User login
Transcatheter tricuspid valve repair effective and safe for regurgitation
NEW ORLEANS – In the first pivotal randomized, controlled trial of a transcatheter device for the repair of severe tricuspid regurgitation, a large reduction in valve dysfunction was associated with substantial improvement in quality of life (QOL) persisting out of 1 year of follow-up, according to results of the TRILUMINATE trial.
Based on the low procedural risks of the repair, the principal investigator, Paul Sorajja, MD, called the results “very clinically meaningful” as he presented the results at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.

Conducted at 65 centers in the United States, Canada, and North America, TRILUMINATE evaluated a transcatheter end-to-end (TEER) repair performed with the TriClip G4 Delivery System (Abbott). The study included two cohorts, both of which will be followed for 5 years. One included patients with very severe tricuspid regurgitation enrolled in a single arm. Data on this cohort is expected later in 2023.
In the randomized portion of the study, 350 patients enrolled with severe tricuspid regurgitation underwent TEER with a clipping device and then were followed on the guideline-directed therapy (GDMT) for heart failure they were receiving at baseline. The control group was managed on GDMT alone.
The primary composite endpoint at 1 year was a composite of death from any cause and/or tricuspid valve surgery, hospitalization for heart failure, and quality of life as measured with the Kansas City Cardiomyopathy questionnaire (KCCQ).
Benefit driven by quality of life
For the primary endpoint, the win ratio, a statistical calculation of those who did relative to those who did not benefit, was 1.48, signifying a 48% advantage (P = .02). This was driven almost entirely by the KCCQ endpoint. There was no significant difference death and/or tricuspid valve surgery, which occurred in about 10% of both groups (P = .75) or heart failure hospitalization, which was occurred in slightly more patients randomized to repair (14.9% vs. 12.1%; P = .41).
For KCCQ, the mean increase at 1 year was 12.3 points in the repair group versus 0.6 points (P < .001) in the control group. With an increase of 5-10 points typically considered to be clinically meaningful, the advantage of repair over GDMT at the threshold of 15 points or greater was highly statistically significant (49.7% vs. 26.4%; P < .0001).
This advantage was attributed to control of regurgitation. The proportion achieving moderate or less regurgitation sustained at 1 year was 87% in the repair group versus 4.8% in the GDMT group (P < .0001).
When assessed independent of treatment, KCCQ benefits at 1 year increased in a stepwise fashion as severity of regurgitation was reduced, climbing from 2 points if there was no improvement to 6 points with one grade in improvement and then to 18 points with at least a two-grade improvement.
For regurgitation, “the repair was extremely effective,” said Dr. Sorajja of Allina Health Minneapolis Heart Institute at Abbott Northwestern Hospital, Minneapolis. He added that the degree of regurgitation control in the TRILUMINATE trial “is the highest ever reported.” With previous trials with other transcatheter devices in development, the improvement so far has been on the order of 70%-80%.
For enrollment in TRILUMINATE, patients were required to have at least an intermediate risk of morbidity or mortality from tricuspid valve surgery. Exclusion criteria included a left ventricular ejection fraction (LVEF) less than 20% and severe pulmonary hypertension.
More than 70% of patients had the highest (torrential) or second highest (massive) category of regurgitation on a five-level scale by echocardiography. Almost all the remaining were at the third level (severe).
Of those enrolled, the average age was roughly 78 years. About 55% were women. Nearly 60% were in New York Heart Association class III or IV heart failure and most had significant comorbidities, including hypertension (> 80%), atrial fibrillation (about 90%), and renal disease (35%). Patients with diabetes (16%), chronic obstructive pulmonary disease (10%), and liver disease (7.5%) were represented in lower numbers.
Surgery is not necessarily an option
All enrolled patients were considered to be at intermediate or greater risk for mortality with surgical replacement of the tricuspid valve, but Dr. Sorajja pointed out that surgery, which involves valve replacement, is not necessarily an alternative to valve repair. Even in fit patients, the high morbidity, mortality, and extended hospital stay associated with surgical valve replacement makes this procedure unattractive.
In this trial, most patients who underwent the transcatheter procedure were discharged within a day. The safety was excellent, Dr. Sorajja said. Only three patients (1.7%) had a major adverse event. This included two cases of new-onset renal failure and one cardiovascular death. There were no cases of endocarditis requiring surgery or any other type of nonelective cardiovascular surgery, including for any device-related issue.
In the sick population enrolled, Dr. Sorajja characterized the number of adverse events over 1 year as “very low.”

These results are important, according to Kendra Grubb, MD, surgical director of the Structural Heart and Valve Center, Emory University, Atlanta. While she expressed surprise that there was no signal of benefit on hard endpoints at 1 year, she emphasized that “these patients feel terrible,” and they are frustrating to manage because surgery is often contraindicated or impractical.
“Finally, we have something for this group,” she said, noting that the mortality from valve replacement surgery even among patients who are fit enough for surgery to be considered is about 10%.
Ajay Kirtane, MD, director of the Cardiac Catheterization Laboratories at Columbia University, New York, was more circumspect. He agreed that the improvement in QOL was encouraging, but cautioned that QOL is a particularly soft outcome in a nonrandomized trial in which patients may feel better just knowing that there regurgitation has been controlled. He found the lack of benefit on hard outcomes not just surprising but “disappointing.”
Still, he agreed the improvement in QOL is potentially meaningful for a procedure that appears to be relatively safe.
Dr. Sorajja reported financial relationships with Boston Scientific, Edwards Lifesciences, Foldax. 4C Medical, Gore Medtronic, Phillips, Siemens, Shifamed, Vdyne, xDot, and Abbott Structural, which provided funding for this trial. Dr. Grubb reported financial relationships with Abbott Vascular, Ancora Heart, Bioventrix, Boston Scientific, Edwards Lifesciences, 4C Medical, JenaValve, and Medtronic. Dr. Kirtane reported financial relationships with Abbott Vascular, Amgen, Boston Scientific, Chiesi, Medtronic, Opsens, Phillips, ReCor, Regeneron, and Zoll.
NEW ORLEANS – In the first pivotal randomized, controlled trial of a transcatheter device for the repair of severe tricuspid regurgitation, a large reduction in valve dysfunction was associated with substantial improvement in quality of life (QOL) persisting out of 1 year of follow-up, according to results of the TRILUMINATE trial.
Based on the low procedural risks of the repair, the principal investigator, Paul Sorajja, MD, called the results “very clinically meaningful” as he presented the results at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
Conducted at 65 centers in the United States, Canada, and North America, TRILUMINATE evaluated a transcatheter end-to-end (TEER) repair performed with the TriClip G4 Delivery System (Abbott). The study included two cohorts, both of which will be followed for 5 years. One included patients with very severe tricuspid regurgitation enrolled in a single arm. Data on this cohort is expected later in 2023.
In the randomized portion of the study, 350 patients enrolled with severe tricuspid regurgitation underwent TEER with a clipping device and then were followed on the guideline-directed therapy (GDMT) for heart failure they were receiving at baseline. The control group was managed on GDMT alone.
The primary composite endpoint at 1 year was a composite of death from any cause and/or tricuspid valve surgery, hospitalization for heart failure, and quality of life as measured with the Kansas City Cardiomyopathy questionnaire (KCCQ).
Benefit driven by quality of life
For the primary endpoint, the win ratio, a statistical calculation of those who did relative to those who did not benefit, was 1.48, signifying a 48% advantage (P = .02). This was driven almost entirely by the KCCQ endpoint. There was no significant difference death and/or tricuspid valve surgery, which occurred in about 10% of both groups (P = .75) or heart failure hospitalization, which was occurred in slightly more patients randomized to repair (14.9% vs. 12.1%; P = .41).
For KCCQ, the mean increase at 1 year was 12.3 points in the repair group versus 0.6 points (P < .001) in the control group. With an increase of 5-10 points typically considered to be clinically meaningful, the advantage of repair over GDMT at the threshold of 15 points or greater was highly statistically significant (49.7% vs. 26.4%; P < .0001).
This advantage was attributed to control of regurgitation. The proportion achieving moderate or less regurgitation sustained at 1 year was 87% in the repair group versus 4.8% in the GDMT group (P < .0001).
When assessed independent of treatment, KCCQ benefits at 1 year increased in a stepwise fashion as severity of regurgitation was reduced, climbing from 2 points if there was no improvement to 6 points with one grade in improvement and then to 18 points with at least a two-grade improvement.
For regurgitation, “the repair was extremely effective,” said Dr. Sorajja of Allina Health Minneapolis Heart Institute at Abbott Northwestern Hospital, Minneapolis. He added that the degree of regurgitation control in the TRILUMINATE trial “is the highest ever reported.” With previous trials with other transcatheter devices in development, the improvement so far has been on the order of 70%-80%.
For enrollment in TRILUMINATE, patients were required to have at least an intermediate risk of morbidity or mortality from tricuspid valve surgery. Exclusion criteria included a left ventricular ejection fraction (LVEF) less than 20% and severe pulmonary hypertension.
More than 70% of patients had the highest (torrential) or second highest (massive) category of regurgitation on a five-level scale by echocardiography. Almost all the remaining were at the third level (severe).
Of those enrolled, the average age was roughly 78 years. About 55% were women. Nearly 60% were in New York Heart Association class III or IV heart failure and most had significant comorbidities, including hypertension (> 80%), atrial fibrillation (about 90%), and renal disease (35%). Patients with diabetes (16%), chronic obstructive pulmonary disease (10%), and liver disease (7.5%) were represented in lower numbers.
Surgery is not necessarily an option
All enrolled patients were considered to be at intermediate or greater risk for mortality with surgical replacement of the tricuspid valve, but Dr. Sorajja pointed out that surgery, which involves valve replacement, is not necessarily an alternative to valve repair. Even in fit patients, the high morbidity, mortality, and extended hospital stay associated with surgical valve replacement makes this procedure unattractive.
In this trial, most patients who underwent the transcatheter procedure were discharged within a day. The safety was excellent, Dr. Sorajja said. Only three patients (1.7%) had a major adverse event. This included two cases of new-onset renal failure and one cardiovascular death. There were no cases of endocarditis requiring surgery or any other type of nonelective cardiovascular surgery, including for any device-related issue.
In the sick population enrolled, Dr. Sorajja characterized the number of adverse events over 1 year as “very low.”
These results are important, according to Kendra Grubb, MD, surgical director of the Structural Heart and Valve Center, Emory University, Atlanta. While she expressed surprise that there was no signal of benefit on hard endpoints at 1 year, she emphasized that “these patients feel terrible,” and they are frustrating to manage because surgery is often contraindicated or impractical.
“Finally, we have something for this group,” she said, noting that the mortality from valve replacement surgery even among patients who are fit enough for surgery to be considered is about 10%.
Ajay Kirtane, MD, director of the Cardiac Catheterization Laboratories at Columbia University, New York, was more circumspect. He agreed that the improvement in QOL was encouraging, but cautioned that QOL is a particularly soft outcome in a nonrandomized trial in which patients may feel better just knowing that there regurgitation has been controlled. He found the lack of benefit on hard outcomes not just surprising but “disappointing.”
Still, he agreed the improvement in QOL is potentially meaningful for a procedure that appears to be relatively safe.
Dr. Sorajja reported financial relationships with Boston Scientific, Edwards Lifesciences, Foldax. 4C Medical, Gore Medtronic, Phillips, Siemens, Shifamed, Vdyne, xDot, and Abbott Structural, which provided funding for this trial. Dr. Grubb reported financial relationships with Abbott Vascular, Ancora Heart, Bioventrix, Boston Scientific, Edwards Lifesciences, 4C Medical, JenaValve, and Medtronic. Dr. Kirtane reported financial relationships with Abbott Vascular, Amgen, Boston Scientific, Chiesi, Medtronic, Opsens, Phillips, ReCor, Regeneron, and Zoll.
NEW ORLEANS – In the first pivotal randomized, controlled trial of a transcatheter device for the repair of severe tricuspid regurgitation, a large reduction in valve dysfunction was associated with substantial improvement in quality of life (QOL) persisting out of 1 year of follow-up, according to results of the TRILUMINATE trial.
Based on the low procedural risks of the repair, the principal investigator, Paul Sorajja, MD, called the results “very clinically meaningful” as he presented the results at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
Conducted at 65 centers in the United States, Canada, and North America, TRILUMINATE evaluated a transcatheter end-to-end (TEER) repair performed with the TriClip G4 Delivery System (Abbott). The study included two cohorts, both of which will be followed for 5 years. One included patients with very severe tricuspid regurgitation enrolled in a single arm. Data on this cohort is expected later in 2023.
In the randomized portion of the study, 350 patients enrolled with severe tricuspid regurgitation underwent TEER with a clipping device and then were followed on the guideline-directed therapy (GDMT) for heart failure they were receiving at baseline. The control group was managed on GDMT alone.
The primary composite endpoint at 1 year was a composite of death from any cause and/or tricuspid valve surgery, hospitalization for heart failure, and quality of life as measured with the Kansas City Cardiomyopathy questionnaire (KCCQ).
Benefit driven by quality of life
For the primary endpoint, the win ratio, a statistical calculation of those who did relative to those who did not benefit, was 1.48, signifying a 48% advantage (P = .02). This was driven almost entirely by the KCCQ endpoint. There was no significant difference death and/or tricuspid valve surgery, which occurred in about 10% of both groups (P = .75) or heart failure hospitalization, which was occurred in slightly more patients randomized to repair (14.9% vs. 12.1%; P = .41).
For KCCQ, the mean increase at 1 year was 12.3 points in the repair group versus 0.6 points (P < .001) in the control group. With an increase of 5-10 points typically considered to be clinically meaningful, the advantage of repair over GDMT at the threshold of 15 points or greater was highly statistically significant (49.7% vs. 26.4%; P < .0001).
This advantage was attributed to control of regurgitation. The proportion achieving moderate or less regurgitation sustained at 1 year was 87% in the repair group versus 4.8% in the GDMT group (P < .0001).
When assessed independent of treatment, KCCQ benefits at 1 year increased in a stepwise fashion as severity of regurgitation was reduced, climbing from 2 points if there was no improvement to 6 points with one grade in improvement and then to 18 points with at least a two-grade improvement.
For regurgitation, “the repair was extremely effective,” said Dr. Sorajja of Allina Health Minneapolis Heart Institute at Abbott Northwestern Hospital, Minneapolis. He added that the degree of regurgitation control in the TRILUMINATE trial “is the highest ever reported.” With previous trials with other transcatheter devices in development, the improvement so far has been on the order of 70%-80%.
For enrollment in TRILUMINATE, patients were required to have at least an intermediate risk of morbidity or mortality from tricuspid valve surgery. Exclusion criteria included a left ventricular ejection fraction (LVEF) less than 20% and severe pulmonary hypertension.
More than 70% of patients had the highest (torrential) or second highest (massive) category of regurgitation on a five-level scale by echocardiography. Almost all the remaining were at the third level (severe).
Of those enrolled, the average age was roughly 78 years. About 55% were women. Nearly 60% were in New York Heart Association class III or IV heart failure and most had significant comorbidities, including hypertension (> 80%), atrial fibrillation (about 90%), and renal disease (35%). Patients with diabetes (16%), chronic obstructive pulmonary disease (10%), and liver disease (7.5%) were represented in lower numbers.
Surgery is not necessarily an option
All enrolled patients were considered to be at intermediate or greater risk for mortality with surgical replacement of the tricuspid valve, but Dr. Sorajja pointed out that surgery, which involves valve replacement, is not necessarily an alternative to valve repair. Even in fit patients, the high morbidity, mortality, and extended hospital stay associated with surgical valve replacement makes this procedure unattractive.
In this trial, most patients who underwent the transcatheter procedure were discharged within a day. The safety was excellent, Dr. Sorajja said. Only three patients (1.7%) had a major adverse event. This included two cases of new-onset renal failure and one cardiovascular death. There were no cases of endocarditis requiring surgery or any other type of nonelective cardiovascular surgery, including for any device-related issue.
In the sick population enrolled, Dr. Sorajja characterized the number of adverse events over 1 year as “very low.”
These results are important, according to Kendra Grubb, MD, surgical director of the Structural Heart and Valve Center, Emory University, Atlanta. While she expressed surprise that there was no signal of benefit on hard endpoints at 1 year, she emphasized that “these patients feel terrible,” and they are frustrating to manage because surgery is often contraindicated or impractical.
“Finally, we have something for this group,” she said, noting that the mortality from valve replacement surgery even among patients who are fit enough for surgery to be considered is about 10%.
Ajay Kirtane, MD, director of the Cardiac Catheterization Laboratories at Columbia University, New York, was more circumspect. He agreed that the improvement in QOL was encouraging, but cautioned that QOL is a particularly soft outcome in a nonrandomized trial in which patients may feel better just knowing that there regurgitation has been controlled. He found the lack of benefit on hard outcomes not just surprising but “disappointing.”
Still, he agreed the improvement in QOL is potentially meaningful for a procedure that appears to be relatively safe.
Dr. Sorajja reported financial relationships with Boston Scientific, Edwards Lifesciences, Foldax. 4C Medical, Gore Medtronic, Phillips, Siemens, Shifamed, Vdyne, xDot, and Abbott Structural, which provided funding for this trial. Dr. Grubb reported financial relationships with Abbott Vascular, Ancora Heart, Bioventrix, Boston Scientific, Edwards Lifesciences, 4C Medical, JenaValve, and Medtronic. Dr. Kirtane reported financial relationships with Abbott Vascular, Amgen, Boston Scientific, Chiesi, Medtronic, Opsens, Phillips, ReCor, Regeneron, and Zoll.
AT ACC 2023
Beware risk of sedatives for respiratory patients
Both asthma and chronic obstructive pulmonary disease can be challenging to diagnose, and medication-driven episodes of sedation or hypoventilation are often overlooked as causes of acute exacerbations in these conditions, according to a letter published in The Lancet Respiratory Medicine.
write Christos V. Chalitsios, PhD, of the University of Nottingham, England, and colleagues.
The authors note that exacerbations are the main complications of both asthma and COPD, and stress the importance of identifying causes and preventive strategies.
Sedatives such as opioids have been shown to depress respiratory drive, reduce muscle tone, and increase the risk of pneumonia, they write. The authors also propose that the risk of sedative-induced aspiration or hypoventilation would be associated with medications including pregabalin, gabapentin, and amitriptyline.
Other mechanisms may be involved in the association between sedatives and exacerbations in asthma and COPD. For example, sedative medications can suppress coughing, which may promote airway mucous compaction and possible infection, the authors write.
Most research involving prevention of asthma and COPD exacerbations has not addressed the potential impact of sedatives taken for reasons outside of obstructive lung disease, the authors say.
“Although the risk of sedation and hypoventilation events are known to be increased by opioids and antipsychotic drugs, there has not been a systematic assessment of commonly prescribed medications with potential respiratory side-effects, including gabapentin, amitriptyline, and pregabalin,” they write.
Polypharmacy is increasingly common and results in many patients with asthma or COPD presenting for treatment of acute exacerbations while on a combination of gabapentin, pregabalin, amitriptyline, and opioids, the authors note; “however, there is little data or disease-specific guidance on how best to manage this problem, which often starts with a prescription in primary care,” they write. Simply stopping sedatives is not an option for many patients given the addictive nature of these drugs and the unlikely resolution of the condition for which the drugs were prescribed, the authors say. However, “cautious dose reduction” of sedatives is possible once patients understand the reason, they add.
Clinicians may be able to suggest reduced doses and alternative treatments to patients with asthma and COPD while highlighting the risk of respiratory depression and polypharmacy – “potentially reducing the number of exacerbations of obstructive lung disease,” the authors conclude.
The study received no outside funding. The authors have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Both asthma and chronic obstructive pulmonary disease can be challenging to diagnose, and medication-driven episodes of sedation or hypoventilation are often overlooked as causes of acute exacerbations in these conditions, according to a letter published in The Lancet Respiratory Medicine.
write Christos V. Chalitsios, PhD, of the University of Nottingham, England, and colleagues.
The authors note that exacerbations are the main complications of both asthma and COPD, and stress the importance of identifying causes and preventive strategies.
Sedatives such as opioids have been shown to depress respiratory drive, reduce muscle tone, and increase the risk of pneumonia, they write. The authors also propose that the risk of sedative-induced aspiration or hypoventilation would be associated with medications including pregabalin, gabapentin, and amitriptyline.
Other mechanisms may be involved in the association between sedatives and exacerbations in asthma and COPD. For example, sedative medications can suppress coughing, which may promote airway mucous compaction and possible infection, the authors write.
Most research involving prevention of asthma and COPD exacerbations has not addressed the potential impact of sedatives taken for reasons outside of obstructive lung disease, the authors say.
“Although the risk of sedation and hypoventilation events are known to be increased by opioids and antipsychotic drugs, there has not been a systematic assessment of commonly prescribed medications with potential respiratory side-effects, including gabapentin, amitriptyline, and pregabalin,” they write.
Polypharmacy is increasingly common and results in many patients with asthma or COPD presenting for treatment of acute exacerbations while on a combination of gabapentin, pregabalin, amitriptyline, and opioids, the authors note; “however, there is little data or disease-specific guidance on how best to manage this problem, which often starts with a prescription in primary care,” they write. Simply stopping sedatives is not an option for many patients given the addictive nature of these drugs and the unlikely resolution of the condition for which the drugs were prescribed, the authors say. However, “cautious dose reduction” of sedatives is possible once patients understand the reason, they add.
Clinicians may be able to suggest reduced doses and alternative treatments to patients with asthma and COPD while highlighting the risk of respiratory depression and polypharmacy – “potentially reducing the number of exacerbations of obstructive lung disease,” the authors conclude.
The study received no outside funding. The authors have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Both asthma and chronic obstructive pulmonary disease can be challenging to diagnose, and medication-driven episodes of sedation or hypoventilation are often overlooked as causes of acute exacerbations in these conditions, according to a letter published in The Lancet Respiratory Medicine.
write Christos V. Chalitsios, PhD, of the University of Nottingham, England, and colleagues.
The authors note that exacerbations are the main complications of both asthma and COPD, and stress the importance of identifying causes and preventive strategies.
Sedatives such as opioids have been shown to depress respiratory drive, reduce muscle tone, and increase the risk of pneumonia, they write. The authors also propose that the risk of sedative-induced aspiration or hypoventilation would be associated with medications including pregabalin, gabapentin, and amitriptyline.
Other mechanisms may be involved in the association between sedatives and exacerbations in asthma and COPD. For example, sedative medications can suppress coughing, which may promote airway mucous compaction and possible infection, the authors write.
Most research involving prevention of asthma and COPD exacerbations has not addressed the potential impact of sedatives taken for reasons outside of obstructive lung disease, the authors say.
“Although the risk of sedation and hypoventilation events are known to be increased by opioids and antipsychotic drugs, there has not been a systematic assessment of commonly prescribed medications with potential respiratory side-effects, including gabapentin, amitriptyline, and pregabalin,” they write.
Polypharmacy is increasingly common and results in many patients with asthma or COPD presenting for treatment of acute exacerbations while on a combination of gabapentin, pregabalin, amitriptyline, and opioids, the authors note; “however, there is little data or disease-specific guidance on how best to manage this problem, which often starts with a prescription in primary care,” they write. Simply stopping sedatives is not an option for many patients given the addictive nature of these drugs and the unlikely resolution of the condition for which the drugs were prescribed, the authors say. However, “cautious dose reduction” of sedatives is possible once patients understand the reason, they add.
