User login
Management of treatment-resistant depression: A review of 3 studies
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.

The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.
The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.
The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.

Antipsychotics and seizures: What are the risks?
Antipsychotics, especially second-generation antipsychotics (SGAs), have been proven effective for treating psychosis as well as mood disorders.1,2 Because antipsychotics can lower the epileptogenic threshold, seizures are a serious potential adverse effect. Antipsychotics can cause isolated EEG abnormalities in 7% of patients with no history of epilepsy, and clinical seizures in .5% to 1.2% of such patients.3 Additionally, the neuropathophysiology underlying epilepsy can predispose patients to psychiatric disorders4; the estimated prevalence of psychosis in patients with epilepsy is approximately 7%.5 This review will shed light on the risk of clinical seizures related to antipsychotics.
Comparing seizure risk among antipsychotics
In a review of the World Health Organization’s adverse drug reactions database, Kumlien and Lundberg6 calculated the ratio of the number of reports of seizures to the total number of reports for each drug. They found that approximately 9% of all adverse drug reaction reports involving clozapine were due to seizures. Equivalent ratios were 5.90% for quetiapine, 4.91% for olanzapine, 3.68% for risperidone, 3.27% for haloperidol, and 2.59% for aripiprazole. Using the database of the Pharmacovigilance Unit of the Basque Country, Lertxundi et al7 reported a 3.2-fold increased risk of seizure with SGAs in comparison with first-generation antipsychotics (FGAs) (95% confidence interval [CI], 2.21 to 4.63), which went down to 2.08 (CI, 1.39 to 3.12) once clozapine was excluded. However, as the authors of both studies noted, the quality and relevance of this data are limited because it relies on spontaneous reporting.
Overall, the evidence regarding the seizure risk associated with antipsychotics is scarce. To the best of our knowledge, only 2 large observational studies have compared the seizure risks associated with different antipsychotics.
Using data from the UK-based Clinical Practice Research Datalink between 1998 and 2013, Bloechlinger et al8 examined the incidence rates of seizures among patients newly diagnosed with schizophrenia, affective disorders, or dementia who were prescribed antipsychotics. They excluded patients with a history of seizures or antiepileptic use. In the cohort of 60,121 patients, the incidence rates of seizures per 10,000 person-years were 11.7 (CI, 10.0 to 13.4) for those who did not use antipsychotics, 12.4 (CI, 10.9 to 13.8) for past users, 115.4 (CI, 50.1 to 180.7) for current users of haloperidol, 48.8 (CI, 30.7 to 66.9) for current users of quetiapine, 25.9 (CI, 11.8 to 40.0) for current users of risperidone, and 19.0 (CI, 8.7 to 29.3) for current users of olanzapine. No data were available about clozapine use.
In subsequent analyses, the authors found that among patients with affective disorders, only current use of medium- to high-potency FGAs (haloperidol, prochlorperazine, and trifluoperazine) was associated with a significantly increased risk of seizures (adjusted odds ratio: 2.51, CI, 1.51 to 4.18) compared with non-users.8 Among patients with dementia, current use of olanzapine or quetiapine and current use of any FGAs were associated with significantly increased odds of seizures. This study suggests that the underlying mental illness might modulate the seizure risk associated with antipsychotics.8
Wu et al9 conducted a study based on the National Health Insurance Research Database in Taiwan. They examined the 1-year incidence of new-onset seizures among patients diagnosed with schizophrenia or mood disorders who were new to antipsychotic treatment, and calculated the risk of seizure associated with each antipsychotic in reference to risperidone. They found that those receiving clozapine, thioridazine, and haloperidol were 2 to 3 times more likely to develop seizures than those treated with risperidone; risks associated with the rest of the FGAs were similar to that of risperidone.
The results of these 2 large cohort studies are somewhat concurrent in indicating that, other than clozapine, SGAs incur similar risks of seizures; furthermore, they specify that, contrary to earlier studies,10 haloperidol is associated with significantly higher odds of seizures. While both of these cohort studies controlled for several sociodemographic and clinical confounders, they have several limitations. First, diagnoses of seizures were based on information available in databases, which might be subject to inaccuracies. Second, neither study evaluated the effect of drug dosage and duration of exposure on new-onset seizures.
Continue to: Most evidence is from case reports
Most evidence is from case reports
Other than these 2 large studies, most of the evidence addressing the relationship between the use of antipsychotics and incidence of seizures is low quality and relies on case reports or expert opinions. Older studies found that, among FGAs, seizure risk is highest with chlorpromazine and promazine, and lowest with thioridazine and haloperidol.10 As for SGAs, case reports have described seizuresassociated with the use of quetiapine, aripiprazole, risperidone, paliperidone, and olanzapine.
Quetiapine. Three case reports published between 2002 and 2010 describe generalized
Aripiprazole. Five case reports described staring spells and tonic-clonic seizures in patients receiving 10 to 15 mg of aripiprazole.15-19 In the New Drug Application (NDA) for aripiprazole, the incidence of seizures was estimated to be .11% (1 of 926 patients) in placebo-controlled trials and .46% (3 of 859 patients) in haloperidol-controlled trials.20
Risperidone’s product labeling suggests the drug should be used with caution in patients with a history of seizures or conditions that could result in a lower seizure threshold. In Phase III placebo-controlled trials, seizures occurred in .3% of patients treated with risperidone, although in some cases, the seizures were induced by electrolyte disturbances such as hyponatremia.21 Gonzalez-Heydrich et al22 and Holzhausen et al23 found no increase in seizure activity among patients with epilepsy who were receiving risperidone. Lane et al24 published a case report of a geriatric woman who presented with a generalized tonic-clonic seizure related to rapid titration of risperidone; however, with slower titration and lower doses, she stopped having seizures without adding any antiepileptic drugs. Komossa et al25 found that risperidone is less epileptogenic than clozapine, with a relative risk of .22.
Paliperidone is the active metabolite of risperidone and does not have pharmacokinetic interactions with drugs metabolized by the cytochrome P450 (CYP) enzymes. Its labeling indicates that the drug should be used with caution in patients with a history of seizures.26 In Phase III placebo-controlled trials of paliperidone, the rate of seizures was .22%.27 Two case reports suggest close monitoring of seizure risk in patients receiving paliperidone.28,29 Liang et al29 reported that co-administration of valproic acid could mask an underlying decrease of the seizure threshold caused by antipsychotics such as paliperidone.
Continue to: Olanzapine
Olanzapine is a thienobenzodiazepine derivative and is chemically related to clozapine.30 The olanzapine NDA31 shows that 23 of 3,139 patients developed seizures, mainly tonic-clonic, with evidence suggesting that the seizures may have been due to confounding factors such as a history of seizures or metabolic abnormalities. There were no statistically significant differences in the rate of seizures associated with olanzapine compared with placebo or haloperidol (P = .252 and .168, respectively).

A literature review for olanzapine yielded 1 case report of repetitive focal seizures and lingual dystonia,32 5 case reports of generalized tonic-clonic seizures and myoclonus,33-37 and 2 case reports of status epilepticus.38,39 Olanzapine’s clearance is 25% to 30% lower in women, and most of these case reports occurred women.40

Details of the above case reports are summarized in Table 1 (aripiprazole15-19), Table 2 (olanzapine32-39), and Table 3 (paliperidone,28,29 quetiapine,11-13 and risperidone22-24).

Ziprasidone. According to the NDA safety database, the seizure rate attributed to ziprasidone was 1.8 per 100 subject-years or 0.54% of participants (12 of 2,588).41 No additional studies have been published regarding its seizure risk.
Clozapine has a black-box warning

To the best of our knowledge, clozapine is the only antipsychotic that carries an FDA “black-box” warning regarding its risk of inducing seizures.42 Devinsky and Pacia43 reported a cumulative risk of 10% after 3.8 years of treatment. The literature has described clozapine-induced generalized tonic-clonic, myoclonic, simple and complex partial, and absence seizures.44 Table 445 lists the estimated frequency of each seizure type based on 101 cases of clozapine-induced seizures. Myoclonic seizures and drop attacks could be precursors/warning signs of grand mal tonic-clonic seizures.46,47 Seizures have been observed at all stages of treatment, but were more common during initiation of clozapine, which emphasizes the importance of a progressive and slow titration.43,48 The incidence of seizures was estimated to be 6% in a sample of 216 patients with schizophrenia with no history of epilepsy who were prescribed clozapine.49
Continue to: Regarding a possible association between...
Regarding a possible association between clozapine dose or clozapine plasma levels and seizure risk, there is a positive linear relationship between the dose of clozapine and its serum concentration over a dosing range of 25 to 800 mg/d.50 However, the plasma concentration is also significantly affected by factors such as smoking, gender, age, drug interactions, and CYP genotypes. Therefore, the same clozapine dose will yield a lower serum concentration in an older male who smokes compared with a younger, non-smoking female.51 Perry et al52 suggested a dosing nomogram to calculate the influence of gender and smoking. Seizure risk, especially for tonic-clonic seizures, has been reported to increase with clozapine doses >600 mg/d,53 and with plasma concentrations exceeding 1,000 to 1,300 mg/L.54 However, in a 2011 regression analysis, Varma et al55 found no statistically significant relationship between seizure risk and clozapine oral dose; there was not enough data to test a correlation between clozapine plasma levels and the incidence of seizures.
How antipsychotics might lower the seizure threshold
Researchers have suggested several possible mechanisms to explain how antipsychotics might lower the seizure threshold. Antagonism of dopamine D4, histamine H1, and acetylcholine-muscarinic receptors seems to induce EEG alterations and increase the risk of seizures.56 Additionally, modulation of the N-methyl-
Watch for pharmacokinetic interactions
The CYP enzymes involved in drug metabolism include CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Most commonly used antiepileptics and antipsychotics are metabolized by CYP enzymes, and may also act as inhibitors or inducers of these enzymes.61 Drug interactions may impair seizure control, which is why monotherapy is preferable to combination treatment in patients with epilepsy.62 Carbamazepine and phenytoin are inducers of both CYP1A2 (which metabolizes olanzapine and clozapine), and CYP3A4 (which metabolizes haloperidol, risperidone, quetiapine, ziprasidone and clozapine). Paliperidone is not metabolized by CYP enzymes.62 Discontinuing an enzyme-inducing agent may result in increased antipsychotic plasma concentrations, which might lead to an increased risk of seizures.
Valproic acid, which is often used to prevent or treat clozapine-induced seizures, has an unclear effect on clozapine plasma concentrations.63 Although valproic acid is known to inhibit clozapine metabolism, 2 reports have suggested that the plasma concentrations of clozapine and its metabolites may decrease after adding valproic acid.64,65 Other studies have found that valproic acid increases plasma concentrations of clozapine while it decreases plasma concentrations of norclozapine; norclozapine is the main clozapine metabolite responsible for inducing seizures.66,67
Steps for minimizing seizure risk
Determining the seizure risk for a patient taking an antipsychotic is challenging because doing so depends not only on the seizurogenic potential of each drug but also on individualized predisposing factors.11,57,68 Choosing the “best” antipsychotic therefore largely depends on each patient’s profile. The predisposing factors consist mainly of the individually inherited seizure threshold (personal history of febrile convulsions or a family history of seizures) and other comorbid seizurogenic conditions, such as a history of head trauma, brain injury, intellectual disability, cerebral arteriosclerosis, neurodegenerative diseases, encephalopathy, chronic renal insufficiency, and hyponatremia. Furthermore, seizure risk depends on the antipsychotic dose administered and the rate of titration.11
Continue to: There is not enough evidence...
There is not enough evidence to recommend performing an EEG in all patients taking antipsychotics. Such testing is recommended only for patients who have predisposing factors for seizures. If an EEG shows any abnormality in a patient taking clozapine, consider decreasing the clozapine dose69,70 or adding an antiepileptic drug such as valproic acid or lamotrigine.44,70
Although clozapine carries a black-box warning of increased risk of causing seizures, there is no consensus regarding the efficacy of co-prescribing an antiepileptic. Some studies have suggested prescribing valproic acid prophylactically,71 after the occurrence of 1 seizure,59 or after 2 seizures.54,72 Others have recommended prescribing prophylactic valproic acid for patients taking ≥600 mg/d of clozapine or whose clozapine plasma levels are >500 mg/L.73 Varma et al55 recommended starting an antiepileptic medication if there are clear epileptiform discharges on EEG, if the patient develops stuttering or speech difficulties, or if seizures occur. Liukkonen et al72 advised initiating an antiepileptic at the start of clozapine treatment in patients who are taking other epileptogenic medications, patients with pre-existing seizure disorder, and patients with neurologic abnormalities. On the other hand, Caetano51 argued against primary prevention of seizures for patients receiving >600 mg/d of clozapine, suggesting that “the risk of seizures would be better managed by close clinical monitoring and measures of clozapine serum concentration rather than adding an anticonvulsant drug.”
Current recommendations for primary and secondary prevention of clozapine-induced seizures are detailed in Table 5.42,44,45,51,55,57,69,74,75

