lack of clinical trials designed to address this issue. That is, studies of triglyceride-lowering therapies such as niacin, fibrates, and omega-3 fatty acids from fish oil have focused on patients who did not have hypertriglyceridemia (mean triglyceride levels were less than 200 mg/dL). Yet in subgroup analyses of patients with triglyceride levels greater than 200 mg/dL or in the upper tertile (often in association with low levels of HDL-C), either a trend toward or a statistically significant reduced risk of cardiovascular disease was observed.17–19
Until results of ongoing randomized controlled trials dictate otherwise, it may be reasonable to consider drug therapy in patients at high risk (eg, those with preexisting cardiovascular disease) whose levels may be insufficiently responsive to lifestyle measures (see discussion below) after the risks of treatment are weighed against the possible benefits.
WHAT HAPPENED TO THE ATP III TARGETS?
In 2002, the Third Adult Treatment Panel recommended that if triglyceride levels were higher than 200 mg/dL, non-HDL cholesterol should become a secondary target of therapy.20 What happened to this recommendation?
The writing committee of the 2013 American College of Cardiology/American Heart Association cholesterol guidelines based its recommendations on clinical evidence available from randomized controlled trials.16 Unfortunately, neither LDL-C nor non-HDL-C targets were the primary or secondary variables of interest when the available trials were designed. Still, aggregate data over the past several decades, combined with our knowledge of the pathophysiology of both LDL-C and triglyceride-rich lipoproteins (ie, remnants), indicate that non-HDL-C levels predict the risk of cardiovascular disease.21
Moreover, a post hoc analysis of the PROVE IT-TIMI 22 trial demonstrated residual cardiovascular disease risk in hypertriglyceridemic patients after an acute coronary syndrome event even though the patients were currently on statin therapy at the time.22
In addition, the ACCORD study also reported a reduction in cardiovascular disease risk in hypertriglyceridemic patients with low HDL-C assigned to triglyceride-lowering in addition to statin therapy.17,19
Finally, data from two large US health care databases of more than 40,000 adults with triglyceride levels above 500 mg/dL at baseline found that patients who had triglyceride levels lower than 200 mg/dL at follow-up had lower rates of pancreatitis and coronary heart disease events.23
DOES ADDING TO STATIN THERAPY HAVE LONG-TERM CLINICAL BENEFIT?
Two randomized clinical outcome trials are currently testing whether supplementing statin therapy in order to lower triglyceride levels is superior to statin therapy alone in reducing the risk of cardiovascular events in high-risk patients.
REDUCE-IT
REDUCE-IT24 is studying whether AMR101 (Vascepa), a purified ethyl ester of eicosapentaenoic acid, reduces risk in patients with hypertriglyceridemia (with a baseline level of 200 to 500 mg/dL) who have cardiovascular disease or are at high risk for it. However, a number of factors in this trial will make it difficult to separate out the clinical benefit directly related to triglyceride-lowering. These factors include associated beneficial effects of the treatment on LDL-C composition, HDL-C remodeling, and remnant accumulation and clearance, as well as other potential benefits on cardiovascular disease risk independent of lipids and lipoproteins, such as inflammation.
Nevertheless, REDUCE-IT should provide valuable insight into whether this therapy may be clinically useful in these high-risk patients. Enrollment of 8,000 patients is nearing completion in this event-driven trial, with an anticipated median treatment and follow-up period of 4 years.24
STRENGTH
The STRENGTH study will enroll 13,000 patients with hypertriglyceridemia (200–500 mg/dL) and low HDL-C to receive a fish-oil preparation or placebo. This large 5-year phase 3 outcomes study began enrollment in late 2014.25
WHAT IS THE RISK OF PANCREATITIS FROM ELEVATED TRIGLYCERIDES?
The premise of screening for very high triglyceride levels (> 500 mg/dL) in the new cholesterol guidelines and superimposed on the 2011 American Heart Association scientific statement on triglycerides and cardiovascular disease is the concern that very high levels predict pancreatitis. The risk of pancreatitis increases as triglyceride levels exceed 1,000 mg/dL, with an approximate overall risk of 20%.26
While there is no absolute proof that treating chylomicronemia reduces the risk of pancreatitis, ample data from case series show that strategies aimed at reducing plasma triglyceride concentrations are also effective in reducing the risk of pancreatitis.27 Conversely, patients with previous triglyceride-induced pancreatitis unquestionably have recurrent episodes of pancreatitis when they develop severe chylomicronemia. The American Heart Association scientific statement provides a list of other factors, including metabolic conditions and medications, associated with increased risk of pancreatitis.1
WHAT ARE THE RECOMMENDATIONS FOR VERY HIGH TRIGLYCERIDES?
Both the American College of Cardiology/American Heart Association cholesterol guidelines16 and the 2011 American Heart Association statement1 reserve pharmacologic therapy for very high triglyceride levels, defined as 500 mg/dL or higher.

While lifestyle recommendations are still an important part of therapy (Table 1), genetically induced hypertriglyceridemia may not respond as well to diet, exercise, and fish oil. In addition to statin therapy, if there is concomitant cardiovascular disease or diabetes, primary triglyceride-lowering therapies include fibrates (which lower triglycerides 30% to 50%), niacin (20%–50%) and omega-3 fatty acids (10%–40%)1 with eicosapentaenoic acid alone, docosahexaenoic acid alone, or the two in combination.
The focus of the American Heart Association statement was to intensify lifestyle therapies in patients with triglyceride levels between 200 and 500 mg/dL because weight loss, aerobic activity, and the addition of marine-derived polyunsaturated fatty acids can be very effective. The most important step is aimed at weight loss through combined caloric restriction and energy expenditure, increasing monounsaturated and polyunsaturated fat intake at the expense of less complex carbohydrates, and adding marine-derived omega-3 fatty acids. Intensive lifestyle therapy can reduce triglyceride levels by 30% to 50%, and by more in some cases.
Of note, without weight loss, a Mediterranean high-fat diet can aggravate hypertriglyceridemia, especially in patients with fasting triglyceride concentrations above 500 mg/dL. This is particularly true for patients with genetically induced states (eg, lipoprotein lipase deficiency) or other significant defects in chylomicron clearance because olive oil and nut oils serve as excellent substrates for chylomicron formation.
- Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 2011; 123:2292–2333.
- Imke C, Rodriguez BL, Grove JS, et al. Are remnant-like particles independent predictors of coronary heart disease incidence? The Honolulu Heart study. Arterioscler Thromb Vasc Biol 2005; 25:1718–1722.
- Kawakami A, Yoshida M. Apolipoprotein CIII links dyslipidemia with atherosclerosis. J Atheroscler Thromb 2009; 16:6–11.
- Cromwell WC, Otvos JD. Low-density lipoprotein particle number and risk for cardiovascular disease. Curr Atheroscler Rep 2004; 6:381–387.
- Skeggs JW, Morton RE. LDL and HDL enriched in triglyceride promote abnormal cholesterol transport. J Lipid Res 2002; 43:1264–1274.
- Liu J, Zeng FF, Liu ZM, Zhang CX, Ling WH, Chen YM. Effects of blood triglycerides on cardiovascular and all-cause mortality: a systematic review and meta-analysis of 61 prospective studies. Lipids Health Dis 2013; 12:159.
- Voors-Pette C, de Bruin TW. Excess coronary heart disease in familial combined hyperlipidemia, in relation to genetic factors and central obesity. Atherosclerosis 2001; 157:481–489.
- Skretteberg PT, Grytten AN, Gjertsen K, et al. Triglycerides-diabetes association in healthy middle-aged men: modified by physical fitness? A long term follow-up of 1962 Norwegian men in the Oslo Ischemia Study. Diabetes Res Clin Pract 2013; 101:201–209.
- Miller M. The epidemiology of triglyceride as a coronary artery disease risk factor. Clin Cardiol 1999; 22(suppl 6):II1–II6.
- Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008; 322:1702–1705.
- TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute; Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371:22–31.
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Miller M, Seidler A, Moalemi A, Pearson TA. Normal triglyceride levels and coronary artery disease events: the Baltimore Coronary Observational Long-Term Study. J Am Coll Cardiol 1998; 31:1252–1257.
- Thomsen M, Varbo A, Tybjærg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem 2014; 60:737–746.
- Miller M, Kim Y, Havas S, Kwiterovich PO, Fazio S. Does low HDL-C increase CHD risk when TG and LDL-C are normal? The Framingham Offspring Study. Presentation number 1278M-364A. J Am Coll Cardiol 2014; 63.
- Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2889–2934.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med 2010; 363:692–694.
- Guyton JR, Slee AE, Anderson T, et al. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1580–1584.
- Grundy SM, Becker D, Clark LT, et al. Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Liu J, Sempos CT, Donahue RP, Dorn J, Trevisan M, Grundy SM. Non-high-density lipoprotein and very-low-density lipoprotein cholesterol and their risk predictive values in coronary heart disease. Am J Cardiol 2006; 98:1363–1368.
- Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E; PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2008; 51:724–730.
- Christian JB, Arondekar B, Buysman EK, Jacobson TA, Snipes RG, Horwitz RI. Determining triglyceride reductions needed for clinical impact in severe hypertriglyceridemia. Am J Med 2014; 127:36–44.e1.
- ClinicalTrials.gov. A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event (REDUCE-IT). www.clinicaltrials.gov/ct2/show/NCT01492361?term=REDUCE+IT&rank=1. Accessed January 13, 2015.
- ClinicalTrials.gov. Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH). www.clinicaltrials.gov/ct2/show/NCT02104817?term=strength&rank=7. Accessed January 13, 2015.
- Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas 2008; 37:13–22.
- Tsuang W, Navaneethan U, Ruiz L, Palascak JB, Gelrud A. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol 2009; 104:984–991.
Triglyceride levels do matter. Not only are they a marker of risk of cardiovascular disease, they may be mechanistically linked to it. Although we still lack evidence that specifically lowering elevated triglyceride levels reduces the risk of cardiovascular disease, the reason is that controlled trials have not yet been done. Such studies are under way to determine whether fish-oil-derived omega-3 preparations added to statin therapy can reduce coronary heart disease risk in high-risk patients with hypertriglyceridemia.
POSSIBLY MECHANISTICALLY LINKED TO CORONARY HEART DISEASE
Hypertriglyceridemia does not cause atherosclerosis directly, but there is evidence that it is mechanistically linked to it.
While lipolysis of triglyceride-rich lipoproteins, chylomicrons, and very-low-density lipoprotein cholesterol serves as a mammalian source of energy, the cholesterol-enriched byproducts are atherogenic.1 The higher the triglyceride level, the greater the likelihood of accumulation of atherogenic remnant particles.2 A high-triglyceride state, defined as a fasting level greater than 200 mg/dL, is associated with several atherogenic factors:
- Higher levels of apolipoprotein C3-containing particles, which promote inflammation and insulin resistance3
- A higher concentration of atherogenic low-density lipoprotein cholesterol (LDL-C) particles4
- Dysfunctional high-density lipoprotein cholesterol (HDL-C) particles.5
In general, the risk of death from cardiovascular disease is 25% higher with triglyceride levels above 200 mg/dL than with levels below 150 mg/dL.6 Hypertriglyceridemic phenotypes, most notably dysbetalipoproteinemia and mixed hyperlipidemia, may be particularly atherogenic in the presence of other risk factors for cardiovascular disease.7
LOW LEVELS ARE BEST
In US adults, the mean age-adjusted triglyceride level is 128 mg/dL in men and 110 mg/dL in women.1 Currently, a “desirable” fasting level is less than 150 mg/dL, with borderline-high levels between 150 and 199 mg/dL. In its 2011 scientific statement on triglycerides and cardiovascular disease,1 the American Heart Association defined a fasting triglyceride level of less than 100 mg/dL as “optimal” in order to define a metric of metabolic health.
Supporting the concept that low levels are best, a study of 1,962 middle-aged Norwegian men found that the risk of incident diabetes over a 7-year period was 2.6 times lower in the lowest triglyceride tertile (mean 69 mg/dL) compared with levels in higher tertiles (mean 177 mg/dL).8
Moreover, in contrast to the atherogenic phenotype of combined or mixed hyperlipidemia, levels below 100 mg/dL seem to pose a low risk of cardiovascular disease as seen in studies of hunter-gatherer populations.9 Recent genetic studies have extended these findings: specifically, mutations in the gene encoding apolipoprotein C3 (APOC3) have been associated with fasting triglyceride levels less than 100 mg/dL, reduced coronary calcification,10 and decreased risk of cardiovascular disease.11,12
The Baltimore Coronary Observational Long-term Study observed a 50% lower rate of recurrent coronary heart disease events in patients whose baseline triglyceride level was less than 100 mg/dL compared with higher levels.13 In the Copenhagen Heart Study,14 levels in the lowest quartile (< 89 mg/dL) were associated with a 41% lower risk of all-cause mortality compared with the highest quartile (> 265 mg/dL).
The Framingham Offspring Study15 recently reported that an isolated low level of HDL-C—ie, below the median of less than 42 mg/dL in men and less than 54 mg/dL in women—was associated with a very low risk of incident coronary heart disease when accompanied by a triglyceride level below 100 mg/dL and an LDL-C level below 100 mg/dL. However, at higher levels of triglyceride and LDL-C, the risk of myocardial infarction or death from cardiovascular disease was more than twice as high after adjustment for other covariates.15 This raises the possibility that “isolated” low HDL-C itself is not an atherogenic lipoprotein phenotype, but rather requires other triggers (eg, an increase in triglyceride-rich and LDL-C particles) to drive the process.
WHERE DO TRIGLYCERIDES FIT IN THE NEW CHOLESTEROL GUIDELINES?
The 2013 joint guidelines of the American College of Cardiology and the American Heart Association on the treatment of blood cholesterol16 provide evidenced-based recommendations from randomized clinical trials. While they recommend measuring the fasting triglyceride level if nonfasting levels exceed 500 mg/dL, there are no recommendations for triglyceride-lowering therapies unless fasting levels exceed 500 mg/dL.
This in no way implies that lowering triglyceride levels may not be beneficial when levels are below 500 mg/dL; rather, it stems from a lack of clinical trials designed to address this issue. That is, studies of triglyceride-lowering therapies such as niacin, fibrates, and omega-3 fatty acids from fish oil have focused on patients who did not have hypertriglyceridemia (mean triglyceride levels were less than 200 mg/dL). Yet in subgroup analyses of patients with triglyceride levels greater than 200 mg/dL or in the upper tertile (often in association with low levels of HDL-C), either a trend toward or a statistically significant reduced risk of cardiovascular disease was observed.17–19
Until results of ongoing randomized controlled trials dictate otherwise, it may be reasonable to consider drug therapy in patients at high risk (eg, those with preexisting cardiovascular disease) whose levels may be insufficiently responsive to lifestyle measures (see discussion below) after the risks of treatment are weighed against the possible benefits.
WHAT HAPPENED TO THE ATP III TARGETS?
In 2002, the Third Adult Treatment Panel recommended that if triglyceride levels were higher than 200 mg/dL, non-HDL cholesterol should become a secondary target of therapy.20 What happened to this recommendation?
The writing committee of the 2013 American College of Cardiology/American Heart Association cholesterol guidelines based its recommendations on clinical evidence available from randomized controlled trials.16 Unfortunately, neither LDL-C nor non-HDL-C targets were the primary or secondary variables of interest when the available trials were designed. Still, aggregate data over the past several decades, combined with our knowledge of the pathophysiology of both LDL-C and triglyceride-rich lipoproteins (ie, remnants), indicate that non-HDL-C levels predict the risk of cardiovascular disease.21
Moreover, a post hoc analysis of the PROVE IT-TIMI 22 trial demonstrated residual cardiovascular disease risk in hypertriglyceridemic patients after an acute coronary syndrome event even though the patients were currently on statin therapy at the time.22
In addition, the ACCORD study also reported a reduction in cardiovascular disease risk in hypertriglyceridemic patients with low HDL-C assigned to triglyceride-lowering in addition to statin therapy.17,19
Finally, data from two large US health care databases of more than 40,000 adults with triglyceride levels above 500 mg/dL at baseline found that patients who had triglyceride levels lower than 200 mg/dL at follow-up had lower rates of pancreatitis and coronary heart disease events.23
DOES ADDING TO STATIN THERAPY HAVE LONG-TERM CLINICAL BENEFIT?
Two randomized clinical outcome trials are currently testing whether supplementing statin therapy in order to lower triglyceride levels is superior to statin therapy alone in reducing the risk of cardiovascular events in high-risk patients.
REDUCE-IT
REDUCE-IT24 is studying whether AMR101 (Vascepa), a purified ethyl ester of eicosapentaenoic acid, reduces risk in patients with hypertriglyceridemia (with a baseline level of 200 to 500 mg/dL) who have cardiovascular disease or are at high risk for it. However, a number of factors in this trial will make it difficult to separate out the clinical benefit directly related to triglyceride-lowering. These factors include associated beneficial effects of the treatment on LDL-C composition, HDL-C remodeling, and remnant accumulation and clearance, as well as other potential benefits on cardiovascular disease risk independent of lipids and lipoproteins, such as inflammation.
Nevertheless, REDUCE-IT should provide valuable insight into whether this therapy may be clinically useful in these high-risk patients. Enrollment of 8,000 patients is nearing completion in this event-driven trial, with an anticipated median treatment and follow-up period of 4 years.24
STRENGTH
The STRENGTH study will enroll 13,000 patients with hypertriglyceridemia (200–500 mg/dL) and low HDL-C to receive a fish-oil preparation or placebo. This large 5-year phase 3 outcomes study began enrollment in late 2014.25
WHAT IS THE RISK OF PANCREATITIS FROM ELEVATED TRIGLYCERIDES?
The premise of screening for very high triglyceride levels (> 500 mg/dL) in the new cholesterol guidelines and superimposed on the 2011 American Heart Association scientific statement on triglycerides and cardiovascular disease is the concern that very high levels predict pancreatitis. The risk of pancreatitis increases as triglyceride levels exceed 1,000 mg/dL, with an approximate overall risk of 20%.26
While there is no absolute proof that treating chylomicronemia reduces the risk of pancreatitis, ample data from case series show that strategies aimed at reducing plasma triglyceride concentrations are also effective in reducing the risk of pancreatitis.27 Conversely, patients with previous triglyceride-induced pancreatitis unquestionably have recurrent episodes of pancreatitis when they develop severe chylomicronemia. The American Heart Association scientific statement provides a list of other factors, including metabolic conditions and medications, associated with increased risk of pancreatitis.1
WHAT ARE THE RECOMMENDATIONS FOR VERY HIGH TRIGLYCERIDES?
Both the American College of Cardiology/American Heart Association cholesterol guidelines16 and the 2011 American Heart Association statement1 reserve pharmacologic therapy for very high triglyceride levels, defined as 500 mg/dL or higher.
While lifestyle recommendations are still an important part of therapy (Table 1), genetically induced hypertriglyceridemia may not respond as well to diet, exercise, and fish oil. In addition to statin therapy, if there is concomitant cardiovascular disease or diabetes, primary triglyceride-lowering therapies include fibrates (which lower triglycerides 30% to 50%), niacin (20%–50%) and omega-3 fatty acids (10%–40%)1 with eicosapentaenoic acid alone, docosahexaenoic acid alone, or the two in combination.
The focus of the American Heart Association statement was to intensify lifestyle therapies in patients with triglyceride levels between 200 and 500 mg/dL because weight loss, aerobic activity, and the addition of marine-derived polyunsaturated fatty acids can be very effective. The most important step is aimed at weight loss through combined caloric restriction and energy expenditure, increasing monounsaturated and polyunsaturated fat intake at the expense of less complex carbohydrates, and adding marine-derived omega-3 fatty acids. Intensive lifestyle therapy can reduce triglyceride levels by 30% to 50%, and by more in some cases.
Of note, without weight loss, a Mediterranean high-fat diet can aggravate hypertriglyceridemia, especially in patients with fasting triglyceride concentrations above 500 mg/dL. This is particularly true for patients with genetically induced states (eg, lipoprotein lipase deficiency) or other significant defects in chylomicron clearance because olive oil and nut oils serve as excellent substrates for chylomicron formation.
Triglyceride levels do matter. Not only are they a marker of risk of cardiovascular disease, they may be mechanistically linked to it. Although we still lack evidence that specifically lowering elevated triglyceride levels reduces the risk of cardiovascular disease, the reason is that controlled trials have not yet been done. Such studies are under way to determine whether fish-oil-derived omega-3 preparations added to statin therapy can reduce coronary heart disease risk in high-risk patients with hypertriglyceridemia.
POSSIBLY MECHANISTICALLY LINKED TO CORONARY HEART DISEASE
Hypertriglyceridemia does not cause atherosclerosis directly, but there is evidence that it is mechanistically linked to it.
While lipolysis of triglyceride-rich lipoproteins, chylomicrons, and very-low-density lipoprotein cholesterol serves as a mammalian source of energy, the cholesterol-enriched byproducts are atherogenic.1 The higher the triglyceride level, the greater the likelihood of accumulation of atherogenic remnant particles.2 A high-triglyceride state, defined as a fasting level greater than 200 mg/dL, is associated with several atherogenic factors:
- Higher levels of apolipoprotein C3-containing particles, which promote inflammation and insulin resistance3
- A higher concentration of atherogenic low-density lipoprotein cholesterol (LDL-C) particles4
- Dysfunctional high-density lipoprotein cholesterol (HDL-C) particles.5
In general, the risk of death from cardiovascular disease is 25% higher with triglyceride levels above 200 mg/dL than with levels below 150 mg/dL.6 Hypertriglyceridemic phenotypes, most notably dysbetalipoproteinemia and mixed hyperlipidemia, may be particularly atherogenic in the presence of other risk factors for cardiovascular disease.7
LOW LEVELS ARE BEST
In US adults, the mean age-adjusted triglyceride level is 128 mg/dL in men and 110 mg/dL in women.1 Currently, a “desirable” fasting level is less than 150 mg/dL, with borderline-high levels between 150 and 199 mg/dL. In its 2011 scientific statement on triglycerides and cardiovascular disease,1 the American Heart Association defined a fasting triglyceride level of less than 100 mg/dL as “optimal” in order to define a metric of metabolic health.
Supporting the concept that low levels are best, a study of 1,962 middle-aged Norwegian men found that the risk of incident diabetes over a 7-year period was 2.6 times lower in the lowest triglyceride tertile (mean 69 mg/dL) compared with levels in higher tertiles (mean 177 mg/dL).8
Moreover, in contrast to the atherogenic phenotype of combined or mixed hyperlipidemia, levels below 100 mg/dL seem to pose a low risk of cardiovascular disease as seen in studies of hunter-gatherer populations.9 Recent genetic studies have extended these findings: specifically, mutations in the gene encoding apolipoprotein C3 (APOC3) have been associated with fasting triglyceride levels less than 100 mg/dL, reduced coronary calcification,10 and decreased risk of cardiovascular disease.11,12
The Baltimore Coronary Observational Long-term Study observed a 50% lower rate of recurrent coronary heart disease events in patients whose baseline triglyceride level was less than 100 mg/dL compared with higher levels.13 In the Copenhagen Heart Study,14 levels in the lowest quartile (< 89 mg/dL) were associated with a 41% lower risk of all-cause mortality compared with the highest quartile (> 265 mg/dL).
The Framingham Offspring Study15 recently reported that an isolated low level of HDL-C—ie, below the median of less than 42 mg/dL in men and less than 54 mg/dL in women—was associated with a very low risk of incident coronary heart disease when accompanied by a triglyceride level below 100 mg/dL and an LDL-C level below 100 mg/dL. However, at higher levels of triglyceride and LDL-C, the risk of myocardial infarction or death from cardiovascular disease was more than twice as high after adjustment for other covariates.15 This raises the possibility that “isolated” low HDL-C itself is not an atherogenic lipoprotein phenotype, but rather requires other triggers (eg, an increase in triglyceride-rich and LDL-C particles) to drive the process.
WHERE DO TRIGLYCERIDES FIT IN THE NEW CHOLESTEROL GUIDELINES?
The 2013 joint guidelines of the American College of Cardiology and the American Heart Association on the treatment of blood cholesterol16 provide evidenced-based recommendations from randomized clinical trials. While they recommend measuring the fasting triglyceride level if nonfasting levels exceed 500 mg/dL, there are no recommendations for triglyceride-lowering therapies unless fasting levels exceed 500 mg/dL.
This in no way implies that lowering triglyceride levels may not be beneficial when levels are below 500 mg/dL; rather, it stems from a lack of clinical trials designed to address this issue. That is, studies of triglyceride-lowering therapies such as niacin, fibrates, and omega-3 fatty acids from fish oil have focused on patients who did not have hypertriglyceridemia (mean triglyceride levels were less than 200 mg/dL). Yet in subgroup analyses of patients with triglyceride levels greater than 200 mg/dL or in the upper tertile (often in association with low levels of HDL-C), either a trend toward or a statistically significant reduced risk of cardiovascular disease was observed.17–19
Until results of ongoing randomized controlled trials dictate otherwise, it may be reasonable to consider drug therapy in patients at high risk (eg, those with preexisting cardiovascular disease) whose levels may be insufficiently responsive to lifestyle measures (see discussion below) after the risks of treatment are weighed against the possible benefits.
WHAT HAPPENED TO THE ATP III TARGETS?
In 2002, the Third Adult Treatment Panel recommended that if triglyceride levels were higher than 200 mg/dL, non-HDL cholesterol should become a secondary target of therapy.20 What happened to this recommendation?
The writing committee of the 2013 American College of Cardiology/American Heart Association cholesterol guidelines based its recommendations on clinical evidence available from randomized controlled trials.16 Unfortunately, neither LDL-C nor non-HDL-C targets were the primary or secondary variables of interest when the available trials were designed. Still, aggregate data over the past several decades, combined with our knowledge of the pathophysiology of both LDL-C and triglyceride-rich lipoproteins (ie, remnants), indicate that non-HDL-C levels predict the risk of cardiovascular disease.21
Moreover, a post hoc analysis of the PROVE IT-TIMI 22 trial demonstrated residual cardiovascular disease risk in hypertriglyceridemic patients after an acute coronary syndrome event even though the patients were currently on statin therapy at the time.22
In addition, the ACCORD study also reported a reduction in cardiovascular disease risk in hypertriglyceridemic patients with low HDL-C assigned to triglyceride-lowering in addition to statin therapy.17,19
Finally, data from two large US health care databases of more than 40,000 adults with triglyceride levels above 500 mg/dL at baseline found that patients who had triglyceride levels lower than 200 mg/dL at follow-up had lower rates of pancreatitis and coronary heart disease events.23
DOES ADDING TO STATIN THERAPY HAVE LONG-TERM CLINICAL BENEFIT?
Two randomized clinical outcome trials are currently testing whether supplementing statin therapy in order to lower triglyceride levels is superior to statin therapy alone in reducing the risk of cardiovascular events in high-risk patients.
REDUCE-IT
REDUCE-IT24 is studying whether AMR101 (Vascepa), a purified ethyl ester of eicosapentaenoic acid, reduces risk in patients with hypertriglyceridemia (with a baseline level of 200 to 500 mg/dL) who have cardiovascular disease or are at high risk for it. However, a number of factors in this trial will make it difficult to separate out the clinical benefit directly related to triglyceride-lowering. These factors include associated beneficial effects of the treatment on LDL-C composition, HDL-C remodeling, and remnant accumulation and clearance, as well as other potential benefits on cardiovascular disease risk independent of lipids and lipoproteins, such as inflammation.
Nevertheless, REDUCE-IT should provide valuable insight into whether this therapy may be clinically useful in these high-risk patients. Enrollment of 8,000 patients is nearing completion in this event-driven trial, with an anticipated median treatment and follow-up period of 4 years.24
STRENGTH
The STRENGTH study will enroll 13,000 patients with hypertriglyceridemia (200–500 mg/dL) and low HDL-C to receive a fish-oil preparation or placebo. This large 5-year phase 3 outcomes study began enrollment in late 2014.25
WHAT IS THE RISK OF PANCREATITIS FROM ELEVATED TRIGLYCERIDES?
The premise of screening for very high triglyceride levels (> 500 mg/dL) in the new cholesterol guidelines and superimposed on the 2011 American Heart Association scientific statement on triglycerides and cardiovascular disease is the concern that very high levels predict pancreatitis. The risk of pancreatitis increases as triglyceride levels exceed 1,000 mg/dL, with an approximate overall risk of 20%.26
While there is no absolute proof that treating chylomicronemia reduces the risk of pancreatitis, ample data from case series show that strategies aimed at reducing plasma triglyceride concentrations are also effective in reducing the risk of pancreatitis.27 Conversely, patients with previous triglyceride-induced pancreatitis unquestionably have recurrent episodes of pancreatitis when they develop severe chylomicronemia. The American Heart Association scientific statement provides a list of other factors, including metabolic conditions and medications, associated with increased risk of pancreatitis.1
WHAT ARE THE RECOMMENDATIONS FOR VERY HIGH TRIGLYCERIDES?
Both the American College of Cardiology/American Heart Association cholesterol guidelines16 and the 2011 American Heart Association statement1 reserve pharmacologic therapy for very high triglyceride levels, defined as 500 mg/dL or higher.
While lifestyle recommendations are still an important part of therapy (Table 1), genetically induced hypertriglyceridemia may not respond as well to diet, exercise, and fish oil. In addition to statin therapy, if there is concomitant cardiovascular disease or diabetes, primary triglyceride-lowering therapies include fibrates (which lower triglycerides 30% to 50%), niacin (20%–50%) and omega-3 fatty acids (10%–40%)1 with eicosapentaenoic acid alone, docosahexaenoic acid alone, or the two in combination.