Clinicians may be able to suggest reduced doses and alternative treatments to patients with asthma and COPD while highlighting the risk of respiratory depression and polypharmacy – “potentially reducing the number of exacerbations of obstructive lung disease,” the authors conclude.
The study received no outside funding. The authors have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New influx of Humira biosimilars may not drive immediate change
Gastroenterologists in 2023 will have more tools in their arsenal to treat patients with Crohn’s disease or ulcerative colitis. As many as 8-10 adalimumab biosimilars are anticipated to come on the market this year, giving mainstay drug Humira some vigorous competition.
Three scenarios will drive adalimumab biosimilar initiation: Insurance preference for the initial treatment of a newly diagnosed condition, a change in a patient’s insurance plan, or an insurance-mandated switch, said Edward C. Oldfield IV, MD, assistant professor at Eastern Virginia Medical School’s division of gastroenterology in Norfolk.
“Outside of these scenarios, I would encourage patients to remain on their current biologic so long as cost and accessibility remain stable,” said Dr. Oldfield.

Many factors will contribute to the success of biosimilars. Will physicians be prescribing them? How are biosimilars placed on formularies and will they be given preferred status? How will manufacturers price their biosimilars? “We have to wait and see to get the answers to these questions,” said Steven Newmark, JD, MPA, chief legal officer and director of policy, Global Healthy Living Foundation/CreakyJoints, a nonprofit advocacy organization based in New York.
Prescribing biosimilars is no different than prescribing originator biologics, so providers should know how to use them, said Mr. Newmark. “Most important will be the availability of patient-friendly resources that providers can share with their patients to provide education about and confidence in using biosimilars,” he added.
Overall, biosimilars are a good thing, said Dr. Oldfield. “In the long run they should bring down costs and increase access to medications for our patients.”
Others are skeptical that the adalimumab biosimilars will save patients much money.
Biosimilar laws were created to lower costs. However, if a patient with insurance pays only $5 a month out of pocket for Humira – a drug that normally costs $7,000 without coverage – it’s unlikely they would want to switch unless there’s comparable savings from the biosimilar, said Stephen B. Hanauer, MD, medical director of the Digestive Health Center and professor of medicine at Northwestern Medicine, Northwestern University, Evanston, Ill.
Like generics, Humira biosimilars may face some initial backlash, said Dr. Hanauer.
2023 broadens scope of adalimumab treatments
The American Gastroenterological Association describes a biosimilar as something that’s “highly similar to, but not an exact copy of, a biologic reference product already approved” by the Food and Drug Administration. Congress under the 2010 Affordable Care Act created a special, abbreviated pathway to approval for biosimilars.
AbbVie’s Humira, the global revenue for which exceeded $20 billion in 2021, has long dominated the U.S. market on injectable treatments for autoimmune diseases. The popular drug faces some competition in 2023, however, following a series of legal settlements that allowed AbbVie competitors to release their own adalimumab biosimilars.
“So far, we haven’t seen biosimilars live up to their potential in the U.S. in the inflammatory space,” said Mr. Newmark. This may change, however. Previously, biosimilars have required infusion, which demanded more time, commitment, and travel from patients. “The new set of forthcoming Humira biosimilars are injectables, an administration method preferred by patients,” he said.
The FDA will approve a biosimilar if it determines that the biological product is highly similar to the reference product, and that there are no clinically meaningful differences between the biological and reference product in terms of the safety, purity, and potency of the product.
The agency to date has approved 8 adalimumab biosimilars. These include: Idacio (adalimumab-aacf, Fresenius Kabi); Amjevita (adalimumab-atto, Amgen); Hadlima (adalimumab-bwwd, Organon); Cyltezo (adalimumab-adbm, Boehringer Ingelheim); Yusimry (adalimumab-aqvh from Coherus BioSciences); Hulio (adalimumab-fkjp; Mylan/Fujifilm Kyowa Kirin Biologics); Hyrimoz (adalimumab-adaz, Sandoz), and Abrilada (adalimumab-afzb, Pfizer).
“While FDA doesn’t formally track when products come to market, we know based on published reports that application holders for many of the currently FDA-approved biosimilars plan to market this year, starting with Amjevita being the first adalimumab biosimilar launched” in January, said Sarah Yim, MD, director of the Office of Therapeutic Biologics and Biosimilars at the agency.
At press time, two other companies (Celltrion and Alvotech/Teva) were awaiting FDA approval for their adalimumab biosimilar drugs.
Among the eight approved drugs, Cyltezo is the only one that has a designation for interchangeability with Humira.
An interchangeable biosimilar may be substituted at the pharmacy without the intervention of the prescriber – much like generics are substituted, depending on state laws, said Dr. Yim. “However, in terms of safety and effectiveness, FDA’s standards for approval mean that biosimilar or interchangeable biosimilar products can be used in place of the reference product they were compared to.”
FDA-approved biosimilars undergo a rigorous evaluation for safety, effectiveness, and quality for their approved conditions of use, she continued. “Therefore, patients and health care providers can rely on a biosimilar to be as safe and effective for its approved uses as the original biological product.”
Remicade as a yard stick
Gastroenterologists dealt with this situation once before, when Remicade (infliximab) biosimilars came on the market in 2016, noted Miguel Regueiro, MD, chair of the Digestive Disease and Surgery Institute at the Cleveland Clinic.

Remicade and Humira are both tumor necrosis factor inhibitors with the same mechanism of action and many of the same indications. “We already had that experience with Remicade and biosimilar switch 2 or 3 years ago. Now we’re talking about Humira,” said Dr. Regueiro.
Most GI doctors have prescribed one of the more common infliximab biosimilars (Inflectra or Renflexis), noted Dr. Oldfield.
Cardinal Health, which recently surveyed 300 gastroenterologists, rheumatologists, and dermatologists about adalimumab biosimilars, found that gastroenterologists had the highest comfort level in prescribing them. Their top concern, however, was changing a patient from adalimumab to an adalimumab biosimilar.
For most patients, Dr. Oldfield sees the Humira reference biologic and biosimilar as equivalent.
However, he said he would change a patient’s drug only if there were a good reason or if his hand was forced by insurance. He would not make the change for a patient who recently began induction with the reference biologic or a patient with highly active clinical disease.
“While there is limited data to support this, I would also have some qualms about changing a patient from reference biologic to a biosimilar if they previously had immune-mediated pharmacokinetic failure due to antibody development with a biologic and were currently doing well on their new biologic,” he said.
Those with a new ulcerative colitis or Crohn’s diagnosis who are initiating a biologic for the first time might consider a biosimilar. If a patient is transitioning from a reference biologic to a biosimilar, “I would want to make that change during a time of stable remission and with the recognition that the switch is not a temporary switch, but a long-term switch,” he continued.
A paper that reviewed 23 observational studies of adalimumab and other biosimilars found that switching biosimilars was safe and effective. But if possible, patients should minimize the number of switches until more robust long-term data are available, added Dr. Oldfield.
If a patient is apprehensive about switching to a new therapy, “one may need to be cognizant of the ‘nocebo’ effect in which there is an unexplained or unfavorable therapeutic effect after switching,” he said.
Other gastroenterologists voiced similar reservations about switching. “I won’t use an adalimumab biosimilar unless the patient requests it, the insurance requires it, or there is a cost advantage for the patient such that they prefer it,” said Doug Wolf, MD, an Atlanta gastroenterologist.
“There is no medical treatment advantage to a biosimilar, especially if switching from Humira,” added Dr. Wolf.
Insurance will guide treatment
Once a drug is approved for use by the FDA, that drug will be available in all 50 states. “Different private insurance formularies, as well as state Medicaid formularies, might affect the actual ability of patients to receive such drugs,” said Mr. Newmark.

Patients should consult with their providers and insurance companies to see what therapies are available, he advised.
Dr. Hanauer anticipates some headaches arising for patients and doctors alike when negotiating for a specific drug.
Cyltezo may be the only biosimilar interchangeable with Humira, but the third-party pharmacy benefit manager (PBM) could negotiate for one of the noninterchangeable ones. “On a yearly basis they could switch their preference,” said Dr. Hanauer.
In the Cardinal Health survey, more than 60% of respondents said they would feel comfortable prescribing an adalimumab biosimilar only with an interchangeability designation.
A PBM may offer a patient Cyltezo if it’s cheaper than Humira. If the patient insists on staying on Humira, then they’ll have to pay more for that drug on their payer’s formulary, said Dr. Hanauer. In a worst-case scenario, a physician may have to appeal on a patient’s behalf to get Humira if the insurer offers only the biosimilar.
Taking that step to appeal is a major hassle for the physician, and leads to extra back door costs as well, said Dr. Hanauer.
Humira manufacturer AbbVie, in turn, may offer discounts and rebates to the PBMs to put Humira on their formulary. “That’s the AbbVie negotiating power. It’s not that the cost is going to be that much different. It’s going to be that there are rebates and discounts that are going to make the cost different,” he added.
As a community physician, Dr. Oldfield has specific concerns about accessibility.
The ever-increasing burden of insurance documentation and prior authorization means it can take weeks or months to get these medications approved. “The addition of new biosimilars is a welcome entrance if it can get patients the medications they need when they need it,” he said.
When it comes to prescribing biologics, many physicians rely on ancillary staff for assistance. It’s a team effort to sift through all the paperwork, observed Dr. Oldfield.
“While many community GI practices have specialized staff to deal with prior authorizations, they are still a far cry from the IBD [inflammatory bowel disease] academic centers where there are often pharmacists, nursing specialists, and home-monitoring programs to check in on patients,” he explained.
Landscape on cost is uncertain
At present, little is known about the cost of the biosimilars and impact on future drug pricing, said Dr. Oldfield.
At least for Medicare, Humira biosimilars will be considered Medicare Part D drugs if used for a medically accepted indication, said a spokesperson for the Centers for Medicare and Medicaid Services.
Part D sponsors (pharmacy and therapeutic committees) “will make the determination as to whether Amjevita and other products will be added to their formularies,” said the spokesperson.
Patients never saw a significant cost savings with Remicade biosimilars. “I imagine the same would be true with biosimilars for Humira,” said Dr. Regueiro. Patients may see greater access to these drugs, however, because the insurance plan or the pharmacy plan will make them more readily available, he added.
The hope is that, as biosimilars are introduced, the price of the originator biologic will go down, said Mr. Newmark. “Therefore, we can expect Humira to be offered at a lower price as it faces competition. Where it will sit in comparison to the forthcoming biosimilars will depend on how much biosimilar companies drop their price and how much pressure will be on PBMs and insurers to cover the lowest list price drug,” he said.
AbbVie did not respond to several requests for comment.
Charitable patient assistance programs for biosimilars or biologics can help offset the price of copayments, Mr. Newmark offered.
Ideally, insurers will offer designated biosimilars at a reduced or even no out-of-pocket expense on their formularies. This should lead to a decreased administrative burden for approval with streamlined (or even removal) of prior authorizations for certain medications, said Dr. Oldfield.
Without insurance or medication assistance programs, the cost of biosimilars is prohibitively expensive, he added.
“Biosimilars have higher research, development, and manufacturing costs than what people conventionally think of [for] a generic medication.”
Educating, advising patients
Dr. Oldfield advised that gastroenterologists refer to biologics by the generic name rather than branded name when initiating therapy unless there is a very specific reason not to. “This approach should make the process more streamlined and less subjected to quick denials for brand-only requests as biosimilars start to assume a larger market share,” he said.
Uptake of the Humira biosimilars also will depend on proper education of physicians and patients and their comfort level with the biosimilars, said Dr. Regueiro. Cleveland Clinic uses a team approach to educate on this topic, relying on pharmacists, clinicians, and nurses to explain that there’s no real difference between the reference drug and its biosimilars, based on efficacy and safety data.
Physicians can also direct patients to patient-friendly resources, said Mr. Newmark. “By starting the conversation early, it ensures that when/if the time comes that your patient is switched to or chooses a biosimilar they will feel more confident because they have the knowledge to make decisions about their care.”
The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars , is a free resource for patients, he added.
It’s important that doctors also understand these products so they can explain to their patients what to expect, said the FDA’s Dr. Yim. The FDA provides educational materials on its website, including a comprehensive curriculum toolkit.
Dr. Hanauer has served as a consultant for AbbVie, Amgen, American College of Gastroenterology, GlaxoSmithKline, American Gastroenterological Association, Pfizer, and a host of other companies . Dr. Regueiro has served on advisory boards and as a consultant for Abbvie, Janssen, UCB, Takeda, Pfizer, BMS, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET Pharma Solutions,Trellis, and Boehringer Ingelheim Pharmaceuticals. Dr. Wolf, Dr. Yim, Dr. Oldfield, and Mr. Newmark have no financial conflicts of interest.
Gastroenterologists in 2023 will have more tools in their arsenal to treat patients with Crohn’s disease or ulcerative colitis. As many as 8-10 adalimumab biosimilars are anticipated to come on the market this year, giving mainstay drug Humira some vigorous competition.
Three scenarios will drive adalimumab biosimilar initiation: Insurance preference for the initial treatment of a newly diagnosed condition, a change in a patient’s insurance plan, or an insurance-mandated switch, said Edward C. Oldfield IV, MD, assistant professor at Eastern Virginia Medical School’s division of gastroenterology in Norfolk.
“Outside of these scenarios, I would encourage patients to remain on their current biologic so long as cost and accessibility remain stable,” said Dr. Oldfield.
Many factors will contribute to the success of biosimilars. Will physicians be prescribing them? How are biosimilars placed on formularies and will they be given preferred status? How will manufacturers price their biosimilars? “We have to wait and see to get the answers to these questions,” said Steven Newmark, JD, MPA, chief legal officer and director of policy, Global Healthy Living Foundation/CreakyJoints, a nonprofit advocacy organization based in New York.
Prescribing biosimilars is no different than prescribing originator biologics, so providers should know how to use them, said Mr. Newmark. “Most important will be the availability of patient-friendly resources that providers can share with their patients to provide education about and confidence in using biosimilars,” he added.
Overall, biosimilars are a good thing, said Dr. Oldfield. “In the long run they should bring down costs and increase access to medications for our patients.”
Others are skeptical that the adalimumab biosimilars will save patients much money.
Biosimilar laws were created to lower costs. However, if a patient with insurance pays only $5 a month out of pocket for Humira – a drug that normally costs $7,000 without coverage – it’s unlikely they would want to switch unless there’s comparable savings from the biosimilar, said Stephen B. Hanauer, MD, medical director of the Digestive Health Center and professor of medicine at Northwestern Medicine, Northwestern University, Evanston, Ill.
Like generics, Humira biosimilars may face some initial backlash, said Dr. Hanauer.
2023 broadens scope of adalimumab treatments
The American Gastroenterological Association describes a biosimilar as something that’s “highly similar to, but not an exact copy of, a biologic reference product already approved” by the Food and Drug Administration. Congress under the 2010 Affordable Care Act created a special, abbreviated pathway to approval for biosimilars.
AbbVie’s Humira, the global revenue for which exceeded $20 billion in 2021, has long dominated the U.S. market on injectable treatments for autoimmune diseases. The popular drug faces some competition in 2023, however, following a series of legal settlements that allowed AbbVie competitors to release their own adalimumab biosimilars.
“So far, we haven’t seen biosimilars live up to their potential in the U.S. in the inflammatory space,” said Mr. Newmark. This may change, however. Previously, biosimilars have required infusion, which demanded more time, commitment, and travel from patients. “The new set of forthcoming Humira biosimilars are injectables, an administration method preferred by patients,” he said.
The FDA will approve a biosimilar if it determines that the biological product is highly similar to the reference product, and that there are no clinically meaningful differences between the biological and reference product in terms of the safety, purity, and potency of the product.
The agency to date has approved 8 adalimumab biosimilars. These include: Idacio (adalimumab-aacf, Fresenius Kabi); Amjevita (adalimumab-atto, Amgen); Hadlima (adalimumab-bwwd, Organon); Cyltezo (adalimumab-adbm, Boehringer Ingelheim); Yusimry (adalimumab-aqvh from Coherus BioSciences); Hulio (adalimumab-fkjp; Mylan/Fujifilm Kyowa Kirin Biologics); Hyrimoz (adalimumab-adaz, Sandoz), and Abrilada (adalimumab-afzb, Pfizer).
“While FDA doesn’t formally track when products come to market, we know based on published reports that application holders for many of the currently FDA-approved biosimilars plan to market this year, starting with Amjevita being the first adalimumab biosimilar launched” in January, said Sarah Yim, MD, director of the Office of Therapeutic Biologics and Biosimilars at the agency.
At press time, two other companies (Celltrion and Alvotech/Teva) were awaiting FDA approval for their adalimumab biosimilar drugs.
Among the eight approved drugs, Cyltezo is the only one that has a designation for interchangeability with Humira.
An interchangeable biosimilar may be substituted at the pharmacy without the intervention of the prescriber – much like generics are substituted, depending on state laws, said Dr. Yim. “However, in terms of safety and effectiveness, FDA’s standards for approval mean that biosimilar or interchangeable biosimilar products can be used in place of the reference product they were compared to.”
FDA-approved biosimilars undergo a rigorous evaluation for safety, effectiveness, and quality for their approved conditions of use, she continued. “Therefore, patients and health care providers can rely on a biosimilar to be as safe and effective for its approved uses as the original biological product.”
Remicade as a yard stick
Gastroenterologists dealt with this situation once before, when Remicade (infliximab) biosimilars came on the market in 2016, noted Miguel Regueiro, MD, chair of the Digestive Disease and Surgery Institute at the Cleveland Clinic.
Remicade and Humira are both tumor necrosis factor inhibitors with the same mechanism of action and many of the same indications. “We already had that experience with Remicade and biosimilar switch 2 or 3 years ago. Now we’re talking about Humira,” said Dr. Regueiro.
Most GI doctors have prescribed one of the more common infliximab biosimilars (Inflectra or Renflexis), noted Dr. Oldfield.
Cardinal Health, which recently surveyed 300 gastroenterologists, rheumatologists, and dermatologists about adalimumab biosimilars, found that gastroenterologists had the highest comfort level in prescribing them. Their top concern, however, was changing a patient from adalimumab to an adalimumab biosimilar.
For most patients, Dr. Oldfield sees the Humira reference biologic and biosimilar as equivalent.
However, he said he would change a patient’s drug only if there were a good reason or if his hand was forced by insurance. He would not make the change for a patient who recently began induction with the reference biologic or a patient with highly active clinical disease.
“While there is limited data to support this, I would also have some qualms about changing a patient from reference biologic to a biosimilar if they previously had immune-mediated pharmacokinetic failure due to antibody development with a biologic and were currently doing well on their new biologic,” he said.
Those with a new ulcerative colitis or Crohn’s diagnosis who are initiating a biologic for the first time might consider a biosimilar. If a patient is transitioning from a reference biologic to a biosimilar, “I would want to make that change during a time of stable remission and with the recognition that the switch is not a temporary switch, but a long-term switch,” he continued.
A paper that reviewed 23 observational studies of adalimumab and other biosimilars found that switching biosimilars was safe and effective. But if possible, patients should minimize the number of switches until more robust long-term data are available, added Dr. Oldfield.
If a patient is apprehensive about switching to a new therapy, “one may need to be cognizant of the ‘nocebo’ effect in which there is an unexplained or unfavorable therapeutic effect after switching,” he said.
Other gastroenterologists voiced similar reservations about switching. “I won’t use an adalimumab biosimilar unless the patient requests it, the insurance requires it, or there is a cost advantage for the patient such that they prefer it,” said Doug Wolf, MD, an Atlanta gastroenterologist.
“There is no medical treatment advantage to a biosimilar, especially if switching from Humira,” added Dr. Wolf.
Insurance will guide treatment
Once a drug is approved for use by the FDA, that drug will be available in all 50 states. “Different private insurance formularies, as well as state Medicaid formularies, might affect the actual ability of patients to receive such drugs,” said Mr. Newmark.
Patients should consult with their providers and insurance companies to see what therapies are available, he advised.
Dr. Hanauer anticipates some headaches arising for patients and doctors alike when negotiating for a specific drug.
Cyltezo may be the only biosimilar interchangeable with Humira, but the third-party pharmacy benefit manager (PBM) could negotiate for one of the noninterchangeable ones. “On a yearly basis they could switch their preference,” said Dr. Hanauer.
In the Cardinal Health survey, more than 60% of respondents said they would feel comfortable prescribing an adalimumab biosimilar only with an interchangeability designation.
A PBM may offer a patient Cyltezo if it’s cheaper than Humira. If the patient insists on staying on Humira, then they’ll have to pay more for that drug on their payer’s formulary, said Dr. Hanauer. In a worst-case scenario, a physician may have to appeal on a patient’s behalf to get Humira if the insurer offers only the biosimilar.
Taking that step to appeal is a major hassle for the physician, and leads to extra back door costs as well, said Dr. Hanauer.
Humira manufacturer AbbVie, in turn, may offer discounts and rebates to the PBMs to put Humira on their formulary. “That’s the AbbVie negotiating power. It’s not that the cost is going to be that much different. It’s going to be that there are rebates and discounts that are going to make the cost different,” he added.
As a community physician, Dr. Oldfield has specific concerns about accessibility.
The ever-increasing burden of insurance documentation and prior authorization means it can take weeks or months to get these medications approved. “The addition of new biosimilars is a welcome entrance if it can get patients the medications they need when they need it,” he said.
When it comes to prescribing biologics, many physicians rely on ancillary staff for assistance. It’s a team effort to sift through all the paperwork, observed Dr. Oldfield.
“While many community GI practices have specialized staff to deal with prior authorizations, they are still a far cry from the IBD [inflammatory bowel disease] academic centers where there are often pharmacists, nursing specialists, and home-monitoring programs to check in on patients,” he explained.
Landscape on cost is uncertain
At present, little is known about the cost of the biosimilars and impact on future drug pricing, said Dr. Oldfield.
At least for Medicare, Humira biosimilars will be considered Medicare Part D drugs if used for a medically accepted indication, said a spokesperson for the Centers for Medicare and Medicaid Services.
Part D sponsors (pharmacy and therapeutic committees) “will make the determination as to whether Amjevita and other products will be added to their formularies,” said the spokesperson.
Patients never saw a significant cost savings with Remicade biosimilars. “I imagine the same would be true with biosimilars for Humira,” said Dr. Regueiro. Patients may see greater access to these drugs, however, because the insurance plan or the pharmacy plan will make them more readily available, he added.
The hope is that, as biosimilars are introduced, the price of the originator biologic will go down, said Mr. Newmark. “Therefore, we can expect Humira to be offered at a lower price as it faces competition. Where it will sit in comparison to the forthcoming biosimilars will depend on how much biosimilar companies drop their price and how much pressure will be on PBMs and insurers to cover the lowest list price drug,” he said.
AbbVie did not respond to several requests for comment.
Charitable patient assistance programs for biosimilars or biologics can help offset the price of copayments, Mr. Newmark offered.
Ideally, insurers will offer designated biosimilars at a reduced or even no out-of-pocket expense on their formularies. This should lead to a decreased administrative burden for approval with streamlined (or even removal) of prior authorizations for certain medications, said Dr. Oldfield.
Without insurance or medication assistance programs, the cost of biosimilars is prohibitively expensive, he added.
“Biosimilars have higher research, development, and manufacturing costs than what people conventionally think of [for] a generic medication.”
Educating, advising patients
Dr. Oldfield advised that gastroenterologists refer to biologics by the generic name rather than branded name when initiating therapy unless there is a very specific reason not to. “This approach should make the process more streamlined and less subjected to quick denials for brand-only requests as biosimilars start to assume a larger market share,” he said.