Studies addressing the seizurogenic potential of SGAs other than clozapine have a low level of evidence and include patients who had comorbid conditions and were taking other medications that could cause seizures. Additionally, clinical trials of SGAs rarely include patients with seizure disorders; this might underestimate the risk of seizures.4
The effect of the mental illness itself on the seizure threshold needs to be considered.43 Bloechlinger et al8 found that dementia might be inherently associated with a higher risk of antipsychotic-related seizures. Moreover, numerous qualitative EEG studies have found abnormalities in 20% to 60% of patients with schizophrenia.56 Other quantitative studies have reported mild and nonspecific EEG abnormalities, such as increased delta and/or theta activity, in many non-medicated patients with schizophrenia.10,76 Additionally, brain tissue analysis of deceased patients who had schizophrenia has shown a significant increase in dopamine concentrations in the left amygdala compared with controls, and this might be responsible for enhanced electrical activity in this region.10 Some studies have described EEG slowing in the frontal brain regions of patients with schizophrenia,77 and was selectively normalized in these areas with antipsychotics.78
As always, start low, go slow
Mounting evidence suggests that antipsychotic medications decrease the seizure threshold. Practitioners should thus be cautious in prescribing antipsychotics and should target reaching the minimal effective dose with slow titration, especially in patients with predisposing factors for epilepsy.
Continue to: Although evidence suggests...
Although evidence suggests antipsychotics can induce different types of epileptic seizures, the quality of this evidence is low. Randomized controlled trials are needed to determine which antipsychotics increase seizure risk and whether there is a dose-effect relationship.
Bottom Line
Among second-generation antipsychotics, clozapine appears to increase the risk of clinical seizure the most. Correlations with dosage and/or plasma levels have not been proven. Psychiatrists should be vigilant for pharmacokinetic interactions between antipsychotics and antiepileptics, notably via CYP1A2 and CYP3A4.
Related Resources
- Druschky K, Bleich S, Grohmann R, et al. Seizure rates under treatment with antipsychotic drugs: Data from the AMSP project. World J Biol Psychiatry. 2018;15:1-10.
- Epilepsy Foundation. For professionals: Antipsychotics. https://www.epilepsy.com/learn/professionals/diagnosistreatment/psychotropic-drugs-developmental-disabilities/comorbid-5.
Drug Brand Names
Aripiprazole • Abilify
Benztropine • Cogentin
Bethanechol • Duvoid
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cimetidine • Tagamet
Ciprofloxacin • Cipro
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Donepezil • Aricept
Enalapril • Vasotec
Erythromycin • Erythrocin
Escitalopram • Lexapro
Flunitrazepam • Rohypnol
Fluvoxamine • Luvox
Gabapentin • Neurontin
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Metformin • Fortamet, Glucophage
Mirtazapine • Remeron
Nitrofurantoin • Furadantin
Olanzapine • Zyprexa
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Prochlorperazine • Compazine
Procyclidine • Kemadrin
Propranolol • Inderal
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Simvastatin • Zocor
Sulfamethoxazole/trimethoprim • Bactrim, Sulfatrim
Topiramate • Topamax
Trifluoperazine • Stelazine
Valproic acid • Depakene, Depakote
Ziprasidone • Geodon
1. Bruijnzeel D, Suryadevara U, Tandon R. Antipsychotic treatment of schizophrenia: an update. Asian J Psychiatr. 2014;11:3-7.
2. Hrdlicka M, Dudova I. Atypical antipsychotics in the treatment of early-onset schizophrenia. Neuropsychiatr Dis Treat. 2015;11:907-913.
3. Koch-Stoecker S. Antipsychotic drugs and epilepsy: indications and treatment guidelines. Epilepsia. 2002;43(suppl 2):19-24.
4. Alper K, Schwartz KA, Kolts RL, et al. Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry. 2007;62(4):345-354.
5. Torta R, Keller R. Behavioral, psychotic, and anxiety disorders in epilepsy: etiology, clinical features, and therapeutic implications. Epilepsia. 1999;40(suppl 10):S2-S20.
6. Kumlien E, Lundberg PO. Seizure risk associated with neuroactive drugs: data from the WHO adverse drug reactions database. Seizure. 2010;19(2):69-73.
7. Lertxundi U, Hernandez R, Medrano J, et al. Antipsychotics and seizures: higher risk with atypicals? Seizure. 2013;22(2):141-143.
8. Bloechliger M, Rüegg S, Jick SS, et al. Antipsychotic drug use and the risk of seizures: follow-up study with a nested case-control analysis. CNS Drugs. 2015;29(7):591-603.
9. Wu CS, Wang SC, Yeh IJ, et al. Comparative risk of seizure with use of first- and second-generation antipsychotics in patients with schizophrenia and mood disorders. J Clin Psychiatry. 2016;77(5):e573-e579.
10. Cold JA, Wells BG, Froemming JH. Seizure activity associated with antipsychotic therapy. [Erratum in DICP. 1990;24(10):1012.] DICP. 1990;24(6):601-606.
11. Hedges DW, Jeppson KG. New-onset seizure associated with quetiapine and olanzapine. Ann Pharmacother. 2002;36(3):437-439.
12. Dogu O, Sevim S, Kaleagasi HS. Seizures associated with quetiapine treatment. Ann Pharmacother. 2003;37(9):1224-1227.
13. Young AC, Kleinschmidt KC, Wax PM. Late-onset seizures associated with quetiapine poisoning. J Med Toxicol. 2009;5(1):24-26.
14. US Food and Drug Administration. Recommendation of approvable action for quetiapine fumarate extended release (Seroquel® XR) for the treatment of schizophrenia. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022047Orig1s000MedR.pdf. April 24, 2007. Accessed January 28, 2019.
15. Malik AR, Ravasia S. Aripiprazole-induced seizure. Can J Psychiatry. 2005;50(3):186.
16. Tsai JF. Aripiprazole-associated seizure. J Clin Psychiatry. 2006;67(6):995-996.
17. Arora M, Arndorfer L. EEG abnormalities in a patient taking aripiprazole. Psychiatry (Edgmont). 2007;4(7):18-19.
18. Yueh CL, Yu SL, Chen HM, et al. Aripiprazole-induced seizure: a second case report. BMJ case reports. 2009;2009:bcr03.2009.1693. doi: 10.1136/bcr.03.2009.1693.
19. Thabet FI, Sweis RT, Joseph SA. Aripiprazole-induced seizure in a 3-year-old child: a case report and literature review. Clin Neuropharmacol. 2013;36(1):29-30.
20. US Food and Drug Administration. Abilify (Aripiprazole) tablets. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-436_Abilify_medr_P2.pdf. Published March 07, 2003. Accessed January 28, 2019.
21. US Food and Drug Administration. Drugs@FDA: FDA approved drug products. Risperdal tablets, Risperdal oral solution & Risperdal M-tab orally disintegrating tablets. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021444_S004_RISPERDAL_TABLETS.pdf. Published September 10, 2003. Accessed January 28, 2019.
22. Gonzalez-Heydrich J, Pandina GJ, Fleisher CA, et al. No seizure exacerbation from risperidone in youth with comorbid epilepsy and psychiatric disorders: a case series. J Child Adolesc Psychopharmacol. 2004;14(2):295-310.
23. Holzhausen SPF, Guerreiro MM, Baccin CE, et al. Use of risperidone in children with epilepsy. Epilepsy Behav. 2007;10(3):412-416.
24. Lane HY, Chang WH, Chou JC. Seizure during risperidone treatment in an elderly woman treated with concomitant medications. J Clinl Psychiatry. 1998;59(2):81-82.
25. Komossa K, Rummel-Kluge C, Schwarz S, et al. Risperidone versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2011;(1):19:CD006626.
26. Paliperidone [package insert]. Mountainville, CA: Janssen Pharmaceuticals, Inc.; 2007.
27. Brugge, MD; US Food and Drug Administration. Paliperidone OROS oral formulation. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021999s000_MedR_Part4.pdf. Accessed January 28, 2019.
28. Schneider RA, Lizer MH. Apparent seizure and atrial fibrillation associated with paliperidone. Am J Health System Pharm. 2008;65(22):2122-2125.
29. Liang CS, Yang FW, Chiang KT. Paliperidone-associated seizure after discontinuation of sodium valproate: a case report. J Clin Psychopharmacol. 2011;31(2):246-247.
30. Fulton B, Goa KL. Olanzapine. A review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs. 1997;53(2):281-298.
31. US Food and Drug Administration. Drugs@FDA: FDA approved drug products: Zyprexa (olanzapine). ORIG-1. http://www.accessdata.fda.gov/drugsatfda_docs/nda/96/020592_Original_Approval_Pkg%20.pdf. Published September 30, 1996. Accessed January 28, 2019.
32. Anzellotti F, Capasso M, Frazzini V, et al. Olanzapine-related repetitive focal seizures with lingual dystonia. Epileptic Disord. 2016;18(1):83-86.
33. Lee JW, Crismon ML, Dorson PG. Seizure associated with olanzapine. Ann Pharmac. 1999;33(5):554-556.
34. Woolley J, Smith S. Lowered seizure threshold on olanzapine. Br J Psychiatry. 2001;178(1):85-86.
35. Behere RV, Anjith D, Rao NP, et al. Olanzapine-induced clinical seizure: a case report. Clin Neuropharmacol. 2009;32(5):297-298.
36. Camacho A, García-Navarro M, Martínez B, et al. Olanzapine-induced myoclonic status. Clin Neuropharmacol. 2005;28(3):145-147.
37. Rosen JB, Milstein MJ, Haut SR. Olanzapine-associated myoclonus. Epilepsy Res. 2012;98(2-3):247-250.
38. Wyderski RJ, Starrett WG, Abou-Saif A. Fatal status epilepticus associated with olanzapine therapy. Ann Pharmacother. 1999;33(7-8):787-789.
39. Spyridi S, Sokolaki S, Nimatoudis J, et al. Status epilepticus in a patient treated with olanzapine and mirtazapine. Int J Clin Pharmacol Ther. 2009;47(2):120-123.
40. Schatzberg AF, Nemeroff CB. Essentials of clinical psychopharmacology. 2nd ed. Arlington, Virginia: American Psychiatric Publishing; 2006.
41. US Food and Drug Administration. Drug approval package: Geodon (Ziprasidone HCI) Capsules. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20-825_Geodan_medr_P2.pdf. Published February 5, 2001. Accessed January 29, 2019.
42. Clozaril [package insert]. East Hanover, NJ: Novartis; 2008.
43. Devinsky O, Pacia SV. Seizures during clozapine therapy. J Clin Psychiatry. 1994;55(suppl B):153-156.
44. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
45. Wong J, Delva N. Clozapine-induced seizures: recognition and treatment. Can J Psychiatry. 2007;52(7):457-463.
46. Berman I, Zalma A, DuRand CJ, et al. Clozapine-induced myoclonic jerks and drop attacks. J Clin Psychiatry. 1992;53(9):329-330.
47. Gouzoulis E, Ozdaglar A, Kasper J. Myoclonic seizures followed by grand mal seizures during clozapine treatment. Am J Psychiatry. 1993;150(7):1128.
48. Sajatovic M, Meltzer HY. Clozapine-induced myoclonus and generalized seizures. Biol Psychiatry. 1996;39(5):367-370.
49. Grover S, Hazari N, Chakrabarti S, et al. Association of clozapine with seizures: a brief report involving 222 patients prescribed clozapine. East Asian Arch Psychiatry. 2015;25(2):73-78.
50. Byerly MJ, DeVane CL. Pharmacokinetics of clozapine and risperidone: a review of recent literature. J Clin Psychopharmacol. 1996;16(2):177-187.
51. Caetano D. Use of anticonvulsants as prophylaxis for seizures in patients on clozapine. Australas Psychiatry. 2014;22(1):78-83.
52. Perry PJ, Bever KA, Arndt S, et al. Relationship between patient variables and plasma clozapine concentrations: a dosing nomogram. Biol Psychiatry.1998;44(8):733-738.
53. Dumortier G, Mahé V, Pons D, et al. Clonic seizure associated with high clozapine plasma level. J Neuropsychiatry Clin Neurosci. 2001;13(2):302-303.
54. Funderburg LG, Vertrees JE, True JE, et al. Seizure following addition of erythromycin to clozapine treatment. Am J Psychiatry. 1994;151(12):1840-1841.
55. Varma S, Bishara D, Besag FMC, et al. Clozapine-related EEG changes and seizures: dose and plasma-level relationships. Ther Adv Psychopharmacol. 2011;1(2):47-66.
56. Amann BL, Pogarell O, Mergl R, et al. EEG abnormalities associated with antipsychotics: a comparison of quetiapine, olanzapine, haloperidol and healthy subjects. Hum Psychopharmacol. 2003;18(8):641-646.
57. Pisani F, Oteri G, Costa C, et al. Effects of psychotropic drugs on seizure threshold. Drug Saf. 2002;25(2):91-110.
58. Maurice T, Phan VL, Urani A, et al. Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jpn J Pharmacol. 1999;81(2):125-155.
59. Haller E, Binder RL. Clozapine and seizures. Am J Psychiatry. 1990;147(8):1069-1071.
60. Torta R, Monaco F. Atypical antipsychotics and serotoninergic antidepressants in patients with epilepsy: pharmacodynamic considerations. Epilepsia. 2002;43(suppl 2):8-13.
61. Spina E. Drug interactions. In: Shorvon S, Perucca E, Engel J Jr, eds. The treatment of epilepsy. 3rd ed. Oxford, UK: Blackwell Publishing; 2009:361-377.
62. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
63. de Leon J, Santoro V, D’Arrigo C, et al. Interactions between antiepileptics and second-generation antipsychotics. Expert Opin Drug Metab Toxicol. 2012;8(3):311-334.
64. Finley P, Warner D. Potential impact of valproic acid therapy on clozapine disposition. Biol Psychiatry. 1994;36(7):487-488.
65. Longo LP, Salzman C. Valproic acid effects on serum concentrations of clozapine and norclozapine. Am J Psychiatry. 1995;152(4):650.
66. Centorrino F, Baldessarini RJ, Kando J, et al. Serum concentrations of clozapine and its major metabolites: effects of cotreatment with fluoxetine or valproate. Am J Psychiatry. 1994;151(1):123-125.
67. Facciolà G, Avenoso A, Scordo MG, et al. Small effects of valproic acid on the plasma concentrations of clozapine and its major metabolites in patients with schizophrenic or affective disorders. Ther Drug Monit. 1999;21(3):341-345.
68. Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23(4):611-622.
69. Muzyk A, Gala G, Kahn DA. Use of lamotrigine in a patient with a clozapine-related seizure. J Psychiatr Pract. 2010;16(2):125-128.
70. Kikuchi YS, Sato W, Ataka K, et al. Clozapine-induced seizures, electroencephalography abnormalities, and clinical responses in Japanese patients with schizophrenia. Neuropsychiatr Dis Treat. 2014;10:1973-1978.
71. Taner E, Coşar B, Işik E. Clozapine-induced myoclonic seizures and valproic acid. Int J Psychiatry Clin Pract. 1998;2(1):53-55.
72. Liukkonen J, Koponen HJ, Nousiainen U. Clinical picture and long-term course of epileptic seizures that occur during clozapine treatment. Psychiatry Res. 1992;44(2):107-112.
73. Devinsky O, Honigfeld G, Patin J. Clozapine-related seizures. Neurology. 1991;41(3):369-371.
74. Foster R, Olajide D. A case of clozapine-induced tonic-clonic seizures managed with valproate: implications for clinical care. J Psychopharmacol. 2005;19(1):93-96.
75. Gandelman-Marton R, Theitler J, Klein C, et al. Phenytoin intoxication in a clozapine-related prolonged seizure. J Emerg Med. 2008;35(4):407-409.
76. Primavera A, Giberti L, Scotto P, et al. Nonconvulsive status epilepticus as a cause of confusion in later life: a report of 5 cases. Neuropsychobiology. 1994;30(2-3):148-152.
77. Boutros NN, Arfken C, Galderisi S, et al. The status of spectral EEG abnormality as a diagnostic test for schizophrenia. Schizophrenia Res. 2008;99(1-3):225-237.
78. Takahashi T, Cho RY, Mizuno T, et al. Antipsychotics reverse abnormal EEG complexity in drug-naïve schizophrenia: a multiscale entropy analysis. Neuroimage. 2010;51(1):173-182.
Antipsychotics, especially second-generation antipsychotics (SGAs), have been proven effective for treating psychosis as well as mood disorders.1,2 Because antipsychotics can lower the epileptogenic threshold, seizures are a serious potential adverse effect. Antipsychotics can cause isolated EEG abnormalities in 7% of patients with no history of epilepsy, and clinical seizures in .5% to 1.2% of such patients.3 Additionally, the neuropathophysiology underlying epilepsy can predispose patients to psychiatric disorders4; the estimated prevalence of psychosis in patients with epilepsy is approximately 7%.5 This review will shed light on the risk of clinical seizures related to antipsychotics.
Comparing seizure risk among antipsychotics
In a review of the World Health Organization’s adverse drug reactions database, Kumlien and Lundberg6 calculated the ratio of the number of reports of seizures to the total number of reports for each drug. They found that approximately 9% of all adverse drug reaction reports involving clozapine were due to seizures. Equivalent ratios were 5.90% for quetiapine, 4.91% for olanzapine, 3.68% for risperidone, 3.27% for haloperidol, and 2.59% for aripiprazole. Using the database of the Pharmacovigilance Unit of the Basque Country, Lertxundi et al7 reported a 3.2-fold increased risk of seizure with SGAs in comparison with first-generation antipsychotics (FGAs) (95% confidence interval [CI], 2.21 to 4.63), which went down to 2.08 (CI, 1.39 to 3.12) once clozapine was excluded. However, as the authors of both studies noted, the quality and relevance of this data are limited because it relies on spontaneous reporting.
Overall, the evidence regarding the seizure risk associated with antipsychotics is scarce. To the best of our knowledge, only 2 large observational studies have compared the seizure risks associated with different antipsychotics.
Using data from the UK-based Clinical Practice Research Datalink between 1998 and 2013, Bloechlinger et al8 examined the incidence rates of seizures among patients newly diagnosed with schizophrenia, affective disorders, or dementia who were prescribed antipsychotics. They excluded patients with a history of seizures or antiepileptic use. In the cohort of 60,121 patients, the incidence rates of seizures per 10,000 person-years were 11.7 (CI, 10.0 to 13.4) for those who did not use antipsychotics, 12.4 (CI, 10.9 to 13.8) for past users, 115.4 (CI, 50.1 to 180.7) for current users of haloperidol, 48.8 (CI, 30.7 to 66.9) for current users of quetiapine, 25.9 (CI, 11.8 to 40.0) for current users of risperidone, and 19.0 (CI, 8.7 to 29.3) for current users of olanzapine. No data were available about clozapine use.
In subsequent analyses, the authors found that among patients with affective disorders, only current use of medium- to high-potency FGAs (haloperidol, prochlorperazine, and trifluoperazine) was associated with a significantly increased risk of seizures (adjusted odds ratio: 2.51, CI, 1.51 to 4.18) compared with non-users.8 Among patients with dementia, current use of olanzapine or quetiapine and current use of any FGAs were associated with significantly increased odds of seizures. This study suggests that the underlying mental illness might modulate the seizure risk associated with antipsychotics.8
