User login
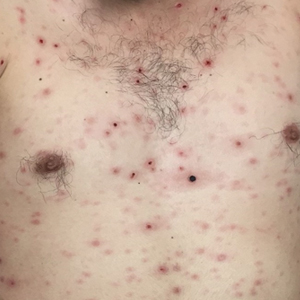
Painless Ulcer on the Areola
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
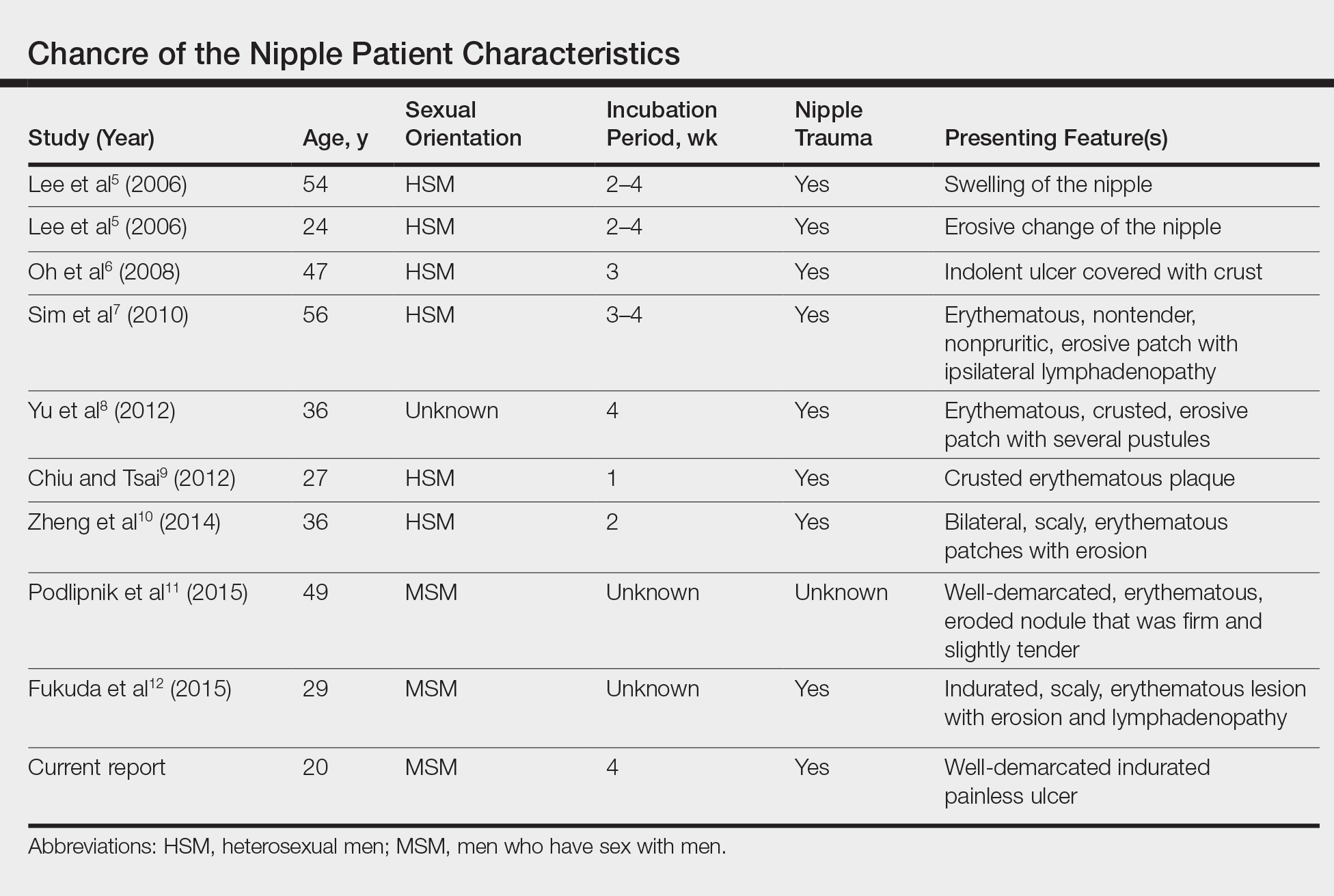
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
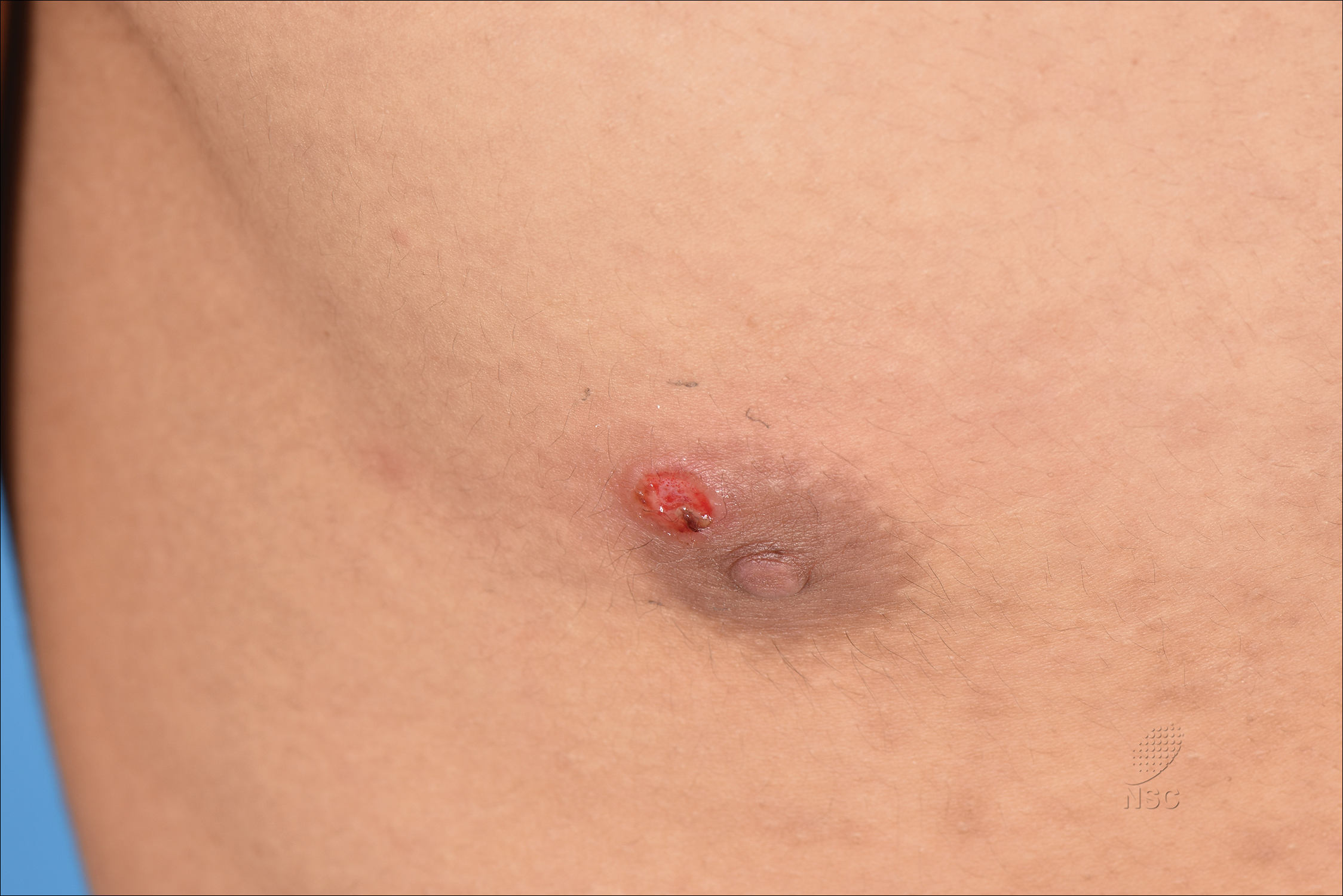
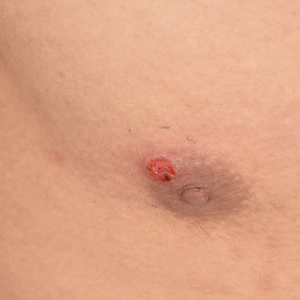
A previously healthy 20-year-old Chinese man presented to our dermatology outpatient clinic with a solitary painless ulcer on the right areola of 1 week's duration. Examination showed a small, slightly indurated ulcer with well-defined borders. No lesions were noted elsewhere. Swabs for pyogenic culture and herpes simplex virus polymerase chain reaction tests were sent, and he was treated empirically with oral cephalexin and tetracycline ointment 3%. At 1-week follow-up the ulcer had dried up and begun to heal, and results from the laboratory investigations were negative.
Slow-growing, Asymptomatic, Annular Plaques on the Bilateral Palms
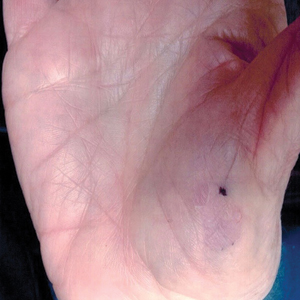
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
(H&E, original magnification ×4). No notable inflammation was evident in the dermis (B)(H&E, original magnification ×10).")