Uptake of the Humira biosimilars also will depend on proper education of physicians and patients and their comfort level with the biosimilars, said Dr. Regueiro. Cleveland Clinic uses a team approach to educate on this topic, relying on pharmacists, clinicians, and nurses to explain that there’s no real difference between the reference drug and its biosimilars, based on efficacy and safety data.
Physicians can also direct patients to patient-friendly resources, said Mr. Newmark. “By starting the conversation early, it ensures that when/if the time comes that your patient is switched to or chooses a biosimilar they will feel more confident because they have the knowledge to make decisions about their care.”
The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars , is a free resource for patients, he added.
It’s important that doctors also understand these products so they can explain to their patients what to expect, said the FDA’s Dr. Yim. The FDA provides educational materials on its website, including a comprehensive curriculum toolkit.
Dr. Hanauer has served as a consultant for AbbVie, Amgen, American College of Gastroenterology, GlaxoSmithKline, American Gastroenterological Association, Pfizer, and a host of other companies . Dr. Regueiro has served on advisory boards and as a consultant for Abbvie, Janssen, UCB, Takeda, Pfizer, BMS, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET Pharma Solutions,Trellis, and Boehringer Ingelheim Pharmaceuticals. Dr. Wolf, Dr. Yim, Dr. Oldfield, and Mr. Newmark have no financial conflicts of interest.
Gastroenterologists in 2023 will have more tools in their arsenal to treat patients with Crohn’s disease or ulcerative colitis. As many as 8-10 adalimumab biosimilars are anticipated to come on the market this year, giving mainstay drug Humira some vigorous competition.
Three scenarios will drive adalimumab biosimilar initiation: Insurance preference for the initial treatment of a newly diagnosed condition, a change in a patient’s insurance plan, or an insurance-mandated switch, said Edward C. Oldfield IV, MD, assistant professor at Eastern Virginia Medical School’s division of gastroenterology in Norfolk.
“Outside of these scenarios, I would encourage patients to remain on their current biologic so long as cost and accessibility remain stable,” said Dr. Oldfield.
Many factors will contribute to the success of biosimilars. Will physicians be prescribing them? How are biosimilars placed on formularies and will they be given preferred status? How will manufacturers price their biosimilars? “We have to wait and see to get the answers to these questions,” said Steven Newmark, JD, MPA, chief legal officer and director of policy, Global Healthy Living Foundation/CreakyJoints, a nonprofit advocacy organization based in New York.
Prescribing biosimilars is no different than prescribing originator biologics, so providers should know how to use them, said Mr. Newmark. “Most important will be the availability of patient-friendly resources that providers can share with their patients to provide education about and confidence in using biosimilars,” he added.
Overall, biosimilars are a good thing, said Dr. Oldfield. “In the long run they should bring down costs and increase access to medications for our patients.”
Others are skeptical that the adalimumab biosimilars will save patients much money.
Biosimilar laws were created to lower costs. However, if a patient with insurance pays only $5 a month out of pocket for Humira – a drug that normally costs $7,000 without coverage – it’s unlikely they would want to switch unless there’s comparable savings from the biosimilar, said Stephen B. Hanauer, MD, medical director of the Digestive Health Center and professor of medicine at Northwestern Medicine, Northwestern University, Evanston, Ill.
Like generics, Humira biosimilars may face some initial backlash, said Dr. Hanauer.
2023 broadens scope of adalimumab treatments
The American Gastroenterological Association describes a biosimilar as something that’s “highly similar to, but not an exact copy of, a biologic reference product already approved” by the Food and Drug Administration. Congress under the 2010 Affordable Care Act created a special, abbreviated pathway to approval for biosimilars.
AbbVie’s Humira, the global revenue for which exceeded $20 billion in 2021, has long dominated the U.S. market on injectable treatments for autoimmune diseases. The popular drug faces some competition in 2023, however, following a series of legal settlements that allowed AbbVie competitors to release their own adalimumab biosimilars.
“So far, we haven’t seen biosimilars live up to their potential in the U.S. in the inflammatory space,” said Mr. Newmark. This may change, however. Previously, biosimilars have required infusion, which demanded more time, commitment, and travel from patients. “The new set of forthcoming Humira biosimilars are injectables, an administration method preferred by patients,” he said.
The FDA will approve a biosimilar if it determines that the biological product is highly similar to the reference product, and that there are no clinically meaningful differences between the biological and reference product in terms of the safety, purity, and potency of the product.
The agency to date has approved 8 adalimumab biosimilars. These include: Idacio (adalimumab-aacf, Fresenius Kabi); Amjevita (adalimumab-atto, Amgen); Hadlima (adalimumab-bwwd, Organon); Cyltezo (adalimumab-adbm, Boehringer Ingelheim); Yusimry (adalimumab-aqvh from Coherus BioSciences); Hulio (adalimumab-fkjp; Mylan/Fujifilm Kyowa Kirin Biologics); Hyrimoz (adalimumab-adaz, Sandoz), and Abrilada (adalimumab-afzb, Pfizer).
“While FDA doesn’t formally track when products come to market, we know based on published reports that application holders for many of the currently FDA-approved biosimilars plan to market this year, starting with Amjevita being the first adalimumab biosimilar launched” in January, said Sarah Yim, MD, director of the Office of Therapeutic Biologics and Biosimilars at the agency.
At press time, two other companies (Celltrion and Alvotech/Teva) were awaiting FDA approval for their adalimumab biosimilar drugs.
Among the eight approved drugs, Cyltezo is the only one that has a designation for interchangeability with Humira.
An interchangeable biosimilar may be substituted at the pharmacy without the intervention of the prescriber – much like generics are substituted, depending on state laws, said Dr. Yim. “However, in terms of safety and effectiveness, FDA’s standards for approval mean that biosimilar or interchangeable biosimilar products can be used in place of the reference product they were compared to.”
FDA-approved biosimilars undergo a rigorous evaluation for safety, effectiveness, and quality for their approved conditions of use, she continued. “Therefore, patients and health care providers can rely on a biosimilar to be as safe and effective for its approved uses as the original biological product.”
Remicade as a yard stick
Gastroenterologists dealt with this situation once before, when Remicade (infliximab) biosimilars came on the market in 2016, noted Miguel Regueiro, MD, chair of the Digestive Disease and Surgery Institute at the Cleveland Clinic.
Remicade and Humira are both tumor necrosis factor inhibitors with the same mechanism of action and many of the same indications. “We already had that experience with Remicade and biosimilar switch 2 or 3 years ago. Now we’re talking about Humira,” said Dr. Regueiro.
Most GI doctors have prescribed one of the more common infliximab biosimilars (Inflectra or Renflexis), noted Dr. Oldfield.
Cardinal Health, which recently surveyed 300 gastroenterologists, rheumatologists, and dermatologists about adalimumab biosimilars, found that gastroenterologists had the highest comfort level in prescribing them. Their top concern, however, was changing a patient from adalimumab to an adalimumab biosimilar.
For most patients, Dr. Oldfield sees the Humira reference biologic and biosimilar as equivalent.
However, he said he would change a patient’s drug only if there were a good reason or if his hand was forced by insurance. He would not make the change for a patient who recently began induction with the reference biologic or a patient with highly active clinical disease.
“While there is limited data to support this, I would also have some qualms about changing a patient from reference biologic to a biosimilar if they previously had immune-mediated pharmacokinetic failure due to antibody development with a biologic and were currently doing well on their new biologic,” he said.
Those with a new ulcerative colitis or Crohn’s diagnosis who are initiating a biologic for the first time might consider a biosimilar. If a patient is transitioning from a reference biologic to a biosimilar, “I would want to make that change during a time of stable remission and with the recognition that the switch is not a temporary switch, but a long-term switch,” he continued.
A paper that reviewed 23 observational studies of adalimumab and other biosimilars found that switching biosimilars was safe and effective. But if possible, patients should minimize the number of switches until more robust long-term data are available, added Dr. Oldfield.
If a patient is apprehensive about switching to a new therapy, “one may need to be cognizant of the ‘nocebo’ effect in which there is an unexplained or unfavorable therapeutic effect after switching,” he said.
Other gastroenterologists voiced similar reservations about switching. “I won’t use an adalimumab biosimilar unless the patient requests it, the insurance requires it, or there is a cost advantage for the patient such that they prefer it,” said Doug Wolf, MD, an Atlanta gastroenterologist.
“There is no medical treatment advantage to a biosimilar, especially if switching from Humira,” added Dr. Wolf.
Insurance will guide treatment
Once a drug is approved for use by the FDA, that drug will be available in all 50 states. “Different private insurance formularies, as well as state Medicaid formularies, might affect the actual ability of patients to receive such drugs,” said Mr. Newmark.
Patients should consult with their providers and insurance companies to see what therapies are available, he advised.
Dr. Hanauer anticipates some headaches arising for patients and doctors alike when negotiating for a specific drug.
Cyltezo may be the only biosimilar interchangeable with Humira, but the third-party pharmacy benefit manager (PBM) could negotiate for one of the noninterchangeable ones. “On a yearly basis they could switch their preference,” said Dr. Hanauer.
In the Cardinal Health survey, more than 60% of respondents said they would feel comfortable prescribing an adalimumab biosimilar only with an interchangeability designation.
A PBM may offer a patient Cyltezo if it’s cheaper than Humira. If the patient insists on staying on Humira, then they’ll have to pay more for that drug on their payer’s formulary, said Dr. Hanauer. In a worst-case scenario, a physician may have to appeal on a patient’s behalf to get Humira if the insurer offers only the biosimilar.
Taking that step to appeal is a major hassle for the physician, and leads to extra back door costs as well, said Dr. Hanauer.
Humira manufacturer AbbVie, in turn, may offer discounts and rebates to the PBMs to put Humira on their formulary. “That’s the AbbVie negotiating power. It’s not that the cost is going to be that much different. It’s going to be that there are rebates and discounts that are going to make the cost different,” he added.
As a community physician, Dr. Oldfield has specific concerns about accessibility.
The ever-increasing burden of insurance documentation and prior authorization means it can take weeks or months to get these medications approved. “The addition of new biosimilars is a welcome entrance if it can get patients the medications they need when they need it,” he said.
When it comes to prescribing biologics, many physicians rely on ancillary staff for assistance. It’s a team effort to sift through all the paperwork, observed Dr. Oldfield.
“While many community GI practices have specialized staff to deal with prior authorizations, they are still a far cry from the IBD [inflammatory bowel disease] academic centers where there are often pharmacists, nursing specialists, and home-monitoring programs to check in on patients,” he explained.
Landscape on cost is uncertain
At present, little is known about the cost of the biosimilars and impact on future drug pricing, said Dr. Oldfield.
At least for Medicare, Humira biosimilars will be considered Medicare Part D drugs if used for a medically accepted indication, said a spokesperson for the Centers for Medicare and Medicaid Services.
Part D sponsors (pharmacy and therapeutic committees) “will make the determination as to whether Amjevita and other products will be added to their formularies,” said the spokesperson.
Patients never saw a significant cost savings with Remicade biosimilars. “I imagine the same would be true with biosimilars for Humira,” said Dr. Regueiro. Patients may see greater access to these drugs, however, because the insurance plan or the pharmacy plan will make them more readily available, he added.
The hope is that, as biosimilars are introduced, the price of the originator biologic will go down, said Mr. Newmark. “Therefore, we can expect Humira to be offered at a lower price as it faces competition. Where it will sit in comparison to the forthcoming biosimilars will depend on how much biosimilar companies drop their price and how much pressure will be on PBMs and insurers to cover the lowest list price drug,” he said.
AbbVie did not respond to several requests for comment.
Charitable patient assistance programs for biosimilars or biologics can help offset the price of copayments, Mr. Newmark offered.
Ideally, insurers will offer designated biosimilars at a reduced or even no out-of-pocket expense on their formularies. This should lead to a decreased administrative burden for approval with streamlined (or even removal) of prior authorizations for certain medications, said Dr. Oldfield.
Without insurance or medication assistance programs, the cost of biosimilars is prohibitively expensive, he added.
“Biosimilars have higher research, development, and manufacturing costs than what people conventionally think of [for] a generic medication.”
Educating, advising patients
Dr. Oldfield advised that gastroenterologists refer to biologics by the generic name rather than branded name when initiating therapy unless there is a very specific reason not to. “This approach should make the process more streamlined and less subjected to quick denials for brand-only requests as biosimilars start to assume a larger market share,” he said.
Uptake of the Humira biosimilars also will depend on proper education of physicians and patients and their comfort level with the biosimilars, said Dr. Regueiro. Cleveland Clinic uses a team approach to educate on this topic, relying on pharmacists, clinicians, and nurses to explain that there’s no real difference between the reference drug and its biosimilars, based on efficacy and safety data.
Physicians can also direct patients to patient-friendly resources, said Mr. Newmark. “By starting the conversation early, it ensures that when/if the time comes that your patient is switched to or chooses a biosimilar they will feel more confident because they have the knowledge to make decisions about their care.”
The Global Healthy Living Foundation’s podcast, Breaking Down Biosimilars , is a free resource for patients, he added.
It’s important that doctors also understand these products so they can explain to their patients what to expect, said the FDA’s Dr. Yim. The FDA provides educational materials on its website, including a comprehensive curriculum toolkit.
Dr. Hanauer has served as a consultant for AbbVie, Amgen, American College of Gastroenterology, GlaxoSmithKline, American Gastroenterological Association, Pfizer, and a host of other companies . Dr. Regueiro has served on advisory boards and as a consultant for Abbvie, Janssen, UCB, Takeda, Pfizer, BMS, Organon, Amgen, Genentech, Gilead, Salix, Prometheus, Lilly, Celgene, TARGET Pharma Solutions,Trellis, and Boehringer Ingelheim Pharmaceuticals. Dr. Wolf, Dr. Yim, Dr. Oldfield, and Mr. Newmark have no financial conflicts of interest.
Eight-week TB treatment strategy shows potential
A strategy for the
“We found that if we use the strategy of a bedaquiline-linezolid five-drug regimen for 8 weeks and then followed patients for 96 weeks, [the regimen] was noninferior, clinically, to the standard regimen in terms of the number of people alive, free of TB disease, and not on treatment,” said lead author Nicholas Paton, MD, of the National University of Singapore, in a press conference held during the Conference on Retroviruses & Opportunistic Infections.
“The total time on treatment was reduced by half – instead of 160 days, it was 85 days for the total duration.”
Commenting on the study, which was published concurrently in the New England Journal of Medicine, Richard E. Chaisson, MD, noted that, although more needs to be understood, the high number of responses is nevertheless encouraging.
“Clinicians will not feel comfortable with the short regimens at this point, but it is remarkable that so many patients did well with shorter treatments,” Dr. Chaisson, who is a professor of medicine, epidemiology, and international health and director of the Johns Hopkins University Center for Tuberculosis Research, Baltimore, said in an interview.
Importantly, the study should help push forward “future studies [that] will stratify patients according to their likelihood of responding to shorter treatments,” he said.
The current global standard for TB treatment, practiced for 4 decades, has been a 6-month rifampin-based regimen. Although the regimen performs well, curing more than 95% of cases in clinical trials, in real-world practice, the prolonged duration can be problematic, with issues of nonadherence and loss of patients to follow-up.
Previous research has shown that shorter regimens have potential, with some studies showing as many as 85% of patients cured with 3- and 4-month regimens, and some promising 2-month regimens showing efficacy specifically for those with smear-negative TB.
These efforts suggest that “the current 6-month regimen may lead to overtreatment in the majority of persons in order to prevent relapse in a minority of persons,” the authors asserted.
TRUNCATE-TB
To investigate a suitable shorter-term alternative, the authors conducted the phase 2-3, prospective, open-label TRUNCATE-TB trial, in which 674 patients with rifampin-susceptible pulmonary TB were enrolled at 18 sites in Asia and Africa.
The patients were randomly assigned to receive either the standard treatment regimen (rifampin and isoniazid for 24 weeks with pyrazinamide and ethambutol for the first 8 weeks; n = 181), or one of four novel five-drug regimens to be administered over 8 weeks, along with extended treatment for persistent clinical disease of up to 12 weeks, if needed, and a plan for retreatment in the case of relapse (n = 493).
Two of the regimens were dropped because of logistic criteria; the two remaining shorter-course groups included in the study involved either high-dose rifampin plus linezolid or bedaquiline plus linezolid, each combined with isoniazid, pyrazinamide, and ethambutol.
Of the patients, 62% were male, and four withdrew or were lost to follow-up by the end of the study at a final follow-up at week 96.
Among patients assigned to the 8-week regimens, 80% stopped at exactly 8 weeks, while 9% wound up having extended treatment to 10 weeks and 3% were extended to 12 weeks.
For the primary endpoint, a composite of death, ongoing treatment, or active disease at week 96, the rate was lowest in the standard 24-week therapy group, occurring in 7 of 181 patients (3.9%), compared with 21 of 184 patients (11.4%) in the rifampin plus linezolid group (adjusted difference, 7.4 percentage points, which did not meet noninferiority criterion), and 11 of 189 (5.8%) in the group in the bedaquiline plus linezolid group (adjusted difference, 0.8 percentage points, meeting noninferiority criterion).
The mean total duration of treatment through week 96 in the standard treatment group was 180 days versus 106 days in the rifampin–linezolid group, and 85 days in the bedaquiline-linezolid group.
The results were consistent across multiple subgroups defined according to baseline characteristics, including some that could be linked to severe disease and a high risk for relapse.
In terms of safety, there were no significant differences between the groups in terms of grade 3 or 4 adverse events.
Of note, only two patients (1.1%) in the bedaquiline plus linezolid group acquired a resistance, which Dr. Paton said was “encouraging,” because of concerns about resistance to that drug.
‘Unfavorable’ composite also evaluated
In an updated analysis of the study that Dr. Paton presented at the meeting, the authors looked at a revised “unfavorable” primary outcome – a composite including treatment failure, relapse, death, or nonattendance at week 96 without evidence of prior disease clearance.
The rate remained lowest in the standard 24-week therapy group (3.9%) versus 25% in the rifampin plus linezolid group, and 13.8% in the bedaquiline plus linezolid group.
Though the lower rate with the standard treatment was expected, Dr. Paton said the results nevertheless hold promise, at least for some patients, for successful treatment with the 8-week bedaquiline plus linezolid strategy.
“What the trial has told us is that even with that 13.8% relapse rate, we can manage patients within this strategy and people can do fine at the end, because with some simple clinical biomarkers, we can pick the people who may have a high chance of achieving a cure.”
Dr. Chaisson expressed concern over the higher unfavorable rates, but said the results help pave the way for refining a workable-shorter term strategy.
“TRUNCATE-TB did find that most patients could be successfully treated in 2 months with the novel regimen of bedaquiline plus linezolid, but the failure rate was still unacceptably high,” he said.
“This regimen will not be widely adapted at this point, but additional analyses may identify subsets of patients who will do well with shorter regimens, and future studies will stratify patients according to their likelihood of responding to shorter treatments.”
The authors of an accompanying editorial further commented that the benefits of a shorter treatment strategy could very well outweigh possible shortcomings.
“Treatment algorithms such as that used in the TRUNCATE-TB trial are fundamental to tuberculosis control,” wrote Véronique Dartois, PhD, Center for Discovery and Innovation, Nutley, N.J., and Eric J. Rubin, MD, PhD, the editor-in-chief of NEJM. “Although implementing them could be a challenge, any added burden might be offset by reduced costs, better adherence, and increased patient satisfaction. Thus, for tuberculosis, a strategy might be more than just a regimen.”
The good news, as summed up by CROI vice-chair Landon Myer, MD, PhD, in the press conference, is that “we’re moving closer and closer to the holy grail of a short, efficacious regimen for TB treatment. We’re getting there slowly, but we’re getting there.”
The study received grant funding from the Singapore National Medical Research Council; a grant from the Department of Health and Social Care; the Foreign, Commonwealth, and Development Office; the Medical Research Council; and Wellcome Trust; as well as a grant from the UK Research and Innovation Medical Research Council. Dr. Dartois reported no relevant financial relationships. Dr. Chaisson had no disclosures to report.
A version of this article originally appeared on Medscape.com.
A strategy for the
“We found that if we use the strategy of a bedaquiline-linezolid five-drug regimen for 8 weeks and then followed patients for 96 weeks, [the regimen] was noninferior, clinically, to the standard regimen in terms of the number of people alive, free of TB disease, and not on treatment,” said lead author Nicholas Paton, MD, of the National University of Singapore, in a press conference held during the Conference on Retroviruses & Opportunistic Infections.
“The total time on treatment was reduced by half – instead of 160 days, it was 85 days for the total duration.”
Commenting on the study, which was published concurrently in the New England Journal of Medicine, Richard E. Chaisson, MD, noted that, although more needs to be understood, the high number of responses is nevertheless encouraging.
“Clinicians will not feel comfortable with the short regimens at this point, but it is remarkable that so many patients did well with shorter treatments,” Dr. Chaisson, who is a professor of medicine, epidemiology, and international health and director of the Johns Hopkins University Center for Tuberculosis Research, Baltimore, said in an interview.
Importantly, the study should help push forward “future studies [that] will stratify patients according to their likelihood of responding to shorter treatments,” he said.
The current global standard for TB treatment, practiced for 4 decades, has been a 6-month rifampin-based regimen. Although the regimen performs well, curing more than 95% of cases in clinical trials, in real-world practice, the prolonged duration can be problematic, with issues of nonadherence and loss of patients to follow-up.
Previous research has shown that shorter regimens have potential, with some studies showing as many as 85% of patients cured with 3- and 4-month regimens, and some promising 2-month regimens showing efficacy specifically for those with smear-negative TB.
These efforts suggest that “the current 6-month regimen may lead to overtreatment in the majority of persons in order to prevent relapse in a minority of persons,” the authors asserted.
TRUNCATE-TB
To investigate a suitable shorter-term alternative, the authors conducted the phase 2-3, prospective, open-label TRUNCATE-TB trial, in which 674 patients with rifampin-susceptible pulmonary TB were enrolled at 18 sites in Asia and Africa.
The patients were randomly assigned to receive either the standard treatment regimen (rifampin and isoniazid for 24 weeks with pyrazinamide and ethambutol for the first 8 weeks; n = 181), or one of four novel five-drug regimens to be administered over 8 weeks, along with extended treatment for persistent clinical disease of up to 12 weeks, if needed, and a plan for retreatment in the case of relapse (n = 493).
Two of the regimens were dropped because of logistic criteria; the two remaining shorter-course groups included in the study involved either high-dose rifampin plus linezolid or bedaquiline plus linezolid, each combined with isoniazid, pyrazinamide, and ethambutol.
Of the patients, 62% were male, and four withdrew or were lost to follow-up by the end of the study at a final follow-up at week 96.
Among patients assigned to the 8-week regimens, 80% stopped at exactly 8 weeks, while 9% wound up having extended treatment to 10 weeks and 3% were extended to 12 weeks.
For the primary endpoint, a composite of death, ongoing treatment, or active disease at week 96, the rate was lowest in the standard 24-week therapy group, occurring in 7 of 181 patients (3.9%), compared with 21 of 184 patients (11.4%) in the rifampin plus linezolid group (adjusted difference, 7.4 percentage points, which did not meet noninferiority criterion), and 11 of 189 (5.8%) in the group in the bedaquiline plus linezolid group (adjusted difference, 0.8 percentage points, meeting noninferiority criterion).