Wu et al9 conducted a study based on the National Health Insurance Research Database in Taiwan. They examined the 1-year incidence of new-onset seizures among patients diagnosed with schizophrenia or mood disorders who were new to antipsychotic treatment, and calculated the risk of seizure associated with each antipsychotic in reference to risperidone. They found that those receiving clozapine, thioridazine, and haloperidol were 2 to 3 times more likely to develop seizures than those treated with risperidone; risks associated with the rest of the FGAs were similar to that of risperidone.
The results of these 2 large cohort studies are somewhat concurrent in indicating that, other than clozapine, SGAs incur similar risks of seizures; furthermore, they specify that, contrary to earlier studies,10 haloperidol is associated with significantly higher odds of seizures. While both of these cohort studies controlled for several sociodemographic and clinical confounders, they have several limitations. First, diagnoses of seizures were based on information available in databases, which might be subject to inaccuracies. Second, neither study evaluated the effect of drug dosage and duration of exposure on new-onset seizures.
Continue to: Most evidence is from case reports
Most evidence is from case reports
Other than these 2 large studies, most of the evidence addressing the relationship between the use of antipsychotics and incidence of seizures is low quality and relies on case reports or expert opinions. Older studies found that, among FGAs, seizure risk is highest with chlorpromazine and promazine, and lowest with thioridazine and haloperidol.10 As for SGAs, case reports have described seizuresassociated with the use of quetiapine, aripiprazole, risperidone, paliperidone, and olanzapine.
Quetiapine. Three case reports published between 2002 and 2010 describe generalized
Aripiprazole. Five case reports described staring spells and tonic-clonic seizures in patients receiving 10 to 15 mg of aripiprazole.15-19 In the New Drug Application (NDA) for aripiprazole, the incidence of seizures was estimated to be .11% (1 of 926 patients) in placebo-controlled trials and .46% (3 of 859 patients) in haloperidol-controlled trials.20
Risperidone’s product labeling suggests the drug should be used with caution in patients with a history of seizures or conditions that could result in a lower seizure threshold. In Phase III placebo-controlled trials, seizures occurred in .3% of patients treated with risperidone, although in some cases, the seizures were induced by electrolyte disturbances such as hyponatremia.21 Gonzalez-Heydrich et al22 and Holzhausen et al23 found no increase in seizure activity among patients with epilepsy who were receiving risperidone. Lane et al24 published a case report of a geriatric woman who presented with a generalized tonic-clonic seizure related to rapid titration of risperidone; however, with slower titration and lower doses, she stopped having seizures without adding any antiepileptic drugs. Komossa et al25 found that risperidone is less epileptogenic than clozapine, with a relative risk of .22.
Paliperidone is the active metabolite of risperidone and does not have pharmacokinetic interactions with drugs metabolized by the cytochrome P450 (CYP) enzymes. Its labeling indicates that the drug should be used with caution in patients with a history of seizures.26 In Phase III placebo-controlled trials of paliperidone, the rate of seizures was .22%.27 Two case reports suggest close monitoring of seizure risk in patients receiving paliperidone.28,29 Liang et al29 reported that co-administration of valproic acid could mask an underlying decrease of the seizure threshold caused by antipsychotics such as paliperidone.
Continue to: Olanzapine
Olanzapine is a thienobenzodiazepine derivative and is chemically related to clozapine.30 The olanzapine NDA31 shows that 23 of 3,139 patients developed seizures, mainly tonic-clonic, with evidence suggesting that the seizures may have been due to confounding factors such as a history of seizures or metabolic abnormalities. There were no statistically significant differences in the rate of seizures associated with olanzapine compared with placebo or haloperidol (P = .252 and .168, respectively).
A literature review for olanzapine yielded 1 case report of repetitive focal seizures and lingual dystonia,32 5 case reports of generalized tonic-clonic seizures and myoclonus,33-37 and 2 case reports of status epilepticus.38,39 Olanzapine’s clearance is 25% to 30% lower in women, and most of these case reports occurred women.40
Details of the above case reports are summarized in Table 1 (aripiprazole15-19), Table 2 (olanzapine32-39), and Table 3 (paliperidone,28,29 quetiapine,11-13 and risperidone22-24).
Ziprasidone. According to the NDA safety database, the seizure rate attributed to ziprasidone was 1.8 per 100 subject-years or 0.54% of participants (12 of 2,588).41 No additional studies have been published regarding its seizure risk.
Clozapine has a black-box warning
To the best of our knowledge, clozapine is the only antipsychotic that carries an FDA “black-box” warning regarding its risk of inducing seizures.42 Devinsky and Pacia43 reported a cumulative risk of 10% after 3.8 years of treatment. The literature has described clozapine-induced generalized tonic-clonic, myoclonic, simple and complex partial, and absence seizures.44 Table 445 lists the estimated frequency of each seizure type based on 101 cases of clozapine-induced seizures. Myoclonic seizures and drop attacks could be precursors/warning signs of grand mal tonic-clonic seizures.46,47 Seizures have been observed at all stages of treatment, but were more common during initiation of clozapine, which emphasizes the importance of a progressive and slow titration.43,48 The incidence of seizures was estimated to be 6% in a sample of 216 patients with schizophrenia with no history of epilepsy who were prescribed clozapine.49
Continue to: Regarding a possible association between...
Regarding a possible association between clozapine dose or clozapine plasma levels and seizure risk, there is a positive linear relationship between the dose of clozapine and its serum concentration over a dosing range of 25 to 800 mg/d.50 However, the plasma concentration is also significantly affected by factors such as smoking, gender, age, drug interactions, and CYP genotypes. Therefore, the same clozapine dose will yield a lower serum concentration in an older male who smokes compared with a younger, non-smoking female.51 Perry et al52 suggested a dosing nomogram to calculate the influence of gender and smoking. Seizure risk, especially for tonic-clonic seizures, has been reported to increase with clozapine doses >600 mg/d,53 and with plasma concentrations exceeding 1,000 to 1,300 mg/L.54 However, in a 2011 regression analysis, Varma et al55 found no statistically significant relationship between seizure risk and clozapine oral dose; there was not enough data to test a correlation between clozapine plasma levels and the incidence of seizures.
How antipsychotics might lower the seizure threshold
Researchers have suggested several possible mechanisms to explain how antipsychotics might lower the seizure threshold. Antagonism of dopamine D4, histamine H1, and acetylcholine-muscarinic receptors seems to induce EEG alterations and increase the risk of seizures.56 Additionally, modulation of the N-methyl-
Watch for pharmacokinetic interactions
The CYP enzymes involved in drug metabolism include CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Most commonly used antiepileptics and antipsychotics are metabolized by CYP enzymes, and may also act as inhibitors or inducers of these enzymes.61 Drug interactions may impair seizure control, which is why monotherapy is preferable to combination treatment in patients with epilepsy.62 Carbamazepine and phenytoin are inducers of both CYP1A2 (which metabolizes olanzapine and clozapine), and CYP3A4 (which metabolizes haloperidol, risperidone, quetiapine, ziprasidone and clozapine). Paliperidone is not metabolized by CYP enzymes.62 Discontinuing an enzyme-inducing agent may result in increased antipsychotic plasma concentrations, which might lead to an increased risk of seizures.
Valproic acid, which is often used to prevent or treat clozapine-induced seizures, has an unclear effect on clozapine plasma concentrations.63 Although valproic acid is known to inhibit clozapine metabolism, 2 reports have suggested that the plasma concentrations of clozapine and its metabolites may decrease after adding valproic acid.64,65 Other studies have found that valproic acid increases plasma concentrations of clozapine while it decreases plasma concentrations of norclozapine; norclozapine is the main clozapine metabolite responsible for inducing seizures.66,67
Steps for minimizing seizure risk
Determining the seizure risk for a patient taking an antipsychotic is challenging because doing so depends not only on the seizurogenic potential of each drug but also on individualized predisposing factors.11,57,68 Choosing the “best” antipsychotic therefore largely depends on each patient’s profile. The predisposing factors consist mainly of the individually inherited seizure threshold (personal history of febrile convulsions or a family history of seizures) and other comorbid seizurogenic conditions, such as a history of head trauma, brain injury, intellectual disability, cerebral arteriosclerosis, neurodegenerative diseases, encephalopathy, chronic renal insufficiency, and hyponatremia. Furthermore, seizure risk depends on the antipsychotic dose administered and the rate of titration.11
Continue to: There is not enough evidence...
There is not enough evidence to recommend performing an EEG in all patients taking antipsychotics. Such testing is recommended only for patients who have predisposing factors for seizures. If an EEG shows any abnormality in a patient taking clozapine, consider decreasing the clozapine dose69,70 or adding an antiepileptic drug such as valproic acid or lamotrigine.44,70
Although clozapine carries a black-box warning of increased risk of causing seizures, there is no consensus regarding the efficacy of co-prescribing an antiepileptic. Some studies have suggested prescribing valproic acid prophylactically,71 after the occurrence of 1 seizure,59 or after 2 seizures.54,72 Others have recommended prescribing prophylactic valproic acid for patients taking ≥600 mg/d of clozapine or whose clozapine plasma levels are >500 mg/L.73 Varma et al55 recommended starting an antiepileptic medication if there are clear epileptiform discharges on EEG, if the patient develops stuttering or speech difficulties, or if seizures occur. Liukkonen et al72 advised initiating an antiepileptic at the start of clozapine treatment in patients who are taking other epileptogenic medications, patients with pre-existing seizure disorder, and patients with neurologic abnormalities. On the other hand, Caetano51 argued against primary prevention of seizures for patients receiving >600 mg/d of clozapine, suggesting that “the risk of seizures would be better managed by close clinical monitoring and measures of clozapine serum concentration rather than adding an anticonvulsant drug.”
Current recommendations for primary and secondary prevention of clozapine-induced seizures are detailed in Table 5.42,44,45,51,55,57,69,74,75
Studies addressing the seizurogenic potential of SGAs other than clozapine have a low level of evidence and include patients who had comorbid conditions and were taking other medications that could cause seizures. Additionally, clinical trials of SGAs rarely include patients with seizure disorders; this might underestimate the risk of seizures.4
The effect of the mental illness itself on the seizure threshold needs to be considered.43 Bloechlinger et al8 found that dementia might be inherently associated with a higher risk of antipsychotic-related seizures. Moreover, numerous qualitative EEG studies have found abnormalities in 20% to 60% of patients with schizophrenia.56 Other quantitative studies have reported mild and nonspecific EEG abnormalities, such as increased delta and/or theta activity, in many non-medicated patients with schizophrenia.10,76 Additionally, brain tissue analysis of deceased patients who had schizophrenia has shown a significant increase in dopamine concentrations in the left amygdala compared with controls, and this might be responsible for enhanced electrical activity in this region.10 Some studies have described EEG slowing in the frontal brain regions of patients with schizophrenia,77 and was selectively normalized in these areas with antipsychotics.78
As always, start low, go slow
Mounting evidence suggests that antipsychotic medications decrease the seizure threshold. Practitioners should thus be cautious in prescribing antipsychotics and should target reaching the minimal effective dose with slow titration, especially in patients with predisposing factors for epilepsy.
Continue to: Although evidence suggests...
Although evidence suggests antipsychotics can induce different types of epileptic seizures, the quality of this evidence is low. Randomized controlled trials are needed to determine which antipsychotics increase seizure risk and whether there is a dose-effect relationship.
Bottom Line
Among second-generation antipsychotics, clozapine appears to increase the risk of clinical seizure the most. Correlations with dosage and/or plasma levels have not been proven. Psychiatrists should be vigilant for pharmacokinetic interactions between antipsychotics and antiepileptics, notably via CYP1A2 and CYP3A4.
Related Resources
- Druschky K, Bleich S, Grohmann R, et al. Seizure rates under treatment with antipsychotic drugs: Data from the AMSP project. World J Biol Psychiatry. 2018;15:1-10.
- Epilepsy Foundation. For professionals: Antipsychotics. https://www.epilepsy.com/learn/professionals/diagnosistreatment/psychotropic-drugs-developmental-disabilities/comorbid-5.
Drug Brand Names
Aripiprazole • Abilify
Benztropine • Cogentin
Bethanechol • Duvoid
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cimetidine • Tagamet
Ciprofloxacin • Cipro
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Donepezil • Aricept
Enalapril • Vasotec
Erythromycin • Erythrocin
Escitalopram • Lexapro
Flunitrazepam • Rohypnol
Fluvoxamine • Luvox
Gabapentin • Neurontin
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Metformin • Fortamet, Glucophage
Mirtazapine • Remeron
Nitrofurantoin • Furadantin
Olanzapine • Zyprexa
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Prochlorperazine • Compazine
Procyclidine • Kemadrin
Propranolol • Inderal
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Simvastatin • Zocor
Sulfamethoxazole/trimethoprim • Bactrim, Sulfatrim
Topiramate • Topamax
Trifluoperazine • Stelazine
Valproic acid • Depakene, Depakote
Ziprasidone • Geodon
Antipsychotics, especially second-generation antipsychotics (SGAs), have been proven effective for treating psychosis as well as mood disorders.1,2 Because antipsychotics can lower the epileptogenic threshold, seizures are a serious potential adverse effect. Antipsychotics can cause isolated EEG abnormalities in 7% of patients with no history of epilepsy, and clinical seizures in .5% to 1.2% of such patients.3 Additionally, the neuropathophysiology underlying epilepsy can predispose patients to psychiatric disorders4; the estimated prevalence of psychosis in patients with epilepsy is approximately 7%.5 This review will shed light on the risk of clinical seizures related to antipsychotics.
Comparing seizure risk among antipsychotics
In a review of the World Health Organization’s adverse drug reactions database, Kumlien and Lundberg6 calculated the ratio of the number of reports of seizures to the total number of reports for each drug. They found that approximately 9% of all adverse drug reaction reports involving clozapine were due to seizures. Equivalent ratios were 5.90% for quetiapine, 4.91% for olanzapine, 3.68% for risperidone, 3.27% for haloperidol, and 2.59% for aripiprazole. Using the database of the Pharmacovigilance Unit of the Basque Country, Lertxundi et al7 reported a 3.2-fold increased risk of seizure with SGAs in comparison with first-generation antipsychotics (FGAs) (95% confidence interval [CI], 2.21 to 4.63), which went down to 2.08 (CI, 1.39 to 3.12) once clozapine was excluded. However, as the authors of both studies noted, the quality and relevance of this data are limited because it relies on spontaneous reporting.
Overall, the evidence regarding the seizure risk associated with antipsychotics is scarce. To the best of our knowledge, only 2 large observational studies have compared the seizure risks associated with different antipsychotics.
Using data from the UK-based Clinical Practice Research Datalink between 1998 and 2013, Bloechlinger et al8 examined the incidence rates of seizures among patients newly diagnosed with schizophrenia, affective disorders, or dementia who were prescribed antipsychotics. They excluded patients with a history of seizures or antiepileptic use. In the cohort of 60,121 patients, the incidence rates of seizures per 10,000 person-years were 11.7 (CI, 10.0 to 13.4) for those who did not use antipsychotics, 12.4 (CI, 10.9 to 13.8) for past users, 115.4 (CI, 50.1 to 180.7) for current users of haloperidol, 48.8 (CI, 30.7 to 66.9) for current users of quetiapine, 25.9 (CI, 11.8 to 40.0) for current users of risperidone, and 19.0 (CI, 8.7 to 29.3) for current users of olanzapine. No data were available about clozapine use.
In subsequent analyses, the authors found that among patients with affective disorders, only current use of medium- to high-potency FGAs (haloperidol, prochlorperazine, and trifluoperazine) was associated with a significantly increased risk of seizures (adjusted odds ratio: 2.51, CI, 1.51 to 4.18) compared with non-users.8 Among patients with dementia, current use of olanzapine or quetiapine and current use of any FGAs were associated with significantly increased odds of seizures. This study suggests that the underlying mental illness might modulate the seizure risk associated with antipsychotics.8
Wu et al9 conducted a study based on the National Health Insurance Research Database in Taiwan. They examined the 1-year incidence of new-onset seizures among patients diagnosed with schizophrenia or mood disorders who were new to antipsychotic treatment, and calculated the risk of seizure associated with each antipsychotic in reference to risperidone. They found that those receiving clozapine, thioridazine, and haloperidol were 2 to 3 times more likely to develop seizures than those treated with risperidone; risks associated with the rest of the FGAs were similar to that of risperidone.