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
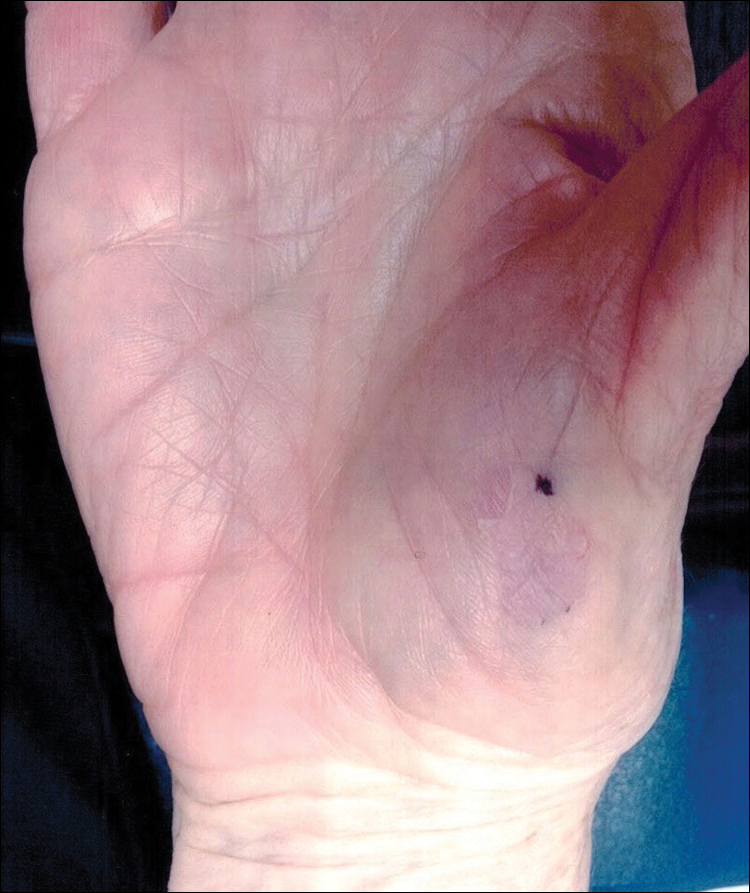
A 77-year-old woman presented with slow-growing, asymptomatic, annular plaques on the bilateral palms of many years' duration. There was no history of trauma or local infection. Prior treatment with over-the-counter creams was unsuccessful. A 3-mm punch biopsy of the lesion on the right palm was performed.
Painful Nonhealing Vulvar and Perianal Erosions
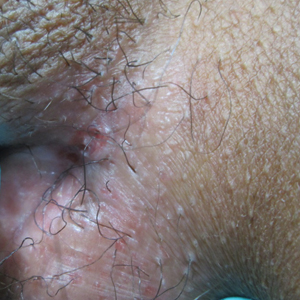
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
(H&E, original magnification ×4) and mixed inflammatory granulomas (B)(H&E, original magnification ×40).")
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
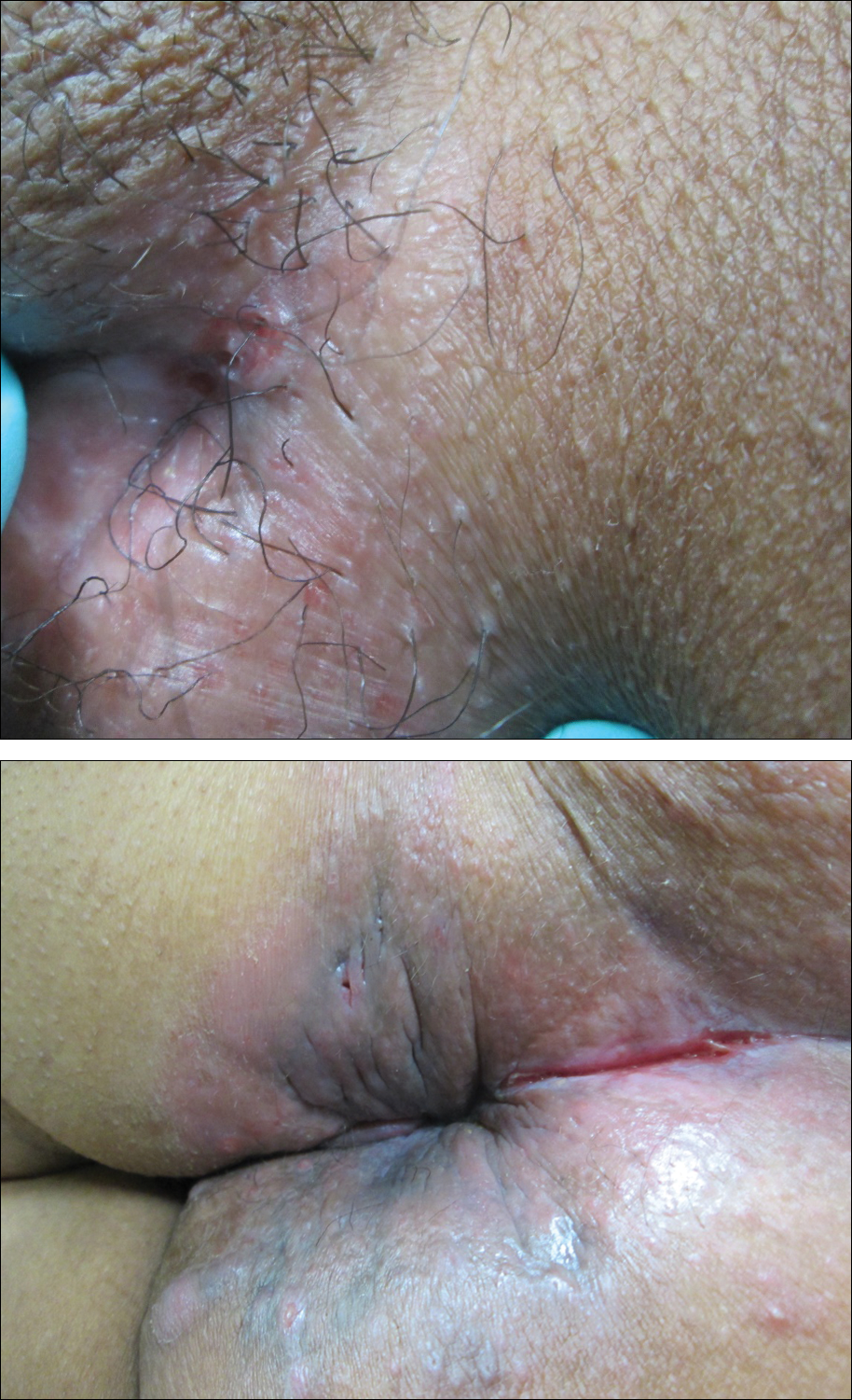
A 38-year-old woman with a history of Crohn disease presented with painful nonhealing vulvar and perianal erosions of 6 months' duration. The erosions developed 4 months after discontinuing adalimumab for a planned surgery. During this time, the patient also had an exacerbation of Crohn colitis and developed an anal fistula. Prior to this break in adalimumab, the patient's Crohn disease was well controlled on adalimumab 40 mg every 2 weeks, azathioprine 100 mg daily, and mesalamine 4.8 g daily. Despite restarting adalimumab and therapy with multiple antibiotics (ie, metronidazole, ciprofloxacin), the erosions persisted. On physical examination erythematous plaques and nodules were present at the vulvar (top) and perianal (bottom) skin. In addition, well-demarcated erosions measuring 20 mm and 80 mm were present on the vulvar and perianal skin, respectively. Human immunodeficiency virus screening and rapid plasma reagin were negative.
Scaly Annular and Concentric Plaques
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
(H&E, original magnification ×4) with neutrophils entrapped in thescale (B)(H&E, original magnification ×20).")
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).

Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
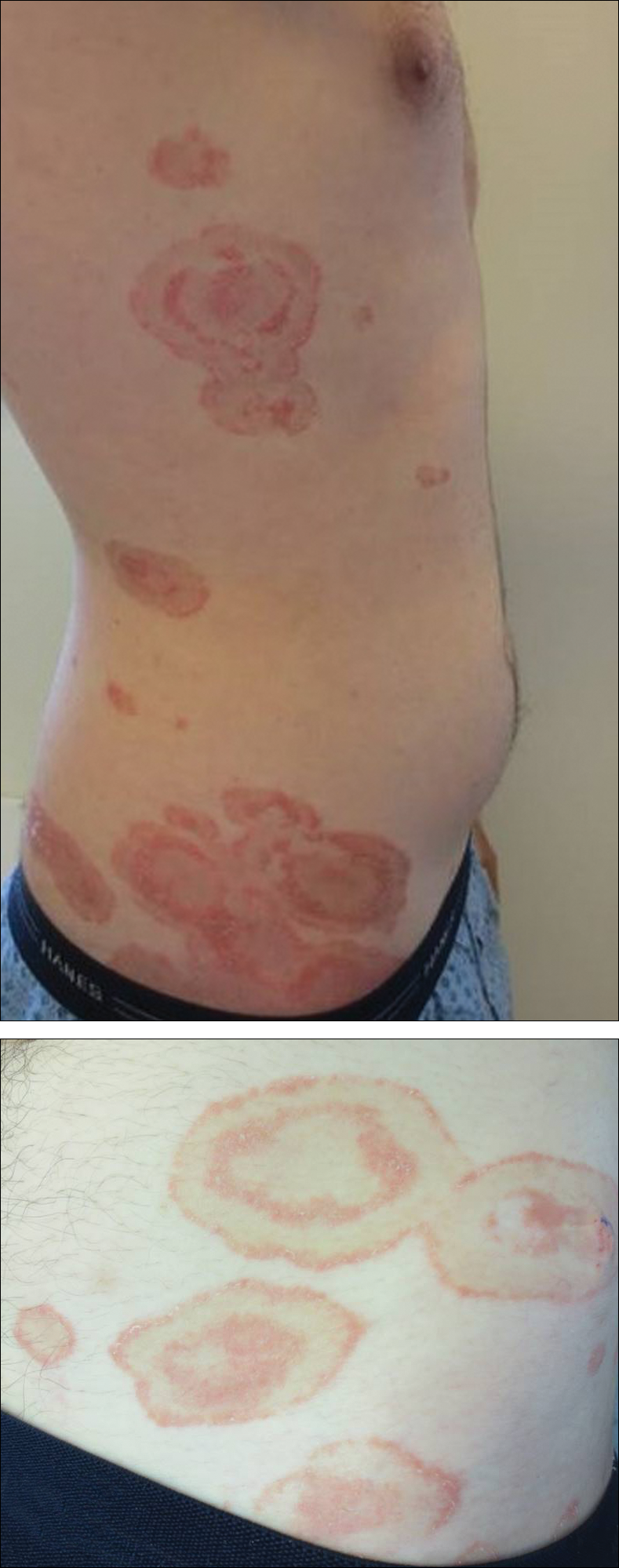
A healthy 23-year-old man presented for evaluation of an enlarging annular pruritic rash of 1.5 years' duration. Treatment with ciclopirox cream 0.77%, calcipotriene cream 0.005%, tacrolimus ointment 0.1%, fluticasone cream 0.05%, and halobetasol cream 0.05% prescribed by an outside physician provided only modest temporary improvement. The patient reported no history of travel outside of western New York, camping, tick bites, or medications. He denied any joint swelling or morning stiffness. Physical examination revealed multiple 4- to 6-cm pink, annular, scaly plaques with central clearing on the abdomen (top) and thighs. A few 1-cm pink scaly patches were present on the back (bottom), and few 2- to 3-mm pink scaly papules were noted on the extensor aspects of the elbows and forearms. A potassium hydroxide examination revealed no hyphal elements or yeast forms.