The mean total duration of treatment through week 96 in the standard treatment group was 180 days versus 106 days in the rifampin–linezolid group, and 85 days in the bedaquiline-linezolid group.
The results were consistent across multiple subgroups defined according to baseline characteristics, including some that could be linked to severe disease and a high risk for relapse.
In terms of safety, there were no significant differences between the groups in terms of grade 3 or 4 adverse events.
Of note, only two patients (1.1%) in the bedaquiline plus linezolid group acquired a resistance, which Dr. Paton said was “encouraging,” because of concerns about resistance to that drug.
‘Unfavorable’ composite also evaluated
In an updated analysis of the study that Dr. Paton presented at the meeting, the authors looked at a revised “unfavorable” primary outcome – a composite including treatment failure, relapse, death, or nonattendance at week 96 without evidence of prior disease clearance.
The rate remained lowest in the standard 24-week therapy group (3.9%) versus 25% in the rifampin plus linezolid group, and 13.8% in the bedaquiline plus linezolid group.
Though the lower rate with the standard treatment was expected, Dr. Paton said the results nevertheless hold promise, at least for some patients, for successful treatment with the 8-week bedaquiline plus linezolid strategy.
“What the trial has told us is that even with that 13.8% relapse rate, we can manage patients within this strategy and people can do fine at the end, because with some simple clinical biomarkers, we can pick the people who may have a high chance of achieving a cure.”
Dr. Chaisson expressed concern over the higher unfavorable rates, but said the results help pave the way for refining a workable-shorter term strategy.
“TRUNCATE-TB did find that most patients could be successfully treated in 2 months with the novel regimen of bedaquiline plus linezolid, but the failure rate was still unacceptably high,” he said.
“This regimen will not be widely adapted at this point, but additional analyses may identify subsets of patients who will do well with shorter regimens, and future studies will stratify patients according to their likelihood of responding to shorter treatments.”
The authors of an accompanying editorial further commented that the benefits of a shorter treatment strategy could very well outweigh possible shortcomings.
“Treatment algorithms such as that used in the TRUNCATE-TB trial are fundamental to tuberculosis control,” wrote Véronique Dartois, PhD, Center for Discovery and Innovation, Nutley, N.J., and Eric J. Rubin, MD, PhD, the editor-in-chief of NEJM. “Although implementing them could be a challenge, any added burden might be offset by reduced costs, better adherence, and increased patient satisfaction. Thus, for tuberculosis, a strategy might be more than just a regimen.”
The good news, as summed up by CROI vice-chair Landon Myer, MD, PhD, in the press conference, is that “we’re moving closer and closer to the holy grail of a short, efficacious regimen for TB treatment. We’re getting there slowly, but we’re getting there.”
The study received grant funding from the Singapore National Medical Research Council; a grant from the Department of Health and Social Care; the Foreign, Commonwealth, and Development Office; the Medical Research Council; and Wellcome Trust; as well as a grant from the UK Research and Innovation Medical Research Council. Dr. Dartois reported no relevant financial relationships. Dr. Chaisson had no disclosures to report.
A version of this article originally appeared on Medscape.com.
A strategy for the
“We found that if we use the strategy of a bedaquiline-linezolid five-drug regimen for 8 weeks and then followed patients for 96 weeks, [the regimen] was noninferior, clinically, to the standard regimen in terms of the number of people alive, free of TB disease, and not on treatment,” said lead author Nicholas Paton, MD, of the National University of Singapore, in a press conference held during the Conference on Retroviruses & Opportunistic Infections.
“The total time on treatment was reduced by half – instead of 160 days, it was 85 days for the total duration.”
Commenting on the study, which was published concurrently in the New England Journal of Medicine, Richard E. Chaisson, MD, noted that, although more needs to be understood, the high number of responses is nevertheless encouraging.
“Clinicians will not feel comfortable with the short regimens at this point, but it is remarkable that so many patients did well with shorter treatments,” Dr. Chaisson, who is a professor of medicine, epidemiology, and international health and director of the Johns Hopkins University Center for Tuberculosis Research, Baltimore, said in an interview.
Importantly, the study should help push forward “future studies [that] will stratify patients according to their likelihood of responding to shorter treatments,” he said.
The current global standard for TB treatment, practiced for 4 decades, has been a 6-month rifampin-based regimen. Although the regimen performs well, curing more than 95% of cases in clinical trials, in real-world practice, the prolonged duration can be problematic, with issues of nonadherence and loss of patients to follow-up.
Previous research has shown that shorter regimens have potential, with some studies showing as many as 85% of patients cured with 3- and 4-month regimens, and some promising 2-month regimens showing efficacy specifically for those with smear-negative TB.
These efforts suggest that “the current 6-month regimen may lead to overtreatment in the majority of persons in order to prevent relapse in a minority of persons,” the authors asserted.
TRUNCATE-TB
To investigate a suitable shorter-term alternative, the authors conducted the phase 2-3, prospective, open-label TRUNCATE-TB trial, in which 674 patients with rifampin-susceptible pulmonary TB were enrolled at 18 sites in Asia and Africa.
The patients were randomly assigned to receive either the standard treatment regimen (rifampin and isoniazid for 24 weeks with pyrazinamide and ethambutol for the first 8 weeks; n = 181), or one of four novel five-drug regimens to be administered over 8 weeks, along with extended treatment for persistent clinical disease of up to 12 weeks, if needed, and a plan for retreatment in the case of relapse (n = 493).
Two of the regimens were dropped because of logistic criteria; the two remaining shorter-course groups included in the study involved either high-dose rifampin plus linezolid or bedaquiline plus linezolid, each combined with isoniazid, pyrazinamide, and ethambutol.
Of the patients, 62% were male, and four withdrew or were lost to follow-up by the end of the study at a final follow-up at week 96.
Among patients assigned to the 8-week regimens, 80% stopped at exactly 8 weeks, while 9% wound up having extended treatment to 10 weeks and 3% were extended to 12 weeks.
For the primary endpoint, a composite of death, ongoing treatment, or active disease at week 96, the rate was lowest in the standard 24-week therapy group, occurring in 7 of 181 patients (3.9%), compared with 21 of 184 patients (11.4%) in the rifampin plus linezolid group (adjusted difference, 7.4 percentage points, which did not meet noninferiority criterion), and 11 of 189 (5.8%) in the group in the bedaquiline plus linezolid group (adjusted difference, 0.8 percentage points, meeting noninferiority criterion).
The mean total duration of treatment through week 96 in the standard treatment group was 180 days versus 106 days in the rifampin–linezolid group, and 85 days in the bedaquiline-linezolid group.
The results were consistent across multiple subgroups defined according to baseline characteristics, including some that could be linked to severe disease and a high risk for relapse.
In terms of safety, there were no significant differences between the groups in terms of grade 3 or 4 adverse events.
Of note, only two patients (1.1%) in the bedaquiline plus linezolid group acquired a resistance, which Dr. Paton said was “encouraging,” because of concerns about resistance to that drug.
‘Unfavorable’ composite also evaluated
In an updated analysis of the study that Dr. Paton presented at the meeting, the authors looked at a revised “unfavorable” primary outcome – a composite including treatment failure, relapse, death, or nonattendance at week 96 without evidence of prior disease clearance.
The rate remained lowest in the standard 24-week therapy group (3.9%) versus 25% in the rifampin plus linezolid group, and 13.8% in the bedaquiline plus linezolid group.
Though the lower rate with the standard treatment was expected, Dr. Paton said the results nevertheless hold promise, at least for some patients, for successful treatment with the 8-week bedaquiline plus linezolid strategy.
“What the trial has told us is that even with that 13.8% relapse rate, we can manage patients within this strategy and people can do fine at the end, because with some simple clinical biomarkers, we can pick the people who may have a high chance of achieving a cure.”
Dr. Chaisson expressed concern over the higher unfavorable rates, but said the results help pave the way for refining a workable-shorter term strategy.
“TRUNCATE-TB did find that most patients could be successfully treated in 2 months with the novel regimen of bedaquiline plus linezolid, but the failure rate was still unacceptably high,” he said.
“This regimen will not be widely adapted at this point, but additional analyses may identify subsets of patients who will do well with shorter regimens, and future studies will stratify patients according to their likelihood of responding to shorter treatments.”
The authors of an accompanying editorial further commented that the benefits of a shorter treatment strategy could very well outweigh possible shortcomings.
“Treatment algorithms such as that used in the TRUNCATE-TB trial are fundamental to tuberculosis control,” wrote Véronique Dartois, PhD, Center for Discovery and Innovation, Nutley, N.J., and Eric J. Rubin, MD, PhD, the editor-in-chief of NEJM. “Although implementing them could be a challenge, any added burden might be offset by reduced costs, better adherence, and increased patient satisfaction. Thus, for tuberculosis, a strategy might be more than just a regimen.”
The good news, as summed up by CROI vice-chair Landon Myer, MD, PhD, in the press conference, is that “we’re moving closer and closer to the holy grail of a short, efficacious regimen for TB treatment. We’re getting there slowly, but we’re getting there.”
The study received grant funding from the Singapore National Medical Research Council; a grant from the Department of Health and Social Care; the Foreign, Commonwealth, and Development Office; the Medical Research Council; and Wellcome Trust; as well as a grant from the UK Research and Innovation Medical Research Council. Dr. Dartois reported no relevant financial relationships. Dr. Chaisson had no disclosures to report.
A version of this article originally appeared on Medscape.com.
FROM CROI 2023
Diabetes drug tied to lower dementia risk
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM NEUROLOGY
Tenecteplase noninferior to alteplase for ischemic stroke: TRACE-2
, a new study has found. “This was a pivotal trial in establishing the safety and efficacy of tenecteplase as an alternative to alteplase in the thrombolytic treatment of acute ischemic stroke within 4.5 hours in Asian patients,” said study author Shuya Li, MD, associate chief physician, department of neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing.
The findings in this all-Chinese population should have an impact on the use of tenecteplase going forward, said Dr. Li. “The results provide further evidence to support a worldwide switch to tenecteplase as the preferred thrombolytic for acute ischemic stroke.”
The findings were presented at the 2023 International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Single bolus
Use of alteplase (tissue plasminogen activator [tPA]) has for years been the main approach to thrombolytic reperfusion therapy for patients with acute stroke, but tenecteplase has recently emerged as a potential successor.
Tenecteplase is a tPA produced by recombinant DNA technology. It has a relatively long half-life and can be delivered in a single bolus instead of requiring an hour-long infusion, as is the case with alteplase.
The phase 3 noninferiority Tenecteplase Reperfusion Therapy in Acute ischemic Cerebrovascular Events (TRACE-2) trial – the first of its kind in an Asian population – included 1,430 adult ischemic stroke patients at 53 Chinese centers. Patients had to have a National Institutes of Health Stroke Scale (NIHSS) score of 5-25 and either not be eligible for or have refused endovascular therapy.
The mean age of study participants was about 66 years, and the percentage of women was about 31%. The mean baseline NIHSS score was 7 in both groups, and the symptom-onset-to-needle time was similar at 180 minutes for the tenecteplase group and 178.5 minutes for the alteplase group.
Researchers randomly assigned patients to receive tenecteplase or alteplase within 4.5 hours of symptom onset.
Those in the tenecteplase group received 0.25 mg/kg of the drug in a single IV bolus (maximum dose, 25 mg). Control group members who were treated with alteplase were given the drug as a 10% bolus, with the remainder given as a 1-hour infusion (0.9 mg/kg with a maximum dose of 90 mg).
Showed noninferiority
The primary efficacy outcome was a modified Rankins scale (mRS) score of 0-1 at 90 days, which is considered excellent function. About 62% of tenecteplase patients and 58% of alteplase patients attained this outcome (risk ratio, 1.09; 95% confidence interval, 1.00-1.18).
The P value was .001 for noninferiority and .06 for superiority, but Dr. Li explained that these values may change when considering the site effect.
There were no statistically significant differences between the two drugs on secondary outcomes of favorable function. For example, 73% of tenecteplase patients and 72% of alteplase patients had an mRS score of 0-2 at 3 months, and 50% in the tenecteplase and 49% in the alteplase group improved by 4 or more points on the NIHSS, or had a score of 1 or less, at 24 hours.
The groups also had comparable scores on the European quality-of-life visual analogue scale and on the Barthel index, which measures functional independence related to personal care and mobility.
Tenecteplase also turned out to be as safe at alteplase. About 2% in both groups had symptomatic intracranial hemorrhage within 36 hours, and both groups had that same percentage for such hemorrhages within 90 days. As well, the groups had a similar rate of any intracranial hemorrhage within 90 days (6% and 7%).
The mortality rate was 7% in the tenecteplase group, compared with 5% in the alteplase group.
Adverse events (AEs) occurred in 86% and 87%, and serious AEs in 16% and 15%, of the tenecteplase and alteplase groups, respectively, again with no statistically significant differences.
The research team aims to test the effectiveness of tenecteplase in other stroke patients, including those with minor strokes, those receiving thrombolysis in a later window, and those receiving endovascular therapy, said Dr. Li.
Strong evidence
Commenting on the study findings, Larry B. Goldstein, MD, professor and chair of neurology, University of Kentucky, Lexington, said it is important to determine the efficacy of tenecteplase among Asians, as they represent “an entirely different population” with unique concerns, such as bleeding complications from anticoagulants.
He noted an advantage of tenecteplase is ease of administration. “You don’t have to go through the loading dose and then the 1-hour infusion,” which poses an “additional hassle” when transferring patients between institutions, he said.
However, he noted that a possible “downside” to having both drugs available in the emergency department is “using the wrong drug at the wrong dose” because of their similar sounding names.
Also commenting on the study, Tudor G. Jovin, MD, professor and chair, department of neurology, Rowan University, Camden, N.J., said he welcomes another trial that confirms that these two drugs are biologically similar.
“I’m very glad this trial was done because it adds another very strong piece of evidence of equivalency.”
But the two drugs are not the same in some important respects, said Dr. Jovin, whose center switched to using tenecteplase almost 3 years ago. That switch has resulted in cutting 17 minutes from the door-to-needle time “which is quite significant,” he said.
“There’s no question that once we used tenecteplase in lieu of tPA, it’s been just so much easier to administer and affects the interhospital transfer protocols, because you’re not transferring the patient with a critical care IV. It’s a win-win situation for everyone.”
The study received funding from the National Science and Technology Major Project, the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences, the National Natural Science Foundation of China, and the China Shijiazhuang Pharmaceutical Company Recomgen Pharmaceutical (Guangzhou). Dr. Li, Dr. Goldstein, and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, a new study has found. “This was a pivotal trial in establishing the safety and efficacy of tenecteplase as an alternative to alteplase in the thrombolytic treatment of acute ischemic stroke within 4.5 hours in Asian patients,” said study author Shuya Li, MD, associate chief physician, department of neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing.
The findings in this all-Chinese population should have an impact on the use of tenecteplase going forward, said Dr. Li. “The results provide further evidence to support a worldwide switch to tenecteplase as the preferred thrombolytic for acute ischemic stroke.”
The findings were presented at the 2023 International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Single bolus
Use of alteplase (tissue plasminogen activator [tPA]) has for years been the main approach to thrombolytic reperfusion therapy for patients with acute stroke, but tenecteplase has recently emerged as a potential successor.
Tenecteplase is a tPA produced by recombinant DNA technology. It has a relatively long half-life and can be delivered in a single bolus instead of requiring an hour-long infusion, as is the case with alteplase.
The phase 3 noninferiority Tenecteplase Reperfusion Therapy in Acute ischemic Cerebrovascular Events (TRACE-2) trial – the first of its kind in an Asian population – included 1,430 adult ischemic stroke patients at 53 Chinese centers. Patients had to have a National Institutes of Health Stroke Scale (NIHSS) score of 5-25 and either not be eligible for or have refused endovascular therapy.
The mean age of study participants was about 66 years, and the percentage of women was about 31%. The mean baseline NIHSS score was 7 in both groups, and the symptom-onset-to-needle time was similar at 180 minutes for the tenecteplase group and 178.5 minutes for the alteplase group.
Researchers randomly assigned patients to receive tenecteplase or alteplase within 4.5 hours of symptom onset.
Those in the tenecteplase group received 0.25 mg/kg of the drug in a single IV bolus (maximum dose, 25 mg). Control group members who were treated with alteplase were given the drug as a 10% bolus, with the remainder given as a 1-hour infusion (0.9 mg/kg with a maximum dose of 90 mg).
Showed noninferiority
The primary efficacy outcome was a modified Rankins scale (mRS) score of 0-1 at 90 days, which is considered excellent function. About 62% of tenecteplase patients and 58% of alteplase patients attained this outcome (risk ratio, 1.09; 95% confidence interval, 1.00-1.18).
The P value was .001 for noninferiority and .06 for superiority, but Dr. Li explained that these values may change when considering the site effect.
There were no statistically significant differences between the two drugs on secondary outcomes of favorable function. For example, 73% of tenecteplase patients and 72% of alteplase patients had an mRS score of 0-2 at 3 months, and 50% in the tenecteplase and 49% in the alteplase group improved by 4 or more points on the NIHSS, or had a score of 1 or less, at 24 hours.
The groups also had comparable scores on the European quality-of-life visual analogue scale and on the Barthel index, which measures functional independence related to personal care and mobility.
Tenecteplase also turned out to be as safe at alteplase. About 2% in both groups had symptomatic intracranial hemorrhage within 36 hours, and both groups had that same percentage for such hemorrhages within 90 days. As well, the groups had a similar rate of any intracranial hemorrhage within 90 days (6% and 7%).
The mortality rate was 7% in the tenecteplase group, compared with 5% in the alteplase group.
Adverse events (AEs) occurred in 86% and 87%, and serious AEs in 16% and 15%, of the tenecteplase and alteplase groups, respectively, again with no statistically significant differences.
The research team aims to test the effectiveness of tenecteplase in other stroke patients, including those with minor strokes, those receiving thrombolysis in a later window, and those receiving endovascular therapy, said Dr. Li.
Strong evidence
Commenting on the study findings, Larry B. Goldstein, MD, professor and chair of neurology, University of Kentucky, Lexington, said it is important to determine the efficacy of tenecteplase among Asians, as they represent “an entirely different population” with unique concerns, such as bleeding complications from anticoagulants.
He noted an advantage of tenecteplase is ease of administration. “You don’t have to go through the loading dose and then the 1-hour infusion,” which poses an “additional hassle” when transferring patients between institutions, he said.
However, he noted that a possible “downside” to having both drugs available in the emergency department is “using the wrong drug at the wrong dose” because of their similar sounding names.
Also commenting on the study, Tudor G. Jovin, MD, professor and chair, department of neurology, Rowan University, Camden, N.J., said he welcomes another trial that confirms that these two drugs are biologically similar.
“I’m very glad this trial was done because it adds another very strong piece of evidence of equivalency.”
But the two drugs are not the same in some important respects, said Dr. Jovin, whose center switched to using tenecteplase almost 3 years ago. That switch has resulted in cutting 17 minutes from the door-to-needle time “which is quite significant,” he said.
“There’s no question that once we used tenecteplase in lieu of tPA, it’s been just so much easier to administer and affects the interhospital transfer protocols, because you’re not transferring the patient with a critical care IV. It’s a win-win situation for everyone.”
The study received funding from the National Science and Technology Major Project, the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences, the National Natural Science Foundation of China, and the China Shijiazhuang Pharmaceutical Company Recomgen Pharmaceutical (Guangzhou). Dr. Li, Dr. Goldstein, and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, a new study has found. “This was a pivotal trial in establishing the safety and efficacy of tenecteplase as an alternative to alteplase in the thrombolytic treatment of acute ischemic stroke within 4.5 hours in Asian patients,” said study author Shuya Li, MD, associate chief physician, department of neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing.
The findings in this all-Chinese population should have an impact on the use of tenecteplase going forward, said Dr. Li. “The results provide further evidence to support a worldwide switch to tenecteplase as the preferred thrombolytic for acute ischemic stroke.”
The findings were presented at the 2023 International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Single bolus
Use of alteplase (tissue plasminogen activator [tPA]) has for years been the main approach to thrombolytic reperfusion therapy for patients with acute stroke, but tenecteplase has recently emerged as a potential successor.
Tenecteplase is a tPA produced by recombinant DNA technology. It has a relatively long half-life and can be delivered in a single bolus instead of requiring an hour-long infusion, as is the case with alteplase.
The phase 3 noninferiority Tenecteplase Reperfusion Therapy in Acute ischemic Cerebrovascular Events (TRACE-2) trial – the first of its kind in an Asian population – included 1,430 adult ischemic stroke patients at 53 Chinese centers. Patients had to have a National Institutes of Health Stroke Scale (NIHSS) score of 5-25 and either not be eligible for or have refused endovascular therapy.
The mean age of study participants was about 66 years, and the percentage of women was about 31%. The mean baseline NIHSS score was 7 in both groups, and the symptom-onset-to-needle time was similar at 180 minutes for the tenecteplase group and 178.5 minutes for the alteplase group.
Researchers randomly assigned patients to receive tenecteplase or alteplase within 4.5 hours of symptom onset.
Those in the tenecteplase group received 0.25 mg/kg of the drug in a single IV bolus (maximum dose, 25 mg). Control group members who were treated with alteplase were given the drug as a 10% bolus, with the remainder given as a 1-hour infusion (0.9 mg/kg with a maximum dose of 90 mg).
Showed noninferiority
The primary efficacy outcome was a modified Rankins scale (mRS) score of 0-1 at 90 days, which is considered excellent function. About 62% of tenecteplase patients and 58% of alteplase patients attained this outcome (risk ratio, 1.09; 95% confidence interval, 1.00-1.18).
The P value was .001 for noninferiority and .06 for superiority, but Dr. Li explained that these values may change when considering the site effect.
There were no statistically significant differences between the two drugs on secondary outcomes of favorable function. For example, 73% of tenecteplase patients and 72% of alteplase patients had an mRS score of 0-2 at 3 months, and 50% in the tenecteplase and 49% in the alteplase group improved by 4 or more points on the NIHSS, or had a score of 1 or less, at 24 hours.
The groups also had comparable scores on the European quality-of-life visual analogue scale and on the Barthel index, which measures functional independence related to personal care and mobility.
Tenecteplase also turned out to be as safe at alteplase. About 2% in both groups had symptomatic intracranial hemorrhage within 36 hours, and both groups had that same percentage for such hemorrhages within 90 days. As well, the groups had a similar rate of any intracranial hemorrhage within 90 days (6% and 7%).
The mortality rate was 7% in the tenecteplase group, compared with 5% in the alteplase group.
Adverse events (AEs) occurred in 86% and 87%, and serious AEs in 16% and 15%, of the tenecteplase and alteplase groups, respectively, again with no statistically significant differences.
The research team aims to test the effectiveness of tenecteplase in other stroke patients, including those with minor strokes, those receiving thrombolysis in a later window, and those receiving endovascular therapy, said Dr. Li.
Strong evidence
Commenting on the study findings, Larry B. Goldstein, MD, professor and chair of neurology, University of Kentucky, Lexington, said it is important to determine the efficacy of tenecteplase among Asians, as they represent “an entirely different population” with unique concerns, such as bleeding complications from anticoagulants.
He noted an advantage of tenecteplase is ease of administration. “You don’t have to go through the loading dose and then the 1-hour infusion,” which poses an “additional hassle” when transferring patients between institutions, he said.
However, he noted that a possible “downside” to having both drugs available in the emergency department is “using the wrong drug at the wrong dose” because of their similar sounding names.