The focus of the American Heart Association statement was to intensify lifestyle therapies in patients with triglyceride levels between 200 and 500 mg/dL because weight loss, aerobic activity, and the addition of marine-derived polyunsaturated fatty acids can be very effective. The most important step is aimed at weight loss through combined caloric restriction and energy expenditure, increasing monounsaturated and polyunsaturated fat intake at the expense of less complex carbohydrates, and adding marine-derived omega-3 fatty acids. Intensive lifestyle therapy can reduce triglyceride levels by 30% to 50%, and by more in some cases.
Of note, without weight loss, a Mediterranean high-fat diet can aggravate hypertriglyceridemia, especially in patients with fasting triglyceride concentrations above 500 mg/dL. This is particularly true for patients with genetically induced states (eg, lipoprotein lipase deficiency) or other significant defects in chylomicron clearance because olive oil and nut oils serve as excellent substrates for chylomicron formation.
- Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 2011; 123:2292–2333.
- Imke C, Rodriguez BL, Grove JS, et al. Are remnant-like particles independent predictors of coronary heart disease incidence? The Honolulu Heart study. Arterioscler Thromb Vasc Biol 2005; 25:1718–1722.
- Kawakami A, Yoshida M. Apolipoprotein CIII links dyslipidemia with atherosclerosis. J Atheroscler Thromb 2009; 16:6–11.
- Cromwell WC, Otvos JD. Low-density lipoprotein particle number and risk for cardiovascular disease. Curr Atheroscler Rep 2004; 6:381–387.
- Skeggs JW, Morton RE. LDL and HDL enriched in triglyceride promote abnormal cholesterol transport. J Lipid Res 2002; 43:1264–1274.
- Liu J, Zeng FF, Liu ZM, Zhang CX, Ling WH, Chen YM. Effects of blood triglycerides on cardiovascular and all-cause mortality: a systematic review and meta-analysis of 61 prospective studies. Lipids Health Dis 2013; 12:159.
- Voors-Pette C, de Bruin TW. Excess coronary heart disease in familial combined hyperlipidemia, in relation to genetic factors and central obesity. Atherosclerosis 2001; 157:481–489.
- Skretteberg PT, Grytten AN, Gjertsen K, et al. Triglycerides-diabetes association in healthy middle-aged men: modified by physical fitness? A long term follow-up of 1962 Norwegian men in the Oslo Ischemia Study. Diabetes Res Clin Pract 2013; 101:201–209.
- Miller M. The epidemiology of triglyceride as a coronary artery disease risk factor. Clin Cardiol 1999; 22(suppl 6):II1–II6.
- Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008; 322:1702–1705.
- TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute; Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371:22–31.
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Miller M, Seidler A, Moalemi A, Pearson TA. Normal triglyceride levels and coronary artery disease events: the Baltimore Coronary Observational Long-Term Study. J Am Coll Cardiol 1998; 31:1252–1257.
- Thomsen M, Varbo A, Tybjærg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem 2014; 60:737–746.
- Miller M, Kim Y, Havas S, Kwiterovich PO, Fazio S. Does low HDL-C increase CHD risk when TG and LDL-C are normal? The Framingham Offspring Study. Presentation number 1278M-364A. J Am Coll Cardiol 2014; 63.
- Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2889–2934.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med 2010; 363:692–694.
- Guyton JR, Slee AE, Anderson T, et al. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1580–1584.
- Grundy SM, Becker D, Clark LT, et al. Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Liu J, Sempos CT, Donahue RP, Dorn J, Trevisan M, Grundy SM. Non-high-density lipoprotein and very-low-density lipoprotein cholesterol and their risk predictive values in coronary heart disease. Am J Cardiol 2006; 98:1363–1368.
- Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E; PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2008; 51:724–730.
- Christian JB, Arondekar B, Buysman EK, Jacobson TA, Snipes RG, Horwitz RI. Determining triglyceride reductions needed for clinical impact in severe hypertriglyceridemia. Am J Med 2014; 127:36–44.e1.
- ClinicalTrials.gov. A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event (REDUCE-IT). www.clinicaltrials.gov/ct2/show/NCT01492361?term=REDUCE+IT&rank=1. Accessed January 13, 2015.
- ClinicalTrials.gov. Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH). www.clinicaltrials.gov/ct2/show/NCT02104817?term=strength&rank=7. Accessed January 13, 2015.
- Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas 2008; 37:13–22.
- Tsuang W, Navaneethan U, Ruiz L, Palascak JB, Gelrud A. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol 2009; 104:984–991.
- Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 2011; 123:2292–2333.
- Imke C, Rodriguez BL, Grove JS, et al. Are remnant-like particles independent predictors of coronary heart disease incidence? The Honolulu Heart study. Arterioscler Thromb Vasc Biol 2005; 25:1718–1722.
- Kawakami A, Yoshida M. Apolipoprotein CIII links dyslipidemia with atherosclerosis. J Atheroscler Thromb 2009; 16:6–11.
- Cromwell WC, Otvos JD. Low-density lipoprotein particle number and risk for cardiovascular disease. Curr Atheroscler Rep 2004; 6:381–387.
- Skeggs JW, Morton RE. LDL and HDL enriched in triglyceride promote abnormal cholesterol transport. J Lipid Res 2002; 43:1264–1274.
- Liu J, Zeng FF, Liu ZM, Zhang CX, Ling WH, Chen YM. Effects of blood triglycerides on cardiovascular and all-cause mortality: a systematic review and meta-analysis of 61 prospective studies. Lipids Health Dis 2013; 12:159.
- Voors-Pette C, de Bruin TW. Excess coronary heart disease in familial combined hyperlipidemia, in relation to genetic factors and central obesity. Atherosclerosis 2001; 157:481–489.
- Skretteberg PT, Grytten AN, Gjertsen K, et al. Triglycerides-diabetes association in healthy middle-aged men: modified by physical fitness? A long term follow-up of 1962 Norwegian men in the Oslo Ischemia Study. Diabetes Res Clin Pract 2013; 101:201–209.
- Miller M. The epidemiology of triglyceride as a coronary artery disease risk factor. Clin Cardiol 1999; 22(suppl 6):II1–II6.
- Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008; 322:1702–1705.
- TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute; Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371:22–31.
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Miller M, Seidler A, Moalemi A, Pearson TA. Normal triglyceride levels and coronary artery disease events: the Baltimore Coronary Observational Long-Term Study. J Am Coll Cardiol 1998; 31:1252–1257.
- Thomsen M, Varbo A, Tybjærg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem 2014; 60:737–746.
- Miller M, Kim Y, Havas S, Kwiterovich PO, Fazio S. Does low HDL-C increase CHD risk when TG and LDL-C are normal? The Framingham Offspring Study. Presentation number 1278M-364A. J Am Coll Cardiol 2014; 63.
- Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:2889–2934.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med 2010; 363:692–694.
- Guyton JR, Slee AE, Anderson T, et al. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1580–1584.
- Grundy SM, Becker D, Clark LT, et al. Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Liu J, Sempos CT, Donahue RP, Dorn J, Trevisan M, Grundy SM. Non-high-density lipoprotein and very-low-density lipoprotein cholesterol and their risk predictive values in coronary heart disease. Am J Cardiol 2006; 98:1363–1368.
- Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E; PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2008; 51:724–730.
- Christian JB, Arondekar B, Buysman EK, Jacobson TA, Snipes RG, Horwitz RI. Determining triglyceride reductions needed for clinical impact in severe hypertriglyceridemia. Am J Med 2014; 127:36–44.e1.
- ClinicalTrials.gov. A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event (REDUCE-IT). www.clinicaltrials.gov/ct2/show/NCT01492361?term=REDUCE+IT&rank=1. Accessed January 13, 2015.
- ClinicalTrials.gov. Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH). www.clinicaltrials.gov/ct2/show/NCT02104817?term=strength&rank=7. Accessed January 13, 2015.
- Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas 2008; 37:13–22.
- Tsuang W, Navaneethan U, Ruiz L, Palascak JB, Gelrud A. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol 2009; 104:984–991.
KEY POINTS
- Triglycerides are an excellent marker of coronary heart disease risk and should be treated when fasting levels exceed 150 mg/dL.
- The cornerstone of therapy for triglyceride levels up to 500 mg/dL is intensive lifestyle therapy aimed at reducing excess weight through diet and aerobic activity.
- Drug therapy with fibrates, niacin, and omega-3 fatty acids is indicated for levels exceeding 500 mg/dL because of concern related to pancreatitis risk.
- It remains to be established whether lowering elevated triglyceride levels in patients with coronary heart disease or at risk of it will translate into clinical benefit. However, two studies are under way.
Left atrial appendage closure: An emerging option in atrial fibrillation when oral anticoagulants are not tolerated
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
KEY POINTS
- Few well-designed studies of surgical closure have been done.
- The Watchman percutaneous device was shown to be noninferior to warfarin in certain patients. Other closure devices demonstrate similar success, though trials have not compared them with warfarin.
- Occlusion of the left atrial appendage is an emerging option for general internists to be aware of when treating those with atrial fibrillation who cannot tolerate oral anticoagulation.
Abnormal calcium level in a psychiatric presentation? Rule out parathyroid disease
In some patients, symptoms of depression, psychosis, delirium, or dementia exist concomitantly with, or as a result of, an abnormal (elevated or low) serum calcium concentration that has been precipitated by an unrecognized endocrinopathy. The apparent psychiatric presentations of such patients might reflect parathyroid pathology—not psychopathology.
Hypercalcemia and hypocalcemia often are related to a distinct spectrum of conditions, such as diseases of the parathyroid glands, kidneys, and various neoplasms including malignancies. Be alert to the possibility of parathyroid disease in patients whose presentation suggests mental illness concurrent with, or as a direct consequence of, an abnormal calcium level, and investigate appropriately.
The Table1-9 illustrates how 3 clinical laboratory tests—serum calcium, serum parathyroid hormone (PTH), and phosphate—can narrow the differential diagnosis when the clinical impression is parathyroid-related illness. Seek endocrinology consultation whenever a parathyroid-associated ailment is discovered or suspected. Serum calcium is routinely assayed in hospitalized patients; when managing a patient with treatment-refractory psychiatric illness, (1) always check the reported result of that test and (2) consider measuring PTH.
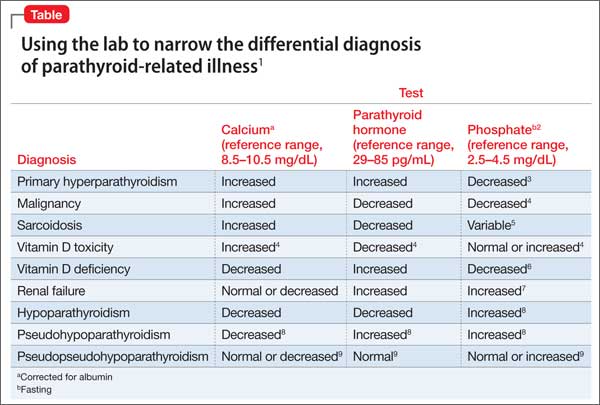
Case reports1
Case 1: Woman with chronic depression. The patient was hospitalized while suicidal. Serial serum calcium levels were 12.5 mg/dL and 15.8 mg/dL (reference range, 8.2–10.2 mg/dL). The PTH level was elevated at 287 pg/mL (reference range, 10–65 pg/mL).
After thyroid imaging, surgery revealed a parathyroid mass, which was resected. Histologic examination confirmed an adenoma.
The calcium concentration declined to 8.6 mg/dL postoperatively and stabilized at 9.2 mg/dL. Psychiatric symptoms resolved fully; she experienced a complete recovery.
Case 2: Man on long-term lithium maintenance. The patient was admitted in a delusional psychotic state. The serum calcium level was 14.3 mg/dL initially, decreasing to 11.5 mg/dL after lithium was discontinued. The PTH level was elevated at 97 pg/mL at admission, consistent with hyperparathyroidism.
A parathyroid adenoma was resected. Serum calcium level normalized at 10.7 mg/dL; psychosis resolved with striking, sustained improvement in mental status.
Full return to mental, physical health
The diagnosis of parathyroid adenoma in these 2 patients, which began with a psychiatric presentation, was properly made after an abnormal serum calcium level was documented. Surgical treatment of the endocrinopathy produced full remission and a return to normal mental and physical health.
Although psychiatric manifestations are associated with an abnormal serum calcium concentration, the severity of those presentations does not correlate with the degree of abnormality of the calcium level.10
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Velasco PJ, Manshadi M, Breen K, et al. Psychiatric aspects of parathyroid disease. Psychosomatics. 1999;40(6):486-490.
2. Harrop JS, Bailey JE, Woodhead JS. Incidence of hypercalcaemia and primary hyperparathyroidism in relation to the biochemical profile. J Clin Pathol. 1982; 35(4):395-400.
3. Assadi F. Hypophosphatemia: an evidence-based problem-solving approach to clinical cases. Iran J Kidney Dis. 2010;4(3):195-201.
4. Ozkhan B, Hatun S, Bereket A. Vitamin D intoxication. Turk J Pediatr. 2012;54(2):93-98.
5. Studdy PR, Bird R, Neville E, et al. Biochemical findings in sarcoidosis. J Clin Pathol. 1980;33(6):528-533.
6. Geller JL, Adam JS. Vitamin D therapy. Curr Osteoporos Rep. 2008;6(1):5-11.
7. Albaaj F, Hutchison A. Hyperphosphatemia in renal failure: causes, consequences and current management. Drugs. 2003;63(6):577-596.
8. Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012;26(4):517-522.
9. Brown H, Englert E, Wallach S. The syndrome of pseudo-pseudohypoparathyroidism. AMA Arch Intern Med. 1956;98(4):517-524.
10. Pfitzenmeyer P, Besancenot JF, Verges B, et al. Primary hyperparathyroidism in very old patients. Eur J Med. 1993;2(8):453-456.
In some patients, symptoms of depression, psychosis, delirium, or dementia exist concomitantly with, or as a result of, an abnormal (elevated or low) serum calcium concentration that has been precipitated by an unrecognized endocrinopathy. The apparent psychiatric presentations of such patients might reflect parathyroid pathology—not psychopathology.
Hypercalcemia and hypocalcemia often are related to a distinct spectrum of conditions, such as diseases of the parathyroid glands, kidneys, and various neoplasms including malignancies. Be alert to the possibility of parathyroid disease in patients whose presentation suggests mental illness concurrent with, or as a direct consequence of, an abnormal calcium level, and investigate appropriately.
The Table1-9 illustrates how 3 clinical laboratory tests—serum calcium, serum parathyroid hormone (PTH), and phosphate—can narrow the differential diagnosis when the clinical impression is parathyroid-related illness. Seek endocrinology consultation whenever a parathyroid-associated ailment is discovered or suspected. Serum calcium is routinely assayed in hospitalized patients; when managing a patient with treatment-refractory psychiatric illness, (1) always check the reported result of that test and (2) consider measuring PTH.
Case reports1
Case 1: Woman with chronic depression. The patient was hospitalized while suicidal. Serial serum calcium levels were 12.5 mg/dL and 15.8 mg/dL (reference range, 8.2–10.2 mg/dL). The PTH level was elevated at 287 pg/mL (reference range, 10–65 pg/mL).
After thyroid imaging, surgery revealed a parathyroid mass, which was resected. Histologic examination confirmed an adenoma.
The calcium concentration declined to 8.6 mg/dL postoperatively and stabilized at 9.2 mg/dL. Psychiatric symptoms resolved fully; she experienced a complete recovery.
Case 2: Man on long-term lithium maintenance. The patient was admitted in a delusional psychotic state. The serum calcium level was 14.3 mg/dL initially, decreasing to 11.5 mg/dL after lithium was discontinued. The PTH level was elevated at 97 pg/mL at admission, consistent with hyperparathyroidism.
A parathyroid adenoma was resected. Serum calcium level normalized at 10.7 mg/dL; psychosis resolved with striking, sustained improvement in mental status.
Full return to mental, physical health
The diagnosis of parathyroid adenoma in these 2 patients, which began with a psychiatric presentation, was properly made after an abnormal serum calcium level was documented. Surgical treatment of the endocrinopathy produced full remission and a return to normal mental and physical health.
Although psychiatric manifestations are associated with an abnormal serum calcium concentration, the severity of those presentations does not correlate with the degree of abnormality of the calcium level.10
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
In some patients, symptoms of depression, psychosis, delirium, or dementia exist concomitantly with, or as a result of, an abnormal (elevated or low) serum calcium concentration that has been precipitated by an unrecognized endocrinopathy. The apparent psychiatric presentations of such patients might reflect parathyroid pathology—not psychopathology.
Hypercalcemia and hypocalcemia often are related to a distinct spectrum of conditions, such as diseases of the parathyroid glands, kidneys, and various neoplasms including malignancies. Be alert to the possibility of parathyroid disease in patients whose presentation suggests mental illness concurrent with, or as a direct consequence of, an abnormal calcium level, and investigate appropriately.
The Table1-9 illustrates how 3 clinical laboratory tests—serum calcium, serum parathyroid hormone (PTH), and phosphate—can narrow the differential diagnosis when the clinical impression is parathyroid-related illness. Seek endocrinology consultation whenever a parathyroid-associated ailment is discovered or suspected. Serum calcium is routinely assayed in hospitalized patients; when managing a patient with treatment-refractory psychiatric illness, (1) always check the reported result of that test and (2) consider measuring PTH.
Case reports1
Case 1: Woman with chronic depression. The patient was hospitalized while suicidal. Serial serum calcium levels were 12.5 mg/dL and 15.8 mg/dL (reference range, 8.2–10.2 mg/dL). The PTH level was elevated at 287 pg/mL (reference range, 10–65 pg/mL).
After thyroid imaging, surgery revealed a parathyroid mass, which was resected. Histologic examination confirmed an adenoma.
The calcium concentration declined to 8.6 mg/dL postoperatively and stabilized at 9.2 mg/dL. Psychiatric symptoms resolved fully; she experienced a complete recovery.
Case 2: Man on long-term lithium maintenance. The patient was admitted in a delusional psychotic state. The serum calcium level was 14.3 mg/dL initially, decreasing to 11.5 mg/dL after lithium was discontinued. The PTH level was elevated at 97 pg/mL at admission, consistent with hyperparathyroidism.
A parathyroid adenoma was resected. Serum calcium level normalized at 10.7 mg/dL; psychosis resolved with striking, sustained improvement in mental status.
Full return to mental, physical health
The diagnosis of parathyroid adenoma in these 2 patients, which began with a psychiatric presentation, was properly made after an abnormal serum calcium level was documented. Surgical treatment of the endocrinopathy produced full remission and a return to normal mental and physical health.
Although psychiatric manifestations are associated with an abnormal serum calcium concentration, the severity of those presentations does not correlate with the degree of abnormality of the calcium level.10
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Velasco PJ, Manshadi M, Breen K, et al. Psychiatric aspects of parathyroid disease. Psychosomatics. 1999;40(6):486-490.
2. Harrop JS, Bailey JE, Woodhead JS. Incidence of hypercalcaemia and primary hyperparathyroidism in relation to the biochemical profile. J Clin Pathol. 1982; 35(4):395-400.
3. Assadi F. Hypophosphatemia: an evidence-based problem-solving approach to clinical cases. Iran J Kidney Dis. 2010;4(3):195-201.
4. Ozkhan B, Hatun S, Bereket A. Vitamin D intoxication. Turk J Pediatr. 2012;54(2):93-98.
5. Studdy PR, Bird R, Neville E, et al. Biochemical findings in sarcoidosis. J Clin Pathol. 1980;33(6):528-533.
6. Geller JL, Adam JS. Vitamin D therapy. Curr Osteoporos Rep. 2008;6(1):5-11.
7. Albaaj F, Hutchison A. Hyperphosphatemia in renal failure: causes, consequences and current management. Drugs. 2003;63(6):577-596.
8. Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012;26(4):517-522.
9. Brown H, Englert E, Wallach S. The syndrome of pseudo-pseudohypoparathyroidism. AMA Arch Intern Med. 1956;98(4):517-524.
10. Pfitzenmeyer P, Besancenot JF, Verges B, et al. Primary hyperparathyroidism in very old patients. Eur J Med. 1993;2(8):453-456.
1. Velasco PJ, Manshadi M, Breen K, et al. Psychiatric aspects of parathyroid disease. Psychosomatics. 1999;40(6):486-490.
2. Harrop JS, Bailey JE, Woodhead JS. Incidence of hypercalcaemia and primary hyperparathyroidism in relation to the biochemical profile. J Clin Pathol. 1982; 35(4):395-400.
3. Assadi F. Hypophosphatemia: an evidence-based problem-solving approach to clinical cases. Iran J Kidney Dis. 2010;4(3):195-201.
4. Ozkhan B, Hatun S, Bereket A. Vitamin D intoxication. Turk J Pediatr. 2012;54(2):93-98.
5. Studdy PR, Bird R, Neville E, et al. Biochemical findings in sarcoidosis. J Clin Pathol. 1980;33(6):528-533.
6. Geller JL, Adam JS. Vitamin D therapy. Curr Osteoporos Rep. 2008;6(1):5-11.
7. Albaaj F, Hutchison A. Hyperphosphatemia in renal failure: causes, consequences and current management. Drugs. 2003;63(6):577-596.
8. Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2012;26(4):517-522.
9. Brown H, Englert E, Wallach S. The syndrome of pseudo-pseudohypoparathyroidism. AMA Arch Intern Med. 1956;98(4):517-524.
10. Pfitzenmeyer P, Besancenot JF, Verges B, et al. Primary hyperparathyroidism in very old patients. Eur J Med. 1993;2(8):453-456.
How to write a suicide risk assessment that’s clinically sound and legally defensible
Suicidologists and legal experts implore clinicians to document their suicide risk assessments (SRAs) thoroughly. It’s difficult, however, to find practical guidance on how to write a clinically sound, legally defensible SRA.
The crux of every SRA is written justification of suicide risk. That justification should reveal your thinking and present a well-reasoned basis for your decision.
Reasoned vs right
It’s more important to provide a justification of suicide risk that’s well-reasoned rather than one that’s right. Suicide is impossible to predict. Instead of prediction, legally we are asked to reasonably anticipate suicide based on clinical facts. In hindsight, especially in the context of a courtroom, decisions might look ill-considered. You need to craft a logical argument, be clear, and avoid jargon.
Convey thoroughness by covering each component of an SRA. Use the mnemonic device CAIPS to help the reader (and you) understand how a conclusion was reached based on the facts of the case.
Chronic and Acute factors. Address the chronic and acute factors that weigh heaviest in your mind. Chronic factors are conditions, past events, and demographics that generally do not change. Acute factors are recent events or conditions that potentially are modifiable. Pay attention to combinations of factors that dramatically elevate risk (eg, previous attempts in the context of acute depression). Avoid repeating every factor, especially when these are documented elsewhere, such as on a checklist.
Imminent warning signs for suicide. Address warning signs (Table 1),1 the nature of current suicidal thoughts (Table 2), and other aspects of mental status (eg, future orientation) that influenced your decision. Use words like “moreover,” “however,” and “in addition” to draw the reader’s attention to the building blocks of your argument.


Protective factors. Discuss the protective factors last; they deserve the least weight because none has been shown to immunize people against suicide. Don’t solely rely on your judgment of what is protective (eg, children in the home). Instead, elicit the patient’s reasons for living and dying. Be concerned if he (she) reports more of the latter.
Summary statement. Make an explicit statement about risk, focusing on imminent risk (ie, the next few hours and days). Avoid a “plot twist,” which is a risk level inconsistent with the preceding evidence, because it suggests an error in judgment. The Box gives an example of a justification that follows the CAIPS method. 
Additional tips
Consider these strategies:
• Bolster your argument by explicitly addressing hopelessness (the strongest psychological correlate of suicide); use quotes from the patient that support your decision; refer to consultation with family members and colleagues; and include pertinent negatives to show completeness2 (ie, “denied suicide plans”).
• Critically resolve discrepancies between what the patient says and behavior that suggests suicidal intent (eg, a patient who minimizes suicidal intent but shopped for a gun yesterday).
• Last, while reviewing your justification, imagine that your patient completed suicide after leaving your office and that you are in court for negligence. In our experience, this exercise reveals dangerous errors of judgment. A clear and reasoned justification will reduce the risk of litigation and help you make prudent treatment plans.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. American Association of Suicidology. Know the warning signs of suicide. http://www.suicidology.org/resources/ warning-signs. Accessed February 9, 2014.
2. Ballas C. How to write a suicide note: practical tips for documenting the evaluation of a suicidal patient. Psychiatric Times. http://www.psychiatrictimes.com/articles/how-write-suicide-note-practical-tips-documenting-evaluation-suicidal-patient. Published May 1, 2007. Accessed July 29, 2013.
Suicidologists and legal experts implore clinicians to document their suicide risk assessments (SRAs) thoroughly. It’s difficult, however, to find practical guidance on how to write a clinically sound, legally defensible SRA.
The crux of every SRA is written justification of suicide risk. That justification should reveal your thinking and present a well-reasoned basis for your decision.
Reasoned vs right
It’s more important to provide a justification of suicide risk that’s well-reasoned rather than one that’s right. Suicide is impossible to predict. Instead of prediction, legally we are asked to reasonably anticipate suicide based on clinical facts. In hindsight, especially in the context of a courtroom, decisions might look ill-considered. You need to craft a logical argument, be clear, and avoid jargon.
Convey thoroughness by covering each component of an SRA. Use the mnemonic device CAIPS to help the reader (and you) understand how a conclusion was reached based on the facts of the case.
Chronic and Acute factors. Address the chronic and acute factors that weigh heaviest in your mind. Chronic factors are conditions, past events, and demographics that generally do not change. Acute factors are recent events or conditions that potentially are modifiable. Pay attention to combinations of factors that dramatically elevate risk (eg, previous attempts in the context of acute depression). Avoid repeating every factor, especially when these are documented elsewhere, such as on a checklist.
Imminent warning signs for suicide. Address warning signs (Table 1),1 the nature of current suicidal thoughts (Table 2), and other aspects of mental status (eg, future orientation) that influenced your decision. Use words like “moreover,” “however,” and “in addition” to draw the reader’s attention to the building blocks of your argument.
Protective factors. Discuss the protective factors last; they deserve the least weight because none has been shown to immunize people against suicide. Don’t solely rely on your judgment of what is protective (eg, children in the home). Instead, elicit the patient’s reasons for living and dying. Be concerned if he (she) reports more of the latter.
Summary statement. Make an explicit statement about risk, focusing on imminent risk (ie, the next few hours and days). Avoid a “plot twist,” which is a risk level inconsistent with the preceding evidence, because it suggests an error in judgment. The Box gives an example of a justification that follows the CAIPS method.
Additional tips
Consider these strategies:
• Bolster your argument by explicitly addressing hopelessness (the strongest psychological correlate of suicide); use quotes from the patient that support your decision; refer to consultation with family members and colleagues; and include pertinent negatives to show completeness2 (ie, “denied suicide plans”).
• Critically resolve discrepancies between what the patient says and behavior that suggests suicidal intent (eg, a patient who minimizes suicidal intent but shopped for a gun yesterday).
• Last, while reviewing your justification, imagine that your patient completed suicide after leaving your office and that you are in court for negligence. In our experience, this exercise reveals dangerous errors of judgment. A clear and reasoned justification will reduce the risk of litigation and help you make prudent treatment plans.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Suicidologists and legal experts implore clinicians to document their suicide risk assessments (SRAs) thoroughly. It’s difficult, however, to find practical guidance on how to write a clinically sound, legally defensible SRA.
The crux of every SRA is written justification of suicide risk. That justification should reveal your thinking and present a well-reasoned basis for your decision.
Reasoned vs right
It’s more important to provide a justification of suicide risk that’s well-reasoned rather than one that’s right. Suicide is impossible to predict. Instead of prediction, legally we are asked to reasonably anticipate suicide based on clinical facts. In hindsight, especially in the context of a courtroom, decisions might look ill-considered. You need to craft a logical argument, be clear, and avoid jargon.
Convey thoroughness by covering each component of an SRA. Use the mnemonic device CAIPS to help the reader (and you) understand how a conclusion was reached based on the facts of the case.
Chronic and Acute factors. Address the chronic and acute factors that weigh heaviest in your mind. Chronic factors are conditions, past events, and demographics that generally do not change. Acute factors are recent events or conditions that potentially are modifiable. Pay attention to combinations of factors that dramatically elevate risk (eg, previous attempts in the context of acute depression). Avoid repeating every factor, especially when these are documented elsewhere, such as on a checklist.
Imminent warning signs for suicide. Address warning signs (Table 1),1 the nature of current suicidal thoughts (Table 2), and other aspects of mental status (eg, future orientation) that influenced your decision. Use words like “moreover,” “however,” and “in addition” to draw the reader’s attention to the building blocks of your argument.
Protective factors. Discuss the protective factors last; they deserve the least weight because none has been shown to immunize people against suicide. Don’t solely rely on your judgment of what is protective (eg, children in the home). Instead, elicit the patient’s reasons for living and dying. Be concerned if he (she) reports more of the latter.
Summary statement. Make an explicit statement about risk, focusing on imminent risk (ie, the next few hours and days). Avoid a “plot twist,” which is a risk level inconsistent with the preceding evidence, because it suggests an error in judgment. The Box gives an example of a justification that follows the CAIPS method.
Additional tips
Consider these strategies:
• Bolster your argument by explicitly addressing hopelessness (the strongest psychological correlate of suicide); use quotes from the patient that support your decision; refer to consultation with family members and colleagues; and include pertinent negatives to show completeness2 (ie, “denied suicide plans”).
• Critically resolve discrepancies between what the patient says and behavior that suggests suicidal intent (eg, a patient who minimizes suicidal intent but shopped for a gun yesterday).
• Last, while reviewing your justification, imagine that your patient completed suicide after leaving your office and that you are in court for negligence. In our experience, this exercise reveals dangerous errors of judgment. A clear and reasoned justification will reduce the risk of litigation and help you make prudent treatment plans.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. American Association of Suicidology. Know the warning signs of suicide. http://www.suicidology.org/resources/ warning-signs. Accessed February 9, 2014.