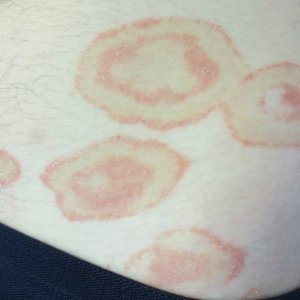
Red-Brown Plaque on the Leg
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.

Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
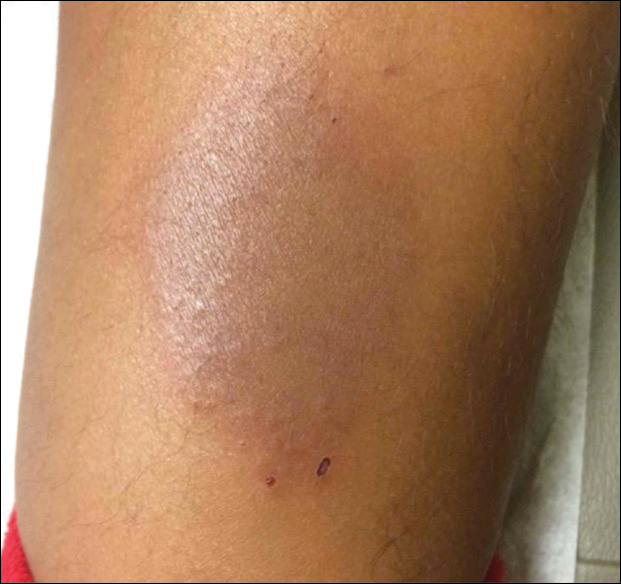
A healthy 7-year-old boy presented with an enlarging hyperpigmented plaque on the anterior aspect of the lower left leg of 2 months' duration. His mother reported onset following a mosquito bite. Clotrimazole was used without improvement. His mother denied recent travel, similar lesions in close contacts, fever, asthma, and arthralgia. Physical examination revealed a 5.2 ×3-cm nonscaly, red-brown, ovoid, thin plaque with a slightly raised border.
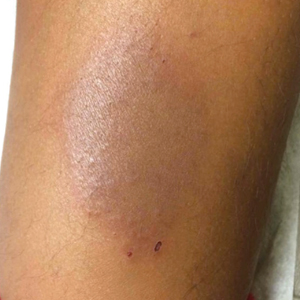
Progressive Widespread Telangiectasias
The Diagnosis: Cutaneous Collagenous Vasculopathy
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
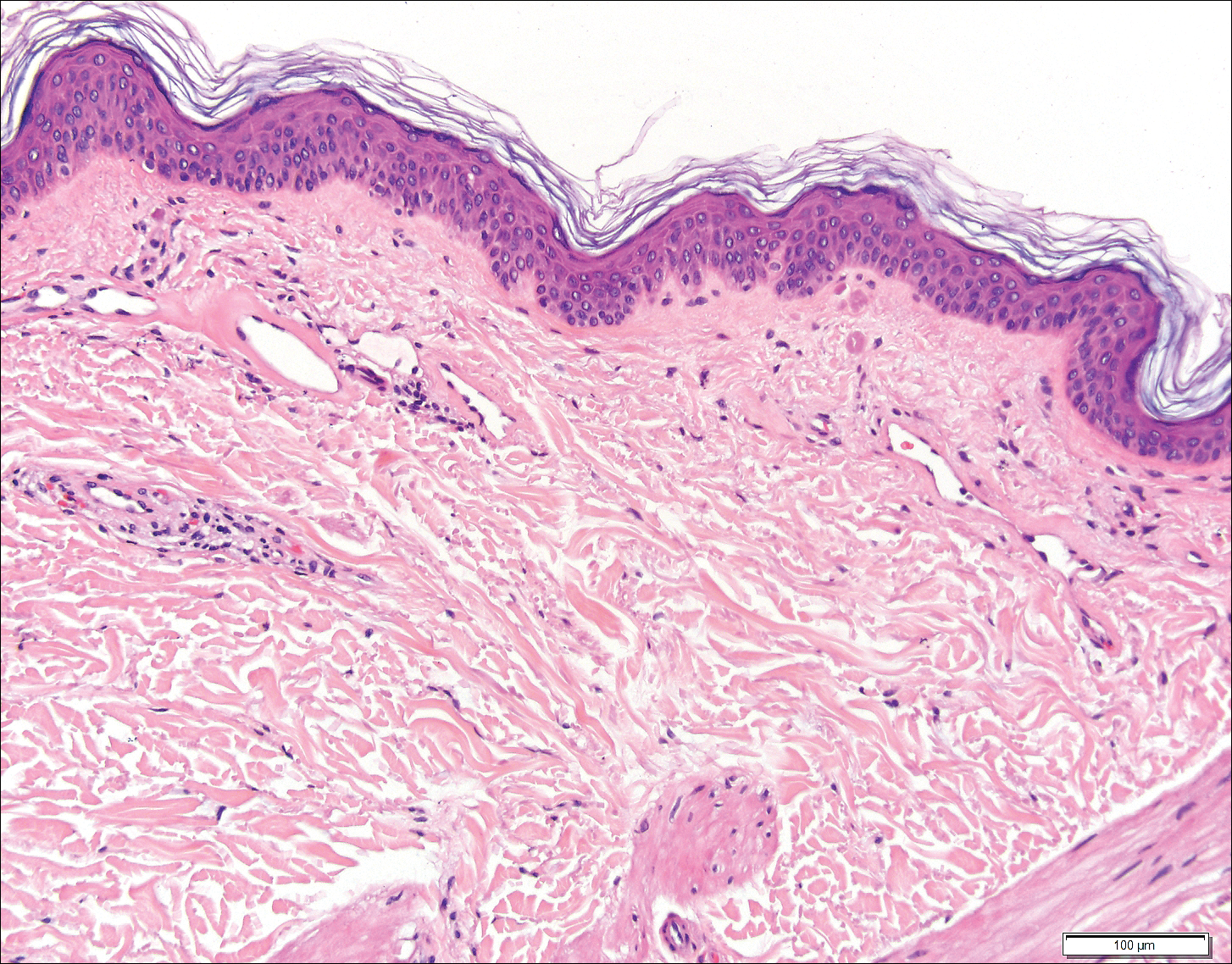
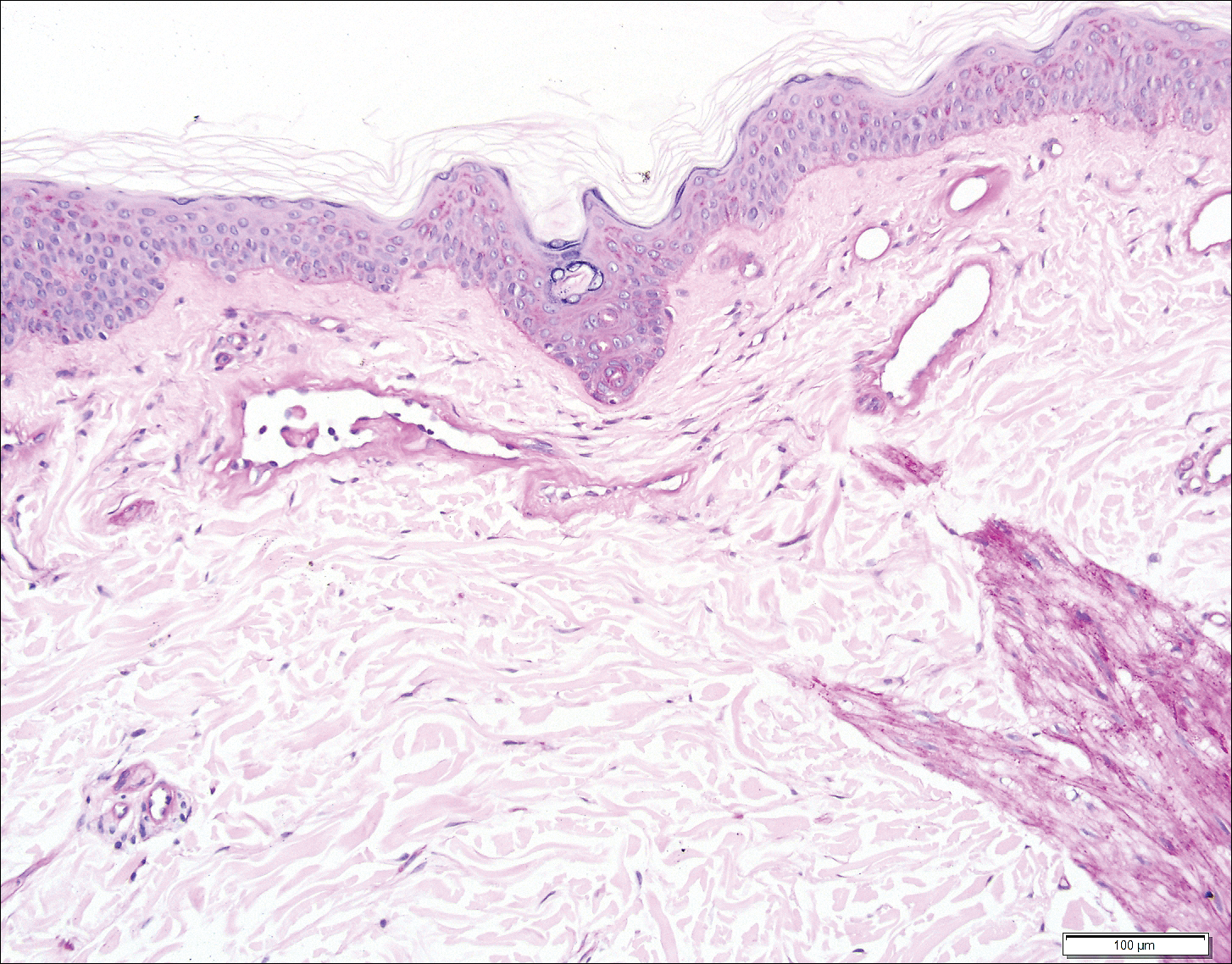
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
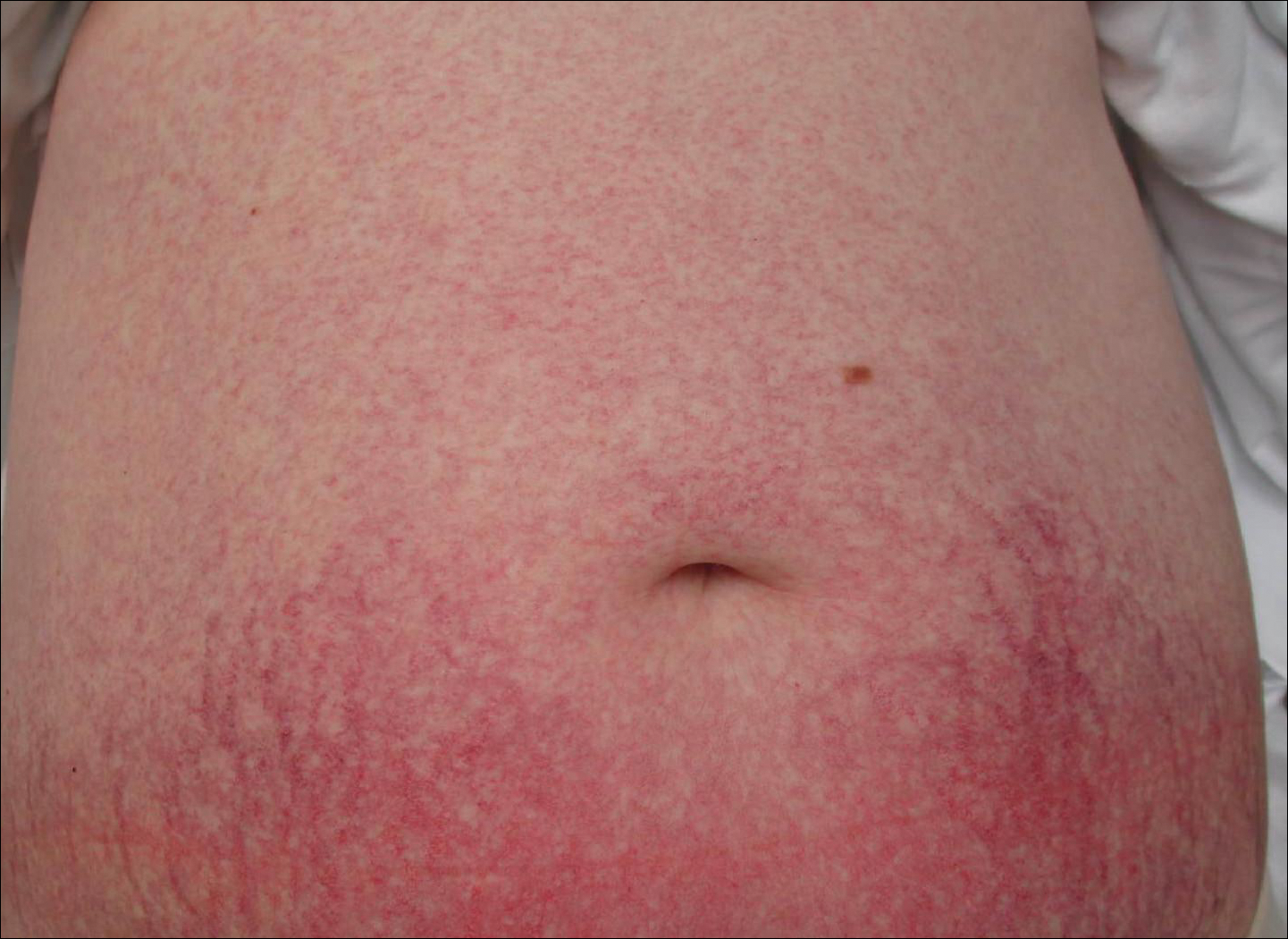