Also commenting on the study, Tudor G. Jovin, MD, professor and chair, department of neurology, Rowan University, Camden, N.J., said he welcomes another trial that confirms that these two drugs are biologically similar.
“I’m very glad this trial was done because it adds another very strong piece of evidence of equivalency.”
But the two drugs are not the same in some important respects, said Dr. Jovin, whose center switched to using tenecteplase almost 3 years ago. That switch has resulted in cutting 17 minutes from the door-to-needle time “which is quite significant,” he said.
“There’s no question that once we used tenecteplase in lieu of tPA, it’s been just so much easier to administer and affects the interhospital transfer protocols, because you’re not transferring the patient with a critical care IV. It’s a win-win situation for everyone.”
The study received funding from the National Science and Technology Major Project, the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences, the National Natural Science Foundation of China, and the China Shijiazhuang Pharmaceutical Company Recomgen Pharmaceutical (Guangzhou). Dr. Li, Dr. Goldstein, and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ISC 2023
Novel neuroprotective agent promising in stroke
preliminary results of a first-in-human study show.
The findings illustrate that it is possible to improve outcomes for stroke patients “not only with reperfusion therapy but with neuroprotectants,” study author Macarena Hernandez, PhD, associate professor, University Complutense, Madrid, told this news organization.
Dr. Hernandez said she hopes these positive results will spur investigation into other neuroprotective agents.
The findings were presented at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Best doses
The study investigated ApTOLL, which blocks the TOLL-like receptor 4 (TLR4) that induces inflammation after a stroke. Previous studies found that ApTOLL protected brain tissue in animal models of stroke.
The phase 1B part of the study found no safety issues and determined the best two doses to be used in phase 2A were 0.05 mg/kg and 0.2 mg/kg.
The analysis included 139 patients at 14 centers in Spain and France (mean age, about 70 years; 42% women) who had a large-vessel occlusion and were eligible for endovascular therapy.
“Our aim was to have a very homogeneous population” to try to replicate in humans what had worked in animals, another study author, Marc Ribó, MD, interventional neurologist, Hospital Vall d’Hebron, Barcelona, told this news organization.
Study participants had an Alberta Stroke Program Early CT Score (ASPECTS) of 5-10, and estimated infarct core volume on CT-perfusion was 5-70 mL. All were treated within 6 hours of stroke onset.
Researchers randomly assigned patients to receive the low dose of the drug, the high dose of the drug, or placebo. The drug was administered intravenously over a 30-minute period just prior to the groin puncture for the thrombectomy procedure.
“So, the drug had already started to work when they underwent the usual standard practice, the thrombectomy,” said Dr. Ribó.
Those who were eligible also received tissue plasminogen activator.
The primary endpoint was safety, including death, symptomatic intracranial hemorrhage (SICH), and recurrent stroke.
Lower mortality
At 90 days, there was a statistically significant lower mortality rate in the high-dose group, compared with the group that received placebo (4.76% vs. 18.18%).
The mortality rate was 26.19% in the low-dose group, but Dr. Ribó stressed that this dose was a quarter of the higher dose and so performed “much more like placebo.”
The higher dose also yielded a better SICH outcome (4.76% of patients vs. 7.27% for placebo and 7.14% for the lower dose). And it was superior in terms of brain edema (2.4% of the population vs. 7.3% for the placebo and 4.8% for the low-dose groups).
About 7.1% of the high-dose group, 3.7% of the placebo group, and 4.8% of the low-dose group had a recurrent transient ischemic attack or stroke.
A secondary efficacy endpoint was infarct volume on MRI at 72 hours. Here, for the higher-dose group, mean infarct volume was reduced, compared with the patients who received placebo (–29.31 cc; 90% confidence interval, –49.28 to –9.34).
This higher dose was also superior for the secondary outcome of National Institutes of Health Stroke Scale score at 72 hours and for the disability outcome on the modified Rankin Score (mRS).
Clear shift in disability
“There was a clear shift toward less disability across levels of the mRS score in the high-dose group at 90 days,” said Dr. Ribó.
He added that he and his colleagues are “very happy” with these results, as they reflect “a consistency” of outcomes.
“We observed that the infarct volumes were lower in the high-dose group, and that led to a significant lower NIH score, meaning less clinical neurological symptoms at 72 hours, and finally, this led to less disability at 90 days.”
These results are “very exciting,” Dr. Hernandez added. “This is the first neuroprotectant that has demonstrated this acute effect in reducing deaths, in reducing the infarct volume and improving functionality long-term in patients treated with the higher dose.”
Dr. Ribó noted the treatment would eventually be used in addition to reperfusion therapy. “It’s not competing with reperfusion treatment; it’s an additional layer” of treatment.
Although it would initially be offered only to patients eligible for thrombectomy, researchers will explore the drug’s effectiveness for other stroke patients, said Dr. Ribó. “We wanted to secure this indication, and from there, progressively expand to other profiles of stroke patients, and even to patients with intracranial hemorrhage.”
The study confirmed the safety of the drug. “There were no safety issues at all,” said Dr. Ribó. “We were initially concerned that an anti-inflammatory in these patients could lead to higher rates of infections, but this was absolutely not the case.”
The next step is to confirm the effects in a larger, multicenter study, which is planned to launch at the end of this year, said Dr. Hernandez.
‘Very robust results’
In a comment, Philip B. Gorelick, MD, professor of neurology, Northwestern University, Chicago, said that, while this was a small early-phase study, the results are “very robust.”
“The authors demonstrated proof of a neuroprotective effect; they showed at 90 days that the death rates were substantially reduced by about four times – 4% vs. 18% – and the size of the damaged tissue at about 72 hours was reduced by 40%,” said Dr. Gorelick, who did not participate in the study.
He also noted that the disability was “less pronounced” at 90 days in the 0.2 mg/kg group.
“So overall, these are very encouraging results,” said Dr. Gorelick. “We have had a lot of difficulty finding neuroprotectant drugs that work, and this drug, in combination with endovascular therapy, seems to be very promising.”
However, he stressed the drug “is not ready for prime-time practice.”
“The proof in the pudding will be in the large-scale main phase 3 trials,” he added.
The study was funded by aptaTargets. Dr. Hernandez is chief scientific officer at aptaTargets. Dr. Ribó is an adviser at AptaTargets; a consultant at Medtronic; has ownership interest in Anaconda and NoraHealth; is a consultant for Cerenovus and Philips; and has stock options at Methink. Dr. Gorelick has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
preliminary results of a first-in-human study show.
The findings illustrate that it is possible to improve outcomes for stroke patients “not only with reperfusion therapy but with neuroprotectants,” study author Macarena Hernandez, PhD, associate professor, University Complutense, Madrid, told this news organization.
Dr. Hernandez said she hopes these positive results will spur investigation into other neuroprotective agents.
The findings were presented at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Best doses
The study investigated ApTOLL, which blocks the TOLL-like receptor 4 (TLR4) that induces inflammation after a stroke. Previous studies found that ApTOLL protected brain tissue in animal models of stroke.
The phase 1B part of the study found no safety issues and determined the best two doses to be used in phase 2A were 0.05 mg/kg and 0.2 mg/kg.
The analysis included 139 patients at 14 centers in Spain and France (mean age, about 70 years; 42% women) who had a large-vessel occlusion and were eligible for endovascular therapy.
“Our aim was to have a very homogeneous population” to try to replicate in humans what had worked in animals, another study author, Marc Ribó, MD, interventional neurologist, Hospital Vall d’Hebron, Barcelona, told this news organization.
Study participants had an Alberta Stroke Program Early CT Score (ASPECTS) of 5-10, and estimated infarct core volume on CT-perfusion was 5-70 mL. All were treated within 6 hours of stroke onset.
Researchers randomly assigned patients to receive the low dose of the drug, the high dose of the drug, or placebo. The drug was administered intravenously over a 30-minute period just prior to the groin puncture for the thrombectomy procedure.
“So, the drug had already started to work when they underwent the usual standard practice, the thrombectomy,” said Dr. Ribó.
Those who were eligible also received tissue plasminogen activator.
The primary endpoint was safety, including death, symptomatic intracranial hemorrhage (SICH), and recurrent stroke.
Lower mortality
At 90 days, there was a statistically significant lower mortality rate in the high-dose group, compared with the group that received placebo (4.76% vs. 18.18%).
The mortality rate was 26.19% in the low-dose group, but Dr. Ribó stressed that this dose was a quarter of the higher dose and so performed “much more like placebo.”
The higher dose also yielded a better SICH outcome (4.76% of patients vs. 7.27% for placebo and 7.14% for the lower dose). And it was superior in terms of brain edema (2.4% of the population vs. 7.3% for the placebo and 4.8% for the low-dose groups).
About 7.1% of the high-dose group, 3.7% of the placebo group, and 4.8% of the low-dose group had a recurrent transient ischemic attack or stroke.
A secondary efficacy endpoint was infarct volume on MRI at 72 hours. Here, for the higher-dose group, mean infarct volume was reduced, compared with the patients who received placebo (–29.31 cc; 90% confidence interval, –49.28 to –9.34).
This higher dose was also superior for the secondary outcome of National Institutes of Health Stroke Scale score at 72 hours and for the disability outcome on the modified Rankin Score (mRS).
Clear shift in disability
“There was a clear shift toward less disability across levels of the mRS score in the high-dose group at 90 days,” said Dr. Ribó.
He added that he and his colleagues are “very happy” with these results, as they reflect “a consistency” of outcomes.
“We observed that the infarct volumes were lower in the high-dose group, and that led to a significant lower NIH score, meaning less clinical neurological symptoms at 72 hours, and finally, this led to less disability at 90 days.”
These results are “very exciting,” Dr. Hernandez added. “This is the first neuroprotectant that has demonstrated this acute effect in reducing deaths, in reducing the infarct volume and improving functionality long-term in patients treated with the higher dose.”
Dr. Ribó noted the treatment would eventually be used in addition to reperfusion therapy. “It’s not competing with reperfusion treatment; it’s an additional layer” of treatment.
Although it would initially be offered only to patients eligible for thrombectomy, researchers will explore the drug’s effectiveness for other stroke patients, said Dr. Ribó. “We wanted to secure this indication, and from there, progressively expand to other profiles of stroke patients, and even to patients with intracranial hemorrhage.”
The study confirmed the safety of the drug. “There were no safety issues at all,” said Dr. Ribó. “We were initially concerned that an anti-inflammatory in these patients could lead to higher rates of infections, but this was absolutely not the case.”
The next step is to confirm the effects in a larger, multicenter study, which is planned to launch at the end of this year, said Dr. Hernandez.
‘Very robust results’
In a comment, Philip B. Gorelick, MD, professor of neurology, Northwestern University, Chicago, said that, while this was a small early-phase study, the results are “very robust.”
“The authors demonstrated proof of a neuroprotective effect; they showed at 90 days that the death rates were substantially reduced by about four times – 4% vs. 18% – and the size of the damaged tissue at about 72 hours was reduced by 40%,” said Dr. Gorelick, who did not participate in the study.
He also noted that the disability was “less pronounced” at 90 days in the 0.2 mg/kg group.
“So overall, these are very encouraging results,” said Dr. Gorelick. “We have had a lot of difficulty finding neuroprotectant drugs that work, and this drug, in combination with endovascular therapy, seems to be very promising.”
However, he stressed the drug “is not ready for prime-time practice.”
“The proof in the pudding will be in the large-scale main phase 3 trials,” he added.
The study was funded by aptaTargets. Dr. Hernandez is chief scientific officer at aptaTargets. Dr. Ribó is an adviser at AptaTargets; a consultant at Medtronic; has ownership interest in Anaconda and NoraHealth; is a consultant for Cerenovus and Philips; and has stock options at Methink. Dr. Gorelick has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
preliminary results of a first-in-human study show.
The findings illustrate that it is possible to improve outcomes for stroke patients “not only with reperfusion therapy but with neuroprotectants,” study author Macarena Hernandez, PhD, associate professor, University Complutense, Madrid, told this news organization.
Dr. Hernandez said she hopes these positive results will spur investigation into other neuroprotective agents.
The findings were presented at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
Best doses
The study investigated ApTOLL, which blocks the TOLL-like receptor 4 (TLR4) that induces inflammation after a stroke. Previous studies found that ApTOLL protected brain tissue in animal models of stroke.
The phase 1B part of the study found no safety issues and determined the best two doses to be used in phase 2A were 0.05 mg/kg and 0.2 mg/kg.
The analysis included 139 patients at 14 centers in Spain and France (mean age, about 70 years; 42% women) who had a large-vessel occlusion and were eligible for endovascular therapy.
“Our aim was to have a very homogeneous population” to try to replicate in humans what had worked in animals, another study author, Marc Ribó, MD, interventional neurologist, Hospital Vall d’Hebron, Barcelona, told this news organization.
Study participants had an Alberta Stroke Program Early CT Score (ASPECTS) of 5-10, and estimated infarct core volume on CT-perfusion was 5-70 mL. All were treated within 6 hours of stroke onset.
Researchers randomly assigned patients to receive the low dose of the drug, the high dose of the drug, or placebo. The drug was administered intravenously over a 30-minute period just prior to the groin puncture for the thrombectomy procedure.
“So, the drug had already started to work when they underwent the usual standard practice, the thrombectomy,” said Dr. Ribó.
Those who were eligible also received tissue plasminogen activator.
The primary endpoint was safety, including death, symptomatic intracranial hemorrhage (SICH), and recurrent stroke.
Lower mortality
At 90 days, there was a statistically significant lower mortality rate in the high-dose group, compared with the group that received placebo (4.76% vs. 18.18%).
The mortality rate was 26.19% in the low-dose group, but Dr. Ribó stressed that this dose was a quarter of the higher dose and so performed “much more like placebo.”
The higher dose also yielded a better SICH outcome (4.76% of patients vs. 7.27% for placebo and 7.14% for the lower dose). And it was superior in terms of brain edema (2.4% of the population vs. 7.3% for the placebo and 4.8% for the low-dose groups).
About 7.1% of the high-dose group, 3.7% of the placebo group, and 4.8% of the low-dose group had a recurrent transient ischemic attack or stroke.
A secondary efficacy endpoint was infarct volume on MRI at 72 hours. Here, for the higher-dose group, mean infarct volume was reduced, compared with the patients who received placebo (–29.31 cc; 90% confidence interval, –49.28 to –9.34).
This higher dose was also superior for the secondary outcome of National Institutes of Health Stroke Scale score at 72 hours and for the disability outcome on the modified Rankin Score (mRS).
Clear shift in disability
“There was a clear shift toward less disability across levels of the mRS score in the high-dose group at 90 days,” said Dr. Ribó.
He added that he and his colleagues are “very happy” with these results, as they reflect “a consistency” of outcomes.
“We observed that the infarct volumes were lower in the high-dose group, and that led to a significant lower NIH score, meaning less clinical neurological symptoms at 72 hours, and finally, this led to less disability at 90 days.”
These results are “very exciting,” Dr. Hernandez added. “This is the first neuroprotectant that has demonstrated this acute effect in reducing deaths, in reducing the infarct volume and improving functionality long-term in patients treated with the higher dose.”
Dr. Ribó noted the treatment would eventually be used in addition to reperfusion therapy. “It’s not competing with reperfusion treatment; it’s an additional layer” of treatment.
Although it would initially be offered only to patients eligible for thrombectomy, researchers will explore the drug’s effectiveness for other stroke patients, said Dr. Ribó. “We wanted to secure this indication, and from there, progressively expand to other profiles of stroke patients, and even to patients with intracranial hemorrhage.”
The study confirmed the safety of the drug. “There were no safety issues at all,” said Dr. Ribó. “We were initially concerned that an anti-inflammatory in these patients could lead to higher rates of infections, but this was absolutely not the case.”
The next step is to confirm the effects in a larger, multicenter study, which is planned to launch at the end of this year, said Dr. Hernandez.
‘Very robust results’
In a comment, Philip B. Gorelick, MD, professor of neurology, Northwestern University, Chicago, said that, while this was a small early-phase study, the results are “very robust.”
“The authors demonstrated proof of a neuroprotective effect; they showed at 90 days that the death rates were substantially reduced by about four times – 4% vs. 18% – and the size of the damaged tissue at about 72 hours was reduced by 40%,” said Dr. Gorelick, who did not participate in the study.
He also noted that the disability was “less pronounced” at 90 days in the 0.2 mg/kg group.
“So overall, these are very encouraging results,” said Dr. Gorelick. “We have had a lot of difficulty finding neuroprotectant drugs that work, and this drug, in combination with endovascular therapy, seems to be very promising.”
However, he stressed the drug “is not ready for prime-time practice.”
“The proof in the pudding will be in the large-scale main phase 3 trials,” he added.
The study was funded by aptaTargets. Dr. Hernandez is chief scientific officer at aptaTargets. Dr. Ribó is an adviser at AptaTargets; a consultant at Medtronic; has ownership interest in Anaconda and NoraHealth; is a consultant for Cerenovus and Philips; and has stock options at Methink. Dr. Gorelick has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ISC 2023
Muscle-Related Adverse Events Associated With PCSK9 Inhibitors in a Veteran Population
HMG-CoA reductase inhibitors (statins) have been shown to effectively reduce low-density lipoprotein cholesterol (LDL-C) as well as morbidity and mortality in patients who have either atherosclerotic cardiovascular disease (ASCVD) or risk factors for ASCVD.1-12 However, research shows that up to 20% of patients are unable to tolerate statin therapy due to muscle-related adverse events (AEs).13 This presents a substantial clinical challenge, as current management strategies for patients with statin-associated muscle symptoms, such as intermittent administration of statins and ezetimibe, seldom achieve the > 50% LDL-C reduction recommended by the 2018 American Heart Association/American College of Cardiology Clinical Practice Guidelines.14 Additionally, statin-intolerant patients who have antihyperlipidemic medication lowered or discontinued are at an increased risk of future cardiovascular events.15 Observational data also show that about 70% of adult patients (primarily with genetic lipid disorders such as heterozygous familial hypercholesterolemia) do not achieve an LDL-C level < 100 mg/dL despite treatment with maximum doses of statins with or without ezetimibe.16,17
PCSK9 inhibitors (PCSK9i) have robust efficacy data to support use in patients who do not meet their LDL-C goal despite maximally tolerated lipid therapy.14 However, long-term safety data for PCSK9i are not as robust as its efficacy data. Specifically, safety data relating to muscle-related AEs, which are the most widely recognized AE associated with statins, have only been reported in a few clinical trials with varying incidence rates, levels of significance, and relatively small study populations. Furthermore, the real-world prevalence of muscle-related PCSK9i AEs is unknown. Clinical guidance for management strategies for muscle-related AEs associated with PCSK9i is largely lacking. For this study, muscle-related AEs were defined as any new or unusual muscle soreness, weakness, cramping, aches, and stiffness that persists, is generally bilateral, and typically affects the large muscles. It is important to note, that muscle-related AEs associated with statins, ezetimibe, and PCSK9i can be attributed to the nocebo effect.
According to the prescribing information for alirocumab and evolocumab, myalgia, muscle spasms, and musculoskeletal pain each occurred in < 5% of the study populations.18,19 From these data, muscle-related PCSK9i AEs are thought to be relatively rare, based on the ODYSSEY-OUTCOME and FOURIER trials, which did not enroll statin-intolerant patients.20,21 However, currently available safety data from 3 small, randomized clinical trials specifically in statin-intolerant patients taking a PCSK9i suggest that muscle-related AEs occur at a rate of 12.2% to 32.5% and discontinuation rates varied from 0% to 15.9%.22-25 As the incidence rates of muscle-related AEs in the prescribing information and clinical trials varied widely, this study will provide quantitative data on the percentage of patients that developed muscle-related PCSK9i AEs in a veteran population to help shed light on a topic that is not well studied.
Methods
This was a single-center, retrospective chart review of patients prescribed a PCSK9i between December 1, 2017, and September 1, 2021, and were managed in a pharmacy-led patient aligned care team (PACT) clinic at the Wilkes-Barre US Department of Veterans Affairs (VA) Medical Center (WBVAMC) in Pennsylvania. This study was approved by the Coatesville VA Medical Center Institutional Review Board, which oversees research conducted at WBVAMC. Veterans aged ≥ 18 years were included in the study. Patients were excluded if they had a history of serious hypersensitivity reaction to a PCSK9i or rhabdomyolysis or did not meet the VA criteria for use.26
The primary outcome was the percentage of patients who developed a muscle-related AE while on a PCSK9i in a PACT clinic. Data were further analyzed based on patients who (1) tolerated a full PCSK9i dose; (2) tolerated alternative PCSK9i following initial intolerance; (3) required a PCSK9i dose reduction, or (4) discontinued PCSK9i. A secondary outcome was the percentage of statin- and/or ezetimibe-intolerant patients in these 4 groups. Another secondary outcome was the management strategies taken for patients who were on a reduced (monthly) dose of PCSK9i who did not reach their LDL-C goal. Management strategies that were assessed included restarting weekly statin, restarting weekly ezetimibe, increasing the dose of the same PCSK9i administered monthly, and switching to an alternative PCSK9i.
Data were collected using the VA Computerized Patient Record System (CPRS) and stored in a secure, locked spreadsheet. Baseline patient demographic characteristics collected included age (at PCSK9i start); sex; race; and PCSK9i name, dose, and frequency. We recorded when a patient switched PCSK9i, whether or not it was due to a muscle-related AE, and the name of the original PCSK9i. Also collected were lipid therapy intolerances prior to PCSK9i initiation (ie, intolerance to statin, ezetimibe, or both).
Patients were considered statin intolerant due to a muscle-related AE in accordance with the VA PCSK9i Criteria for Use, which requires trial of at least 3 statins, one of which was trialed at the lowest dosage approved by the US Food and Drug Administration (FDA) and resulted in intolerable skeletal muscle AEs that worsened during treatment and resolved when the statin was stopped. For our study purposes, patients taking alternative day dosing of statins due to muscle-related AEs (ie, 2- or 3-times weekly dosing) were not considered statin intolerant; however, patients taking once-weekly statin dosing were considered statin intolerant. Patients were considered ezetimibe intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when ezetimibe was stopped. Patients were considered PCSK9i intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when the PCSK9i was stopped. Patients with non–muscle-related intolerances to statins, ezetimibe, and PCSK9i were not considered statin, ezetimibe, and PCSK9i intolerant.
Alirocumab was initiated at 75 mg subcutaneous (SQ) once every 2 weeks or evolocumab 140 mg SQ once every 2 weeks in our study. The protocol allowed for a dose reduction of alirocumab 75 mg SQ once monthly if a patient experienced AEs, but this dose reduction strategy was not used for any patients on evolocumab in this study. Of note, alirocumab 75 mg SQ once monthly is not an FDA-approved dosing strategy. However, it is similar in concept to the alternative statin dosing (ie, alternate day dosing, once-weekly dosing) and may avoid the need to discontinue PCSK9i therapy altogether.
A review of the CPRS also documented whether a muscle-related AE occurred while the patient was on a PCSK9i (if yes, the specific AE was recorded), the result of PCSK9i therapy (tolerated full dose, required a dose reduction, switched medication, or discontinued), and management strategies taken for patients who did not meet their LDL-C goal while on a reduced (monthly) PCSK9i dose. Prior lipid therapy intolerances, PCSK9i-related AEs, results of PCSK9i therapy, and management strategies for patients who did not meet LDL-C goal while on a reduced PCSK9i dose were obtained by reviewing the PACT pharmacist’s clinic notes and assessment, along with clinic notes and medication history listed within the CPRS.