The results of these 2 large cohort studies are somewhat concurrent in indicating that, other than clozapine, SGAs incur similar risks of seizures; furthermore, they specify that, contrary to earlier studies,10 haloperidol is associated with significantly higher odds of seizures. While both of these cohort studies controlled for several sociodemographic and clinical confounders, they have several limitations. First, diagnoses of seizures were based on information available in databases, which might be subject to inaccuracies. Second, neither study evaluated the effect of drug dosage and duration of exposure on new-onset seizures.
Continue to: Most evidence is from case reports
Most evidence is from case reports
Other than these 2 large studies, most of the evidence addressing the relationship between the use of antipsychotics and incidence of seizures is low quality and relies on case reports or expert opinions. Older studies found that, among FGAs, seizure risk is highest with chlorpromazine and promazine, and lowest with thioridazine and haloperidol.10 As for SGAs, case reports have described seizuresassociated with the use of quetiapine, aripiprazole, risperidone, paliperidone, and olanzapine.
Quetiapine. Three case reports published between 2002 and 2010 describe generalized
Aripiprazole. Five case reports described staring spells and tonic-clonic seizures in patients receiving 10 to 15 mg of aripiprazole.15-19 In the New Drug Application (NDA) for aripiprazole, the incidence of seizures was estimated to be .11% (1 of 926 patients) in placebo-controlled trials and .46% (3 of 859 patients) in haloperidol-controlled trials.20
Risperidone’s product labeling suggests the drug should be used with caution in patients with a history of seizures or conditions that could result in a lower seizure threshold. In Phase III placebo-controlled trials, seizures occurred in .3% of patients treated with risperidone, although in some cases, the seizures were induced by electrolyte disturbances such as hyponatremia.21 Gonzalez-Heydrich et al22 and Holzhausen et al23 found no increase in seizure activity among patients with epilepsy who were receiving risperidone. Lane et al24 published a case report of a geriatric woman who presented with a generalized tonic-clonic seizure related to rapid titration of risperidone; however, with slower titration and lower doses, she stopped having seizures without adding any antiepileptic drugs. Komossa et al25 found that risperidone is less epileptogenic than clozapine, with a relative risk of .22.
Paliperidone is the active metabolite of risperidone and does not have pharmacokinetic interactions with drugs metabolized by the cytochrome P450 (CYP) enzymes. Its labeling indicates that the drug should be used with caution in patients with a history of seizures.26 In Phase III placebo-controlled trials of paliperidone, the rate of seizures was .22%.27 Two case reports suggest close monitoring of seizure risk in patients receiving paliperidone.28,29 Liang et al29 reported that co-administration of valproic acid could mask an underlying decrease of the seizure threshold caused by antipsychotics such as paliperidone.
Continue to: Olanzapine
Olanzapine is a thienobenzodiazepine derivative and is chemically related to clozapine.30 The olanzapine NDA31 shows that 23 of 3,139 patients developed seizures, mainly tonic-clonic, with evidence suggesting that the seizures may have been due to confounding factors such as a history of seizures or metabolic abnormalities. There were no statistically significant differences in the rate of seizures associated with olanzapine compared with placebo or haloperidol (P = .252 and .168, respectively).
A literature review for olanzapine yielded 1 case report of repetitive focal seizures and lingual dystonia,32 5 case reports of generalized tonic-clonic seizures and myoclonus,33-37 and 2 case reports of status epilepticus.38,39 Olanzapine’s clearance is 25% to 30% lower in women, and most of these case reports occurred women.40
Details of the above case reports are summarized in Table 1 (aripiprazole15-19), Table 2 (olanzapine32-39), and Table 3 (paliperidone,28,29 quetiapine,11-13 and risperidone22-24).
Ziprasidone. According to the NDA safety database, the seizure rate attributed to ziprasidone was 1.8 per 100 subject-years or 0.54% of participants (12 of 2,588).41 No additional studies have been published regarding its seizure risk.
Clozapine has a black-box warning
To the best of our knowledge, clozapine is the only antipsychotic that carries an FDA “black-box” warning regarding its risk of inducing seizures.42 Devinsky and Pacia43 reported a cumulative risk of 10% after 3.8 years of treatment. The literature has described clozapine-induced generalized tonic-clonic, myoclonic, simple and complex partial, and absence seizures.44 Table 445 lists the estimated frequency of each seizure type based on 101 cases of clozapine-induced seizures. Myoclonic seizures and drop attacks could be precursors/warning signs of grand mal tonic-clonic seizures.46,47 Seizures have been observed at all stages of treatment, but were more common during initiation of clozapine, which emphasizes the importance of a progressive and slow titration.43,48 The incidence of seizures was estimated to be 6% in a sample of 216 patients with schizophrenia with no history of epilepsy who were prescribed clozapine.49
Continue to: Regarding a possible association between...
Regarding a possible association between clozapine dose or clozapine plasma levels and seizure risk, there is a positive linear relationship between the dose of clozapine and its serum concentration over a dosing range of 25 to 800 mg/d.50 However, the plasma concentration is also significantly affected by factors such as smoking, gender, age, drug interactions, and CYP genotypes. Therefore, the same clozapine dose will yield a lower serum concentration in an older male who smokes compared with a younger, non-smoking female.51 Perry et al52 suggested a dosing nomogram to calculate the influence of gender and smoking. Seizure risk, especially for tonic-clonic seizures, has been reported to increase with clozapine doses >600 mg/d,53 and with plasma concentrations exceeding 1,000 to 1,300 mg/L.54 However, in a 2011 regression analysis, Varma et al55 found no statistically significant relationship between seizure risk and clozapine oral dose; there was not enough data to test a correlation between clozapine plasma levels and the incidence of seizures.
How antipsychotics might lower the seizure threshold
Researchers have suggested several possible mechanisms to explain how antipsychotics might lower the seizure threshold. Antagonism of dopamine D4, histamine H1, and acetylcholine-muscarinic receptors seems to induce EEG alterations and increase the risk of seizures.56 Additionally, modulation of the N-methyl-
Watch for pharmacokinetic interactions
The CYP enzymes involved in drug metabolism include CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Most commonly used antiepileptics and antipsychotics are metabolized by CYP enzymes, and may also act as inhibitors or inducers of these enzymes.61 Drug interactions may impair seizure control, which is why monotherapy is preferable to combination treatment in patients with epilepsy.62 Carbamazepine and phenytoin are inducers of both CYP1A2 (which metabolizes olanzapine and clozapine), and CYP3A4 (which metabolizes haloperidol, risperidone, quetiapine, ziprasidone and clozapine). Paliperidone is not metabolized by CYP enzymes.62 Discontinuing an enzyme-inducing agent may result in increased antipsychotic plasma concentrations, which might lead to an increased risk of seizures.
Valproic acid, which is often used to prevent or treat clozapine-induced seizures, has an unclear effect on clozapine plasma concentrations.63 Although valproic acid is known to inhibit clozapine metabolism, 2 reports have suggested that the plasma concentrations of clozapine and its metabolites may decrease after adding valproic acid.64,65 Other studies have found that valproic acid increases plasma concentrations of clozapine while it decreases plasma concentrations of norclozapine; norclozapine is the main clozapine metabolite responsible for inducing seizures.66,67
Steps for minimizing seizure risk
Determining the seizure risk for a patient taking an antipsychotic is challenging because doing so depends not only on the seizurogenic potential of each drug but also on individualized predisposing factors.11,57,68 Choosing the “best” antipsychotic therefore largely depends on each patient’s profile. The predisposing factors consist mainly of the individually inherited seizure threshold (personal history of febrile convulsions or a family history of seizures) and other comorbid seizurogenic conditions, such as a history of head trauma, brain injury, intellectual disability, cerebral arteriosclerosis, neurodegenerative diseases, encephalopathy, chronic renal insufficiency, and hyponatremia. Furthermore, seizure risk depends on the antipsychotic dose administered and the rate of titration.11
Continue to: There is not enough evidence...
There is not enough evidence to recommend performing an EEG in all patients taking antipsychotics. Such testing is recommended only for patients who have predisposing factors for seizures. If an EEG shows any abnormality in a patient taking clozapine, consider decreasing the clozapine dose69,70 or adding an antiepileptic drug such as valproic acid or lamotrigine.44,70
Although clozapine carries a black-box warning of increased risk of causing seizures, there is no consensus regarding the efficacy of co-prescribing an antiepileptic. Some studies have suggested prescribing valproic acid prophylactically,71 after the occurrence of 1 seizure,59 or after 2 seizures.54,72 Others have recommended prescribing prophylactic valproic acid for patients taking ≥600 mg/d of clozapine or whose clozapine plasma levels are >500 mg/L.73 Varma et al55 recommended starting an antiepileptic medication if there are clear epileptiform discharges on EEG, if the patient develops stuttering or speech difficulties, or if seizures occur. Liukkonen et al72 advised initiating an antiepileptic at the start of clozapine treatment in patients who are taking other epileptogenic medications, patients with pre-existing seizure disorder, and patients with neurologic abnormalities. On the other hand, Caetano51 argued against primary prevention of seizures for patients receiving >600 mg/d of clozapine, suggesting that “the risk of seizures would be better managed by close clinical monitoring and measures of clozapine serum concentration rather than adding an anticonvulsant drug.”
Current recommendations for primary and secondary prevention of clozapine-induced seizures are detailed in Table 5.42,44,45,51,55,57,69,74,75
Studies addressing the seizurogenic potential of SGAs other than clozapine have a low level of evidence and include patients who had comorbid conditions and were taking other medications that could cause seizures. Additionally, clinical trials of SGAs rarely include patients with seizure disorders; this might underestimate the risk of seizures.4
The effect of the mental illness itself on the seizure threshold needs to be considered.43 Bloechlinger et al8 found that dementia might be inherently associated with a higher risk of antipsychotic-related seizures. Moreover, numerous qualitative EEG studies have found abnormalities in 20% to 60% of patients with schizophrenia.56 Other quantitative studies have reported mild and nonspecific EEG abnormalities, such as increased delta and/or theta activity, in many non-medicated patients with schizophrenia.10,76 Additionally, brain tissue analysis of deceased patients who had schizophrenia has shown a significant increase in dopamine concentrations in the left amygdala compared with controls, and this might be responsible for enhanced electrical activity in this region.10 Some studies have described EEG slowing in the frontal brain regions of patients with schizophrenia,77 and was selectively normalized in these areas with antipsychotics.78
As always, start low, go slow
Mounting evidence suggests that antipsychotic medications decrease the seizure threshold. Practitioners should thus be cautious in prescribing antipsychotics and should target reaching the minimal effective dose with slow titration, especially in patients with predisposing factors for epilepsy.
Continue to: Although evidence suggests...
Although evidence suggests antipsychotics can induce different types of epileptic seizures, the quality of this evidence is low. Randomized controlled trials are needed to determine which antipsychotics increase seizure risk and whether there is a dose-effect relationship.
Bottom Line
Among second-generation antipsychotics, clozapine appears to increase the risk of clinical seizure the most. Correlations with dosage and/or plasma levels have not been proven. Psychiatrists should be vigilant for pharmacokinetic interactions between antipsychotics and antiepileptics, notably via CYP1A2 and CYP3A4.
Related Resources
- Druschky K, Bleich S, Grohmann R, et al. Seizure rates under treatment with antipsychotic drugs: Data from the AMSP project. World J Biol Psychiatry. 2018;15:1-10.
- Epilepsy Foundation. For professionals: Antipsychotics. https://www.epilepsy.com/learn/professionals/diagnosistreatment/psychotropic-drugs-developmental-disabilities/comorbid-5.
Drug Brand Names
Aripiprazole • Abilify
Benztropine • Cogentin
Bethanechol • Duvoid
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cimetidine • Tagamet
Ciprofloxacin • Cipro
Citalopram • Celexa
Clonazepam • Klonopin
Clozapine • Clozaril
Donepezil • Aricept
Enalapril • Vasotec
Erythromycin • Erythrocin
Escitalopram • Lexapro
Flunitrazepam • Rohypnol
Fluvoxamine • Luvox
Gabapentin • Neurontin
Haloperidol • Haldol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Metformin • Fortamet, Glucophage
Mirtazapine • Remeron
Nitrofurantoin • Furadantin
Olanzapine • Zyprexa
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Prochlorperazine • Compazine
Procyclidine • Kemadrin
Propranolol • Inderal
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Simvastatin • Zocor
Sulfamethoxazole/trimethoprim • Bactrim, Sulfatrim
Topiramate • Topamax
Trifluoperazine • Stelazine
Valproic acid • Depakene, Depakote
Ziprasidone • Geodon
1. Bruijnzeel D, Suryadevara U, Tandon R. Antipsychotic treatment of schizophrenia: an update. Asian J Psychiatr. 2014;11:3-7.
2. Hrdlicka M, Dudova I. Atypical antipsychotics in the treatment of early-onset schizophrenia. Neuropsychiatr Dis Treat. 2015;11:907-913.
3. Koch-Stoecker S. Antipsychotic drugs and epilepsy: indications and treatment guidelines. Epilepsia. 2002;43(suppl 2):19-24.
4. Alper K, Schwartz KA, Kolts RL, et al. Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry. 2007;62(4):345-354.
5. Torta R, Keller R. Behavioral, psychotic, and anxiety disorders in epilepsy: etiology, clinical features, and therapeutic implications. Epilepsia. 1999;40(suppl 10):S2-S20.
6. Kumlien E, Lundberg PO. Seizure risk associated with neuroactive drugs: data from the WHO adverse drug reactions database. Seizure. 2010;19(2):69-73.
7. Lertxundi U, Hernandez R, Medrano J, et al. Antipsychotics and seizures: higher risk with atypicals? Seizure. 2013;22(2):141-143.
8. Bloechliger M, Rüegg S, Jick SS, et al. Antipsychotic drug use and the risk of seizures: follow-up study with a nested case-control analysis. CNS Drugs. 2015;29(7):591-603.
9. Wu CS, Wang SC, Yeh IJ, et al. Comparative risk of seizure with use of first- and second-generation antipsychotics in patients with schizophrenia and mood disorders. J Clin Psychiatry. 2016;77(5):e573-e579.
10. Cold JA, Wells BG, Froemming JH. Seizure activity associated with antipsychotic therapy. [Erratum in DICP. 1990;24(10):1012.] DICP. 1990;24(6):601-606.
11. Hedges DW, Jeppson KG. New-onset seizure associated with quetiapine and olanzapine. Ann Pharmacother. 2002;36(3):437-439.
12. Dogu O, Sevim S, Kaleagasi HS. Seizures associated with quetiapine treatment. Ann Pharmacother. 2003;37(9):1224-1227.
13. Young AC, Kleinschmidt KC, Wax PM. Late-onset seizures associated with quetiapine poisoning. J Med Toxicol. 2009;5(1):24-26.
14. US Food and Drug Administration. Recommendation of approvable action for quetiapine fumarate extended release (Seroquel® XR) for the treatment of schizophrenia. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022047Orig1s000MedR.pdf. April 24, 2007. Accessed January 28, 2019.
15. Malik AR, Ravasia S. Aripiprazole-induced seizure. Can J Psychiatry. 2005;50(3):186.
16. Tsai JF. Aripiprazole-associated seizure. J Clin Psychiatry. 2006;67(6):995-996.
17. Arora M, Arndorfer L. EEG abnormalities in a patient taking aripiprazole. Psychiatry (Edgmont). 2007;4(7):18-19.
18. Yueh CL, Yu SL, Chen HM, et al. Aripiprazole-induced seizure: a second case report. BMJ case reports. 2009;2009:bcr03.2009.1693. doi: 10.1136/bcr.03.2009.1693.
19. Thabet FI, Sweis RT, Joseph SA. Aripiprazole-induced seizure in a 3-year-old child: a case report and literature review. Clin Neuropharmacol. 2013;36(1):29-30.
20. US Food and Drug Administration. Abilify (Aripiprazole) tablets. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-436_Abilify_medr_P2.pdf. Published March 07, 2003. Accessed January 28, 2019.
21. US Food and Drug Administration. Drugs@FDA: FDA approved drug products. Risperdal tablets, Risperdal oral solution & Risperdal M-tab orally disintegrating tablets. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021444_S004_RISPERDAL_TABLETS.pdf. Published September 10, 2003. Accessed January 28, 2019.
22. Gonzalez-Heydrich J, Pandina GJ, Fleisher CA, et al. No seizure exacerbation from risperidone in youth with comorbid epilepsy and psychiatric disorders: a case series. J Child Adolesc Psychopharmacol. 2004;14(2):295-310.
23. Holzhausen SPF, Guerreiro MM, Baccin CE, et al. Use of risperidone in children with epilepsy. Epilepsy Behav. 2007;10(3):412-416.
24. Lane HY, Chang WH, Chou JC. Seizure during risperidone treatment in an elderly woman treated with concomitant medications. J Clinl Psychiatry. 1998;59(2):81-82.
25. Komossa K, Rummel-Kluge C, Schwarz S, et al. Risperidone versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2011;(1):19:CD006626.
26. Paliperidone [package insert]. Mountainville, CA: Janssen Pharmaceuticals, Inc.; 2007.
27. Brugge, MD; US Food and Drug Administration. Paliperidone OROS oral formulation. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021999s000_MedR_Part4.pdf. Accessed January 28, 2019.
28. Schneider RA, Lizer MH. Apparent seizure and atrial fibrillation associated with paliperidone. Am J Health System Pharm. 2008;65(22):2122-2125.
29. Liang CS, Yang FW, Chiang KT. Paliperidone-associated seizure after discontinuation of sodium valproate: a case report. J Clin Psychopharmacol. 2011;31(2):246-247.