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
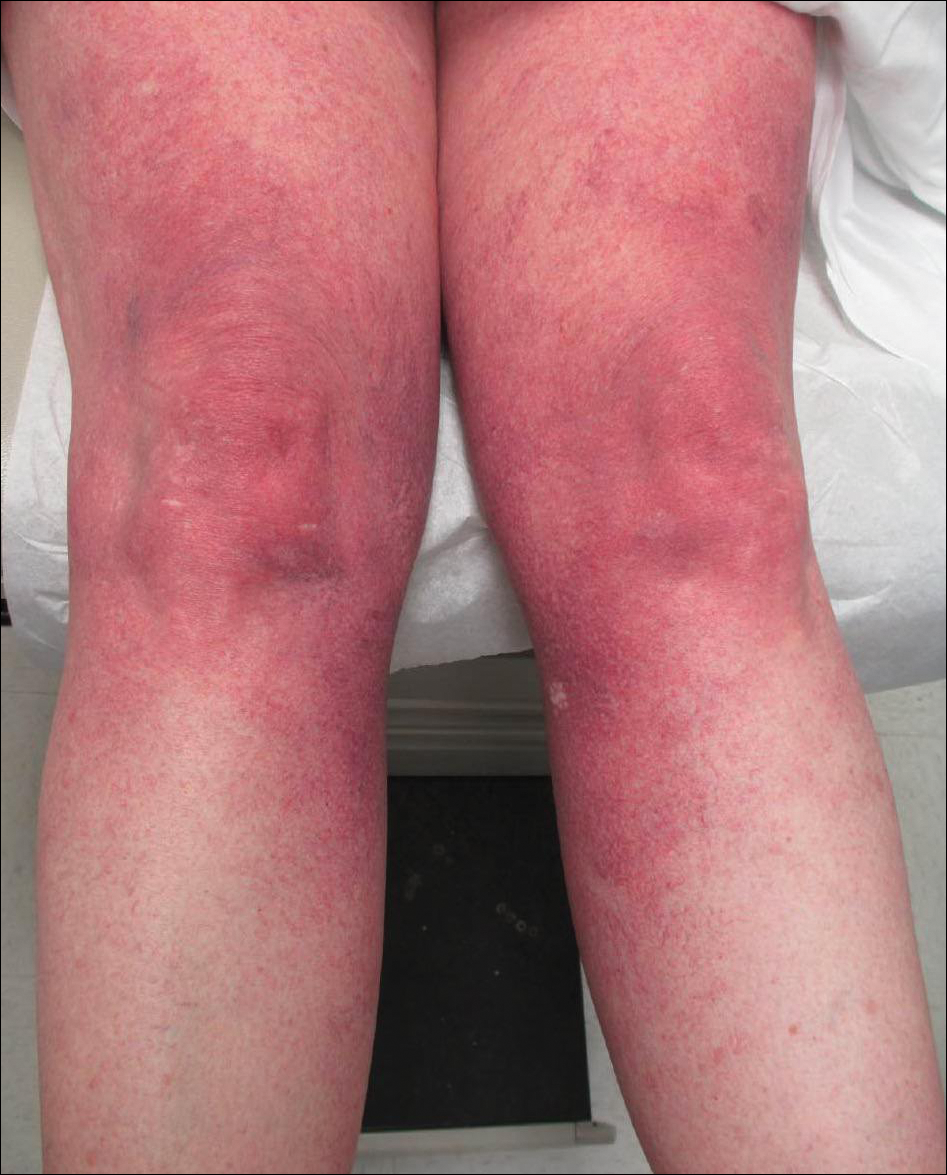
A 55-year-old woman presented for evaluation of widespread asymptomatic telangiectasias of several years' duration that first appeared on the legs and steadily progressed to involve the trunk and arms. A review of systems was remarkable for episodic glossitis and oral erosions that developed at the same time as the eruption. The patient had no history of bleeding diasthesis, and her family history was unremarkable. A laboratory workup (including autoimmune screening) and a malignancy workup were negative. Physical examination revealed confluent sheets of erythematous and purple blanching telangiectasias scattered symmetrically on the trunk, bilateral arms and legs, buttocks, and dorsal aspects of the feet with sparing of the palms, soles, and head and neck regions. A small, shallow erosion was present on the lateral aspect of the tongue. A 4-mm punch biopsy of a thigh lesion revealed ectatic blood vessels with hyalinized walls.