2. Ballas C. How to write a suicide note: practical tips for documenting the evaluation of a suicidal patient. Psychiatric Times. http://www.psychiatrictimes.com/articles/how-write-suicide-note-practical-tips-documenting-evaluation-suicidal-patient. Published May 1, 2007. Accessed July 29, 2013.
1. American Association of Suicidology. Know the warning signs of suicide. http://www.suicidology.org/resources/ warning-signs. Accessed February 9, 2014.
2. Ballas C. How to write a suicide note: practical tips for documenting the evaluation of a suicidal patient. Psychiatric Times. http://www.psychiatrictimes.com/articles/how-write-suicide-note-practical-tips-documenting-evaluation-suicidal-patient. Published May 1, 2007. Accessed July 29, 2013.
Lisdexamfetamine for binge eating disorder: New indication
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.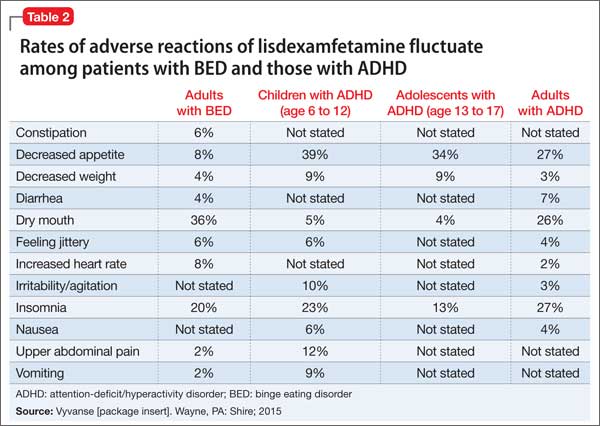
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Telepsychiatry: Ready to consider a different kind of practice?
Too few psychiatrists. A growing number of patients. A new federal law, technological advances, and a generational shift in the way people communicate. Add them together and you have the perfect environment for telepsychiatry—the remote practice of psychiatry by means of telemedicine—to take root (Box 1). Although telepsychiatry has, in various forms, been around since the 1950s,1 only recently has it expanded into almost all areas of psychiatric practice.
Here are some observations from my daily work on why I see this method of delivering mental health care is poised to expand in 2015 and beyond. Does telepsychiatry make sense for you?
Lack of supply is a big driver
There are simply not enough psychiatrists where they are needed, which is the primary driver of the expansion of telepsychiatry. With 77% of counties in the United States reporting a shortage of psychiatrists2 and the “graying” of the psychiatric workforce,3 a more efficient way to make use of a psychiatrist’s time is needed. Telepsychiatry eliminates travel time and allows psychiatrists to visit distant sites virtually.
The shortage of psychiatric practitioners that we see today is only going to become worse. The Patient Protection and Affordable Care Act of 2010 includes mental health care and substance abuse treatment among its 10 essential benefits; just as important, new rules arising from the Mental Health Parity and Addiction Equity Act of 2008 limit restrictions on access to mental health care when insurance provides such coverage.4 These legislative initiatives likely will lead to increased demand for psychiatrists in all care settings—from outpatient consults to acute inpatient admissions.
Why so attractive an option?
The shortage of psychiatrists creates limitations on access to care. Fortunately, telemedicine has entered a new age, ushered in by widely available teleconferencing technology. Specialists from dermatology to surgery currently are using telemedicine; psychiatry is a good fit for telemedicine because of (1) the limited amount of “touch” required to make a psychiatric assessment, (2) significant improvements in video quality in recent years, and (3) a decrease in the stigma associated with visiting a psychiatrist.
A generation raised on the Internet is entering the health care marketplace. These consumers and clinicians are accustomed to using video for many daily activities, and they seek health information from the Web. Visiting a psychiatrist through teleconferencing isn’t strange or alienating to this generation; their comfort with technology allows them to have intimate exchanges on video.
Subspecialty particulars
The earliest adopters, not surprisingly, are in areas where the strain of shortage has been felt most, with pediatric, geriatric, and correctional psychiatrists leading the way. In these fields, a substantial literature supports the use of telepsychiatry from a number of practice perspectives.
Pediatric psychiatry. The literature shows that children, families, and clinicians are, on the whole, satisfied with telepsychiatry.5 Children and adolescents who have been shown to benefit from telepsychiatry include those with depression,6 posttraumatic stress disorder, and eating disorders.7 Based on a case series, some authors have asserted that telepsychiatry might be preferable to in-person treatment (Box 2).8

Geriatric psychiatry. Research shows that geriatric patients, who are most likely to feel threatened by new technology, accept telepsychiatry visits.9 For psychiatrists treating geriatric patients, telepsychiatry can significantly lower costs by cutting commuting10 and make more accessible for patients whose age makes them unable to drive.
Correctional psychiatry. Clinicians working in correctional psychiatry have been at the forefront of experimentation with telepsychiatry. The technology is a natural fit for this setting:
• Prisons often are located in remote locations.
• Psychiatrists can be reluctant to provide on-site services because of safety concerns.
With correctional telepsychiatry, not only are patient outcomes comparable with in-person psychiatry, but the cost of delivering care can be significantly lower.11 With the U.S. Department of Justice reporting that 50% of inmates have a diagnosable mental disorder, including substance abuse,12 the need for access to a psychiatrist in the correctional system is acute.
Telepsychiatry can confidently be provided in a number of settings:
• emergency rooms
• nursing homes
• offices of primary care physicians
• in-home care.
Clinical services in these settings have been offered, studied, and reviewed.13
Can confidentiality and security be assured?
As with any new medical tool, the risk and benefits must be weighed care fully. The most obvious risk is to privacy. Telepsychiatry visits, like all patient encounters, must be secure and confidential. Given the growing suspicion among the public and professionals who use computers that all data are at risk, clinicians must take appropriate cautions and, at the same time, warn patients of the risks. Readily available videoconferencing software, such as Skype, does not provide the level of security that patients expect from health care providers.14
Other common concerns about telepsychiatry are stable access to videoconferencing and the safety from hackers of necessary hardware. Medical device companies have created hardware and software for use in telepsychiatry that provide a Health Insurance Portability and Accountability Act-compliant high-quality, stable, videoconferencing visit.
Do patients benefit?
Clinically, patients have fared well when they receive care through telepsychiatry. In some studies, however, clinicians have expressed some dissatisfaction with the technology13— understandable, given the value that psychiatry traditionally has put on sitting with the patient. As Knoedler15 described it, making the switch to telepsychiatry from in-person contact can engender loneliness in some physicians; not only is patient contact shifted to videoconferencing, but the psychiatrist loses the supportive environment of a busy clinical practice. Knoedler also pointed out that, on the other hand, telepsychiatry offers practitioners the opportunity to evaluate and treat people who otherwise would not have mental health care.
Obstacles—practical, knotty ones
Reimbursement and licensing. These are 2 pressing problems of telepsychiatry, although recent policy developments will help expand telepsychiatry and make it more appealing to physicians:
• Medicare reimburses for telepsychiatry in non-metropolitan areas.
• In 41 states, Medicaid has included telepsychiatry as a benefit.16
• Nine states offer a specific medical license for practicing telepsychiatry17 (in the remaining states, a full medical license must be obtained before one can provide telemedicine services).
• The Joint Commission has included language in its regulations that could expedite privileging of telepsychiatrists.18
Even with such advancements, problems with licensure, credentialing, privacy, security, confidentiality, informed consent, and professional liability remain.19 I urge you to do your research on these key areas before plunging in.
Changes to models of care. The risk that telepsychiatry poses to various models of care has to be considered. Telepsychiatry is a dramatic innovation, but it should be used to support only high-quality, evidence-based care to which patients are entitled.20 With new technology—as with new medications—use must be carefully monitored and scrutinized.
Although evidence of the value of telepsychiatry is growing, many methods of long-distance practice are still in their infancy. Data must be collected and poor outcomes assessed honestly to ensure that the “more-good-than-harm” mandate is met.
Good reasons to call this shift ‘inevitable’
The future of telepsychiatry includes expansion into new areas of practice. The move to providing services to patients where they happen to be—at work or home— seems inevitable:
• In rural areas, practitioners can communicate with patients so that they are cared for in their homes, without the expense of transportation.
• Employers can invest in workplace health clinics that use telemedicine services to reduce absenteeism.
• For psychiatrists, the ability to provide services to patients across a wide region, from a single convenient location, and at lower cost is an attractive prospect.
To conclude: telepsychiatry holds potential to provide greater reimbursement and improved quality of life for psychiatrists and patients: It allows physicians to choose where they live and work, and limits the number of unreimbursed commutes, and gives patients access to psychiatric care locally, without disruptive travel and delays.
Bottom Line
The exchange of medical information from 1 site to another by means of electronic communication has great potential to improve the health of patients and to alleviate the shortage of psychiatric practitioners across regions and settings. Pediatric, geriatric, and correctional psychiatry stand to benefit because of the nature of the patients and locations.
Related Resources
• American Telemedicine Association. Practice guidelines for video-based online mental health services. http://www. americantelemed.org/docs/default-source/standards/practice-guidelines-for-video-based-online-mental-health-services. pdf?sfvrsn=6. Published May 2013. Accessed February 10, 2015.
• Freudenberg N, Yellowlees PM. Telepsychiatry as part of a comprehensive care plan. Virtual Mentor. 2014;16(12):964-968.
• Kornbluh R. Telepsychiatry is a tool that we must exploit. Clinical Psychiatry News. August 7, 2014. http://www. clinicalpsychiatrynews.com/home/article/telepsychiatry-is-a-tool-that-we-must-exploit/28c87bec298e0aa208309fa 9bc48dedc.html.
• University of Colorado Denver. Telemental Health Guide. http:// www.tmhguide.org.
Disclosure
Dr. Kornbluh reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shore JH. Telepsychiatry: videoconferencing in the delivery of psychiatric care. Am J Psychiatry. 2013;170(3):256-262.
2. Konrad TR, Ellis AR, Thomas KC, et al. County-level estimates of need for mental health professionals in the United States. Psychiatr Serv. 2009;60(10):1307-1314.
3. Vernon DJ, Salsberg E, Erikson C, et al. Planning the future mental health workforce: with progress on coverage, what role will psychiatrists play? Acad Psychiatry. 2009;33(3):187-192.
4. Carrns A. Understanding new rules that widen mental health coverage. The New York Times. http://www. nytimes.com/2014/01/10/your-money/understanding-new-rules-that-widen-mental-health-coverage.html. Published January 9, 2014. Accessed February 10, 2015.
5. Myers KM, Valentine JM, Melzer SM. Feasibility, acceptability, and sustainability of telepsychiatry for children and adolescents. Psychiatr Serv. 2007;58(11):1493-1496.
6. Nelson EL, Barnard M, Cain S. Treating childhood depression over videoconferencing. Telemed J E Health. 2003;9(1):49-55.
7. Boydell KM, Hodgins M, Pignatiello A, et al. Using technology to deliver mental health services to children and youth: a scoping review. J Can Acad Child Adolesc Psychiatry. 2014;23(2):87-99.
8. Pakyurek M, Yellowlees P, Hilty D. The child and adolescent telepsychiatry consultation: can it be a more effective clinical process for certain patients than conventional practice? Telemed J E Health. 2010;16(3):289-292.
9. Poon P, Hui E, Dai D, et al. Cognitive intervention for community-dwelling older persons with memory problems: telemedicine versus face-to-face treatment. Int J Geriatr Psychiatry. 2005;20(3):285-286.
10. Rabinowitz T, Murphy KM, Amour JL, et al. Benefits of a telepsychiatry consultation service for rural nursing home residents. Telemed J E Health. 2010;16(1):34-40.
11. Deslich SA, Thistlethwaite T, Coustasse A. Telepsychiatry in correctional facilities: using technology to improve access and decrease costs of mental health care in underserved populations. Perm J. 2013;17(3):80-86.
12. James DJ, Glaze LE. Mental health problems of prison and jail inmates. U.S. Department of Justice, Office of Justice Programs. http://www.bjs.gov/content/pub/pdf/mhppji. pdf. Updated December 14, 2006. Accessed February 10, 2015.
13. Hilty DN, Ferrer DC, Parish MB, et al. The effectiveness of telemental health: a 2013 review. Telemed J E Health. 2013;19(6):444-454.
14. Maheu MM, Mcmenamin J. Telepsychiatry: the perils of using skype. Psychiatric Times. http://www. psychiatrictimes.com/blog/telepsychiatry-perils-using-skype. Published March 28, 2013. Accessed February 10, 2015.
15. Knoedler DW. Telepsychiatry: first week in the trenches. Psychiatric Times. http://www.psychiatrictimes.com/ blogs/couch-crisis/telepsychiatry-first-week-trenches. Published January 22, 2014. Accessed February 15, 2015.
16. Secure Telehealth. Medicaid reimburses for telehealth in 41 states. http://www.securetelehealth.com/medicaid-reimbursement.html. Updated January 15, 2015. Accessed February 10, 2015.
17. Federation of State Medical Boards. Telemedicine overview: Board-by-Board approach. http://library.fsmb.org/pdf/ grpol_telemedicine_licensure.pdf. Updated June 2013. Accessed February 10, 2015.
18. Joint Commission Perspectives. Accepted: final revisions to telemedicine standards. http://www.jointcommission. org/assets/1/6/Revisions_telemedicine_standards.pdf. Published January 2012. Accessed February 10, 2015.
19. Hyler SE, Gangure DP. Legal and ethical challenges in telepsychiatry. J Psychiatr Pract. 2004;10(4):272-276.
20. Kornbluh RA. Staying true to the mission: adapting telepsychiatry to a new environment. CNS Spectr. 2014;19(6):482-483.
Too few psychiatrists. A growing number of patients. A new federal law, technological advances, and a generational shift in the way people communicate. Add them together and you have the perfect environment for telepsychiatry—the remote practice of psychiatry by means of telemedicine—to take root (Box 1). Although telepsychiatry has, in various forms, been around since the 1950s,1 only recently has it expanded into almost all areas of psychiatric practice.
Here are some observations from my daily work on why I see this method of delivering mental health care is poised to expand in 2015 and beyond. Does telepsychiatry make sense for you?
Lack of supply is a big driver
There are simply not enough psychiatrists where they are needed, which is the primary driver of the expansion of telepsychiatry. With 77% of counties in the United States reporting a shortage of psychiatrists2 and the “graying” of the psychiatric workforce,3 a more efficient way to make use of a psychiatrist’s time is needed. Telepsychiatry eliminates travel time and allows psychiatrists to visit distant sites virtually.
The shortage of psychiatric practitioners that we see today is only going to become worse. The Patient Protection and Affordable Care Act of 2010 includes mental health care and substance abuse treatment among its 10 essential benefits; just as important, new rules arising from the Mental Health Parity and Addiction Equity Act of 2008 limit restrictions on access to mental health care when insurance provides such coverage.4 These legislative initiatives likely will lead to increased demand for psychiatrists in all care settings—from outpatient consults to acute inpatient admissions.
Why so attractive an option?
The shortage of psychiatrists creates limitations on access to care. Fortunately, telemedicine has entered a new age, ushered in by widely available teleconferencing technology. Specialists from dermatology to surgery currently are using telemedicine; psychiatry is a good fit for telemedicine because of (1) the limited amount of “touch” required to make a psychiatric assessment, (2) significant improvements in video quality in recent years, and (3) a decrease in the stigma associated with visiting a psychiatrist.
A generation raised on the Internet is entering the health care marketplace. These consumers and clinicians are accustomed to using video for many daily activities, and they seek health information from the Web. Visiting a psychiatrist through teleconferencing isn’t strange or alienating to this generation; their comfort with technology allows them to have intimate exchanges on video.
Subspecialty particulars
The earliest adopters, not surprisingly, are in areas where the strain of shortage has been felt most, with pediatric, geriatric, and correctional psychiatrists leading the way. In these fields, a substantial literature supports the use of telepsychiatry from a number of practice perspectives.
Pediatric psychiatry. The literature shows that children, families, and clinicians are, on the whole, satisfied with telepsychiatry.5 Children and adolescents who have been shown to benefit from telepsychiatry include those with depression,6 posttraumatic stress disorder, and eating disorders.7 Based on a case series, some authors have asserted that telepsychiatry might be preferable to in-person treatment (Box 2).8
Geriatric psychiatry. Research shows that geriatric patients, who are most likely to feel threatened by new technology, accept telepsychiatry visits.9 For psychiatrists treating geriatric patients, telepsychiatry can significantly lower costs by cutting commuting10 and make more accessible for patients whose age makes them unable to drive.
Correctional psychiatry. Clinicians working in correctional psychiatry have been at the forefront of experimentation with telepsychiatry. The technology is a natural fit for this setting:
• Prisons often are located in remote locations.
• Psychiatrists can be reluctant to provide on-site services because of safety concerns.
With correctional telepsychiatry, not only are patient outcomes comparable with in-person psychiatry, but the cost of delivering care can be significantly lower.11 With the U.S. Department of Justice reporting that 50% of inmates have a diagnosable mental disorder, including substance abuse,12 the need for access to a psychiatrist in the correctional system is acute.
Telepsychiatry can confidently be provided in a number of settings:
• emergency rooms
• nursing homes
• offices of primary care physicians
• in-home care.
Clinical services in these settings have been offered, studied, and reviewed.13
Can confidentiality and security be assured?
As with any new medical tool, the risk and benefits must be weighed care fully. The most obvious risk is to privacy. Telepsychiatry visits, like all patient encounters, must be secure and confidential. Given the growing suspicion among the public and professionals who use computers that all data are at risk, clinicians must take appropriate cautions and, at the same time, warn patients of the risks. Readily available videoconferencing software, such as Skype, does not provide the level of security that patients expect from health care providers.14
Other common concerns about telepsychiatry are stable access to videoconferencing and the safety from hackers of necessary hardware. Medical device companies have created hardware and software for use in telepsychiatry that provide a Health Insurance Portability and Accountability Act-compliant high-quality, stable, videoconferencing visit.
Do patients benefit?
Clinically, patients have fared well when they receive care through telepsychiatry. In some studies, however, clinicians have expressed some dissatisfaction with the technology13— understandable, given the value that psychiatry traditionally has put on sitting with the patient. As Knoedler15 described it, making the switch to telepsychiatry from in-person contact can engender loneliness in some physicians; not only is patient contact shifted to videoconferencing, but the psychiatrist loses the supportive environment of a busy clinical practice. Knoedler also pointed out that, on the other hand, telepsychiatry offers practitioners the opportunity to evaluate and treat people who otherwise would not have mental health care.
Obstacles—practical, knotty ones
Reimbursement and licensing. These are 2 pressing problems of telepsychiatry, although recent policy developments will help expand telepsychiatry and make it more appealing to physicians:
• Medicare reimburses for telepsychiatry in non-metropolitan areas.
• In 41 states, Medicaid has included telepsychiatry as a benefit.16
• Nine states offer a specific medical license for practicing telepsychiatry17 (in the remaining states, a full medical license must be obtained before one can provide telemedicine services).
• The Joint Commission has included language in its regulations that could expedite privileging of telepsychiatrists.18
Even with such advancements, problems with licensure, credentialing, privacy, security, confidentiality, informed consent, and professional liability remain.19 I urge you to do your research on these key areas before plunging in.
Changes to models of care. The risk that telepsychiatry poses to various models of care has to be considered. Telepsychiatry is a dramatic innovation, but it should be used to support only high-quality, evidence-based care to which patients are entitled.20 With new technology—as with new medications—use must be carefully monitored and scrutinized.
Although evidence of the value of telepsychiatry is growing, many methods of long-distance practice are still in their infancy. Data must be collected and poor outcomes assessed honestly to ensure that the “more-good-than-harm” mandate is met.
Good reasons to call this shift ‘inevitable’
The future of telepsychiatry includes expansion into new areas of practice. The move to providing services to patients where they happen to be—at work or home— seems inevitable:
• In rural areas, practitioners can communicate with patients so that they are cared for in their homes, without the expense of transportation.
• Employers can invest in workplace health clinics that use telemedicine services to reduce absenteeism.
• For psychiatrists, the ability to provide services to patients across a wide region, from a single convenient location, and at lower cost is an attractive prospect.
To conclude: telepsychiatry holds potential to provide greater reimbursement and improved quality of life for psychiatrists and patients: It allows physicians to choose where they live and work, and limits the number of unreimbursed commutes, and gives patients access to psychiatric care locally, without disruptive travel and delays.
Bottom Line
The exchange of medical information from 1 site to another by means of electronic communication has great potential to improve the health of patients and to alleviate the shortage of psychiatric practitioners across regions and settings. Pediatric, geriatric, and correctional psychiatry stand to benefit because of the nature of the patients and locations.
Related Resources
• American Telemedicine Association. Practice guidelines for video-based online mental health services. http://www. americantelemed.org/docs/default-source/standards/practice-guidelines-for-video-based-online-mental-health-services. pdf?sfvrsn=6. Published May 2013. Accessed February 10, 2015.
• Freudenberg N, Yellowlees PM. Telepsychiatry as part of a comprehensive care plan. Virtual Mentor. 2014;16(12):964-968.
• Kornbluh R. Telepsychiatry is a tool that we must exploit. Clinical Psychiatry News. August 7, 2014. http://www. clinicalpsychiatrynews.com/home/article/telepsychiatry-is-a-tool-that-we-must-exploit/28c87bec298e0aa208309fa 9bc48dedc.html.
• University of Colorado Denver. Telemental Health Guide. http:// www.tmhguide.org.
Disclosure
Dr. Kornbluh reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Too few psychiatrists. A growing number of patients. A new federal law, technological advances, and a generational shift in the way people communicate. Add them together and you have the perfect environment for telepsychiatry—the remote practice of psychiatry by means of telemedicine—to take root (Box 1). Although telepsychiatry has, in various forms, been around since the 1950s,1 only recently has it expanded into almost all areas of psychiatric practice.
Here are some observations from my daily work on why I see this method of delivering mental health care is poised to expand in 2015 and beyond. Does telepsychiatry make sense for you?
Lack of supply is a big driver
There are simply not enough psychiatrists where they are needed, which is the primary driver of the expansion of telepsychiatry. With 77% of counties in the United States reporting a shortage of psychiatrists2 and the “graying” of the psychiatric workforce,3 a more efficient way to make use of a psychiatrist’s time is needed. Telepsychiatry eliminates travel time and allows psychiatrists to visit distant sites virtually.
The shortage of psychiatric practitioners that we see today is only going to become worse. The Patient Protection and Affordable Care Act of 2010 includes mental health care and substance abuse treatment among its 10 essential benefits; just as important, new rules arising from the Mental Health Parity and Addiction Equity Act of 2008 limit restrictions on access to mental health care when insurance provides such coverage.4 These legislative initiatives likely will lead to increased demand for psychiatrists in all care settings—from outpatient consults to acute inpatient admissions.
Why so attractive an option?
The shortage of psychiatrists creates limitations on access to care. Fortunately, telemedicine has entered a new age, ushered in by widely available teleconferencing technology. Specialists from dermatology to surgery currently are using telemedicine; psychiatry is a good fit for telemedicine because of (1) the limited amount of “touch” required to make a psychiatric assessment, (2) significant improvements in video quality in recent years, and (3) a decrease in the stigma associated with visiting a psychiatrist.
A generation raised on the Internet is entering the health care marketplace. These consumers and clinicians are accustomed to using video for many daily activities, and they seek health information from the Web. Visiting a psychiatrist through teleconferencing isn’t strange or alienating to this generation; their comfort with technology allows them to have intimate exchanges on video.
Subspecialty particulars
The earliest adopters, not surprisingly, are in areas where the strain of shortage has been felt most, with pediatric, geriatric, and correctional psychiatrists leading the way. In these fields, a substantial literature supports the use of telepsychiatry from a number of practice perspectives.
Pediatric psychiatry. The literature shows that children, families, and clinicians are, on the whole, satisfied with telepsychiatry.5 Children and adolescents who have been shown to benefit from telepsychiatry include those with depression,6 posttraumatic stress disorder, and eating disorders.7 Based on a case series, some authors have asserted that telepsychiatry might be preferable to in-person treatment (Box 2).8
Geriatric psychiatry. Research shows that geriatric patients, who are most likely to feel threatened by new technology, accept telepsychiatry visits.9 For psychiatrists treating geriatric patients, telepsychiatry can significantly lower costs by cutting commuting10 and make more accessible for patients whose age makes them unable to drive.
Correctional psychiatry. Clinicians working in correctional psychiatry have been at the forefront of experimentation with telepsychiatry. The technology is a natural fit for this setting:
• Prisons often are located in remote locations.
• Psychiatrists can be reluctant to provide on-site services because of safety concerns.
With correctional telepsychiatry, not only are patient outcomes comparable with in-person psychiatry, but the cost of delivering care can be significantly lower.11 With the U.S. Department of Justice reporting that 50% of inmates have a diagnosable mental disorder, including substance abuse,12 the need for access to a psychiatrist in the correctional system is acute.
Telepsychiatry can confidently be provided in a number of settings:
• emergency rooms
• nursing homes
• offices of primary care physicians
• in-home care.
Clinical services in these settings have been offered, studied, and reviewed.13
Can confidentiality and security be assured?
As with any new medical tool, the risk and benefits must be weighed care fully. The most obvious risk is to privacy. Telepsychiatry visits, like all patient encounters, must be secure and confidential. Given the growing suspicion among the public and professionals who use computers that all data are at risk, clinicians must take appropriate cautions and, at the same time, warn patients of the risks. Readily available videoconferencing software, such as Skype, does not provide the level of security that patients expect from health care providers.14
Other common concerns about telepsychiatry are stable access to videoconferencing and the safety from hackers of necessary hardware. Medical device companies have created hardware and software for use in telepsychiatry that provide a Health Insurance Portability and Accountability Act-compliant high-quality, stable, videoconferencing visit.
Do patients benefit?
Clinically, patients have fared well when they receive care through telepsychiatry. In some studies, however, clinicians have expressed some dissatisfaction with the technology13— understandable, given the value that psychiatry traditionally has put on sitting with the patient. As Knoedler15 described it, making the switch to telepsychiatry from in-person contact can engender loneliness in some physicians; not only is patient contact shifted to videoconferencing, but the psychiatrist loses the supportive environment of a busy clinical practice. Knoedler also pointed out that, on the other hand, telepsychiatry offers practitioners the opportunity to evaluate and treat people who otherwise would not have mental health care.
Obstacles—practical, knotty ones
Reimbursement and licensing. These are 2 pressing problems of telepsychiatry, although recent policy developments will help expand telepsychiatry and make it more appealing to physicians:
• Medicare reimburses for telepsychiatry in non-metropolitan areas.
• In 41 states, Medicaid has included telepsychiatry as a benefit.16
• Nine states offer a specific medical license for practicing telepsychiatry17 (in the remaining states, a full medical license must be obtained before one can provide telemedicine services).
• The Joint Commission has included language in its regulations that could expedite privileging of telepsychiatrists.18
Even with such advancements, problems with licensure, credentialing, privacy, security, confidentiality, informed consent, and professional liability remain.19 I urge you to do your research on these key areas before plunging in.
Changes to models of care. The risk that telepsychiatry poses to various models of care has to be considered. Telepsychiatry is a dramatic innovation, but it should be used to support only high-quality, evidence-based care to which patients are entitled.20 With new technology—as with new medications—use must be carefully monitored and scrutinized.
Although evidence of the value of telepsychiatry is growing, many methods of long-distance practice are still in their infancy. Data must be collected and poor outcomes assessed honestly to ensure that the “more-good-than-harm” mandate is met.
Good reasons to call this shift ‘inevitable’
The future of telepsychiatry includes expansion into new areas of practice. The move to providing services to patients where they happen to be—at work or home— seems inevitable:
• In rural areas, practitioners can communicate with patients so that they are cared for in their homes, without the expense of transportation.
• Employers can invest in workplace health clinics that use telemedicine services to reduce absenteeism.
• For psychiatrists, the ability to provide services to patients across a wide region, from a single convenient location, and at lower cost is an attractive prospect.
To conclude: telepsychiatry holds potential to provide greater reimbursement and improved quality of life for psychiatrists and patients: It allows physicians to choose where they live and work, and limits the number of unreimbursed commutes, and gives patients access to psychiatric care locally, without disruptive travel and delays.
Bottom Line
The exchange of medical information from 1 site to another by means of electronic communication has great potential to improve the health of patients and to alleviate the shortage of psychiatric practitioners across regions and settings. Pediatric, geriatric, and correctional psychiatry stand to benefit because of the nature of the patients and locations.
Related Resources
• American Telemedicine Association. Practice guidelines for video-based online mental health services. http://www. americantelemed.org/docs/default-source/standards/practice-guidelines-for-video-based-online-mental-health-services. pdf?sfvrsn=6. Published May 2013. Accessed February 10, 2015.
• Freudenberg N, Yellowlees PM. Telepsychiatry as part of a comprehensive care plan. Virtual Mentor. 2014;16(12):964-968.
• Kornbluh R. Telepsychiatry is a tool that we must exploit. Clinical Psychiatry News. August 7, 2014. http://www. clinicalpsychiatrynews.com/home/article/telepsychiatry-is-a-tool-that-we-must-exploit/28c87bec298e0aa208309fa 9bc48dedc.html.
• University of Colorado Denver. Telemental Health Guide. http:// www.tmhguide.org.
Disclosure
Dr. Kornbluh reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Shore JH. Telepsychiatry: videoconferencing in the delivery of psychiatric care. Am J Psychiatry. 2013;170(3):256-262.
2. Konrad TR, Ellis AR, Thomas KC, et al. County-level estimates of need for mental health professionals in the United States. Psychiatr Serv. 2009;60(10):1307-1314.