Statistical Analysis
Descriptive statistics were used for the demographic characteristics of study patients. The primary outcome was calculated as a binary measure (yes/no) of whether the patient developed a muscle-related AE while on a PCSK9i. The secondary outcome of statin, ezetimibe, or statin and ezetimibe intolerances in subgroups also was calculated as a binary measure.
Results
For the study, 156 charts were reviewed and 137 patients were included (Figure). 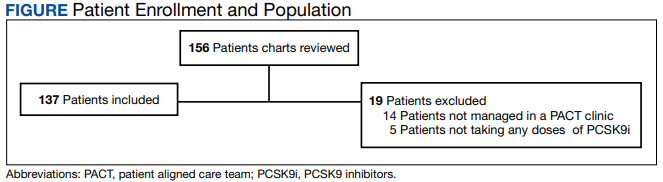
For the secondary results, 4 patients (2.9%) tolerated an alternate PCSK9i (evolocumab 140 mg SQ every 2 weeks) after initial intolerance to PCSK9i, 16 (11.7%) required a dose reduction, and 6 (4.4%) discontinued PCSK9i due to a muscle-related AE.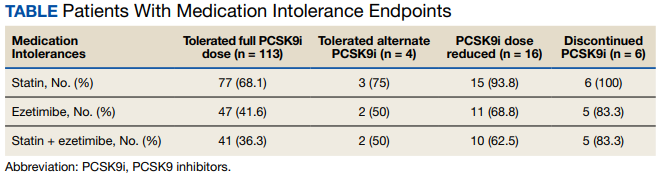
Statin intolerance was most common in all groups, followed by ezetimibe intolerance, and intolerance to statins + ezetimibe. Of the 113 patients who tolerated a full dose of PCSK9i, 77 (68.1%) had intolerance to statin, 47 (41.6%) to ezetimibe, and 41 (36.3%) to both statins and ezetimibe. Of the 6 patients who discontinued PCSK9i, all had intolerance to statins, 5 (83.3%) to ezetimibe, and 5 (83.3%) to statins and ezetimibe.
For patients who were on a reduced (monthly) dose of a PCSK9i who did not reach their LDL-C goal, we found that 16 patients (11.7%) required a PCSK9i dose reduction following muscle-related AEs. Of the patients who had their dose of PCSK9i reduced to monthly dosing, 5 (31%) met their LDL-C goal. For the 11 patients who did not meet their LDL-C goal, different management strategies were taken. Lifestyle modifications were made in 6 patients (54%), the monthly PCSK9i dose was increased to alirocumab 150 mg SQ monthly in 4 patients (36%), and 1 patient (9.1%) was switched to an alternative PCSK9i. There were no identified muscle-related AEs recorded in patients whose dose was increased to alirocumab 150 mg SQ monthly.
Discussion
This retrospective study found 17.5% of patients experienced muscle-related PCSK9i AEs. These occurred at a higher rate than reported in the prescribing information (< 5%) and were similar to the incidence rates reported in the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE clinical trials (12.0%-32.5%), which is what we hypothesized.18,19,22-25 It is important to note that the incidence rates of muscle-related AEs reported in the prescribing information for alirocumab and evolocumab were based on trials that did not include statin- and/or ezetimibe-intolerant patients; whereas many patients in our study and patients in the clinical trials were statin and/or ezetimibe intolerant.
Additionally, a new study by Donald and colleagues found an incidence rate of 32% to 36% for muscle-related PCSK9i AEs.27 Collectively, the data from clinical trials and our study indicate that patients with prior intolerances to statin and/or ezetimibe appear to have a higher likelihood of developing a muscle-related PCSK9i intolerance. In our study, 23 of 24 patients who developed a muscle-related PCSK9i AE had a prior history of statin and/or ezetimibe intolerances. This should alert clinicians prescribing PCSK9i in patients with a history of statin and/or ezetimibe intolerance to counsel their patients on the possibility of muscle-related PCSK9i AEs and management strategies. However, it is important to note that there was a substantial number of patients in our study who were statin and/or ezetimibe intolerant due to a prior muscle-related AE who tolerated the full dose of PCSK9i.
To our knowledge, this was the first trial to evaluate muscle-related PCSK9i AEs in a veteran population. Additionally, our study appears to be the first to use 2 PCSK9i dosing strategies that are not FDA approved: Dose reduction for patients who experienced a muscle-related AE on alirocumab 75 mg SQ every 2 weeks and dose escalation for patients who did not meet their LDL-C goal on alirocumab 75 mg SQ monthly following an initial intolerance to 2-week dosing. The dose-reduction strategy allowed patients who experienced a muscle-related AE to alirocumab 75 mg to reduce administration from every 2 weeks to monthly.
This strategy was only performed with alirocumab, the preferred PCSK9i at WBVAMC, but the same dose-reduction strategy can theoretically be used with evolocumab as well. Reduced monthly dosing of alirocumab allowed patients with a prior intolerance to remain on a lower dosage without discontinuation. This is important because as noted by Myers and colleagues, individuals without access to PCSK9i were found to have a significantly higher incidence ratio of cardiovascular events compared with those taking PCSK9i.15 Also of note, > 30% of patients on the reduced monthly dose of alirocumab still met their LDL-C goal. Therefore, using this dose-reduction strategy (instead of patients discontinuing therapy altogether due to a muscle-related intolerance) can lessen the risk of major adverse cardiovascular events (MACE) as well as mitigate muscle-related AEs that occurred while on 2-week PCSK9i dosing regimens. While we acknowledge that this reduced monthly dose of either alirocumab or evolocumab is not FDA approved, it is similar to alternative statin dosing that also is not FDA approved but may minimize the need to discontinue PCSK9i therapy. It would be beneficial if these dosing strategies were investigated by future research.
The dose-escalation strategy for patients who did not meet their LDL-C goal while on the reduced, monthly dose of alirocumab also was unique. Alirocumab was increased from 75 mg SQ once monthly to 150 mg SQ once monthly. Interestingly, we found that through the end of the chart review period, all patients tolerated the increase well, despite having an initial muscle-related AE to alirocumab 75 mg every 2 weeks, which is the same total monthly dosage. This approach is similar to that of once-weekly statin dosing or a drug holiday and may be explained by the long half-life of PCSK9i. Regardless of the mechanism, this finding suggests that an increased monthly dose of PCSK9i is a potential alternative for patients who cannot tolerate the FDA-approved dose. However, the ability for patients to achieve goal LDL-C on the monthly dosage requires future study.
In our study, only 6 patients (4.4%) discontinued PCSK9i therapy. This low discontinuation rate is largely attributable to our unique study design, which allowed for a dose reduction in patients who experienced muscle-related AEs. The earlier ODYSSEY-ALTERNATIVE trial evaluated the safety and efficacy of alirocumab compared with ezetimibe in confirmed statin-intolerant subjects after 24 weeks. This trial did not use a dose-reduction strategy and found 15.9% of patients discontinued alirocumab due to a muscle-related AE.24 This is notably higher than our discontinuation rate of 4.4%. If patients with a muscle-related AE discontinued PCKS9i instead of reducing the dose, they would likely return to their baseline LDL-C, which would increase the risk of MACE.
In general, myalgias due to antihyperlipidemic medications are not completely understood. One possible mechanism for statin-induced myalgias is the depletion of ubiquinone. However, this theory cannot explain muscle-related AEs associated with PCSK9i or ezetimibe, which have not been shown to deplete ubiquinone. We also found that the onset of muscle-related AEs associated with PCSK9i tends to appear later in therapy than what we know about statin therapy. Our study showed that the onset of a muscle-related PCSK9i AEs occurred a mean (SD) 8 (5.3) months after initiation (range, 1-19). Statin muscle-related AEs typically occur within the initial 4 to 8 weeks of treatment, although they can occur at any time.28
Limitations
The results of this study should be considered with the following limitations. First, this was a retrospective chart review performed over a prespecified period. Any muscle-related AEs or LDL-C lowering effects from PCSK9i that occurred outside the review period were not captured. Our study was small and only included 137 patients, though it was similar in size to the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE trials.22-24 Additionally, the study was primarily composed of White men and may not be representative of other populations. Some muscle-related PCSK9i AEs may be attributed to the nocebo. Last, our study did not capture patients on a PCSK9i who were not followed in the PACT clinic.
Conclusions
We found that muscle-related PCSK9i AEs occurred at a similar rate as those reported in previous clinical trials and exceeded the incidence rate reported in the prescribing information for alirocumab and evolocumab. It appears that patients who have a prior muscle-related intolerance to a statin and/or ezetimibe had a higher likelihood of developing a muscle-related PCSK9i AE. In our study, only 1 patient developed a muscle-related PCSK9i AE who did not have a prior history of muscle-related intolerance to either a statin or ezetimibe. However, in our study, a substantial percentage of patients with statin and/or ezetimibe intolerances tolerated the full PCSK9i dose well, proving that PCSK9i are still a reasonable alternative for patients with prior intolerances to statins and/or ezetimibe.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the US Department of Veterans Affairs Medical Center, Wilkes-Barre, Pennsylvania.
1. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383-1389.
2. Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335(14):1001-1009. doi:10.1056/NEJM199610033351401
3. Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339(19):1349-1357. doi:10.1056/NEJM199811053391902.
4. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7-22. doi:10.1016/S0140-6736(02)09327-3
5. Koren MJ, Hunninghake DB; ALLIANCE Investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics: the alliance study. J Am Coll Cardiol. 2004;44(9):1772-1779. doi:10.1016/j.jacc.2004.07.053
6. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622. doi:10.1001/jama.279.20.1615
7. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA. 2002;288(23):2998-3007. doi:10.1001/jama.288.23.2998
8. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158. doi:10.1016/S0140-6736(03)12948-0
9. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
10. Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet. 2006;368(9542):1155-1163. doi:10.1016/S0140-6736(06)69472-5
11. Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623-1630. doi:10.1016/s0140-6736(02)11600-x
12. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307. doi:10.1056/NEJM199511163332001

13. Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36(17):1012-1022. doi:10.1093/eurheartj/ehv043
14. Grundy SM, Stone NJ, Bailey AL, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24) e285-350. doi:10.1016/j.jacc.2018.11003
15. Myers KD, Farboodi N, Mwamburi M, et al. Effect of access to prescribed PCSK9 inhibitors on cardiovascular outcomes. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005404. doi:10.1161/CIRCOUTCOMES.118.005404
16. Wong ND, Chuang J, Zhao Y, Rosenblit PD. Residual dyslipidemia according to low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B among statin-treated US adults: National Health and Nutrition Examination Survey 2009-2010. J Clin Lipidol. 2015;9(4):525-532. doi:10.1016/j.jacl.2015.05.003
17. Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther. 2016;164:183-194. doi:10.1016/j.pharmthera.2016.04.011
18. Praluent (alirocumab) injection. Prescribing information. Regeneron Pharmaceuticals; 2021.
19. Repatha (evolocumab) injection. Prescribing information. Amgen; 2021.
20. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
21. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
22. Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541-2548. doi:10.1016/j.jacc.2014.03.019
23. Nissen SE, Stroes E, Dent-Acosta RE, et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: the GAUSS-3 randomized clinical trial. JAMA. 2016;315(15):1580-1590. doi:10.1001/jama.2016.3608
24. Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9(6):758-769. doi:10.1016/j.jacl.2015.08.006
25. Mesi O, Lin C, Ahmed H, Cho LS. Statin intolerance and new lipid-lowering treatments. Cleve Clin J Med. 2021;88(7):381-387. Published 2021 Jul 1. doi:10.3949/ccjm.88a.20165
26. US Department of Veterans Affairs. Clinical Guidance - Criteria For Use. September 2016. Accessed January 23, 2023. https://www.pbm.va.gov/clinicalguidance/criteriaforuse.asp
27. Donald DR, Reynolds VW, Hall N, DeClercq J, Choi L. Exploring rates of PCSK9 inhibitor persistence and reasons for treatment non-persistence in an integrated specialty pharmacy model. J Clin Lipidol. 2022;16(3):315-324. doi:10.1016/j.jacl.2022.03.004
28. Warden BA, Guyton JR, Kovacs AC, et al. Assessment and management of statin-associated muscle symptoms: A clinical perspective from the National Lipid Association. J Clin Lipidol. Published online September 10, 2022. doi:10.1016/j.jacl.2022.09.001
HMG-CoA reductase inhibitors (statins) have been shown to effectively reduce low-density lipoprotein cholesterol (LDL-C) as well as morbidity and mortality in patients who have either atherosclerotic cardiovascular disease (ASCVD) or risk factors for ASCVD.1-12 However, research shows that up to 20% of patients are unable to tolerate statin therapy due to muscle-related adverse events (AEs).13 This presents a substantial clinical challenge, as current management strategies for patients with statin-associated muscle symptoms, such as intermittent administration of statins and ezetimibe, seldom achieve the > 50% LDL-C reduction recommended by the 2018 American Heart Association/American College of Cardiology Clinical Practice Guidelines.14 Additionally, statin-intolerant patients who have antihyperlipidemic medication lowered or discontinued are at an increased risk of future cardiovascular events.15 Observational data also show that about 70% of adult patients (primarily with genetic lipid disorders such as heterozygous familial hypercholesterolemia) do not achieve an LDL-C level < 100 mg/dL despite treatment with maximum doses of statins with or without ezetimibe.16,17
PCSK9 inhibitors (PCSK9i) have robust efficacy data to support use in patients who do not meet their LDL-C goal despite maximally tolerated lipid therapy.14 However, long-term safety data for PCSK9i are not as robust as its efficacy data. Specifically, safety data relating to muscle-related AEs, which are the most widely recognized AE associated with statins, have only been reported in a few clinical trials with varying incidence rates, levels of significance, and relatively small study populations. Furthermore, the real-world prevalence of muscle-related PCSK9i AEs is unknown. Clinical guidance for management strategies for muscle-related AEs associated with PCSK9i is largely lacking. For this study, muscle-related AEs were defined as any new or unusual muscle soreness, weakness, cramping, aches, and stiffness that persists, is generally bilateral, and typically affects the large muscles. It is important to note, that muscle-related AEs associated with statins, ezetimibe, and PCSK9i can be attributed to the nocebo effect.
According to the prescribing information for alirocumab and evolocumab, myalgia, muscle spasms, and musculoskeletal pain each occurred in < 5% of the study populations.18,19 From these data, muscle-related PCSK9i AEs are thought to be relatively rare, based on the ODYSSEY-OUTCOME and FOURIER trials, which did not enroll statin-intolerant patients.20,21 However, currently available safety data from 3 small, randomized clinical trials specifically in statin-intolerant patients taking a PCSK9i suggest that muscle-related AEs occur at a rate of 12.2% to 32.5% and discontinuation rates varied from 0% to 15.9%.22-25 As the incidence rates of muscle-related AEs in the prescribing information and clinical trials varied widely, this study will provide quantitative data on the percentage of patients that developed muscle-related PCSK9i AEs in a veteran population to help shed light on a topic that is not well studied.
Methods
This was a single-center, retrospective chart review of patients prescribed a PCSK9i between December 1, 2017, and September 1, 2021, and were managed in a pharmacy-led patient aligned care team (PACT) clinic at the Wilkes-Barre US Department of Veterans Affairs (VA) Medical Center (WBVAMC) in Pennsylvania. This study was approved by the Coatesville VA Medical Center Institutional Review Board, which oversees research conducted at WBVAMC. Veterans aged ≥ 18 years were included in the study. Patients were excluded if they had a history of serious hypersensitivity reaction to a PCSK9i or rhabdomyolysis or did not meet the VA criteria for use.26
The primary outcome was the percentage of patients who developed a muscle-related AE while on a PCSK9i in a PACT clinic. Data were further analyzed based on patients who (1) tolerated a full PCSK9i dose; (2) tolerated alternative PCSK9i following initial intolerance; (3) required a PCSK9i dose reduction, or (4) discontinued PCSK9i. A secondary outcome was the percentage of statin- and/or ezetimibe-intolerant patients in these 4 groups. Another secondary outcome was the management strategies taken for patients who were on a reduced (monthly) dose of PCSK9i who did not reach their LDL-C goal. Management strategies that were assessed included restarting weekly statin, restarting weekly ezetimibe, increasing the dose of the same PCSK9i administered monthly, and switching to an alternative PCSK9i.
Data were collected using the VA Computerized Patient Record System (CPRS) and stored in a secure, locked spreadsheet. Baseline patient demographic characteristics collected included age (at PCSK9i start); sex; race; and PCSK9i name, dose, and frequency. We recorded when a patient switched PCSK9i, whether or not it was due to a muscle-related AE, and the name of the original PCSK9i. Also collected were lipid therapy intolerances prior to PCSK9i initiation (ie, intolerance to statin, ezetimibe, or both).
Patients were considered statin intolerant due to a muscle-related AE in accordance with the VA PCSK9i Criteria for Use, which requires trial of at least 3 statins, one of which was trialed at the lowest dosage approved by the US Food and Drug Administration (FDA) and resulted in intolerable skeletal muscle AEs that worsened during treatment and resolved when the statin was stopped. For our study purposes, patients taking alternative day dosing of statins due to muscle-related AEs (ie, 2- or 3-times weekly dosing) were not considered statin intolerant; however, patients taking once-weekly statin dosing were considered statin intolerant. Patients were considered ezetimibe intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when ezetimibe was stopped. Patients were considered PCSK9i intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when the PCSK9i was stopped. Patients with non–muscle-related intolerances to statins, ezetimibe, and PCSK9i were not considered statin, ezetimibe, and PCSK9i intolerant.
Alirocumab was initiated at 75 mg subcutaneous (SQ) once every 2 weeks or evolocumab 140 mg SQ once every 2 weeks in our study. The protocol allowed for a dose reduction of alirocumab 75 mg SQ once monthly if a patient experienced AEs, but this dose reduction strategy was not used for any patients on evolocumab in this study. Of note, alirocumab 75 mg SQ once monthly is not an FDA-approved dosing strategy. However, it is similar in concept to the alternative statin dosing (ie, alternate day dosing, once-weekly dosing) and may avoid the need to discontinue PCSK9i therapy altogether.
A review of the CPRS also documented whether a muscle-related AE occurred while the patient was on a PCSK9i (if yes, the specific AE was recorded), the result of PCSK9i therapy (tolerated full dose, required a dose reduction, switched medication, or discontinued), and management strategies taken for patients who did not meet their LDL-C goal while on a reduced (monthly) PCSK9i dose. Prior lipid therapy intolerances, PCSK9i-related AEs, results of PCSK9i therapy, and management strategies for patients who did not meet LDL-C goal while on a reduced PCSK9i dose were obtained by reviewing the PACT pharmacist’s clinic notes and assessment, along with clinic notes and medication history listed within the CPRS.
Statistical Analysis
Descriptive statistics were used for the demographic characteristics of study patients. The primary outcome was calculated as a binary measure (yes/no) of whether the patient developed a muscle-related AE while on a PCSK9i. The secondary outcome of statin, ezetimibe, or statin and ezetimibe intolerances in subgroups also was calculated as a binary measure.
Results
For the study, 156 charts were reviewed and 137 patients were included (Figure).
For the secondary results, 4 patients (2.9%) tolerated an alternate PCSK9i (evolocumab 140 mg SQ every 2 weeks) after initial intolerance to PCSK9i, 16 (11.7%) required a dose reduction, and 6 (4.4%) discontinued PCSK9i due to a muscle-related AE.
Statin intolerance was most common in all groups, followed by ezetimibe intolerance, and intolerance to statins + ezetimibe. Of the 113 patients who tolerated a full dose of PCSK9i, 77 (68.1%) had intolerance to statin, 47 (41.6%) to ezetimibe, and 41 (36.3%) to both statins and ezetimibe. Of the 6 patients who discontinued PCSK9i, all had intolerance to statins, 5 (83.3%) to ezetimibe, and 5 (83.3%) to statins and ezetimibe.
For patients who were on a reduced (monthly) dose of a PCSK9i who did not reach their LDL-C goal, we found that 16 patients (11.7%) required a PCSK9i dose reduction following muscle-related AEs. Of the patients who had their dose of PCSK9i reduced to monthly dosing, 5 (31%) met their LDL-C goal. For the 11 patients who did not meet their LDL-C goal, different management strategies were taken. Lifestyle modifications were made in 6 patients (54%), the monthly PCSK9i dose was increased to alirocumab 150 mg SQ monthly in 4 patients (36%), and 1 patient (9.1%) was switched to an alternative PCSK9i. There were no identified muscle-related AEs recorded in patients whose dose was increased to alirocumab 150 mg SQ monthly.
Discussion
This retrospective study found 17.5% of patients experienced muscle-related PCSK9i AEs. These occurred at a higher rate than reported in the prescribing information (< 5%) and were similar to the incidence rates reported in the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE clinical trials (12.0%-32.5%), which is what we hypothesized.18,19,22-25 It is important to note that the incidence rates of muscle-related AEs reported in the prescribing information for alirocumab and evolocumab were based on trials that did not include statin- and/or ezetimibe-intolerant patients; whereas many patients in our study and patients in the clinical trials were statin and/or ezetimibe intolerant.
Additionally, a new study by Donald and colleagues found an incidence rate of 32% to 36% for muscle-related PCSK9i AEs.27 Collectively, the data from clinical trials and our study indicate that patients with prior intolerances to statin and/or ezetimibe appear to have a higher likelihood of developing a muscle-related PCSK9i intolerance. In our study, 23 of 24 patients who developed a muscle-related PCSK9i AE had a prior history of statin and/or ezetimibe intolerances. This should alert clinicians prescribing PCSK9i in patients with a history of statin and/or ezetimibe intolerance to counsel their patients on the possibility of muscle-related PCSK9i AEs and management strategies. However, it is important to note that there was a substantial number of patients in our study who were statin and/or ezetimibe intolerant due to a prior muscle-related AE who tolerated the full dose of PCSK9i.
To our knowledge, this was the first trial to evaluate muscle-related PCSK9i AEs in a veteran population. Additionally, our study appears to be the first to use 2 PCSK9i dosing strategies that are not FDA approved: Dose reduction for patients who experienced a muscle-related AE on alirocumab 75 mg SQ every 2 weeks and dose escalation for patients who did not meet their LDL-C goal on alirocumab 75 mg SQ monthly following an initial intolerance to 2-week dosing. The dose-reduction strategy allowed patients who experienced a muscle-related AE to alirocumab 75 mg to reduce administration from every 2 weeks to monthly.
This strategy was only performed with alirocumab, the preferred PCSK9i at WBVAMC, but the same dose-reduction strategy can theoretically be used with evolocumab as well. Reduced monthly dosing of alirocumab allowed patients with a prior intolerance to remain on a lower dosage without discontinuation. This is important because as noted by Myers and colleagues, individuals without access to PCSK9i were found to have a significantly higher incidence ratio of cardiovascular events compared with those taking PCSK9i.15 Also of note, > 30% of patients on the reduced monthly dose of alirocumab still met their LDL-C goal. Therefore, using this dose-reduction strategy (instead of patients discontinuing therapy altogether due to a muscle-related intolerance) can lessen the risk of major adverse cardiovascular events (MACE) as well as mitigate muscle-related AEs that occurred while on 2-week PCSK9i dosing regimens. While we acknowledge that this reduced monthly dose of either alirocumab or evolocumab is not FDA approved, it is similar to alternative statin dosing that also is not FDA approved but may minimize the need to discontinue PCSK9i therapy. It would be beneficial if these dosing strategies were investigated by future research.