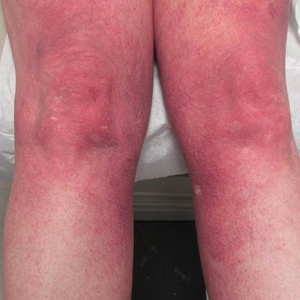
Reticular Hyperpigmented Patches With Indurated Subcutaneous Plaques
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
The Diagnosis: Superficial Migratory Thrombophlebitis
On initial presentation, the differential diagnosis included livedoid vasculopathy, cutaneous polyarteritis nodosa, erythema ab igne, cholesterol embolism, and livedo reticularis. Laboratory investigation included antiphospholipid antibody syndrome (APS), antinuclear antibody, rheumatoid factor, antineutrophil cytoplasmic antibody, serum protein electrophoresis, and coagulation tests. Pertinent findings included transient low total complement activity but normal complement protein C2, C3, and C5 levels and negative cryoglobulins. Additional laboratory testing revealed elevated antiphosphatidylserine IgG, which remained elevated 12 weeks later.
New lesions continued to appear over the next several months as painful, erythematous, linear, pruritic nodules that resolved as hyperpigmented linear patches, which intersected to form a livedo reticularis-like pattern that covered the lower legs. Biopsy of an erythematous nodule on the right leg revealed fibrin occlusion of a medium-sized vein in the subcutaneous fat. Direct immunofluorescence was not specific. Venous duplex ultrasonography demonstrated chronic superficial thrombophlebitis and was crucial to the diagnosis. Ultimately, the patient's history, clinical presentation, laboratory results, venous studies, and histopathologic analysis were consistent with a diagnosis of superficial migratory thrombophlebitis (SMT) with resultant postinflammatory hyperpigmentation presenting in a reticular pattern that mimicked livedoid vasculopathy, livedo reticularis, or erythema ab igne.
Superficial migratory thrombophlebitis, also known as thrombophlebitis migrans, is defined as the recurrent formation of thrombi within superficial veins.1 The presence of a thrombus in a superficial vein evokes an inflammatory response, resulting in swelling, tenderness, erythema, and warmth in the affected area. Superficial migratory thrombophlebitis has been associated with several etiologies, including pregnancy, oral contraceptive use, APS, vasculitic disorders, and malignancies (eg, pancreas, lung, breast), as well as infections such as secondary syphilis.1
When SMT is associated with an occult malignancy, it is known as Trousseau syndrome. Common malignancies found in association with Trousseau syndrome include pancreatic, lung, and breast cancers.2 A systematic review from 2008 evaluated the utility of extensive cancer screening strategies in patients with newly diagnosed, unprovoked venous thromboembolic events.3 Using a wide screening strategy that included computed tomography of the abdomen and pelvis, the investigators detected a considerable number of formerly undiagnosed cancers, increasing detection rates from 49.4% to 69.7%. After the diagnosis of SMT was made in our patient, computed tomography of the chest, abdomen, and pelvis was performed, but the findings were unremarkable.
Because occult malignancy was excluded in our patient, the likely etiology of SMT was APS, an acquired autoimmune condition diagnosed based on the presence of a vascular thrombosis and/or pregnancy failure in women as well as elevation of at least one antiphospholipid antibody laboratory marker (eg, lupus anticoagulant, anticardiolipin antibody, and anti-β2 glycoprotein I antibody) on 2 or more occasions at least 12 weeks apart.4 Other antibodies such as those directed against negatively charged phospholipids (eg, antiphosphatidylserine [which was elevated in our patient], phosphatidylinositol, phosphatidic acid) have been reported in patients with APS, although their diagnostic use is controversial.5 For example, the presence of antiphosphatidylserine antibodies has been considered common but not specific in patients with APS.4 However, a recent observational study demonstrated that antiphosphatidylserine antibodies are highly specific (87%) and useful in diagnosing clinical APS cases in the presence of other negative markers.6
In our patient, diagnosis of SMT with resultant postinflammatory hyperpigmentation in a reticular pattern was based on the patient's medical history, clinical examination, and histopathologic findings, as well as laboratory results and venous studies. However, it is important to note that a livedo reticularis-like pattern also is a very common finding in APS and must be included in the differential diagnosis of a reticular network on the skin.7 Moreover, differentiating livedo reticularis from SMT has prognostic importance since SMT may be associated with underlying malignancies while livedo reticularis may be associated with Sneddon syndrome, a disorder in which neurologic vascular events (eg, cerebrovascular accidents) are present.8 While this distinction is important, there are no pathognomonic histologic findings seen in livedo reticularis, and consideration of the clinical picture and additional testing is critical.4,8
Livedo vasculopathy was excluded in our patient due to the lack of diagnostic histopathologic findings, such as fibrin deposition and thrombus formation involving the upper- and mid-dermal capillaries.9 Furthermore, characteristic direct immunofluorescence findings of a homogenous or granular deposition in the vessel wall consisting of immune complexes, complement, and fibrin were absent in our patient.9 Our patient also lacked common clinical findings found in livedo vasculopathy such as small ulcerations or atrophic, porcelain-white scars on the lower legs. Erythema ab igne also was excluded in our patient due to the absence of heat exposure and presence of fibrin occlusion in the superficial leg veins. Physiologic livedo reticularis, defined as a livedoid pattern due to physiologic changes in the skin in response to cold exposure,10 also was excluded, as our patient's cutaneous changes included an alteration in pigmentation with a brown reticular pattern and no blanching, erythematous or violaceous hue, warmth, or tenderness.
In conclusion, SMT is a disorder with multiple associations that may clinically mimic livedo reticularis and livedoid vasculopathy when postinflammatory hyperpigmentation has a lacelike or livedoid pattern. While nontraditional antibodies may be useful in diagnosis in patients suspected of having APS with otherwise negative markers, standardized assays and further studies are needed to determine the specificity and value of these antibodies, particularly when used in isolation. Our patient's elevated antiphosphatidylserine IgG may have been the cause of her hypercoagulable state causing the SMT. A livedoid pattern is a common finding in APS and also was seen in our patient with SMT, but the differentiation of the brown pigmentary change and more active erythema was critical to the appropriate clinical workup of our patient.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. secondary hypercoagulable states. J Am Acad Dermatol. 1990;23:1-18.
- Rigdon EE. Trousseau's syndrome and acute arterial thrombosis. Cardiovasc Surg. 2000;8:214-218.
- Carrier M, Le Gal G, Wells PS, et al. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149:323-333.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Bertolaccini ML, Amengual O, Atsumi T, et al. 'Non-criteria' aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus. 2011;20:191-205.
- Khogeer H, Alfattani A, Al Kaff M, et al. Antiphosphatidylserine antibodies as diagnostic indicators of antiphospholipid syndrome. Lupus. 2015;24:186-190.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol. 1997;36(6 pt 1):970-982.
- Francès C, Papo T, Wechsler B, et al. Sneddon syndrome with or without antiphospholipid antibodies. a comparative study in 46 patients. Medicine (Baltimore). 1999;78:209-219.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478-488.
- James WD, Berger TG, Elston DM. Andrews' Diseases Of The Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: Elsevier Saunders; 2006.

A 32-year-old woman presented for evaluation of small, tender, erythematous nodules on the lower legs of 1 year's duration that had started to spread to the thighs over several months prior to presentation. The patient reported no history of ulceration or other cutaneous findings. On physical examination, a hyperpigmented, linear to reticular pattern was noted on the lower legs with a few 1-cm, erythematous, mildly indurated and tender subcutaneous nodules. The patient denied any recent medical procedures, history of malignancy or cardiovascular disease, use of tobacco or illicit drugs, prolonged contact with a heat source, recent unintentional weight loss, fevers, or night sweats. Her medical history was notable for asthma and migraines, which were treated with albuterol, fluticasone, and topiramate.