3. Vernon DJ, Salsberg E, Erikson C, et al. Planning the future mental health workforce: with progress on coverage, what role will psychiatrists play? Acad Psychiatry. 2009;33(3):187-192.
4. Carrns A. Understanding new rules that widen mental health coverage. The New York Times. http://www. nytimes.com/2014/01/10/your-money/understanding-new-rules-that-widen-mental-health-coverage.html. Published January 9, 2014. Accessed February 10, 2015.
5. Myers KM, Valentine JM, Melzer SM. Feasibility, acceptability, and sustainability of telepsychiatry for children and adolescents. Psychiatr Serv. 2007;58(11):1493-1496.
6. Nelson EL, Barnard M, Cain S. Treating childhood depression over videoconferencing. Telemed J E Health. 2003;9(1):49-55.
7. Boydell KM, Hodgins M, Pignatiello A, et al. Using technology to deliver mental health services to children and youth: a scoping review. J Can Acad Child Adolesc Psychiatry. 2014;23(2):87-99.
8. Pakyurek M, Yellowlees P, Hilty D. The child and adolescent telepsychiatry consultation: can it be a more effective clinical process for certain patients than conventional practice? Telemed J E Health. 2010;16(3):289-292.
9. Poon P, Hui E, Dai D, et al. Cognitive intervention for community-dwelling older persons with memory problems: telemedicine versus face-to-face treatment. Int J Geriatr Psychiatry. 2005;20(3):285-286.
10. Rabinowitz T, Murphy KM, Amour JL, et al. Benefits of a telepsychiatry consultation service for rural nursing home residents. Telemed J E Health. 2010;16(1):34-40.
11. Deslich SA, Thistlethwaite T, Coustasse A. Telepsychiatry in correctional facilities: using technology to improve access and decrease costs of mental health care in underserved populations. Perm J. 2013;17(3):80-86.
12. James DJ, Glaze LE. Mental health problems of prison and jail inmates. U.S. Department of Justice, Office of Justice Programs. http://www.bjs.gov/content/pub/pdf/mhppji. pdf. Updated December 14, 2006. Accessed February 10, 2015.
13. Hilty DN, Ferrer DC, Parish MB, et al. The effectiveness of telemental health: a 2013 review. Telemed J E Health. 2013;19(6):444-454.
14. Maheu MM, Mcmenamin J. Telepsychiatry: the perils of using skype. Psychiatric Times. http://www. psychiatrictimes.com/blog/telepsychiatry-perils-using-skype. Published March 28, 2013. Accessed February 10, 2015.
15. Knoedler DW. Telepsychiatry: first week in the trenches. Psychiatric Times. http://www.psychiatrictimes.com/ blogs/couch-crisis/telepsychiatry-first-week-trenches. Published January 22, 2014. Accessed February 15, 2015.
16. Secure Telehealth. Medicaid reimburses for telehealth in 41 states. http://www.securetelehealth.com/medicaid-reimbursement.html. Updated January 15, 2015. Accessed February 10, 2015.
17. Federation of State Medical Boards. Telemedicine overview: Board-by-Board approach. http://library.fsmb.org/pdf/ grpol_telemedicine_licensure.pdf. Updated June 2013. Accessed February 10, 2015.
18. Joint Commission Perspectives. Accepted: final revisions to telemedicine standards. http://www.jointcommission. org/assets/1/6/Revisions_telemedicine_standards.pdf. Published January 2012. Accessed February 10, 2015.
19. Hyler SE, Gangure DP. Legal and ethical challenges in telepsychiatry. J Psychiatr Pract. 2004;10(4):272-276.
20. Kornbluh RA. Staying true to the mission: adapting telepsychiatry to a new environment. CNS Spectr. 2014;19(6):482-483.
1. Shore JH. Telepsychiatry: videoconferencing in the delivery of psychiatric care. Am J Psychiatry. 2013;170(3):256-262.
2. Konrad TR, Ellis AR, Thomas KC, et al. County-level estimates of need for mental health professionals in the United States. Psychiatr Serv. 2009;60(10):1307-1314.
3. Vernon DJ, Salsberg E, Erikson C, et al. Planning the future mental health workforce: with progress on coverage, what role will psychiatrists play? Acad Psychiatry. 2009;33(3):187-192.
4. Carrns A. Understanding new rules that widen mental health coverage. The New York Times. http://www. nytimes.com/2014/01/10/your-money/understanding-new-rules-that-widen-mental-health-coverage.html. Published January 9, 2014. Accessed February 10, 2015.
5. Myers KM, Valentine JM, Melzer SM. Feasibility, acceptability, and sustainability of telepsychiatry for children and adolescents. Psychiatr Serv. 2007;58(11):1493-1496.
6. Nelson EL, Barnard M, Cain S. Treating childhood depression over videoconferencing. Telemed J E Health. 2003;9(1):49-55.
7. Boydell KM, Hodgins M, Pignatiello A, et al. Using technology to deliver mental health services to children and youth: a scoping review. J Can Acad Child Adolesc Psychiatry. 2014;23(2):87-99.
8. Pakyurek M, Yellowlees P, Hilty D. The child and adolescent telepsychiatry consultation: can it be a more effective clinical process for certain patients than conventional practice? Telemed J E Health. 2010;16(3):289-292.
9. Poon P, Hui E, Dai D, et al. Cognitive intervention for community-dwelling older persons with memory problems: telemedicine versus face-to-face treatment. Int J Geriatr Psychiatry. 2005;20(3):285-286.
10. Rabinowitz T, Murphy KM, Amour JL, et al. Benefits of a telepsychiatry consultation service for rural nursing home residents. Telemed J E Health. 2010;16(1):34-40.
11. Deslich SA, Thistlethwaite T, Coustasse A. Telepsychiatry in correctional facilities: using technology to improve access and decrease costs of mental health care in underserved populations. Perm J. 2013;17(3):80-86.
12. James DJ, Glaze LE. Mental health problems of prison and jail inmates. U.S. Department of Justice, Office of Justice Programs. http://www.bjs.gov/content/pub/pdf/mhppji. pdf. Updated December 14, 2006. Accessed February 10, 2015.
13. Hilty DN, Ferrer DC, Parish MB, et al. The effectiveness of telemental health: a 2013 review. Telemed J E Health. 2013;19(6):444-454.
14. Maheu MM, Mcmenamin J. Telepsychiatry: the perils of using skype. Psychiatric Times. http://www. psychiatrictimes.com/blog/telepsychiatry-perils-using-skype. Published March 28, 2013. Accessed February 10, 2015.
15. Knoedler DW. Telepsychiatry: first week in the trenches. Psychiatric Times. http://www.psychiatrictimes.com/ blogs/couch-crisis/telepsychiatry-first-week-trenches. Published January 22, 2014. Accessed February 15, 2015.
16. Secure Telehealth. Medicaid reimburses for telehealth in 41 states. http://www.securetelehealth.com/medicaid-reimbursement.html. Updated January 15, 2015. Accessed February 10, 2015.
17. Federation of State Medical Boards. Telemedicine overview: Board-by-Board approach. http://library.fsmb.org/pdf/ grpol_telemedicine_licensure.pdf. Updated June 2013. Accessed February 10, 2015.
18. Joint Commission Perspectives. Accepted: final revisions to telemedicine standards. http://www.jointcommission. org/assets/1/6/Revisions_telemedicine_standards.pdf. Published January 2012. Accessed February 10, 2015.
19. Hyler SE, Gangure DP. Legal and ethical challenges in telepsychiatry. J Psychiatr Pract. 2004;10(4):272-276.
20. Kornbluh RA. Staying true to the mission: adapting telepsychiatry to a new environment. CNS Spectr. 2014;19(6):482-483.

Clearing up confusion
“Mr. Smith seems somewhat confused today” is one of the most serious and concerning pre-visit reports you can receive from your staff or the patient’s family. Such a descriptor can be confusing—pardon the pun—not only for the patient, but to even seasoned mental health providers.
The term confusion can be code for diagnoses ranging from deliriuma to a progressive neurocognitive disorder (NCD) such as major NCD due to Alzheimer’s disease (AD), or even a more challenging problem such as beclouded dementia (delirium superimposed on dementia/NCD). It is essential for all mental health professionals to have an evidence-based approach when encountering signs or symptoms of confusion.
aICD-10 code R41.0 encompasses Confusion, Other Specified Delirium, or Unspecified Delirium.
CASE REPORT
Ms. T, age 62, has hypothyroidism and bipolar I disorder, most recently depressed, with comorbid generalized anxiety disorder. She has been taking lithium, 600 mg/d, to control her mood symptoms. Her daughter-in-law reports that Ms. T has been exhibiting increasing signs of confusion. During the office evaluation, Ms. T minimizes her symptoms, only describing mild issues with forgetfulness while cooking and concern over increasing anxiety. Her daughter-in-law plays a voicemail message from earlier in the week, in which Ms. T’s speech is halting, disorganized, and in a word, confused. I decide to use the mnemonic decision chart MR. MIND (Table 1) to get to the bottom of her recent confusion.

Measure cognition
It is nice to receive advanced warning about a cognitive change or a change in activities of daily living; however, many patients present with subtle, sub-acute changes that are more difficult to assess. When encountering a broad symptom such as “confusion”—which has an equally broad differential diagnosis—systematic assessment of the current cognitive state compared with the patient’s baseline becomes the first order of business. However, this requires that the patient has had a baseline cognitive assessment.
In my practice, I often administer one of the validated neurocognitive screening instruments when a patient first begins care—even a brief test such as the Mini- Cog (3-item recall plus clock drawing test), which is comparable to longer screening tests at least for NCD/dementia.1 During a presentation for confusion, a more detailed neurocognitive assessment instrument would be recommended, allowing one to marry the clinical impression with a validated, objective measure. Formal neuropsychological testing by a clinical neuropsychologist is the gold standard, but such testing is time-consuming and expensive and often not readily available. The screening instrument I use for a more thorough evaluation depends on the clinical scenario.
The Six-Item Screener is used in some emergency settings because it is short but boasts a higher sensitivity than the Mini- Cog (94% vs 75%) with similar specificity when screening for cognitive impairment.2 The Mini-Mental State Examination (MMSE) is a valuable instrument, although, recently, the Saint Louis University Mental Status Examination has been thought to be better at detecting mild NCD than the MMSE; more data are needed to substantiate this claim.3 The Montreal Cognitive Assessment is another validated screening tool that has been shown to be superior to the MMSE in terms of screening for mild cognitive impairment.4 The best delirium-specific assessment tool is the Confusion Assessment Method (Table 2).5
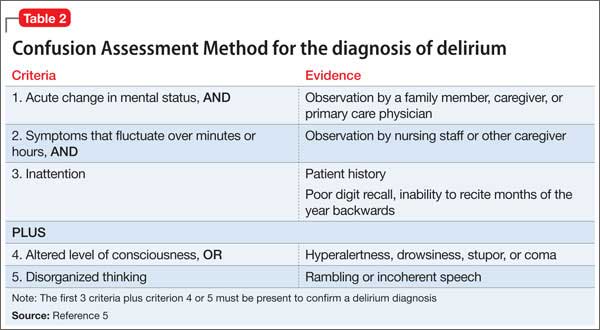
Ms. T’s MMSE score was 26/30, down from 29/30 at baseline. Her score fell below the cutoff score of 27 for mild cognitive impairment for someone with at least 8 years of completed education. Her results were abnormal mainly in the memory domain (3-item recall), raising the question of a possible prodromal state of AD although the acute nature of the change made delirium or mild NCD high in the differential.
Review medications
A review of the medication list is not just a Joint Commission mandate (medication reconciliation during each encounter) but is important whenever confusion is noted. Polypharmacy can be a concern, but is not as concerning as the class of medication prescribed, particularly anticholinergic and sedative medications in patients age >65. The Drug Burden Index can be helpful in assessing this risk.6 Medications such as the benzodiazepine-receptor agonists, tricyclic antidepressants, and antipsychotics should be discontinued if possible, keeping in mind that the addition or subtraction of medications must be done prudently and only after reviewing the evidence and in consultation with the patient. A detailed medication review is as important for confused outpatients as it is for an inpatient case (steps 2 and 3 of the inpatient algorithm outlined in Table 3).7
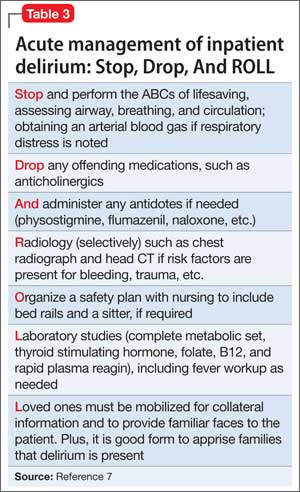
In Ms. T’s case, the primary concern on her medication list was that her medical team was prescribing levothyroxine, 112 mcg/d, and desiccated thyroid (combination thyroxine and triiodothyronine in the form of 20 mg Armour Thyroid), despite a lack of data for such combination therapy. Earlier, I had discontinued lorazepam, leaving lithium, 600 mg/d, quetiapine, 400 mg/d, and escitalopram, 10 mg/d, as her remaining psychotropics. Her other medications included atorvastatin, 40 mg/d, for hyper-lipidemia and metformin, 750 mg/d, for type 2 diabetes mellitus.
Medical illness
An organic basis must rank high in the differential diagnosis if medications are not the culprit. There are myriad medical disorders that can lead to confusion (Table 4).8 In an outpatient psychiatric setting, laboratory and radiology testing might not be readily available. It then becomes important to collaborate with a patient’s medical team if any of the following are met:
•there is high suspicion of a medical cause
•there could be delays in performing a medical workup
•a physical examination is needed.
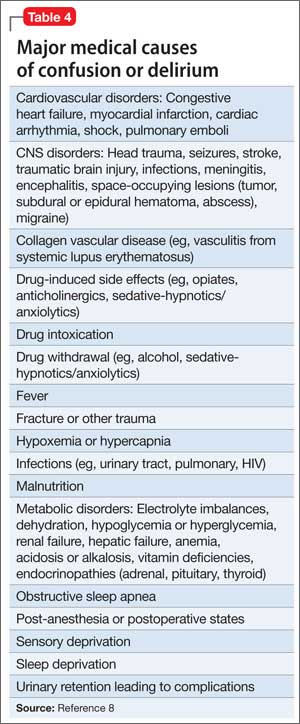
Laboratory work-up should include:
•comprehensive metabolic panel (CMP) to assess for electrolyte derangements and liver or kidney disease
•urinalysis if there are signs of urinary tract infection (low threshold for testing in patients age >65 even if they are asymptomatic)
•urine drug screen or serum alcohol level if substance use is suspected
•complete blood count (CBC) if there are reports of infection (white blood cell count) or blood loss/bruising to ensure that anemia or thrombocytopenia is not playing a role
•thyroid-stimulating hormone (TSH) because thyroid disorders can cause neuropsychiatric as well as somatic symptoms.9
Other laboratory testing could be valuable depending on the clinical scenario. These include tests such as:
•drug level monitoring (lithium, valproic acid, etc.) to assess for toxicity
•HIV and rapid plasma reagin for suspected sexually transmitted infections
•vitamin levels in patients with poor nutrition or post bariatric surgery
•erythrocyte sedimentation rate or C-reactive protein, or both, if there are signs of inflammation
•bacterial culture if blood or tissue infection is a concern.
Esoteric tests include ceruloplasmin (Wilson’s disease), heavy metals screen, and even tests such as anti-gliadin antibodies because the prevalence of gluten sensitivity and celiac disease appear to be on the rise and have been associated with neuropsychiatric problems including encephalopathy.10
Brain imaging is an important consideration when a medical differential diagnosis for confusion is formulated. Unfortunately, there is little evidence-based guidance as to when brain imaging should be performed, often leading to overuse of tests such as CT, especially in emergency settings when confusion is noted. From a clinical standpoint, a head CT scan often is best ordered for patients who demonstrate an acute change in mental status, are age >70, are receiving anticoagulation, or have sustained trauma to the head. The key concern would be intracranial hemorrhage. However, some data suggest that the best use of head CT is for patients who have an impaired level of consciousness or a new focal neurologic deficit.11
Apart from more acute changes, a brain MRI study is more helpful than a head CT when evaluating the brain parenchyma for more sub-acute diagnoses such as multiple sclerosis or a brain tumor. T2-weighted hyperintensities seen on an MRI are thought to predict an increased risk of stroke, dementia, and death.
Their discovery should prompt a detailed evaluation for risk factors of stroke and dementia/NCD.12
In Ms. T’s case, she was taking lithium, so it was logical to obtain a trough lithium level 12 hours after the last dose and to check kidney function (serum creatinine to estimate the glomerular filtration rate), which were in the therapeutic/normal range. Her serum lithium level was 0.7 mEq/L. Brain imaging was not ordered, but several other labs (CMP, CBC, hemoglobin A1c [HgbA1c], and TSH) were drawn. These labs were notable for HgbA1c of 5.1% (normal <5.7%) and TSH of 0.5 mIU/L (normal level, 1.5 mIU/L), which is low for someone taking thyroid replacement.
I requested that Ms. T stop Armour Thyroid to address the suppressed TSH. I also requested that she stop metformin because, although hypoglycemia from metformin monotherapy is uncommon, it can happen in older patients. Hypoglycemia associated with metformin also can occur in situations when caloric intake is deficient or when metformin is used in combination with other drugs such as sulfonylureas (ie, glipizide), beta-adrenergic blocking drugs, angiotensin-converting enzyme inhibitors, or even nonsteroidal anti-inflammatory drugs.13
Identifying overlapping psychiatric (or psychological) illness
Symptoms of depression, anxiety, psychosis, and even dissociation can present as confusion. The term pseudodementia describes patients who exhibit cognitive symptoms consistent with NCD but could improve once the underlying mood, thought, anxiety, or personality disorder is treated.
For example, a patient with depression typically exhibits neurovegetative symptoms—such as poor sleep or appetite— amotivation, and low energy. All of these can lead to abrupt-onset cognitive changes, which are a hallmark of pseudodementia rather than the more insidious pattern of mild NCD. In cases of pseudodementia, neurocognitive testing will show impairment that often rapidly improves after the primary psychiatric (or psychological) issue is rectified. Making a diagnosis of pseudodementia at the initial presentation is difficult because neurocognitive tests such as the MMSE often fail to separate depression from true cognitive changes.14 Such a diagnosis typically requires hindsight. Yet, one must also keep in mind that pseudodementia may be part of a NCD prodrome.15
Conversion disorder as well as the dissociative disorders and substance-related disorders are notorious for causing confusion. In Ms. T’s case, pseudodementia stemming from her underlying bipolar disorder and anxiety figured prominently in the differential diagnosis, but she did not have any other overt psychopathology, personality disorder, or signs of malingering to further complicate her picture.
Notebook. I recommend that my patients keep a small notebook to record medical data ranging from blood pressure and glycemic measurements to details about sleep and dietary intake. Such data comprise the necessary metrics to properly assess target conditions and then track changes once treatment is initiated. This exercise not only yields much-needed detail about the patient’s condition for the clinician; the act of journaling also can be therapeutic for the writer through a process known as experimental disclosure, in which writing down one’s thoughts and observations has a positive impact on the writer’s physical health and psychology.16
Diagnosis. The first rule in medicine (perhaps the second, behind primum non nocere) is to determine what you are treating before beginning treatment (decernite quid tractemus, prius cura ministrandi, for Latin buffs). This means trying to fashion the best diagnostic label, even if it is merely a place-holder, while assessment of the confused state continues. DSM-5 has attempted to remove stigma from several neuropsychiatric disorders. On the cognition front, the new name for dementia is “neurocognitive disorder (NCD),” the umbrella term that focuses on the decline from a previous level of cognitive functioning. NCD has been divided into mild or major cognitive impairment headings either “with” or “without behavioral disturbance” subspecifiers.17
Aside from NCD, there are several other diagnoses in the differential for confusion. Delirium remains the most prominent and focuses on disturbances in attention and orientation that develops over a short period of time, with a change seen in an additional cognitive domain, such as memory, but not in the context of a severely reduced level of arousal such as coma. Subjective cognitive impairment (SCI) is when subjective complaints of cognitive impairment are hallmark compared with objective findings—with evidence suggesting that the presence of SCI could predict a 4.5 times higher rate of developing mild cognitive impairment (MCI) over 7 years.18 MCI was originally used to describe the early prodrome of AD, minus functional decline.
Treatment
After even a provisional diagnosis comes the final, all-important challenge: treating the neuropsychiatric symptoms (NPS) of the confused patient. NPS are nearly universal in NCD/delirium throughout the course of illness. There are no FDA-approved treatments for the NPS associated with these conditions. In terms of treating delirium, the best approach is to treat the underlying medical condition. For control of behavior, which can range from agitated to psychotic to hypoactive, nonpharmacotherapeutic interventions are paramount; they include making sure that the patient is at the appropriate level of care, which, for the confused outpatient, could mean hospitalization. Ensuring proper nutrition, hydration, sensory care (hearing aids, glasses, etc.), and stability in ambulation must be done before considering pharmacotherapy.
Antipsychotic use has been the mainstay of drug treatment of behavioral dyscontrol. Haloperidol has been the traditional go-to medication because there is no evidence that low-dose haloperidol (<3 mg/d) has any different efficacy compared with the atypical antipsychotics or has a greater frequency of adverse drug effects. However, high-dose haloperidol (>4.5 mg/d) was associated with a greater incidence of adverse effects, mainly parkinsonism, than atypical antipsychotics.19 Neither the typical nor atypical antipsychotics have shown mortality benefit—the real outcome measure of interest.
In terms of treating major (or minor) NCD, there are only 2 FDA-approved medication classes: cholinesterase inhibitors (donepezil, galantamine, rivastigmine, etc.) and memantine. However, these medication classes—even when combined together—have only shown marginal benefit in terms of improving cognition. Worse, even when given early in the course of illness they do not reduce the rate of NCD. For pseudodementia, selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors tend to form the mainstay of treating underlying depression or anxiety leading to cognitive changes. Preliminary data suggest that some SSRIs might improve cognition in terms of processing speed, verbal learning, and memory.20 More studies are needed before definitive conclusions can be drawn.
For the confused patient, a personalized therapeutic program, in which multiple interventions are considered at once (targeting all areas of the patient’s life) is gaining research traction. For example, a novel, comprehensive program involving multiple modalities designed to achieve metabolic enhancement for neurodegeneration (MEND) recently has shown robust benefit for patients with AD, MCI, and SCI.21 Using an individual approach to improve diet, activity, sleep, metabolic status including body mass index, and several other markers that affect neural plasticity, researchers demonstrated symptom improvement in 9 of 10 study patients.
Yet, some of the interventions, such as the use of statins for hyperlipidemia, remain controversial, with some studies suggesting that they help cognition,22,23 and others showing no association.24 The researchers caution that further research is warranted before costly dementia prevention trials with statins are undertaken. It does not appear that there are current MEND-type research projects in delirium but it’s to be hoped that we will see these in the future.
In the case of Ms. T, the cause of delirium vs mild NCD was thought to be multifactorial. Discontinuing Armour Thyroid and metformin—symptoms of hypoglycemia emerged as a leading concern—were simple adjustments that led to resolution of the most concerning elements of her confusion. She continued her other psychotropics, although there might be mild residual cognitive issues that warrant close observation.
Related Resources
• Lin JS, O’Connor E, Rossum RC, et al. Screening for cognitive impairment in older adults: an evidence update for the U.S. Preventive Services Task Force. Rockville, MD: Agency for Healthcare Research and Quality (US); 2013.
• Grover S, Kate N. Assessment scales for delirium: a review. World J Psychiatry. 2012;2(4):58-70.
Drug Brand Names
Atorvastatin • Lipitor Lithium • Eskalith, Lithobid
Donepezil • Aricept Lorazepam • Ativan
Escitalopram • Lexapro Memantine • Namenda
Flumazenil • Romazicon Metformin • Glucophage
Galantamine • Razadyne Naloxone • Narcan
Glipizide • Glucotrol Physostigmine • Antilirium
Haloperidol • Haldol Quetiapine • Seroquel
Levothyroxine • Levoxyl, Synthroid Rivastigmine • Exelon
Lithium • Eskalith, Lithobid Valproic acid • Depakene
Disclosure
Dr. Raj is a speaker for Actavis Pharmaceuticals, AstraZeneca, and Merck.
1. Borson S, Scanlan JM, Chen P, et al. The Mini-Cog as a screen for dementia: validation in a population-based sample. J Am Geriatr Soc. 2003;51(10):1451-1454.
2. Wilber ST, Lofgren SD, Mager TG, et al. An evaluation of two screening tools for cognitive impairment in older emergency department patients. Acad Emerg Med. 2005;12(7):612-616.
3. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
4. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695- 699.
5. Inouye S, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Int Med. 1990;113(12):941-948.
6. Hillmer SN, Mager DE, Simonsick EM, et al. A drug burden index to define the functional burden of medications in older people. Arch Intern Med. 2007;167(8):781-787.
7. Raj YP. Psychiatric emergencies. In: Jiang W, Gagliardi JP, Krishnan KR, eds. Clinician’s guide to psychiatric care. New York, NY: Oxford University Press; 2009:33-40.
8. Liptzin B. Clinical diagnosis and management of delirium. In: Stoudemire A, Fogel BS, Greenberg DB, eds. Psychiatric care of the medical patient. 2nd ed. New York, NY: Oxford University Press; 2000:581-596.
9. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
10. Poloni N, Vender S, Bolla E, et al. Gluten encephalopathy with psychiatric onset: case report. Clin Pract Epidemiol Ment Health. 2009;5:16.
11. Naughton BJ, Moran M, Ghaly Y, et al. Computed tomography scanning and delirium in elder patients. Acad Emerg Med. 1997;4(12):1107-1110.
12. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. doi: 10.1136/bmj.c3666.
13. Zitzmann S, Reimann IR, Schmechel H. Severe hypoglycemia in an elderly patient treated with metformin. Int J Clin Pharmacol Ther. 2002;40(3):108-110.
14. Benson AD, Slavin MJ, Tran TT, et al. Screening for early Alzheimer’s Disease: is there still a role for the Mini-Mental State Examination? Prim Care Companion J Clin Psychiatry. 2005;7(2):62-69.
15. Brown WA. Pseudodementia: issues in diagnosis. Psychiatric Times. http://www.psychiatrictimes.com/ pseudodementia-issues-diagnosis. Published April 9, 2005. Accessed February 2, 2015.
16. Frattaroli J. Experimental disclosure and its moderators: a meta-analysis. Psychol Bull. 2006;132(6):823-865.
17. Stetka BS, Correll CU. A guide to DSM-5: neurocognitive disorder. Medscape. http://www.medscape.com/ viewarticle/803884_13. Published May 21, 2013. Accessed October 30, 2014.
18. Reisberg B, Sulman MD, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
19. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.
20. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Intern Clin Psychopharmacol. 2012;27(4):215-223.
21. Bredesen DE. Reversal of cognitive decline: a novel therapeutic program. Aging (Albany NY). 2014;6(9):707-717.
22. Sparks DL, Kryscio RJ, Sabbagh MN, et al. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr Alzheimer Res. 2008;5(4):416-421.
23. Andrade C, Radhakrishnan R. The prevention and treatment of cognitive decline and dementia: an overview of recent research on experimental treatments. Indian J Psychiatry. 2009;51(1):12-25.
24. Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry. 2005;62(2):217-224.
“Mr. Smith seems somewhat confused today” is one of the most serious and concerning pre-visit reports you can receive from your staff or the patient’s family. Such a descriptor can be confusing—pardon the pun—not only for the patient, but to even seasoned mental health providers.
The term confusion can be code for diagnoses ranging from deliriuma to a progressive neurocognitive disorder (NCD) such as major NCD due to Alzheimer’s disease (AD), or even a more challenging problem such as beclouded dementia (delirium superimposed on dementia/NCD). It is essential for all mental health professionals to have an evidence-based approach when encountering signs or symptoms of confusion.
aICD-10 code R41.0 encompasses Confusion, Other Specified Delirium, or Unspecified Delirium.
CASE REPORT
Ms. T, age 62, has hypothyroidism and bipolar I disorder, most recently depressed, with comorbid generalized anxiety disorder. She has been taking lithium, 600 mg/d, to control her mood symptoms. Her daughter-in-law reports that Ms. T has been exhibiting increasing signs of confusion. During the office evaluation, Ms. T minimizes her symptoms, only describing mild issues with forgetfulness while cooking and concern over increasing anxiety. Her daughter-in-law plays a voicemail message from earlier in the week, in which Ms. T’s speech is halting, disorganized, and in a word, confused. I decide to use the mnemonic decision chart MR. MIND (Table 1) to get to the bottom of her recent confusion.
Measure cognition
It is nice to receive advanced warning about a cognitive change or a change in activities of daily living; however, many patients present with subtle, sub-acute changes that are more difficult to assess. When encountering a broad symptom such as “confusion”—which has an equally broad differential diagnosis—systematic assessment of the current cognitive state compared with the patient’s baseline becomes the first order of business. However, this requires that the patient has had a baseline cognitive assessment.
In my practice, I often administer one of the validated neurocognitive screening instruments when a patient first begins care—even a brief test such as the Mini- Cog (3-item recall plus clock drawing test), which is comparable to longer screening tests at least for NCD/dementia.1 During a presentation for confusion, a more detailed neurocognitive assessment instrument would be recommended, allowing one to marry the clinical impression with a validated, objective measure. Formal neuropsychological testing by a clinical neuropsychologist is the gold standard, but such testing is time-consuming and expensive and often not readily available. The screening instrument I use for a more thorough evaluation depends on the clinical scenario.