30. Fulton B, Goa KL. Olanzapine. A review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs. 1997;53(2):281-298.
31. US Food and Drug Administration. Drugs@FDA: FDA approved drug products: Zyprexa (olanzapine). ORIG-1. http://www.accessdata.fda.gov/drugsatfda_docs/nda/96/020592_Original_Approval_Pkg%20.pdf. Published September 30, 1996. Accessed January 28, 2019.
32. Anzellotti F, Capasso M, Frazzini V, et al. Olanzapine-related repetitive focal seizures with lingual dystonia. Epileptic Disord. 2016;18(1):83-86.
33. Lee JW, Crismon ML, Dorson PG. Seizure associated with olanzapine. Ann Pharmac. 1999;33(5):554-556.
34. Woolley J, Smith S. Lowered seizure threshold on olanzapine. Br J Psychiatry. 2001;178(1):85-86.
35. Behere RV, Anjith D, Rao NP, et al. Olanzapine-induced clinical seizure: a case report. Clin Neuropharmacol. 2009;32(5):297-298.
36. Camacho A, García-Navarro M, Martínez B, et al. Olanzapine-induced myoclonic status. Clin Neuropharmacol. 2005;28(3):145-147.
37. Rosen JB, Milstein MJ, Haut SR. Olanzapine-associated myoclonus. Epilepsy Res. 2012;98(2-3):247-250.
38. Wyderski RJ, Starrett WG, Abou-Saif A. Fatal status epilepticus associated with olanzapine therapy. Ann Pharmacother. 1999;33(7-8):787-789.
39. Spyridi S, Sokolaki S, Nimatoudis J, et al. Status epilepticus in a patient treated with olanzapine and mirtazapine. Int J Clin Pharmacol Ther. 2009;47(2):120-123.
40. Schatzberg AF, Nemeroff CB. Essentials of clinical psychopharmacology. 2nd ed. Arlington, Virginia: American Psychiatric Publishing; 2006.
41. US Food and Drug Administration. Drug approval package: Geodon (Ziprasidone HCI) Capsules. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20-825_Geodan_medr_P2.pdf. Published February 5, 2001. Accessed January 29, 2019.
42. Clozaril [package insert]. East Hanover, NJ: Novartis; 2008.
43. Devinsky O, Pacia SV. Seizures during clozapine therapy. J Clin Psychiatry. 1994;55(suppl B):153-156.
44. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
45. Wong J, Delva N. Clozapine-induced seizures: recognition and treatment. Can J Psychiatry. 2007;52(7):457-463.
46. Berman I, Zalma A, DuRand CJ, et al. Clozapine-induced myoclonic jerks and drop attacks. J Clin Psychiatry. 1992;53(9):329-330.
47. Gouzoulis E, Ozdaglar A, Kasper J. Myoclonic seizures followed by grand mal seizures during clozapine treatment. Am J Psychiatry. 1993;150(7):1128.
48. Sajatovic M, Meltzer HY. Clozapine-induced myoclonus and generalized seizures. Biol Psychiatry. 1996;39(5):367-370.
49. Grover S, Hazari N, Chakrabarti S, et al. Association of clozapine with seizures: a brief report involving 222 patients prescribed clozapine. East Asian Arch Psychiatry. 2015;25(2):73-78.
50. Byerly MJ, DeVane CL. Pharmacokinetics of clozapine and risperidone: a review of recent literature. J Clin Psychopharmacol. 1996;16(2):177-187.
51. Caetano D. Use of anticonvulsants as prophylaxis for seizures in patients on clozapine. Australas Psychiatry. 2014;22(1):78-83.
52. Perry PJ, Bever KA, Arndt S, et al. Relationship between patient variables and plasma clozapine concentrations: a dosing nomogram. Biol Psychiatry.1998;44(8):733-738.
53. Dumortier G, Mahé V, Pons D, et al. Clonic seizure associated with high clozapine plasma level. J Neuropsychiatry Clin Neurosci. 2001;13(2):302-303.
54. Funderburg LG, Vertrees JE, True JE, et al. Seizure following addition of erythromycin to clozapine treatment. Am J Psychiatry. 1994;151(12):1840-1841.
55. Varma S, Bishara D, Besag FMC, et al. Clozapine-related EEG changes and seizures: dose and plasma-level relationships. Ther Adv Psychopharmacol. 2011;1(2):47-66.
56. Amann BL, Pogarell O, Mergl R, et al. EEG abnormalities associated with antipsychotics: a comparison of quetiapine, olanzapine, haloperidol and healthy subjects. Hum Psychopharmacol. 2003;18(8):641-646.
57. Pisani F, Oteri G, Costa C, et al. Effects of psychotropic drugs on seizure threshold. Drug Saf. 2002;25(2):91-110.
58. Maurice T, Phan VL, Urani A, et al. Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jpn J Pharmacol. 1999;81(2):125-155.
59. Haller E, Binder RL. Clozapine and seizures. Am J Psychiatry. 1990;147(8):1069-1071.
60. Torta R, Monaco F. Atypical antipsychotics and serotoninergic antidepressants in patients with epilepsy: pharmacodynamic considerations. Epilepsia. 2002;43(suppl 2):8-13.
61. Spina E. Drug interactions. In: Shorvon S, Perucca E, Engel J Jr, eds. The treatment of epilepsy. 3rd ed. Oxford, UK: Blackwell Publishing; 2009:361-377.
62. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
63. de Leon J, Santoro V, D’Arrigo C, et al. Interactions between antiepileptics and second-generation antipsychotics. Expert Opin Drug Metab Toxicol. 2012;8(3):311-334.
64. Finley P, Warner D. Potential impact of valproic acid therapy on clozapine disposition. Biol Psychiatry. 1994;36(7):487-488.
65. Longo LP, Salzman C. Valproic acid effects on serum concentrations of clozapine and norclozapine. Am J Psychiatry. 1995;152(4):650.
66. Centorrino F, Baldessarini RJ, Kando J, et al. Serum concentrations of clozapine and its major metabolites: effects of cotreatment with fluoxetine or valproate. Am J Psychiatry. 1994;151(1):123-125.
67. Facciolà G, Avenoso A, Scordo MG, et al. Small effects of valproic acid on the plasma concentrations of clozapine and its major metabolites in patients with schizophrenic or affective disorders. Ther Drug Monit. 1999;21(3):341-345.
68. Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23(4):611-622.
69. Muzyk A, Gala G, Kahn DA. Use of lamotrigine in a patient with a clozapine-related seizure. J Psychiatr Pract. 2010;16(2):125-128.
70. Kikuchi YS, Sato W, Ataka K, et al. Clozapine-induced seizures, electroencephalography abnormalities, and clinical responses in Japanese patients with schizophrenia. Neuropsychiatr Dis Treat. 2014;10:1973-1978.
71. Taner E, Coşar B, Işik E. Clozapine-induced myoclonic seizures and valproic acid. Int J Psychiatry Clin Pract. 1998;2(1):53-55.
72. Liukkonen J, Koponen HJ, Nousiainen U. Clinical picture and long-term course of epileptic seizures that occur during clozapine treatment. Psychiatry Res. 1992;44(2):107-112.
73. Devinsky O, Honigfeld G, Patin J. Clozapine-related seizures. Neurology. 1991;41(3):369-371.
74. Foster R, Olajide D. A case of clozapine-induced tonic-clonic seizures managed with valproate: implications for clinical care. J Psychopharmacol. 2005;19(1):93-96.
75. Gandelman-Marton R, Theitler J, Klein C, et al. Phenytoin intoxication in a clozapine-related prolonged seizure. J Emerg Med. 2008;35(4):407-409.
76. Primavera A, Giberti L, Scotto P, et al. Nonconvulsive status epilepticus as a cause of confusion in later life: a report of 5 cases. Neuropsychobiology. 1994;30(2-3):148-152.
77. Boutros NN, Arfken C, Galderisi S, et al. The status of spectral EEG abnormality as a diagnostic test for schizophrenia. Schizophrenia Res. 2008;99(1-3):225-237.
78. Takahashi T, Cho RY, Mizuno T, et al. Antipsychotics reverse abnormal EEG complexity in drug-naïve schizophrenia: a multiscale entropy analysis. Neuroimage. 2010;51(1):173-182.
1. Bruijnzeel D, Suryadevara U, Tandon R. Antipsychotic treatment of schizophrenia: an update. Asian J Psychiatr. 2014;11:3-7.
2. Hrdlicka M, Dudova I. Atypical antipsychotics in the treatment of early-onset schizophrenia. Neuropsychiatr Dis Treat. 2015;11:907-913.
3. Koch-Stoecker S. Antipsychotic drugs and epilepsy: indications and treatment guidelines. Epilepsia. 2002;43(suppl 2):19-24.
4. Alper K, Schwartz KA, Kolts RL, et al. Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry. 2007;62(4):345-354.
5. Torta R, Keller R. Behavioral, psychotic, and anxiety disorders in epilepsy: etiology, clinical features, and therapeutic implications. Epilepsia. 1999;40(suppl 10):S2-S20.
6. Kumlien E, Lundberg PO. Seizure risk associated with neuroactive drugs: data from the WHO adverse drug reactions database. Seizure. 2010;19(2):69-73.
7. Lertxundi U, Hernandez R, Medrano J, et al. Antipsychotics and seizures: higher risk with atypicals? Seizure. 2013;22(2):141-143.
8. Bloechliger M, Rüegg S, Jick SS, et al. Antipsychotic drug use and the risk of seizures: follow-up study with a nested case-control analysis. CNS Drugs. 2015;29(7):591-603.
9. Wu CS, Wang SC, Yeh IJ, et al. Comparative risk of seizure with use of first- and second-generation antipsychotics in patients with schizophrenia and mood disorders. J Clin Psychiatry. 2016;77(5):e573-e579.
10. Cold JA, Wells BG, Froemming JH. Seizure activity associated with antipsychotic therapy. [Erratum in DICP. 1990;24(10):1012.] DICP. 1990;24(6):601-606.
11. Hedges DW, Jeppson KG. New-onset seizure associated with quetiapine and olanzapine. Ann Pharmacother. 2002;36(3):437-439.
12. Dogu O, Sevim S, Kaleagasi HS. Seizures associated with quetiapine treatment. Ann Pharmacother. 2003;37(9):1224-1227.
13. Young AC, Kleinschmidt KC, Wax PM. Late-onset seizures associated with quetiapine poisoning. J Med Toxicol. 2009;5(1):24-26.
14. US Food and Drug Administration. Recommendation of approvable action for quetiapine fumarate extended release (Seroquel® XR) for the treatment of schizophrenia. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022047Orig1s000MedR.pdf. April 24, 2007. Accessed January 28, 2019.
15. Malik AR, Ravasia S. Aripiprazole-induced seizure. Can J Psychiatry. 2005;50(3):186.
16. Tsai JF. Aripiprazole-associated seizure. J Clin Psychiatry. 2006;67(6):995-996.
17. Arora M, Arndorfer L. EEG abnormalities in a patient taking aripiprazole. Psychiatry (Edgmont). 2007;4(7):18-19.
18. Yueh CL, Yu SL, Chen HM, et al. Aripiprazole-induced seizure: a second case report. BMJ case reports. 2009;2009:bcr03.2009.1693. doi: 10.1136/bcr.03.2009.1693.
19. Thabet FI, Sweis RT, Joseph SA. Aripiprazole-induced seizure in a 3-year-old child: a case report and literature review. Clin Neuropharmacol. 2013;36(1):29-30.
20. US Food and Drug Administration. Abilify (Aripiprazole) tablets. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-436_Abilify_medr_P2.pdf. Published March 07, 2003. Accessed January 28, 2019.
21. US Food and Drug Administration. Drugs@FDA: FDA approved drug products. Risperdal tablets, Risperdal oral solution & Risperdal M-tab orally disintegrating tablets. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021444_S004_RISPERDAL_TABLETS.pdf. Published September 10, 2003. Accessed January 28, 2019.
22. Gonzalez-Heydrich J, Pandina GJ, Fleisher CA, et al. No seizure exacerbation from risperidone in youth with comorbid epilepsy and psychiatric disorders: a case series. J Child Adolesc Psychopharmacol. 2004;14(2):295-310.
23. Holzhausen SPF, Guerreiro MM, Baccin CE, et al. Use of risperidone in children with epilepsy. Epilepsy Behav. 2007;10(3):412-416.
24. Lane HY, Chang WH, Chou JC. Seizure during risperidone treatment in an elderly woman treated with concomitant medications. J Clinl Psychiatry. 1998;59(2):81-82.
25. Komossa K, Rummel-Kluge C, Schwarz S, et al. Risperidone versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2011;(1):19:CD006626.
26. Paliperidone [package insert]. Mountainville, CA: Janssen Pharmaceuticals, Inc.; 2007.
27. Brugge, MD; US Food and Drug Administration. Paliperidone OROS oral formulation. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021999s000_MedR_Part4.pdf. Accessed January 28, 2019.
28. Schneider RA, Lizer MH. Apparent seizure and atrial fibrillation associated with paliperidone. Am J Health System Pharm. 2008;65(22):2122-2125.
29. Liang CS, Yang FW, Chiang KT. Paliperidone-associated seizure after discontinuation of sodium valproate: a case report. J Clin Psychopharmacol. 2011;31(2):246-247.
30. Fulton B, Goa KL. Olanzapine. A review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs. 1997;53(2):281-298.
31. US Food and Drug Administration. Drugs@FDA: FDA approved drug products: Zyprexa (olanzapine). ORIG-1. http://www.accessdata.fda.gov/drugsatfda_docs/nda/96/020592_Original_Approval_Pkg%20.pdf. Published September 30, 1996. Accessed January 28, 2019.
32. Anzellotti F, Capasso M, Frazzini V, et al. Olanzapine-related repetitive focal seizures with lingual dystonia. Epileptic Disord. 2016;18(1):83-86.
33. Lee JW, Crismon ML, Dorson PG. Seizure associated with olanzapine. Ann Pharmac. 1999;33(5):554-556.
34. Woolley J, Smith S. Lowered seizure threshold on olanzapine. Br J Psychiatry. 2001;178(1):85-86.
35. Behere RV, Anjith D, Rao NP, et al. Olanzapine-induced clinical seizure: a case report. Clin Neuropharmacol. 2009;32(5):297-298.
36. Camacho A, García-Navarro M, Martínez B, et al. Olanzapine-induced myoclonic status. Clin Neuropharmacol. 2005;28(3):145-147.
37. Rosen JB, Milstein MJ, Haut SR. Olanzapine-associated myoclonus. Epilepsy Res. 2012;98(2-3):247-250.
38. Wyderski RJ, Starrett WG, Abou-Saif A. Fatal status epilepticus associated with olanzapine therapy. Ann Pharmacother. 1999;33(7-8):787-789.
39. Spyridi S, Sokolaki S, Nimatoudis J, et al. Status epilepticus in a patient treated with olanzapine and mirtazapine. Int J Clin Pharmacol Ther. 2009;47(2):120-123.
40. Schatzberg AF, Nemeroff CB. Essentials of clinical psychopharmacology. 2nd ed. Arlington, Virginia: American Psychiatric Publishing; 2006.
41. US Food and Drug Administration. Drug approval package: Geodon (Ziprasidone HCI) Capsules. Medical Review Part 2. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20-825_Geodan_medr_P2.pdf. Published February 5, 2001. Accessed January 29, 2019.
42. Clozaril [package insert]. East Hanover, NJ: Novartis; 2008.
43. Devinsky O, Pacia SV. Seizures during clozapine therapy. J Clin Psychiatry. 1994;55(suppl B):153-156.
44. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
45. Wong J, Delva N. Clozapine-induced seizures: recognition and treatment. Can J Psychiatry. 2007;52(7):457-463.
46. Berman I, Zalma A, DuRand CJ, et al. Clozapine-induced myoclonic jerks and drop attacks. J Clin Psychiatry. 1992;53(9):329-330.
47. Gouzoulis E, Ozdaglar A, Kasper J. Myoclonic seizures followed by grand mal seizures during clozapine treatment. Am J Psychiatry. 1993;150(7):1128.
48. Sajatovic M, Meltzer HY. Clozapine-induced myoclonus and generalized seizures. Biol Psychiatry. 1996;39(5):367-370.
49. Grover S, Hazari N, Chakrabarti S, et al. Association of clozapine with seizures: a brief report involving 222 patients prescribed clozapine. East Asian Arch Psychiatry. 2015;25(2):73-78.
50. Byerly MJ, DeVane CL. Pharmacokinetics of clozapine and risperidone: a review of recent literature. J Clin Psychopharmacol. 1996;16(2):177-187.
51. Caetano D. Use of anticonvulsants as prophylaxis for seizures in patients on clozapine. Australas Psychiatry. 2014;22(1):78-83.
52. Perry PJ, Bever KA, Arndt S, et al. Relationship between patient variables and plasma clozapine concentrations: a dosing nomogram. Biol Psychiatry.1998;44(8):733-738.
53. Dumortier G, Mahé V, Pons D, et al. Clonic seizure associated with high clozapine plasma level. J Neuropsychiatry Clin Neurosci. 2001;13(2):302-303.
54. Funderburg LG, Vertrees JE, True JE, et al. Seizure following addition of erythromycin to clozapine treatment. Am J Psychiatry. 1994;151(12):1840-1841.
55. Varma S, Bishara D, Besag FMC, et al. Clozapine-related EEG changes and seizures: dose and plasma-level relationships. Ther Adv Psychopharmacol. 2011;1(2):47-66.
56. Amann BL, Pogarell O, Mergl R, et al. EEG abnormalities associated with antipsychotics: a comparison of quetiapine, olanzapine, haloperidol and healthy subjects. Hum Psychopharmacol. 2003;18(8):641-646.
57. Pisani F, Oteri G, Costa C, et al. Effects of psychotropic drugs on seizure threshold. Drug Saf. 2002;25(2):91-110.
58. Maurice T, Phan VL, Urani A, et al. Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jpn J Pharmacol. 1999;81(2):125-155.
59. Haller E, Binder RL. Clozapine and seizures. Am J Psychiatry. 1990;147(8):1069-1071.
60. Torta R, Monaco F. Atypical antipsychotics and serotoninergic antidepressants in patients with epilepsy: pharmacodynamic considerations. Epilepsia. 2002;43(suppl 2):8-13.
61. Spina E. Drug interactions. In: Shorvon S, Perucca E, Engel J Jr, eds. The treatment of epilepsy. 3rd ed. Oxford, UK: Blackwell Publishing; 2009:361-377.
62. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
63. de Leon J, Santoro V, D’Arrigo C, et al. Interactions between antiepileptics and second-generation antipsychotics. Expert Opin Drug Metab Toxicol. 2012;8(3):311-334.
64. Finley P, Warner D. Potential impact of valproic acid therapy on clozapine disposition. Biol Psychiatry. 1994;36(7):487-488.
65. Longo LP, Salzman C. Valproic acid effects on serum concentrations of clozapine and norclozapine. Am J Psychiatry. 1995;152(4):650.
66. Centorrino F, Baldessarini RJ, Kando J, et al. Serum concentrations of clozapine and its major metabolites: effects of cotreatment with fluoxetine or valproate. Am J Psychiatry. 1994;151(1):123-125.
67. Facciolà G, Avenoso A, Scordo MG, et al. Small effects of valproic acid on the plasma concentrations of clozapine and its major metabolites in patients with schizophrenic or affective disorders. Ther Drug Monit. 1999;21(3):341-345.
68. Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23(4):611-622.
69. Muzyk A, Gala G, Kahn DA. Use of lamotrigine in a patient with a clozapine-related seizure. J Psychiatr Pract. 2010;16(2):125-128.
70. Kikuchi YS, Sato W, Ataka K, et al. Clozapine-induced seizures, electroencephalography abnormalities, and clinical responses in Japanese patients with schizophrenia. Neuropsychiatr Dis Treat. 2014;10:1973-1978.
71. Taner E, Coşar B, Işik E. Clozapine-induced myoclonic seizures and valproic acid. Int J Psychiatry Clin Pract. 1998;2(1):53-55.
72. Liukkonen J, Koponen HJ, Nousiainen U. Clinical picture and long-term course of epileptic seizures that occur during clozapine treatment. Psychiatry Res. 1992;44(2):107-112.
73. Devinsky O, Honigfeld G, Patin J. Clozapine-related seizures. Neurology. 1991;41(3):369-371.
74. Foster R, Olajide D. A case of clozapine-induced tonic-clonic seizures managed with valproate: implications for clinical care. J Psychopharmacol. 2005;19(1):93-96.
75. Gandelman-Marton R, Theitler J, Klein C, et al. Phenytoin intoxication in a clozapine-related prolonged seizure. J Emerg Med. 2008;35(4):407-409.
76. Primavera A, Giberti L, Scotto P, et al. Nonconvulsive status epilepticus as a cause of confusion in later life: a report of 5 cases. Neuropsychobiology. 1994;30(2-3):148-152.
77. Boutros NN, Arfken C, Galderisi S, et al. The status of spectral EEG abnormality as a diagnostic test for schizophrenia. Schizophrenia Res. 2008;99(1-3):225-237.
78. Takahashi T, Cho RY, Mizuno T, et al. Antipsychotics reverse abnormal EEG complexity in drug-naïve schizophrenia: a multiscale entropy analysis. Neuroimage. 2010;51(1):173-182.