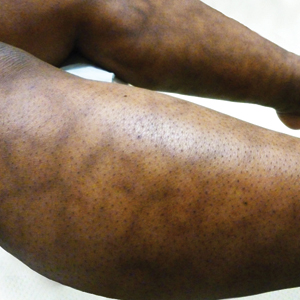
Pigmented Peduncule on the Leg
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
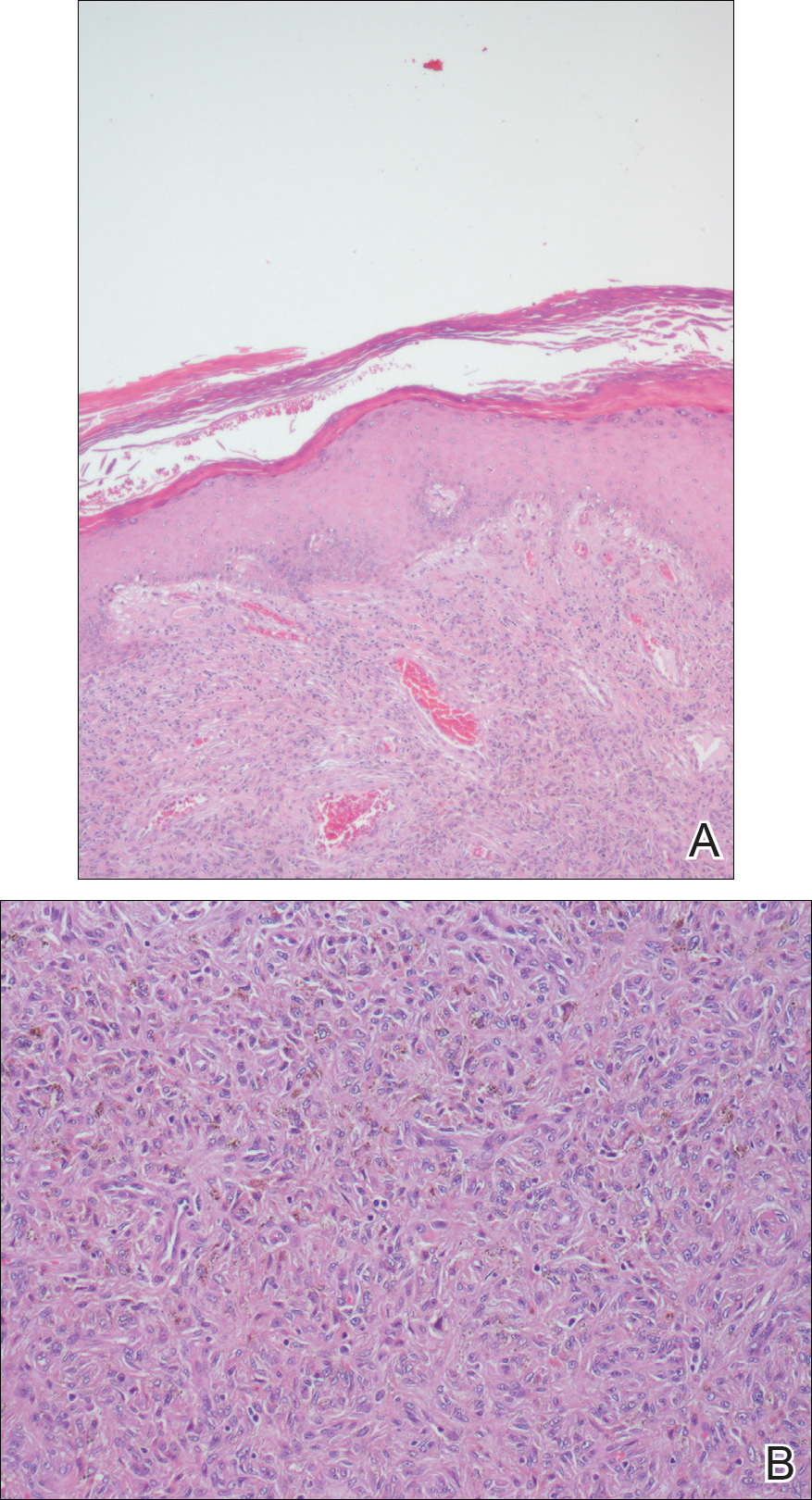
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
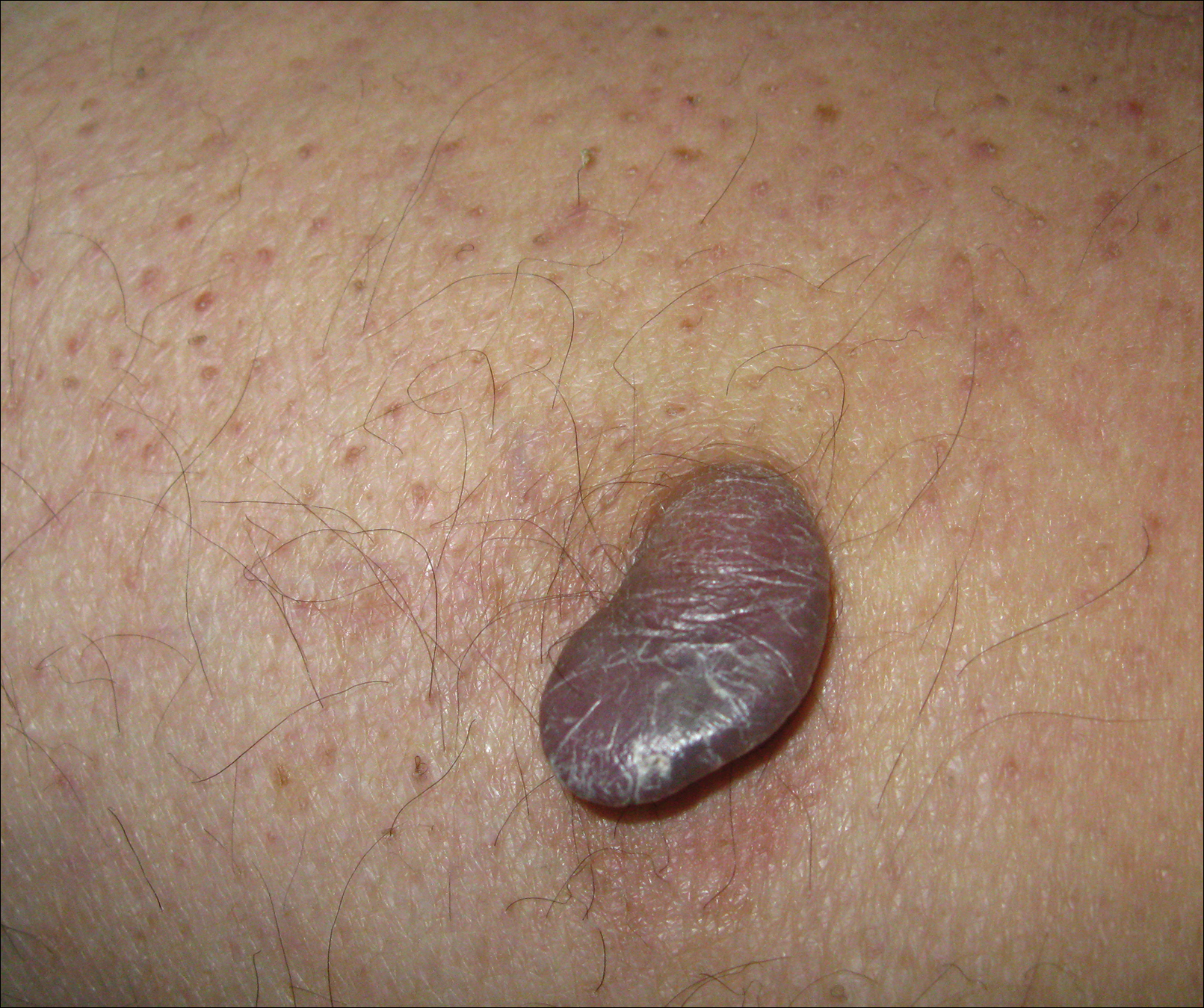
A 68-year-old man with a history of type 2 diabetes mellitus and hypercholesterolemia presented to the dermatology department with a cutaneous lesion on the posterior aspect of the right thigh of 2 years' duration. The lesion had become larger during the 4 months prior to presentation and was mostly asymptomatic but became tender when subjected to trauma. Physical examination revealed a firm, 2-cm, slightly pigmented peduncule on the posterior right thigh. No lymphadenopathies were noted. The lesion was completely excised for histologic examination.
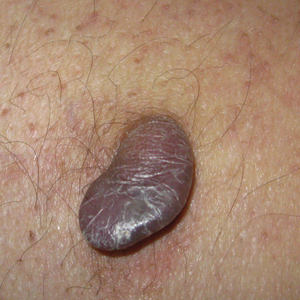
Painful Violaceous Nodule With Peripheral Hyperpigmentation
The Diagnosis: Aneurysmal Dermatofibroma
Biopsy of the lesion revealed a circumscribed dermal nodule comprised of storiform arrangements of enlarged, plump, fibrohistiocytic cells punctuated by variably sized clefts and large cystic spaces filled with blood that lacked an endothelial lining. No bizarre nuclear pleomorphism, atypical mitoses, or tumor necrosis were identified. The overlying epidermis exhibited mild acanthosis with broadening of the rete ridges. Proliferative spindled cells entrapped dermal collagen bundles at the periphery. Hemosiderin-laden macrophages were present throughout the proliferation and in the adjacent dermis (Figure). These findings supported the diagnosis of aneurysmal dermatofibroma (ADF).