The Six-Item Screener is used in some emergency settings because it is short but boasts a higher sensitivity than the Mini- Cog (94% vs 75%) with similar specificity when screening for cognitive impairment.2 The Mini-Mental State Examination (MMSE) is a valuable instrument, although, recently, the Saint Louis University Mental Status Examination has been thought to be better at detecting mild NCD than the MMSE; more data are needed to substantiate this claim.3 The Montreal Cognitive Assessment is another validated screening tool that has been shown to be superior to the MMSE in terms of screening for mild cognitive impairment.4 The best delirium-specific assessment tool is the Confusion Assessment Method (Table 2).5
Ms. T’s MMSE score was 26/30, down from 29/30 at baseline. Her score fell below the cutoff score of 27 for mild cognitive impairment for someone with at least 8 years of completed education. Her results were abnormal mainly in the memory domain (3-item recall), raising the question of a possible prodromal state of AD although the acute nature of the change made delirium or mild NCD high in the differential.
Review medications
A review of the medication list is not just a Joint Commission mandate (medication reconciliation during each encounter) but is important whenever confusion is noted. Polypharmacy can be a concern, but is not as concerning as the class of medication prescribed, particularly anticholinergic and sedative medications in patients age >65. The Drug Burden Index can be helpful in assessing this risk.6 Medications such as the benzodiazepine-receptor agonists, tricyclic antidepressants, and antipsychotics should be discontinued if possible, keeping in mind that the addition or subtraction of medications must be done prudently and only after reviewing the evidence and in consultation with the patient. A detailed medication review is as important for confused outpatients as it is for an inpatient case (steps 2 and 3 of the inpatient algorithm outlined in Table 3).7
In Ms. T’s case, the primary concern on her medication list was that her medical team was prescribing levothyroxine, 112 mcg/d, and desiccated thyroid (combination thyroxine and triiodothyronine in the form of 20 mg Armour Thyroid), despite a lack of data for such combination therapy. Earlier, I had discontinued lorazepam, leaving lithium, 600 mg/d, quetiapine, 400 mg/d, and escitalopram, 10 mg/d, as her remaining psychotropics. Her other medications included atorvastatin, 40 mg/d, for hyper-lipidemia and metformin, 750 mg/d, for type 2 diabetes mellitus.
Medical illness
An organic basis must rank high in the differential diagnosis if medications are not the culprit. There are myriad medical disorders that can lead to confusion (Table 4).8 In an outpatient psychiatric setting, laboratory and radiology testing might not be readily available. It then becomes important to collaborate with a patient’s medical team if any of the following are met:
•there is high suspicion of a medical cause
•there could be delays in performing a medical workup
•a physical examination is needed.
Laboratory work-up should include:
•comprehensive metabolic panel (CMP) to assess for electrolyte derangements and liver or kidney disease
•urinalysis if there are signs of urinary tract infection (low threshold for testing in patients age >65 even if they are asymptomatic)
•urine drug screen or serum alcohol level if substance use is suspected
•complete blood count (CBC) if there are reports of infection (white blood cell count) or blood loss/bruising to ensure that anemia or thrombocytopenia is not playing a role
•thyroid-stimulating hormone (TSH) because thyroid disorders can cause neuropsychiatric as well as somatic symptoms.9
Other laboratory testing could be valuable depending on the clinical scenario. These include tests such as:
•drug level monitoring (lithium, valproic acid, etc.) to assess for toxicity
•HIV and rapid plasma reagin for suspected sexually transmitted infections
•vitamin levels in patients with poor nutrition or post bariatric surgery
•erythrocyte sedimentation rate or C-reactive protein, or both, if there are signs of inflammation
•bacterial culture if blood or tissue infection is a concern.
Esoteric tests include ceruloplasmin (Wilson’s disease), heavy metals screen, and even tests such as anti-gliadin antibodies because the prevalence of gluten sensitivity and celiac disease appear to be on the rise and have been associated with neuropsychiatric problems including encephalopathy.10
Brain imaging is an important consideration when a medical differential diagnosis for confusion is formulated. Unfortunately, there is little evidence-based guidance as to when brain imaging should be performed, often leading to overuse of tests such as CT, especially in emergency settings when confusion is noted. From a clinical standpoint, a head CT scan often is best ordered for patients who demonstrate an acute change in mental status, are age >70, are receiving anticoagulation, or have sustained trauma to the head. The key concern would be intracranial hemorrhage. However, some data suggest that the best use of head CT is for patients who have an impaired level of consciousness or a new focal neurologic deficit.11
Apart from more acute changes, a brain MRI study is more helpful than a head CT when evaluating the brain parenchyma for more sub-acute diagnoses such as multiple sclerosis or a brain tumor. T2-weighted hyperintensities seen on an MRI are thought to predict an increased risk of stroke, dementia, and death.
Their discovery should prompt a detailed evaluation for risk factors of stroke and dementia/NCD.12
In Ms. T’s case, she was taking lithium, so it was logical to obtain a trough lithium level 12 hours after the last dose and to check kidney function (serum creatinine to estimate the glomerular filtration rate), which were in the therapeutic/normal range. Her serum lithium level was 0.7 mEq/L. Brain imaging was not ordered, but several other labs (CMP, CBC, hemoglobin A1c [HgbA1c], and TSH) were drawn. These labs were notable for HgbA1c of 5.1% (normal <5.7%) and TSH of 0.5 mIU/L (normal level, 1.5 mIU/L), which is low for someone taking thyroid replacement.
I requested that Ms. T stop Armour Thyroid to address the suppressed TSH. I also requested that she stop metformin because, although hypoglycemia from metformin monotherapy is uncommon, it can happen in older patients. Hypoglycemia associated with metformin also can occur in situations when caloric intake is deficient or when metformin is used in combination with other drugs such as sulfonylureas (ie, glipizide), beta-adrenergic blocking drugs, angiotensin-converting enzyme inhibitors, or even nonsteroidal anti-inflammatory drugs.13
Identifying overlapping psychiatric (or psychological) illness
Symptoms of depression, anxiety, psychosis, and even dissociation can present as confusion. The term pseudodementia describes patients who exhibit cognitive symptoms consistent with NCD but could improve once the underlying mood, thought, anxiety, or personality disorder is treated.
For example, a patient with depression typically exhibits neurovegetative symptoms—such as poor sleep or appetite— amotivation, and low energy. All of these can lead to abrupt-onset cognitive changes, which are a hallmark of pseudodementia rather than the more insidious pattern of mild NCD. In cases of pseudodementia, neurocognitive testing will show impairment that often rapidly improves after the primary psychiatric (or psychological) issue is rectified. Making a diagnosis of pseudodementia at the initial presentation is difficult because neurocognitive tests such as the MMSE often fail to separate depression from true cognitive changes.14 Such a diagnosis typically requires hindsight. Yet, one must also keep in mind that pseudodementia may be part of a NCD prodrome.15
Conversion disorder as well as the dissociative disorders and substance-related disorders are notorious for causing confusion. In Ms. T’s case, pseudodementia stemming from her underlying bipolar disorder and anxiety figured prominently in the differential diagnosis, but she did not have any other overt psychopathology, personality disorder, or signs of malingering to further complicate her picture.
Notebook. I recommend that my patients keep a small notebook to record medical data ranging from blood pressure and glycemic measurements to details about sleep and dietary intake. Such data comprise the necessary metrics to properly assess target conditions and then track changes once treatment is initiated. This exercise not only yields much-needed detail about the patient’s condition for the clinician; the act of journaling also can be therapeutic for the writer through a process known as experimental disclosure, in which writing down one’s thoughts and observations has a positive impact on the writer’s physical health and psychology.16
Diagnosis. The first rule in medicine (perhaps the second, behind primum non nocere) is to determine what you are treating before beginning treatment (decernite quid tractemus, prius cura ministrandi, for Latin buffs). This means trying to fashion the best diagnostic label, even if it is merely a place-holder, while assessment of the confused state continues. DSM-5 has attempted to remove stigma from several neuropsychiatric disorders. On the cognition front, the new name for dementia is “neurocognitive disorder (NCD),” the umbrella term that focuses on the decline from a previous level of cognitive functioning. NCD has been divided into mild or major cognitive impairment headings either “with” or “without behavioral disturbance” subspecifiers.17
Aside from NCD, there are several other diagnoses in the differential for confusion. Delirium remains the most prominent and focuses on disturbances in attention and orientation that develops over a short period of time, with a change seen in an additional cognitive domain, such as memory, but not in the context of a severely reduced level of arousal such as coma. Subjective cognitive impairment (SCI) is when subjective complaints of cognitive impairment are hallmark compared with objective findings—with evidence suggesting that the presence of SCI could predict a 4.5 times higher rate of developing mild cognitive impairment (MCI) over 7 years.18 MCI was originally used to describe the early prodrome of AD, minus functional decline.
Treatment
After even a provisional diagnosis comes the final, all-important challenge: treating the neuropsychiatric symptoms (NPS) of the confused patient. NPS are nearly universal in NCD/delirium throughout the course of illness. There are no FDA-approved treatments for the NPS associated with these conditions. In terms of treating delirium, the best approach is to treat the underlying medical condition. For control of behavior, which can range from agitated to psychotic to hypoactive, nonpharmacotherapeutic interventions are paramount; they include making sure that the patient is at the appropriate level of care, which, for the confused outpatient, could mean hospitalization. Ensuring proper nutrition, hydration, sensory care (hearing aids, glasses, etc.), and stability in ambulation must be done before considering pharmacotherapy.
Antipsychotic use has been the mainstay of drug treatment of behavioral dyscontrol. Haloperidol has been the traditional go-to medication because there is no evidence that low-dose haloperidol (<3 mg/d) has any different efficacy compared with the atypical antipsychotics or has a greater frequency of adverse drug effects. However, high-dose haloperidol (>4.5 mg/d) was associated with a greater incidence of adverse effects, mainly parkinsonism, than atypical antipsychotics.19 Neither the typical nor atypical antipsychotics have shown mortality benefit—the real outcome measure of interest.
In terms of treating major (or minor) NCD, there are only 2 FDA-approved medication classes: cholinesterase inhibitors (donepezil, galantamine, rivastigmine, etc.) and memantine. However, these medication classes—even when combined together—have only shown marginal benefit in terms of improving cognition. Worse, even when given early in the course of illness they do not reduce the rate of NCD. For pseudodementia, selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors tend to form the mainstay of treating underlying depression or anxiety leading to cognitive changes. Preliminary data suggest that some SSRIs might improve cognition in terms of processing speed, verbal learning, and memory.20 More studies are needed before definitive conclusions can be drawn.
For the confused patient, a personalized therapeutic program, in which multiple interventions are considered at once (targeting all areas of the patient’s life) is gaining research traction. For example, a novel, comprehensive program involving multiple modalities designed to achieve metabolic enhancement for neurodegeneration (MEND) recently has shown robust benefit for patients with AD, MCI, and SCI.21 Using an individual approach to improve diet, activity, sleep, metabolic status including body mass index, and several other markers that affect neural plasticity, researchers demonstrated symptom improvement in 9 of 10 study patients.
Yet, some of the interventions, such as the use of statins for hyperlipidemia, remain controversial, with some studies suggesting that they help cognition,22,23 and others showing no association.24 The researchers caution that further research is warranted before costly dementia prevention trials with statins are undertaken. It does not appear that there are current MEND-type research projects in delirium but it’s to be hoped that we will see these in the future.
In the case of Ms. T, the cause of delirium vs mild NCD was thought to be multifactorial. Discontinuing Armour Thyroid and metformin—symptoms of hypoglycemia emerged as a leading concern—were simple adjustments that led to resolution of the most concerning elements of her confusion. She continued her other psychotropics, although there might be mild residual cognitive issues that warrant close observation.
Related Resources
• Lin JS, O’Connor E, Rossum RC, et al. Screening for cognitive impairment in older adults: an evidence update for the U.S. Preventive Services Task Force. Rockville, MD: Agency for Healthcare Research and Quality (US); 2013.
• Grover S, Kate N. Assessment scales for delirium: a review. World J Psychiatry. 2012;2(4):58-70.
Drug Brand Names
Atorvastatin • Lipitor Lithium • Eskalith, Lithobid
Donepezil • Aricept Lorazepam • Ativan
Escitalopram • Lexapro Memantine • Namenda
Flumazenil • Romazicon Metformin • Glucophage
Galantamine • Razadyne Naloxone • Narcan
Glipizide • Glucotrol Physostigmine • Antilirium
Haloperidol • Haldol Quetiapine • Seroquel
Levothyroxine • Levoxyl, Synthroid Rivastigmine • Exelon
Lithium • Eskalith, Lithobid Valproic acid • Depakene
Disclosure
Dr. Raj is a speaker for Actavis Pharmaceuticals, AstraZeneca, and Merck.
“Mr. Smith seems somewhat confused today” is one of the most serious and concerning pre-visit reports you can receive from your staff or the patient’s family. Such a descriptor can be confusing—pardon the pun—not only for the patient, but to even seasoned mental health providers.
The term confusion can be code for diagnoses ranging from deliriuma to a progressive neurocognitive disorder (NCD) such as major NCD due to Alzheimer’s disease (AD), or even a more challenging problem such as beclouded dementia (delirium superimposed on dementia/NCD). It is essential for all mental health professionals to have an evidence-based approach when encountering signs or symptoms of confusion.
aICD-10 code R41.0 encompasses Confusion, Other Specified Delirium, or Unspecified Delirium.
CASE REPORT
Ms. T, age 62, has hypothyroidism and bipolar I disorder, most recently depressed, with comorbid generalized anxiety disorder. She has been taking lithium, 600 mg/d, to control her mood symptoms. Her daughter-in-law reports that Ms. T has been exhibiting increasing signs of confusion. During the office evaluation, Ms. T minimizes her symptoms, only describing mild issues with forgetfulness while cooking and concern over increasing anxiety. Her daughter-in-law plays a voicemail message from earlier in the week, in which Ms. T’s speech is halting, disorganized, and in a word, confused. I decide to use the mnemonic decision chart MR. MIND (Table 1) to get to the bottom of her recent confusion.
Measure cognition
It is nice to receive advanced warning about a cognitive change or a change in activities of daily living; however, many patients present with subtle, sub-acute changes that are more difficult to assess. When encountering a broad symptom such as “confusion”—which has an equally broad differential diagnosis—systematic assessment of the current cognitive state compared with the patient’s baseline becomes the first order of business. However, this requires that the patient has had a baseline cognitive assessment.
In my practice, I often administer one of the validated neurocognitive screening instruments when a patient first begins care—even a brief test such as the Mini- Cog (3-item recall plus clock drawing test), which is comparable to longer screening tests at least for NCD/dementia.1 During a presentation for confusion, a more detailed neurocognitive assessment instrument would be recommended, allowing one to marry the clinical impression with a validated, objective measure. Formal neuropsychological testing by a clinical neuropsychologist is the gold standard, but such testing is time-consuming and expensive and often not readily available. The screening instrument I use for a more thorough evaluation depends on the clinical scenario.
The Six-Item Screener is used in some emergency settings because it is short but boasts a higher sensitivity than the Mini- Cog (94% vs 75%) with similar specificity when screening for cognitive impairment.2 The Mini-Mental State Examination (MMSE) is a valuable instrument, although, recently, the Saint Louis University Mental Status Examination has been thought to be better at detecting mild NCD than the MMSE; more data are needed to substantiate this claim.3 The Montreal Cognitive Assessment is another validated screening tool that has been shown to be superior to the MMSE in terms of screening for mild cognitive impairment.4 The best delirium-specific assessment tool is the Confusion Assessment Method (Table 2).5
Ms. T’s MMSE score was 26/30, down from 29/30 at baseline. Her score fell below the cutoff score of 27 for mild cognitive impairment for someone with at least 8 years of completed education. Her results were abnormal mainly in the memory domain (3-item recall), raising the question of a possible prodromal state of AD although the acute nature of the change made delirium or mild NCD high in the differential.
Review medications
A review of the medication list is not just a Joint Commission mandate (medication reconciliation during each encounter) but is important whenever confusion is noted. Polypharmacy can be a concern, but is not as concerning as the class of medication prescribed, particularly anticholinergic and sedative medications in patients age >65. The Drug Burden Index can be helpful in assessing this risk.6 Medications such as the benzodiazepine-receptor agonists, tricyclic antidepressants, and antipsychotics should be discontinued if possible, keeping in mind that the addition or subtraction of medications must be done prudently and only after reviewing the evidence and in consultation with the patient. A detailed medication review is as important for confused outpatients as it is for an inpatient case (steps 2 and 3 of the inpatient algorithm outlined in Table 3).7
In Ms. T’s case, the primary concern on her medication list was that her medical team was prescribing levothyroxine, 112 mcg/d, and desiccated thyroid (combination thyroxine and triiodothyronine in the form of 20 mg Armour Thyroid), despite a lack of data for such combination therapy. Earlier, I had discontinued lorazepam, leaving lithium, 600 mg/d, quetiapine, 400 mg/d, and escitalopram, 10 mg/d, as her remaining psychotropics. Her other medications included atorvastatin, 40 mg/d, for hyper-lipidemia and metformin, 750 mg/d, for type 2 diabetes mellitus.
Medical illness
An organic basis must rank high in the differential diagnosis if medications are not the culprit. There are myriad medical disorders that can lead to confusion (Table 4).8 In an outpatient psychiatric setting, laboratory and radiology testing might not be readily available. It then becomes important to collaborate with a patient’s medical team if any of the following are met:
•there is high suspicion of a medical cause
•there could be delays in performing a medical workup
•a physical examination is needed.
Laboratory work-up should include:
•comprehensive metabolic panel (CMP) to assess for electrolyte derangements and liver or kidney disease
•urinalysis if there are signs of urinary tract infection (low threshold for testing in patients age >65 even if they are asymptomatic)
•urine drug screen or serum alcohol level if substance use is suspected
•complete blood count (CBC) if there are reports of infection (white blood cell count) or blood loss/bruising to ensure that anemia or thrombocytopenia is not playing a role
•thyroid-stimulating hormone (TSH) because thyroid disorders can cause neuropsychiatric as well as somatic symptoms.9
Other laboratory testing could be valuable depending on the clinical scenario. These include tests such as:
•drug level monitoring (lithium, valproic acid, etc.) to assess for toxicity
•HIV and rapid plasma reagin for suspected sexually transmitted infections
•vitamin levels in patients with poor nutrition or post bariatric surgery
•erythrocyte sedimentation rate or C-reactive protein, or both, if there are signs of inflammation
•bacterial culture if blood or tissue infection is a concern.
Esoteric tests include ceruloplasmin (Wilson’s disease), heavy metals screen, and even tests such as anti-gliadin antibodies because the prevalence of gluten sensitivity and celiac disease appear to be on the rise and have been associated with neuropsychiatric problems including encephalopathy.10
Brain imaging is an important consideration when a medical differential diagnosis for confusion is formulated. Unfortunately, there is little evidence-based guidance as to when brain imaging should be performed, often leading to overuse of tests such as CT, especially in emergency settings when confusion is noted. From a clinical standpoint, a head CT scan often is best ordered for patients who demonstrate an acute change in mental status, are age >70, are receiving anticoagulation, or have sustained trauma to the head. The key concern would be intracranial hemorrhage. However, some data suggest that the best use of head CT is for patients who have an impaired level of consciousness or a new focal neurologic deficit.11
Apart from more acute changes, a brain MRI study is more helpful than a head CT when evaluating the brain parenchyma for more sub-acute diagnoses such as multiple sclerosis or a brain tumor. T2-weighted hyperintensities seen on an MRI are thought to predict an increased risk of stroke, dementia, and death.
Their discovery should prompt a detailed evaluation for risk factors of stroke and dementia/NCD.12
In Ms. T’s case, she was taking lithium, so it was logical to obtain a trough lithium level 12 hours after the last dose and to check kidney function (serum creatinine to estimate the glomerular filtration rate), which were in the therapeutic/normal range. Her serum lithium level was 0.7 mEq/L. Brain imaging was not ordered, but several other labs (CMP, CBC, hemoglobin A1c [HgbA1c], and TSH) were drawn. These labs were notable for HgbA1c of 5.1% (normal <5.7%) and TSH of 0.5 mIU/L (normal level, 1.5 mIU/L), which is low for someone taking thyroid replacement.
I requested that Ms. T stop Armour Thyroid to address the suppressed TSH. I also requested that she stop metformin because, although hypoglycemia from metformin monotherapy is uncommon, it can happen in older patients. Hypoglycemia associated with metformin also can occur in situations when caloric intake is deficient or when metformin is used in combination with other drugs such as sulfonylureas (ie, glipizide), beta-adrenergic blocking drugs, angiotensin-converting enzyme inhibitors, or even nonsteroidal anti-inflammatory drugs.13
Identifying overlapping psychiatric (or psychological) illness
Symptoms of depression, anxiety, psychosis, and even dissociation can present as confusion. The term pseudodementia describes patients who exhibit cognitive symptoms consistent with NCD but could improve once the underlying mood, thought, anxiety, or personality disorder is treated.
For example, a patient with depression typically exhibits neurovegetative symptoms—such as poor sleep or appetite— amotivation, and low energy. All of these can lead to abrupt-onset cognitive changes, which are a hallmark of pseudodementia rather than the more insidious pattern of mild NCD. In cases of pseudodementia, neurocognitive testing will show impairment that often rapidly improves after the primary psychiatric (or psychological) issue is rectified. Making a diagnosis of pseudodementia at the initial presentation is difficult because neurocognitive tests such as the MMSE often fail to separate depression from true cognitive changes.14 Such a diagnosis typically requires hindsight. Yet, one must also keep in mind that pseudodementia may be part of a NCD prodrome.15
Conversion disorder as well as the dissociative disorders and substance-related disorders are notorious for causing confusion. In Ms. T’s case, pseudodementia stemming from her underlying bipolar disorder and anxiety figured prominently in the differential diagnosis, but she did not have any other overt psychopathology, personality disorder, or signs of malingering to further complicate her picture.
Notebook. I recommend that my patients keep a small notebook to record medical data ranging from blood pressure and glycemic measurements to details about sleep and dietary intake. Such data comprise the necessary metrics to properly assess target conditions and then track changes once treatment is initiated. This exercise not only yields much-needed detail about the patient’s condition for the clinician; the act of journaling also can be therapeutic for the writer through a process known as experimental disclosure, in which writing down one’s thoughts and observations has a positive impact on the writer’s physical health and psychology.16
Diagnosis. The first rule in medicine (perhaps the second, behind primum non nocere) is to determine what you are treating before beginning treatment (decernite quid tractemus, prius cura ministrandi, for Latin buffs). This means trying to fashion the best diagnostic label, even if it is merely a place-holder, while assessment of the confused state continues. DSM-5 has attempted to remove stigma from several neuropsychiatric disorders. On the cognition front, the new name for dementia is “neurocognitive disorder (NCD),” the umbrella term that focuses on the decline from a previous level of cognitive functioning. NCD has been divided into mild or major cognitive impairment headings either “with” or “without behavioral disturbance” subspecifiers.17
Aside from NCD, there are several other diagnoses in the differential for confusion. Delirium remains the most prominent and focuses on disturbances in attention and orientation that develops over a short period of time, with a change seen in an additional cognitive domain, such as memory, but not in the context of a severely reduced level of arousal such as coma. Subjective cognitive impairment (SCI) is when subjective complaints of cognitive impairment are hallmark compared with objective findings—with evidence suggesting that the presence of SCI could predict a 4.5 times higher rate of developing mild cognitive impairment (MCI) over 7 years.18 MCI was originally used to describe the early prodrome of AD, minus functional decline.
Treatment
After even a provisional diagnosis comes the final, all-important challenge: treating the neuropsychiatric symptoms (NPS) of the confused patient. NPS are nearly universal in NCD/delirium throughout the course of illness. There are no FDA-approved treatments for the NPS associated with these conditions. In terms of treating delirium, the best approach is to treat the underlying medical condition. For control of behavior, which can range from agitated to psychotic to hypoactive, nonpharmacotherapeutic interventions are paramount; they include making sure that the patient is at the appropriate level of care, which, for the confused outpatient, could mean hospitalization. Ensuring proper nutrition, hydration, sensory care (hearing aids, glasses, etc.), and stability in ambulation must be done before considering pharmacotherapy.
Antipsychotic use has been the mainstay of drug treatment of behavioral dyscontrol. Haloperidol has been the traditional go-to medication because there is no evidence that low-dose haloperidol (<3 mg/d) has any different efficacy compared with the atypical antipsychotics or has a greater frequency of adverse drug effects. However, high-dose haloperidol (>4.5 mg/d) was associated with a greater incidence of adverse effects, mainly parkinsonism, than atypical antipsychotics.19 Neither the typical nor atypical antipsychotics have shown mortality benefit—the real outcome measure of interest.
In terms of treating major (or minor) NCD, there are only 2 FDA-approved medication classes: cholinesterase inhibitors (donepezil, galantamine, rivastigmine, etc.) and memantine. However, these medication classes—even when combined together—have only shown marginal benefit in terms of improving cognition. Worse, even when given early in the course of illness they do not reduce the rate of NCD. For pseudodementia, selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors tend to form the mainstay of treating underlying depression or anxiety leading to cognitive changes. Preliminary data suggest that some SSRIs might improve cognition in terms of processing speed, verbal learning, and memory.20 More studies are needed before definitive conclusions can be drawn.
For the confused patient, a personalized therapeutic program, in which multiple interventions are considered at once (targeting all areas of the patient’s life) is gaining research traction. For example, a novel, comprehensive program involving multiple modalities designed to achieve metabolic enhancement for neurodegeneration (MEND) recently has shown robust benefit for patients with AD, MCI, and SCI.21 Using an individual approach to improve diet, activity, sleep, metabolic status including body mass index, and several other markers that affect neural plasticity, researchers demonstrated symptom improvement in 9 of 10 study patients.
Yet, some of the interventions, such as the use of statins for hyperlipidemia, remain controversial, with some studies suggesting that they help cognition,22,23 and others showing no association.24 The researchers caution that further research is warranted before costly dementia prevention trials with statins are undertaken. It does not appear that there are current MEND-type research projects in delirium but it’s to be hoped that we will see these in the future.
In the case of Ms. T, the cause of delirium vs mild NCD was thought to be multifactorial. Discontinuing Armour Thyroid and metformin—symptoms of hypoglycemia emerged as a leading concern—were simple adjustments that led to resolution of the most concerning elements of her confusion. She continued her other psychotropics, although there might be mild residual cognitive issues that warrant close observation.
Related Resources
• Lin JS, O’Connor E, Rossum RC, et al. Screening for cognitive impairment in older adults: an evidence update for the U.S. Preventive Services Task Force. Rockville, MD: Agency for Healthcare Research and Quality (US); 2013.
• Grover S, Kate N. Assessment scales for delirium: a review. World J Psychiatry. 2012;2(4):58-70.
Drug Brand Names
Atorvastatin • Lipitor Lithium • Eskalith, Lithobid
Donepezil • Aricept Lorazepam • Ativan
Escitalopram • Lexapro Memantine • Namenda
Flumazenil • Romazicon Metformin • Glucophage
Galantamine • Razadyne Naloxone • Narcan
Glipizide • Glucotrol Physostigmine • Antilirium
Haloperidol • Haldol Quetiapine • Seroquel
Levothyroxine • Levoxyl, Synthroid Rivastigmine • Exelon
Lithium • Eskalith, Lithobid Valproic acid • Depakene
Disclosure
Dr. Raj is a speaker for Actavis Pharmaceuticals, AstraZeneca, and Merck.
1. Borson S, Scanlan JM, Chen P, et al. The Mini-Cog as a screen for dementia: validation in a population-based sample. J Am Geriatr Soc. 2003;51(10):1451-1454.
2. Wilber ST, Lofgren SD, Mager TG, et al. An evaluation of two screening tools for cognitive impairment in older emergency department patients. Acad Emerg Med. 2005;12(7):612-616.
3. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
4. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695- 699.
5. Inouye S, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Int Med. 1990;113(12):941-948.
6. Hillmer SN, Mager DE, Simonsick EM, et al. A drug burden index to define the functional burden of medications in older people. Arch Intern Med. 2007;167(8):781-787.
7. Raj YP. Psychiatric emergencies. In: Jiang W, Gagliardi JP, Krishnan KR, eds. Clinician’s guide to psychiatric care. New York, NY: Oxford University Press; 2009:33-40.
8. Liptzin B. Clinical diagnosis and management of delirium. In: Stoudemire A, Fogel BS, Greenberg DB, eds. Psychiatric care of the medical patient. 2nd ed. New York, NY: Oxford University Press; 2000:581-596.
9. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
10. Poloni N, Vender S, Bolla E, et al. Gluten encephalopathy with psychiatric onset: case report. Clin Pract Epidemiol Ment Health. 2009;5:16.
11. Naughton BJ, Moran M, Ghaly Y, et al. Computed tomography scanning and delirium in elder patients. Acad Emerg Med. 1997;4(12):1107-1110.
12. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. doi: 10.1136/bmj.c3666.
13. Zitzmann S, Reimann IR, Schmechel H. Severe hypoglycemia in an elderly patient treated with metformin. Int J Clin Pharmacol Ther. 2002;40(3):108-110.
14. Benson AD, Slavin MJ, Tran TT, et al. Screening for early Alzheimer’s Disease: is there still a role for the Mini-Mental State Examination? Prim Care Companion J Clin Psychiatry. 2005;7(2):62-69.
15. Brown WA. Pseudodementia: issues in diagnosis. Psychiatric Times. http://www.psychiatrictimes.com/ pseudodementia-issues-diagnosis. Published April 9, 2005. Accessed February 2, 2015.
16. Frattaroli J. Experimental disclosure and its moderators: a meta-analysis. Psychol Bull. 2006;132(6):823-865.
17. Stetka BS, Correll CU. A guide to DSM-5: neurocognitive disorder. Medscape. http://www.medscape.com/ viewarticle/803884_13. Published May 21, 2013. Accessed October 30, 2014.
18. Reisberg B, Sulman MD, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
19. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.
20. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Intern Clin Psychopharmacol. 2012;27(4):215-223.
21. Bredesen DE. Reversal of cognitive decline: a novel therapeutic program. Aging (Albany NY). 2014;6(9):707-717.
22. Sparks DL, Kryscio RJ, Sabbagh MN, et al. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr Alzheimer Res. 2008;5(4):416-421.
23. Andrade C, Radhakrishnan R. The prevention and treatment of cognitive decline and dementia: an overview of recent research on experimental treatments. Indian J Psychiatry. 2009;51(1):12-25.
24. Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry. 2005;62(2):217-224.
1. Borson S, Scanlan JM, Chen P, et al. The Mini-Cog as a screen for dementia: validation in a population-based sample. J Am Geriatr Soc. 2003;51(10):1451-1454.
2. Wilber ST, Lofgren SD, Mager TG, et al. An evaluation of two screening tools for cognitive impairment in older emergency department patients. Acad Emerg Med. 2005;12(7):612-616.
3. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
4. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695- 699.
5. Inouye S, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Int Med. 1990;113(12):941-948.
6. Hillmer SN, Mager DE, Simonsick EM, et al. A drug burden index to define the functional burden of medications in older people. Arch Intern Med. 2007;167(8):781-787.
7. Raj YP. Psychiatric emergencies. In: Jiang W, Gagliardi JP, Krishnan KR, eds. Clinician’s guide to psychiatric care. New York, NY: Oxford University Press; 2009:33-40.
8. Liptzin B. Clinical diagnosis and management of delirium. In: Stoudemire A, Fogel BS, Greenberg DB, eds. Psychiatric care of the medical patient. 2nd ed. New York, NY: Oxford University Press; 2000:581-596.
9. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
10. Poloni N, Vender S, Bolla E, et al. Gluten encephalopathy with psychiatric onset: case report. Clin Pract Epidemiol Ment Health. 2009;5:16.
11. Naughton BJ, Moran M, Ghaly Y, et al. Computed tomography scanning and delirium in elder patients. Acad Emerg Med. 1997;4(12):1107-1110.
12. Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. doi: 10.1136/bmj.c3666.
13. Zitzmann S, Reimann IR, Schmechel H. Severe hypoglycemia in an elderly patient treated with metformin. Int J Clin Pharmacol Ther. 2002;40(3):108-110.
14. Benson AD, Slavin MJ, Tran TT, et al. Screening for early Alzheimer’s Disease: is there still a role for the Mini-Mental State Examination? Prim Care Companion J Clin Psychiatry. 2005;7(2):62-69.
15. Brown WA. Pseudodementia: issues in diagnosis. Psychiatric Times. http://www.psychiatrictimes.com/ pseudodementia-issues-diagnosis. Published April 9, 2005. Accessed February 2, 2015.
16. Frattaroli J. Experimental disclosure and its moderators: a meta-analysis. Psychol Bull. 2006;132(6):823-865.
17. Stetka BS, Correll CU. A guide to DSM-5: neurocognitive disorder. Medscape. http://www.medscape.com/ viewarticle/803884_13. Published May 21, 2013. Accessed October 30, 2014.
18. Reisberg B, Sulman MD, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
19. Lonergan E, Britton AM, Luxenberg J, et al. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;(2):CD005594.
20. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Intern Clin Psychopharmacol. 2012;27(4):215-223.
21. Bredesen DE. Reversal of cognitive decline: a novel therapeutic program. Aging (Albany NY). 2014;6(9):707-717.
22. Sparks DL, Kryscio RJ, Sabbagh MN, et al. Reduced risk of incident AD with elective statin use in a clinical trial cohort. Curr Alzheimer Res. 2008;5(4):416-421.
23. Andrade C, Radhakrishnan R. The prevention and treatment of cognitive decline and dementia: an overview of recent research on experimental treatments. Indian J Psychiatry. 2009;51(1):12-25.
24. Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry. 2005;62(2):217-224.

Cystic lung disease: Systematic, stepwise diagnosis
Air-filled pulmonary lesions commonly detected on chest computed tomography. Cystic lung lesions should be distinguished from other air-filled lesions to facilitate diagnosis. Primary care physicians play an integral role in the recognition of cystic lung disease.
The differential diagnosis of cystic lung disease is broad and includes isolated pulmonary, systemic, infectious, and congenital etiologies.
Here, we aim to provide a systematic, stepwise approach to help differentiate among the various cystic lung diseases and devise an algorithm for diagnosis. In doing so, we will discuss the clinical and radiographic features of many of these diseases:
- Lymphangioleiomyomatosis
- Birt-Hogg-Dubé syndrome
- Pulmonary Langerhans cell histiocytosis
- Interstitial pneumonia (desquamative interstitial pneumonia, lymphocytic interstitial pneumonia)
- Congenital cystic lung disease (congenital pulmonary airway malformation, pulmonary sequestration, bronchogenic cyst) Pulmonary infection
- Systemic disease (amyloidosis, light chain deposition disease, neurofibromatosis type 1).
STEP 1: RULE OUT CYST-MIMICS
A pulmonary cyst is a round, circumscribed space surrounded by an epithelial or fibrous wall of variable thickness.1 On chest radiography and computed tomography, a cyst appears as a round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.1 Cysts vary in wall thickness but usually have a thin wall (< 2 mm) and occur without associated pulmonary emphysema.1 They typically contain air but occasionally contain fluid or solid material.
A pulmonary cyst can be categorized as a bulla, bleb, or pneumatocele.
Bullae are larger than 1 cm in diameter, sharply demarcated by a thin wall, and usually accompanied by emphysematous changes in the adjacent lung.1
Blebs are no larger than 1 cm in diameter, are located within the visceral pleura or the subpleural space, and appear on computed tomography as thin-walled air spaces that are contiguous with the pleura.1 The distinction between a bleb and a bulla is of little clinical importance, and is often unnecessary.
Pneumatoceles are cysts that are frequently caused by acute pneumonia, trauma, or aspiration of hydrocarbon fluid, and are usually transient.1
Mimics of pulmonary cysts include pulmonary cavities, emphysema, loculated pneumothoraces, honeycomb lung, and bronchiectasis (Figure 1).2
Pulmonary cavities differ from cysts in that their walls are typically thicker (usually > 4 mm).3
Emphysema differs from cystic lung disease as it typically leads to focal areas or regions of decreased lung attenuation that do not have defined walls.1
Honeycombing refers to a cluster or row of cysts, 1 to 3 mm in wall thickness and typically 3 to 10 mm in diameter, that are associated with end-stage lung fibrosis.1 They are typically subpleural in distribution and are accompanied by fibrotic features such as reticulation and traction bronchiectasis.1
Bronchiectasis is dilation and distortion of bronchi and bronchioles and can be mistaken for cysts when viewed en face.1
Loculated pneumothoraces can also mimic pulmonary cysts, but they typically fail to adhere to a defined anatomic unit and are subpleural in distribution.
STEP 2: CHARACTERIZE THE CLINICAL PRESENTATION
Clinical signs and symptoms of cystic lung disease play a key role in diagnosis (Table 1). For instance, spontaneous pneumothorax is commonly associated with diffuse cystic lung disease (lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome), while insidious dyspnea, with or without associated pneumothorax, is usually associated with the interstitial pneumonias (lymphocytic interstitial pneumonia and desquamative interstitial pneumonia).
In addition, congenital abnormalities of the lung can lead to cyst formation. These abnormalities, especially when associated with other congenital abnormalities, are often diagnosed in the prenatal and perinatal periods. However, some remain undetected until incidentally found later in adulthood or if superimposing infection develops.
Primary pulmonary infections can also cause parenchymal necrosis, which in turn cavitates or forms cysts.4
Lastly, cystic lung diseases can occur as part of a multiorgan or systemic illness in which the lung is one of the organs involved. Although usually diagnosed before the discovery of cysts or manifestations of pulmonary symptoms, they can present as a diagnostic challenge, especially when lung cysts are the initial presentation.bsence of amyloid fibrils.
In view of the features of the different types of cystic lung disease, adults with cystic lung disease can be grouped according to their typical clinical presentations (Table 2):
- Insidious dyspnea or spontaneous pneumothorax
- Incidentally found cysts or recurrent pneumonia
- Signs and symptoms of primary pulmonary infection
- Signs and symptoms that are primarily nonpulmonary.
Insidious dyspnea or spontaneous pneumothorax
Insidious dyspnea or spontaneous pneumothorax can be manifestations of lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, or lymphocytic interstitial pneumonia.
Lymphangioleiomyomatosis is characterized by abnormal cellular proliferation within the lung, kidney, lymphatic system, or any combination.5 The peak prevalence is in the third to fourth decades of life, and most patients are women of childbearing age.6 In addition to progressive dyspnea on exertion and pneumothorax, other signs and symptoms include hemoptysis, nonproductive cough, chylous pleural effusion, and ascites.7,8
Birt-Hogg-Dubé syndrome is caused by germline mutations in the folliculin (FLCN) gene.9 It is characterized by skin fibrofolliculomas, pulmonary cysts, spontaneous pneumothorax, and renal cancer.10
Pulmonary Langerhans cell histiocytosis is part of the spectrum of Langerhans cell histiocytosis that, in addition to the lungs, can also involve the bone, pituitary gland, thyroid, skin, lymph nodes, and liver.11 It occurs almost exclusively in smokers, affecting individuals in their 20s and 30s, with no gender predilection.12,13 In addition to nonproductive cough and dyspnea, patients can also present with fever, anorexia, and weight loss,13 but approximately 25% of patients are asymptomatic.14
Desquamative interstitial pneumonia is an idiopathic interstitial pneumonia that, like pulmonary Langerhans cell histiocytosis, is seen almost exclusively in current or former smokers, who account for about 90% of patients with this disease. It affects almost twice as many men as women.15,16 The mean age at onset is 42 to 46.15,16 In addition to insidious cough and dyspnea, digital clubbing develops in 26% to 40% of patients.16,17
Lymphocytic interstitial pneumonia is another rare idiopathic pneumonia, usually associated with connective tissue disease, Sjögren syndrome, immunodeficiencies, and viral infections.18–21 It is more common in women, presenting between the 4th and 7th decades of life, with a mean age at diagnosis of 50 to 56.18,22 In addition to progressive dyspnea and cough, other symptoms include weight loss, pleuritic pain, arthralgias, fatigue, night sweats, and fever.23
In summary, in this clinical group, lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome should be considered when patients present with spontaneous pneumothorax; those with Birt-Hogg-Dubé syndrome also present with skin lesions or renal cancer. In patients with progressive dyspnea and cough, lymphocytic interstitial pneumonia should be considered in those with a known history of connective tissue disease or immunodeficiency. Pulmonary Langerhans cell histiocytosis typically presents at a younger age (20 to 30 years old) than desquamative interstitial pneumonia (smokers in their 40s). Making the distinction, however, will likely require imaging with computed tomography.
Incidentally found cysts or recurrent pneumonia
Incidentally found cysts or recurrent pneumonia can be manifestations of congenital pulmonary airway malformation, pulmonary sequestration, or bronchogenic cyst.
Congenital pulmonary airway malformation, of which there are five types, is the most common pulmonary congenital abnormality. It accounts for up to 95% of cases of congenital cystic lung disease.24,25 About 85% of cases are detected in the prenatal or perinatal periods.26 Late-onset congenital pulmonary airway malformation (arising in childhood to adulthood) presents with recurrent pneumonia in about 75% of cases and can be misdiagnosed as lung abscess, pulmonary tuberculosis, or bronchiectasis.27
Pulmonary sequestration, the second most common pulmonary congenital abnormality, is characterized by a portion of lung that does not connect to the tracheobronchial tree and has its own systemic arterial supply.24 Intralobar sequestration, which shares the pleural investment with normal lung, accounts for about 80% of cases of pulmonary sequestration.28–30 In addition to signs or symptoms of pulmonary infection, patients with pulmonary sequestration can remain asymp-
tomatic (about 25% of cases), or can present with hemoptysis or hemothorax.28–30 In adults, the typical age at presentation is between 20 and 25.29,30
Bronchogenic cyst is usually life-threatening in children. In adults, it commonly causes cough and chest pain.31 Hemoptysis, dysphagia, hoarseness, and diaphragmatic paralysis can also occur.32,33 The mean age at diagnosis in adults is 35 to 40.31,32
In summary, most cases of recurrent pneumonia with cysts are due to congenital pulmonary airway malformation. Pulmonary sequestration is the second most common cause of cystic lung disease in this group. Bronchogenic cyst is usually fatal in fetal development; smaller cysts can go unnoticed during the earlier years and are later found incidentally as imaging abnormalities in adults.
Signs and symptoms of primary pulmonary infections
Signs and symptoms of primary pulmonary infections can be due to Pneumocystis jirovecii pneumonia or echinococcal infections.
P jirovecii pneumonia commonly develops in patients with human immunodeficiency virus infection and low CD4 counts, recipients of hematologic or solid-organ transplants, and those receiving immunosuppressive therapy (eg, glucocorticoids or chemotherapy).
Echinococcal infections (with Echinococcus granulosus or multilocularis species) are more common in less-developed countries such as those in South America or the Middle East, in China, or in patients who have traveled to endemic areas.34
In summary, cystic lung disease in patients with primary pulmonary infections can be diagnosed by the patient’s clinical history and risk factors for infections. Those with human immunodeficiency virus infection and other causes of immunodeficiency are predisposed to P jirovecii pneumonia. Echinococcal infections occur in those with a history of travel to an endemic area.
Primarily nonpulmonary signs and symptoms
If the patient has primarily nonpulmonary signs and symptoms, think about pulmonary amyloidosis, light chain deposition disease, and neurofibromatosis type 1.
Pulmonary amyloidosis has a variety of manifestations, including tracheobronchial disease, nodular parenchymal disease, diffuse or alveolar septal pattern, pleural disease, lymphadenopathy, and pulmonary cysts.4
Light chain deposition disease shares some clinical features with amyloidosis. However, the light chain fragments in this disease do not form amyloid fibrils and therefore do not stain positively with Congo red. The kidney is the most commonly involved organ.4
Neurofibromatosis type 1 is characterized by collections of neurofibromas, café-au-lait spots, and pigmented hamartomas in the iris (Lisch nodules).35
In summary, patients in this group typically present with complications related to systemic involvement. Those with neurofibromatosis type 1 present with ophthalmologic, dermatologic, and neurologic manifestations. Amyloidosis and light chain deposition disease most commonly involve the renal system; their distinction will likely require tissue biopsy and Congo-red staining.
STEP 3: CHARACTERIZE THE RADIOGRAPHIC FEATURES
Characterization of pulmonary cysts and their distribution plays a key role in the diagnosis. Radiographically, cystic lung diseases can be subclassified into two major categories according to their cystic distribution:
- Discrete (focal or multifocal)
- Diffuse (unilobular or panlobular).2,3
Discrete cystic lung diseases include congenital abnormalities, infectious diseases, and interstitial pneumonias.2,3
Diffuse, panlobular cystic lung diseases include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dubé syndrome, amyloidosis, light chain deposition disease, and neurofibromatosis type 1.7,13,36–39
In addition, other associated radiographic findings play a major role in diagnosis.
Cysts in patients presenting with insidious dyspnea or spontaneous pneumothorax
Lymphangioleiomyomatosis. Cysts are seen in nearly all cases of advanced lymphangioleiomyomatosis, typically in a diffuse pattern, varying from 2 mm to 40 mm in diameter, and uniform in shape (Figure 2A).7,8,40–42
Other radiographic features include vessels located at the periphery of the cysts (in contrast to the centrilobular pattern seen with emphysema), and chylous pleural effusions (in about 22% of patients).40 Nodules are typically not seen with lymphangioleiomyomatosis, and if found represent type 2 pneumocyte hyperplasia.
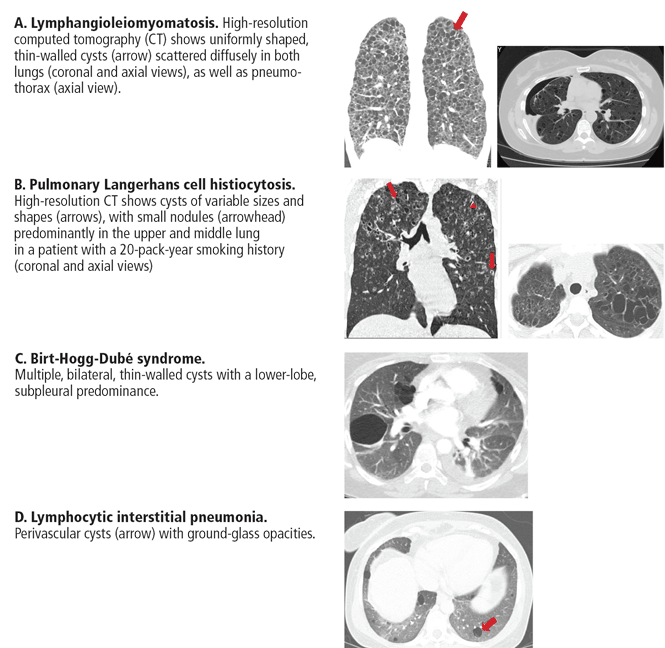
Pulmonary Langerhans cell histiocytosis. Nodules measuring 1 to 10 mm in diameter and favoring a centrilobular location are often seen on computed tomography. Pulmonary cysts occur in about 61% of patients.13,43 Cysts are variable in size and shape (Figure 2B), in contrast to their uniform appearance in lymphangioleiomyomatosis. Most cysts are less than 10 mm in diameter; however, they can be up to 80 mm.13,43 Early in its course, nodules may predominate in the upper and middle lobes. Over time, diffuse cysts become more common and can be difficult to differentiate from advanced smoking-induced emphysema.44
Birt-Hogg-Dubé syndrome. Approximately 70% to 100% of patients with Birt-Hogg-Dubé syndrome will have multiple pulmonary cysts detected on computed tomography. These cysts are characteristically basal and subpleural in location, with varying sizes and irregular shapes in otherwise normal lung parenchyma (Figure 2C).36,45,46
Desquamative interstitial pneumonia. Pulmonary cysts are present on computed tomography in about 32% of patients.47 They are usually round and less than 20 mm in diameter.48 Ground-glass opacity is present in almost all cases of desquamative interstitial pneumonia, with a diffuse pattern in 25% to 44% of patients.16,17,47
Pulmonary cysts occur in up to two-thirds of those with lymphocytic interstitial pneumonia. Cysts are usually multifocal and perivascular in distribution and have varying sizes and shapes (Figure 2D).22 Ground-glass opacity and poorly defined centrilobular nodules are also frequently seen. Other computed tomographic findings include thickening of the bronchovascular bundles, focal consolidation, interseptal lobular thickening, pleural thickening, and lymph node enlargement.22
In summary, in this group of patients, diffuse panlobular cysts are due to lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, or Birt-Hogg-Dubé syndrome. Cysts due to lymphangioleiomyomatosis have a diffuse distribution, while those due to pulmonary Langerhans cell histiocytosis tend to be upper-lobe-predominant and in the early stages are associated with stellate centrilobular nodules. Cysts in Birt-Hogg-Dubé syndrome tend to be subpleural and those due to lymphocytic interstitial pneumonia are perivascular in distribution.
Cysts that are incidentally found or occur in patients with recurrent pneumonia
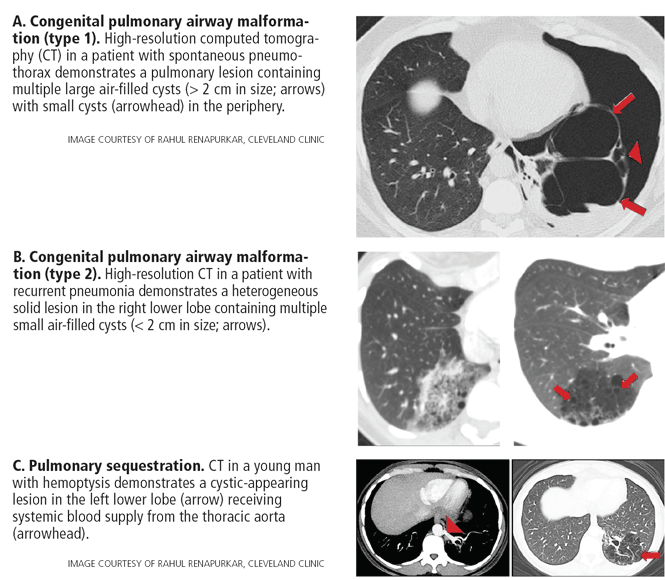
Congenital pulmonary airway malformation types 1, 2, and 4 (Figure 3A, 3B). Cysts are typically discrete and focal or multifocal in distribution, but cases of multilobar and bilateral distribution have also been reported.27,49 The lower lobes are more often involved.49 Cysts vary in size and shape and can contain air, fluid, or both.27,49 Up to 50% of cases can occur in conjunction with pulmonary sequestration.50
Pulmonary sequestration displays an anomalous arterial supply on computed tomography (Figure 3C). Other imaging findings include mass lesions (49%), cystic lesions (29%), cavitary lesions (12%), and bronchiectasis.30 Air trapping can be seen in the adjacent lung. Lower lobe involvement accounts for more than 95% of total cases of sequestration.30 The cysts are usually discrete or focal in distribution. Misdiagnosis of pulmonary sequestration is common, and can include pulmonary abscess, pneumonia, bronchiectasis, and lung cancer.30
Bronchogenic cyst. Cyst contents generally demonstrate water attenuation, or higher attenuation if filled with proteinaceous/mucoid material or calcium deposits; air-fluid levels are seen in infected cysts.32 Intrapulmonary cysts have a predilection for the lower lobes and are usually discrete or focal in distribution.31,32 Mediastinal cysts are usually homogeneous, solitary, and located in the middle mediastinum.32 Cysts vary in size from 20 to 90 mm, with a mean diameter of 40 mm.31
In summary, in this group of cystic lung diseases, characteristic computed tomographic findings will suggest the diagnosis—air-filled cysts of varying sizes for congenital pulmonary airway malformation and anomalous vascular supply for pulmonary sequestration. Bronchogenic cysts will tend to have water or higher-than-water attenuation due to proteinaceous-mucoid material or calcium deposits.
Cysts in patients with signs and symptoms of primary pulmonary infections
P jirovecii pneumonia. Between 10% and 15% of patients have cysts, and about 18% present with spontaneous pneumothorax.51 Cysts in P jirovecii pneumonia vary in size from 15 to 85 mm in diameter and tend to occur in the upper lobes (Figure 4A).51,52
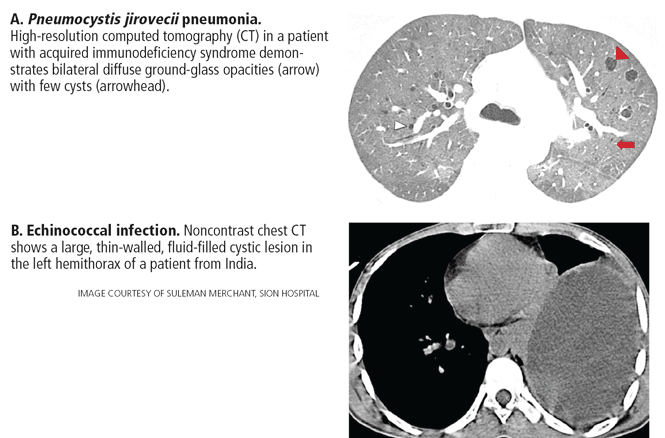
Echinococcal infection. Echinococcal pulmonary cysts typically are single and located more often in the lower lobes (Figure 4B).53,54 Cysts can be complicated by air-fluid levels, hydropneumothorax, or pneumothorax, or they can turn into cavitary lesions.
The diagnoses of these pulmonary infections are usually made by clinical and computed tomographic findings and depend less on detecting and characterizing lung cysts. Patients with P jirovecii pneumonia tend to have bilateral perihilar ground-glass opacities, while air-fluid levels suggest echinococcal infections. Cysts in this group of patients tend to be discrete or focal or multifocal in distribution, and vary in size.
Cysts in patients with primarily nonpulmonary signs and symptoms
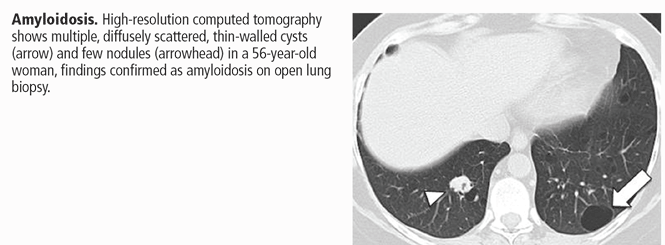
Amyloidosis. Cyst formation is rare in amyloidosis.4 When present, cysts can be diffuse and scattered in distribution, in varying sizes (usually < 30 mm in diameter) and irregular shapes (Figure 5).55,56
Pulmonary light chain deposition disease usually presents as linear opacities and small nodules on chest computed tomography. Numerous cysts that are diffuse in distribution and have no topographic predominance can also be present. They can progress in number and size and coalesce to form irregular shapes.57
Neurofibromatosis type 1. In neurofibromatosis type 1, the most common radiographic presentations are bibasilar reticular opacities (50%), bullae (50%), and ground glass opacities (37%).58 Well-formed cysts occur in up to 25% of patients and tend to be diffuse and smaller (2 to 18 mm in diameter), with upper lobe predominance.58,59
In summary, in this group of patients, bibasilar reticular and ground-glass opacities suggest neurofibromatosis type 1, while nodules and linear opacities suggest amyloidosis or light chain deposition disease. Cysts tend to be diffuse with varying sizes.
STEP 4: PUT IT ALL TOGETHER
Diagnosis in insidious dyspnea or spontaneous pneumothorax

For patients who present with insidious dyspnea or spontaneous pneumothorax, the diagnosis of cystic lung disease can be made by characterizing the distribution, size, and shape of the cysts (Table 3).
Diffuse, panlobular distribution. Cystic lung diseases with this pattern include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, and Birt-Hogg-Dubé syndrome. In this group, cysts that are uniform in size and regular in shape are invariably due to lymphangioleiomyomatosis. Those with variable size and irregular shapes can be due to pulmonary Langerhans cell histiocytosis or Birt-Hogg-Dubé syndrome. Patients with pulmonary Langerhans cell histiocytosis tend to be smokers and their cysts tend to be upper- lobe-predominant. Those with Birt-Hogg-Dubé syndrome will likely have renal cancer or skin lesions; their cysts tend to be basilar and subpleural in distribution.
Cysts that are focal or multifocal and unilobular are due to lymphocytic interstitial pneumonia or desquamative interstitial pneumonia. Patients with lymphocytic interstitial pneumonia tend to have underlying connective tissue disease; those with desquamative interstitial pneumonia are almost always smokers. The definitive diagnosis for lymphocytic interstitial pneumonia or desquamative interstitial pneumonia can require a tissue biopsy.
Diagnosis in patients with incidentally found cysts or recurrent pneumonia
In those who present with incidentally found cysts or recurrent pneumonia, suspicion for a congenital lung malformation should be raised. Patients with a type 1, 2, or 4 congenital pulmonary airway malformation typically have air-filled cysts in varying sizes; those with pulmonary sequestration have an anomalous arterial supply in addition to cysts that are usually located in the lower lobes. Bronchogenic cysts tend to be larger, with attenuation equal to or greater than that of water, and distinguishing them from congenital pulmonary airway malformation will likely require surgical examination.
Diagnosis in patients with signs and symptoms of pulmonary infections
Patients with signs and symptoms of pulmonary infections should be investigated according to clinical risk factors for P jirovecii pneumonia or echinococcal infections.
Diagnosis in patients with primarily nonpulmonary presentations
The distinction between amyloidosis and neurofibromatosis type 1 can be made by the history and the clinical examination. However, a definitive diagnosis of amyloidosis or light chain deposition disease requires tissue examination for the presence or absence of amyloid fibrils.
- Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology 2008; 246:697–722.
- Cosgrove GP, Frankel SK, Brown KK. Challenges in pulmonary fibrosis. 3: cystic lung disease. Thorax 2007; 62:820–829.
- Ryu JH, Swensen SJ. Cystic and cavitary lung diseases: focal and diffuse. Mayo Clin Proc 2003; 78:744–752.
- Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med 2013; 7:316–327.
- McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest 2008; 133:507–516.
- Johnson SR, Cordier JF, Lazor R, et al; Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010; 35:14–26.
- Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990; 323:1254–1260.
- Chu SC, Horiba K, Usuki J. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115:1041–1052.
- Graham RB, Nolasco M, Peterlin B, Garcia CK. Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 2005; 172:39–44.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977; 113:1674–1677.
- Sundar KM, Gosselin MV, Chung HL, Cahill BC. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology, and clinical evolution of disease. Chest 2003; 123:1673–1683.
- Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000; 342:1969–1978.
- Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346:484–490.
- Mendez JL, Nadrous HF, Vassallo R, Decker PA, Ryu JH. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125:1028–1032.
- Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med 1978; 298:801–809.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology 2005; 236:10–21.
- Strimlan CV, Rosenow EC 3rd, Weiland LH, Brown LR. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med 1978; 88:616–621.
- Arish N, Eldor R, Fellig Y, et al. Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax 2006; 61:1096–1097.
- Schooley RT, Carey RW, Miller G, et al. Chronic Epstein-Barr virus infection associated with fever and interstitial pneumonitis. Clinical and serologic features and response to antiviral chemotherapy. Ann Intern Med 1986; 104:636–643.
- Kramer MR, Saldana MJ, Ramos M, Pitchenik AE. High titers of Epstein-Barr virus antibodies in adult patients with lymphocytic interstitial pneumonitis associated with AIDS. Respir Med 1992; 86:49–52.
- Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999; 212:567–572.
- Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest 2002; 122:2150–2164.
- Biyyam DR, Chapman T, Ferguson MR, Deutsch G, Dighe MK. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics 2010; 30:1721–1738.