What’s new in transcranial magnetic stimulation
Therapeutic neuromodulation takes advantage of the brain’s electrochemical makeup. This allows for treatment devices that modulate neurocircuits relevant to behaviors disrupted in disorders such as major depressive disorder (MDD) (eg, sleep quality, appetite, cognitive, and executive functions). The default mode network (comprised of structures such as the medial prefrontal cortex [MPFC], the posterior cingulate cortex, the hippocampus, and their functional connectivity) serves as a prime example of circuitry that can be targeted by this approach.1
For 80 years, electroconvulsive therapy (ECT) has been an important neuromodulation option for patients with more severe illness. Recently, additional neuromodulatory approaches have been FDA-cleared, including transcranial magnetic stimulation (TMS), vagus nerve stimulation (VNS), and deep brain stimulation (DBS). Another approach, transcranial direct current stimulation (tDCS), has been extensively studied for its potential clinical utility but is not FDA-cleared. The Table provides descriptions of these therapies.

Since being cleared by the FDA in 2008, TMS has arguably made the greatest strides in providing an alternate neuromodulation treatment option for patients with MDD, with >1,000 centers nationally and 7 TMS devices FDA-cleared for treatment of depression. In this article, we review recent developments in TMS.
An evolving therapeutic option
While primarily studied as a monotherapy for MDD, in clinical practice TMS (Box) is typically used as an adjunct to medication and psychotherapy.2,3 In this context, it has demonstrated efficacy for more difficult-to-treat mood disorders with an excellent safety and tolerability profile whether used with or without medication.4-6
To further improve the efficiency and efficacy of TMS while maintaining its safety and tolerability, researchers and clinicians have been exploring a few initiatives.
Box
- Transcranial magnetic stimulation (TMS) utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression
- Randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression
- Clinical availability of TMS has grown steadily over the past 10 years as >1,000 centers have been opened and additional devices have been FDA-cleared
- TMS has the potential to avoid safety and tolerability concerns associated with antidepressant pharmacotherapy (eg, weight gain, sexual dysfunction) and electroconvulsive therapy (eg, cognitive deficits)
- Greater sophistication in the choice of stimulation parameters, as well as other ongoing efforts to optimize the benefits of TMS, are yielding better clinical outcomes
Altered treatment parameters
One initiative is assessing the feasibility of altering various treatment parameters, such as the total number of treatment sessions (30 to 60 sessions); the frequency of sessions (eg, more than once daily); the total number of magnetic pulses per session (eg, >3,000); the stimulation coil localization (eg, left vs right dorsal lateral prefrontal cortex [DLPFC]; MPFC; and various methods to determine optimal coil placement (eg, EEG F3 coordinate or MRI-guided neuro-navigational methods). Such refinements offer the potential for enhanced efficacy, shorter treatment sessions, and/or improved tolerability. For example, lower frequency right DLPFC stimulations (eg, 1 Hz) can decrease the risk of seizures and improve overall tolerability. While this has not been studied as extensively as higher frequency left DLPFC stimulations (eg, 5 to 20 Hz), existing evidence supports similar efficacy between these 2 approaches.7
Theta burst stimulation. Some TMS devices can be adapted to deliver theta burst stimulation (TBS). This produces trains of triple, 50 Hz, pulsed bursts (usually with 200 ms inter-burst intervals occurring at a rate of 5 Hz; at 80% MT) to model naturally occurring theta rhythms. These bursts can be administered in stimulation protocols using intermittent TBS (iTBS) (eg, 10 bursts of triplets over 2 seconds every 10 seconds; 30 pulses per burst; for approximately 3 minutes; totaling 600 pulses) or continuous TBS (cTBS) bursts given in an uninterrupted train (eg, 40 seconds, 600 pulses). Evidence indicates these protocols facilitate long-term potentiation (ie, iTBS) and long-term depression (ie, cTBS), which in turn can modulate synaptic plasticity.
Continue to: While some clinicians are using...
While some clinicians are using TBS off-label, a recent non-inferiority trial (N = 395) reported similar efficacy and safety comparing standard 10 Hz TMS to an iTBS protocol at 120% of resting motor threshold (both over the left DLPFC).8 This has led to FDA clearance of the TMS device adapted to provide iTBS in this trial.8
From a more practical perspective, TBS has the potential to reduce the number of pulses (eg, 600 vs 3,000) and the total number of sessions required, as well as the duration of treatment sessions (eg, 37.5 minutes to <5 minutes). This can accelerate the time to response and decrease patient and staff commitment, with resulting cost savings.9 Despite this recent progress, ongoing research still needs to clarify issues such as the risk/benefit profile, particularly in younger and older populations, as well as assessment of duration of initial benefit and appropriate maintenance strategies.
New devices
Another initiative is the development of alternative TMS equipment. For example, newer coil designs with enhanced cooling ability allow for a substantial decrease in the required inter-train interval duration between stimulation trains, thus shortening the total session duration by approximately 50% (eg, from 37.5 to 19 minutes). The use of different coil arrays (eg, the H-coil capable of deeper vs surface stimulation) may allow for more direct stimulation of relevant neurocircuitry (eg, cingulate cortex), possibly improving efficacy and shortening time to onset of benefit. However, in head-to-head comparisons with single-coil devices, enhanced efficacy for depression has not been clearly demonstrated. One caveat is that the increase in depth of magnetic field penetration results in a loss of focality, resulting in the stimulation of larger brain areas. This might increase the risk of adverse effects such as seizures.
Increasing durability of effect
Because high relapse and recurrence rates compromise the initial benefit of any antidepressant therapy, appropriate maintenance strategies are essential. Several studies have evaluated strategies to maintain the acute benefit of TMS for treatment-resistant depression.
One was a 6-month, open-label TMS durability of effect trial for acute responders (n = 99) in the pivotal registration study.5 During this study, all participants were given antidepressant medication monotherapy. In addition, with early indication of relapse, patients received a reintroduction of TMS sessions (32/99 patients; mean number of sessions = 14.3). With this protocol, approximately 84% re-achieved their response status. The overall relapse rate was approximately 13%.5
Continue to: In a 1-year naturalistic study...
In a 1-year naturalistic study, 63% of patients (75/120) who met response or remission criteria after an acute course of TMS still met response criteria after 12 months. These patients received clinician-determined maintenance treatment that included reintroduction of TMS when indicated.3
In a prospective, 12-month, multisite, randomized pilot study, 67 patients with treatment-resistant MDD underwent an antidepressant medication washout and then received 30 sessions of TMS monotherapy.10 Those who met criteria for improvement (n = 49) were then randomized to once-monthly TMS or observation only. All patients remained medication-free but could receive TMS re-introduction if they deteriorated. At the end of the study, both groups demonstrated comparable outcomes, with a trend to a longer time before relapse among participants who received once-monthly TMS. Although these results are preliminary, they suggest that some patients could be treated both acutely and then maintained with TMS alone.
Re-introducing TMS in patients who show early signs of relapse after having an initial response achieves rates of sustained improvement that compare favorably with those of other strategies used to manage patients with treatment-resistant depression.
TMS vs ECT
The question often arises as to whether TMS is a viable alternate treatment to ECT. I believe the answer is unequivocally yes and no. By this, I mean some patients who in the past only had ECT as their next option when medications and psychotherapy were insufficient may now consider TMS. In support, there is evidence of comparable efficacy between TMS and ECT in a subgroup of patients who were considered clinically appropriate for ECT.11-13
How to best identify this group remains unclear, but investigators are exploring predictive biomarkers. For example, a large study (N = 1,188), with functional magnetic resonance imaging (fMRI) reported that depressed patients could be divided into 4 neurophysiological “biotypes” based on different patterns of aberrant connectivity in limbic and fronto-striatal networks.14 The authors further noted that such distinctions were helpful in predicting response in a subgroup of patients (n = 154) who received TMS.
Continue to: For now...
For now, experience indicates certain clinical factors may provide some guidance. Patients are usually better served by ECT if they:
- have depressive episodes of longer duration (eg, >3 years)
- have a high risk of suicide
- have psychotic or catatonic features associated with their depression
- have difficulty maintaining their physical well-being
- have bipolar depression.
Although existing evidence supports a possible benefit with TMS for bipolar depression (used in combination with a mood stabilizer), the lack of a definitive trial (precluding FDA clearance for this indication) and the lack of insurance coverage both limit the routine use of TMS for this indication.15
One potential advantage of TMS over ECT is a lower cost.13 Transcranial magnetic stimulation also may make it possible to achieve similar efficacy as ECT with fewer cognitive adverse effects when used in combination with ECT to reduce the number of acute ECT treatments required or as part of a maintenance strategy after a patient experiences an acute response to ECT.13
Magnetic seizure therapy (MST) vs ECT. An experimental treatment, MST uses a TMS device capable of producing more intense magnetic fields sufficient to induce a seizure.16 The advantage of MST over ECT-induced seizures is better control of intra-cerebral current path and density, thus avoiding deeper cortical areas associated with memory (eg, hippocampus) and minimizing cognitive adverse effects. As with ECT, however, anesthesia and muscle relaxation are required. Presently, MST remains investigational.
Other potential indications
In addition to MDD, TMS is also being studied as a potential treatment for other neuropsychiatric disorders.
Continue to: Obsessive-compulsive disorder
Obsessive-compulsive disorder (OCD). A recent double-blind study that evaluated a deep TMS (DTMS) device reported a significantly better outcome based on the Yale-Brown Obsessive-Compulsive Scale score with active high-frequency (20 Hz) DTMS (n = 18) vs a sham control (n = 15).17 The initial benefit persisted up to 1 month after the end of treatment. The authors speculated that this benefit may be due to direct modulation of the anterior cingulate cortex. These results led to the first FDA clearance of a deep TMS device for treating OCD.
Cognition. Because TMS does not require a seizure to produce its antidepressant effect and does not require anesthesia, the risk of neurocognitive disruption is low. In fact, evidence suggests TMS may have beneficial cognitive effects.18
In an effort to take advantage of this benefit, researchers have explored providing psychoeducation and psychotherapy sessions (eg, behavioral activation) during TMS treatments (“online”).19,20 The rationale is that neurocircuitry subserving various cognitive functions may be in a heightened state of receptivity during a TMS treatment, which would allow patients to assimilate and better utilize the therapeutic information provided.19,20
Researchers are also looking at the use of TMS to treat patients with mild cognitive impairment or early dementia. These patients often experience comorbid depression, and TMS could potentially improve memory via both its pro-cognitive and antidepressant effects.1 The lack of effective treatments for dementia supports pursuing TMS as a therapeutic option for these patients.
Other neuropsychiatric disorders. In addition to early-onset cognitive problems, other neurologic indications with promising data for TMS include chronic pain syndromes, Parkinson’s disease, tinnitus, and migraine headaches (a hand-held FDA-cleared device is now available for treating migraines). In addition to OCD and bipolar depression, other psychiatric indications with promising data include schizophrenia (eg, refractory auditory hallucinations, negative symptoms), posttraumatic stress disorder, and various addictive disorders.21 Because results have been mixed for most of these disorders, definitive trials are needed to clearly characterize the potential role of TMS.
Continue to: An ongoing evolution
An ongoing evolution
Neuromodulation is undergoing a renaissance spurred on by the need for more effective treatments to manage some of our most challenging illnesses. Transcranial magnetic stimulation and other forms of therapeutic neuromodulation are welcome additions for managing treatment-resistant depression, OCD, and possibly other disorders. But perhaps their greatest value is as a bellwether for what’s to come. In addition to the ongoing refinements to existing neuromodulation devices, newer modulation approaches (eg, temporal interference stimulation) and the search for reliable biomarkers may dramatically expand and enhance our clinical options.14,22
Bottom Line
Transcranial magnetic stimulation (TMS) continues to evolve as a nonpharmacologic treatment for mood disorders, obsessive-compulsive disorder, and potentially for other indications. Recent developments, including altered treatment parameters, new devices, and strategies for increasing the durability of antidepressant effects, have enhanced the benefits of TMS.
Related Resources
- Ziemann U. Thirty years of transcranial magnetic stimulation: where do we stand? Exp Brain Res. 2017;235(4):973-984.
- Janicak PG, Sackett V, Kudrna K, et al. Transcranial magnetic stimulation for the treatment of major depression: an update on recent advances. Current Psychiatry. 2016:15(6):49-56.
1. Koch G, Bonnì S, Pellicciari MC, et al. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage. 2018;169: 302-310.
2. O’Reardon JP, Solvason B, Janicak PG, et al. Efficacy and safety of repetitive transcranial magnetic stimulation (rTMS) in the acute treatment of major depression: results of a multicenter randomized controlled trial. Biol Psychiatry. 2007;62(11):1208-1216.
3. Dunner DL, Aaronson ST, Sackheim HA, et al. A multisite, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a one-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
4. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction treatment. J Clin Psychiatry. 2008;69:222-232.
5. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
6. Janicak PG. Risk management issues in transcranial magnetic stimulation for treatment of major depression. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
7. Chen J, Zhou C, Wu B, et al. Left versus right repetitive transcranial magnetic stimulation in treating major depression: a meta-analysis of randomised controlled trials. Psychiatry Res. 2013;210(3):1260-1264.
8. Blumberger DM, Vila-Rodriguez F, Thorpe KE, et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet. 2018;391(10131):1683-1692.
9. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
10. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
11. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prop Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
12. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depressive: preliminary results of a randomized trial. Biol Psychiatry. 2002;51(8):659-667.
13. Lanocha K, Janicak PG. TMS for depression: relationship to ECT and other therapeutic neuromodulation approaches. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
14. Drysdale AT, Grosenick L, Downar J, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med. 2017;23(1):28-38.
15. Aaronson ST, Croarkin PE. Transcranial magnetic stimulation for the treatment of other mood disorders. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
16. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
17. Carmi L, Alyagon U, Barnea-Ygael N, et al. Clinical and electrophysiological outcomes of deep TMS over the medial prefrontal and anterior cingulate cortices in OCD patients. Brain Stimul. 2018;11(1):158-165.
18. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114:1125-1132.
19. Donse L, Padberg F, Sack AT, et al. Simultaneous rTMS and psychotherapy in major depressive disorder: Clinical outcomes and predictors from a large naturalistic study. Brain Stimul. 2018;11(2):337-345.
20. Russo GB, Tirrell E, Busch A, et al. Behavioral activation therapy during transcranial magnetic stimulation for major depressive disorder. J Affect Disord. 2018;236:101-104.
21. Pannu J, DE Souza DD, Samara Z, et al. Transcranial magnetic stimulation for disorders other than depression. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
22. Grossman N. Modulation without surgical intervention. Science. 2018;361:461-462.
Therapeutic neuromodulation takes advantage of the brain’s electrochemical makeup. This allows for treatment devices that modulate neurocircuits relevant to behaviors disrupted in disorders such as major depressive disorder (MDD) (eg, sleep quality, appetite, cognitive, and executive functions). The default mode network (comprised of structures such as the medial prefrontal cortex [MPFC], the posterior cingulate cortex, the hippocampus, and their functional connectivity) serves as a prime example of circuitry that can be targeted by this approach.1
For 80 years, electroconvulsive therapy (ECT) has been an important neuromodulation option for patients with more severe illness. Recently, additional neuromodulatory approaches have been FDA-cleared, including transcranial magnetic stimulation (TMS), vagus nerve stimulation (VNS), and deep brain stimulation (DBS). Another approach, transcranial direct current stimulation (tDCS), has been extensively studied for its potential clinical utility but is not FDA-cleared. The Table provides descriptions of these therapies.
Since being cleared by the FDA in 2008, TMS has arguably made the greatest strides in providing an alternate neuromodulation treatment option for patients with MDD, with >1,000 centers nationally and 7 TMS devices FDA-cleared for treatment of depression. In this article, we review recent developments in TMS.
An evolving therapeutic option
While primarily studied as a monotherapy for MDD, in clinical practice TMS (Box) is typically used as an adjunct to medication and psychotherapy.2,3 In this context, it has demonstrated efficacy for more difficult-to-treat mood disorders with an excellent safety and tolerability profile whether used with or without medication.4-6
To further improve the efficiency and efficacy of TMS while maintaining its safety and tolerability, researchers and clinicians have been exploring a few initiatives.
Box
- Transcranial magnetic stimulation (TMS) utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression
- Randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression
- Clinical availability of TMS has grown steadily over the past 10 years as >1,000 centers have been opened and additional devices have been FDA-cleared
- TMS has the potential to avoid safety and tolerability concerns associated with antidepressant pharmacotherapy (eg, weight gain, sexual dysfunction) and electroconvulsive therapy (eg, cognitive deficits)
- Greater sophistication in the choice of stimulation parameters, as well as other ongoing efforts to optimize the benefits of TMS, are yielding better clinical outcomes
Altered treatment parameters
One initiative is assessing the feasibility of altering various treatment parameters, such as the total number of treatment sessions (30 to 60 sessions); the frequency of sessions (eg, more than once daily); the total number of magnetic pulses per session (eg, >3,000); the stimulation coil localization (eg, left vs right dorsal lateral prefrontal cortex [DLPFC]; MPFC; and various methods to determine optimal coil placement (eg, EEG F3 coordinate or MRI-guided neuro-navigational methods). Such refinements offer the potential for enhanced efficacy, shorter treatment sessions, and/or improved tolerability. For example, lower frequency right DLPFC stimulations (eg, 1 Hz) can decrease the risk of seizures and improve overall tolerability. While this has not been studied as extensively as higher frequency left DLPFC stimulations (eg, 5 to 20 Hz), existing evidence supports similar efficacy between these 2 approaches.7
Theta burst stimulation. Some TMS devices can be adapted to deliver theta burst stimulation (TBS). This produces trains of triple, 50 Hz, pulsed bursts (usually with 200 ms inter-burst intervals occurring at a rate of 5 Hz; at 80% MT) to model naturally occurring theta rhythms. These bursts can be administered in stimulation protocols using intermittent TBS (iTBS) (eg, 10 bursts of triplets over 2 seconds every 10 seconds; 30 pulses per burst; for approximately 3 minutes; totaling 600 pulses) or continuous TBS (cTBS) bursts given in an uninterrupted train (eg, 40 seconds, 600 pulses). Evidence indicates these protocols facilitate long-term potentiation (ie, iTBS) and long-term depression (ie, cTBS), which in turn can modulate synaptic plasticity.
Continue to: While some clinicians are using...
While some clinicians are using TBS off-label, a recent non-inferiority trial (N = 395) reported similar efficacy and safety comparing standard 10 Hz TMS to an iTBS protocol at 120% of resting motor threshold (both over the left DLPFC).8 This has led to FDA clearance of the TMS device adapted to provide iTBS in this trial.8
From a more practical perspective, TBS has the potential to reduce the number of pulses (eg, 600 vs 3,000) and the total number of sessions required, as well as the duration of treatment sessions (eg, 37.5 minutes to <5 minutes). This can accelerate the time to response and decrease patient and staff commitment, with resulting cost savings.9 Despite this recent progress, ongoing research still needs to clarify issues such as the risk/benefit profile, particularly in younger and older populations, as well as assessment of duration of initial benefit and appropriate maintenance strategies.
New devices
Another initiative is the development of alternative TMS equipment. For example, newer coil designs with enhanced cooling ability allow for a substantial decrease in the required inter-train interval duration between stimulation trains, thus shortening the total session duration by approximately 50% (eg, from 37.5 to 19 minutes). The use of different coil arrays (eg, the H-coil capable of deeper vs surface stimulation) may allow for more direct stimulation of relevant neurocircuitry (eg, cingulate cortex), possibly improving efficacy and shortening time to onset of benefit. However, in head-to-head comparisons with single-coil devices, enhanced efficacy for depression has not been clearly demonstrated. One caveat is that the increase in depth of magnetic field penetration results in a loss of focality, resulting in the stimulation of larger brain areas. This might increase the risk of adverse effects such as seizures.
Increasing durability of effect
Because high relapse and recurrence rates compromise the initial benefit of any antidepressant therapy, appropriate maintenance strategies are essential. Several studies have evaluated strategies to maintain the acute benefit of TMS for treatment-resistant depression.
One was a 6-month, open-label TMS durability of effect trial for acute responders (n = 99) in the pivotal registration study.5 During this study, all participants were given antidepressant medication monotherapy. In addition, with early indication of relapse, patients received a reintroduction of TMS sessions (32/99 patients; mean number of sessions = 14.3). With this protocol, approximately 84% re-achieved their response status. The overall relapse rate was approximately 13%.5
Continue to: In a 1-year naturalistic study...
In a 1-year naturalistic study, 63% of patients (75/120) who met response or remission criteria after an acute course of TMS still met response criteria after 12 months. These patients received clinician-determined maintenance treatment that included reintroduction of TMS when indicated.3
In a prospective, 12-month, multisite, randomized pilot study, 67 patients with treatment-resistant MDD underwent an antidepressant medication washout and then received 30 sessions of TMS monotherapy.10 Those who met criteria for improvement (n = 49) were then randomized to once-monthly TMS or observation only. All patients remained medication-free but could receive TMS re-introduction if they deteriorated. At the end of the study, both groups demonstrated comparable outcomes, with a trend to a longer time before relapse among participants who received once-monthly TMS. Although these results are preliminary, they suggest that some patients could be treated both acutely and then maintained with TMS alone.
Re-introducing TMS in patients who show early signs of relapse after having an initial response achieves rates of sustained improvement that compare favorably with those of other strategies used to manage patients with treatment-resistant depression.
TMS vs ECT
The question often arises as to whether TMS is a viable alternate treatment to ECT. I believe the answer is unequivocally yes and no. By this, I mean some patients who in the past only had ECT as their next option when medications and psychotherapy were insufficient may now consider TMS. In support, there is evidence of comparable efficacy between TMS and ECT in a subgroup of patients who were considered clinically appropriate for ECT.11-13