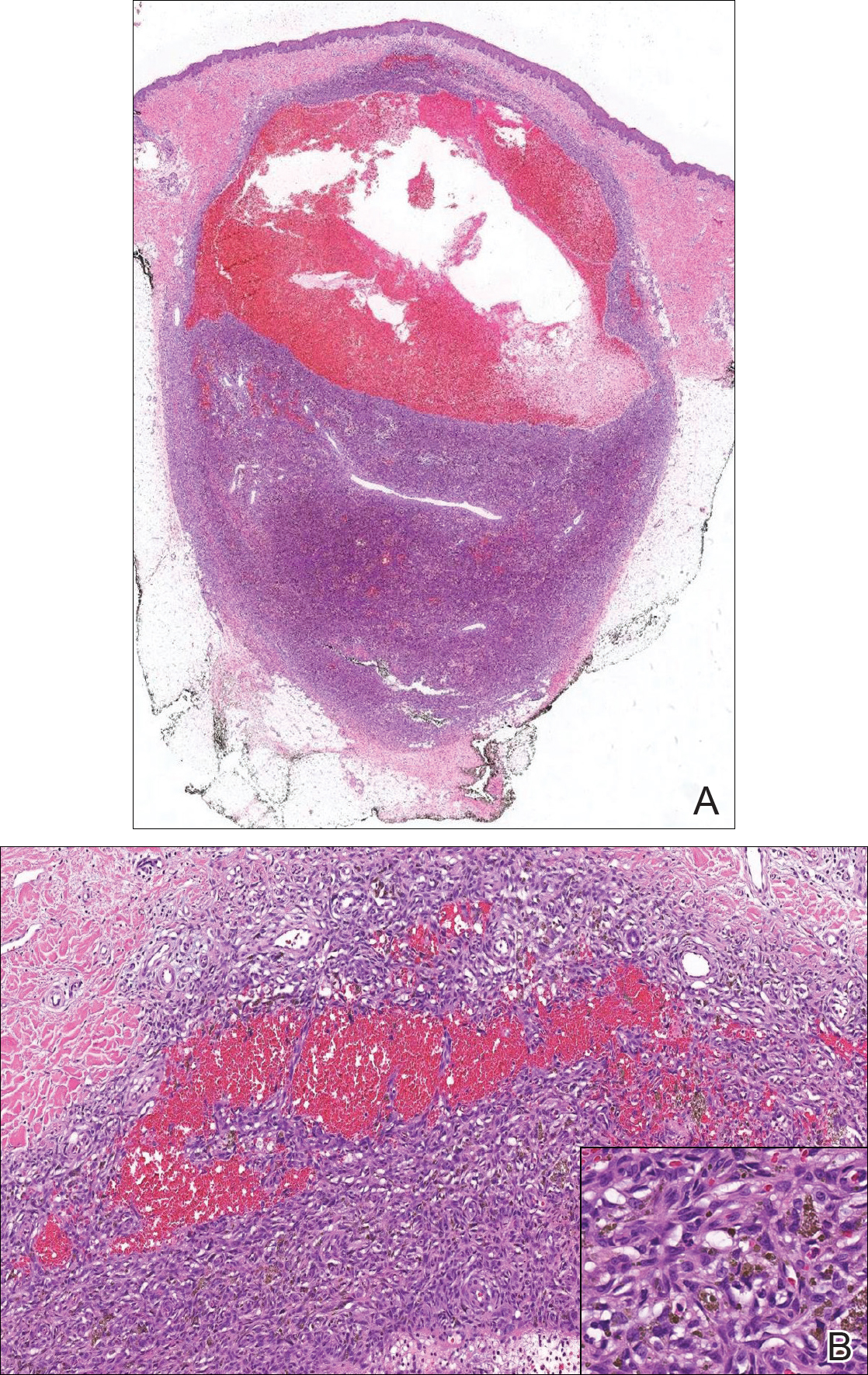
Aneurysmal dermatofibroma, also known as aneurysmal fibrous histiocytoma, is a rare variant of dermatofibroma that was described by Santa Cruz et al1 in 1981 and represents 2% to 6% of dermatofibromas.1,2 Aneurysmal dermatofibromas often lack the characteristic clinical and dermoscopic findings of conventional dermatofibromas, creating a diagnostic challenge for the clinician.3 Incomplete excision of this benign tumor was associated with a local recurrence rate of 19% (5/26) in one study,4 in contrast with the exceedingly low rate of local recurrence (<2%) attributed to conventional dermatofibromas.2,4
Clinically, ADFs commonly appear as blue-brown nodules on the arms and legs, often with a history of rapid and sometimes painful growth.1 Clinically, an ADF can have vascular, cystic, or melanocytic features that, in the context of lacking typical clinical findings of a dermatofibroma, can complicate clinical diagnosis; for example, ADFs can demonstrate several melanomalike features including atypical vessels, chrysalis structures, blue-white structures, a pinkish-white veil, irregular brown globulelike structures, an atypical pigment network, color variegation, a multicomponent pattern, and ulceration.3 Alternatively, ADFs can present with a vascular tumor-like pattern consisting of white areas and globular blue-red areas or a polymorphous vascular pattern with a peripheral collarette.
Our case illustrates the classic histologic appearance of an ADF. Large cavities and slitlike spaces filled with blood distinguish this entity from conventional dermatofibroma and other dermatofibroma variants; for example, cellular dermatofibroma is a benign variant of dermatofibroma that exhibits crowded fascicular architecture without an increase in vascular spaces. Aneurysmal dermatofibromas also should be distinguished from angiomatoid fibrous histiocytoma, which has intermediate malignant potential despite a similar-sounding name and a similar nodular appearance with large blood-filled spaces; however, many cases are located predominantly in the subcutis with epithelioid morphology, desmin immunohistochemical reactivity, and prominent tumor-associated lymphoid proliferation that can be mistaken for a lymph node.5 Furthermore, in contrast with vascular tumors, the blood-filled spaces of ADFs do not have an endothelial lining.
In summary, ADF is a rare dermatofibroma variant that has a variety of clinical presentations, often masquerading as a cyst, vascular tumor, or melanocytic neoplasm. The classic histopathologic features confirm the diagnosis. Although ADFs can be painful and have a tendency to recur, these lesions have a benign clinical course.
- Santa Cruz DJ, Kyriakos M. Aneurysmal ("angiomatoid") fibrous histiocytoma of the skin. Cancer. 1981;47:2053-2061.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Ferrari A, Argenziano G, Buccini P, et al. Typical and atypical dermoscopic presentations of dermatofibroma. J Eur Acad Dermatol Venereol. 2013;27:1375-1380.
- Calonje E, Fletcher CD. Aneurysmal benign fibrous histiocytoma: clinicopathological analysis of 40 cases of a tumour frequently misdiagnosed as a vascular neoplasm. Histopathology. 1995;26:323-331.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
The Diagnosis: Aneurysmal Dermatofibroma
Biopsy of the lesion revealed a circumscribed dermal nodule comprised of storiform arrangements of enlarged, plump, fibrohistiocytic cells punctuated by variably sized clefts and large cystic spaces filled with blood that lacked an endothelial lining. No bizarre nuclear pleomorphism, atypical mitoses, or tumor necrosis were identified. The overlying epidermis exhibited mild acanthosis with broadening of the rete ridges. Proliferative spindled cells entrapped dermal collagen bundles at the periphery. Hemosiderin-laden macrophages were present throughout the proliferation and in the adjacent dermis (Figure). These findings supported the diagnosis of aneurysmal dermatofibroma (ADF).
Aneurysmal dermatofibroma, also known as aneurysmal fibrous histiocytoma, is a rare variant of dermatofibroma that was described by Santa Cruz et al1 in 1981 and represents 2% to 6% of dermatofibromas.1,2 Aneurysmal dermatofibromas often lack the characteristic clinical and dermoscopic findings of conventional dermatofibromas, creating a diagnostic challenge for the clinician.3 Incomplete excision of this benign tumor was associated with a local recurrence rate of 19% (5/26) in one study,4 in contrast with the exceedingly low rate of local recurrence (<2%) attributed to conventional dermatofibromas.2,4
Clinically, ADFs commonly appear as blue-brown nodules on the arms and legs, often with a history of rapid and sometimes painful growth.1 Clinically, an ADF can have vascular, cystic, or melanocytic features that, in the context of lacking typical clinical findings of a dermatofibroma, can complicate clinical diagnosis; for example, ADFs can demonstrate several melanomalike features including atypical vessels, chrysalis structures, blue-white structures, a pinkish-white veil, irregular brown globulelike structures, an atypical pigment network, color variegation, a multicomponent pattern, and ulceration.3 Alternatively, ADFs can present with a vascular tumor-like pattern consisting of white areas and globular blue-red areas or a polymorphous vascular pattern with a peripheral collarette.
Our case illustrates the classic histologic appearance of an ADF. Large cavities and slitlike spaces filled with blood distinguish this entity from conventional dermatofibroma and other dermatofibroma variants; for example, cellular dermatofibroma is a benign variant of dermatofibroma that exhibits crowded fascicular architecture without an increase in vascular spaces. Aneurysmal dermatofibromas also should be distinguished from angiomatoid fibrous histiocytoma, which has intermediate malignant potential despite a similar-sounding name and a similar nodular appearance with large blood-filled spaces; however, many cases are located predominantly in the subcutis with epithelioid morphology, desmin immunohistochemical reactivity, and prominent tumor-associated lymphoid proliferation that can be mistaken for a lymph node.5 Furthermore, in contrast with vascular tumors, the blood-filled spaces of ADFs do not have an endothelial lining.
In summary, ADF is a rare dermatofibroma variant that has a variety of clinical presentations, often masquerading as a cyst, vascular tumor, or melanocytic neoplasm. The classic histopathologic features confirm the diagnosis. Although ADFs can be painful and have a tendency to recur, these lesions have a benign clinical course.
The Diagnosis: Aneurysmal Dermatofibroma
Biopsy of the lesion revealed a circumscribed dermal nodule comprised of storiform arrangements of enlarged, plump, fibrohistiocytic cells punctuated by variably sized clefts and large cystic spaces filled with blood that lacked an endothelial lining. No bizarre nuclear pleomorphism, atypical mitoses, or tumor necrosis were identified. The overlying epidermis exhibited mild acanthosis with broadening of the rete ridges. Proliferative spindled cells entrapped dermal collagen bundles at the periphery. Hemosiderin-laden macrophages were present throughout the proliferation and in the adjacent dermis (Figure). These findings supported the diagnosis of aneurysmal dermatofibroma (ADF).