The dose-escalation strategy for patients who did not meet their LDL-C goal while on the reduced, monthly dose of alirocumab also was unique. Alirocumab was increased from 75 mg SQ once monthly to 150 mg SQ once monthly. Interestingly, we found that through the end of the chart review period, all patients tolerated the increase well, despite having an initial muscle-related AE to alirocumab 75 mg every 2 weeks, which is the same total monthly dosage. This approach is similar to that of once-weekly statin dosing or a drug holiday and may be explained by the long half-life of PCSK9i. Regardless of the mechanism, this finding suggests that an increased monthly dose of PCSK9i is a potential alternative for patients who cannot tolerate the FDA-approved dose. However, the ability for patients to achieve goal LDL-C on the monthly dosage requires future study.
In our study, only 6 patients (4.4%) discontinued PCSK9i therapy. This low discontinuation rate is largely attributable to our unique study design, which allowed for a dose reduction in patients who experienced muscle-related AEs. The earlier ODYSSEY-ALTERNATIVE trial evaluated the safety and efficacy of alirocumab compared with ezetimibe in confirmed statin-intolerant subjects after 24 weeks. This trial did not use a dose-reduction strategy and found 15.9% of patients discontinued alirocumab due to a muscle-related AE.24 This is notably higher than our discontinuation rate of 4.4%. If patients with a muscle-related AE discontinued PCKS9i instead of reducing the dose, they would likely return to their baseline LDL-C, which would increase the risk of MACE.
In general, myalgias due to antihyperlipidemic medications are not completely understood. One possible mechanism for statin-induced myalgias is the depletion of ubiquinone. However, this theory cannot explain muscle-related AEs associated with PCSK9i or ezetimibe, which have not been shown to deplete ubiquinone. We also found that the onset of muscle-related AEs associated with PCSK9i tends to appear later in therapy than what we know about statin therapy. Our study showed that the onset of a muscle-related PCSK9i AEs occurred a mean (SD) 8 (5.3) months after initiation (range, 1-19). Statin muscle-related AEs typically occur within the initial 4 to 8 weeks of treatment, although they can occur at any time.28
Limitations
The results of this study should be considered with the following limitations. First, this was a retrospective chart review performed over a prespecified period. Any muscle-related AEs or LDL-C lowering effects from PCSK9i that occurred outside the review period were not captured. Our study was small and only included 137 patients, though it was similar in size to the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE trials.22-24 Additionally, the study was primarily composed of White men and may not be representative of other populations. Some muscle-related PCSK9i AEs may be attributed to the nocebo. Last, our study did not capture patients on a PCSK9i who were not followed in the PACT clinic.
Conclusions
We found that muscle-related PCSK9i AEs occurred at a similar rate as those reported in previous clinical trials and exceeded the incidence rate reported in the prescribing information for alirocumab and evolocumab. It appears that patients who have a prior muscle-related intolerance to a statin and/or ezetimibe had a higher likelihood of developing a muscle-related PCSK9i AE. In our study, only 1 patient developed a muscle-related PCSK9i AE who did not have a prior history of muscle-related intolerance to either a statin or ezetimibe. However, in our study, a substantial percentage of patients with statin and/or ezetimibe intolerances tolerated the full PCSK9i dose well, proving that PCSK9i are still a reasonable alternative for patients with prior intolerances to statins and/or ezetimibe.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the US Department of Veterans Affairs Medical Center, Wilkes-Barre, Pennsylvania.
HMG-CoA reductase inhibitors (statins) have been shown to effectively reduce low-density lipoprotein cholesterol (LDL-C) as well as morbidity and mortality in patients who have either atherosclerotic cardiovascular disease (ASCVD) or risk factors for ASCVD.1-12 However, research shows that up to 20% of patients are unable to tolerate statin therapy due to muscle-related adverse events (AEs).13 This presents a substantial clinical challenge, as current management strategies for patients with statin-associated muscle symptoms, such as intermittent administration of statins and ezetimibe, seldom achieve the > 50% LDL-C reduction recommended by the 2018 American Heart Association/American College of Cardiology Clinical Practice Guidelines.14 Additionally, statin-intolerant patients who have antihyperlipidemic medication lowered or discontinued are at an increased risk of future cardiovascular events.15 Observational data also show that about 70% of adult patients (primarily with genetic lipid disorders such as heterozygous familial hypercholesterolemia) do not achieve an LDL-C level < 100 mg/dL despite treatment with maximum doses of statins with or without ezetimibe.16,17
PCSK9 inhibitors (PCSK9i) have robust efficacy data to support use in patients who do not meet their LDL-C goal despite maximally tolerated lipid therapy.14 However, long-term safety data for PCSK9i are not as robust as its efficacy data. Specifically, safety data relating to muscle-related AEs, which are the most widely recognized AE associated with statins, have only been reported in a few clinical trials with varying incidence rates, levels of significance, and relatively small study populations. Furthermore, the real-world prevalence of muscle-related PCSK9i AEs is unknown. Clinical guidance for management strategies for muscle-related AEs associated with PCSK9i is largely lacking. For this study, muscle-related AEs were defined as any new or unusual muscle soreness, weakness, cramping, aches, and stiffness that persists, is generally bilateral, and typically affects the large muscles. It is important to note, that muscle-related AEs associated with statins, ezetimibe, and PCSK9i can be attributed to the nocebo effect.
According to the prescribing information for alirocumab and evolocumab, myalgia, muscle spasms, and musculoskeletal pain each occurred in < 5% of the study populations.18,19 From these data, muscle-related PCSK9i AEs are thought to be relatively rare, based on the ODYSSEY-OUTCOME and FOURIER trials, which did not enroll statin-intolerant patients.20,21 However, currently available safety data from 3 small, randomized clinical trials specifically in statin-intolerant patients taking a PCSK9i suggest that muscle-related AEs occur at a rate of 12.2% to 32.5% and discontinuation rates varied from 0% to 15.9%.22-25 As the incidence rates of muscle-related AEs in the prescribing information and clinical trials varied widely, this study will provide quantitative data on the percentage of patients that developed muscle-related PCSK9i AEs in a veteran population to help shed light on a topic that is not well studied.
Methods
This was a single-center, retrospective chart review of patients prescribed a PCSK9i between December 1, 2017, and September 1, 2021, and were managed in a pharmacy-led patient aligned care team (PACT) clinic at the Wilkes-Barre US Department of Veterans Affairs (VA) Medical Center (WBVAMC) in Pennsylvania. This study was approved by the Coatesville VA Medical Center Institutional Review Board, which oversees research conducted at WBVAMC. Veterans aged ≥ 18 years were included in the study. Patients were excluded if they had a history of serious hypersensitivity reaction to a PCSK9i or rhabdomyolysis or did not meet the VA criteria for use.26
The primary outcome was the percentage of patients who developed a muscle-related AE while on a PCSK9i in a PACT clinic. Data were further analyzed based on patients who (1) tolerated a full PCSK9i dose; (2) tolerated alternative PCSK9i following initial intolerance; (3) required a PCSK9i dose reduction, or (4) discontinued PCSK9i. A secondary outcome was the percentage of statin- and/or ezetimibe-intolerant patients in these 4 groups. Another secondary outcome was the management strategies taken for patients who were on a reduced (monthly) dose of PCSK9i who did not reach their LDL-C goal. Management strategies that were assessed included restarting weekly statin, restarting weekly ezetimibe, increasing the dose of the same PCSK9i administered monthly, and switching to an alternative PCSK9i.
Data were collected using the VA Computerized Patient Record System (CPRS) and stored in a secure, locked spreadsheet. Baseline patient demographic characteristics collected included age (at PCSK9i start); sex; race; and PCSK9i name, dose, and frequency. We recorded when a patient switched PCSK9i, whether or not it was due to a muscle-related AE, and the name of the original PCSK9i. Also collected were lipid therapy intolerances prior to PCSK9i initiation (ie, intolerance to statin, ezetimibe, or both).
Patients were considered statin intolerant due to a muscle-related AE in accordance with the VA PCSK9i Criteria for Use, which requires trial of at least 3 statins, one of which was trialed at the lowest dosage approved by the US Food and Drug Administration (FDA) and resulted in intolerable skeletal muscle AEs that worsened during treatment and resolved when the statin was stopped. For our study purposes, patients taking alternative day dosing of statins due to muscle-related AEs (ie, 2- or 3-times weekly dosing) were not considered statin intolerant; however, patients taking once-weekly statin dosing were considered statin intolerant. Patients were considered ezetimibe intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when ezetimibe was stopped. Patients were considered PCSK9i intolerant due to a muscle-related AE if the intolerance was due to skeletal muscle concerns that worsened during treatment and resolved when the PCSK9i was stopped. Patients with non–muscle-related intolerances to statins, ezetimibe, and PCSK9i were not considered statin, ezetimibe, and PCSK9i intolerant.
Alirocumab was initiated at 75 mg subcutaneous (SQ) once every 2 weeks or evolocumab 140 mg SQ once every 2 weeks in our study. The protocol allowed for a dose reduction of alirocumab 75 mg SQ once monthly if a patient experienced AEs, but this dose reduction strategy was not used for any patients on evolocumab in this study. Of note, alirocumab 75 mg SQ once monthly is not an FDA-approved dosing strategy. However, it is similar in concept to the alternative statin dosing (ie, alternate day dosing, once-weekly dosing) and may avoid the need to discontinue PCSK9i therapy altogether.
A review of the CPRS also documented whether a muscle-related AE occurred while the patient was on a PCSK9i (if yes, the specific AE was recorded), the result of PCSK9i therapy (tolerated full dose, required a dose reduction, switched medication, or discontinued), and management strategies taken for patients who did not meet their LDL-C goal while on a reduced (monthly) PCSK9i dose. Prior lipid therapy intolerances, PCSK9i-related AEs, results of PCSK9i therapy, and management strategies for patients who did not meet LDL-C goal while on a reduced PCSK9i dose were obtained by reviewing the PACT pharmacist’s clinic notes and assessment, along with clinic notes and medication history listed within the CPRS.
Statistical Analysis
Descriptive statistics were used for the demographic characteristics of study patients. The primary outcome was calculated as a binary measure (yes/no) of whether the patient developed a muscle-related AE while on a PCSK9i. The secondary outcome of statin, ezetimibe, or statin and ezetimibe intolerances in subgroups also was calculated as a binary measure.
Results
For the study, 156 charts were reviewed and 137 patients were included (Figure).
For the secondary results, 4 patients (2.9%) tolerated an alternate PCSK9i (evolocumab 140 mg SQ every 2 weeks) after initial intolerance to PCSK9i, 16 (11.7%) required a dose reduction, and 6 (4.4%) discontinued PCSK9i due to a muscle-related AE.
Statin intolerance was most common in all groups, followed by ezetimibe intolerance, and intolerance to statins + ezetimibe. Of the 113 patients who tolerated a full dose of PCSK9i, 77 (68.1%) had intolerance to statin, 47 (41.6%) to ezetimibe, and 41 (36.3%) to both statins and ezetimibe. Of the 6 patients who discontinued PCSK9i, all had intolerance to statins, 5 (83.3%) to ezetimibe, and 5 (83.3%) to statins and ezetimibe.
For patients who were on a reduced (monthly) dose of a PCSK9i who did not reach their LDL-C goal, we found that 16 patients (11.7%) required a PCSK9i dose reduction following muscle-related AEs. Of the patients who had their dose of PCSK9i reduced to monthly dosing, 5 (31%) met their LDL-C goal. For the 11 patients who did not meet their LDL-C goal, different management strategies were taken. Lifestyle modifications were made in 6 patients (54%), the monthly PCSK9i dose was increased to alirocumab 150 mg SQ monthly in 4 patients (36%), and 1 patient (9.1%) was switched to an alternative PCSK9i. There were no identified muscle-related AEs recorded in patients whose dose was increased to alirocumab 150 mg SQ monthly.
Discussion
This retrospective study found 17.5% of patients experienced muscle-related PCSK9i AEs. These occurred at a higher rate than reported in the prescribing information (< 5%) and were similar to the incidence rates reported in the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE clinical trials (12.0%-32.5%), which is what we hypothesized.18,19,22-25 It is important to note that the incidence rates of muscle-related AEs reported in the prescribing information for alirocumab and evolocumab were based on trials that did not include statin- and/or ezetimibe-intolerant patients; whereas many patients in our study and patients in the clinical trials were statin and/or ezetimibe intolerant.
Additionally, a new study by Donald and colleagues found an incidence rate of 32% to 36% for muscle-related PCSK9i AEs.27 Collectively, the data from clinical trials and our study indicate that patients with prior intolerances to statin and/or ezetimibe appear to have a higher likelihood of developing a muscle-related PCSK9i intolerance. In our study, 23 of 24 patients who developed a muscle-related PCSK9i AE had a prior history of statin and/or ezetimibe intolerances. This should alert clinicians prescribing PCSK9i in patients with a history of statin and/or ezetimibe intolerance to counsel their patients on the possibility of muscle-related PCSK9i AEs and management strategies. However, it is important to note that there was a substantial number of patients in our study who were statin and/or ezetimibe intolerant due to a prior muscle-related AE who tolerated the full dose of PCSK9i.
To our knowledge, this was the first trial to evaluate muscle-related PCSK9i AEs in a veteran population. Additionally, our study appears to be the first to use 2 PCSK9i dosing strategies that are not FDA approved: Dose reduction for patients who experienced a muscle-related AE on alirocumab 75 mg SQ every 2 weeks and dose escalation for patients who did not meet their LDL-C goal on alirocumab 75 mg SQ monthly following an initial intolerance to 2-week dosing. The dose-reduction strategy allowed patients who experienced a muscle-related AE to alirocumab 75 mg to reduce administration from every 2 weeks to monthly.
This strategy was only performed with alirocumab, the preferred PCSK9i at WBVAMC, but the same dose-reduction strategy can theoretically be used with evolocumab as well. Reduced monthly dosing of alirocumab allowed patients with a prior intolerance to remain on a lower dosage without discontinuation. This is important because as noted by Myers and colleagues, individuals without access to PCSK9i were found to have a significantly higher incidence ratio of cardiovascular events compared with those taking PCSK9i.15 Also of note, > 30% of patients on the reduced monthly dose of alirocumab still met their LDL-C goal. Therefore, using this dose-reduction strategy (instead of patients discontinuing therapy altogether due to a muscle-related intolerance) can lessen the risk of major adverse cardiovascular events (MACE) as well as mitigate muscle-related AEs that occurred while on 2-week PCSK9i dosing regimens. While we acknowledge that this reduced monthly dose of either alirocumab or evolocumab is not FDA approved, it is similar to alternative statin dosing that also is not FDA approved but may minimize the need to discontinue PCSK9i therapy. It would be beneficial if these dosing strategies were investigated by future research.
The dose-escalation strategy for patients who did not meet their LDL-C goal while on the reduced, monthly dose of alirocumab also was unique. Alirocumab was increased from 75 mg SQ once monthly to 150 mg SQ once monthly. Interestingly, we found that through the end of the chart review period, all patients tolerated the increase well, despite having an initial muscle-related AE to alirocumab 75 mg every 2 weeks, which is the same total monthly dosage. This approach is similar to that of once-weekly statin dosing or a drug holiday and may be explained by the long half-life of PCSK9i. Regardless of the mechanism, this finding suggests that an increased monthly dose of PCSK9i is a potential alternative for patients who cannot tolerate the FDA-approved dose. However, the ability for patients to achieve goal LDL-C on the monthly dosage requires future study.
In our study, only 6 patients (4.4%) discontinued PCSK9i therapy. This low discontinuation rate is largely attributable to our unique study design, which allowed for a dose reduction in patients who experienced muscle-related AEs. The earlier ODYSSEY-ALTERNATIVE trial evaluated the safety and efficacy of alirocumab compared with ezetimibe in confirmed statin-intolerant subjects after 24 weeks. This trial did not use a dose-reduction strategy and found 15.9% of patients discontinued alirocumab due to a muscle-related AE.24 This is notably higher than our discontinuation rate of 4.4%. If patients with a muscle-related AE discontinued PCKS9i instead of reducing the dose, they would likely return to their baseline LDL-C, which would increase the risk of MACE.
In general, myalgias due to antihyperlipidemic medications are not completely understood. One possible mechanism for statin-induced myalgias is the depletion of ubiquinone. However, this theory cannot explain muscle-related AEs associated with PCSK9i or ezetimibe, which have not been shown to deplete ubiquinone. We also found that the onset of muscle-related AEs associated with PCSK9i tends to appear later in therapy than what we know about statin therapy. Our study showed that the onset of a muscle-related PCSK9i AEs occurred a mean (SD) 8 (5.3) months after initiation (range, 1-19). Statin muscle-related AEs typically occur within the initial 4 to 8 weeks of treatment, although they can occur at any time.28
Limitations
The results of this study should be considered with the following limitations. First, this was a retrospective chart review performed over a prespecified period. Any muscle-related AEs or LDL-C lowering effects from PCSK9i that occurred outside the review period were not captured. Our study was small and only included 137 patients, though it was similar in size to the GAUSS-2, GAUSS-3, and ODYSSEY-ALTERNATIVE trials.22-24 Additionally, the study was primarily composed of White men and may not be representative of other populations. Some muscle-related PCSK9i AEs may be attributed to the nocebo. Last, our study did not capture patients on a PCSK9i who were not followed in the PACT clinic.
Conclusions
We found that muscle-related PCSK9i AEs occurred at a similar rate as those reported in previous clinical trials and exceeded the incidence rate reported in the prescribing information for alirocumab and evolocumab. It appears that patients who have a prior muscle-related intolerance to a statin and/or ezetimibe had a higher likelihood of developing a muscle-related PCSK9i AE. In our study, only 1 patient developed a muscle-related PCSK9i AE who did not have a prior history of muscle-related intolerance to either a statin or ezetimibe. However, in our study, a substantial percentage of patients with statin and/or ezetimibe intolerances tolerated the full PCSK9i dose well, proving that PCSK9i are still a reasonable alternative for patients with prior intolerances to statins and/or ezetimibe.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the US Department of Veterans Affairs Medical Center, Wilkes-Barre, Pennsylvania.
1. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383-1389.
2. Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335(14):1001-1009. doi:10.1056/NEJM199610033351401
3. Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339(19):1349-1357. doi:10.1056/NEJM199811053391902.
4. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7-22. doi:10.1016/S0140-6736(02)09327-3
5. Koren MJ, Hunninghake DB; ALLIANCE Investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics: the alliance study. J Am Coll Cardiol. 2004;44(9):1772-1779. doi:10.1016/j.jacc.2004.07.053
6. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622. doi:10.1001/jama.279.20.1615
7. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA. 2002;288(23):2998-3007. doi:10.1001/jama.288.23.2998
8. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158. doi:10.1016/S0140-6736(03)12948-0
9. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
10. Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet. 2006;368(9542):1155-1163. doi:10.1016/S0140-6736(06)69472-5
11. Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623-1630. doi:10.1016/s0140-6736(02)11600-x
12. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307. doi:10.1056/NEJM199511163332001
13. Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36(17):1012-1022. doi:10.1093/eurheartj/ehv043
14. Grundy SM, Stone NJ, Bailey AL, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24) e285-350. doi:10.1016/j.jacc.2018.11003
15. Myers KD, Farboodi N, Mwamburi M, et al. Effect of access to prescribed PCSK9 inhibitors on cardiovascular outcomes. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005404. doi:10.1161/CIRCOUTCOMES.118.005404
16. Wong ND, Chuang J, Zhao Y, Rosenblit PD. Residual dyslipidemia according to low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B among statin-treated US adults: National Health and Nutrition Examination Survey 2009-2010. J Clin Lipidol. 2015;9(4):525-532. doi:10.1016/j.jacl.2015.05.003
17. Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther. 2016;164:183-194. doi:10.1016/j.pharmthera.2016.04.011
18. Praluent (alirocumab) injection. Prescribing information. Regeneron Pharmaceuticals; 2021.
19. Repatha (evolocumab) injection. Prescribing information. Amgen; 2021.
20. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
21. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
22. Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541-2548. doi:10.1016/j.jacc.2014.03.019
23. Nissen SE, Stroes E, Dent-Acosta RE, et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: the GAUSS-3 randomized clinical trial. JAMA. 2016;315(15):1580-1590. doi:10.1001/jama.2016.3608
24. Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9(6):758-769. doi:10.1016/j.jacl.2015.08.006
25. Mesi O, Lin C, Ahmed H, Cho LS. Statin intolerance and new lipid-lowering treatments. Cleve Clin J Med. 2021;88(7):381-387. Published 2021 Jul 1. doi:10.3949/ccjm.88a.20165
26. US Department of Veterans Affairs. Clinical Guidance - Criteria For Use. September 2016. Accessed January 23, 2023. https://www.pbm.va.gov/clinicalguidance/criteriaforuse.asp
27. Donald DR, Reynolds VW, Hall N, DeClercq J, Choi L. Exploring rates of PCSK9 inhibitor persistence and reasons for treatment non-persistence in an integrated specialty pharmacy model. J Clin Lipidol. 2022;16(3):315-324. doi:10.1016/j.jacl.2022.03.004
28. Warden BA, Guyton JR, Kovacs AC, et al. Assessment and management of statin-associated muscle symptoms: A clinical perspective from the National Lipid Association. J Clin Lipidol. Published online September 10, 2022. doi:10.1016/j.jacl.2022.09.001
1. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383-1389.
2. Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335(14):1001-1009. doi:10.1056/NEJM199610033351401
3. Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339(19):1349-1357. doi:10.1056/NEJM199811053391902.
4. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):7-22. doi:10.1016/S0140-6736(02)09327-3
5. Koren MJ, Hunninghake DB; ALLIANCE Investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics: the alliance study. J Am Coll Cardiol. 2004;44(9):1772-1779. doi:10.1016/j.jacc.2004.07.053
6. Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279(20):1615-1622. doi:10.1001/jama.279.20.1615
7. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA. 2002;288(23):2998-3007. doi:10.1001/jama.288.23.2998
8. Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158. doi:10.1016/S0140-6736(03)12948-0
9. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
10. Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet. 2006;368(9542):1155-1163. doi:10.1016/S0140-6736(06)69472-5
11. Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623-1630. doi:10.1016/s0140-6736(02)11600-x
12. Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301-1307. doi:10.1056/NEJM199511163332001
13. Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J. 2015;36(17):1012-1022. doi:10.1093/eurheartj/ehv043
14. Grundy SM, Stone NJ, Bailey AL, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24) e285-350. doi:10.1016/j.jacc.2018.11003
15. Myers KD, Farboodi N, Mwamburi M, et al. Effect of access to prescribed PCSK9 inhibitors on cardiovascular outcomes. Circ Cardiovasc Qual Outcomes. 2019;12(8):e005404. doi:10.1161/CIRCOUTCOMES.118.005404
16. Wong ND, Chuang J, Zhao Y, Rosenblit PD. Residual dyslipidemia according to low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B among statin-treated US adults: National Health and Nutrition Examination Survey 2009-2010. J Clin Lipidol. 2015;9(4):525-532. doi:10.1016/j.jacl.2015.05.003
17. Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther. 2016;164:183-194. doi:10.1016/j.pharmthera.2016.04.011
18. Praluent (alirocumab) injection. Prescribing information. Regeneron Pharmaceuticals; 2021.
19. Repatha (evolocumab) injection. Prescribing information. Amgen; 2021.
20. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
21. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
22. Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541-2548. doi:10.1016/j.jacc.2014.03.019
23. Nissen SE, Stroes E, Dent-Acosta RE, et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: the GAUSS-3 randomized clinical trial. JAMA. 2016;315(15):1580-1590. doi:10.1001/jama.2016.3608
24. Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9(6):758-769. doi:10.1016/j.jacl.2015.08.006
25. Mesi O, Lin C, Ahmed H, Cho LS. Statin intolerance and new lipid-lowering treatments. Cleve Clin J Med. 2021;88(7):381-387. Published 2021 Jul 1. doi:10.3949/ccjm.88a.20165
26. US Department of Veterans Affairs. Clinical Guidance - Criteria For Use. September 2016. Accessed January 23, 2023. https://www.pbm.va.gov/clinicalguidance/criteriaforuse.asp
27. Donald DR, Reynolds VW, Hall N, DeClercq J, Choi L. Exploring rates of PCSK9 inhibitor persistence and reasons for treatment non-persistence in an integrated specialty pharmacy model. J Clin Lipidol. 2022;16(3):315-324. doi:10.1016/j.jacl.2022.03.004
28. Warden BA, Guyton JR, Kovacs AC, et al. Assessment and management of statin-associated muscle symptoms: A clinical perspective from the National Lipid Association. J Clin Lipidol. Published online September 10, 2022. doi:10.1016/j.jacl.2022.09.001
No spike in overdose deaths from relaxed buprenorphine regulations
Researchers say the data add weight to the argument for permanently adopting the pandemic-era prescribing regulations for buprenorphine, a treatment for opioid use disorder.
