User login
Treating negative symptoms of schizophrenia
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
In schizophrenia, negative symptoms such as social withdrawal, avoidance, lack of spontaneity and flow of conversation, reduced initiative, anhedonia, and blunted affect are among the most challenging to treat. These symptoms commonly persist after positive symptoms such as hallucinations, delusions, and paranoia have subsided. In an analysis of 20 pivotal placebo-controlled trials of second-generation antipsychotics (SGAs), almost 45% of patients who completed 6 weeks of treatment still had at least 1 residual negative symptom of at least moderate severity, and approximately 25% had 2 or more.1 Negative symptoms are viewed as being intrinsic to schizophrenia, and also as the result of extrapyramidal symptoms, depression, and psychosis.
Nearly a decade ago, the Schizophrenia Patient Outcomes Research Team (PORT) published its recommendations for psychopharmacologic and psychosocial treatments of schizophrenia. Unfortunately, due to insufficient evidence, there is still no proven treatment for negative symptoms.2-4 This is particularly problematic because negative symptoms are a major determinant of the poor social and vocational abilities of patients with schizophrenia.
This review focuses on treatments for negative symptoms of schizophrenia that have been evaluated since the PORT treatment recommendations were published and highlights those approaches that show promise.
_
The limitations of antipsychotics
Antipsychotics can both worsen and alleviate negative symptoms by reducing psychotic symptoms. Double-blind, placebo-controlled trials have found that most, if not all, antipsychotics are superior to placebo for treating negative symptoms in patients with acute psychosis.4 However, because these improvements occur in the early stages of treatment, concomitantly with improvement of psychotic symptoms, antipsychotics generally are not viewed as being very effective in the treatment of primary negative symptoms.4 Indeed, an examination of patients with prominent negative symptoms without prominent positive symptoms in the NEWMEDS cohort, which was extracted from 20 pivotal placebo-controlled trials of SGAs, revealed no clinically meaningful treatment effect on negative symptoms.1
There is evidence that antipsychotics can contribute to the development of apathy, flat affect, and other negative symptoms.5 Dopamine (D2)-blocking antipsychotics produce secondary negative symptoms that are not always easy to distinguish from primary negative symptoms.6 In a double-blind, placebo-controlled trial of single doses of risperidone, haloperidol, or placebo in healthy participants, the antipsychotics increased negative symptoms, particularly avolition/apathy.7 Another study found that chronic treatment with antipsychotics did not necessarily affect motivation in patients with schizophrenia.8
Adverse effects, such as anhedonia, often produce and enhance negative symptoms and therefore can limit the use of pharmacologic treatment options. Other adverse effects associated with specific antipsychotics include extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Continue to: Seeking new pharmacologic options
Seeking new pharmacologic options
The years since the PORT review have been filled with initial promise, a series of bitter disappointments, and a renewed spark of hope in the quest to treat negative symptoms in schizophrenia.
Compounds that have been abandoned. Since PORT, researchers have evaluated 5 major compounds that mainly targeted cognition and negative symptoms in patients with schizophrenia (Box9-17). Unfortunately, 4 of them failed to provide significant superiority over placebo, and 1 was withdrawn due to safety concerns.
Box
Since the Schizophrenia Patient Outcomes Research Team (PORT) treatment recommendations were published in 2010, many compounds have been investigated for treating negative symptoms of schizophrenia. Based on the findings of early research, further development of 5 of these has been abandoned.
Encenicline and TC-56199 were both α-7 nicotinic acetylcholine receptor agonists10; bitopertin and AMG 74711 were glycine reuptake inhibitors12; and pomaglumetad methionil13 was an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor.
Encenicline showed a treatment effect on negative symptoms in an add-on phase II study,14 but not in 2 subsequent phase III trials (NCT01716975, NCT01714661). TC-5619 showed a treatment effect in a 12-week phase II study of participants with persistent negative symptoms,15 but then failed in a subsequent study.9 Bitopertin showed a treatment effect on negative symptoms in 1 clinical trial,16 but the results were not replicated in a subsequent large multi-center trial.17 The AMG 747 development program was halted due to safety concerns.11 Finally, pomaglumetad methionil failed to meet its primary endpoint in a study of prominent negative symptoms and to show a treatment effect on psychotic symptoms in 2 pivotal phase III trials.13
Initial favorable results. Registered, robust trials of other compounds have had some initial favorable results that need to be replicated. These agents include:
- MIN-101 is a novel cyclic amide derivative.18 In a phase IIb 12-week study of MIN-101 monotherapy (32 mg, n = 78; 64 mg, n = 83) vs placebo (n = 83), both dose groups had significantly more improvement on the Positive and Negative Syndrome Scale (PANSS) negative factor score, which was the primary outcome measure, than placebo (32 mg/d; effect size = .45, P < .02, 64 mg/d; effect size = .57, P < .004) as well as on PANSS negative symptom score and other measures of negative symptoms.18
- Cariprazine is a D2 and D3 receptor partial agonist with high selectivity towards the D3 receptor19
- Minocycline is a broad-spectrum tetracyclic antibiotic displaying neuroprotective properties18,20,21
- Raloxifene is a selective estrogen receptor modulator for postmenopausal women22,23
- Pimavanserin, which was FDA-approved in 2016 for the treatment of Parkinson’s disease psychosis, is being tested in a large trial for adjunctive treatment of patients with negative symptoms of schizophrenia. This medication is a nondopaminergic antipsychotic that acts as a selective serotonin inverse agonist that preferentially targets 5-HT2A receptors while avoiding activity at common targets such as dopamine.24
All of these compounds except MIN-101 are currently available in the U.S. but have not been approved for the treatment of negative symptoms in patients with schizophrenia. MIN-101 is in phase III testing (NCT03397134).
Continue to: Nonpharmacologic treatments
Nonpharmacologic treatments
Recent studies of nonpharmacologic treatments for negative symptoms, including psychosocial approaches and noninvasive electromagnetic neurostimulation, have also been performed. The major psychosocial approaches that have been studied include social skills training (SST), cognitive-behavioral therapy (CBT) for psychosis, cognitive remediation, and family intervention. Some positive findings have been reported. A recent review of psychosocial treatments for negative symptoms in schizophrenia concluded that CBT and SST have the most empirical support, with some evidence even suggesting that gains from CBT are maintained as long as 6 months after treatment.25 Another review found that CBT was significantly more efficacious for reducing positive symptoms and SST in reducing negative symptoms.26
It remains unclear if a combined treatment approach provides improvements above and beyond those associated with each individual treatment modality. Motivation and Enhancement therapy (MOVE) is a potentially promising approach that combines environmental support, CBT, skills training, and other components in an attempt to address all domains of negative symptoms.27 Preliminary results from a randomized controlled trial examining 51 patients with clinically meaningful negative symptoms suggested that MOVE improves negative symptoms. However, the group differences were not significant until after 9 months of treatment and not for all negative symptom scales. A follow-up study has been completed, but the results are not yet available.28
Some small studies have suggested improvement of negative symptoms after noninvasive electromagnetic neurostimulation,29-31 but this has not been replicated in larger studies.32 In the last few years, there were several studies underway that could help clarify if there is a role for noninvasive electromagnetic neurostimulation in the treatment of negative symptoms in schizophrenia; however, results have not been reported at this time.33-35
_
Social skills training and combined interventions
Taken together, the data suggest that treating negative symptoms in schizophrenia remains a major challenge. Patients with negative symptoms are difficult to engage and motivate for treatment and there are no well-supported treatment options. Given the lack of evidence, it is not possible to synthesize this data into clear treatment recommendations. Because many of the negative symptoms are social in nature, it is perhaps not surprising that some evidence has emerged supporting the role of psychosocial approaches. Studies have pointed to the potential role of SST. It is believed to be beneficial as it targets participants’ social functioning by training verbal and nonverbal communication alongside perception and responses to social cues.36 Some evidence suggests that treatment packages that combine several psychosocial interventions (eg, family psychoeducation and skill training) achieve better outcomes than standalone interventions.37 Thus, psychosocial approaches appear to be potentially effective for the treatment of negative symptoms in patients with schizophrenia. In addition, because some antipsychotics has been shown to be associated with fewer negative symptoms than others, another treatment strategy could be to attempt the use of a different antipsychotic, or to revisit whether continued antipsychotic treatment is needed in the absence of positive symptoms.
Bottom Line
Treating negative symptoms in schizophrenia remains a major challenge. There is a lack of evidence for pharmacologic treatments; psychosocial approaches may be beneficial due to the social nature of many negative symptoms. Further, some evidence suggests that treatment packages that combine several psychosocial interventions may achieve better outcomes than standalone interventions.
Related Resource
Tandon R, Jibson M. Negative symptoms of schizophrenia: How to treat them most effectively. Current Psychiatry. 2002;1(9):36-42.
Drug Brand Names
Cariprazine • Vraylar
Haloperidol • Haldol
Minocycline • Dynacin, Minocin
Pimavanserin • Nuplazid
Raloxifene • Evista
Risperidone • Risperdal
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.
1. Rabinowitz J, Werbeloff N, Caers I, et al. Negative symptoms in schizophrenia--the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res. 2013;150(2-3):334-338.
2. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al. The schizophrenia patient outcomes research team (PORT): updated treatment recommendations 2009. Schizophrenia bulletin. 2010;36(1):94-103.
3. Veerman SRT, Schulte PFJ, de Haan L. Treatment for negative symptoms in schizophrenia: a comprehensive review. Drugs. 2017.
4. Aleman A, Lincoln TM, Bruggeman R, et al. Treatment of negative symptoms: Where do we stand, and where do we go? Schizophr Res. 2017;186:55-62.
5. Awad AG. Subjective tolerability of antipsychotic medications and the emerging science of subjective tolerability disorders. Expert Rev Pharmacoecon Outcomes Res. 2010;10(1):1-4.
6. Kirkpatrick B. Recognizing primary vs secondary negative symptoms and apathy vs expression domains. J Clin Psychiatry. 2014;75(4):e09.
7. Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488-493.
8. Fervaha G, Takeuchi H, Lee J, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40(6):1539-1548.
9. Walling D, Marder SR, Kane J, et al. Phase 2 Trial of an alpha-7 nicotinic receptor agonist (TC-5619) in negative and cognitive symptoms of schizophrenia. Schizophr Bull. 2016;42(2):335-343.
10. Haig GM, Bain EE, Robieson WZ, et al. A randomized trial to assess the efficacy and safety of ABT-126, a selective alpha7 nicotinic acetylcholine receptor agonist, in the treatment of cognitive impairment in schizophrenia. Am J Psychiatry. 2016;173(8):827-835.
11. U.S. National Library of Medicing. ClinicalTrials.gov. 20110165: Study to evaluate the effect of AMG 747 on schizophrenia negative symptoms (study 165). https://clinicaltrials.gov/ct2/show/NCT01568229. Accessed July 1, 2017.
12. Bugarski-Kirola D, Blaettler T, Arango C, et al. Bitopertin in negative symptoms of schizophrenia-results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82(1):8-16.
13. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2-3):434-441.
14. Keefe RS, Meltzer HA, Dgetluck N, et al. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40(13):3053-3060.
15. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
16. Umbricht D, Alberati D, Martin-Facklam M, et al. Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry. 2014;71(6):637-646.
17. Kingwell K. Schizophrenia drug gets negative results for negative symptoms. Nat Rev Drug Discov. 2014;13(4):244-245.
18. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of MIN-101: a 12-week randomized, double-blind, placebo-controlled trial of a new drug in development for the treatment of negative symptoms in schizophrenia. Am J Psychiatry. 2017;172(12):1195-1202.
19. Nemeth G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
20. Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138-149.
21. Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacology. 2012;26(9):1185-1193.
22. Usall J, Huerta-Ramos E, Labad J, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a 24-week double-blind, randomized, parallel, placebo-controlled trial. Schizophr Bull. 2016;42(2):309-317.
23. Usall J, Huerta-Ramos E, Iniesta R, et al. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2011;72(11):1552-1557.
24. Acadia Pharmaceuticals. Pimavanserin - schizophrenia negative symptoms. http://www.acadia-pharm.com/pipeline/pimavanserin-schizophrenia-negative-symptoms/. Accessed July 23, 2017.
25. Elis O, Caponigro JM, Kring AM. Psychosocial treatments for negative symptoms in schizophrenia: current practices and future directions. Clin Psychol Rev. 2013;33(8):914-928.
26. Turner DT, van der Gaag M, Karyotaki E, et al. Psychological interventions for psychosis: a meta-analysis of comparative outcome studies. Am J Psychiatry. 2014;171(5):523-538.
27. Velligan DI, Roberts D, Mintz J, et al. A randomized pilot study of MOtiVation and Enhancement (MOVE) Training for negative symptoms in schizophrenia. Schizophr Res. 2015;165(2-3):175-180.
28. U.S. National Library of Medicing. ClinicalTrials.gov. Treatment Development Targeting Severe and Persistent Negative Symptoms (MOVE). https://clinicaltrials.gov/ct2/show/NCT01550666. Accessed July 20, 2017.
29. Rabany L, Deutsch L, Levkovitz Y. Double-blind, randomized sham controlled study of deep-TMS add-on treatment for negative symptoms and cognitive deficits in schizophrenia. J Psychopharmacology. 2014;28(7):686-690.
30. Bation R, Brunelin J, Saoud M, et al. Intermittent theta burst stimulation of the left dorsolateral prefrontal cortex for the treatment of persistent negative symptoms in schizophrenia. European Neuropsychopharmacology. 2015;25:S329-S30.
31. Li Z, Yin M, Lyu XL, et al. Delayed effect of repetitive transcranial magnetic stimulation (rTMS) on negative symptoms of schizophrenia: findings from a randomized controlled trial. Psychiatry Res. 2016;240:333-335.
32. Wobrock T, Guse B, Cordes J, et al. Left prefrontal high-frequency repetitive transcranial magnetic stimulation for the treatment of schizophrenia with predominant negative symptoms: a sham-controlled, randomized multicenter trial. Biol Psychiatry. 2015;77(11):979-988.
33. U.S. National Library of Medicing. ClinicalTrials.gov. Repetitive transcranial magnetic stimulation and intermittent theta burst (iTBS) in schizophrenia phase 2. https://clinicaltrials.gov/ct2/show/NCT01315587. Accessed July 18, 2017.
34. Treatment of Negative Symptoms and Schizophrenia (STICCS) Phase 1/2. https://clinicaltrials.gov/ct2/show/NCT02204787. Accessed July 15, 2017.
35. U.S. National Library of Medicing. ClinicalTrials.gov. Schizophrenia TreAtment With electRic Transcranial Stimulation (STARTS). https://clinicaltrials.gov/ct2/show/NCT02535676. Accessed July 10, 2017.
36. Bellack AS, Mueser KT, Gingerich S, Agresta J. Social skills training for schizophrenia. A step-by-step guide. New York, NY: Guilford Press; 1997:20-30.
37. Hogarty GE, Anderson CM, Reiss DJ, et al. Family psychoeducation, social skills training, and maintenance chemotherapy in the aftercare treatment of schizophrenia. I. one-year effects of a controlled study on relapse and expressed emotion. Arch Gen Psychiatry. 1986;43(7):633-642.

Treatment-resistant OCD: There’s more we can do
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”

Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
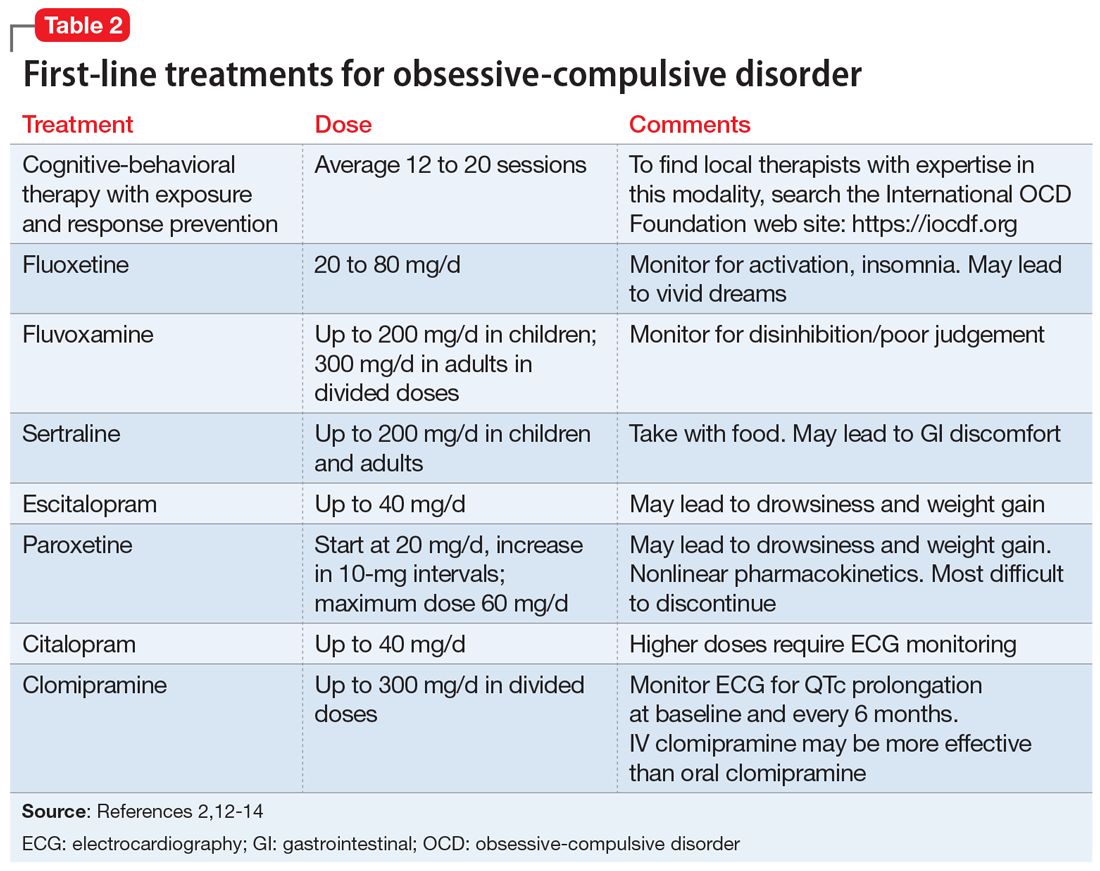
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.

Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
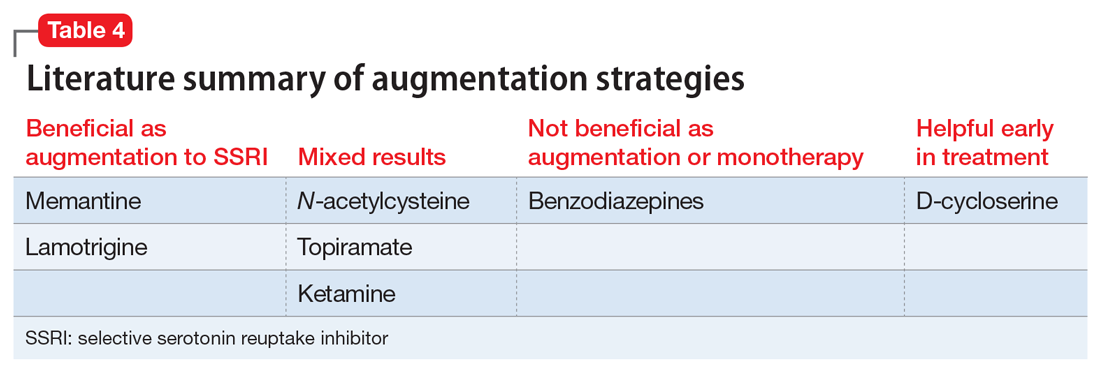
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
Treatment-resistant OCD can be a debilitating condition. Diagnostic clarity is crucial to fully elicit symptoms and identify comorbid conditions in order to develop practical, evidence-based treatment strategies and improve the patient’s and family’s quality of life. In this article, we delineate first-line strategies for treatment-resistant OCD and then review augmentation strategies, with an emphasis on glutamate-modulating agents.
Making the diagnosis
The diagnosis of OCD is made when a patient meets DSM-5 criteria for the presence of obsessions and/or compulsions, which are defined as unwanted, distressing, intrusive, recurrent thoughts or images (obsessions) and repetitive behaviors or mental acts (compulsions).1 OCD is considered a chronic waxing and waning disorder; stress and lack of sleep lead to worsening symptoms. The hidden nature of symptoms and the reinforcement provided by the reduction in anxiety after performing a compulsion contribute to sustained illness. Eliciting symptoms from patients may be challenging due to the shame they may feel. When reviewing symptoms on the Y-BOCS, it is helpful to preface questions with statements such as “Many people report excessive concern or disgust with…” to help the patient feel understood and less anxious, rather than using direct queries, such as “Are you bothered by…?”
Consider comorbid conditions
After making the initial diagnosis of OCD, it is important to assess whether the symptoms are better accounted for by another condition, and whether comorbid conditions are present (Table 1).
CASE CONTINUED
Ruling out other diagnoses
_
Initial treatment: CBT
Cognitive-behavioral therapy with exposures and response prevention (from here on referred to as CBT) has been established as a first-line, evidence-based treatment for OCD in both children and adults.2,3 For patients with treatment-resistant OCD, intensive daily CBT in a partial hospitalization or inpatient setting that is a tailor-made, patient-specific program is one of the most effective treatments, with response rates of up to 70%4-8 CBT’s advantages over medication include lower relapse rates and no known adverse effects. Unfortunately, CBT is underused9-11 due in part to a shortage of trained clinicians, and because patients may favor the ease of taking medication over the time, effort, and cost involved in CBT.
First-line pharmacologic options for treating OCD are SSRIs and clomipramine, as supported by multiple randomized controlled trials (RCTs), meta-analyses, expert guidelines, and consensus statements (Table 22,12-14). No significant difference has been found among SSRIs for the treatment of OCD in a review of 17 studies that included more than 3,000 patients.15 Treatment with SSRIs or clomipramine is effective for 50% to 60% of patients.16 Many clinicians view the combination of an SSRI and CBT as the treatment of choice for OCD.2
Continue to: Reluctance to engage in CBT
CASE CONTINUED
Reluctance to engage in CBT
To determine the next course of action, you review Mr. S’s treatment history. He has received adequate doses of 2 SSRIs and currently is taking clomipramine, 100 mg twice daily. He recently began CBT, which includes homework to help face his fears; however, Mr. S is reluctant to complete the exposure assignments, and after pausing for a few seconds as he tries to resist sending an apology email to his coworkers, he then returns to his compulsive behavior.
Facing treatment resistance
Although currently there isn’t a cure to resolve all traces of OCD, the goal of treatment is to decrease distress, interference, and the frequency of symptoms to a minimal level such that only the patients themselves are aware of symptoms. In broad terms, “response” has been defined as a decrease in symptoms, and “remission” has been defined as minimal symptoms after treatment.
Close to half of adults treated for OCD respond well to standard-of-care treatment (CBT and/or an SSRI), while the other 50% are considered partial responders or nonresponders.2 For patients with OCD, researchers often define “treatment response” as a ≥25% reduction in symptom severity score on the Y-BOCS. Approximately 30% of adults with OCD do not respond substantially to the first-line treatments, and even those who are defined as “responders” in research studies typically continue to have significant symptoms that impact their quality of life.2 In children, a clinical definition for treatment-refractory OCD has been presented as failing to achieve adequate symptom relief despite receiving an adequate course of CBT and at least 2 adequate trials of an SSRI or clomipramine.17 In the Pediatric OCD Treatment Study (POTS) trial, >46% of youth did not achieve remission from their OCD symptoms, even after receiving evidence-based care provided by experienced clinicians (combined treatment with CBT and an SSRI).18
_
Challenges in psychotherapy
Compassion is a key element in developing rapport with patients to help them face increasingly more challenging exposures. Making OCD the problem, not the person, is an essential element in helping patients move forward. Some clinicians may become frustrated with patients when treatment is not moving along well, referring to resistance, denial, or sabotage. According to March and Mulle,19 these terms lack the recognition and compassion that exposures are inherently difficult.19
Another challenge for therapists is if the patient’s presenting symptoms are personally offensive or a sensitive topic. For example, a therapist who is disgusted by public restrooms will find it difficult to tolerate the risks associated with exposure to germs and support a patient in touching objects in the restroom. Therapists also may be challenged when the patient’s fears align with the therapist’s religious beliefs. In these situations, consider transferring care to another therapist.
Family members need to learn about the nature of the illness and their roles in helping patients improve. Family members may unknowingly enable symptoms or criticize patients for their lack of motivation, which can lead to conflict in the home. Family dysfunction can in turn worsen OCD symptoms.
The most likely cause of lack of response to therapy is inexpert CBT.19 Deep breathing and relaxation training have been used as an active placebo in studies20; in a meta-analysis examining the effective components of CBT, studies that added relaxation training were not more effective than those that employed exposures alone.21 Patients receiving CBT should be able to articulate the hierarchical approach used to gradually face their fears.
Continue to: Pharmacologic augmentation strategies
Pharmacologic augmentation strategies
Selective serotonin reuptake inhibitors. While most OCD research trials have assessed SSRIs in 12-week studies, clinicians may consider extending SSRI treatment for an additional 12 weeks for nonresponders because some patients will continue to make gains. In the past, it was generally believed that higher doses of SSRIs are needed for treating OCD than for treating major depressive disorder. For instance, greater improvement was seen with 250 to 400 mg/d of sertraline compared with 200 mg/d22 and with escitalopram after an increase of dose up to 50 mg/d.23 However, more recently, this notion of higher doses being necessary for treatment response has been called into question. For example, a study of escitalopram found similar responses to 10 mg/d vs 20 mg/d after 24 weeks.24 A meta-analysis of adult studies of SSRIs for OCD supported higher doses as being more effective, but noted that the drop-out rate from treatment was greater in patients treated with higher doses.25 As a note of caution, long-term, high-dose maintenance therapy increases the risk of adverse reactions.26
Following a failed treatment with a first SSRI, it remains debatable as to what ought to be the second pharmacologic treatment. Although clomipramine is often reserved for treatment after 2 failed trials of an SSRI due to its greater risk of adverse effects, in an open-label study, switching from an SSRI to clomipramine led to greater response than switching from one SSRI to another.27 On the other hand, while meta-analyses have reported greater treatment effect for oral clomipramine than for SSRIs, direct head-to-head comparisons have not supported this notion.28 To get the best of both worlds, some clinicians employ a strategy of combining clomipramine with an SSRI, while monitoring for adverse effects and interactions such as serotonin syndrome.29-31
Benzodiazepines. Although benzodiazepines are useful for brief treatment of an anxiety disorder (eg, for a person with a fear of heights who needs to take an airplane),32 they have not been shown to be effective for OCD33 or as augmentation to an SSRI.34
N-acetylcysteine (NAC). Two RCTs of adults with OCD who received adjunctive NAC, 3 g/d in divided doses, found no significant difference in the treatment arms by the conclusion of 16 weeks—either both groups improved, or both groups failed to improve.35,36 In a 10-week study of patients with moderate to severe OCD symptoms, NAC, 2 g/d, as augmentation to fluvoxamine, 200 mg/d, showed a significant time x interaction in the treatment group.37 No follow-up information is available, however.
In a multicenter RCT of NAC given to children and adolescents with OCD as augmentation to citalopram, symptoms decreased and the quality-of-life score improved, with a large treatment effect size in the NAC group.38 However, in a study aimed at examining NAC in youth with Tourette syndrome, OCD symptoms were measured as a secondary outcome and there was no benefit of NAC over placebo.39
Memantine. Four 8- to 12-week RCTs in adults with OCD favored adjunctive memantine, 20 mg/d, taken with an SSRI, over placebo.40-43 A small study suggests that patients with OCD may be more likely to respond to memantine than patients with generalized anxiety disorder.44 Case reports have noted that memantine has been beneficial for pediatric patients with refractory OCD.45
Continue to: Topiramate
Topiramate. Three 12-week RCTs examined topiramate augmentation at 100 to 400 mg/d in patients with OCD who had failed at least 1 previous trial of an SSRI. The earliest study was most encouraging: Y-BOCS scores decreased by 32% in the topiramate group but by only 2.4% in the placebo group.46 However, the other 2 studies found no difference in the final OCD symptom severity score between active treatment and placebo groups,47,48 and the use of topiramate, particularly at higher doses, was limited by its adverse effects.
Lamotrigine. Initially, lamotrigine augmentation of SSRIs in OCD did not appear to be helpful.49 More recently, several case studies reported that lamotrigine, 100 to 200 mg/d, added to paroxetine or clomipramine, resulted in dramatic improvement in Y-BOCS scores for patients with long-standing refractory symptoms.50,51 In a retrospective review of 22 patients who received augmentation with lamotrigine, 150 mg/d, 20 had a significant response; the mean decrease in Y-BOCS score was 67%.52 Finally, in a 16-week RCT, lamotrigine, 100 mg/d, added to an SSRI led to a significant decrease in both Y-BOCS score and depressive symptoms while also improving semantic fluency.53
Ketamine. Ketamine is drawing increased attention for its nearly instantaneous antidepressant effect that lasts for up to 2 weeks after a single infusion.54 In a study of 15 medication-free adults with continuous intrusive obsessions, 4 of 8 patients who received a single IV infusion of ketamine, 0.5 mg/kg, met the criteria for treatment response (>35% reduction in Y-BOCS score measured 1 week later); none of the patients who received a placebo infusion of saline met this criteria.55 A small open-label trial of 10 treatment-refractory patients found that an infusion of ketamine, 0.5 mg/kg, was beneficial for comorbid depression but had only a minimal effect on OCD symptoms measured 3 days post-infusion.56 A short-term follow-up on these patients revealed dysphoria in some responders.57
D-cycloserine. The idea of using a pharmacologic agent to increase the speed or efficacy of behavioral therapy is intriguing. Proof of concept was demonstrated in a study that found that giving D-cycloserine prior to computerized exposure therapy significantly improved clinical response in patients with acrophobia.58 However, using this approach to treating OCD netted mixed results; D-cycloserine was found to be most helpful during early stages of treatment.59,60
Table 3 outlines the mechanisms of action and common uses for NAC, memantine, ketamine, topiramate, lamotrigine, and D-cycloserine. Table 4 summarizes the literature on the efficacy of some of the augmentation strategies for treating OCD described in this article.
Continue to: Alternative strategies
Alternative strategies
Augmentation strategies with neuroleptics,61 transcranial magnetic stimulation,62 and deep brain stimulation63 have recently been reviewed. Space limitations preclude a comprehensive review of these strategies, but in a cross-sectional study of augmentation strategies in OCD, no difference was found in terms of symptom severity between those prescribed SSRI monotherapy or augmentation with neuroleptics, benzodiazepines, or antidepressants.64
CASE CONTINUED
Progress in CBT
Mr. S agrees to a trial of NAC as an augmentation strategy, but after 8 weeks of treatment with NAC, 600 mg twice daily, his Y-BOCS had declined by only 2 points. He also complains of nausea and does not want to increase the dose. You discontinue NAC and opt to further explore his reaction to CBT. Mr. S shares that he has been seeing his psychologist only once every 3 weeks because he does not want to miss work. You encourage him to increase to weekly CBT sessions, and you obtain his permission to contact his therapist and his family members. Fortunately, his therapist is highly qualified, but unfortunately, Mr. S’s father has been sending him multiple critical emails about not advancing at his job and for being “lazy” at work. You schedule a session with Mr. S and his father. Great progress is made after Mr. S and his father both share their frustrations and come to understand and appreciate each other’s struggles. Four weeks later, after weekly CBT appointments, Mr. S has a Y-BOCS of 18 and spends <2 hours/d checking emails for errors and apologizing.
Bottom Line
It is unrealistic to expect OCD symptoms to be cured. Many ‘treatment-resistant’ patients have not received properly delivered cognitive-behavioral therapy, and this first-line treatment modality should be considered in every eligible patient, and augmented with a selective serotonin reuptake inhibitor (SSRI) when needed. Glutamatergic agents, in turn, can augment SSRIs.
Related Resources
- Yale-Brown Obsessive-Compulsive Scale. https://iocdf.org/ wp-content/uploads/2014/08/Assessment-Tools.pdf.
- The International OCD Foundation. https://iocdf.org.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Ketamine • Ketalar
Lamotrigine • Lamictal
Memantine • Namenda
Paroxetine • Paxil
Sertraline • Zoloft
Topiramate • Topomax
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Koran LM, Hanna GL, Hollander E, et al. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry; 2007;164(suppl 7):5-53.
3. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
4. Bystritsky A, Munford PR, Rosen RM, et al. A preliminary study of partial hospital management of severe obsessive-compulsive disorder. Psychiatr Serv. 1996;47(2):170-174.
5. Calvocoressi L, McDougle CI, Wasylink S, et al. Inpatient treatment of patients with severe obsessive-compulsive disorder. Hosp Community Psychiatry. 1993;44(12):1150-1154.
6. Eddy KT, Dutra L, Bradley R, et al. A multidimensional meta-analysis of psychotherapy and pharmacotherapy for obsessive-compulsive disorder. Clin Psychol Rev. 2004;24(8):1011-1030.
7. Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407-416.
8. Simpson HB, Huppert JD, Petkova E, et al. Response versus remission in obsessive-compulsive disorder. J Clin Psychiatry. 2006;67(2):269-276.
9. Marques L, LeBlanc NJ, Weingarden HM, et al. Barriers to treatment and service utilization in an internet sample of individuals with obsessive-compulsive symptoms. Depress Anxiety. 2010;27(5):470-475.
10. Goodwin R, Koenen KC, Hellman F, et al. Helpseeking and access to mental health treatment for obsessive-compulsive disorder. Acta Psychiatr Scand. 2002;106(2):143-149.
11. Kohn R, Saxena S, Levav I, et al. The treatment gap in mental health care. Bull World Health Organ. 2004;82(11):858-866.
12. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
13. Lovell K, Bee P. Implementing the NICE OCD/BDD guidelines. Psychol Psychother. 2008;81(Pt 4):365-376.
14. Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive-compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77-84.
15. Soomro GM, Altman D, Rajagopal S, et al. Selective serotonin re-uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database Syst Rev. 2008;(1):CD001765.
16. Pittenger C, Bloch MH. Pharmacological treatment of obsessive-compulsive disorder. Psychiatr Clin North Am. 2014;37(3):375-391.
17. Bloch MH, Storch EA. Assessment and management of treatment-refractory obsessive-compulsive disorder in children. J Am Acad Child Adolesc Psychiatry. 2015;54(4):251-262.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
19. March JS, Mulle K. OCD in children and adolescents: a cognitive-behavioral treatment manual. New York, NY: Guilford Press; 1998.
20. Marks IM. Fears, phobias, and rituals: Panic, anxiety, and their disorders. 1987, New York, NY: Oxford University Press; 1987.
21. Ale CM, McCarthy DM, Rothschild LM, et al. Components of cognitive behavioral therapy related to outcome in childhood anxiety disorders. Clin Child Fam Psychol Rev. 2015;18(3):240-251.
22. Ninan PT, Koran LM, Kiev A, et al. High-dose sertraline strategy for nonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry. 2006;67(1):15-22.
23. Rabinowitz I, Baruch Y, Barak Y. High-dose escitalopram for the treatment of obsessive-compulsive disorder. Int Clin Psychopharmacol. 2008;23(1):49-53.
24. Stein DJ, Andersen EW, Tonnoir B, et al. Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin. 2007;23(4):701-711.
25. Bloch MH, McGuire J, Landeros-Weisenberger A, et al. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol Psychiatry. 2010;15(8):850-855.
26. Sayyah M, Majzoob S, Sayyah M. Metabolic and toxicological considerations for obsessive-compulsive disorder drug therapy. Expert Opin Drug Metab Toxicol. 2013;9(6):657-673.
27. Hollander E, Bienstock CA, Koran LM, et al. Refractory obsessive-compulsive disorder: state-of-the-art treatment. J Clin Psychiatry. 2002;63(suppl 6):20-29.
28. Fineberg NA, Gale TM. Evidence-based pharmacotherapy of obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2005;8(1):107-129.
29. Marazziti D, Golia F, Consoli G, et al. Effectiveness of long-term augmentation with citalopram to clomipramine in treatment-resistant OCD patients. CNS Spectr. 2008;13(11):971-976.
30. Browne M, Horn E, Jones TT. The benefits of clomipramine-fluoxetine combination in obsessive compulsive disorder. Can J Psychiatry. 1993;38(4):242-243.
31. Ravizza L, Barzega G, Bellino S, et al. Drug treatment of obsessive-compulsive disorder (OCD): long-term trial with clomipramine and selective serotonin reuptake inhibitors (SSRIs). Psychopharmacol Bull. 1996;32(1):167-173.
32. Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci. 2011;13(4):423-437.
33. Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
34. Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam with sertraline in obsessive-compulsive disorder. Ann Clin Psychiatry. 2004;16(3):127-132.
35. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of n-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766-e773.
36. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801-809.
37. Paydary K, Akamaloo A, Ahmadipour A, et al. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214-219.
38. Ghanizadeh A, Mohammadi MR, Bahraini S, et al. Efficacy of N-acetylcysteine augmentation on obsessive compulsive disorder: a multicenter randomized double blind placebo controlled clinical trial. Iran J Psychiatry. 2017;12(2):134-141.
39. Bloch MH, Panza KE, Yaffa A, et al. N-acetylcysteine in the treatment of pediatric tourette syndrome: randomized, double-blind, placebo-controlled add-on trial. J Child Adolesc Psychopharmacol. 2016;26(4):327-334.
40. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175-180.
41. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34-39.
42. Modarresi A, Sayyah M, Razooghi S, et al. Memantine augmentation improves symptoms in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder: a randomized controlled trial. Pharmacopsychiatry. 2017. doi: 10.1055/s-0043-120268. [Epub ahead of print].
43. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633-640.
44. Feusner JD, Kerwin L, Saxena S, et al. Differential efficacy of memantine for obsessive-compulsive disorder vs. generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81-93.
45. Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237.
46. Mowla A, Khajeian AM, Sahraian A, et al. topiramate augmentation in resistant ocd: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613-617.
47. Berlin H, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716-721.
48. Afshar H, Akuchekian S, Mahaky B, et al. Topiramate augmentation in refractory obsessive-compulsive disorder: A randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976-981.
49. Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-528.
50. Arrojo-Romero M, Tajes Alonso M, de Leon J. Lamotrigine augmentation of serotonin reuptake inhibitors in severe and long-term treatment-resistant obsessive-compulsive disorder. Case Rep Psychiatry. 2013;2013:612459.
51. Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425-427.
52. Hussain A, Dar MA, Wani RA, et al. Role of lamotrigine augmentation in treatment-resistant obsessive compulsive disorder: a retrospective case review from South Asia. Indian J Psychol Med. 2015;37(2):154-158.
53. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-1462.
54. Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):113311-41.
55. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475-2483.
56. Bloch MH, Wasylink S, Landeros-Weisenberger A,, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964-970.
57. Niciu MJ, Grunschel BD, Corlett PR, et al. Two cases of delayed-onset suicidal ideation, dysphoria and anxiety after ketamine infusion in patients with obsessive-compulsive disorder and a history of major depressive disorder. J Psychopharmacol. 2013;27(7):651-654.
58. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61(11):1136-1144.
59. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
60. Xia J, Du Y, Han J, et al. D-cycloserine augmentation in behavioral therapy for obsessive-compulsive disorder: a meta-analysis. Drug Des Devel Ther. 2015;9:2101-2117.
61. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and meta-analysis. BMC Psychiatry. 2014;14:317.
62. Guo Q, Li C, Wang J. Updated review on the clinical use of repetitive transcranial magnetic stimulation in psychiatric disorders. Neurosci Bull. 2017;33(6):747-756.
63. Naesström, M, Blomstedt P, Bodlund O. A systematic review of psychiatric indications for deep brain stimulation, with focus on major depressive and obsessive-compulsive disorder. Nord J Psychiatry. 2016;70(7):483-491.
64. Van Ameringen M, Simpson W, Patterson B, et al. Pharmacological treatment strategies in obsessive compulsive disorder: A cross-sectional view in nine international OCD centers. J Psychopharmacol, 2014;28(6):596-602.

Disruptive mood dysregulation disorder: A better understanding
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
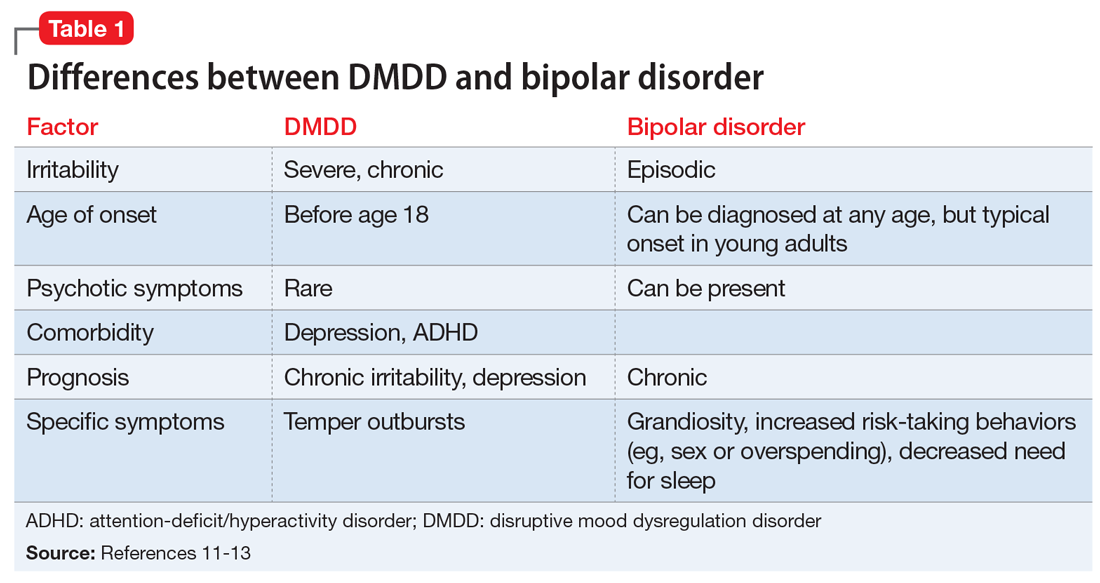
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
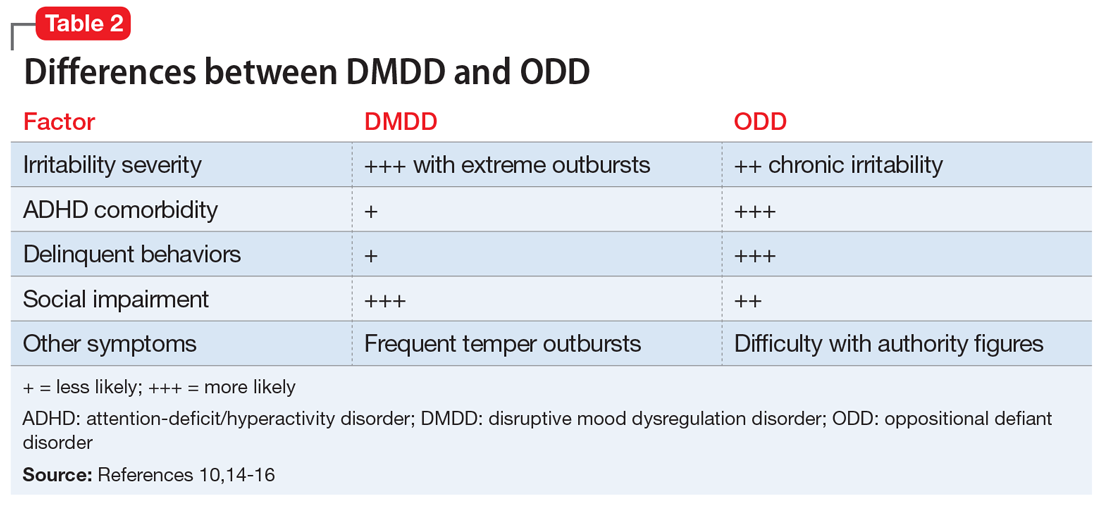
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
Disruptive mood dysregulation disorder (DMDD)—a childhood condition of extreme irritability, anger, and frequent, intense temper outbursts—has been a source of controversy among clinicians in the field of pediatric mental health. Before DSM-5 was published, the validity of DMDD had been questioned because DMDD had failed a field trial; agreement between clinicians on the diagnosis of DMDD was poor.1 Axelson2 and Birmaher et al3 examined its validity in their COBY (Course and Outcome of Bipolar Youth) sample. They concluded that only 19% met the criteria for DMDD in 3 times of follow-up. Furthermore, most DMDD criteria overlap with those of other common pediatric psychiatric disorders, including oppositional defiant disorder (ODD), attention-deficit/hyperactivity disorder (ADHD), and pediatric bipolar disorder (BD). Because diagnosis of pediatric BD increased drastically from 2.9% to 15.1% between 1990 and 2000,4 it was believed that introducing DMDD as a diagnosis might lessen the overdiagnosis of pediatric BD by identifying children with chronic irritability and temper tantrums who previously would have been diagnosed with BD.
It is important to recognize that in pediatric patients, mood disorders present differently than they do in adults.5 In children/adolescents, mood disorders are less likely to present as distinct episodes (narrow band), but more likely to present as chronic, broad symptoms. Also, irritability is a common presentation in many pediatric psychiatric disorders, such as ODD, BD (irritability without elation),6 and depression. Thus, for many clinicians, determining the correct mood disorder diagnosis in pediatric patients can be challenging.
This article describes the diagnosis of DMDD, and how its presentation is similar to—and different from—those of other common pediatric psychiatric disorders.
_
The origin of DMDD
Many researchers have investigated the broadband phenotypical presentation of pediatric mood disorders, which have been mostly diagnosed in the psychiatric community as pediatric BD. Leibenluft7 identified a subtype of mood disorder that they termed “severe mood dysregulation” (SMD). Compared with the narrow-band, clearly episodic BD, SMD has a different trajectory, outcome, and findings on brain imaging. SMD is characterized by chronic irritability with abnormal mood (anger or sadness) at least half of the day on most days, with 3 hyperarousal symptoms, including pressured speech, racing thoughts or flight of ideas, intrusiveness, distractibility, insomnia, and agitation.8 Eventually, SMD became the foundation of the development of DMDD.
DSM-5 diagnostic criteria for DMDD include severe recurrent temper outbursts that are out of proportion to the situation, inconsistent with developmental level, and occurring on average ≥3 times per week, plus persistently irritable or angry mood for most of the day nearly every day.9 Additional criteria include the presence of symptoms for at least 12 months (without a symptom-free period of at least 3 consecutive months) in ≥2 settings (at home, at school, or with peers) with onset before age 10. The course of DMDD typically is chronic with accompanying severe temperament. The estimated 6-month to 1-year prevalence is 2% to 5%; the diagnosis is more common among males and school-age children than it is in females and adolescents.9,10
_
DMDD or bipolar disorder?
A patient cannot be dually diagnosed with both disorders. If a patient exhibits a manic episode for more than 1 day, that would null and void the DMDD diagnosis. However, in a study to evaluate BD in pediatric patients, researchers divided BD symptoms into BD-specific categories (elevated mood, grandiosity, and increased goal-directed activity) and nonspecific symptoms such as irritability and talkativeness, distractibility, and flight of ideas or racing thoughts. They found that in the absence of specific symptoms, a diagnosis of BD is unlikely to be the correct diagnosis.11 Hence, as a nonspecific symptom, chronic irritability should be attributed to the symptom count for DMDD, rather than BD. Most epidemiologic studies have concluded that depression and anxiety, and not irritability, are typically the preceeding presentations prior to the development of BD in young adults.12 Chronic irritability, however, predicts major depressive disorder and anxiety disorders in later adolescence and one’s early twenties.13 Furthermore, BD commonly presents with infrequent and discrete episodes and a later age of onset, while DMDD presents with chronic and frequent, severe temper outbursts. Some differences between DMDD and BD are illustrated in Table 1.11-13
Continue to: CASE 1
CASE 1
Irritable and taking risks
Ms. N, age 16, is brought to the outpatient psychiatry clinic by her parents for evaluation of mood symptoms, including irritability. Her mother claims her daughter was an introverted, anxious, shy child, but by the beginning of middle school, she began to feel irritable and frequently stayed up at night with little sleep. In high school, Ms. N had displayed several episodes of risk-taking behaviors, including taking her father’s vehicle for a drive despite not having a driver’s permit, running away for 2 days, and having unprotected sex.
During her assessment, Ms. N is pleasant and claims she usually has a great mood. She fought with her mother several times this year, which led her to run away. Her parents had divorced when Ms. N was 5 years old and have shared custody. Ms. N is doing well in school despite her parents’ concerns.
Diagnosis. The most likely diagnosis is emerging BD. Notice that Ms. N may have had anxiety symptoms before she developed irritability. She had a relatively late onset of symptoms that were episodic in nature, which further supports a diagnosis of BD.
_
>
DMDD or oppositional defiant disorder?
DMDD and ODD cannot be dually diagnosed. However, if a patient meets the criteria for both DMDD and ODD, only the DMDD diagnosis should be considered. One of many issues of DMDD is its similarity to ODD. In fact, more than 70% of patients with DMDD also meet the diagnostic criteria for ODD.10,14 Some researchers have conceptualized DMDD as a severe form of ODD. However, there are a few differences that clinicians can use to distinguish the 2 disorders.
Compared with patients with ODD, those with DMDD more frequently experience severe irritability.15 Patients with ODD may present with delinquent behaviors and trouble with authority figures. Moreover, comorbidity with ADHD is twice as common in ODD; more than 65% of patients with ADHD have ODD vs 30% who have DMDD.10,16 Finally, in general, children with DMDD have more social impairments compared with those with ODD. Differences between DMDD and BD are illustrated in Table 2.10,14-16
Continue to: CASE 2
CASE 2
Angry and defiant
Mr. R, age 14, is brought to the emergency department (ED) by his parents after becoming very aggressive with them. He punched a wall and vandalized his room after his parents grounded him because of his previous defiant behavior. He had been suspended from school that day for disrespecting his teacher after he was caught fighting another student.
His parents describe Mr. R as a strong-willed, stubborn child. He has difficulty with rules and refuses to follow them. He is grouchy and irritable around adults, including the ED staff. Mr. R enjoys being with his friends and playing video games. He had been diagnosed with ADHD when he was in kindergarten, when his teacher noticed he had a poor attention span and could not sit still. According to his parents, Mr. R has “blown up” a few times before, smashing items in his room and shouting obscenities. Mr. R’s parents noticed that he is more defiant in concurrence with discontinuing his ADHD stimulant medication.
Diagnosis. The most likely diagnosis for Mr. R is ODD. Notice the comorbidity of ADHD, which is more commonly associated with ODD. The frequency and severity of his outbursts and irritability symptoms were lower than that typically associated with DMDD.
_
Treatment strategies for DMDD
Management of DMDD should focus on helping children and adolescents improve their emotional dysregulation.
Clinicians should always consider behavioral therapy as a first-line intervention. The behavioral planning team may include (but is not limited to) a behavior specialist, child psychiatrist, psychologist, therapist, skills trainer, teachers, and the caregiver(s). The plan should be implemented across all settings, including home and school. Furthermore, social skills training is necessary for many children with DMDD, who may require intensive behavioral modification planning. Comorbidity with ADHD should be addressed with a combination of behavioral planning and stimulant medications.17 If available, parent training and parent-child interactive therapy can help to improve defiant behavior.
Pharmacotherapy
Currently, no medications are FDA-approved for treating DMDD. Most pharmacologic trials that included patients with DMDD focused on managing chronic irritability and/or stabilizing comorbid disorders (ie, ADHD, depression, and anxiety).
Continue to: Stimulants
Stimulants. Previous trials examined the benefit of CNS stimulant medications, alone or in conjunction with behavioral therapy, in treating DMDD and comorbid ADHD. Methylphenidate results in a significant reduction in aggression18 with a dosing recommendation range from 1 to 1.2 mg/kg/d. CNS stimulants should be considered as first-line pharmacotherapy for DMDD, especially for patients with comorbid ADHD.
Anticonvulsants. Divalproex sodium is superior to placebo in treating aggression in children and adolescents.19 Trials found that divalproex sodium reduces irritability and aggression whether it is prescribed as monotherapy or combined with stimulant medications.19
Lithium is one of the main treatment options for mania in BD. The benefits of lithium for controlling aggression in DMDD are still under investigation. Earlier studies found that lithium significantly improves aggressive behavior in hospitalized pediatric with conduct disorder.20,21 However, a later study that evaluated lithium vs placebo for children with SMD (which arguably is phenotypically related to the DMDD) found there were no significant differences in improvement of irritability symptoms between groups.22 More research is needed to determine if lithium may play a role in treating patients with DMDD.
Antipsychotics. Aripiprazole and risperidone are FDA-approved for treating irritability in autism. A 2017 meta-analysis found both medications were effective in controlling irritability and aggression in other diagnoses as well.23 Other antipsychotic medications did not show sufficient benefits in treating irritability.23 When considering antipsychotics, clinicians should weigh the risks of metabolic adverse effects and follow practice guidelines.
Antidepressants. A systematic review did not find that selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors effectively reduce irritability.24 However, in most of the studies evaluated, irritability was not the primary outcome measure.24
Other medications. Alpha-2 agonists (guanfacine, clonidine), and atomoxetine may help irritability indirectly by improving ADHD symptoms.25
Bottom Line
Disruptive mood dysregulation disorder (DMDD), bipolar disorder, and oppositional defiant disorder have similar presentations and diagnostic criteria. The frequency and severity of irritability can be a distinguishing factor. Behavioral therapy is a first-line treatment. No medications are FDA-approved for treating DMDD, but pharmacotherapy may help reduce irritability and aggression.
Related Resources
- Rao U. DSM-5: disruptive mood dysregulation disorder. Asian J Psychiatr. 2014;11:119-123.
- Roy AK, Lopes V, Klein RG. Disruptive mood dysregulation disorder: a new diagnostic approach to chronic irritability in youth. Am J Psychiatry. 2014;171(9):918-924.
Drug Brand Names
Aripiprazole • Abilify
Atomoxetine • Strattera
Clonidine • Catapres
Divalproex sodium • Depakote, Depakote ER
Guanfacine • Intuniv, Tenex
Lithium • Eskalith, Lithobid
Methylphenidate • Concerta, Ritalin
Risperidone • Risperdal
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.
1. Regier DA, Narrow WE, Clarke DE, et al. DSM-5 field trials in the United States and Canada, Part II: test-retest reliability of selected categorical diagnoses. Am J Psychiatry. 2013;170(1):59-70.
2. Axelson D. Taking disruptive mood dysregulation disorder out for a test drive. Am J Psychiatry. 2013;170(2):136-139.
3. Birmaher B, Axelson D, Goldstein B, et al. Four-year longitudinal course of children and adolescents with bipolar spectrum disorders: the Course and Outcome of Bipolar Youth (COBY) study. Am J Psychiatry. 2009;166(7):795-804.
4. Case BG, Olfson M, Marcus SC, et al. Trends in the inpatient mental health treatment of children and adolescents in US community hospitals between 1990 and 2000. Arch Gen Psychiatry. 2007;64(1):89-96.
5. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.
6. Hunt J, Birmaher B, Leonard H, et al. Irritability without elation in a large bipolar youth sample: frequency and clinical description. J Am Acad Child Adolesc Psychiatry. 2009;48(7):730-739.
7. Leibenluft E. Severe mood dysregulation, irritability, and the diagnostic boundaries of bipolar disorder in youths. Am J Psychiatry. 2011;168(2):129-142.
8. Rich BA, Carver FW, Holroyd T, et al. Different neural pathways to negative affect in youth with pediatric bipolar disorder and severe mood dysregulation. J Psychiatr Res. 2011;45(10):1283-1294.
9. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
10. Copeland WE, Angold A, Costello EJ, et al. Prevalence, comorbidity, and correlates of DSM-5 proposed disruptive mood dysregulation disorder. Am J Psychiatry. 2013;170(2):173-179.
11. Elmaadawi AZ, Jensen PS, Arnold LE, et al. Risk for emerging bipolar disorder, variants, and symptoms in children with attention deficit hyperactivity disorder, now grown up. World J Psychiatry. 2015;5(4):412-424.
12. Duffy A. The early natural history of bipolar disorder: what we have learned from longitudinal high-risk research. Can J Psychiatry. 2010;55(8):477-485.
13. Stringaris A, Cohen P, Pine DS, et al. Adult outcomes of youth irritability: a 20-year prospective community-based study. Am J Psychiatry. 2009;166(9):1048-1054.
14. Mayes SD, Waxmonsky JD, Calhoun SL, et al. Disruptive mood dysregulation disorder symptoms and association with oppositional defiant and other disorders in a general population child sample. J Child Adolesc Psychopharmacol. 2016;26(2):101-106.
15. Stringaris A, Vidal-Ribas P, Brotman MA, et al. Practitioner review: definition, recognition, and treatment challenges of irritability in young people. J Child Psychol Psychiatry. 2018;59(7):721-739.
16. Angold A, Costello EJ, Erkanli A. Comorbidity. J Child Psychol Psychiatry. 1999;40(1):57-87.
17. Fernandez de la Cruz L, Simonoff E, McGough JJ, et al. Treatment of children with attention-deficit/hyperactivity disorder (ADHD) and irritability: results from the multimodal treatment study of children with ADHD (MTA). J Am Acad Child Adolesc Psychiatry. 2015;54(1):62-70.
18. Pappadopulos E, Woolston S, Chait A, et al. Pharmacotherapy of aggression in children and adolescents: efficacy and effect size. J Can Acad Child Adolesc Psychiatry. 2006;15(1):27-39.
19. Donovan SJ, Stewart JW, Nunes EV, et al. Divalproex treatment for youth with explosive temper and mood lability: a double-blind, placebo-controlled crossover design. Am J Psychiatry. 2000;157(5):818-820.
20. Campbell M, Adams PB, Small AM, et al. Lithium in hospitalized aggressive children with conduct disorder: a double-blind and placebo-controlled study. J Am Acad Child Adolesc Psychiatry. 1995;34(4):445-453.
21. Malone RP, Delaney MA, Luebbert JF, et al. A double-blind placebo-controlled study of lithium in hospitalized aggressive children and adolescents with conduct disorder. Arch Gen Psychiatry. 2000;57(7):649-654.
22. Dickstein DP, Towbin KE, Van Der Veen JW, et al. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J Child Adolesc Psychopharmacol. 2009;19(1):61-73.
23. van Schalkwyk GI, Lewis AS, Beyer C, et al. Efficacy of antipsychotics for irritability and aggression in children: a meta-analysis. Expert Rev Neurother. 2017;17(10):1045-1053.
24. Kim S, Boylan K. Effectiveness of antidepressant medications for symptoms of irritability and disruptive behaviors in children and adolescents. J Child Adolesc Psychopharmacol. 2016;26(8):694-704.
25. Scahill L, Chappell PB, Kim YS, et al. A placebo-controlled study of guanfacine in the treatment of children with tic disorders and attention deficit hyperactivity disorder. Am J Psychiatry. 2001;158(7):1067-1074.

Bright light therapy for bipolar depression
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
Bright light therapy (BLT) refers to the use of bright light to treat symptoms of depression. BLT was initially prescribed as a treatment for patients with seasonal affective disorder.1 It was later found helpful for nonseasonal depression,2 premenstrual dysphoric disorder, postpartum depression, and phase shift circadian disorders, including for patients with dementia whose cognitive function improved after treatment with BLT.3 More recent studies suggest year-round benefit for nonseasonal depression.2 The American Psychiatric Association practice guidelines for the treatment of depression list BLT as an alternative and/or addition to pharmacologic and psychological treatment.4 BLT also may be beneficial for patients who are in the depressive phase of bipolar illness.
This article describes the evidence, rationale for use, mechanism of action, benefits, and safety profile of BLT for treating patients with bipolar depression.
Circadian rhythm disruption in bipolar disorder
Clinical manifestation. Patients with bipolar disorder (BD) spend more time in depression than in mania.5 Sleep disturbance is a core symptom of BD; patients typically have little need for sleep during a manic episode, and excess sleepiness during a depressive episode. Sleep complaints can be both precipitating factors and consequences of mood disorders. Patients with seasonal depression have excess sleepiness and weight gain in the winter followed by hypomanic-like symptoms in the spring, including decreased need for sleep and weight loss with psychomotor activation. In a recent review of sleep problems in patients with BD, Steinan et al6 reported that 20% of patients with euthymic mood in bipolar disorder experience a sleep disorder. Circadian disruption and “eveningness” (being more active during the evening) have been associated with mood episodes, functional impairment, poor quality of life, and treatment resistance.7-10
Pathophysiology. Existing hypotheses for the biological mechanism underlying dysregulation of circadian rhythm in BD include changes in melatonin levels, expression of melatonin receptors in the CNS, and daily cortisol profiles.11 Genetic evidence also links circadian rhythm dysregulation with BD. Two polymorphisms on the circadian locomotor output cycles kaput (CLOCK) gene that control circadian rhythm—aryl hydrocarbon receptor nuclear translocator-like (ARNTL) and timeless circadian clock (TIMELESS)—have been linked to lithium responsiveness in BD.12 In addition, Per2, Cry1, and Rev-Erbα expression—all components of the circadian clock—have been found to increase individual susceptibility to the therapeutic effects of lithium in mice.13 In addition, circadian rhythm dysregulation is associated with metabolic problems encountered by patients with BD, including weight gain, diabetes mellitus, and cardiovascular disease.14
Rationale for use
Regulation of a patient’s circadian rhythm disruption is a potential treatment for BD. Hashimoto et al15 demonstrated that midday bright light exposure can phase advance and increase the amplitude of nocturnal melatonin production in healthy individuals. Morning light therapy has been shown to increase blood serotonin throughout the day in both healthy individuals and in patients with nonseasonal depression; the effect was apparent with light intensities as low as 50 lux.16 Lithium may exert its therapeutic effect through its influence on the retino-hypothalamic-pineal tract and thus its effect on melatonin secretion.17
BLT is a logical choice to treat the depression phase of BD when exposure to sunlight is not feasible due to geographical location, season, or other factor. For patients who live in areas that receive frequent sunshine, an outside stroll for half an hour will likely achieve similar benefit to BLT.
The precise mechanism of action of BLT for bipolar depression has not yet been determined. It may be attributed to a phase-resetting effect via melanopsin and the suprachiasmatic nucleus (Box18-24).
Box
Bright light therapy: How it works
The mechanism of action of bright light therapy is yet to be elucidated. The suprachiasmatic nucleus (SCN) in the hypothalamus is the center of circadian rhythm regulation and receives direct input from the retina through the retinohypothalamic tract.18 Melanopsin, a short-wavelength, light-sensitive G-protein–coupled receptor located in human retinal ganglion cells, is known to transduce short-wavelength light signals into neural signals.19 Since melanopsin is primarily responsible for resetting the timing of the SCN, suppressing pineal gland melatonin secretion and improving alertness and electroencephalogram-derived correlates of arousal,20 short-wavelength light with a low light intensity might be a better stimulator for melanopsin-containing retinal ganglion cells and the behaviors mediated via this photoreceptor system.21,22 Whether the antidepressant effect of light is also related to its alerting property is unclear.23 However, the acute alerting and performance-enhancing effects of light are increasingly taken into account for the design of indoor light standards in office environments.24 Response to light therapy is thus attributed to its phase-resetting effect.
Continue to: BLT for BD...
BLT for BD: What’s the evidence?
Several studies and case reports have evaluated the use of BLT for bipolar depression. The number of participants in these studies is small, and there is no uniformity of methodology or patient selection.
Dauphinais et al (2012)25 randomly assigned 44 patients with bipolar depression to BLT or a high-density or low-density negative ion generator for 8 weeks. They reported no difference in outcome between the various groups (50% vs 55.6%, remission and response rate). Only one patient in each group showed a switch to hypomania.
Carmadese et al (2015)26 reported an open-label study of adjunctive BLT in 31 difficult-to-treat patients with depression (16 unipolar and 15 bipolar). Significant improvement was noted within 3 weeks and was sustained in 1 patient with bipolar depression 5 weeks after cessation of BLT.
Papatheodorou and Kutcher (1995)27 treated 7 adolescents with bipolar depression with adjunctive BLT (10,000 lux twice per day). Three patients showed a marked response (>70% decrease from baseline Beck Depression Inventory and Symptom Check List scores). Two patients had a moderate response (40% to 47% decrease) and 2 patients obtained mild to no response. There were no reported adverse effects.
Benedetti et al (2014)28 studied 141 patients with treatment-resistant bipolar depression. Approximately one-quarter (23%) had a history of attempted suicide, and 83% had a history of drug resistance. The authors found a combination of total sleep deprivation, BLT, and lithium rapidly decreased suicidality and improved patients’ depressive symptoms.
Liebenluft et al (1995)29 administered 13 trials of BLT to 9 patients with rapid-cycling BD during a 3-month period. Five patients received the treatment in the morning, 5 around midday, and 3 in the evening. Patients who received BLT at midday had the best outcome, while 3 of the 5 patients who received morning BLT developed unstable mood. The authors recommended titrating the duration of light exposure so that patients could skip a treatment if their mood was trending toward hypomania.
Sit et al (2007)30 evaluated BLT in a case series of 9 women with bipolar I or II disorder in the depression phase. Patients were exposed to 50 lux of red light for 2 weeks, and then they received 7,000 lux BLT for 15, 30, and 45 minutes daily for 2 weeks (4 patients received morning light and 5 received midday light). Mood was assessed using the Structured Interview Guide for the Hamilton Depression Rating Scale with Atypical Depression Supplement and the Mania Rating Scale. Of the 4 patients receiving morning BLT, one patient had a full response and the other 3 developed hypomania. Of the 5 patients who received midday BLT, 2 achieved full response, 2 showed early improvement but required a dose increase, and one remained depressed but had a full response when she was switched to morning BLT.
Tseng et al (2016)31 reported a meta-analysis of BLT for bipolar depression that included a total of 567 patients from 11 studies. They reported significant improvement with BLT alone or in combination with antidepressants or total sleep deprivation. They also reported significant improvement with BLT in 130 patients who were not receiving other treatments. There was no difference in the frequency of mood shifts between patients on BLT alone or in combination with other modalities. The authors reported no mood shift in any of the patients receiving concurrent mood stabilizers. They also reported no difference with the color of light, gender, or duration of illness.
Yorguner et al (2017)32 conducted a 2-week randomized, single-blind study of BLT as an add-on treatment for 32 patients with bipolar depression. Patients were randomly assigned to BLT or dim light, which they were administered each morning for 30 mins for 2 weeks. Sixteen patients who received BLT showed a significantly greater reduction in Hamilton Depression Rating Scale scores (mean score of 24 at baseline down to 12) compared with 16 patients who received dim light (mean score of 24 at baseline down to 18). The authors also reported remission in 4 out of 4 patients who had seasonal depression, compared with 3 out of 12 who did not have seasonal depression (the other 9 showed response but not remission).
Zhou et al (2018)33 conducted a multi-center, randomized, single-blind clinical trial of 63 patients with bipolar depression. Thirty-three patients received morning BLT, and 30 received dim red light therapy (control group). The authors reported a significantly higher response rate in the BLT group (78%) compared with the control group (43%).
Sit et al (2018)34 conducted a 6-week randomized, double-blind, placebo-controlled trial of BLT vs dim red light in patients with bipolar I or II depression. Twenty-three patients were administered 7,000 lux bright white light, and 23 patients received 50 lux dim red light, at midday 5 days a week. The light dose was increased by 15 minutes every week up to 60 minutes by Week 4, unless the patient achieved remission. Patients were maintained on their usual medications, which included mood stabilizers and/or antidepressants. At Week 6, the group randomized to BLT had a significantly higher remission rate (68%) compared with patients who received dim red light (22%). Improvement was noted by Week 4. Patients receiving BLT also had significantly fewer depressive symptoms, and no mood polarity switch was noted.
Prescribing bright light therapy
Light box selection criteria. When selecting a light box or related BLT treatment apparatus, the Center for Environmental Therapeutics recommends consideration of the following factors35:
- clinical efficacy
- ocular and dermatologic safety
- visual comfort.
Selecting a dose. The dose received is determined by the intensity emitted from the light source, distance from the light box, and duration of exposure.36 Begin with midday light therapy between 12 noon and 2
Monitor for adverse effects. Generally, BLT is well tolerated.37 Adverse effects are rare; the most common ones include headache, eyestrain, nausea, and agitation.38 One study found no adverse ocular effects from light therapy after 5 years of treatment.39 Adverse effects tend to remit spontaneously or after dose reduction.35 Evening administration of BLT may increase the incidence of sleep disturbances.40 Like other biologic treatments for bipolar depression, BLT can precipitate manic/hypomanic and mixed states in susceptible patients, although the light dose can be titrated against emergent symptoms of hypomania.41
Bottom Line
Evidence suggests that bright light therapy is an effective, well tolerated, and affordable adjunct treatment for bipolar depression. Exposure to 5,000 to 7,000 lux around noon for 15 to 60 minutes will enhance the remission rate.
Related Resource
Mostert M, Dubovsky S. When bipolar treatment fails: what’s your next step? Current Psychiatry. 2008;7(1):39-46.
Drug Brand Name
Lithium • Eskalith, Lithobid
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.
1. Pjrek E, Winkler D, Stastny J, et al. Bright light therapy in seasonal affective disorder--does it suffice? Eur Neuropsychopharmacol. 2004.14(4):347-351.
2. Al-Karawi D, Jubair L. Bright light therapy for nonseasonal depression: meta-analysis of clinical trials. J Affect Disord. 2016;198:64-71.
3. Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer’s type dementia: a case series. Psychogeriatrics, 2017;17(5):275-281.
4. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice guideline for the treatment of patients with major depressive disorder, third edition. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf American Psychiatric Association. 2010. Accessed August, 10, 2017.
5. Kupka RW, Altshuler LL, Nolen WA, et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007;9(5):531-535.
6. Steinan MK, Krane-Gartiser K, Morken G, et al. Sleep problems in euthymic bipolar disorders: a review of clinical studies. Current Psychiatry Reviews. 2015;11:235-243.
7. Cudney LE, Frey BN, Streiner D, et al. Biological rhythms are independently associated with quality of life in bipolar disorder. Int J Bipolar Disord. 2016;4(1):9.
8. Duarte FA, Cardoso TA, Campos MT, et al. Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord, 2015;186:145-148.
9. Pinho M, Sehmbi M, Cudney LE, et al. The association between biological rhythms, depression, and functioning in bipolar disorder: a large multi-center study. Acta Psychiatr Scand. 2015:133(2);102-108.
10. Ng TH, Chung KF, Lee CT, et al. Eveningness and its associated impairments in remitted bipolar disorder. Behav Sleep Med. 2016:14(6):650-664.
11. Wu YH, Ursinus J, Zahn JN, et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J Affect Disord, 2013:148(2-3):357-367.
12. Rybakowski JK, Dmitrzak-Weglar M, Kliwicki S, et al. Polymorphism of circadian clock genes and prophylactic lithium response. Bipolar Disord. 2014;16(2):151-158.
13. Schnell A, Sandrelli F, Ranc V, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32(8):1075-1089.
14. Kim Y, Santos R, Gage FH, et al. Molecular mechanisms of bipolar disorder: progress made and future challenges. Front Cell Neurosci. 2017;11:30.
15. Hashimoto S, Kohsaka M, Nakamura K. Midday exposure to bright light changes the circadian organization of plasma melatonin rhythm in humans. Neurosci Lett. 1997;221(2-3):
89-92.
16. Rao ML, Müller-Oerlinghausen B, Mackert A, et al. The influence of phototherapy on serotonin and melatonin in non-seasonal depression. Pharmacopsychiatry.1990;23(3):155-158.
17. Moreira J, Geoffroy PA. Lithium and bipolar disorder: impacts from molecular to behavioural circadian rhythms. Chronobiol Int. 2016;33(4):351-373.
18. Oldham MA, Ciraulo DA. Bright light therapy for depression: a review of its effects on chronobiology and the autonomic nervous system. Chronobiol Int. 2014;31(3):305-319.
19. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070-1073.
20. Peirson S, Foster RG. Melanopsin: another way of signaling light. Neuron. 2006;49(3):331-339.
21. Anderson JL, Glod CA, Dai J, et al. Lux vs. wavelength in light treatment of seasonal affective disorder. Acta Psychiatr Scand. 2009;120(3):203-212.
22. Wirz-Justice A, Graw P, Kräuchi K, et al. Effect of light on unmasked circadian rhythms in winter depression. In: Wetterberg L, ed. Light and biological rhythms in man. Oxford, United Kingdom:Pergamon Press;1993:385-393.
23. Cajochen C. Alerting effects of light. Sleep Med Rev. 2007;11(6):453-464.
24. Aries MBC. Human lighting demands: healthy lighting in an office environment. Eindhoven, Eindhoven University Press. 2005;158. doi:10.6100/IR594257.
25. Dauphinais DR, Rosenthal JZ, Terman M, et al. Controlled trial of safety and efficacy of bright light therapy vs. negative air ions in patients with bipolar depression. Psychiatry Res. 2012;196(1):57-61.
26. Camardese G, Leone B, Serrani R, et al. Augmentation of light therapy in difficult-to-treat depressed patients: an open-label trial in both unipolar and bipolar patients. Neuropsychiatr Dis Treat. 2015;11:2331-2338.
27. Papatheodorou G, Kutcher S. The effect of adjunctive light therapy on ameliorating breakthrough depressive symptoms in adolescent-onset bipolar disorder.
J Psychiatry Neurosci. 1995;20(3):226-232.
28. Benedetti F, Riccaboni R, Locatelli C, et al. Rapid treatment response of suicidal symptoms to lithium, sleep deprivation, and light therapy (chronotherapeutics) in drug-resistant bipolar depression. J Clin Psychiatry. 2014;75(2):133-140.
29. Liebenluft E, Turner EH, Felman-Naim S, et al. Light therapy in patients with rapid cycling bipolar disorder: preliminary results. Psychopharmacol Bull. 1995;31(4):
705-710.
30. Sit DK, Wisner KL, Hanusa BH, et al. Light therapy for bipolar disorder: a case series in women. Bipolar Disord. 2007;9(8):918-927.
31. Tseng PT, Chen YW, Tu KY, et al. Light therapy in the treatment of patients with bipolar depression: a meta-analytic study. Eur Neuropsychopharmacol. 2016;26(6):
1037-1047.
32. Yorguner KN, Bulut NS, Carkaxhiu BG, et al. Efficacy of bright light therapy in bipolar depression. Psychiatry Res. 2017;260:432-438.
33. Zhou TH, Dang WM, Ma YT, et al. Clinical efficacy, onset time and safety of bright light therapy in acute bipolar depression as an adjunctive therapy: a randomized controlled trial. J Affect Disord. 2018;227:90-96.
34. Sit DK, McGowan J, Wiltrout C, et al. Adjunctive bright light therapy for bipolar depression: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2018;175(2):
131-139.
35. Center for Environmental Therapeutics. https://www.cet.org/. Center for Environmental Therapeutics. Accessed November 15, 2017.
36. Lam RW, Levitt AJ. Canadian consensus guidelines for the treatment of seasonal affective disorder. https://mdsc.ca/documents/Consumer%20and%20Family%20Support/CCG_on_Seasonal_Affective_Disorder.pdf. 1999. Accessed August 2, 2017.
37. Terman M, Terman JS. Bright light therapy: side effects and benefits across the symptom spectrum. J Clin Psychiatry. 1999; 60(11):799-808;quiz 809.
38. Labbate LA, et al. Side effects induced by bright light treatment for seasonal affective disorder. J Clin Psychiatry. 1994; 55(5):189-191.
39. Gallin PF, et al. Ophthalmologic examination of patients with seasonal affective disorder, before and after bright light therapy. Am J Ophthalmol. 1995;119(2):202-210.
40. Chan PK, Lam RW, Perry KF. Mania precipitated by light therapy for patients with SAD. J Clin Psychiatry. 1994;55(10):454.
41. Kripke DF. Timing of phototherapy and occurrence of mania. Biol Psychiatry. 1991; 29(11):1156-1157.

Antipsychotics for patients with dementia: The road less traveled
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
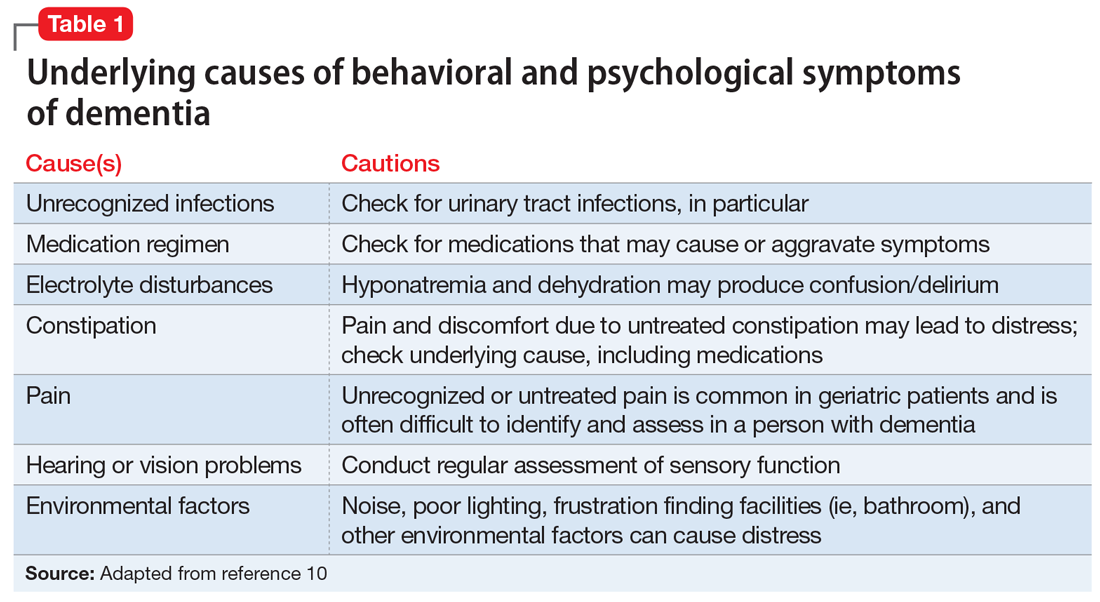
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.

Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
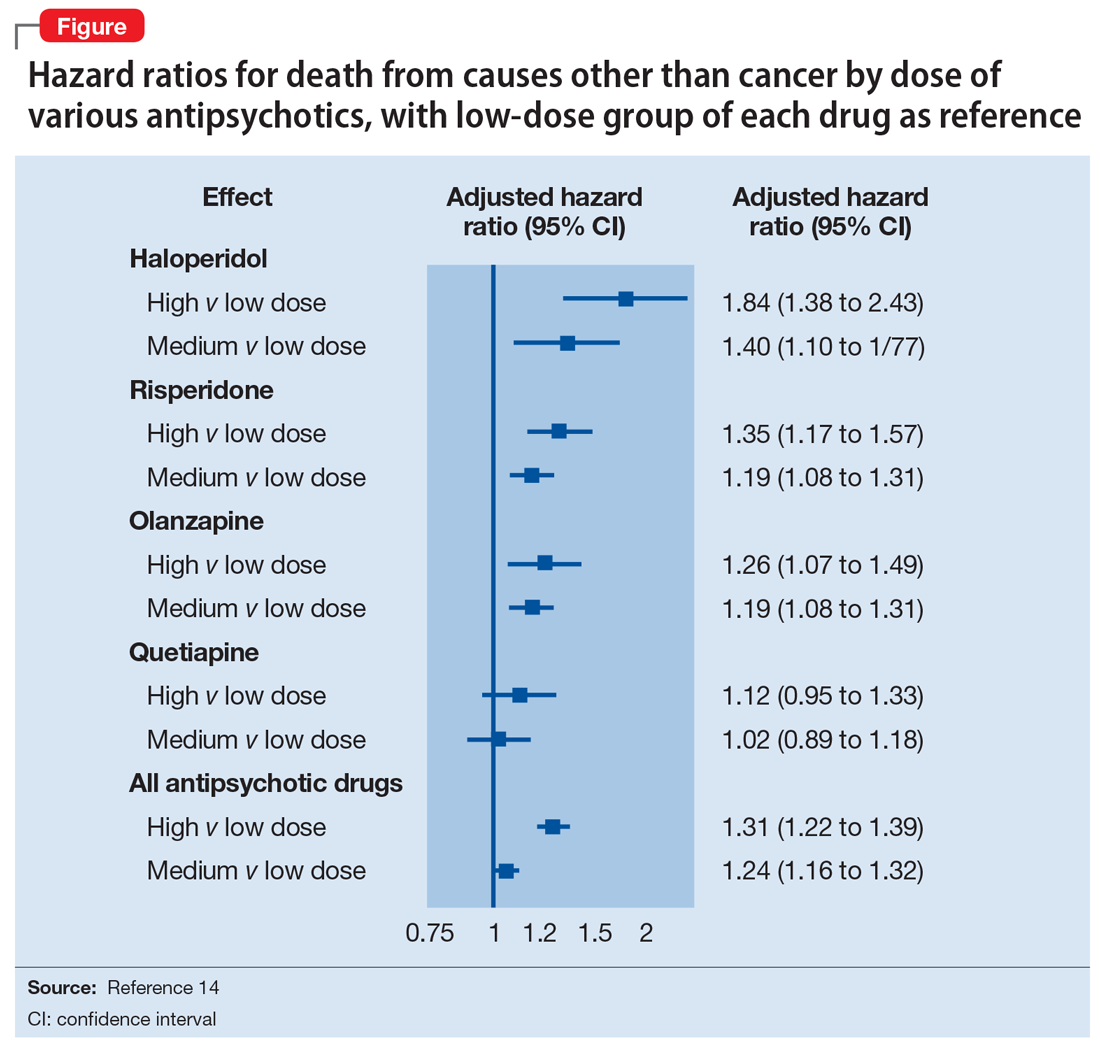
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10

Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing

When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.
Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10
Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing
When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
As psychiatrists treating an aging population, we frequently face the daunting challenges of managing medically complex and behaviorally unstable patients whose fragile condition tests the brightest among us. As our population enters late life, not only are physicians confronted with aging patients whose bodies have decreased renal and hepatic function, but we also face the challenges of the aging brain, severed neuronal networks, and neurotransmitter diminution. These physiological changes can alter treatment response, increase the frequency of adverse effects, and increase the likelihood of emergence of behavioral and psychological symptoms.
During the past decade, the number of people reaching age 65 has dramatically increased. As life expectancy improves, the “oldest old”—those age 85 and older—are the fastest-growing segment of the population. The prevalence of cognitive impairment, including mild cognitive impairment and dementia, in this cohort is >40%.1 Roughly 90% of patients with dementia will develop clinically significant behavioral problems at some point in the course of their illness.2
Behavioral and psychological symptoms of dementia (BPSD) have a tremendous impact on the quality of life for both patients and their caregivers. We are experts in understanding these behaviors and crafting nonpharmacologic treatment plans to manage them. Understanding the context in which behaviors emerge allows us to modify the environment, communication strategies, and other potential triggers, in turn reducing the need for pharmacologic intervention.
However, when nonpharmacologic interventions have been exhausted, what are the options? Antipsychotics have been one of the approaches used to address the challenges of behavioral disturbances and psychosis occurring in dementia. Unfortunately, there is conflicting evidence regarding the risks and benefits associated with the use of antipsychotics in this population. In this article, we provide a roadmap for the judicious use of antipsychotics for patients with dementia.
Weighing the risks and benefits of antipsychotics
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important but limited role in the treatment of behavioral disturbances in dementia. Although safety risks exist, they can be minimized through the careful selection of appropriate patients for treatment, close monitoring, and effective communication with patients and caregivers before and during treatment.
Several studies examining the efficacy of antipsychotics in the treatment of BPSD have demonstrated an increased risk of cerebrovascular events, including stroke and death due to any cause.3 This evidence prompted the FDA to issue a “black-box” warning in 2005 to highlight the increased risk of mortality for patients with dementia who are treated with SGAs.4 Both first-generation antipsychotics (FGAs) and SGAs have been associated with higher rates of mortality than most other psychotropic classes, except anticonvulsants. This increased mortality risk has been shown to persist for at least 6 to 12 months.5,6 FGAs appear to be associated with a greater mortality risk compared with SGAs. As a result, if antipsychotic treatment is necessary, the use of FGAs in this population is not recommended.
The potential mechanisms leading to stroke and death remain unclear. They could include orthostatic hypotension, anticholinergic adverse effects, QT prolongation, platelet aggregation effects, and venous thromboembolism. The presence of cardiovascular and vascular risk factors, electrolyte imbalances, cardiac arrhythmias, and concomitant use of medications that prolong the QTc interval may confer additional risks.
Continued to: Although the use of antipsychotics for patients with dementia...
Although the use of antipsychotics for patients with dementia may increase the risk of mortality, the absolute increased risk to a given individual, at least with short-term treatment, is likely small. The risk may also vary depending on the choice of SGA. Patients who were treated with quetiapine had a slightly lower risk of death than those who were treated risperidone.5 Death rates among patients prescribed aripiprazole, olanzapine, and ziprasidone were similar to the death rates of patients who were treated with risperidone. Compared with patients who were treated with risperidone, patients who were treated with the FGA haloperidol were twice as likely to die during a subsequent 6-month observation period. The largest number of deaths occurred during the first 40 days of treatment.5
While this increased risk of mortality is an important factor to discuss with patients and caregivers when deciding whether to initiate antipsychotic treatment, it is also important to put it into perspective. For example, the risk of suddenly dying from a stroke or heart attack for a person with dementia who is not taking an antipsychotic is approximately 2%. When an individual is started on one of these agents, that risk increases to approximately 4%. While the mortality risk is doubled, it remains relatively small.4 When faced with verbal or physical assaults, hostility, paranoid ideations, or other psychotic symptoms, many families feel that this relatively low risk does not outweigh the potential benefits of reducing caregiver and patient distress. If nonpharmacologic and/or other pharmacologic interventions have failed, the treatment has reached a point of no good alternatives and therapy should then focus on minimizing risk.
Informed consent is essential. A discussion of risks and benefits with the patient, family, or other decision-makers should focus on the risk of stroke, potential metabolic effects, and mortality, as well as potential worsening of cognitive decline associated with antipsychotic treatment. This should be weighed together with the evidence that suggests psychosis and agitation are associated with earlier nursing home admission and death.7,8 Families should be given ample time and opportunity to ask questions. Alternatives to immediate initiation of antipsychotics should be thoroughly reviewed.
Despite the above-noted risks, expert consensus suggests that the use of antipsychotics in the treatment of individuals with dementia can be appropriate, particularly in individuals with dangerous agitation or psychosis.9 These agents can minimize the risk of violence, reduce patient distress, improve the patient’s quality of life, and reduce caregiver burden. In clinical trials, the benefits of antipsychotics have been modest. Nevertheless, evidence has shown that these agents can reduce psychosis, agitation, aggression, hostility, and suspiciousness, which makes them a valid option when other interventions have proven insufficient.
Target specific symptoms
Despite this article’s focus on the appropriate use of antipsychotics for patients with BPSD, it is important to emphasize that the first-line approach to the management of BPSD in this population should always be a person-centered, psychosocial, multidisciplinary, nonpharmacologic approach that focuses on identifying triggers and treating potentially modifiable contributors to behavioral symptoms. Table 110 outlines common underlying causes of BPSD in dementia that should be assessed before prescribing an antipsychotic.
Continued to: Alternative psychopharmacologic treatments...
Alternative psychopharmacologic treatments based on a psychobehavioral metaphor should also be considered (Table 211). This approach matches the dominant target symptoms to the most relevant medication class.11 For example, in the case of a verbally and physically agitated patient who is also irritable, negative, socially withdrawn, and appears dysphoric, we might first undertake a trial of an antidepressant. Conversely, if the patient shows agitation in the context of increased motor activity, loud and rapid speech, and affective lability, we might consider the use of a mood stabilizer. Pharmacologic treatment should be aimed at the modification of clearly identified and documented target behaviors.
Indications to use antipsychotics for patients with dementia include:
- severe agitation and aggression associated with risk of harm
- delusions and hallucinations
- comorbid preexisting mental health conditions (eg, bipolar disorder, schizophrenia, treatment-resistant depression, etc.).
Symptoms that do not usually respond to an antipsychotic include wandering, social withdrawal, shouting, pacing, touching, cognitive defects, and incontinence.12 These symptoms may respond to interventions such as changes to the environment.
Continued to: Choosing an antipsychotic
Choosing an antipsychotic
Once you have identified that an antipsychotic is truly indicated, the choice of an agent will focus on patient-related factors. Considerations such as frailty, comorbid medical conditions including diabetes, history of falls, hepatic insufficiency, cardiac arrhythmias, and cerebrovascular risk factors, should all be analyzed prior to initiating an antipsychotic. The presence of these conditions will increase the likelihood that adverse effects may occur. It will also guide the dose trajectory and the target dose for discontinuation. Antipsychotics differ with respect to their efficacy and adverse effect profile. For practical purposes, adverse effects typically guide the selection of these agents when used for patients with dementia.
Continued to: Gradual structural changes occur...
Gradual structural changes occur in the dopaminergic system with age and increase the propensity for antipsychotic adverse effects. The number of dopaminergic neurons and D2 receptors decreases approximately 10% per decade. In order to avoid the development of adverse effects related to extrapyramidal symptoms, approximately 20% of receptors need to be free. FGAs tend to block approximately 90% of D2 receptors, whereas SGAs block less than 70% to 80% and dissociate more rapidly from D2 receptors.13 FGAs should therefore be avoided, as they have been associated with numerous adverse effects, including parkinsonism, tardive dyskinesia, akathisia, sedation, peripheral and central anticholinergic effects, postural hypotension, cardiac conduction defects, and falls. As noted above, they have been linked to a greater risk of mortality (Figure14 ).
When the decision to use an antipsychotic agent is made for a person with dementia, SGAs appear to be a better choice. There appear to be modest differences within the class of SGAs in terms of effectiveness, tolerability, and adverse effect profile. Although the association between the dose of an antipsychotic and the risk of mortality or stroke remains undefined, other common adverse effects, such as sedation, extrapyramidal symptoms, and risk of falls, can be reduced by starting at the lowest dose possible and titrating slowly.
Dosing considerations
Dose increments should be modest and, in a nonemergent setting, may be adjusted at weekly intervals depending on response. Prior to starting a treatment trial, it is advisable to estimate what will constitute a worthwhile clinical response, the duration of treatment, and the maximum dose. Avoid high doses or prolonged use of antipsychotics that have not significantly improved the target behavior.
When the decision to use a SGA is made, choosing the initial starting dose is challenging given that none of these medications has an indication for use in this population. We propose doses that have been used in completed randomized trials that reflect the best information available about the dose likely to maximize benefit and minimize risk. On the basis of those trials, reasonable starting doses would be15-22:
- quetiapine 25 to 50 mg/d
- risperidone 0.5 to 1 mg/d
- aripiprazole 2 to 10 mg/d
- olanzapine 2.5 to 5 mg/d
- ziprasidone 20 mg/d
Continued to: The highest doses tested...
The highest doses tested for each of these compounds in randomized clinical trials for this population were: risperidone 2 mg/d, olanzapine 10 mg/d, and aripiprazole 15 mg/d. A wide variety of maximum doses of quetiapine were studied in clinical trials, with a top dose of 200 mg being most common. It is worth noting that doses higher than these have been used for other indications.15-22
Quetiapine. One of the most commonly prescribed antipsychotics for the treatment of BPSD in individuals with memory disorders is quetiapine. The reasons for this preference include a low risk of extrapyramidal adverse effects, flexibility of dosing, ability to use lower dosages, and evidence of the lower risk of mortality when compared with other second-generation agents.5,15 If an antipsychotic is indicated, quetiapine should be considered as a first-line antipsychotic therapy. Quetiapine has well-established effects on mood, anxiety, and sleep, all of which can be disrupted in dementia and can act as drivers for agitation.5,15 Starting quetiapine may mitigate the need for separate agents to treat insomnia, loss of appetite, or anxiety, although it is not FDA-indicated for these comorbid conditions. Quetiapine is also less likely to exacerbate motor symptoms compared with other SGAs but has the potential to increase the risk of falls, and orthostasis, and carries a considerable anticholinergic burden.5,15
Risperidone has been shown to provide modest improvements in some people exhibiting symptoms of aggression, agitation, and psychosis.5,15 There is no evidence that risperidone is any more effective than other SGAs, but it has been tested on more geriatric patients than other SGAs. The fact that it is also available in an orally disintegrating tablet makes it a practical treatment in certain populations of patients, such as those who have difficulty swallowing. Risperidone carries the highest extrapyramidal symptom burden among the SGAs due to its potent D2 receptor binding. 5,15
Aripiprazole. There have been several studies of aripiprazole for the treatment of psychosis and agitation in Alzheimer’s dementia.15 This medication showed modest effect and was generally well tolerated. Aripiprazole appears to have less associated weight gain, which may be pertinent for some patients. It also appears to be less sedating than many of the other SGAs. However, some patients may experience activation or insomnia with this agent, particularly with doses <15 mg/d. This activating effect may be beneficial for treating comorbid depressive symptoms, although lower doses could theoretically worsen psychosis due to the activating effects.
Aripiprazole has also been studied in Parkinson’s disease. While some patients had favorable responses with improvement in psychosis and behavioral disturbances, this medication was also associated with worsening of motor symptoms. Certain individuals also experienced a worsening of their psychosis.23 For this reason, it is unlikely to be a useful agent for patients displaying evidence of parkinsonism, Parkinson’s dementia, or dementia with Lewy bodies.
Olanzapine. Several studies have shown that low-dose olanzapine has been modestly effective in decreasing agitation and aggression in patients suffering from Alzheimer’s and vascular dementias.24 The medication is also available in an orally disintegrating form, which may be beneficial when treating individuals whose swallowing abilities are compromised. Olanzapine also has been associated with significant weight gain and metabolic syndrome.24
Continued to: Ziprasidone
Ziprasidone. There are no specific studies of ziprasidone for geriatric patients and none for patients with dementia. However, case reports have suggested both oral and injectable forms of the medication may be well tolerated and have some benefit in treating agitation in this population.25 Based on evidence from younger populations, ziprasidone is less likely to be associated with weight gain or orthostatic hypotension. Medication has been associated with QTc prolongation and should be used with caution and monitored with an ECG.
The initial dosing and potential adverse effects of quetiapine, risperidone, aripiprazole, olanzapine, and ziprasidone are highlighted in Table 3.10
Other SGAs. Newer antipsychotics have recently become available and may serve as additional tools for managing BPSD in the future. Unfortunately, there are currently no available studies regarding their efficacy in the treatment of agitation and psychosis in dementia. One notable exception is pimavaserin, a serotonin 2A receptor inverse agonist. This medication has recently been FDA-approved for the treatment of Parkinson’s disease psychosis. The medication was extensively studied in older patients. It appeared to be effective in reducing delusions and hallucinations while not impairing motor function or causing sedation or hypotension.23 Additional studies are currently ongoing for the treatment of Alzheimer’s dementia psychosis.
Monitor treatment, consider discontinuation
American Psychiatric Association guidelines on the use of antipsychotics to treat agitation or psychosis in patients with dementia currently recommend that clinicians use a quantitative measure to track symptoms and response to treatment.26 These measures may be formal, such as an overall assessment of symptom severity on a Likert scale, or as simple as monitoring the changes in the frequency of periods of agitation.
After starting an antipsychotic, a follow-up appointment should typically take place within 1 month. If the patient is at high risk for developing adverse effects, or if the symptoms are severe, a follow-up appointment for monitoring the response to treatment and potential adverse effects should occur within 1 week. At a minimum, expert consensus suggests follow-up visits should occur every 3 months.
If there is no clinical response after 4 weeks of adequate dosing of an antipsychotic, the medication should be tapered and withdrawn. Switching to an alternative agent may be appropriate.
Many patients will have only partial remission of target symptoms. Therefore, increasing the dose or switching to an alternative agent may be necessary. Concurrent use of multiple antipsychotic agents should be avoided.
Continued to: Maintenance treatment may be appropriate
Maintenance treatment may be appropriate for patients who have demonstrated a clear benefit from antipsychotic treatment without undue adverse effects, and in whom a trial dose reduction has resulted in reappearance of the target symptoms. A formal monitoring plan to assess changes in response and the significance of adverse effects should be in place. Review the target behavior, changes in function, and significance of adverse effects at least every 3 months.
How to approach discontinuation
Behavioral and psychological symptoms of dementia are frequently temporary. If the patient has been stable, gradual dose reduction and eventual discontinuation of antipsychotics should be attempted every 3 months. Studies have reported that most patients who were taken off antipsychotics for treating BPSD showed no worsening of behavioral symptoms.27
Discontinuation of antipsychotics should be done gradually by reducing the dose by 50% every 2 weeks, and then stopping after 2 weeks on the minimum dose, with monitoring for recurrence of target symptoms or emergence of new ones. The longer a medication has been prescribed, the slower the withdrawal occurs. Thus, the possibility of emerging symptoms related to drug withdrawal will lessen.
A roadmap for judicious prescribing
When underlying treatable or reversible causes of BPSD in dementia have been ruled out or nonpharmacologic treatments have failed, a trial of an antipsychotic may be indicated. The choice of agent should focus on patient-related factors and on clearly identified target behaviors. Treatment should be started at a low dose and titrated cautiously to the lowest effective dose.
Behavioral and psychological symptoms of dementia are frequently temporary. Therefore, a gradual reduction and eventual withdrawal of antipsychotic medications should be attempted every 3 months. Studies indicate that most patients are able to tolerate elimination of antipsychotic medications with no worsening of behavioral symptoms.
Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Bottom Line
Until better treatment options become available, second-generation antipsychotics (SGAs) continue to have an important, albeit limited, role in the treatment of behavioral disturbances in dementia. Despite the limitations of treatment, SGAs remain a valid consideration when other interventions have proven insufficient. However, judicious use of these agents remains the cornerstone of therapy.
Related Resources
- Kales HC, Mulsant BH, Sajatovic MS. Prescribing antipsychotics in geriatric patients: Focus on dementia. Third of 3 parts. Current Psychiatry. 2017;16(12):24-30.
- Meeks TW, Jeste DV. Antipsychotics in dementia: Beyond ‘black-box’ warnings. Current Psychiatry. 2008;7(6):51-52, 55-58, 64-65.
Drug Brand Names
Aripiprazole • Abilify
Haloperidol • Haldol
Olanzapine • Zyprexa
Pimavanserin • Nuplazid
Risperidone • Risperdal
Quetiapine • Seroquel
Ziprasidone • Geodon
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.
1. Gardner RC, Valcour V, Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimers Res Ther. 2013;5(4):27.
2. Tariot PN, Blazina L. The psychopathology of dementia. In: Morris JC, ed. Handbook of dementing illnesses. New York, NY: Marcel Dekker Inc.; 1993:461-475.
3. Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934-1943.
4. Lenzer J. FDA warns about using antipsychotic drugs for dementia. BMJ. 2005;330(7497):922.
5. Kales HC, Valenstein M, Kim HM, et al. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am J Psychiatry. 2007;164(10):1568-1576; quiz 1623.
6. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
7. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study. J Am Geriatr Soc. 2011;59:473-481.
8. Banerjee S, Murray J, Foley B, et al. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry. 2003;74:1315-1316.
9. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series. Treatment of dementia and its behavioral disturbances. Introduction: methods, commentary, and summary. Postgrad Med. 2005;Spec No:6-22.
10. Burke AD, Hall G, Yaari R, et al. Pocket reference to Alzheimer’s disease management. Philadelphia, PA: Springer Healthcare Communications; 2015:39-46
11. Burke AD, Burke WJ, Tariot PN. Drug treatments for the behavioural and psychiatric symptoms of dementia. In: Ames D, O’Brien JT, Burns A, eds. Dementia, 5th ed. Boca Raton, FL: CRC Press; 2016:231-252.
12. Royal Australian and New Zealand College of Psychiatrists. Antipsychotics in dementia: best practice guide. https://bpac.org.nz/a4d/resources/docs/bpac_A4D_best_practice_guide.pdf. Accessed September 4, 2018.
13. Nyberg L, Backman L. Cognitive aging: a view from brain imaging. In: Dixon RA, Backman L, Nilsson LG, eds. New frontiers in cognitive aging. Oxford: Oxford Univ Press; 2004:135-60.
14. Huybrechts KF, Gerhard T, Crystal S, et al. Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: population based cohort study. BMJ. 2012;344:e977. doi: 10.1136/bmj.e977.
15. Burke AD, Tariot PN. Atypical antipsychotics in the elderly: a review of therapeutic trends and clinical outcomes. Expert Opin Pharmacother. 2009;10(15):2407-2414.
16. De Deyn PP, Rabheru K, Rasmussen A, et al. A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology.1999;53(5):946-955.
17. De Deyn PP, Jeste DV, Auby P, et al. Aripiprazole in dementia of the Alzheimer’s type. Poster presented at: 16th Annual Meeting of American Association for Geriatric Psychiatry; March 1-4, 2003; Honolulu, HI.
18. Lopez OL, Becker JT, Chang YF, et al. The long-term effects of conventional and atypical antipsychotics in patients with probable Alzheimer’s disease. Am J Psychiatry. 2013;170(9):1051-1058.
19. Mintzer J, Weiner M, Greenspan A, et al. Efficacy and safety of a flexible dose of risperidone versus placebo in the treatment of psychosis of Alzheimer’s disease. In: International College of Geriatric Psychopharmacology. Basel, Switzerland; 2004.
20. Mintzer JE, Tune LE, Breder CD, et al. Aripiprazole for the treatment of psychoses in institutionalized patients with Alzheimer dementia: a multicenter, randomized, double-blind, placebo-controlled assessment of three fixed doses. Am J Geriatr Psychiatry. 2007;15(11):918-931.
21. Sultzer DL, Davis SM, Tariot PN, et al; CATIE-AD Study Group. Clinical symptom responses to atypical antipsychotic medications in Alzheimer’s disease: phase 1 outcomes from the CATIE-AD effectiveness trial. Am J Psychiatry. 2008;165(7):844-854.
22. Zhong KX, Tariot PN, Mintzer J, et al. Quetiapine to treat agitation in dementia: a randomized, double-blind, placebo-controlled study. Curr Alzheimer Res. 2007;4(1):81-93.
23. Bozymski KM, Lowe DK, Pasternak KM, et al. Pimavanserin: a novel antipsychotic for Parkinson’s disease psychosis. Ann Pharmacother. 2017;51(6):479-487.
24. Moretti R, Torre R, Antonello T, et al. Olanzapine as a possible treatment of behavioral symptoms in vascular dementia: risks of cerebrovascular events. J Neurol. 2005;252:1186.
25. Cole SA, Saleem R, Shea WP, et al. Ziprasidone for agitation or psychosis in dementia: four cases. Int J Psychiatry Med. 2005;35(1):91-98.
26. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
27. Horwitz GJ, Tariot PN, Mead K, et al. Discontinuation of antipsychotics in nursing home patients with dementia. Am J Geriatr Psychiatry. 1995;3(4):290-299.

Psychiatric considerations in menopause
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan

Continued to: Why menopause?
Why menopause?

Perimenopausal mood disorders

However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.

Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan
Continued to: Why menopause?
Why menopause?
Perimenopausal mood disorders
However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.
Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
Mrs. J, age 49, presents to your psychiatric clinic. For the last few years, she has been experiencing night sweats and hot flashes, which she has attributed to being perimenopausal. Over the last year, she has noticed that her mood has declined; however, she has suffered several life events that she feels have contributed. Her mother was diagnosed with Alzheimer’s disease and had to move into a nursing home, which Mrs. J found very stressful. At the same time, her daughter left home for college, and her son is exploring his college options. Recently, Mrs. J has not been able to work due to her mood, and she is afraid she may lose her job as a consequence. She has struggled to talk to her husband about how she is feeling, and feels increasingly isolated. Over the last month, she has had increased problems sleeping and less energy; some days she struggles to get out of bed. She is finding it difficult to concentrate and is more forgetful. She has lost interest in her hobbies and is no longer meeting with her friends. She has no history of depression or anxiety, although she recalls feeling very low in mood for months after the birth of each of her children.
Are Mrs. J’s symptoms related to menopause or depression? What further investigations are necessary? Would you modify your treatment plan because of her menopausal status?
Women are at elevated risk of developing psychiatric symptoms and disorders throughout their reproductive lives, including during menopause. Menopause is a time of life transition, when women may experience multiple physical symptoms, including vasomotor symptoms (night sweats and hot flashes), sexual symptoms, and sleep difficulties. Depressive symptoms occur more frequently during menopause, and symptoms of schizophrenia may worsen.
Estrogen plays a role in mental illness throughout a woman’s life. In menopause, decreasing estrogen levels may correlate with increased mood symptoms, physical symptoms, and psychotic symptoms. As such, psychiatrists should consider whether collaboration regarding adjunctive hormone replacement therapy would be beneficial, and whether the benefits outweigh the potential risks. Otherwise, treatment of depression in menopause is similar to treatment outside of the menopausal transition, though serotonergic antidepressants may help target vasomotor symptoms while therapy may focus on role transition and loss. In this article, we review why women are at increased risk for mental illness during menopause, the role of estrogen, and treatment of mood and psychotic disorders during this phase of a woman’s life.
Increased vulnerability across the lifespan
Continued to: Why menopause?
Why menopause?
Perimenopausal mood disorders
However, one should keep in mind that new-onset mania in menopause is rare and should trigger a medical work-up and a dementia evaluation.13 Table 414 provides recommendations for evaluation of women undergoing menopause.
Menopause and serious mental illnesses
A study of 91 perimenopausal and postmenopausal women (age 45 to 55) who were diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or major depressive disorder (MDD) found that women with severe mental illness experienced significant vasomotor, physical, sexual, and psychosocial symptoms related to menopause.15 Furthermore, on 7 of 29 items on the Menopause Specific Quality of Life Scale, including hot flashes, women diagnosed with MDD reported problems significantly more often than women with other serious mental illnesses.15
Women with serious mental illness often have deficits in their knowledge about menopause.3 More than half of the 91 women in the study diagnosed with schizophrenia/schizoaffective disorder, bipolar disorder, or MDD felt more stressed related to menopause, and reported that menopause had a negative effect on their mental health.3 These women rated their top 5 symptoms potentially related to menopause as feeling depressed, anxious, or tired; lacking energy; and experiencing poor memory.3
Continued to: Role of estrogen on mood and psychosis
Role of estrogen on mood and psychosis
Women are at higher risk throughout their reproductive life than are men for MDD, anxiety disorders, and trauma-related disorders.12 Factors associated with depression during the menopause transition are reproductive hormonal changes (rise of follicle-stimulating hormone [FSH] and luteinizing hormone levels, and variability in estrogen [E2] and FSH levels); menopausal symptoms, particularly vasomotor symptoms; prior depression; psychosocial factors (adverse life events, financial strain, poor social supports); high body mass index, smoking, and poor physical health.6,7 Decreasing estrogen in the menopause transition may increase susceptibility to depression in some women.16 The Box17,18 provides more information on the relationship between estrogen and brain function.
Box
Estrogen and brain function
Numerous molecular and clinical studies have established the role of 17-beta estradiol in modulating brain functions via alterations in neurotransmission.17 Estrogen increases serotonin availability in the synapse by various pathways. It increases the rate of degradation of monoamine oxidase; monoamine oxidase enzymes are responsible for catabolizing serotonin, dopamine, and norepinephrine. Estrogen also increases tryptophan hydroxylase expression (rate-limiting enzyme in serotonin synthesis) and promotes intraneuronal serotonin transport in brain regions associated with affect regulation by increasing gene expression of the serotonin reuptake transporter. Studies have linked brain-derived neurotropic factor (BDNF) to increased serotonin turnover and proposed that estrogen may influence depression by increasing BDNF levels within the brain.18
Depressive disorders, including premenstrual dysphoric disorder, postpartum depression, and perimenopausal depression, have been linked to changes in hormonal status in women. Symptomatic menopause transition occurs in at least 20% of women, and a retrospective cohort study suggests that symptomatic menopause transition might increase the risk of new-onset depressive disorders, bipolar disorders, anxiety disorders, and sleep disorders.19 Symptomatic menopause transition also is a vulnerable time for relapse of MDD. Among women experiencing menopausal symptoms, including hot flashes, one-third also report depression—which correlates with a poorer quality of life, less work productivity, and greater use of health care services.9
Women who undergo surgical menopause are at greater risk for depression.8,10,11 This may be due to abrupt deprivation of estrogen—or related to a psychological reaction to the loss of fertility.
The observation that hormonal fluctuations related to women’s reproductive cycle have a significant impact on psychotic symptomatology has resulted in the “hypo-estrogenism hypothesis,” which proposes that gonadal dysfunction may increase vulnerability to schizophrenia, or that schizophrenia may lead to gonadal dysfunction.20 The “estrogen protection hypothesis” proposes that estrogen may protect women from schizophrenia, and may be a factor in the delayed onset of schizophrenia compared with men, less severe psychopathology, better outcomes, and premenstrual and postmenopausal deterioration in women. Many women of reproductive age with schizophrenia experience improvement in symptoms during the high estrogen phase of their menstrual cycle.
Pope et al21 have suggested that a hormone sensitivity syndrome may underlie why some women experience physical, psychological, and emotional symptoms at times of hormonal shifts such as menopause. This may represent a critical window of vulnerability, and also an opportunity to consider E2 as a therapeutic intervention.
Continued to: Treating mental illness in menopause
Treating mental illness in menopause
Changes to drug pharmacokinetics occur because some metabolising enzymes are estrogen-dependent and their levels decline after menopause, which leads to greater variability in drug response, particularly for oral medications. Other factors that can contribute to variability in medication response are polypharmacy, alcohol, illicit drugs, liver mass, smoking, caffeine, and nutritional intake.
While antidepressants are the first-line treatment for MDD and anxiety disorders, some patients remain unresponsive or inadequately responsive to currently available medications. In perimenopausal women with MDD, there may be an indication for adjunctive therapy with transdermal E2 in refractory cases; estrogen may augment the effects of selective serotonin reuptake inhibitor (SSRI) antidepressants as well as hasten the onset of antidepressant action.22 Estrogen also may be worth considering in women with mild depressive symptoms. For MDD, SSRIs plus estrogen may be more beneficial in improving mood than either agent alone. The effectiveness of E2 is less certain in postmenopausal depression.
Hormonal therapy for mental health disorders has equivocal evidence. The individual’s history and risk factors (eg, cardiovascular and osteoporosis risks) must be considered. A recent trial found that treatment with either venlafaxine or low-dose estrogen improved quality of life in menopausal women with vasomotor symptoms.23 Venlafaxine improved the psychosocial domain, while estrogen improved quality of life in other domains. Escitalopram, duloxetine, and citalopram have also been identified as having a possible positive impact on menopausal symptoms.22 SSRIs and serotonin-norepinephrine reuptake inhibitors may help reduce hot flashes and improve sleep.11
Regarding schizophrenia and estrogen, there may be improved symptoms during the high estrogen phase of the menstrual cycle, followed by a premenstrual aggravation of symptoms. Recall that women have a second peak of onset of schizophrenia after age 45, around the age of the onset of menopause.24 In a study of geropsychiatric hospital admissions, women were overrepresented among those with schizophrenia and schizoaffective disorder, compared with other psychiatric disorders.25 Postmenopausally, some women experience a decreased responsiveness to antipsychotics and worsening symptoms. In menopausal women with schizophrenia, check prolactin levels to help determine whether they are experiencing a natural menopause or medication-induced amenorrhea. Gender differences in pharmacotherapy responses and the decreasing response to antipsychotics in women older than age 50 have been observed26 and have led to exploration of the role of estrogen for treating schizophrenia in menopausal women. There have been contradictory results regarding use of estrogen as an adjunct to antipsychotics, with some reports finding this approach is effective and results in lower average doses of antipsychotics. Kulkarni et al27,28 have reported improvements in positive symptoms of treatment-resistant schizophrenia with transdermal use of E2, 200 mcg, as an adjunct to antipsychotics in women of childbearing age. However, they expressed caution regarding the health risks associated with prolonged use of E2. Long-term risks of high-dose estrogen therapies include thromboembolism, endometrial hyperplasia, and breast cancer, and individual factors should be considered before starting any form of hormone therapy. Selective estrogen receptor modulators (SERMs), such as raloxifene, which can cause activation of E2 receptors in a tissue-specific fashion and have less estrogen-related adverse effects, offer hope for future development in this field.27,28 While the use of adjunctive hormone therapy to manage psychotic symptoms in menopause is not routinely advised, the dosages of previously effective antipsychotics may need to be reviewed, or long-acting depot routes considered.29 Increased risk of prolonged QTc interval and tardive dyskinesia in geriatric women also should be considered in decisions regarding changes to antipsychotics or dosages.30
There are no guidelines regarding change in dosage of either individual antidepressants or antipsychotics in women at the time of menopause for managing pre-existing conditions. This may be due to the high variability in the effect of menopause on mental health and recognition that menopause is also a time for deterioration in physical health, as well as psychosocial changes for women, and thus other forms of intervention need to be considered.
Continued to: The biopsychosocial approach to treatment...
The biopsychosocial approach to treatment is particularly important in menopause.11 Common transitions in midlife include changes in relationships, employment, and financial status, and illness or death of family and friends.31 Therapy may focus on accepting a role transition and coping with loss of fertility. Cognitive-behavioral therapy may be helpful for menopausal symptoms, including hot flashes,4 as well as depressive symptoms.11
Although there are overlapping symptoms with both MDD and the perimenopause, these are typically restricted to impaired energy, sleep, and concentration, or changes in libido and weight.32 Therefore, it is vital to obtain a clear history and explore these symptoms in greater depth, as well as collect further information related to additional criteria such as appetite, agitation, feelings of worthlessness or guilt, and suicidal ideation.
Starting an antidepressant
On evaluation, Mrs. J discloses that she had experienced thoughts of wanting to end her life by overdose, although she had not acted on these thoughts. She appears subdued with poor eye contact, latency of response, and a slowed thought process. Mrs. J has blood tests to rule out thyroid abnormality or anemia. FSH and LH levels also are measured; these could provide a useful reference for later.
After a discussion with Mrs. J, she agrees to start an antidepressant. She also plans to speak to her gynecologist about the possibility of hormone replacement therapy. She is referred for psychotherapy to help support her with current life stressors. Mrs. J is started on escitalopram, 10 mg/d, and, after a month, she notices some improvement in her mood, psychomotor symptoms, sleep, and energy levels.
Bottom Line
Menopause is an important transition in our patients’ lives—both biologically and psychosocially. Women’s symptom patterns and medication needs may change during menopause.
Related Resource
- The North American Menopause Society. Depression & menopause. https://www.menopause.org/for-women/menopauseflashes/mental-health-at-menopause/depressionmenopause.
Drug Brand Names
Citalopram • Celexa
Duloxetine • Cymbalta
Escitalopram • Lexapro
Raloxifene • Evista
Venlafaxine • Effexor
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.
1. Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of women’s health across the nation (SWAN) over 10 years. Obstet Gynecol Clin North Am. 2011;38(3):609-625.
2. Almeida OP, Marsh K, Flicker L, et al. Depressive symptoms in midlife: the role of reproductive stage. Menopause. 2016;23(6):669-765.
3. Sajatovic M, Friedman SH, Schuermeyer IN, et al. Menopause knowledge and subjective experience among peri- and postmenopausal women with bipolar disorder, schizophrenia and major depression. J Nerv Ment Dis. 2006;194(3):173-178.
4. Ayers BN, Forshaw MJ, Hunter MS. The menopause. The Psychologist. 2011;24:348-353.
5. Bromberger JT, Kravitz HM, Chang YF, et al. Major depression during and after the menopausal transition: Study of Women’s Health Across the Nation (SWAN). Psychol Med. 2011;41(9):1879-1888.
6. Cohen LS, Soares CN, Vitonis AF, et al. Risk for new onset of depression during the menopausal transition: the Harvard study of moods and cycles. Arch Gen Psychiatry. 2006;63(4):385-390.
7. Freeman EW, Sammel MD, Lin H, et al. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatry. 2006;63(4):375-382.
8. Georgakis MK, Thomopoulos TP, Diamantaras AA, et al. Association of age at menopause and duration of reproductive period with depression after menopause: a systematic review and meta-analysis. JAMA Psychiatry 2016;73(2):139-149.
9. DiBonaventura MC, Wagner JS, Alvir J, et al. Depression, quality of life, work productivity, resource use, and costs among women experiencing menopause and hot flashes: a cross-sectional study [published online November 1, 2012]. Prim Care Companion CNS Disord. 2012;14(6): pii: PCC.12m01410. doi: 10.4088/PCC.12m01410.
10. Llaneza P, Garcia-Portilla MP, Llaneza-Suárez D, et al. Depressive disorders and the menopause transition. Maturitas. 2012;71(2):120-130.
11. Vivian-Taylor J, Hickey M. Menopause and depression: is there a link? Maturitas. 2014;79(2):142-146.
12. Kessler RC, McGonagle KA, Swartz M, et al. Sex and depression in the National Comorbidity Survey. 1: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29(2-3):85-96.
13. Friedman SH, Stankowski JE, Sajatovic M. Bipolar disorder in women. The Female Patient. 2007;32:15-24.
14. Soares C, Cohen L. The perimenopause, depressive disorders, and hormonal variability. Sao Paulo Med J. 2001;119(2):78-83.
15. Friedman SH, Sajatovic M, Schuermeyer IN, et al. Menopause-related quality of life in chronically mentally ill women. Int J Psychiatry Med. 2005;35(3):259-271.
16. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
17. Carretti N, Florio P, Bertolin A et al. Serum fluctuations of total and free tryptophan levels during the menstrual cycle are related to gonadotrophins and reflect brain serotonin utilization. Hum Reprod. 2005;20(6):1548-1553.
18. Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13-25.
19. Hu LY, Shen CC, Hung JH et al. Risk of psychiatric disorders following symptomatic menopausal transition: a nationwide population-based retrospective cohort study. Medicine (Baltimore). 2016;95(6):e2800. doi: 10.1097/MD.0000000000002800.
20. Riecher-Rossler AW. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rossler A, eds. Estrogen effects in psychiatric disorders. Wien, Austria: Springer-Verlag Wien; 2005:31-52.
21. Pope CJ, Oinonen K, Mazmanian D, et al. The hormonal sensitivity hypothesis: a review and new findings. Med Hypotheses. 2017;102:69-77.
22. Dennerstein L, Soares CN. The unique challenges of managing depression in mid-life women. World Psychiatry. 2008;7(3):137-142.
23. Caan B, LaCroix AZ, Joffe H, et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: a placebo-controlled randomized trial. Menopause. 2015;22(6):607-615.
24. Seeman MV. Psychosis in women: Consider midlife medical and psychological triggers. Current Psychiatry. 2010;9(2):64-68,75-76.
25. Sajatovic M, Friedman SH, Sabharwal J, et al. Clinical characteristics and length of hospital stay among older adults with bipolar disorder, schizophrenia or schizoaffective disorder, depression, and dementia. J Geriatr Psychiatry Neurol. 2004;17(1):3-8.
26. Grover S, Talwar P, Baghel R, et al. Genetic variability in estrogen disposition: potential clinical implications for neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(8):1391-1410.
27. Kulkarni J, Gavrilidis E, Wang W, et al. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol Psychiatry. 2015;20(6):695-702.
28. Kulkarni J, Gavrilidis E, Gwini SM, et al. Effect of adjunctive raloxifene therapy on severity of refractory schizophrenia in women: a randomized clinical trial. JAMA Psychiatry. 2016;73(9):947-954.
29. Brzezinski A, Brzezinski-Sinai NA, Seeman MV. Treating schizophrenia during menopause. Menopause. 2017;24(5):582-588.
30. Lange B, Mueller JK, Leweke FM, et al. How gender affects the pharmacotherapeutic approach to treating psychosis - a systematic review. Expert Opin Pharmacother. 2017;18(4):351-362.
31. Ballard KD, Kuh DJ, Wadsworth MEJ. The role of the menopause in women’s experiences of the ‘change of life.’ Sociology of Health & Illness. 2001;23(4):397-424.
32. Clayton AH, Ninan PT. Depression or menopause? Presentation and management of major depressive disorder in perimenopausal and postmenopausal women. Prim Care Companion J Clin Psychiatry. 2010;12(1):PCC.08r00747. doi: 10.4088/PCC.08r00747blu.

Caring for patients with autism spectrum disorder
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
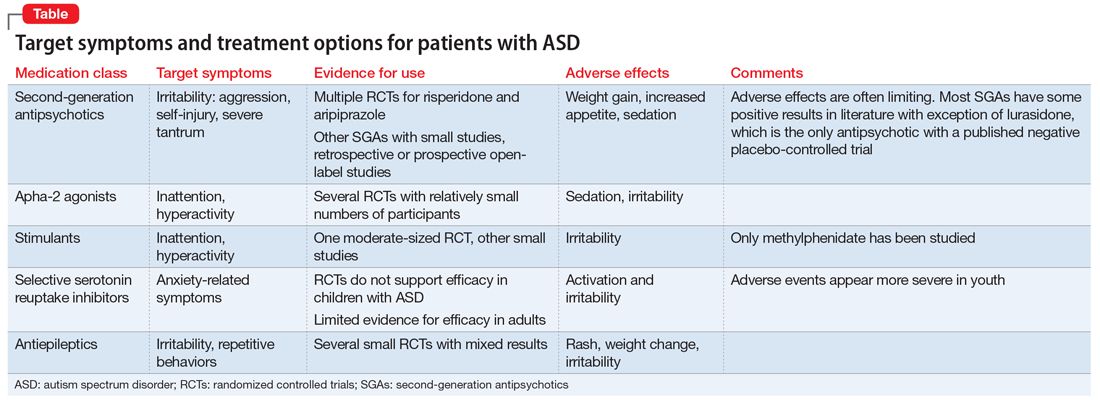
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
Autism spectrum disorder (ASD) is an umbrella term used to describe lifelong neurodevelopmental disorders characterized by impairment in social interactions and communication coupled with restricted, repetitive patterns of behaviors or interests that appear to share a common developmental course.1 In this article, we examine psychiatric care of patients with ASD and the most common symptom clusters treated with pharmacotherapy: irritability, anxiety, and hyperactivity/inattention.
First step: Keep the diagnosis in mind
Prior to 2013, ASD was comprised of 3 separate disorders distinguished by language delay and overall severity: autistic disorder, Asperger’s disorder, and pervasive developmental disorder, not otherwise specified.2 With the release of DSM-5 in 2013, these disorders were essentially collapsed into a single ASD.3 ASD prevalence is estimated to be 1 in 59 children,4 which represents a 20- to 30-fold increase since the 1960s.
In order to provide adequate psychiatric care for individuals with ASD, the first step is to remember the diagnosis; keep it in mind. This may be particularly important for clinicians who primarily care for adults, because such clinicians often receive limited training in disorders first manifesting in childhood and may not consider ASD in patients who have not been previously diagnosed. However, ASD diagnostic criteria have become broader, and public knowledge of the diagnosis has grown. DSM-5 acknowledges that although symptoms begin in early childhood, they may become more recognizable later in life with increasing social demand. The result is that many adults are likely undiagnosed. The estimated prevalence of ASD in adult psychiatric settings range from 1.5% to 4%.5-7 These patients have different treatment needs and unfortunately are often misdiagnosed with other psychiatric conditions.
A recent study in a state psychiatric facility found that 10% of patients in this setting met criteria for ASD.8 Almost all of those patients had been misdiagnosed with some form of schizophrenia, including one patient who had been previously diagnosed with autism by the father of autism himself, Leo Kanner, MD. Through the years, this patient’s autism diagnosis had fallen away, and at the time of the study, the patient carried a diagnosis of undifferentiated schizophrenia and was prescribed 8 psychotropic medications. The patient had repeatedly denied auditory or visual hallucinations; however, his stereotypies and odd behaviors were taken as evidence that he was responding to internal stimuli. This case highlights the importance of keeping the ASD diagnosis in mind when evaluating and treating patients.
Addressing 3 key symptom clusters
Even for patients with an established ASD diagnosis, comprehensive treatment is complex. It typically involves a multimodal approach that includes speech therapy, occupational therapy, applied behavioral analysis (ABA), and vocational training and support as well as management of associated medical conditions. Because medical comorbidities may play an important role in exacerbation of severe behaviors in ASD, often leading to acute behavioral regression and psychiatric admission, it is essential that they not be overlooked during evaluations.9,10
There are no effective pharmacologic treatments for the core social deficits seen in ASD. Novel pharmacotherapies to improve social impairment are in the early stages of research,11,12 but currently social impairment is best addressed through behavioral therapy and social skills training. Our role as psychiatrists is most often to treat co-occurring psychiatric symptoms so that individuals with ASD can fully participate in behavioral and school-based treatments that lead to improved social skills, activities of daily living, and quality of life. Three of the most common of these symptoms are irritability, anxiety, and hyperactivity/inattention.
Irritability
Irritability, marked by aggression, self-injury, and severe tantrums, causes serious distress for both patients and families, and this behavior cluster is the most frequently reported comorbid symptom in ASD.13-15 Nonpharmacologic treatment of irritability often involves ABA-based therapy and communication training.
Continued to: ABA includes an initial functional behavior assessment...
ABA includes an initial functional behavior assessment (FBA) of maladaptive behavior followed by the application of specific schedules of reinforcement for positive behavior. The FBA allows the therapist to determine what desirable consequences maintain a behavior. Without this knowledge, there is the risk of inadvertently rewarding a maladaptive behavior. For instance, if you are recommending a time-out for escape-motivated aggression, the result will likely be an increase rather than decrease in aggression.
Communication training teaches the patient to use communicative means to request a desired outcome to reduce inappropriate behaviors and improve independent functioning. Communication training can include speech therapy, teaching sign language, using picture exchange programs, or navigating communication devices. Consideration of nonpharmacologic management is vital in treatment planning. Continual inadvertent reward of behaviors will limit the effects of medications. Evidence suggests that pharmacotherapy is more effective when it occurs in the context of appropriate behavioral management techniques.16
Irritability has been the focus of significant pharmacotherapy research in ASD. Second-generation antipsychotics (SGAs) are first-line pharmacotherapy for severe irritability. Risperidone and aripiprazole are both FDA-approved for addressing irritability in youth with ASD. Their efficacy has been established in several large, placebo-controlled trials.17-23
Given issues with tolerability and cases refractory to the use of first-line agents,24 other SGAs are frequently used off-label for this indication with limited safety or efficacy data. Olanzapine demonstrated high response rates in early open-label studies,25,26 followed by efficacy over an 8-week double-blind placebo-controlled trial, although with significant weight gain.27 No other SGAs have been examined in double-blind placebo-controlled trials. Paliperidone demonstrated a particularly high response rate (84%) in a prospective open-label study of 25 adolescents and young adults with ASD.28 In a retrospective study of ziprasidone in 42 youth with ASD and irritability, we reported a response rate of 40%, which is lower than that seen for some other SGAs; however, ziprasidone can be an appealing option for patients for whom SGA-associated weight gain has been significant, because it is much more likely to be weight-neutral.29,30 Open-label studies with quetiapine in ASD have generally revealed only minimal efficacy for aggression,31,32 although sleep improvement may be more substantial.32 The safety and tolerability of lurasidone in treating irritability in youth with ASD has yet to be established.33 It is the only SGA with a published negative placebo-controlled trial in ASD.34 Use of SGAs may be limited by adverse effects, including weight gain, increased appetite, sedation, enuresis, and elevated prolactin. Monitoring of body mass index and metabolic profiles is indicated with all SGAs.
Haloperidol is the only first-generation antipsychotic with significant evidence (from multiple studies dating back to 1978) to support its use for ASD-associated irritability.35 However, due to the high incidence of dyskinesias and potential dystonias, use of haloperidol is reserved for severe treatment-refractory symptoms that have often not improved after multiple SGA trials.
Continued to: When severe self-inury and aggression fail to improve...
When severe self-injury and aggression fail to improve with multiple medication trials, the next steps include combination treatment with multiple antipsychotics,36 followed by clozapine, often as a last option.37 Research suggests that clozapine is effective and well-tolerated in ASD38-42; however, it has many potential severe adverse effects, including cardiomyopathy, lowered seizure threshold, severe constipation, weight gain, and agranulocytosis; due to risk of the latter, patients require regular blood draws for monitoring.
There is very little evidence to support the use of antiepileptic medications (AEDs) and mood stabilizers for irritability in ASD.43 Placebo-controlled trials have had mixed results. Some evidence suggests that AEDS may have more utility in individuals with ASD and abnormal EEGs without epilepsy44 or as an adjunct to SGA treatment.45 One study found that lithium may be beneficial for patients with ASD whose clinical presentation includes 2 or more mood symptoms.46
Anxiety
Anxiety is a significant issue for many individuals with ASD.47 Anxiety symptoms and disorders, including specific phobias, obsessive-compulsive disorder (OCD), social anxiety, and generalized anxiety disorder, are commonly seen in persons with ASD.48 Anxiety is often combined with restricted, repetitive behaviors (RBs) in ASD literature. Some evidence suggests that in individuals with ASD, sameness behaviors may limit sensory input and modulate anxiety.49 However, the core RBs symptom domain may not be related solely to anxiety, but rather represents deficits in executive processes that include cognitive flexibility and inhibitory control seen across multiple disorders with prominent RBs.50-54 Research indicates that anxiety is an independent and separable construct in ASD.55
Studies of treatments for both RBs and anxiety have focused primarily on selective serotonin reuptake inhibitors (SSRIs), hoping that the promising results for anxiety and OCD behaviors seen in neurotypical patients would translate to patients with ASD.56 Unfortunately, there is little evidence for effective pharmacologic management of ASD-associated anxiety.57 Large, randomized controlled trials (RCTs) are lacking. A Cochrane Database review of SSRIs for ASD58 examined 9 RCTs with a total of 320 patients. The authors concluded that there is no evidence to support the use of SSRIs for children with ASD, and limited evidence of utility in adults. Youth with ASD are particularly vulnerable to adverse effects from SSRIs, specifically impulsivity and agitation.57,59 However, SSRIs are among the most commonly prescribed medications for youth with ASD. Because there is limited evidence supporting SSRIs’ efficacy for this indication and issues with tolerability, there is significant concern for the overprescribing of SSRIs to patients with ASD. In comparison, there is some compelling evidence of efficacy for modified cognitive-behavioral therapy (CBT) for patients with high-functioning ASD. Seven RCTs have shown that CBT is superior to treatment as usual and waiting list control groups, with most effect sizes >0.8 and with no treatment-associated adverse effects.57
Risperidone has been shown to reduce RBs17,60 and anxiety17 in patients with ASD. In young children with co-occurring irritability, risperidone monotherapy is likely best to address both symptoms. When anxiety occurs in isolation and is severe, clinical experience suggests that SSRIs can be effective in a limited percentage of cases, though we recommend starting at low doses with frequent monitoring for activation and irritability. Treatment of anxiety is further complicated by the significant challenges presented by the diagnosis of true anxiety in the context of ASD.
Continued to: Hyperactivity and impulsivity
Hyperactivity and impulsivity
Hyperactivity and impulsivity are common among patients with ASD, with rates estimated from 41% to 78%.61 Hyperactivity and inattention are treated with a variety of medications. Research examining methylphenidate in ASD has demonstrated modest effects compared with placebo, though with frequent adverse effects, such as increased irritability and insomnia62,63 Other smaller studies have confirmed these results.64-66 One additional study found improvements not only in hyperactivity but also in joint attention and self-regulation of affective state following stimulant treatment.67 There is limited data on the efficacy and tolerability of amphetamine for treating hyperactivity and impulsivity in ASD. Stimulant medications often are avoided as the first-line treatment for hyperactivity because of concerns about increased irritability. Alpha-2 adrenergic receptor agonists often are used before stimulants because of their relatively benign adverse effect profile. Clonidine, guanfacine, and guanfacine ER all have demonstrated effectiveness in double-blind, placebo-controls trials in patients with ASD.68-70 In these trails, sedation was the most common adverse effect, although some studies have reported increased irritability with guanfacine.70,71
The Table provides a summary of the target symptoms and their treatment options for patients with ASD.
Improved diagnosis, but few evidence-based treatments
The rise in ASD cases observed over the past 20 years can be explained in part by a broader diagnostic algorithm and increased awareness. We are better at identifying ASD; however, there are still considerable gaps in identifying ASD in high-functioning patients and adults. One percent of the population has ASD,72,73 and this group is overrepresented in psychiatric clinic and hospital settings.74 Therefore, we must be aware of and understand the diagnosis.
Medication treatments are often less effective and less tolerable in patients with ASD than in patients without neurodevelopmental disability. There are differences in pharmacotherapy response and tolerability across development in ASD and limited evidence to guide prescribing in adults with ASD. SGAs appear to be effective across multiple symptom domains, but carry the risk of significant adverse effects. For anxiety and irritability, there is compelling evidence supporting the use of nonpharmacologic treatments.
Bottom Line
A subset of patients seen in psychiatry will have undiagnosed autism spectrum disorder (ASD). When evaluating worsening behaviors, first rule out organic causes. Second-generation antipsychotics have the most evidence for efficacy in ASD across multiple symptom domains. To sustain improvement in symptoms, it is vital to incorporate nonpharmacologic treatments.
Related Resources
- National Institute of Mental Health. Autism spectrum disorder. https://www.nimh.nih.gov/health/publications/autismspectrum-disorder/index.shtml.
- Centers for Disease Control and Prevention. Autism spectrum disorder (ASD). https://www.cdc.gov/ncbddd/ autism/index.html.
Drug Brand Names
Aripiprazole • Abilify
Clonidine • Catapres
Clozapine • Clozaril
Guanfacine • Tenex
Guanfacine Extended Release • Intuniv
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Methylphenidate • Ritalin
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Ziprasidone • Geodon
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.
1. Volkmar FR, Lord C, Bailey A, et al. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. 2004;45(1):135-170.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Baio J, Wiggins L, Christensen DL, et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill Summ 2018;67(6):1-23.
5. Scragg P, Shah A. Prevalence of Asperger’s syndrome in a secure hospital. Br J Psychiatry. 1994;165(5):679-682.
6. Hare DJ, Gould J, Mills R, et al. A preliminary study of individuals with autistic spectrum disorders in three special hospitals in England. London, UK: National Autistic Society; 1999.
7. Shah A, Holmes N, Wing L. Prevalence of autism and related conditions in adults in a mental handicap hospital. Appl Res Ment Retard. 1982;3(3):303-317.
8. Mandell DS, Lawer LJ, Branch K, et al. Prevalence and correlates of autism in a state psychiatric hospital. Autism. 2012;16(6):557-567.
9. Guinchat V, Cravero C, Diaz L, et al. Acute behavioral crises in psychiatric inpatients with autism spectrum disorder (ASD): recognition of concomitant medical or non-ASD psychiatric conditions predicts enhanced improvement. Res Devel Disabil. 2015;38:242-255.
10. Perisse D, Amiet C, Consoli A, et al. Risk factors of acute behavioral regression in psychiatrically hospitalized adolescents with autism. J Can Acad Child Adolesc Psychiatry. 2010;19(2):100-108.
11. Canitano R. New experimental treatments for core social domain in autism spectrum disorders. Front Pediatr. 2014;2:61.
12. Wink LK, Plawecki MH, Erickson CA, et al. Emerging drugs for the treatment of symptoms associated with autism spectrum disorders. Expert Opin Emerg Drugs. 2010;15(3):481-494.
13. Fitzpatrick SE, Srivorakiat L, Wink LK, et al. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525-1538.
14. Lecavalier L, Leone S, Wiltz J. The impact of behaviour problems on caregiver stress in young people with autism spectrum disorders. J Intellect Disabil Res. 2006;50(pt 3):172-183.
15. Mills R, Wing L. Researching interventions in ASD and priorities for research: surveying the membership of the NAS. London, UK: National Autistic Society; 2005.
16. Aman MG, McDougle CJ, Scahill L, et al. Medication and parent training in children with pervasive developmental disorders and serious behavior problems: results from a randomized clinical trial. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1143-1154.
17. McDougle CJ, Holmes JP, Carlson DC, et al. A double-blind, placebo-controlled study of risperidone in adults with autistic disorder and other pervasive developmental disorders. Arch Gen Psychiatry. 1998;55(7):633-641.
18. Research Units on Pediatric Psychopharmacology Autism Network. Risperidone treatment of autistic disorder: longer-term benefits and blinded discontinuation after 6 months. Am J Psychiatry. 2005;162(7):1361-1369.
19. Shea S, Turgay A, Carroll A, et al. Risperidone in the treatment of disruptive behavioral symptoms in children with autistic and other pervasive developmental disorders. Pediatrics. 2004;114(5):e634-e641.
20. Zuddas A, Zanni R, Usala T. Second generation antipsychotics (SGAs) for non-psychotic disorders in children and adolescents: a review of the randomized controlled studies. Eur Neuropsychopharmacol. 2011;21(8):600-620.
21. Benton TD. Aripiprazole to treat irritability associated with autism: a placebo-controlled, fixed-dose trial. Curr Psychiatry Rep. 2011;13(2):77-79.
22. Marcus RN, Owen R, Kamen L, et al. A placebo-controlled, fixed-dose study of aripiprazole in children and adolescents with irritability associated with autistic disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(11):1110-1119.
23. Owen R, Sikich L, Marcus RN, et al. Aripiprazole in the treatment of irritability in children and adolescents with autistic disorder. Pediatrics. 2009;124(6):1533-1540.
24. Adler BA, Wink LK, Early M, et al. Drug-refractory aggression, self-injurious behavior, and severe tantrums in autism spectrum disorders: a chart review study. Autism. 2015;19(1):102-106.
25. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry. 2001;40(8):887-894.
26. Potenza MN, Holmes JP, Kanes SJ, et al. Olanzapine treatment of children, adolescents, and adults with pervasive developmental disorders: an open-label pilot study. J Clin Psychopharmacol. 1999;19(1):37-44.
27. Hollander E, Wasserman S, Swanson EN, et al. A double-blind placebo-controlled pilot study of olanzapine in childhood/adolescent pervasive developmental disorder. J Child Adolesc Psychopharmacol. 2006;16(5):541-548.
28. Stigler KA, Erickson CA, Mullett JE, et al. Paliperidone for irritability in autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):75-78.
29. Dominick K, Wink LK, McDougle CJ, et al. A retrospective naturalistic study of ziprasidone for irritability in youth with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2015;25(5):397-401.
30. Malone RP, Delaney MA, Hyman SB, et al. Ziprasidone in adolescents with autism: an open-label pilot study. J Child Adolesc Psychopharmacol. 2007;17(6):779-790.
31. Findling RL, McNamara NK, Gracious BL, et al. Quetiapine in nine youths with autistic disorder. J Child Adolesc Psychopharmacol. 2004;14(2):287-294.
32. Golubchik P, Sever J, Weizman A. Low-dose quetiapine for adolescents with autistic spectrum disorder and aggressive behavior: open-label trial. Clin Neuropharmacol. 2011;34(6):216-219.
33. McClellan L, Dominick KC, Pedapati EV, et al. Lurasidone for the treatment of irritability and anger in autism spectrum disorders. Expert Opin Investig Drugs. 2017;26(8):985-989.
34. Loebel A, Brams M, Goldman RS, et al. Lurasidone for the treatment of irritability associated with autistic disorder. J Autism Dev Disord. 2016;46(4):1153-1163.
35. Campbell M, Anderson LT, Meier M, et al. A comparison of haloperidol and behavior therapy and their interaction in autistic children. J Am Acad Child Psychiatry. 1978;17(4):640-655.
36. Wink LK, Pedapati EV, Horn PS, et al. Multiple antipsychotic medication use in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2017;27(1):91-94.
37. Wink LK, Badran I, Pedapati EV, et al. Clozapine for drug-refractory irritability in individuals with developmental disability. J Child Adolesc Psychopharmacol. 2016;26(9):843-846.
38. Chen NC, Bedair HS, McKay B, et al. Clozapine in the treatment of aggression in an adolescent with autistic disorder. J Clin Psychiatry. 2001;62(6):479-480.
39. Gobbi G, Pulvirenti L. Long-term treatment with clozapine in an adult with autistic disorder accompanied by aggressive behaviour. J Psychiatry Neurosci. 2001;26(4):340-341.
40. Lambrey S, Falissard B, Martin-Barrero M, et al. Effectiveness of clozapine for the treatment of aggression in an adolescent with autistic disorder. J Child Adolesc Psychopharmacol. 2010;20(1):79-80.
41. Yalcin O, Kaymak G, Erdogan A, et al. a retrospective investigation of clozapine treatment in autistic and nonautistic children and adolescents in an inpatient clinic in Turkey. J Child Adolesc Psychopharmacol. 2016;26(9):815-821.
42. Beherec L, Lambrey S, Quilici G, et al. Retrospective review of clozapine in the treatment of patients with autism spectrum disorder and severe disruptive behaviors. J Clin Psychopharmacol. 2011;31(3):341-344.
43. Hirota T, Veenstra-Vanderweele J, Hollander E, et al, Antiepileptic medications in autism spectrum disorder: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):948-957.
44. Hollander E, Chaplin W, Soorya L, et al. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacology. 2010;35(4):990-998.
45. Rezaei V, Mohammadi MR, Ghanizadeh A, et al. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(7):1269-1272.
46. Siegel M, Beresford CA, Bunker M, et al. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2014;24(7):399-402.
47. Costello EJ, Egger HL, Angold A. The developmental epidemiology of anxiety disorders: phenomenology, prevalence, and comorbidity. Child Adolesc Psychiatr Clin N Am. 2005;14(4):631-648,vii.
48. van Steensel FJ, Deutschman AA, Bogels SM. Examining the Screen for Child Anxiety-Related Emotional Disorder-71 as an assessment tool for anxiety in children with high-functioning autism spectrum disorders. Autism. 2013;17(6):681-692.
49. Lidstone J, Uljarevic M, Sullivan J, et al. Relations among restricted and repetitive behaviors, anxiety and sensory features in children with autism spectrum disorder. Research in Autism Spectrum Disorders. 2014;8(2):82-92.
50. Turner M. Annotation: Repetitive behaviour in autism: a review of psychological research. J Child Psychol Psychiatry. 1999;40(6):839-849.
51. Kuelz AK, Hohagen F, Voderholzer U. Neuropsychological performance in obsessive-compulsive disorder: a critical review. Biol Psychol. 2004;65(3):185-236.
52. Olley A, Malhi G, Sachdev P. Memory and executive functioning in obsessive-compulsive disorder: a selective review. J Affect Disord. 2007;104(1-3):15-23.
53. Channon S, Gunning A, Frankl J, et al. Tourette’s syndrome (TS): cognitive performance in adults with uncomplicated TS. Neuropsychology. 2006;20(1):58-65.
54. Crawford S, Channon S, Robertson MM. Tourette’s syndrome: performance on tests of behavioural inhibition, working memory and gambling. J Child Psychol Psychiatry. 2005;46(12):1327-1336.
55. Renno P, Wood JJ. Discriminant and convergent validity of the anxiety construct in children with autism spectrum disorders. J Autism Dev Disord. 2013;43(9):2135-2146.
56. Wink LK, Erickson CA, Stigler KA, et al. Riluzole in autistic disorder. J Child Adolesc Psychopharmacol. 2011;21(4):375-379.
57. Vasa RA, Carroll LM, Nozzolillo AA, et al. A systematic review of treatments for anxiety in youth with autism spectrum disorders. J Autism Dev Disord. 2014;44(12):3215-3229.
58. Williams K, Brignell A, Randall M, et al. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;(8):CD004677.
59. Wink LK, Erickson CA, McDougle CJ. Pharmacologic treatment of behavioral symptoms associated with autism and other pervasive developmental disorders. Curr Treat Options Neurol. 2010;12(6):529-538.
60. McDougle CJ, Scahill L, Aman MG, et al. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry. 2005;162(6):1142-1148.
61. Murray MJ, Attention-deficit/hyperactivity disorder in the context of autism spectrum disorders. Curr Psychiatry Rep. 2010;12(5):382-388.
62. Research Units on Pediatric Psychopharmacology Autism Network. Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch Gen Psychiatry. 2005;62(11):1266-1274.
63. Posey DJ, Aman MG, McCracken JT, et al. Positive effects of methylphenidate on inattention and hyperactivity in pervasive developmental disorders: an analysis of secondary measures. Biol Psychiatry. 2007;61(4):538-544.
64. Aman MG, Langworthy KS. Pharmacotherapy for hyperactivity in children with autism and other pervasive developmental disorders. J Autism Dev Disord. 2000;30(5):451-459.
65. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord. 2000;30(3):245-255.
66. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord. 1995;25(3):283-294.
67. Jahromi LB, Kasari CL, McCracken JT, et al. Positive effects of methylphenidate on social communication and self-regulation in children with pervasive developmental disorders and hyperactivity. J Autism Dev Disord. 2009;39(3):395-404.
68. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry. 1992;53(3):77-82.
69. Scahill L, McCracken JT, King BH, et al. Extended-release guanfacine for hyperactivity in children with autism spectrum disorder. Am J Psychiatry. 2015;172(12):1197-1206.
70. Handen BL, Sahl R, Hardan AY. Guanfacine in children with autism and/or intellectual disabilities. J Dev Behav Pediatr. 2008;29(4):303-308.
71. Scahill L, Aman MG, McDougle CJ, et al. A prospective open trial of guanfacine in children with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16(5):589-598.
72. Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1-21.
73. Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68(5):459-465.
74. Mandell DS, Psychiatric hospitalization among children with autism spectrum disorders. J Autism Dev Disord. 2008;38(6):1059-1065.