Aneurysmal dermatofibroma, also known as aneurysmal fibrous histiocytoma, is a rare variant of dermatofibroma that was described by Santa Cruz et al1 in 1981 and represents 2% to 6% of dermatofibromas.1,2 Aneurysmal dermatofibromas often lack the characteristic clinical and dermoscopic findings of conventional dermatofibromas, creating a diagnostic challenge for the clinician.3 Incomplete excision of this benign tumor was associated with a local recurrence rate of 19% (5/26) in one study,4 in contrast with the exceedingly low rate of local recurrence (<2%) attributed to conventional dermatofibromas.2,4
Clinically, ADFs commonly appear as blue-brown nodules on the arms and legs, often with a history of rapid and sometimes painful growth.1 Clinically, an ADF can have vascular, cystic, or melanocytic features that, in the context of lacking typical clinical findings of a dermatofibroma, can complicate clinical diagnosis; for example, ADFs can demonstrate several melanomalike features including atypical vessels, chrysalis structures, blue-white structures, a pinkish-white veil, irregular brown globulelike structures, an atypical pigment network, color variegation, a multicomponent pattern, and ulceration.3 Alternatively, ADFs can present with a vascular tumor-like pattern consisting of white areas and globular blue-red areas or a polymorphous vascular pattern with a peripheral collarette.
Our case illustrates the classic histologic appearance of an ADF. Large cavities and slitlike spaces filled with blood distinguish this entity from conventional dermatofibroma and other dermatofibroma variants; for example, cellular dermatofibroma is a benign variant of dermatofibroma that exhibits crowded fascicular architecture without an increase in vascular spaces. Aneurysmal dermatofibromas also should be distinguished from angiomatoid fibrous histiocytoma, which has intermediate malignant potential despite a similar-sounding name and a similar nodular appearance with large blood-filled spaces; however, many cases are located predominantly in the subcutis with epithelioid morphology, desmin immunohistochemical reactivity, and prominent tumor-associated lymphoid proliferation that can be mistaken for a lymph node.5 Furthermore, in contrast with vascular tumors, the blood-filled spaces of ADFs do not have an endothelial lining.
In summary, ADF is a rare dermatofibroma variant that has a variety of clinical presentations, often masquerading as a cyst, vascular tumor, or melanocytic neoplasm. The classic histopathologic features confirm the diagnosis. Although ADFs can be painful and have a tendency to recur, these lesions have a benign clinical course.
- Santa Cruz DJ, Kyriakos M. Aneurysmal ("angiomatoid") fibrous histiocytoma of the skin. Cancer. 1981;47:2053-2061.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Ferrari A, Argenziano G, Buccini P, et al. Typical and atypical dermoscopic presentations of dermatofibroma. J Eur Acad Dermatol Venereol. 2013;27:1375-1380.
- Calonje E, Fletcher CD. Aneurysmal benign fibrous histiocytoma: clinicopathological analysis of 40 cases of a tumour frequently misdiagnosed as a vascular neoplasm. Histopathology. 1995;26:323-331.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santa Cruz DJ, Kyriakos M. Aneurysmal ("angiomatoid") fibrous histiocytoma of the skin. Cancer. 1981;47:2053-2061.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Ferrari A, Argenziano G, Buccini P, et al. Typical and atypical dermoscopic presentations of dermatofibroma. J Eur Acad Dermatol Venereol. 2013;27:1375-1380.
- Calonje E, Fletcher CD. Aneurysmal benign fibrous histiocytoma: clinicopathological analysis of 40 cases of a tumour frequently misdiagnosed as a vascular neoplasm. Histopathology. 1995;26:323-331.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
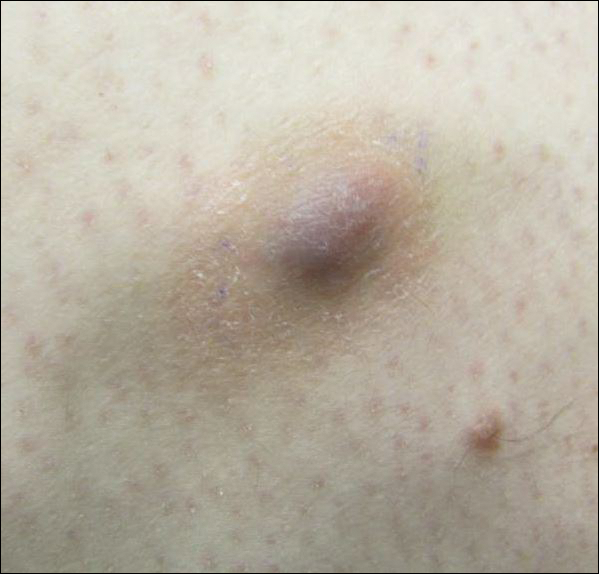
A 30-year-old man presented for evaluation of a painful lesion on the left thigh of 3 to 4 years' duration. Pain was exacerbated on physical exertion and was relieved by application of ice packs and use of over-the-counter analgesics. The patient denied any bleeding from the lesion. No other medical comorbidities were present. Physical examination demonstrated a pink, scaly, 3.2 ×2-cm patch with peripheral hyperpigmentation overlying a central, moderately firm, violaceous, 10.2 ×15-mm nodule on the left anteromedial thigh. The lesion was excised and sent to pathology.
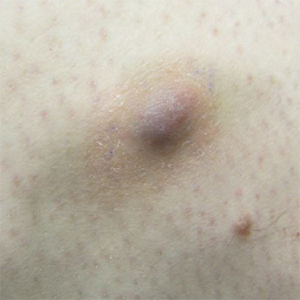
Disseminated Vesicles and Necrotic Papules
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
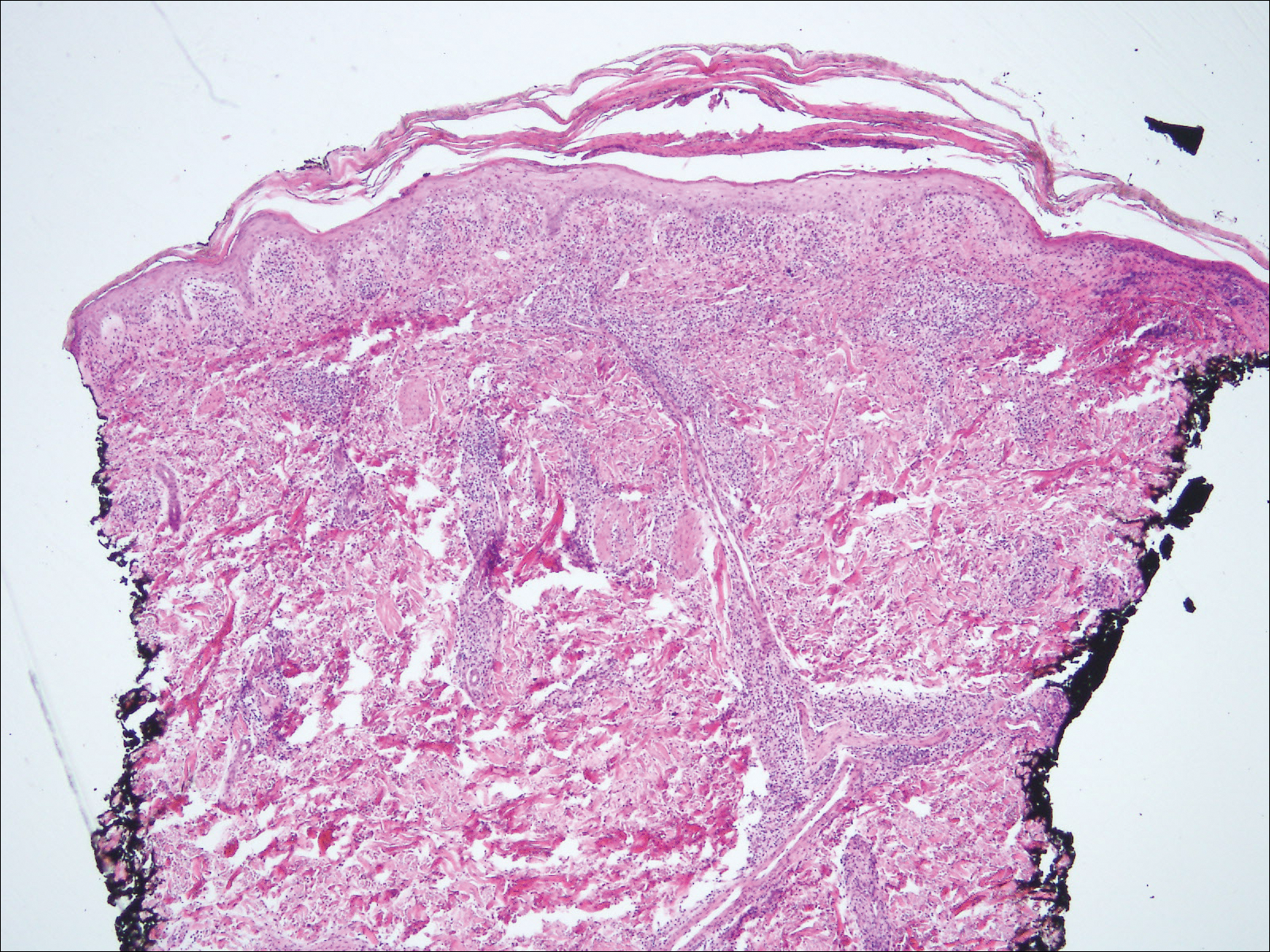
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7

Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7
Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7
Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.

A 30-year-old man who had contracted human immunodeficiency virus from a male sexual partner 4 years prior presented to the emergency department with fevers, chills, night sweats, and rhinorrhea of 2 weeks' duration. He reported that he had been off highly active antiretroviral therapy for 2 years. Physical examination revealed numerous erythematous, papulonecrotic, crusted lesions on the face, neck, chest, back, arms, and legs that had developed over the past 4 days. Fluid-filled vesicles also were noted on the arms and legs, while erythematous, indurated nodules with overlying scaling were noted on the bilateral palms and soles. The patient reported that he had been vaccinated for varicella-zoster virus as a child without primary infection.