“We saw no evidence that increased availability of buprenorphine through the loosening of rules around prescribing and dispensing of buprenorphine during the pandemic increased overdose deaths,” investigator Wilson Compton, MD, deputy director of the National Institute on Drug Abuse, told this news organization.
“This is reassuring that, even when we opened up the doors to easier access to buprenorphine, we didn’t see that most serious consequence,” Dr. Compton said.
The findings were published online in JAMA Network Open .
Cause and effect
Federal agencies relaxed prescribing regulations for buprenorphine in March 2020 to make it easier for clinicians to prescribe the drug via telemedicine and for patients to take the medication at home.
The number of buprenorphine prescriptions has increased since that change, with more than 1 million people receiving the medication in 2021 from retail pharmacies in the United States.
However, questions remained about whether increased access would lead to an increase in buprenorphine-involved overdose.
Researchers with NIDA and the Centers for Disease Control and Prevention analyzed data from the State Unintentional Drug Overdose Reporting System, a CDC database that combines medical examiner and coroner reports and postmortem toxicology testing.
The study included information about overdose deaths from July 2019 to June 2021 in 46 states and the District of Columbia.
Between July 2019 and June 2021, there were 1,955 buprenorphine-involved overdose deaths, which accounted for 2.2% of all drug overdose deaths and 2.6% of opioid-involved overdose deaths.
However, researchers went beyond overall numbers and evaluated details from coroner’s and medical examiner reports, something they had not done before.
“For the first time we looked at the characteristics of decedents from buprenorphine because this has not been studied in this type of detail with a near-national sample,” Dr. Compton said.
“That allowed us to look at patterns of use of other substances as well as the circumstances that are recorded at the death scene that are in the data set,” he added.
Important insights
Reports from nearly all buprenorphine-involved deaths included the presence of at least one other drug, compared with opioid overdose deaths that typically involved only one drug.
“This is consistent with the pharmacology of buprenorphine being a partial agonist, so it may not be as fatal all by itself as some of the other opioids,” Dr. Compton said.
Deaths involving buprenorphine were less likely to include illicitly manufactured fentanyls, and other prescription medications were more often found on the scene, such as antidepressants.
Compared with opioid decedents, buprenorphine decedents were more likely to be women, age 35-44, White, and receiving treatment for mental health conditions, including for substance use disorder (SUD).
These kinds of characteristics provide important insights about potential ways to improve safety and clinical outcomes, Dr. Compton noted.
“When we see things like a little higher rate of SUD treatment and this evidence of other prescription drugs on the scene, and some higher rates of antidepressants in these decedents than I might have expected, I’m very curious about their use of other medical services outside of substance use treatment, because that might be a place where some interventions could be implemented,” he said.
A similar study showed pandemic-era policy changes that allowed methadone to be taken at home was followed by a decrease in methadone-related overdose deaths.
The new findings are consistent with those results, Dr. Compton said.
‘Chipping away’ at stigma
Commenting on the study, O. Trent Hall, DO, assistant professor of addiction medicine, Department of Psychiatry and Behavioral Health, Ohio State University Wexner Medical Center, Columbus, said that, although he welcomed the findings, they aren’t unexpected.
“Buprenorphine is well established as a safe and effective medication for opioid use disorder and as a physician who routinely cares for patients in the hospital after opioid overdose, I am not at all surprised by these results,” said Dr. Hall, who was not involved with the research.
“When my patients leave the hospital with a buprenorphine prescription, they are much less likely to return with another overdose or serious opioid-related medical problem,” he added.
U.S. drug overdose deaths topped 100,000 for the first time in 2021, and most were opioid-related. Although the latest data from the CDC shows drug overdose deaths have been declining slowly since early 2022, the numbers remain high.
Buprenorphine is one of only two drugs known to reduce the risk of opioid overdose. While prescriptions have increased since 2020, the medication remains underutilized, despite its known effectiveness in treating opioid use disorder.
Dr. Hall noted that research such as the new study could help increase buprenorphine’s use.
“Studies like this one chip away at the stigma that has been misapplied to buprenorphine,” he said. “I hope this article will encourage more providers to offer buprenorphine to patients with opioid use disorder.”
The study was funded internally by NIDA and the CDC. Dr. Compton reported owning stock in General Electric, 3M, and Pfizer outside the submitted work. Dr. Hall has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers say the data add weight to the argument for permanently adopting the pandemic-era prescribing regulations for buprenorphine, a treatment for opioid use disorder.
“We saw no evidence that increased availability of buprenorphine through the loosening of rules around prescribing and dispensing of buprenorphine during the pandemic increased overdose deaths,” investigator Wilson Compton, MD, deputy director of the National Institute on Drug Abuse, told this news organization.
“This is reassuring that, even when we opened up the doors to easier access to buprenorphine, we didn’t see that most serious consequence,” Dr. Compton said.
The findings were published online in JAMA Network Open .
Cause and effect
Federal agencies relaxed prescribing regulations for buprenorphine in March 2020 to make it easier for clinicians to prescribe the drug via telemedicine and for patients to take the medication at home.
The number of buprenorphine prescriptions has increased since that change, with more than 1 million people receiving the medication in 2021 from retail pharmacies in the United States.
However, questions remained about whether increased access would lead to an increase in buprenorphine-involved overdose.
Researchers with NIDA and the Centers for Disease Control and Prevention analyzed data from the State Unintentional Drug Overdose Reporting System, a CDC database that combines medical examiner and coroner reports and postmortem toxicology testing.
The study included information about overdose deaths from July 2019 to June 2021 in 46 states and the District of Columbia.
Between July 2019 and June 2021, there were 1,955 buprenorphine-involved overdose deaths, which accounted for 2.2% of all drug overdose deaths and 2.6% of opioid-involved overdose deaths.
However, researchers went beyond overall numbers and evaluated details from coroner’s and medical examiner reports, something they had not done before.
“For the first time we looked at the characteristics of decedents from buprenorphine because this has not been studied in this type of detail with a near-national sample,” Dr. Compton said.
“That allowed us to look at patterns of use of other substances as well as the circumstances that are recorded at the death scene that are in the data set,” he added.
Important insights
Reports from nearly all buprenorphine-involved deaths included the presence of at least one other drug, compared with opioid overdose deaths that typically involved only one drug.
“This is consistent with the pharmacology of buprenorphine being a partial agonist, so it may not be as fatal all by itself as some of the other opioids,” Dr. Compton said.
Deaths involving buprenorphine were less likely to include illicitly manufactured fentanyls, and other prescription medications were more often found on the scene, such as antidepressants.
Compared with opioid decedents, buprenorphine decedents were more likely to be women, age 35-44, White, and receiving treatment for mental health conditions, including for substance use disorder (SUD).
These kinds of characteristics provide important insights about potential ways to improve safety and clinical outcomes, Dr. Compton noted.
“When we see things like a little higher rate of SUD treatment and this evidence of other prescription drugs on the scene, and some higher rates of antidepressants in these decedents than I might have expected, I’m very curious about their use of other medical services outside of substance use treatment, because that might be a place where some interventions could be implemented,” he said.
A similar study showed pandemic-era policy changes that allowed methadone to be taken at home was followed by a decrease in methadone-related overdose deaths.
The new findings are consistent with those results, Dr. Compton said.
‘Chipping away’ at stigma
Commenting on the study, O. Trent Hall, DO, assistant professor of addiction medicine, Department of Psychiatry and Behavioral Health, Ohio State University Wexner Medical Center, Columbus, said that, although he welcomed the findings, they aren’t unexpected.
“Buprenorphine is well established as a safe and effective medication for opioid use disorder and as a physician who routinely cares for patients in the hospital after opioid overdose, I am not at all surprised by these results,” said Dr. Hall, who was not involved with the research.
“When my patients leave the hospital with a buprenorphine prescription, they are much less likely to return with another overdose or serious opioid-related medical problem,” he added.
U.S. drug overdose deaths topped 100,000 for the first time in 2021, and most were opioid-related. Although the latest data from the CDC shows drug overdose deaths have been declining slowly since early 2022, the numbers remain high.
Buprenorphine is one of only two drugs known to reduce the risk of opioid overdose. While prescriptions have increased since 2020, the medication remains underutilized, despite its known effectiveness in treating opioid use disorder.
Dr. Hall noted that research such as the new study could help increase buprenorphine’s use.
“Studies like this one chip away at the stigma that has been misapplied to buprenorphine,” he said. “I hope this article will encourage more providers to offer buprenorphine to patients with opioid use disorder.”
The study was funded internally by NIDA and the CDC. Dr. Compton reported owning stock in General Electric, 3M, and Pfizer outside the submitted work. Dr. Hall has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers say the data add weight to the argument for permanently adopting the pandemic-era prescribing regulations for buprenorphine, a treatment for opioid use disorder.
“We saw no evidence that increased availability of buprenorphine through the loosening of rules around prescribing and dispensing of buprenorphine during the pandemic increased overdose deaths,” investigator Wilson Compton, MD, deputy director of the National Institute on Drug Abuse, told this news organization.
“This is reassuring that, even when we opened up the doors to easier access to buprenorphine, we didn’t see that most serious consequence,” Dr. Compton said.
The findings were published online in JAMA Network Open .
Cause and effect
Federal agencies relaxed prescribing regulations for buprenorphine in March 2020 to make it easier for clinicians to prescribe the drug via telemedicine and for patients to take the medication at home.
The number of buprenorphine prescriptions has increased since that change, with more than 1 million people receiving the medication in 2021 from retail pharmacies in the United States.
However, questions remained about whether increased access would lead to an increase in buprenorphine-involved overdose.
Researchers with NIDA and the Centers for Disease Control and Prevention analyzed data from the State Unintentional Drug Overdose Reporting System, a CDC database that combines medical examiner and coroner reports and postmortem toxicology testing.
The study included information about overdose deaths from July 2019 to June 2021 in 46 states and the District of Columbia.
Between July 2019 and June 2021, there were 1,955 buprenorphine-involved overdose deaths, which accounted for 2.2% of all drug overdose deaths and 2.6% of opioid-involved overdose deaths.
However, researchers went beyond overall numbers and evaluated details from coroner’s and medical examiner reports, something they had not done before.
“For the first time we looked at the characteristics of decedents from buprenorphine because this has not been studied in this type of detail with a near-national sample,” Dr. Compton said.
“That allowed us to look at patterns of use of other substances as well as the circumstances that are recorded at the death scene that are in the data set,” he added.
Important insights
Reports from nearly all buprenorphine-involved deaths included the presence of at least one other drug, compared with opioid overdose deaths that typically involved only one drug.
“This is consistent with the pharmacology of buprenorphine being a partial agonist, so it may not be as fatal all by itself as some of the other opioids,” Dr. Compton said.
Deaths involving buprenorphine were less likely to include illicitly manufactured fentanyls, and other prescription medications were more often found on the scene, such as antidepressants.
Compared with opioid decedents, buprenorphine decedents were more likely to be women, age 35-44, White, and receiving treatment for mental health conditions, including for substance use disorder (SUD).
These kinds of characteristics provide important insights about potential ways to improve safety and clinical outcomes, Dr. Compton noted.
“When we see things like a little higher rate of SUD treatment and this evidence of other prescription drugs on the scene, and some higher rates of antidepressants in these decedents than I might have expected, I’m very curious about their use of other medical services outside of substance use treatment, because that might be a place where some interventions could be implemented,” he said.
A similar study showed pandemic-era policy changes that allowed methadone to be taken at home was followed by a decrease in methadone-related overdose deaths.
The new findings are consistent with those results, Dr. Compton said.
‘Chipping away’ at stigma
Commenting on the study, O. Trent Hall, DO, assistant professor of addiction medicine, Department of Psychiatry and Behavioral Health, Ohio State University Wexner Medical Center, Columbus, said that, although he welcomed the findings, they aren’t unexpected.
“Buprenorphine is well established as a safe and effective medication for opioid use disorder and as a physician who routinely cares for patients in the hospital after opioid overdose, I am not at all surprised by these results,” said Dr. Hall, who was not involved with the research.
“When my patients leave the hospital with a buprenorphine prescription, they are much less likely to return with another overdose or serious opioid-related medical problem,” he added.
U.S. drug overdose deaths topped 100,000 for the first time in 2021, and most were opioid-related. Although the latest data from the CDC shows drug overdose deaths have been declining slowly since early 2022, the numbers remain high.
Buprenorphine is one of only two drugs known to reduce the risk of opioid overdose. While prescriptions have increased since 2020, the medication remains underutilized, despite its known effectiveness in treating opioid use disorder.
Dr. Hall noted that research such as the new study could help increase buprenorphine’s use.
“Studies like this one chip away at the stigma that has been misapplied to buprenorphine,” he said. “I hope this article will encourage more providers to offer buprenorphine to patients with opioid use disorder.”
The study was funded internally by NIDA and the CDC. Dr. Compton reported owning stock in General Electric, 3M, and Pfizer outside the submitted work. Dr. Hall has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Gene test may offer insights into treatment response in advanced NSCLC
therapy despite their poor status, researchers reported.
Positive findings on the test, known as DetermaIO, were “associated with efficacy of response to ICI therapy in advanced NSCLC patients,” Matthew G. Varga, PhD, manager of scientific affairs at Oncocyte, said in an interview. “These data suggest that DetermaIO warrants further study in poor performance status patients as it has the potential to identify likely responders to ICI therapy.”
Oncocyte, which is developing the test, presented the findings in a poster at the annual meeting of the Society for Immunotherapy of Cancer.
According to Dr. Varga, “DetermaIO is an RT-qPCR test that can be applied to FFPE [formalin-fixed, paraffin-embedded] tissue specimens to quantify the relative gene expression of 27 genes and subsequently applies our proprietary algorithm to generate an IO score based on the gene expression profile. The DetermaIO score is a binary IO+ or IO– score, representing likely responder or nonresponder, respectively.”
The test was originally developed for triple negative breast cancer, Dr. Varga said, and it’s been validated in non–small cell lung cancer, metastatic urothelial carcinoma, and metastatic colorectal carcinoma.
For the study, the researchers retrospectively tracked associations between DetermaIO score and either progression-free survival (PFS) or overall survival (OS) in 147 patients in Canada with NSCLC who were treated with ICI monotherapy. All had programmed death-ligand 1 (PD-L1) ≥ 50%.
Overall, outcomes were poor: The median survival was 12.7 months, and median PFS was 7.0 months. These outcomes were even worse in those who underwent therapy as a second- line treatment: The median survival was 9.7 months, and median PFS was 4.4 months.
“DetermaIO was significantly associated with PFS at hazard ratio [HR] = 0.55, 95% [confidence interval] CI, 0.32-0.94, P = .028. In our analyses, a hazard ratio less than 1 suggests lower risk – i.e, that DetermaIO+ patients have lower risk of an event – death or progression – compared to a DetermaIO– patient,” Dr. Varga said. “The association for overall survival was not statistically significant, but it was suggestive of clinically meaningful benefit.”
He added that “we could identify likely responders from nonresponders, suggesting that the DetermaIO score adds both independent and incremental data to the existing gold standard biomarker. The objective response rate for all first-line patients – n = 78 – was 44.9%. Twenty-two DetermaIO– tumors had a 23% response rate (5 partial responses) whereas of the 56 DetermaIO+ patients, the response rate was 54% (2 complete response and 28 partial responses).”
A score on the test, he said, was not associated with OS or PFS in patients who received second-line or later treatment.
The study was not designed to evaluate the predictive power of the test. “For a biomarker to be defined as predictive requires a formal test of interaction between a treatment group (ICI monotherapy, for example) vs. a control group (chemo-only or other regimen),” Dr. Varga explained. “In our analysis, there was no group of patients who did not receive ICI monotherapy. Thus a test for interaction and a predictive claim cannot be made.”
The test is available for at no cost via an early access program, Dr. Varga said, and Oncocyte is getting ready to seek Medicare coverage. The ultimate cost of the test, he said, is unknown.
Oncocyte funded this study. Dr. Varga and several other study authors are Oncocyte employees, and another author is a paid consultant to the company.
therapy despite their poor status, researchers reported.
Positive findings on the test, known as DetermaIO, were “associated with efficacy of response to ICI therapy in advanced NSCLC patients,” Matthew G. Varga, PhD, manager of scientific affairs at Oncocyte, said in an interview. “These data suggest that DetermaIO warrants further study in poor performance status patients as it has the potential to identify likely responders to ICI therapy.”
Oncocyte, which is developing the test, presented the findings in a poster at the annual meeting of the Society for Immunotherapy of Cancer.
According to Dr. Varga, “DetermaIO is an RT-qPCR test that can be applied to FFPE [formalin-fixed, paraffin-embedded] tissue specimens to quantify the relative gene expression of 27 genes and subsequently applies our proprietary algorithm to generate an IO score based on the gene expression profile. The DetermaIO score is a binary IO+ or IO– score, representing likely responder or nonresponder, respectively.”
The test was originally developed for triple negative breast cancer, Dr. Varga said, and it’s been validated in non–small cell lung cancer, metastatic urothelial carcinoma, and metastatic colorectal carcinoma.
For the study, the researchers retrospectively tracked associations between DetermaIO score and either progression-free survival (PFS) or overall survival (OS) in 147 patients in Canada with NSCLC who were treated with ICI monotherapy. All had programmed death-ligand 1 (PD-L1) ≥ 50%.
Overall, outcomes were poor: The median survival was 12.7 months, and median PFS was 7.0 months. These outcomes were even worse in those who underwent therapy as a second- line treatment: The median survival was 9.7 months, and median PFS was 4.4 months.
“DetermaIO was significantly associated with PFS at hazard ratio [HR] = 0.55, 95% [confidence interval] CI, 0.32-0.94, P = .028. In our analyses, a hazard ratio less than 1 suggests lower risk – i.e, that DetermaIO+ patients have lower risk of an event – death or progression – compared to a DetermaIO– patient,” Dr. Varga said. “The association for overall survival was not statistically significant, but it was suggestive of clinically meaningful benefit.”
He added that “we could identify likely responders from nonresponders, suggesting that the DetermaIO score adds both independent and incremental data to the existing gold standard biomarker. The objective response rate for all first-line patients – n = 78 – was 44.9%. Twenty-two DetermaIO– tumors had a 23% response rate (5 partial responses) whereas of the 56 DetermaIO+ patients, the response rate was 54% (2 complete response and 28 partial responses).”
A score on the test, he said, was not associated with OS or PFS in patients who received second-line or later treatment.
The study was not designed to evaluate the predictive power of the test. “For a biomarker to be defined as predictive requires a formal test of interaction between a treatment group (ICI monotherapy, for example) vs. a control group (chemo-only or other regimen),” Dr. Varga explained. “In our analysis, there was no group of patients who did not receive ICI monotherapy. Thus a test for interaction and a predictive claim cannot be made.”
The test is available for at no cost via an early access program, Dr. Varga said, and Oncocyte is getting ready to seek Medicare coverage. The ultimate cost of the test, he said, is unknown.
Oncocyte funded this study. Dr. Varga and several other study authors are Oncocyte employees, and another author is a paid consultant to the company.
therapy despite their poor status, researchers reported.
Positive findings on the test, known as DetermaIO, were “associated with efficacy of response to ICI therapy in advanced NSCLC patients,” Matthew G. Varga, PhD, manager of scientific affairs at Oncocyte, said in an interview. “These data suggest that DetermaIO warrants further study in poor performance status patients as it has the potential to identify likely responders to ICI therapy.”
Oncocyte, which is developing the test, presented the findings in a poster at the annual meeting of the Society for Immunotherapy of Cancer.
According to Dr. Varga, “DetermaIO is an RT-qPCR test that can be applied to FFPE [formalin-fixed, paraffin-embedded] tissue specimens to quantify the relative gene expression of 27 genes and subsequently applies our proprietary algorithm to generate an IO score based on the gene expression profile. The DetermaIO score is a binary IO+ or IO– score, representing likely responder or nonresponder, respectively.”
The test was originally developed for triple negative breast cancer, Dr. Varga said, and it’s been validated in non–small cell lung cancer, metastatic urothelial carcinoma, and metastatic colorectal carcinoma.
For the study, the researchers retrospectively tracked associations between DetermaIO score and either progression-free survival (PFS) or overall survival (OS) in 147 patients in Canada with NSCLC who were treated with ICI monotherapy. All had programmed death-ligand 1 (PD-L1) ≥ 50%.
Overall, outcomes were poor: The median survival was 12.7 months, and median PFS was 7.0 months. These outcomes were even worse in those who underwent therapy as a second- line treatment: The median survival was 9.7 months, and median PFS was 4.4 months.
“DetermaIO was significantly associated with PFS at hazard ratio [HR] = 0.55, 95% [confidence interval] CI, 0.32-0.94, P = .028. In our analyses, a hazard ratio less than 1 suggests lower risk – i.e, that DetermaIO+ patients have lower risk of an event – death or progression – compared to a DetermaIO– patient,” Dr. Varga said. “The association for overall survival was not statistically significant, but it was suggestive of clinically meaningful benefit.”
He added that “we could identify likely responders from nonresponders, suggesting that the DetermaIO score adds both independent and incremental data to the existing gold standard biomarker. The objective response rate for all first-line patients – n = 78 – was 44.9%. Twenty-two DetermaIO– tumors had a 23% response rate (5 partial responses) whereas of the 56 DetermaIO+ patients, the response rate was 54% (2 complete response and 28 partial responses).”
A score on the test, he said, was not associated with OS or PFS in patients who received second-line or later treatment.
The study was not designed to evaluate the predictive power of the test. “For a biomarker to be defined as predictive requires a formal test of interaction between a treatment group (ICI monotherapy, for example) vs. a control group (chemo-only or other regimen),” Dr. Varga explained. “In our analysis, there was no group of patients who did not receive ICI monotherapy. Thus a test for interaction and a predictive claim cannot be made.”
The test is available for at no cost via an early access program, Dr. Varga said, and Oncocyte is getting ready to seek Medicare coverage. The ultimate cost of the test, he said, is unknown.
Oncocyte funded this study. Dr. Varga and several other study authors are Oncocyte employees, and another author is a paid consultant to the company.
FROM SITC 2022