How to best identify this group remains unclear, but investigators are exploring predictive biomarkers. For example, a large study (N = 1,188), with functional magnetic resonance imaging (fMRI) reported that depressed patients could be divided into 4 neurophysiological “biotypes” based on different patterns of aberrant connectivity in limbic and fronto-striatal networks.14 The authors further noted that such distinctions were helpful in predicting response in a subgroup of patients (n = 154) who received TMS.
Continue to: For now...
For now, experience indicates certain clinical factors may provide some guidance. Patients are usually better served by ECT if they:
- have depressive episodes of longer duration (eg, >3 years)
- have a high risk of suicide
- have psychotic or catatonic features associated with their depression
- have difficulty maintaining their physical well-being
- have bipolar depression.
Although existing evidence supports a possible benefit with TMS for bipolar depression (used in combination with a mood stabilizer), the lack of a definitive trial (precluding FDA clearance for this indication) and the lack of insurance coverage both limit the routine use of TMS for this indication.15
One potential advantage of TMS over ECT is a lower cost.13 Transcranial magnetic stimulation also may make it possible to achieve similar efficacy as ECT with fewer cognitive adverse effects when used in combination with ECT to reduce the number of acute ECT treatments required or as part of a maintenance strategy after a patient experiences an acute response to ECT.13
Magnetic seizure therapy (MST) vs ECT. An experimental treatment, MST uses a TMS device capable of producing more intense magnetic fields sufficient to induce a seizure.16 The advantage of MST over ECT-induced seizures is better control of intra-cerebral current path and density, thus avoiding deeper cortical areas associated with memory (eg, hippocampus) and minimizing cognitive adverse effects. As with ECT, however, anesthesia and muscle relaxation are required. Presently, MST remains investigational.
Other potential indications
In addition to MDD, TMS is also being studied as a potential treatment for other neuropsychiatric disorders.
Continue to: Obsessive-compulsive disorder
Obsessive-compulsive disorder (OCD). A recent double-blind study that evaluated a deep TMS (DTMS) device reported a significantly better outcome based on the Yale-Brown Obsessive-Compulsive Scale score with active high-frequency (20 Hz) DTMS (n = 18) vs a sham control (n = 15).17 The initial benefit persisted up to 1 month after the end of treatment. The authors speculated that this benefit may be due to direct modulation of the anterior cingulate cortex. These results led to the first FDA clearance of a deep TMS device for treating OCD.
Cognition. Because TMS does not require a seizure to produce its antidepressant effect and does not require anesthesia, the risk of neurocognitive disruption is low. In fact, evidence suggests TMS may have beneficial cognitive effects.18
In an effort to take advantage of this benefit, researchers have explored providing psychoeducation and psychotherapy sessions (eg, behavioral activation) during TMS treatments (“online”).19,20 The rationale is that neurocircuitry subserving various cognitive functions may be in a heightened state of receptivity during a TMS treatment, which would allow patients to assimilate and better utilize the therapeutic information provided.19,20
Researchers are also looking at the use of TMS to treat patients with mild cognitive impairment or early dementia. These patients often experience comorbid depression, and TMS could potentially improve memory via both its pro-cognitive and antidepressant effects.1 The lack of effective treatments for dementia supports pursuing TMS as a therapeutic option for these patients.
Other neuropsychiatric disorders. In addition to early-onset cognitive problems, other neurologic indications with promising data for TMS include chronic pain syndromes, Parkinson’s disease, tinnitus, and migraine headaches (a hand-held FDA-cleared device is now available for treating migraines). In addition to OCD and bipolar depression, other psychiatric indications with promising data include schizophrenia (eg, refractory auditory hallucinations, negative symptoms), posttraumatic stress disorder, and various addictive disorders.21 Because results have been mixed for most of these disorders, definitive trials are needed to clearly characterize the potential role of TMS.
Continue to: An ongoing evolution
An ongoing evolution
Neuromodulation is undergoing a renaissance spurred on by the need for more effective treatments to manage some of our most challenging illnesses. Transcranial magnetic stimulation and other forms of therapeutic neuromodulation are welcome additions for managing treatment-resistant depression, OCD, and possibly other disorders. But perhaps their greatest value is as a bellwether for what’s to come. In addition to the ongoing refinements to existing neuromodulation devices, newer modulation approaches (eg, temporal interference stimulation) and the search for reliable biomarkers may dramatically expand and enhance our clinical options.14,22
Bottom Line
Transcranial magnetic stimulation (TMS) continues to evolve as a nonpharmacologic treatment for mood disorders, obsessive-compulsive disorder, and potentially for other indications. Recent developments, including altered treatment parameters, new devices, and strategies for increasing the durability of antidepressant effects, have enhanced the benefits of TMS.
Related Resources
- Ziemann U. Thirty years of transcranial magnetic stimulation: where do we stand? Exp Brain Res. 2017;235(4):973-984.
- Janicak PG, Sackett V, Kudrna K, et al. Transcranial magnetic stimulation for the treatment of major depression: an update on recent advances. Current Psychiatry. 2016:15(6):49-56.
Therapeutic neuromodulation takes advantage of the brain’s electrochemical makeup. This allows for treatment devices that modulate neurocircuits relevant to behaviors disrupted in disorders such as major depressive disorder (MDD) (eg, sleep quality, appetite, cognitive, and executive functions). The default mode network (comprised of structures such as the medial prefrontal cortex [MPFC], the posterior cingulate cortex, the hippocampus, and their functional connectivity) serves as a prime example of circuitry that can be targeted by this approach.1
For 80 years, electroconvulsive therapy (ECT) has been an important neuromodulation option for patients with more severe illness. Recently, additional neuromodulatory approaches have been FDA-cleared, including transcranial magnetic stimulation (TMS), vagus nerve stimulation (VNS), and deep brain stimulation (DBS). Another approach, transcranial direct current stimulation (tDCS), has been extensively studied for its potential clinical utility but is not FDA-cleared. The Table provides descriptions of these therapies.
Since being cleared by the FDA in 2008, TMS has arguably made the greatest strides in providing an alternate neuromodulation treatment option for patients with MDD, with >1,000 centers nationally and 7 TMS devices FDA-cleared for treatment of depression. In this article, we review recent developments in TMS.
An evolving therapeutic option
While primarily studied as a monotherapy for MDD, in clinical practice TMS (Box) is typically used as an adjunct to medication and psychotherapy.2,3 In this context, it has demonstrated efficacy for more difficult-to-treat mood disorders with an excellent safety and tolerability profile whether used with or without medication.4-6
To further improve the efficiency and efficacy of TMS while maintaining its safety and tolerability, researchers and clinicians have been exploring a few initiatives.
Box
- Transcranial magnetic stimulation (TMS) utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression
- Randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression
- Clinical availability of TMS has grown steadily over the past 10 years as >1,000 centers have been opened and additional devices have been FDA-cleared
- TMS has the potential to avoid safety and tolerability concerns associated with antidepressant pharmacotherapy (eg, weight gain, sexual dysfunction) and electroconvulsive therapy (eg, cognitive deficits)
- Greater sophistication in the choice of stimulation parameters, as well as other ongoing efforts to optimize the benefits of TMS, are yielding better clinical outcomes
Altered treatment parameters
One initiative is assessing the feasibility of altering various treatment parameters, such as the total number of treatment sessions (30 to 60 sessions); the frequency of sessions (eg, more than once daily); the total number of magnetic pulses per session (eg, >3,000); the stimulation coil localization (eg, left vs right dorsal lateral prefrontal cortex [DLPFC]; MPFC; and various methods to determine optimal coil placement (eg, EEG F3 coordinate or MRI-guided neuro-navigational methods). Such refinements offer the potential for enhanced efficacy, shorter treatment sessions, and/or improved tolerability. For example, lower frequency right DLPFC stimulations (eg, 1 Hz) can decrease the risk of seizures and improve overall tolerability. While this has not been studied as extensively as higher frequency left DLPFC stimulations (eg, 5 to 20 Hz), existing evidence supports similar efficacy between these 2 approaches.7
Theta burst stimulation. Some TMS devices can be adapted to deliver theta burst stimulation (TBS). This produces trains of triple, 50 Hz, pulsed bursts (usually with 200 ms inter-burst intervals occurring at a rate of 5 Hz; at 80% MT) to model naturally occurring theta rhythms. These bursts can be administered in stimulation protocols using intermittent TBS (iTBS) (eg, 10 bursts of triplets over 2 seconds every 10 seconds; 30 pulses per burst; for approximately 3 minutes; totaling 600 pulses) or continuous TBS (cTBS) bursts given in an uninterrupted train (eg, 40 seconds, 600 pulses). Evidence indicates these protocols facilitate long-term potentiation (ie, iTBS) and long-term depression (ie, cTBS), which in turn can modulate synaptic plasticity.
Continue to: While some clinicians are using...
While some clinicians are using TBS off-label, a recent non-inferiority trial (N = 395) reported similar efficacy and safety comparing standard 10 Hz TMS to an iTBS protocol at 120% of resting motor threshold (both over the left DLPFC).8 This has led to FDA clearance of the TMS device adapted to provide iTBS in this trial.8
From a more practical perspective, TBS has the potential to reduce the number of pulses (eg, 600 vs 3,000) and the total number of sessions required, as well as the duration of treatment sessions (eg, 37.5 minutes to <5 minutes). This can accelerate the time to response and decrease patient and staff commitment, with resulting cost savings.9 Despite this recent progress, ongoing research still needs to clarify issues such as the risk/benefit profile, particularly in younger and older populations, as well as assessment of duration of initial benefit and appropriate maintenance strategies.
New devices
Another initiative is the development of alternative TMS equipment. For example, newer coil designs with enhanced cooling ability allow for a substantial decrease in the required inter-train interval duration between stimulation trains, thus shortening the total session duration by approximately 50% (eg, from 37.5 to 19 minutes). The use of different coil arrays (eg, the H-coil capable of deeper vs surface stimulation) may allow for more direct stimulation of relevant neurocircuitry (eg, cingulate cortex), possibly improving efficacy and shortening time to onset of benefit. However, in head-to-head comparisons with single-coil devices, enhanced efficacy for depression has not been clearly demonstrated. One caveat is that the increase in depth of magnetic field penetration results in a loss of focality, resulting in the stimulation of larger brain areas. This might increase the risk of adverse effects such as seizures.
Increasing durability of effect
Because high relapse and recurrence rates compromise the initial benefit of any antidepressant therapy, appropriate maintenance strategies are essential. Several studies have evaluated strategies to maintain the acute benefit of TMS for treatment-resistant depression.
One was a 6-month, open-label TMS durability of effect trial for acute responders (n = 99) in the pivotal registration study.5 During this study, all participants were given antidepressant medication monotherapy. In addition, with early indication of relapse, patients received a reintroduction of TMS sessions (32/99 patients; mean number of sessions = 14.3). With this protocol, approximately 84% re-achieved their response status. The overall relapse rate was approximately 13%.5
Continue to: In a 1-year naturalistic study...
In a 1-year naturalistic study, 63% of patients (75/120) who met response or remission criteria after an acute course of TMS still met response criteria after 12 months. These patients received clinician-determined maintenance treatment that included reintroduction of TMS when indicated.3
In a prospective, 12-month, multisite, randomized pilot study, 67 patients with treatment-resistant MDD underwent an antidepressant medication washout and then received 30 sessions of TMS monotherapy.10 Those who met criteria for improvement (n = 49) were then randomized to once-monthly TMS or observation only. All patients remained medication-free but could receive TMS re-introduction if they deteriorated. At the end of the study, both groups demonstrated comparable outcomes, with a trend to a longer time before relapse among participants who received once-monthly TMS. Although these results are preliminary, they suggest that some patients could be treated both acutely and then maintained with TMS alone.
Re-introducing TMS in patients who show early signs of relapse after having an initial response achieves rates of sustained improvement that compare favorably with those of other strategies used to manage patients with treatment-resistant depression.
TMS vs ECT
The question often arises as to whether TMS is a viable alternate treatment to ECT. I believe the answer is unequivocally yes and no. By this, I mean some patients who in the past only had ECT as their next option when medications and psychotherapy were insufficient may now consider TMS. In support, there is evidence of comparable efficacy between TMS and ECT in a subgroup of patients who were considered clinically appropriate for ECT.11-13
How to best identify this group remains unclear, but investigators are exploring predictive biomarkers. For example, a large study (N = 1,188), with functional magnetic resonance imaging (fMRI) reported that depressed patients could be divided into 4 neurophysiological “biotypes” based on different patterns of aberrant connectivity in limbic and fronto-striatal networks.14 The authors further noted that such distinctions were helpful in predicting response in a subgroup of patients (n = 154) who received TMS.
Continue to: For now...
For now, experience indicates certain clinical factors may provide some guidance. Patients are usually better served by ECT if they:
- have depressive episodes of longer duration (eg, >3 years)
- have a high risk of suicide
- have psychotic or catatonic features associated with their depression
- have difficulty maintaining their physical well-being
- have bipolar depression.
Although existing evidence supports a possible benefit with TMS for bipolar depression (used in combination with a mood stabilizer), the lack of a definitive trial (precluding FDA clearance for this indication) and the lack of insurance coverage both limit the routine use of TMS for this indication.15
One potential advantage of TMS over ECT is a lower cost.13 Transcranial magnetic stimulation also may make it possible to achieve similar efficacy as ECT with fewer cognitive adverse effects when used in combination with ECT to reduce the number of acute ECT treatments required or as part of a maintenance strategy after a patient experiences an acute response to ECT.13
Magnetic seizure therapy (MST) vs ECT. An experimental treatment, MST uses a TMS device capable of producing more intense magnetic fields sufficient to induce a seizure.16 The advantage of MST over ECT-induced seizures is better control of intra-cerebral current path and density, thus avoiding deeper cortical areas associated with memory (eg, hippocampus) and minimizing cognitive adverse effects. As with ECT, however, anesthesia and muscle relaxation are required. Presently, MST remains investigational.
Other potential indications
In addition to MDD, TMS is also being studied as a potential treatment for other neuropsychiatric disorders.
Continue to: Obsessive-compulsive disorder
Obsessive-compulsive disorder (OCD). A recent double-blind study that evaluated a deep TMS (DTMS) device reported a significantly better outcome based on the Yale-Brown Obsessive-Compulsive Scale score with active high-frequency (20 Hz) DTMS (n = 18) vs a sham control (n = 15).17 The initial benefit persisted up to 1 month after the end of treatment. The authors speculated that this benefit may be due to direct modulation of the anterior cingulate cortex. These results led to the first FDA clearance of a deep TMS device for treating OCD.
Cognition. Because TMS does not require a seizure to produce its antidepressant effect and does not require anesthesia, the risk of neurocognitive disruption is low. In fact, evidence suggests TMS may have beneficial cognitive effects.18
In an effort to take advantage of this benefit, researchers have explored providing psychoeducation and psychotherapy sessions (eg, behavioral activation) during TMS treatments (“online”).19,20 The rationale is that neurocircuitry subserving various cognitive functions may be in a heightened state of receptivity during a TMS treatment, which would allow patients to assimilate and better utilize the therapeutic information provided.19,20
Researchers are also looking at the use of TMS to treat patients with mild cognitive impairment or early dementia. These patients often experience comorbid depression, and TMS could potentially improve memory via both its pro-cognitive and antidepressant effects.1 The lack of effective treatments for dementia supports pursuing TMS as a therapeutic option for these patients.
Other neuropsychiatric disorders. In addition to early-onset cognitive problems, other neurologic indications with promising data for TMS include chronic pain syndromes, Parkinson’s disease, tinnitus, and migraine headaches (a hand-held FDA-cleared device is now available for treating migraines). In addition to OCD and bipolar depression, other psychiatric indications with promising data include schizophrenia (eg, refractory auditory hallucinations, negative symptoms), posttraumatic stress disorder, and various addictive disorders.21 Because results have been mixed for most of these disorders, definitive trials are needed to clearly characterize the potential role of TMS.
Continue to: An ongoing evolution
An ongoing evolution
Neuromodulation is undergoing a renaissance spurred on by the need for more effective treatments to manage some of our most challenging illnesses. Transcranial magnetic stimulation and other forms of therapeutic neuromodulation are welcome additions for managing treatment-resistant depression, OCD, and possibly other disorders. But perhaps their greatest value is as a bellwether for what’s to come. In addition to the ongoing refinements to existing neuromodulation devices, newer modulation approaches (eg, temporal interference stimulation) and the search for reliable biomarkers may dramatically expand and enhance our clinical options.14,22
Bottom Line
Transcranial magnetic stimulation (TMS) continues to evolve as a nonpharmacologic treatment for mood disorders, obsessive-compulsive disorder, and potentially for other indications. Recent developments, including altered treatment parameters, new devices, and strategies for increasing the durability of antidepressant effects, have enhanced the benefits of TMS.
Related Resources
- Ziemann U. Thirty years of transcranial magnetic stimulation: where do we stand? Exp Brain Res. 2017;235(4):973-984.
- Janicak PG, Sackett V, Kudrna K, et al. Transcranial magnetic stimulation for the treatment of major depression: an update on recent advances. Current Psychiatry. 2016:15(6):49-56.
1. Koch G, Bonnì S, Pellicciari MC, et al. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage. 2018;169: 302-310.
2. O’Reardon JP, Solvason B, Janicak PG, et al. Efficacy and safety of repetitive transcranial magnetic stimulation (rTMS) in the acute treatment of major depression: results of a multicenter randomized controlled trial. Biol Psychiatry. 2007;62(11):1208-1216.
3. Dunner DL, Aaronson ST, Sackheim HA, et al. A multisite, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a one-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
4. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction treatment. J Clin Psychiatry. 2008;69:222-232.
5. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
6. Janicak PG. Risk management issues in transcranial magnetic stimulation for treatment of major depression. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
7. Chen J, Zhou C, Wu B, et al. Left versus right repetitive transcranial magnetic stimulation in treating major depression: a meta-analysis of randomised controlled trials. Psychiatry Res. 2013;210(3):1260-1264.
8. Blumberger DM, Vila-Rodriguez F, Thorpe KE, et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet. 2018;391(10131):1683-1692.
9. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
10. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
11. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prop Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
12. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depressive: preliminary results of a randomized trial. Biol Psychiatry. 2002;51(8):659-667.
13. Lanocha K, Janicak PG. TMS for depression: relationship to ECT and other therapeutic neuromodulation approaches. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
14. Drysdale AT, Grosenick L, Downar J, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med. 2017;23(1):28-38.
15. Aaronson ST, Croarkin PE. Transcranial magnetic stimulation for the treatment of other mood disorders. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
16. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
17. Carmi L, Alyagon U, Barnea-Ygael N, et al. Clinical and electrophysiological outcomes of deep TMS over the medial prefrontal and anterior cingulate cortices in OCD patients. Brain Stimul. 2018;11(1):158-165.
18. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114:1125-1132.
19. Donse L, Padberg F, Sack AT, et al. Simultaneous rTMS and psychotherapy in major depressive disorder: Clinical outcomes and predictors from a large naturalistic study. Brain Stimul. 2018;11(2):337-345.
20. Russo GB, Tirrell E, Busch A, et al. Behavioral activation therapy during transcranial magnetic stimulation for major depressive disorder. J Affect Disord. 2018;236:101-104.
21. Pannu J, DE Souza DD, Samara Z, et al. Transcranial magnetic stimulation for disorders other than depression. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
22. Grossman N. Modulation without surgical intervention. Science. 2018;361:461-462.
1. Koch G, Bonnì S, Pellicciari MC, et al. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage. 2018;169: 302-310.
2. O’Reardon JP, Solvason B, Janicak PG, et al. Efficacy and safety of repetitive transcranial magnetic stimulation (rTMS) in the acute treatment of major depression: results of a multicenter randomized controlled trial. Biol Psychiatry. 2007;62(11):1208-1216.
3. Dunner DL, Aaronson ST, Sackheim HA, et al. A multisite, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a one-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
4. Janicak PG, O’Reardon JP, Sampson SM, et al. Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction treatment. J Clin Psychiatry. 2008;69:222-232.
5. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
6. Janicak PG. Risk management issues in transcranial magnetic stimulation for treatment of major depression. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
7. Chen J, Zhou C, Wu B, et al. Left versus right repetitive transcranial magnetic stimulation in treating major depression: a meta-analysis of randomised controlled trials. Psychiatry Res. 2013;210(3):1260-1264.
8. Blumberger DM, Vila-Rodriguez F, Thorpe KE, et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet. 2018;391(10131):1683-1692.
9. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
10. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
11. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prop Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
12. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depressive: preliminary results of a randomized trial. Biol Psychiatry. 2002;51(8):659-667.
13. Lanocha K, Janicak PG. TMS for depression: relationship to ECT and other therapeutic neuromodulation approaches. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
14. Drysdale AT, Grosenick L, Downar J, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med. 2017;23(1):28-38.
15. Aaronson ST, Croarkin PE. Transcranial magnetic stimulation for the treatment of other mood disorders. In: Bermudes R, Lanocha K, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
16. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
17. Carmi L, Alyagon U, Barnea-Ygael N, et al. Clinical and electrophysiological outcomes of deep TMS over the medial prefrontal and anterior cingulate cortices in OCD patients. Brain Stimul. 2018;11(1):158-165.
18. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114:1125-1132.
19. Donse L, Padberg F, Sack AT, et al. Simultaneous rTMS and psychotherapy in major depressive disorder: Clinical outcomes and predictors from a large naturalistic study. Brain Stimul. 2018;11(2):337-345.
20. Russo GB, Tirrell E, Busch A, et al. Behavioral activation therapy during transcranial magnetic stimulation for major depressive disorder. J Affect Disord. 2018;236:101-104.
21. Pannu J, DE Souza DD, Samara Z, et al. Transcranial magnetic stimulation for disorders other than depression. In: Bermudes RA, Lanocha KI, Janicak PG (eds). Transcranial magnetic stimulation: clinical applications for psychiatric practice. Washington, DC: American Psychiatric Association Publishing; 2018.
22. Grossman N. Modulation without surgical intervention. Science. 2018;361:461-462.