- Cloutier MM, Schaeffer DA, Hight D. Congenital cystic adenomatoid malformation. Chest 1993; 103:761–764.
- Luján M, Bosque M, Mirapeix RM, Marco MT, Asensio O, Domingo C. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration 2002; 69:148–154.
- Oh BJ, Lee JS, Kim JS, Lim CM, Koh Y. Congenital cystic adenomatoid malformation of the lung in adults: clinical and CT evaluation of seven patients. Respirology 2006; 11:496–501.
- Tsolakis CC, Kollias VD, Panayotopoulos PP. Pulmonary sequestration. Experience with eight consecutive cases. Scand Cardiovasc J 1997; 31:229–232.
- Sauvanet A, Regnard JF, Calanducci F, Rojas-Miranda A, Dartevelle P, Levasseur P. Pulmonary sequestration. Surgical aspects based on 61 cases. Rev Pneumol Clin 1991; 47:126–132. Article in French.
- Wei Y, Li F. Pulmonary sequestration: a retrospective analysis of 2,625 cases in China. Eur J Cardiothorac Surg 2011; 40:e39–e42.
- Patel SR, Meeker DP, Biscotti CV, Kirby TJ, Rice TW. Presentation and management of bronchogenic cysts in the adult. Chest 1994; 106:79–85.
- Limaïem F, Ayadi-Kaddour A, Djilani H, Kilani T, El Mezni F. Pulmonary and mediastinal bronchogenic cysts: a clinicopathologic study of 33 cases. Lung 2008; 186:55–61.
- Liu HS, Li SQ, Cao ZL, Zhang ZY, Ren H. Clinical features and treatment of bronchogenic cyst in adults. Chin Med Sci J 2009; 24:60–63.
- Jenkins DJ, Romig T, Thompson RC. Emergence/re-emergence of Echinococcus spp.—a global update. Int J Parasitol 2005; 35:1205–1219.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med 1981; 305:1617–1627.
- Toro JR, Pautler SE, Stewart L, et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 2007; 175:1044–1053.
- Biko DM, Schwartz M, Anupindi SA, Altes TA. Subpleural lung cysts in Down syndrome: prevalence and association with coexisting diagnoses. Pediatr Radiol 2008; 38:280–284.
- Colombat M, Stern M, Groussard O, et al. Pulmonary cystic disorder related to light chain deposition disease. Am J Respir Crit Care Med 2006; 173:777–780.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in the UK. Thorax 2000; 55:1052–1057.
- Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med 1995; 151:527–533.
- Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 1999; 78:321–337.
- Schönfeld N, Frank W, Wenig S, et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993; 60:38–44.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Kluger N, Giraud S, Coupier I, et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol 2010; 162:527–537.
- Tobino K, Gunji Y, Kurihara M, et al. Characteristics of pulmonary cysts in Birt-Hogg-Dubé syndrome: thin-section CT findings of the chest in 12 patients. Eur J Radiol 2011; 77:403–409.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Koyama M, Johkoh T, Honda O, et al. Chronic cystic lung disease: diagnostic accuracy of high-resolution CT in 92 patients. AJR Am J Roentgenol 2003; 180:827–835.
- Patz EF Jr, Müller NL, Swensen SJ, Dodd LG. Congenital cystic adenomatoid malformation in adults: CT findings. J Comput Assist Tomogr 1995; 19:361–364.
- Conran RM, Stocker JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol 1999; 2:454–463.
- Kennedy CA, Goetz MB. Atypical roentgenographic manifestations of Pneumocystis carinii pneumonia. Arch Intern Med 1992; 152:1390–1398.
- Sandhu JS, Goodman PC. Pulmonary cysts associated with Pneumocystis carinii pneumonia in patients with AIDS. Radiology 1989; 173:33–35.
- Doğan R, Yüksel M, Cetin G, et al. Surgical treatment of hydatid cysts of the lung: report on 1,055 patients. Thorax 1989; 44:192–199.
- Salih OK, Topcuoğlu MS, Celik SK, Ulus T, Tokcan A. Surgical treatment of hydatid cysts of the lung: analysis of 405 patients. Can J Surg 1998; 41:131–135.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Sakai M, Yamaoka M, Kawaguchi M, Hizawa N, Sato Y. Multiple cystic pulmonary amyloidosis. Ann Thorac Surg 2011; 92:e109.
- Colombat M, Caudroy S, Lagonotte E, et al. Pathomechanisms of cyst formation in pulmonary light chain deposition disease. Eur Respir J 2008; 32:1399–1403.
- Zamora AC, Collard HR, Wolters PJ, Webb WR, King TE. Neurofibromatosis-associated lung disease: a case series and literature review. Eur Respir J 2007; 29:210–214.
- Oikonomou A, Vadikolias K, Birbilis T, Bouros D, Prassopoulos P. HRCT findings in the lungs of non-smokers with neurofibromatosis. Eur J Radiol 2011; 80:e520–e523.
Air-filled pulmonary lesions commonly detected on chest computed tomography. Cystic lung lesions should be distinguished from other air-filled lesions to facilitate diagnosis. Primary care physicians play an integral role in the recognition of cystic lung disease.
The differential diagnosis of cystic lung disease is broad and includes isolated pulmonary, systemic, infectious, and congenital etiologies.
Here, we aim to provide a systematic, stepwise approach to help differentiate among the various cystic lung diseases and devise an algorithm for diagnosis. In doing so, we will discuss the clinical and radiographic features of many of these diseases:
- Lymphangioleiomyomatosis
- Birt-Hogg-Dubé syndrome
- Pulmonary Langerhans cell histiocytosis
- Interstitial pneumonia (desquamative interstitial pneumonia, lymphocytic interstitial pneumonia)
- Congenital cystic lung disease (congenital pulmonary airway malformation, pulmonary sequestration, bronchogenic cyst) Pulmonary infection
- Systemic disease (amyloidosis, light chain deposition disease, neurofibromatosis type 1).
STEP 1: RULE OUT CYST-MIMICS
A pulmonary cyst is a round, circumscribed space surrounded by an epithelial or fibrous wall of variable thickness.1 On chest radiography and computed tomography, a cyst appears as a round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.1 Cysts vary in wall thickness but usually have a thin wall (< 2 mm) and occur without associated pulmonary emphysema.1 They typically contain air but occasionally contain fluid or solid material.
A pulmonary cyst can be categorized as a bulla, bleb, or pneumatocele.
Bullae are larger than 1 cm in diameter, sharply demarcated by a thin wall, and usually accompanied by emphysematous changes in the adjacent lung.1
Blebs are no larger than 1 cm in diameter, are located within the visceral pleura or the subpleural space, and appear on computed tomography as thin-walled air spaces that are contiguous with the pleura.1 The distinction between a bleb and a bulla is of little clinical importance, and is often unnecessary.
Pneumatoceles are cysts that are frequently caused by acute pneumonia, trauma, or aspiration of hydrocarbon fluid, and are usually transient.1
Mimics of pulmonary cysts include pulmonary cavities, emphysema, loculated pneumothoraces, honeycomb lung, and bronchiectasis (Figure 1).2
Pulmonary cavities differ from cysts in that their walls are typically thicker (usually > 4 mm).3
Emphysema differs from cystic lung disease as it typically leads to focal areas or regions of decreased lung attenuation that do not have defined walls.1
Honeycombing refers to a cluster or row of cysts, 1 to 3 mm in wall thickness and typically 3 to 10 mm in diameter, that are associated with end-stage lung fibrosis.1 They are typically subpleural in distribution and are accompanied by fibrotic features such as reticulation and traction bronchiectasis.1
Bronchiectasis is dilation and distortion of bronchi and bronchioles and can be mistaken for cysts when viewed en face.1
Loculated pneumothoraces can also mimic pulmonary cysts, but they typically fail to adhere to a defined anatomic unit and are subpleural in distribution.
STEP 2: CHARACTERIZE THE CLINICAL PRESENTATION
Clinical signs and symptoms of cystic lung disease play a key role in diagnosis (Table 1). For instance, spontaneous pneumothorax is commonly associated with diffuse cystic lung disease (lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome), while insidious dyspnea, with or without associated pneumothorax, is usually associated with the interstitial pneumonias (lymphocytic interstitial pneumonia and desquamative interstitial pneumonia).
In addition, congenital abnormalities of the lung can lead to cyst formation. These abnormalities, especially when associated with other congenital abnormalities, are often diagnosed in the prenatal and perinatal periods. However, some remain undetected until incidentally found later in adulthood or if superimposing infection develops.
Primary pulmonary infections can also cause parenchymal necrosis, which in turn cavitates or forms cysts.4
Lastly, cystic lung diseases can occur as part of a multiorgan or systemic illness in which the lung is one of the organs involved. Although usually diagnosed before the discovery of cysts or manifestations of pulmonary symptoms, they can present as a diagnostic challenge, especially when lung cysts are the initial presentation.bsence of amyloid fibrils.
In view of the features of the different types of cystic lung disease, adults with cystic lung disease can be grouped according to their typical clinical presentations (Table 2):
- Insidious dyspnea or spontaneous pneumothorax
- Incidentally found cysts or recurrent pneumonia
- Signs and symptoms of primary pulmonary infection
- Signs and symptoms that are primarily nonpulmonary.
Insidious dyspnea or spontaneous pneumothorax
Insidious dyspnea or spontaneous pneumothorax can be manifestations of lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, or lymphocytic interstitial pneumonia.
Lymphangioleiomyomatosis is characterized by abnormal cellular proliferation within the lung, kidney, lymphatic system, or any combination.5 The peak prevalence is in the third to fourth decades of life, and most patients are women of childbearing age.6 In addition to progressive dyspnea on exertion and pneumothorax, other signs and symptoms include hemoptysis, nonproductive cough, chylous pleural effusion, and ascites.7,8
Birt-Hogg-Dubé syndrome is caused by germline mutations in the folliculin (FLCN) gene.9 It is characterized by skin fibrofolliculomas, pulmonary cysts, spontaneous pneumothorax, and renal cancer.10
Pulmonary Langerhans cell histiocytosis is part of the spectrum of Langerhans cell histiocytosis that, in addition to the lungs, can also involve the bone, pituitary gland, thyroid, skin, lymph nodes, and liver.11 It occurs almost exclusively in smokers, affecting individuals in their 20s and 30s, with no gender predilection.12,13 In addition to nonproductive cough and dyspnea, patients can also present with fever, anorexia, and weight loss,13 but approximately 25% of patients are asymptomatic.14
Desquamative interstitial pneumonia is an idiopathic interstitial pneumonia that, like pulmonary Langerhans cell histiocytosis, is seen almost exclusively in current or former smokers, who account for about 90% of patients with this disease. It affects almost twice as many men as women.15,16 The mean age at onset is 42 to 46.15,16 In addition to insidious cough and dyspnea, digital clubbing develops in 26% to 40% of patients.16,17
Lymphocytic interstitial pneumonia is another rare idiopathic pneumonia, usually associated with connective tissue disease, Sjögren syndrome, immunodeficiencies, and viral infections.18–21 It is more common in women, presenting between the 4th and 7th decades of life, with a mean age at diagnosis of 50 to 56.18,22 In addition to progressive dyspnea and cough, other symptoms include weight loss, pleuritic pain, arthralgias, fatigue, night sweats, and fever.23
In summary, in this clinical group, lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome should be considered when patients present with spontaneous pneumothorax; those with Birt-Hogg-Dubé syndrome also present with skin lesions or renal cancer. In patients with progressive dyspnea and cough, lymphocytic interstitial pneumonia should be considered in those with a known history of connective tissue disease or immunodeficiency. Pulmonary Langerhans cell histiocytosis typically presents at a younger age (20 to 30 years old) than desquamative interstitial pneumonia (smokers in their 40s). Making the distinction, however, will likely require imaging with computed tomography.
Incidentally found cysts or recurrent pneumonia
Incidentally found cysts or recurrent pneumonia can be manifestations of congenital pulmonary airway malformation, pulmonary sequestration, or bronchogenic cyst.
Congenital pulmonary airway malformation, of which there are five types, is the most common pulmonary congenital abnormality. It accounts for up to 95% of cases of congenital cystic lung disease.24,25 About 85% of cases are detected in the prenatal or perinatal periods.26 Late-onset congenital pulmonary airway malformation (arising in childhood to adulthood) presents with recurrent pneumonia in about 75% of cases and can be misdiagnosed as lung abscess, pulmonary tuberculosis, or bronchiectasis.27
Pulmonary sequestration, the second most common pulmonary congenital abnormality, is characterized by a portion of lung that does not connect to the tracheobronchial tree and has its own systemic arterial supply.24 Intralobar sequestration, which shares the pleural investment with normal lung, accounts for about 80% of cases of pulmonary sequestration.28–30 In addition to signs or symptoms of pulmonary infection, patients with pulmonary sequestration can remain asymp-
tomatic (about 25% of cases), or can present with hemoptysis or hemothorax.28–30 In adults, the typical age at presentation is between 20 and 25.29,30
Bronchogenic cyst is usually life-threatening in children. In adults, it commonly causes cough and chest pain.31 Hemoptysis, dysphagia, hoarseness, and diaphragmatic paralysis can also occur.32,33 The mean age at diagnosis in adults is 35 to 40.31,32
In summary, most cases of recurrent pneumonia with cysts are due to congenital pulmonary airway malformation. Pulmonary sequestration is the second most common cause of cystic lung disease in this group. Bronchogenic cyst is usually fatal in fetal development; smaller cysts can go unnoticed during the earlier years and are later found incidentally as imaging abnormalities in adults.
Signs and symptoms of primary pulmonary infections
Signs and symptoms of primary pulmonary infections can be due to Pneumocystis jirovecii pneumonia or echinococcal infections.
P jirovecii pneumonia commonly develops in patients with human immunodeficiency virus infection and low CD4 counts, recipients of hematologic or solid-organ transplants, and those receiving immunosuppressive therapy (eg, glucocorticoids or chemotherapy).
Echinococcal infections (with Echinococcus granulosus or multilocularis species) are more common in less-developed countries such as those in South America or the Middle East, in China, or in patients who have traveled to endemic areas.34
In summary, cystic lung disease in patients with primary pulmonary infections can be diagnosed by the patient’s clinical history and risk factors for infections. Those with human immunodeficiency virus infection and other causes of immunodeficiency are predisposed to P jirovecii pneumonia. Echinococcal infections occur in those with a history of travel to an endemic area.
Primarily nonpulmonary signs and symptoms
If the patient has primarily nonpulmonary signs and symptoms, think about pulmonary amyloidosis, light chain deposition disease, and neurofibromatosis type 1.
Pulmonary amyloidosis has a variety of manifestations, including tracheobronchial disease, nodular parenchymal disease, diffuse or alveolar septal pattern, pleural disease, lymphadenopathy, and pulmonary cysts.4
Light chain deposition disease shares some clinical features with amyloidosis. However, the light chain fragments in this disease do not form amyloid fibrils and therefore do not stain positively with Congo red. The kidney is the most commonly involved organ.4
Neurofibromatosis type 1 is characterized by collections of neurofibromas, café-au-lait spots, and pigmented hamartomas in the iris (Lisch nodules).35
In summary, patients in this group typically present with complications related to systemic involvement. Those with neurofibromatosis type 1 present with ophthalmologic, dermatologic, and neurologic manifestations. Amyloidosis and light chain deposition disease most commonly involve the renal system; their distinction will likely require tissue biopsy and Congo-red staining.
STEP 3: CHARACTERIZE THE RADIOGRAPHIC FEATURES
Characterization of pulmonary cysts and their distribution plays a key role in the diagnosis. Radiographically, cystic lung diseases can be subclassified into two major categories according to their cystic distribution:
- Discrete (focal or multifocal)
- Diffuse (unilobular or panlobular).2,3
Discrete cystic lung diseases include congenital abnormalities, infectious diseases, and interstitial pneumonias.2,3
Diffuse, panlobular cystic lung diseases include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dubé syndrome, amyloidosis, light chain deposition disease, and neurofibromatosis type 1.7,13,36–39
In addition, other associated radiographic findings play a major role in diagnosis.
Cysts in patients presenting with insidious dyspnea or spontaneous pneumothorax
Lymphangioleiomyomatosis. Cysts are seen in nearly all cases of advanced lymphangioleiomyomatosis, typically in a diffuse pattern, varying from 2 mm to 40 mm in diameter, and uniform in shape (Figure 2A).7,8,40–42
Other radiographic features include vessels located at the periphery of the cysts (in contrast to the centrilobular pattern seen with emphysema), and chylous pleural effusions (in about 22% of patients).40 Nodules are typically not seen with lymphangioleiomyomatosis, and if found represent type 2 pneumocyte hyperplasia.
Pulmonary Langerhans cell histiocytosis. Nodules measuring 1 to 10 mm in diameter and favoring a centrilobular location are often seen on computed tomography. Pulmonary cysts occur in about 61% of patients.13,43 Cysts are variable in size and shape (Figure 2B), in contrast to their uniform appearance in lymphangioleiomyomatosis. Most cysts are less than 10 mm in diameter; however, they can be up to 80 mm.13,43 Early in its course, nodules may predominate in the upper and middle lobes. Over time, diffuse cysts become more common and can be difficult to differentiate from advanced smoking-induced emphysema.44
Birt-Hogg-Dubé syndrome. Approximately 70% to 100% of patients with Birt-Hogg-Dubé syndrome will have multiple pulmonary cysts detected on computed tomography. These cysts are characteristically basal and subpleural in location, with varying sizes and irregular shapes in otherwise normal lung parenchyma (Figure 2C).36,45,46
Desquamative interstitial pneumonia. Pulmonary cysts are present on computed tomography in about 32% of patients.47 They are usually round and less than 20 mm in diameter.48 Ground-glass opacity is present in almost all cases of desquamative interstitial pneumonia, with a diffuse pattern in 25% to 44% of patients.16,17,47
Pulmonary cysts occur in up to two-thirds of those with lymphocytic interstitial pneumonia. Cysts are usually multifocal and perivascular in distribution and have varying sizes and shapes (Figure 2D).22 Ground-glass opacity and poorly defined centrilobular nodules are also frequently seen. Other computed tomographic findings include thickening of the bronchovascular bundles, focal consolidation, interseptal lobular thickening, pleural thickening, and lymph node enlargement.22
In summary, in this group of patients, diffuse panlobular cysts are due to lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, or Birt-Hogg-Dubé syndrome. Cysts due to lymphangioleiomyomatosis have a diffuse distribution, while those due to pulmonary Langerhans cell histiocytosis tend to be upper-lobe-predominant and in the early stages are associated with stellate centrilobular nodules. Cysts in Birt-Hogg-Dubé syndrome tend to be subpleural and those due to lymphocytic interstitial pneumonia are perivascular in distribution.
Cysts that are incidentally found or occur in patients with recurrent pneumonia
Congenital pulmonary airway malformation types 1, 2, and 4 (Figure 3A, 3B). Cysts are typically discrete and focal or multifocal in distribution, but cases of multilobar and bilateral distribution have also been reported.27,49 The lower lobes are more often involved.49 Cysts vary in size and shape and can contain air, fluid, or both.27,49 Up to 50% of cases can occur in conjunction with pulmonary sequestration.50
Pulmonary sequestration displays an anomalous arterial supply on computed tomography (Figure 3C). Other imaging findings include mass lesions (49%), cystic lesions (29%), cavitary lesions (12%), and bronchiectasis.30 Air trapping can be seen in the adjacent lung. Lower lobe involvement accounts for more than 95% of total cases of sequestration.30 The cysts are usually discrete or focal in distribution. Misdiagnosis of pulmonary sequestration is common, and can include pulmonary abscess, pneumonia, bronchiectasis, and lung cancer.30
Bronchogenic cyst. Cyst contents generally demonstrate water attenuation, or higher attenuation if filled with proteinaceous/mucoid material or calcium deposits; air-fluid levels are seen in infected cysts.32 Intrapulmonary cysts have a predilection for the lower lobes and are usually discrete or focal in distribution.31,32 Mediastinal cysts are usually homogeneous, solitary, and located in the middle mediastinum.32 Cysts vary in size from 20 to 90 mm, with a mean diameter of 40 mm.31
In summary, in this group of cystic lung diseases, characteristic computed tomographic findings will suggest the diagnosis—air-filled cysts of varying sizes for congenital pulmonary airway malformation and anomalous vascular supply for pulmonary sequestration. Bronchogenic cysts will tend to have water or higher-than-water attenuation due to proteinaceous-mucoid material or calcium deposits.
Cysts in patients with signs and symptoms of primary pulmonary infections
P jirovecii pneumonia. Between 10% and 15% of patients have cysts, and about 18% present with spontaneous pneumothorax.51 Cysts in P jirovecii pneumonia vary in size from 15 to 85 mm in diameter and tend to occur in the upper lobes (Figure 4A).51,52
Echinococcal infection. Echinococcal pulmonary cysts typically are single and located more often in the lower lobes (Figure 4B).53,54 Cysts can be complicated by air-fluid levels, hydropneumothorax, or pneumothorax, or they can turn into cavitary lesions.
The diagnoses of these pulmonary infections are usually made by clinical and computed tomographic findings and depend less on detecting and characterizing lung cysts. Patients with P jirovecii pneumonia tend to have bilateral perihilar ground-glass opacities, while air-fluid levels suggest echinococcal infections. Cysts in this group of patients tend to be discrete or focal or multifocal in distribution, and vary in size.
Cysts in patients with primarily nonpulmonary signs and symptoms
Amyloidosis. Cyst formation is rare in amyloidosis.4 When present, cysts can be diffuse and scattered in distribution, in varying sizes (usually < 30 mm in diameter) and irregular shapes (Figure 5).55,56
Pulmonary light chain deposition disease usually presents as linear opacities and small nodules on chest computed tomography. Numerous cysts that are diffuse in distribution and have no topographic predominance can also be present. They can progress in number and size and coalesce to form irregular shapes.57
Neurofibromatosis type 1. In neurofibromatosis type 1, the most common radiographic presentations are bibasilar reticular opacities (50%), bullae (50%), and ground glass opacities (37%).58 Well-formed cysts occur in up to 25% of patients and tend to be diffuse and smaller (2 to 18 mm in diameter), with upper lobe predominance.58,59
In summary, in this group of patients, bibasilar reticular and ground-glass opacities suggest neurofibromatosis type 1, while nodules and linear opacities suggest amyloidosis or light chain deposition disease. Cysts tend to be diffuse with varying sizes.
STEP 4: PUT IT ALL TOGETHER
Diagnosis in insidious dyspnea or spontaneous pneumothorax
For patients who present with insidious dyspnea or spontaneous pneumothorax, the diagnosis of cystic lung disease can be made by characterizing the distribution, size, and shape of the cysts (Table 3).
Diffuse, panlobular distribution. Cystic lung diseases with this pattern include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, and Birt-Hogg-Dubé syndrome. In this group, cysts that are uniform in size and regular in shape are invariably due to lymphangioleiomyomatosis. Those with variable size and irregular shapes can be due to pulmonary Langerhans cell histiocytosis or Birt-Hogg-Dubé syndrome. Patients with pulmonary Langerhans cell histiocytosis tend to be smokers and their cysts tend to be upper- lobe-predominant. Those with Birt-Hogg-Dubé syndrome will likely have renal cancer or skin lesions; their cysts tend to be basilar and subpleural in distribution.
Cysts that are focal or multifocal and unilobular are due to lymphocytic interstitial pneumonia or desquamative interstitial pneumonia. Patients with lymphocytic interstitial pneumonia tend to have underlying connective tissue disease; those with desquamative interstitial pneumonia are almost always smokers. The definitive diagnosis for lymphocytic interstitial pneumonia or desquamative interstitial pneumonia can require a tissue biopsy.
Diagnosis in patients with incidentally found cysts or recurrent pneumonia
In those who present with incidentally found cysts or recurrent pneumonia, suspicion for a congenital lung malformation should be raised. Patients with a type 1, 2, or 4 congenital pulmonary airway malformation typically have air-filled cysts in varying sizes; those with pulmonary sequestration have an anomalous arterial supply in addition to cysts that are usually located in the lower lobes. Bronchogenic cysts tend to be larger, with attenuation equal to or greater than that of water, and distinguishing them from congenital pulmonary airway malformation will likely require surgical examination.
Diagnosis in patients with signs and symptoms of pulmonary infections
Patients with signs and symptoms of pulmonary infections should be investigated according to clinical risk factors for P jirovecii pneumonia or echinococcal infections.
Diagnosis in patients with primarily nonpulmonary presentations
The distinction between amyloidosis and neurofibromatosis type 1 can be made by the history and the clinical examination. However, a definitive diagnosis of amyloidosis or light chain deposition disease requires tissue examination for the presence or absence of amyloid fibrils.
Air-filled pulmonary lesions commonly detected on chest computed tomography. Cystic lung lesions should be distinguished from other air-filled lesions to facilitate diagnosis. Primary care physicians play an integral role in the recognition of cystic lung disease.
The differential diagnosis of cystic lung disease is broad and includes isolated pulmonary, systemic, infectious, and congenital etiologies.
Here, we aim to provide a systematic, stepwise approach to help differentiate among the various cystic lung diseases and devise an algorithm for diagnosis. In doing so, we will discuss the clinical and radiographic features of many of these diseases:
- Lymphangioleiomyomatosis
- Birt-Hogg-Dubé syndrome
- Pulmonary Langerhans cell histiocytosis
- Interstitial pneumonia (desquamative interstitial pneumonia, lymphocytic interstitial pneumonia)
- Congenital cystic lung disease (congenital pulmonary airway malformation, pulmonary sequestration, bronchogenic cyst) Pulmonary infection
- Systemic disease (amyloidosis, light chain deposition disease, neurofibromatosis type 1).
STEP 1: RULE OUT CYST-MIMICS
A pulmonary cyst is a round, circumscribed space surrounded by an epithelial or fibrous wall of variable thickness.1 On chest radiography and computed tomography, a cyst appears as a round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.1 Cysts vary in wall thickness but usually have a thin wall (< 2 mm) and occur without associated pulmonary emphysema.1 They typically contain air but occasionally contain fluid or solid material.
A pulmonary cyst can be categorized as a bulla, bleb, or pneumatocele.
Bullae are larger than 1 cm in diameter, sharply demarcated by a thin wall, and usually accompanied by emphysematous changes in the adjacent lung.1
Blebs are no larger than 1 cm in diameter, are located within the visceral pleura or the subpleural space, and appear on computed tomography as thin-walled air spaces that are contiguous with the pleura.1 The distinction between a bleb and a bulla is of little clinical importance, and is often unnecessary.
Pneumatoceles are cysts that are frequently caused by acute pneumonia, trauma, or aspiration of hydrocarbon fluid, and are usually transient.1
Mimics of pulmonary cysts include pulmonary cavities, emphysema, loculated pneumothoraces, honeycomb lung, and bronchiectasis (Figure 1).2
Pulmonary cavities differ from cysts in that their walls are typically thicker (usually > 4 mm).3
Emphysema differs from cystic lung disease as it typically leads to focal areas or regions of decreased lung attenuation that do not have defined walls.1
Honeycombing refers to a cluster or row of cysts, 1 to 3 mm in wall thickness and typically 3 to 10 mm in diameter, that are associated with end-stage lung fibrosis.1 They are typically subpleural in distribution and are accompanied by fibrotic features such as reticulation and traction bronchiectasis.1
Bronchiectasis is dilation and distortion of bronchi and bronchioles and can be mistaken for cysts when viewed en face.1
Loculated pneumothoraces can also mimic pulmonary cysts, but they typically fail to adhere to a defined anatomic unit and are subpleural in distribution.
STEP 2: CHARACTERIZE THE CLINICAL PRESENTATION
Clinical signs and symptoms of cystic lung disease play a key role in diagnosis (Table 1). For instance, spontaneous pneumothorax is commonly associated with diffuse cystic lung disease (lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome), while insidious dyspnea, with or without associated pneumothorax, is usually associated with the interstitial pneumonias (lymphocytic interstitial pneumonia and desquamative interstitial pneumonia).
In addition, congenital abnormalities of the lung can lead to cyst formation. These abnormalities, especially when associated with other congenital abnormalities, are often diagnosed in the prenatal and perinatal periods. However, some remain undetected until incidentally found later in adulthood or if superimposing infection develops.
Primary pulmonary infections can also cause parenchymal necrosis, which in turn cavitates or forms cysts.4
Lastly, cystic lung diseases can occur as part of a multiorgan or systemic illness in which the lung is one of the organs involved. Although usually diagnosed before the discovery of cysts or manifestations of pulmonary symptoms, they can present as a diagnostic challenge, especially when lung cysts are the initial presentation.bsence of amyloid fibrils.
In view of the features of the different types of cystic lung disease, adults with cystic lung disease can be grouped according to their typical clinical presentations (Table 2):
- Insidious dyspnea or spontaneous pneumothorax
- Incidentally found cysts or recurrent pneumonia
- Signs and symptoms of primary pulmonary infection
- Signs and symptoms that are primarily nonpulmonary.
Insidious dyspnea or spontaneous pneumothorax
Insidious dyspnea or spontaneous pneumothorax can be manifestations of lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, or lymphocytic interstitial pneumonia.