Older-age bipolar disorder: A case series
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
")
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.

A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
")
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.

Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.

The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.

Antidepressants for chronic pain
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.

Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88

Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.

Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.

Injectable extended-release naltrexone for opioid dependence: 3 studies
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).

1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).
1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
Death by drug overdose is the number one cause of death in Americans 50 years of age and younger.1 In 2016, there were 63,632 drug overdose deaths in the United States2 Opioids were involved in 42,249 of these deaths, which represents 66.4% of all drug overdose deaths.2 From 2015 to 2016, the age-adjusted rate of overdose deaths increased significantly by 21.5% from 16.3 per 100,000 to 19.8 per 100,000.2 This means that every day, more than 115 people in the United States die after overdosing on opioids. The misuse of and addiction to opioids—including prescription pain relievers, heroin, and synthetic opioids such as fentanyl—is a serious national crisis that affects public health as well as social and economic welfare.
The gold standard treatment is medication-assisted treatment (MAT)—the use of FDA-approved medications, in combination with counseling and behavioral therapies, to provide a “whole-patient” approach.3 When it comes to MAT options for opioid use disorder (OUD), there are 3 medications, each with its own caveats.
Methadone is an opioid mu-receptor full agonist that prevents withdrawal but does not block other narcotics. It requires daily dosing as a liquid formulation that is dispensed only in regulated clinics.
Buprenorphine is a mu-receptor high affinity partial agonist/antagonist that blocks the majority of other narcotics while reducing withdrawal risk. It requires daily dosing as either a dissolving tablet or cheek film. Recently it has also become available as a 6-month implant as well as a 1-month subcutaneous injection. Buprenorphine is also available as a combined medication with naloxone; naloxone is an opioid antagonist
Naltrexone is a mu-receptor antagonist that blocks the effects of most narcotics. It does not lead to dependence, and is administered daily as a pill or monthly as a deep IM injection of its extended-release formulation.
The first 2 medications are tightly regulated options that are not available in many areas of the United States. Naltrexone, when provided as a daily pill, has adherence issues. As with any illness, lack of adherence to treatment is problematic; in the case of patients with OUD, this includes a high risk of overdose and death.
The use of injectable extended-release naltrexone (XR-NTX) may be a way to address nonadherence and thus prevent relapse. One of the challenges limiting naltrexone’s applicability has been the length of time required for an “opioid washout” of the mu receptors prior to administering naltrexone, which is a mu blocker. The washout can take as long as 7 to 10 days. This interval is not feasible for patients receiving inpatient treatment, and patients receiving treatment as outpatients are vulnerable to relapse during this time. Recently, there have been several attempts to shorten this gap through various experimental protocols based on incremental doses of NTX to facilitate withdrawal while managing symptoms.
Continue to: When selecting appropriate candidates for NTX treatment...
When selecting appropriate candidates for NTX treatment, clinicians should consider individuals who are:
- not interested in or able to receive agonist maintenance treatment (ie, patients who do not have access to an appropriate clinic in their area, or who are restricted to agonist treatment by probation/parole)
- highly abstinence-oriented (eg, active in a 12-step program)
- in professions where agonists are controversial (eg, healthcare and airlines)
- detoxified and abstinent but at risk for relapse.
Individuals who have failed agonist treatment (eg, who experience cravings for opioids and use opioids while receiving it, or are nonadherent or diverting/misusing the medication), who have a less severe form of OUD (short history and low level of use), or who use sporadically are also optimal candidates for NTX. Aside from the relapse-vulnerable washout gap prior to induction, one of the concerns with antagonist treatments is treatment retention; anecdotal clinical reports suggest that individuals often discontinue antagonists in favor of agonists.
Several studies have investigated this by comparing XR-NTX with buprenorphine-naloxone (BUP-NX). Here we summarize 3 studies4-6 to describe which patients might be optimal candidates for XR-NTX, its success in comparison with BUP-NX, and challenges in induction of NTX, with a focus on emerging protocols (Table).
1. Tanum l, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: a randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
This study aimed to determine whether XR-NTX was not inferior to BUP-NX in the treatment of OUD.
Study design
- N = 159, multicenter, randomized, 12-week outpatient study in Norway
- After detoxification, participants were randomized to receive BUP-NX, 4 to 24 mg/d, or XR-NTX, 380 mg/month.
Continue to: Outcomes
Outcomes
- Comparable treatment retention between groups
- Comparable opioid-negative urine drug screens (UDS)
- Significantly lower opioid use in the XR-NTX group.
Conclusion
- XR-NTX was as effective as BUP-NX in maintaining short-term abstinence from heroin and other illicit opioids, and thus should be considered as a treatment option for opioid-dependent individuals.
While this study showed similar efficacy for XR-NTX and BUP-NX, it is important to note that the randomization occurred after patients were detoxified. As a full opioid antagonist, XR-NTX can precipitate severe withdrawal, so patients need to be completely detoxified before starting XR-NTX, in contrast to BUP-NX, which patients can start even while still in mild withdrawal. Additional studies are needed in which individuals are randomized before detoxification, which would make it possible to measure the success of induction.
2. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
This study evaluated XR-NTX vs BUP-NX among adults with OUD who were actively using heroin at baseline and were admitted to community detoxification and treatment programs. Although the study began on inpatient units, it aimed to replicate usual community outpatient conditions across a 24-week outpatient treatment phase of this open-label, comparative effectiveness trial. Researchers assessed the effects on relapse-free survival, opioid use rates, and overdose events.
Study design
- N = 570, multicenter, randomized, 24-week study in the United States
- Detoxification methods: no opioids (clonidine or adjunctive medications), 3- to 5-day methadone taper, and 3- to 14-day BUP taper
- Protocol requirement: opioid-negative UDS before XR-NTX induction
- XR-NTX induction success ranged from 50% at a short-methadone-taper unit to 95% at an extended-opioid-free inpatient program. Nearly all induction failures quickly relapsed
- More participants inducted on BUP-NX group than XR-NTX group (94% vs 72%, respectively).
Continue to: Outcomes
Outcomes (once successfully inducted to treatment [n = 474])
- Comparable relapse events
- Comparable opioid-negative urine drug screens and opioid-abstinent days
- Opioid craving initially less with XR-NTX.
Conclusion
- It was more difficult to initiate patients on XR-NTX than BUP-NX, which negatively affected overall relapse rates. However, once initiated, both medications were equally safe and effective. Future work should focus on facilitating induction to XR-NTX and on improving treatment retention for both medications.
Regarding induction on NTX, patients must be detoxified and opioid-free for at least 7 days. If this medication is given to patients who are physically dependent and/or have opioids in their system, NTX will displace opioids off the receptor and precipitate a severe withdrawal (rather than a slow and gradual spontaneous withdrawal).
Several studies have examined the severity of opioid withdrawal (using Self Opioid Withdrawal Scale scoring) of patients undergoing detoxification with symptomatic management (eg, clonidine, loperamide, etc.), agonist-managed (eg, with a BUP taper), and without any assistance. As expected, the latter yielded the highest scoring and most uncomfortable experiences. Using scores from the first 2 groups, a threshold of symptom tolerability was established where patients remained somewhat comfortable during the process. During detoxification from heroin, administering any dose of NTX during the first 48 to 72 hours after the last use placed patients in a withdrawal of a magnitude above the limit of tolerability. At 48 to 72 hours, however, a very low NTX dose (3 to 6 mg) was found to be well tolerated, and withdrawal symptoms were easily managed supportively to accelerate the detoxification process. Several studies have attempted to devise protocols based on these findings in order to facilitate rapid induction onto NTX. The following study offers encouragement:
Continue to: 3. Sullivan M, Bisaga A, Pavlicova M...
3. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Study design
- N = 150 adults with OUD, randomized to outpatient opioid detoxification
- Patients were randomized to BUP- or NTX-facilitated detoxification, followed by XR-NTX
- BUP detoxification group underwent a 7-day BUP taper followed by a opioid-free week
- NTX group received a 1-day BUP dose followed by 6 days of ascending doses of oral NTX, along with clonidine and other adjunctive medications.
Outcomes
- NTX-assisted detoxification was significantly more successful for XR-NTX induction (56.1% vs 32.7%).
Conclusion
- Compared with the BUP-assisted detoxification group, NTX-assisted detoxification appears to make it significantly more likely for patients to be successfully inducted to XR-NTX.
The evidence discussed here holds promise in addressing some of the major issues surrounding MAT. For suitable candidates, XR-NTX seems to be as efficacious an option as agonist (BUP) MAT, and its induction limitations could be overcome by using NTX-facilitated detoxification protocols.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
1. Rudd RA, Seth P, David F, et al. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452.
2. Centers for Disease Control and Prevention. Drug overdose death data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Updated December 19, 2017. Accessed October 24, 2018.
3. Substance Abuse and Mental Health Services Administration. Medication-assisted treatment (MAT). https://www.samhsa.gov/medication-assisted-treatment. Updated February 7, 2018. Accessed October 23, 2018.
4. Tanum L, Solli KK, Latif ZH, et al. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry. 2017;74(12):1197-1205.
5. Lee JD, Nunes, EV, Novo P, et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): a multicentre, open-label, randomised controlled trial. Lancet. 2018;391(10118):309-318.
6. Sullivan M, Bisaga A, Pavlicova M, et al. Long-acting injectable naltrexone induction: a randomized trial of outpatient opioid detoxification with naltrexone versus buprenorphine. Am J Psychiatry. 2017;174:459-467.
Can lifestyle modifications delay or prevent Alzheimer’s disease?
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4

Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75

On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.

Abuse of psychiatric medications: Not just stimulants and benzodiazepines
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15

Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40

Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73

These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.

With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15
Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40
Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73
These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.
With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15
Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40
Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73
These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.
With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.

Botulinum toxin: Emerging psychiatric indications
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22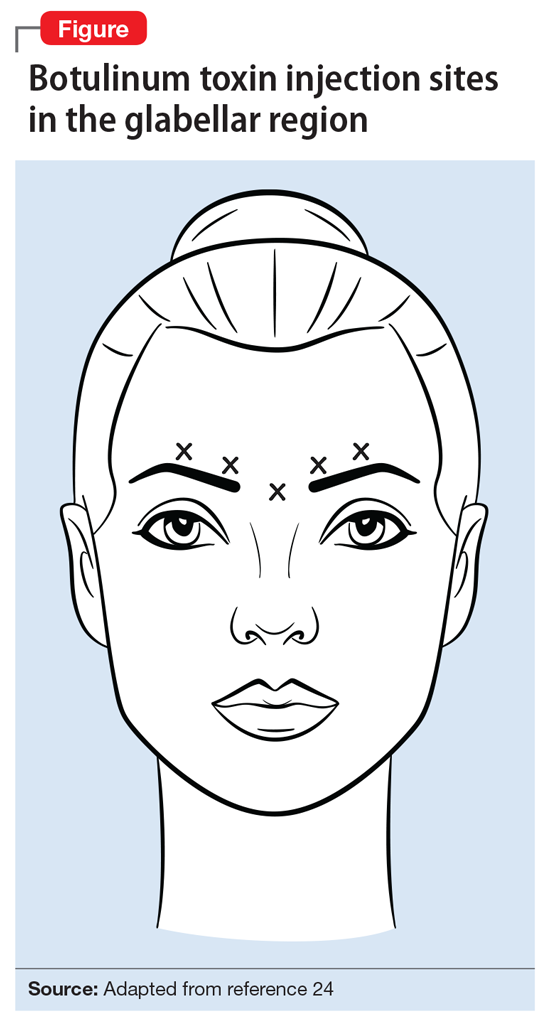
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
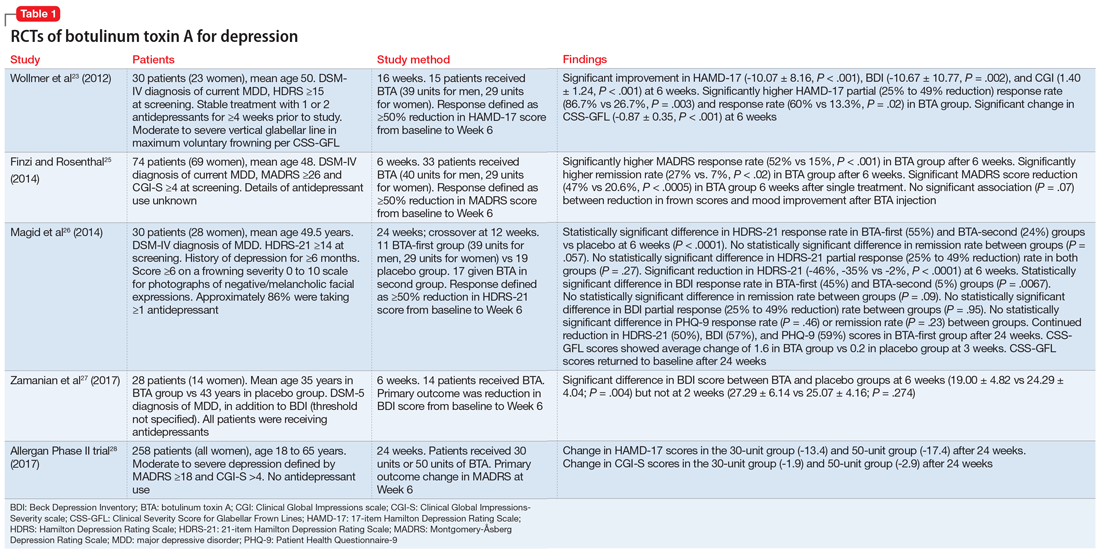
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
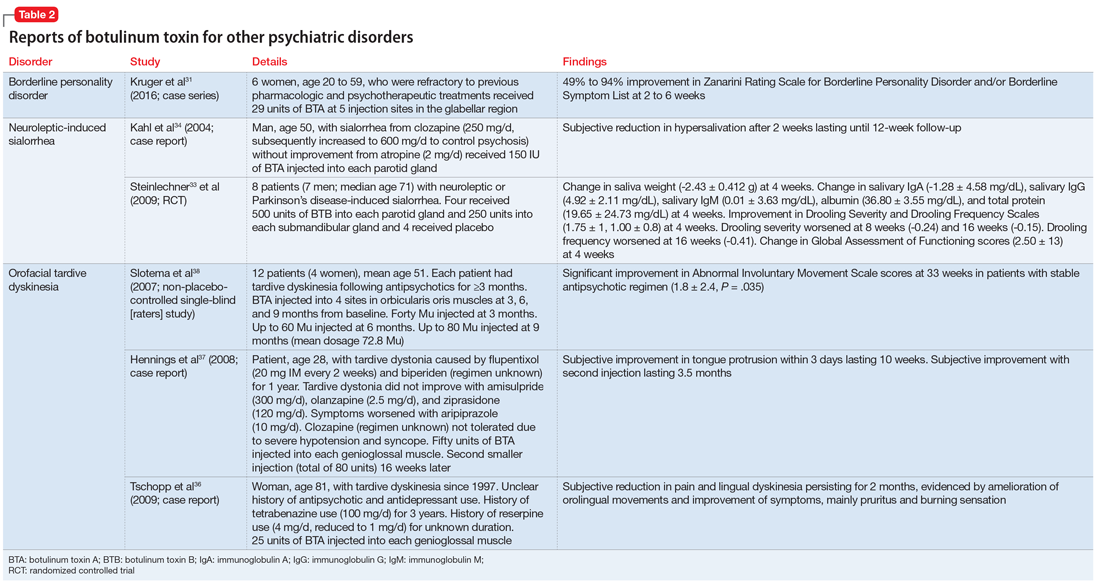
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.