Lymphangioleiomyomatosis is characterized by abnormal cellular proliferation within the lung, kidney, lymphatic system, or any combination.5 The peak prevalence is in the third to fourth decades of life, and most patients are women of childbearing age.6 In addition to progressive dyspnea on exertion and pneumothorax, other signs and symptoms include hemoptysis, nonproductive cough, chylous pleural effusion, and ascites.7,8
Birt-Hogg-Dubé syndrome is caused by germline mutations in the folliculin (FLCN) gene.9 It is characterized by skin fibrofolliculomas, pulmonary cysts, spontaneous pneumothorax, and renal cancer.10
Pulmonary Langerhans cell histiocytosis is part of the spectrum of Langerhans cell histiocytosis that, in addition to the lungs, can also involve the bone, pituitary gland, thyroid, skin, lymph nodes, and liver.11 It occurs almost exclusively in smokers, affecting individuals in their 20s and 30s, with no gender predilection.12,13 In addition to nonproductive cough and dyspnea, patients can also present with fever, anorexia, and weight loss,13 but approximately 25% of patients are asymptomatic.14
Desquamative interstitial pneumonia is an idiopathic interstitial pneumonia that, like pulmonary Langerhans cell histiocytosis, is seen almost exclusively in current or former smokers, who account for about 90% of patients with this disease. It affects almost twice as many men as women.15,16 The mean age at onset is 42 to 46.15,16 In addition to insidious cough and dyspnea, digital clubbing develops in 26% to 40% of patients.16,17
Lymphocytic interstitial pneumonia is another rare idiopathic pneumonia, usually associated with connective tissue disease, Sjögren syndrome, immunodeficiencies, and viral infections.18–21 It is more common in women, presenting between the 4th and 7th decades of life, with a mean age at diagnosis of 50 to 56.18,22 In addition to progressive dyspnea and cough, other symptoms include weight loss, pleuritic pain, arthralgias, fatigue, night sweats, and fever.23
In summary, in this clinical group, lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome should be considered when patients present with spontaneous pneumothorax; those with Birt-Hogg-Dubé syndrome also present with skin lesions or renal cancer. In patients with progressive dyspnea and cough, lymphocytic interstitial pneumonia should be considered in those with a known history of connective tissue disease or immunodeficiency. Pulmonary Langerhans cell histiocytosis typically presents at a younger age (20 to 30 years old) than desquamative interstitial pneumonia (smokers in their 40s). Making the distinction, however, will likely require imaging with computed tomography.
Incidentally found cysts or recurrent pneumonia
Incidentally found cysts or recurrent pneumonia can be manifestations of congenital pulmonary airway malformation, pulmonary sequestration, or bronchogenic cyst.
Congenital pulmonary airway malformation, of which there are five types, is the most common pulmonary congenital abnormality. It accounts for up to 95% of cases of congenital cystic lung disease.24,25 About 85% of cases are detected in the prenatal or perinatal periods.26 Late-onset congenital pulmonary airway malformation (arising in childhood to adulthood) presents with recurrent pneumonia in about 75% of cases and can be misdiagnosed as lung abscess, pulmonary tuberculosis, or bronchiectasis.27
Pulmonary sequestration, the second most common pulmonary congenital abnormality, is characterized by a portion of lung that does not connect to the tracheobronchial tree and has its own systemic arterial supply.24 Intralobar sequestration, which shares the pleural investment with normal lung, accounts for about 80% of cases of pulmonary sequestration.28–30 In addition to signs or symptoms of pulmonary infection, patients with pulmonary sequestration can remain asymp-
tomatic (about 25% of cases), or can present with hemoptysis or hemothorax.28–30 In adults, the typical age at presentation is between 20 and 25.29,30
Bronchogenic cyst is usually life-threatening in children. In adults, it commonly causes cough and chest pain.31 Hemoptysis, dysphagia, hoarseness, and diaphragmatic paralysis can also occur.32,33 The mean age at diagnosis in adults is 35 to 40.31,32
In summary, most cases of recurrent pneumonia with cysts are due to congenital pulmonary airway malformation. Pulmonary sequestration is the second most common cause of cystic lung disease in this group. Bronchogenic cyst is usually fatal in fetal development; smaller cysts can go unnoticed during the earlier years and are later found incidentally as imaging abnormalities in adults.
Signs and symptoms of primary pulmonary infections
Signs and symptoms of primary pulmonary infections can be due to Pneumocystis jirovecii pneumonia or echinococcal infections.
P jirovecii pneumonia commonly develops in patients with human immunodeficiency virus infection and low CD4 counts, recipients of hematologic or solid-organ transplants, and those receiving immunosuppressive therapy (eg, glucocorticoids or chemotherapy).
Echinococcal infections (with Echinococcus granulosus or multilocularis species) are more common in less-developed countries such as those in South America or the Middle East, in China, or in patients who have traveled to endemic areas.34
In summary, cystic lung disease in patients with primary pulmonary infections can be diagnosed by the patient’s clinical history and risk factors for infections. Those with human immunodeficiency virus infection and other causes of immunodeficiency are predisposed to P jirovecii pneumonia. Echinococcal infections occur in those with a history of travel to an endemic area.
Primarily nonpulmonary signs and symptoms
If the patient has primarily nonpulmonary signs and symptoms, think about pulmonary amyloidosis, light chain deposition disease, and neurofibromatosis type 1.
Pulmonary amyloidosis has a variety of manifestations, including tracheobronchial disease, nodular parenchymal disease, diffuse or alveolar septal pattern, pleural disease, lymphadenopathy, and pulmonary cysts.4
Light chain deposition disease shares some clinical features with amyloidosis. However, the light chain fragments in this disease do not form amyloid fibrils and therefore do not stain positively with Congo red. The kidney is the most commonly involved organ.4
Neurofibromatosis type 1 is characterized by collections of neurofibromas, café-au-lait spots, and pigmented hamartomas in the iris (Lisch nodules).35
In summary, patients in this group typically present with complications related to systemic involvement. Those with neurofibromatosis type 1 present with ophthalmologic, dermatologic, and neurologic manifestations. Amyloidosis and light chain deposition disease most commonly involve the renal system; their distinction will likely require tissue biopsy and Congo-red staining.
STEP 3: CHARACTERIZE THE RADIOGRAPHIC FEATURES
Characterization of pulmonary cysts and their distribution plays a key role in the diagnosis. Radiographically, cystic lung diseases can be subclassified into two major categories according to their cystic distribution:
- Discrete (focal or multifocal)
- Diffuse (unilobular or panlobular).2,3
Discrete cystic lung diseases include congenital abnormalities, infectious diseases, and interstitial pneumonias.2,3
Diffuse, panlobular cystic lung diseases include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dubé syndrome, amyloidosis, light chain deposition disease, and neurofibromatosis type 1.7,13,36–39
In addition, other associated radiographic findings play a major role in diagnosis.
Cysts in patients presenting with insidious dyspnea or spontaneous pneumothorax
Lymphangioleiomyomatosis. Cysts are seen in nearly all cases of advanced lymphangioleiomyomatosis, typically in a diffuse pattern, varying from 2 mm to 40 mm in diameter, and uniform in shape (Figure 2A).7,8,40–42
Other radiographic features include vessels located at the periphery of the cysts (in contrast to the centrilobular pattern seen with emphysema), and chylous pleural effusions (in about 22% of patients).40 Nodules are typically not seen with lymphangioleiomyomatosis, and if found represent type 2 pneumocyte hyperplasia.
Pulmonary Langerhans cell histiocytosis. Nodules measuring 1 to 10 mm in diameter and favoring a centrilobular location are often seen on computed tomography. Pulmonary cysts occur in about 61% of patients.13,43 Cysts are variable in size and shape (Figure 2B), in contrast to their uniform appearance in lymphangioleiomyomatosis. Most cysts are less than 10 mm in diameter; however, they can be up to 80 mm.13,43 Early in its course, nodules may predominate in the upper and middle lobes. Over time, diffuse cysts become more common and can be difficult to differentiate from advanced smoking-induced emphysema.44
Birt-Hogg-Dubé syndrome. Approximately 70% to 100% of patients with Birt-Hogg-Dubé syndrome will have multiple pulmonary cysts detected on computed tomography. These cysts are characteristically basal and subpleural in location, with varying sizes and irregular shapes in otherwise normal lung parenchyma (Figure 2C).36,45,46
Desquamative interstitial pneumonia. Pulmonary cysts are present on computed tomography in about 32% of patients.47 They are usually round and less than 20 mm in diameter.48 Ground-glass opacity is present in almost all cases of desquamative interstitial pneumonia, with a diffuse pattern in 25% to 44% of patients.16,17,47
Pulmonary cysts occur in up to two-thirds of those with lymphocytic interstitial pneumonia. Cysts are usually multifocal and perivascular in distribution and have varying sizes and shapes (Figure 2D).22 Ground-glass opacity and poorly defined centrilobular nodules are also frequently seen. Other computed tomographic findings include thickening of the bronchovascular bundles, focal consolidation, interseptal lobular thickening, pleural thickening, and lymph node enlargement.22
In summary, in this group of patients, diffuse panlobular cysts are due to lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, or Birt-Hogg-Dubé syndrome. Cysts due to lymphangioleiomyomatosis have a diffuse distribution, while those due to pulmonary Langerhans cell histiocytosis tend to be upper-lobe-predominant and in the early stages are associated with stellate centrilobular nodules. Cysts in Birt-Hogg-Dubé syndrome tend to be subpleural and those due to lymphocytic interstitial pneumonia are perivascular in distribution.
Cysts that are incidentally found or occur in patients with recurrent pneumonia
Congenital pulmonary airway malformation types 1, 2, and 4 (Figure 3A, 3B). Cysts are typically discrete and focal or multifocal in distribution, but cases of multilobar and bilateral distribution have also been reported.27,49 The lower lobes are more often involved.49 Cysts vary in size and shape and can contain air, fluid, or both.27,49 Up to 50% of cases can occur in conjunction with pulmonary sequestration.50
Pulmonary sequestration displays an anomalous arterial supply on computed tomography (Figure 3C). Other imaging findings include mass lesions (49%), cystic lesions (29%), cavitary lesions (12%), and bronchiectasis.30 Air trapping can be seen in the adjacent lung. Lower lobe involvement accounts for more than 95% of total cases of sequestration.30 The cysts are usually discrete or focal in distribution. Misdiagnosis of pulmonary sequestration is common, and can include pulmonary abscess, pneumonia, bronchiectasis, and lung cancer.30
Bronchogenic cyst. Cyst contents generally demonstrate water attenuation, or higher attenuation if filled with proteinaceous/mucoid material or calcium deposits; air-fluid levels are seen in infected cysts.32 Intrapulmonary cysts have a predilection for the lower lobes and are usually discrete or focal in distribution.31,32 Mediastinal cysts are usually homogeneous, solitary, and located in the middle mediastinum.32 Cysts vary in size from 20 to 90 mm, with a mean diameter of 40 mm.31
In summary, in this group of cystic lung diseases, characteristic computed tomographic findings will suggest the diagnosis—air-filled cysts of varying sizes for congenital pulmonary airway malformation and anomalous vascular supply for pulmonary sequestration. Bronchogenic cysts will tend to have water or higher-than-water attenuation due to proteinaceous-mucoid material or calcium deposits.
Cysts in patients with signs and symptoms of primary pulmonary infections
P jirovecii pneumonia. Between 10% and 15% of patients have cysts, and about 18% present with spontaneous pneumothorax.51 Cysts in P jirovecii pneumonia vary in size from 15 to 85 mm in diameter and tend to occur in the upper lobes (Figure 4A).51,52
Echinococcal infection. Echinococcal pulmonary cysts typically are single and located more often in the lower lobes (Figure 4B).53,54 Cysts can be complicated by air-fluid levels, hydropneumothorax, or pneumothorax, or they can turn into cavitary lesions.
The diagnoses of these pulmonary infections are usually made by clinical and computed tomographic findings and depend less on detecting and characterizing lung cysts. Patients with P jirovecii pneumonia tend to have bilateral perihilar ground-glass opacities, while air-fluid levels suggest echinococcal infections. Cysts in this group of patients tend to be discrete or focal or multifocal in distribution, and vary in size.
Cysts in patients with primarily nonpulmonary signs and symptoms
Amyloidosis. Cyst formation is rare in amyloidosis.4 When present, cysts can be diffuse and scattered in distribution, in varying sizes (usually < 30 mm in diameter) and irregular shapes (Figure 5).55,56
Pulmonary light chain deposition disease usually presents as linear opacities and small nodules on chest computed tomography. Numerous cysts that are diffuse in distribution and have no topographic predominance can also be present. They can progress in number and size and coalesce to form irregular shapes.57
Neurofibromatosis type 1. In neurofibromatosis type 1, the most common radiographic presentations are bibasilar reticular opacities (50%), bullae (50%), and ground glass opacities (37%).58 Well-formed cysts occur in up to 25% of patients and tend to be diffuse and smaller (2 to 18 mm in diameter), with upper lobe predominance.58,59
In summary, in this group of patients, bibasilar reticular and ground-glass opacities suggest neurofibromatosis type 1, while nodules and linear opacities suggest amyloidosis or light chain deposition disease. Cysts tend to be diffuse with varying sizes.
STEP 4: PUT IT ALL TOGETHER
Diagnosis in insidious dyspnea or spontaneous pneumothorax
For patients who present with insidious dyspnea or spontaneous pneumothorax, the diagnosis of cystic lung disease can be made by characterizing the distribution, size, and shape of the cysts (Table 3).
Diffuse, panlobular distribution. Cystic lung diseases with this pattern include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, and Birt-Hogg-Dubé syndrome. In this group, cysts that are uniform in size and regular in shape are invariably due to lymphangioleiomyomatosis. Those with variable size and irregular shapes can be due to pulmonary Langerhans cell histiocytosis or Birt-Hogg-Dubé syndrome. Patients with pulmonary Langerhans cell histiocytosis tend to be smokers and their cysts tend to be upper- lobe-predominant. Those with Birt-Hogg-Dubé syndrome will likely have renal cancer or skin lesions; their cysts tend to be basilar and subpleural in distribution.
Cysts that are focal or multifocal and unilobular are due to lymphocytic interstitial pneumonia or desquamative interstitial pneumonia. Patients with lymphocytic interstitial pneumonia tend to have underlying connective tissue disease; those with desquamative interstitial pneumonia are almost always smokers. The definitive diagnosis for lymphocytic interstitial pneumonia or desquamative interstitial pneumonia can require a tissue biopsy.
Diagnosis in patients with incidentally found cysts or recurrent pneumonia
In those who present with incidentally found cysts or recurrent pneumonia, suspicion for a congenital lung malformation should be raised. Patients with a type 1, 2, or 4 congenital pulmonary airway malformation typically have air-filled cysts in varying sizes; those with pulmonary sequestration have an anomalous arterial supply in addition to cysts that are usually located in the lower lobes. Bronchogenic cysts tend to be larger, with attenuation equal to or greater than that of water, and distinguishing them from congenital pulmonary airway malformation will likely require surgical examination.
Diagnosis in patients with signs and symptoms of pulmonary infections
Patients with signs and symptoms of pulmonary infections should be investigated according to clinical risk factors for P jirovecii pneumonia or echinococcal infections.
Diagnosis in patients with primarily nonpulmonary presentations
The distinction between amyloidosis and neurofibromatosis type 1 can be made by the history and the clinical examination. However, a definitive diagnosis of amyloidosis or light chain deposition disease requires tissue examination for the presence or absence of amyloid fibrils.
- Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology 2008; 246:697–722.
- Cosgrove GP, Frankel SK, Brown KK. Challenges in pulmonary fibrosis. 3: cystic lung disease. Thorax 2007; 62:820–829.
- Ryu JH, Swensen SJ. Cystic and cavitary lung diseases: focal and diffuse. Mayo Clin Proc 2003; 78:744–752.
- Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med 2013; 7:316–327.
- McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest 2008; 133:507–516.
- Johnson SR, Cordier JF, Lazor R, et al; Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010; 35:14–26.
- Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990; 323:1254–1260.
- Chu SC, Horiba K, Usuki J. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115:1041–1052.
- Graham RB, Nolasco M, Peterlin B, Garcia CK. Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 2005; 172:39–44.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977; 113:1674–1677.
- Sundar KM, Gosselin MV, Chung HL, Cahill BC. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology, and clinical evolution of disease. Chest 2003; 123:1673–1683.
- Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000; 342:1969–1978.
- Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346:484–490.
- Mendez JL, Nadrous HF, Vassallo R, Decker PA, Ryu JH. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125:1028–1032.
- Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med 1978; 298:801–809.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology 2005; 236:10–21.
- Strimlan CV, Rosenow EC 3rd, Weiland LH, Brown LR. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med 1978; 88:616–621.
- Arish N, Eldor R, Fellig Y, et al. Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax 2006; 61:1096–1097.
- Schooley RT, Carey RW, Miller G, et al. Chronic Epstein-Barr virus infection associated with fever and interstitial pneumonitis. Clinical and serologic features and response to antiviral chemotherapy. Ann Intern Med 1986; 104:636–643.
- Kramer MR, Saldana MJ, Ramos M, Pitchenik AE. High titers of Epstein-Barr virus antibodies in adult patients with lymphocytic interstitial pneumonitis associated with AIDS. Respir Med 1992; 86:49–52.
- Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999; 212:567–572.
- Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest 2002; 122:2150–2164.
- Biyyam DR, Chapman T, Ferguson MR, Deutsch G, Dighe MK. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics 2010; 30:1721–1738.
- Cloutier MM, Schaeffer DA, Hight D. Congenital cystic adenomatoid malformation. Chest 1993; 103:761–764.
- Luján M, Bosque M, Mirapeix RM, Marco MT, Asensio O, Domingo C. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration 2002; 69:148–154.
- Oh BJ, Lee JS, Kim JS, Lim CM, Koh Y. Congenital cystic adenomatoid malformation of the lung in adults: clinical and CT evaluation of seven patients. Respirology 2006; 11:496–501.
- Tsolakis CC, Kollias VD, Panayotopoulos PP. Pulmonary sequestration. Experience with eight consecutive cases. Scand Cardiovasc J 1997; 31:229–232.
- Sauvanet A, Regnard JF, Calanducci F, Rojas-Miranda A, Dartevelle P, Levasseur P. Pulmonary sequestration. Surgical aspects based on 61 cases. Rev Pneumol Clin 1991; 47:126–132. Article in French.
- Wei Y, Li F. Pulmonary sequestration: a retrospective analysis of 2,625 cases in China. Eur J Cardiothorac Surg 2011; 40:e39–e42.
- Patel SR, Meeker DP, Biscotti CV, Kirby TJ, Rice TW. Presentation and management of bronchogenic cysts in the adult. Chest 1994; 106:79–85.
- Limaïem F, Ayadi-Kaddour A, Djilani H, Kilani T, El Mezni F. Pulmonary and mediastinal bronchogenic cysts: a clinicopathologic study of 33 cases. Lung 2008; 186:55–61.
- Liu HS, Li SQ, Cao ZL, Zhang ZY, Ren H. Clinical features and treatment of bronchogenic cyst in adults. Chin Med Sci J 2009; 24:60–63.
- Jenkins DJ, Romig T, Thompson RC. Emergence/re-emergence of Echinococcus spp.—a global update. Int J Parasitol 2005; 35:1205–1219.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med 1981; 305:1617–1627.
- Toro JR, Pautler SE, Stewart L, et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 2007; 175:1044–1053.
- Biko DM, Schwartz M, Anupindi SA, Altes TA. Subpleural lung cysts in Down syndrome: prevalence and association with coexisting diagnoses. Pediatr Radiol 2008; 38:280–284.
- Colombat M, Stern M, Groussard O, et al. Pulmonary cystic disorder related to light chain deposition disease. Am J Respir Crit Care Med 2006; 173:777–780.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in the UK. Thorax 2000; 55:1052–1057.
- Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med 1995; 151:527–533.
- Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 1999; 78:321–337.
- Schönfeld N, Frank W, Wenig S, et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993; 60:38–44.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Kluger N, Giraud S, Coupier I, et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol 2010; 162:527–537.
- Tobino K, Gunji Y, Kurihara M, et al. Characteristics of pulmonary cysts in Birt-Hogg-Dubé syndrome: thin-section CT findings of the chest in 12 patients. Eur J Radiol 2011; 77:403–409.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Koyama M, Johkoh T, Honda O, et al. Chronic cystic lung disease: diagnostic accuracy of high-resolution CT in 92 patients. AJR Am J Roentgenol 2003; 180:827–835.
- Patz EF Jr, Müller NL, Swensen SJ, Dodd LG. Congenital cystic adenomatoid malformation in adults: CT findings. J Comput Assist Tomogr 1995; 19:361–364.
- Conran RM, Stocker JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol 1999; 2:454–463.
- Kennedy CA, Goetz MB. Atypical roentgenographic manifestations of Pneumocystis carinii pneumonia. Arch Intern Med 1992; 152:1390–1398.
- Sandhu JS, Goodman PC. Pulmonary cysts associated with Pneumocystis carinii pneumonia in patients with AIDS. Radiology 1989; 173:33–35.
- Doğan R, Yüksel M, Cetin G, et al. Surgical treatment of hydatid cysts of the lung: report on 1,055 patients. Thorax 1989; 44:192–199.
- Salih OK, Topcuoğlu MS, Celik SK, Ulus T, Tokcan A. Surgical treatment of hydatid cysts of the lung: analysis of 405 patients. Can J Surg 1998; 41:131–135.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Sakai M, Yamaoka M, Kawaguchi M, Hizawa N, Sato Y. Multiple cystic pulmonary amyloidosis. Ann Thorac Surg 2011; 92:e109.
- Colombat M, Caudroy S, Lagonotte E, et al. Pathomechanisms of cyst formation in pulmonary light chain deposition disease. Eur Respir J 2008; 32:1399–1403.
- Zamora AC, Collard HR, Wolters PJ, Webb WR, King TE. Neurofibromatosis-associated lung disease: a case series and literature review. Eur Respir J 2007; 29:210–214.
- Oikonomou A, Vadikolias K, Birbilis T, Bouros D, Prassopoulos P. HRCT findings in the lungs of non-smokers with neurofibromatosis. Eur J Radiol 2011; 80:e520–e523.
- Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology 2008; 246:697–722.
- Cosgrove GP, Frankel SK, Brown KK. Challenges in pulmonary fibrosis. 3: cystic lung disease. Thorax 2007; 62:820–829.
- Ryu JH, Swensen SJ. Cystic and cavitary lung diseases: focal and diffuse. Mayo Clin Proc 2003; 78:744–752.
- Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med 2013; 7:316–327.
- McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest 2008; 133:507–516.
- Johnson SR, Cordier JF, Lazor R, et al; Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010; 35:14–26.
- Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990; 323:1254–1260.
- Chu SC, Horiba K, Usuki J. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115:1041–1052.
- Graham RB, Nolasco M, Peterlin B, Garcia CK. Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 2005; 172:39–44.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977; 113:1674–1677.
- Sundar KM, Gosselin MV, Chung HL, Cahill BC. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology, and clinical evolution of disease. Chest 2003; 123:1673–1683.
- Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans’-cell histiocytosis. N Engl J Med 2000; 342:1969–1978.
- Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med 2002; 346:484–490.
- Mendez JL, Nadrous HF, Vassallo R, Decker PA, Ryu JH. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125:1028–1032.
- Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med 1978; 298:801–809.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology 2005; 236:10–21.
- Strimlan CV, Rosenow EC 3rd, Weiland LH, Brown LR. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med 1978; 88:616–621.
- Arish N, Eldor R, Fellig Y, et al. Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax 2006; 61:1096–1097.
- Schooley RT, Carey RW, Miller G, et al. Chronic Epstein-Barr virus infection associated with fever and interstitial pneumonitis. Clinical and serologic features and response to antiviral chemotherapy. Ann Intern Med 1986; 104:636–643.
- Kramer MR, Saldana MJ, Ramos M, Pitchenik AE. High titers of Epstein-Barr virus antibodies in adult patients with lymphocytic interstitial pneumonitis associated with AIDS. Respir Med 1992; 86:49–52.
- Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999; 212:567–572.
- Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review. Chest 2002; 122:2150–2164.
- Biyyam DR, Chapman T, Ferguson MR, Deutsch G, Dighe MK. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics 2010; 30:1721–1738.
- Cloutier MM, Schaeffer DA, Hight D. Congenital cystic adenomatoid malformation. Chest 1993; 103:761–764.
- Luján M, Bosque M, Mirapeix RM, Marco MT, Asensio O, Domingo C. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration 2002; 69:148–154.
- Oh BJ, Lee JS, Kim JS, Lim CM, Koh Y. Congenital cystic adenomatoid malformation of the lung in adults: clinical and CT evaluation of seven patients. Respirology 2006; 11:496–501.
- Tsolakis CC, Kollias VD, Panayotopoulos PP. Pulmonary sequestration. Experience with eight consecutive cases. Scand Cardiovasc J 1997; 31:229–232.
- Sauvanet A, Regnard JF, Calanducci F, Rojas-Miranda A, Dartevelle P, Levasseur P. Pulmonary sequestration. Surgical aspects based on 61 cases. Rev Pneumol Clin 1991; 47:126–132. Article in French.
- Wei Y, Li F. Pulmonary sequestration: a retrospective analysis of 2,625 cases in China. Eur J Cardiothorac Surg 2011; 40:e39–e42.
- Patel SR, Meeker DP, Biscotti CV, Kirby TJ, Rice TW. Presentation and management of bronchogenic cysts in the adult. Chest 1994; 106:79–85.
- Limaïem F, Ayadi-Kaddour A, Djilani H, Kilani T, El Mezni F. Pulmonary and mediastinal bronchogenic cysts: a clinicopathologic study of 33 cases. Lung 2008; 186:55–61.
- Liu HS, Li SQ, Cao ZL, Zhang ZY, Ren H. Clinical features and treatment of bronchogenic cyst in adults. Chin Med Sci J 2009; 24:60–63.
- Jenkins DJ, Romig T, Thompson RC. Emergence/re-emergence of Echinococcus spp.—a global update. Int J Parasitol 2005; 35:1205–1219.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med 1981; 305:1617–1627.
- Toro JR, Pautler SE, Stewart L, et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 2007; 175:1044–1053.
- Biko DM, Schwartz M, Anupindi SA, Altes TA. Subpleural lung cysts in Down syndrome: prevalence and association with coexisting diagnoses. Pediatr Radiol 2008; 38:280–284.
- Colombat M, Stern M, Groussard O, et al. Pulmonary cystic disorder related to light chain deposition disease. Am J Respir Crit Care Med 2006; 173:777–780.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Johnson SR, Tattersfield AE. Clinical experience of lymphangioleiomyomatosis in the UK. Thorax 2000; 55:1052–1057.
- Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med 1995; 151:527–533.
- Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 1999; 78:321–337.
- Schönfeld N, Frank W, Wenig S, et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993; 60:38–44.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Kluger N, Giraud S, Coupier I, et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol 2010; 162:527–537.
- Tobino K, Gunji Y, Kurihara M, et al. Characteristics of pulmonary cysts in Birt-Hogg-Dubé syndrome: thin-section CT findings of the chest in 12 patients. Eur J Radiol 2011; 77:403–409.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Koyama M, Johkoh T, Honda O, et al. Chronic cystic lung disease: diagnostic accuracy of high-resolution CT in 92 patients. AJR Am J Roentgenol 2003; 180:827–835.
- Patz EF Jr, Müller NL, Swensen SJ, Dodd LG. Congenital cystic adenomatoid malformation in adults: CT findings. J Comput Assist Tomogr 1995; 19:361–364.
- Conran RM, Stocker JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol 1999; 2:454–463.
- Kennedy CA, Goetz MB. Atypical roentgenographic manifestations of Pneumocystis carinii pneumonia. Arch Intern Med 1992; 152:1390–1398.
- Sandhu JS, Goodman PC. Pulmonary cysts associated with Pneumocystis carinii pneumonia in patients with AIDS. Radiology 1989; 173:33–35.
- Doğan R, Yüksel M, Cetin G, et al. Surgical treatment of hydatid cysts of the lung: report on 1,055 patients. Thorax 1989; 44:192–199.
- Salih OK, Topcuoğlu MS, Celik SK, Ulus T, Tokcan A. Surgical treatment of hydatid cysts of the lung: analysis of 405 patients. Can J Surg 1998; 41:131–135.
- Ohdama S, Akagawa S, Matsubara O, Yoshizawa Y. Primary diffuse alveolar septal amyloidosis with multiple cysts and calcification. Eur Respir J 1996; 9:1569–1571.
- Sakai M, Yamaoka M, Kawaguchi M, Hizawa N, Sato Y. Multiple cystic pulmonary amyloidosis. Ann Thorac Surg 2011; 92:e109.
- Colombat M, Caudroy S, Lagonotte E, et al. Pathomechanisms of cyst formation in pulmonary light chain deposition disease. Eur Respir J 2008; 32:1399–1403.
- Zamora AC, Collard HR, Wolters PJ, Webb WR, King TE. Neurofibromatosis-associated lung disease: a case series and literature review. Eur Respir J 2007; 29:210–214.
- Oikonomou A, Vadikolias K, Birbilis T, Bouros D, Prassopoulos P. HRCT findings in the lungs of non-smokers with neurofibromatosis. Eur J Radiol 2011; 80:e520–e523.
KEY POINTS
- Pulmonary cysts should be differentiated from cyst-mimics.
- Adults with cystic lung disease can be grouped by the clinical presentation: ie, insidious dyspnea or spontaneous pneumothorax; incidentally found cysts or recurrent pneumonia; signs and symptoms of primary pulmonary infection; or signs and symptoms that are primarily nonpulmonary.
- Characterization of pulmonary cysts and their distribution plays a key role in diagnosis. Radiographically, cystic lung disease can be subclassified into two major categories according to the distribution of cysts: discrete (focal or multifocal) and diffuse (unilobular or panlobular).
Genetics and hepatitis C: It’s good to be ‘CC’
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).

Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
KEY POINTS
- In IL28B, the rs12979860 location can be occupied by either cytosine (C) or thymine (T). The CC genotype is more favorable than the CT or TT genotype.
- Testing for the IL28B polymorphism is currently available and allows for better outcomes through proper selection of treatment, particularly with interferon-based treatment.
- Although newer therapies have shifted toward regimens that do not use interferon, the IL28B polymorphism remains clinically significant, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated.