User login
Epileptic and depressed
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
Psychotic and in pain
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Depressed and delusional
Mrs. P, age 58, is a retired art teacher who presents for inpatient psychiatric admission after an 8-month depressive and psychotic illness. She reports profound feelings of worthlessness, anhedonia, psychomotor retardation, daily spontaneous crying spells, and worsening suicidal ideation. She is unkempt, disheveled, and makes limited eye contact. She is floridly psychotic, exhibits hebephrenia at times, and appears to be having conversations with people who are not there. Mrs. P reports derogatory intracranial auditory hallucinations of her brother’s and father’s voices. She also describes a complex delusional system relating to sexual trauma she experienced as a child perpetrated by her brother. Her family corroborates some details of the trauma; however, she says her father, neighbors, pastor, and outpatient psychiatrist are involved. Mrs. P believes these individuals are members of a cult, she has been the victim of a satanic sexual rite, and a television news personality knows about this conspiracy and has been attempting to contact her.
Mrs. P suffers from severe, debilitating chronic pain experienced as shock-like pain lasting for several minutes that starts in her throat and radiates to her left ear. Her pain began several years ago and prompted a neurologic workup, including MRI of the head and somatosensory evoked potentials of the glossopharyngeal nerve. She was diagnosed with “probable” glossopharyngeal neuralgia and failed multiple medication trials, including carbamazepine, phenytoin, gabapentin, and amitriptyline. She underwent microvascular decompression surgery 3 years ago. The operation, which has an 80% to 90% success rate for neuralgias,1,2 offered only brief symptomatic relief. She was maintained on immediate-release opiates until the pain became “unbearable” 8 months ago. This prompted a second neurologic workup, which was unremarkable. Mrs. P was diagnosed with pain disorder associated with psychological factors and a general medical condition.
Ten years ago she had 2 major depressive episodes with inpatient hospitalization and 2 suicide attempts within 1 year, but no history of psychosis before 8 months ago. Mrs. P’s husband says his wife has no history of manic or hypomanic episodes. Her medications are ziprasidone, 20 mg/d, thiothixene, 10 mg/d, benztropine, 3 mg/d, and escitalopram, 30 mg/d. She also receives oxycodone/acetaminophen, 5 mg/325 mg as needed for facial pain and headaches, and clonazepam, 1 mg as needed for panic attacks.
The authors’ observations
Psychosis can be a feature of any of the disorders listed in Table 13; however, several features of Mrs. P’s illness led us to diagnose MDD, recurrent, severe with psychotic features.4 Mrs. P and her husband described several discreet episodes of major debilitating depression without alternating periods of hypomanic or manic symptoms (Table 2).4 Comorbid depressive symptoms and a timeline indicating persistence of psychotic symptoms make a brief psychotic episode less likely. Although uncommon, patients can develop psychotic or mood disorders as a result of opiate abuse or dependence. However, Mrs. P was taking opiates as prescribed and not asking for early refills, which makes substance abuse an unlikely cause of her psychosis. In addition, because Mrs. P had 2 major depressive episodes in the absence of opiate use, a primary mood disorder seemed the more appropriate diagnosis. Schizophrenia is ruled out based on history. Although Mrs. P was suffering from complex delusional constructs, auditory hallucinations, and grossly disorganized behavior, these symptoms occurred only within the context of her depressive episode. New-onset delusional guilt relating to her childhood sexual trauma and hypochondriacal preoccupations within the context of pain complaints make psychotic depression more likely.5
Table 1
Psychiatric diseases in which patients may present with psychotic symptoms
| Bipolar depression |
| Borderline personality disorder |
| Brief psychotic disorder |
| Delirium |
| Delusional disorder |
| Dementia |
| Major depressive disorder |
| Psychotic disorder due to a general medical condition |
| Schizoaffective disorder |
| Schizophrenia |
| Shared psychotic disorder |
| Substance-induced psychosis |
| Source: Reference 3 |
Table 2
DSM-IV-TR criteria for major depressive episode
|
| Source: Reference 4 |
Depression, psychosis, and pain
From the beginning of Mrs. P’s treatment, we considered psychotic depression worsened—if not completely explained—her pain. Her somatic complaints appeared to be subtly woven into her delusional constructs. For instance, she complained that a device had been implanted in her head and she had the scar to prove it, pointing to the scar from her microvascular decompression surgery. Research indicates that depressive illness and chronic pain syndromes are highly comorbid and depressive illness can worsen pain syndromes.6,7 In addition, Mrs. P failed several medical and 1 surgical interventions for her pain condition that had high success rates. Her husband notes that when her outpatient psychiatrist started olanzapine 3 months ago for emerging psychotic symptoms, her pain complaints initially decreased with her psychotic symptoms, and she used less opiate medication during that time. Several months later Mrs. P’s pain complaints increased as her psychotic symptoms worsened. Second-generation antipsychotics have been evaluated as treatment for chronic pain syndromes, and may exert a primary analgesic effect.8,9 However, because of the correlation between her fluctuating psychotic symptoms and pain complaints, the more plausible explanation for olanzapine’s initial efficacy in treating Mrs. P’s pain is a secondary analgesic effect from decreased psychotic somatic preoccupation.
TREATMENT: ECT
Mrs. P is admitted to the inpatient psychiatric unit and placed on suicide precautions. Oxycodone/acetaminophen and clonazepam are tapered and limited to twice daily as needed. Escitalopram is tapered and discontinued. Thiothixene is tapered and replaced by olanzapine, 5 mg/d. Mrs. P receives 3 bifrontal, brief pulse-width ECT treatments. These result in marked improvement in her depressive and psychotic symptoms. In addition, her pain complaints become minimal. She becomes less preoccupied with her sexual trauma and grows to trust many staff members whom she previously believed were part of her traumatic childhood events. Mrs. P is no longer suicidal and asks to continue ECT treatments as an outpatient. She is discharged on olanzapine, 5 mg/d, trazodone, 100 mg/d for insomnia, benztropine, 2 mg/d, clonazepam 0.5 mg twice daily as needed for panic attacks, and oxycodone/acetaminophen, 5 mg/325 mg twice daily as needed for pain.
The authors’ observations
According to the Harvard South Shore Algorithm, treatment strategies for psychotic depression include antidepressant and antipsychotic combinations, lithium augmentation, clozapine, and ECT.10 Several factors made ECT the best option for Mrs. P. She had failed multiple treatment strategies and was suicidal. ECT is an effective treatment for MDD with psychotic features, single or recurrent episode.11 ECT can be used as a primary treatment before psychotropic medications or secondarily when there has been lack of clinical response to medications, intolerable side effects, deterioration in psychiatric condition, or suicidality.11,12 In addition, when treated with ECT, psychotic depression has a significantly higher remission rate than major depression without psychosis.12 Delusional guilt, psychomotor retardation, hypochondriacal preoccupations, loss of insight, paranoia, and obsessive-compulsive symptoms predict a favorable response.12 ECT also has demonstrated efficacy for treating pain secondary to psychotic depression or melancholic depression.13 In addition, ECT has been shown to have analgesic properties beyond treating underlying depression.14 Our primary focus was not to treat Mrs. P’s pain syndrome with ECT; however, in treating her psychotic depression we had hoped that her pain tolerance would improve and she would rely less on opiates.
OUTCOME: Pain relief
As an outpatient, Mrs. P receives 11 bifrontal ECT treatments in her initial series, followed by 7 bifrontal maintenance treatments. Her speech is more spontaneous, her grooming and hygiene improve, and she exhibits a brighter and more reactive affect. Suicidal ideation has resolved. Pain improves from a “10 out of 10” to a “2 out of 10.” Mrs. P consistently requires less oxycodone/acetaminophen. She relates better to her family and begins exploring new hobbies such as pottery. In addition to monthly maintenance bifrontal ECT treatments, she is stable on citalopram, 60 mg/d, and trazodone, 50 mg/d as needed for insomnia.
The authors’ observations
The relationship between depressive illness and chronic pain is complex. Treating a primary depressive illness can lead to improved functional outcomes and decreased disability from chronic pain complaints.15 Patients with comorbid chronic pain and depressive illness are more likely to suffer from unremitting pain despite compliance with evidence-based treatment strategies.16 Mrs. P had 2 co-occurring disorders: psychotic depression and chronic pain disorder secondary to glossopharyngeal neuralgia. Our opinion is that Mrs. P’s psychotic depression worsened her experience of pain.
Treatment strategies that address both depressive symptoms and chronic pain are ideal.17 These treatment modalities include psychotherapeutic techniques such as cognitive-behavioral therapy, medications, and somatic treatments such as ECT.18 In Mrs. P’s case, ECT was an effective treatment that caused remission of psychotic depressive symptoms, which lead to improved pain control and restored social and occupational functioning.
Related Resources
- Schreiber S, Shmueli D, Grunhaus L, et al. The influence of electroconvulsive therapy on pain threshold and pain tolerance in major depression patients before, during and after treatment. Eur J Pain. 2003;7(5):419-424.
- Suzuki K, Ebina Y, Shindo T, et al. Repeated electroconvulsive therapy courses improved chronic regional pain with depression caused by failed back syndrome. Med Sci Monit. 2009;15(4):CS77-CS79.
- Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52(5):1577-1584.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Gabapentin • Neurontin
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Oxycodone/ acetaminophen • Vicodin
- Phenytoin • Dilantin
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosures
Dr. Kugler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Magid receives NARSAD grant support.
1. Kawashima M, Matsushima T, Inoue T, et al. Microvascular decompression for glossopharyngeal neuralgia through the transcondylar fossa (supracondylar transjugular tubercle) approach. Neurosurgery. 2010;66(6 suppl operative):275-280.
2. Ferroli P, Fioravanti A, Schiariti M, et al. Microvascular decompression for glossopharyngeal neuralgia: a long-term retrospective review of the Milan-Bologna experience in 31 consecutive cases. Acta Neurochir (Wien). 2009;151(10):1245-1250.
3. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York NY: Cambridge University Press; 2008.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev.Washington DC: American Psychiatric Association; 2000.
5. Rothschild AJ. Diagnosis and assessment. In: Rothschild AJ. Clinical manual for diagnosis and treatment of psychotic depression. Arlington VA: American Psychiatric Publishing Inc.; 2009:57-71.
6. Tunks ER, Crook J, Weir R. Epidemiology of chronic pain with psychological comorbidity: prevalence risk, course and prognosis. Can J Psychiatry. 2008;53(4):235-242.
7. Hooten MW, Shi Y, Gazelka HM, et al. The effects of depression and smoking on pain severity and opioid use in patients with chronic pain. Pain. 2011;152(1):223-229.
8. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
9. Seidel S, Aigner M, Ossege M, et al. Antipsychotics for acute and chronic pain in adults. J Pain Symptom Manage. 2010;39(4):768-778.
10. Hamoda HM, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on psychotic depression. Harv Rev Psychiatry. 2008;16(4):235-247.
11. American Psychiatric Association. Committee on Electroconvulsive Therapy, Weiner RD, eds. The practice of electroconvulsive therapy: recommendations for treatment, training and privileging. 2nd ed. Washington DC: American Psychiatric Association; 2001.
12. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus nonpsychotic depressed patients: a report from CORE. J ECT. 2001;17(4):244-253.
13. Rasmussen KG, Rummans TA. Electroconvulsive therapy in the management of chronic pain. Curr Pain Headache Rep. 2002;6(1):17-22.
14. Wasan AD, Artin K, Clark MR. A case-matching study of the analgesic properties of electroconvulsive therapy. Pain Med. 2004;5(1):50-58.
15. Teh FC, Zaslavsky AM, Reynolds CF, 3rd, et al. Effect of depression treatment on chronic pain outcomes. Psychosom Med. 2010;72(1):61-67.
16. Sertel Berk HO. The biopsychosocial factors that serve as predictors of the outcome of surgical modalities for chronic pain. Agri. 2010;22(3):93-97.
17. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity. Arch Intern Med. 2003;163(20):2433-2445.
18. Veehof MM, Oskam MJ, Schreurs KM, et al. Acceptance-based interventions for the treatment of chronic pain: a systematic review and meta-analysis. Pain. 2011;152(3):533-542.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Depressed and delusional
Mrs. P, age 58, is a retired art teacher who presents for inpatient psychiatric admission after an 8-month depressive and psychotic illness. She reports profound feelings of worthlessness, anhedonia, psychomotor retardation, daily spontaneous crying spells, and worsening suicidal ideation. She is unkempt, disheveled, and makes limited eye contact. She is floridly psychotic, exhibits hebephrenia at times, and appears to be having conversations with people who are not there. Mrs. P reports derogatory intracranial auditory hallucinations of her brother’s and father’s voices. She also describes a complex delusional system relating to sexual trauma she experienced as a child perpetrated by her brother. Her family corroborates some details of the trauma; however, she says her father, neighbors, pastor, and outpatient psychiatrist are involved. Mrs. P believes these individuals are members of a cult, she has been the victim of a satanic sexual rite, and a television news personality knows about this conspiracy and has been attempting to contact her.
Mrs. P suffers from severe, debilitating chronic pain experienced as shock-like pain lasting for several minutes that starts in her throat and radiates to her left ear. Her pain began several years ago and prompted a neurologic workup, including MRI of the head and somatosensory evoked potentials of the glossopharyngeal nerve. She was diagnosed with “probable” glossopharyngeal neuralgia and failed multiple medication trials, including carbamazepine, phenytoin, gabapentin, and amitriptyline. She underwent microvascular decompression surgery 3 years ago. The operation, which has an 80% to 90% success rate for neuralgias,1,2 offered only brief symptomatic relief. She was maintained on immediate-release opiates until the pain became “unbearable” 8 months ago. This prompted a second neurologic workup, which was unremarkable. Mrs. P was diagnosed with pain disorder associated with psychological factors and a general medical condition.
Ten years ago she had 2 major depressive episodes with inpatient hospitalization and 2 suicide attempts within 1 year, but no history of psychosis before 8 months ago. Mrs. P’s husband says his wife has no history of manic or hypomanic episodes. Her medications are ziprasidone, 20 mg/d, thiothixene, 10 mg/d, benztropine, 3 mg/d, and escitalopram, 30 mg/d. She also receives oxycodone/acetaminophen, 5 mg/325 mg as needed for facial pain and headaches, and clonazepam, 1 mg as needed for panic attacks.
The authors’ observations
Psychosis can be a feature of any of the disorders listed in Table 13; however, several features of Mrs. P’s illness led us to diagnose MDD, recurrent, severe with psychotic features.4 Mrs. P and her husband described several discreet episodes of major debilitating depression without alternating periods of hypomanic or manic symptoms (Table 2).4 Comorbid depressive symptoms and a timeline indicating persistence of psychotic symptoms make a brief psychotic episode less likely. Although uncommon, patients can develop psychotic or mood disorders as a result of opiate abuse or dependence. However, Mrs. P was taking opiates as prescribed and not asking for early refills, which makes substance abuse an unlikely cause of her psychosis. In addition, because Mrs. P had 2 major depressive episodes in the absence of opiate use, a primary mood disorder seemed the more appropriate diagnosis. Schizophrenia is ruled out based on history. Although Mrs. P was suffering from complex delusional constructs, auditory hallucinations, and grossly disorganized behavior, these symptoms occurred only within the context of her depressive episode. New-onset delusional guilt relating to her childhood sexual trauma and hypochondriacal preoccupations within the context of pain complaints make psychotic depression more likely.5
Table 1
Psychiatric diseases in which patients may present with psychotic symptoms
| Bipolar depression |
| Borderline personality disorder |
| Brief psychotic disorder |
| Delirium |
| Delusional disorder |
| Dementia |
| Major depressive disorder |
| Psychotic disorder due to a general medical condition |
| Schizoaffective disorder |
| Schizophrenia |
| Shared psychotic disorder |
| Substance-induced psychosis |
| Source: Reference 3 |
Table 2
DSM-IV-TR criteria for major depressive episode
|
| Source: Reference 4 |
Depression, psychosis, and pain
From the beginning of Mrs. P’s treatment, we considered psychotic depression worsened—if not completely explained—her pain. Her somatic complaints appeared to be subtly woven into her delusional constructs. For instance, she complained that a device had been implanted in her head and she had the scar to prove it, pointing to the scar from her microvascular decompression surgery. Research indicates that depressive illness and chronic pain syndromes are highly comorbid and depressive illness can worsen pain syndromes.6,7 In addition, Mrs. P failed several medical and 1 surgical interventions for her pain condition that had high success rates. Her husband notes that when her outpatient psychiatrist started olanzapine 3 months ago for emerging psychotic symptoms, her pain complaints initially decreased with her psychotic symptoms, and she used less opiate medication during that time. Several months later Mrs. P’s pain complaints increased as her psychotic symptoms worsened. Second-generation antipsychotics have been evaluated as treatment for chronic pain syndromes, and may exert a primary analgesic effect.8,9 However, because of the correlation between her fluctuating psychotic symptoms and pain complaints, the more plausible explanation for olanzapine’s initial efficacy in treating Mrs. P’s pain is a secondary analgesic effect from decreased psychotic somatic preoccupation.
TREATMENT: ECT
Mrs. P is admitted to the inpatient psychiatric unit and placed on suicide precautions. Oxycodone/acetaminophen and clonazepam are tapered and limited to twice daily as needed. Escitalopram is tapered and discontinued. Thiothixene is tapered and replaced by olanzapine, 5 mg/d. Mrs. P receives 3 bifrontal, brief pulse-width ECT treatments. These result in marked improvement in her depressive and psychotic symptoms. In addition, her pain complaints become minimal. She becomes less preoccupied with her sexual trauma and grows to trust many staff members whom she previously believed were part of her traumatic childhood events. Mrs. P is no longer suicidal and asks to continue ECT treatments as an outpatient. She is discharged on olanzapine, 5 mg/d, trazodone, 100 mg/d for insomnia, benztropine, 2 mg/d, clonazepam 0.5 mg twice daily as needed for panic attacks, and oxycodone/acetaminophen, 5 mg/325 mg twice daily as needed for pain.
The authors’ observations
According to the Harvard South Shore Algorithm, treatment strategies for psychotic depression include antidepressant and antipsychotic combinations, lithium augmentation, clozapine, and ECT.10 Several factors made ECT the best option for Mrs. P. She had failed multiple treatment strategies and was suicidal. ECT is an effective treatment for MDD with psychotic features, single or recurrent episode.11 ECT can be used as a primary treatment before psychotropic medications or secondarily when there has been lack of clinical response to medications, intolerable side effects, deterioration in psychiatric condition, or suicidality.11,12 In addition, when treated with ECT, psychotic depression has a significantly higher remission rate than major depression without psychosis.12 Delusional guilt, psychomotor retardation, hypochondriacal preoccupations, loss of insight, paranoia, and obsessive-compulsive symptoms predict a favorable response.12 ECT also has demonstrated efficacy for treating pain secondary to psychotic depression or melancholic depression.13 In addition, ECT has been shown to have analgesic properties beyond treating underlying depression.14 Our primary focus was not to treat Mrs. P’s pain syndrome with ECT; however, in treating her psychotic depression we had hoped that her pain tolerance would improve and she would rely less on opiates.
OUTCOME: Pain relief
As an outpatient, Mrs. P receives 11 bifrontal ECT treatments in her initial series, followed by 7 bifrontal maintenance treatments. Her speech is more spontaneous, her grooming and hygiene improve, and she exhibits a brighter and more reactive affect. Suicidal ideation has resolved. Pain improves from a “10 out of 10” to a “2 out of 10.” Mrs. P consistently requires less oxycodone/acetaminophen. She relates better to her family and begins exploring new hobbies such as pottery. In addition to monthly maintenance bifrontal ECT treatments, she is stable on citalopram, 60 mg/d, and trazodone, 50 mg/d as needed for insomnia.
The authors’ observations
The relationship between depressive illness and chronic pain is complex. Treating a primary depressive illness can lead to improved functional outcomes and decreased disability from chronic pain complaints.15 Patients with comorbid chronic pain and depressive illness are more likely to suffer from unremitting pain despite compliance with evidence-based treatment strategies.16 Mrs. P had 2 co-occurring disorders: psychotic depression and chronic pain disorder secondary to glossopharyngeal neuralgia. Our opinion is that Mrs. P’s psychotic depression worsened her experience of pain.
Treatment strategies that address both depressive symptoms and chronic pain are ideal.17 These treatment modalities include psychotherapeutic techniques such as cognitive-behavioral therapy, medications, and somatic treatments such as ECT.18 In Mrs. P’s case, ECT was an effective treatment that caused remission of psychotic depressive symptoms, which lead to improved pain control and restored social and occupational functioning.
Related Resources
- Schreiber S, Shmueli D, Grunhaus L, et al. The influence of electroconvulsive therapy on pain threshold and pain tolerance in major depression patients before, during and after treatment. Eur J Pain. 2003;7(5):419-424.
- Suzuki K, Ebina Y, Shindo T, et al. Repeated electroconvulsive therapy courses improved chronic regional pain with depression caused by failed back syndrome. Med Sci Monit. 2009;15(4):CS77-CS79.
- Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52(5):1577-1584.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Gabapentin • Neurontin
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Oxycodone/ acetaminophen • Vicodin
- Phenytoin • Dilantin
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosures
Dr. Kugler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Magid receives NARSAD grant support.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Depressed and delusional
Mrs. P, age 58, is a retired art teacher who presents for inpatient psychiatric admission after an 8-month depressive and psychotic illness. She reports profound feelings of worthlessness, anhedonia, psychomotor retardation, daily spontaneous crying spells, and worsening suicidal ideation. She is unkempt, disheveled, and makes limited eye contact. She is floridly psychotic, exhibits hebephrenia at times, and appears to be having conversations with people who are not there. Mrs. P reports derogatory intracranial auditory hallucinations of her brother’s and father’s voices. She also describes a complex delusional system relating to sexual trauma she experienced as a child perpetrated by her brother. Her family corroborates some details of the trauma; however, she says her father, neighbors, pastor, and outpatient psychiatrist are involved. Mrs. P believes these individuals are members of a cult, she has been the victim of a satanic sexual rite, and a television news personality knows about this conspiracy and has been attempting to contact her.
Mrs. P suffers from severe, debilitating chronic pain experienced as shock-like pain lasting for several minutes that starts in her throat and radiates to her left ear. Her pain began several years ago and prompted a neurologic workup, including MRI of the head and somatosensory evoked potentials of the glossopharyngeal nerve. She was diagnosed with “probable” glossopharyngeal neuralgia and failed multiple medication trials, including carbamazepine, phenytoin, gabapentin, and amitriptyline. She underwent microvascular decompression surgery 3 years ago. The operation, which has an 80% to 90% success rate for neuralgias,1,2 offered only brief symptomatic relief. She was maintained on immediate-release opiates until the pain became “unbearable” 8 months ago. This prompted a second neurologic workup, which was unremarkable. Mrs. P was diagnosed with pain disorder associated with psychological factors and a general medical condition.
Ten years ago she had 2 major depressive episodes with inpatient hospitalization and 2 suicide attempts within 1 year, but no history of psychosis before 8 months ago. Mrs. P’s husband says his wife has no history of manic or hypomanic episodes. Her medications are ziprasidone, 20 mg/d, thiothixene, 10 mg/d, benztropine, 3 mg/d, and escitalopram, 30 mg/d. She also receives oxycodone/acetaminophen, 5 mg/325 mg as needed for facial pain and headaches, and clonazepam, 1 mg as needed for panic attacks.
The authors’ observations
Psychosis can be a feature of any of the disorders listed in Table 13; however, several features of Mrs. P’s illness led us to diagnose MDD, recurrent, severe with psychotic features.4 Mrs. P and her husband described several discreet episodes of major debilitating depression without alternating periods of hypomanic or manic symptoms (Table 2).4 Comorbid depressive symptoms and a timeline indicating persistence of psychotic symptoms make a brief psychotic episode less likely. Although uncommon, patients can develop psychotic or mood disorders as a result of opiate abuse or dependence. However, Mrs. P was taking opiates as prescribed and not asking for early refills, which makes substance abuse an unlikely cause of her psychosis. In addition, because Mrs. P had 2 major depressive episodes in the absence of opiate use, a primary mood disorder seemed the more appropriate diagnosis. Schizophrenia is ruled out based on history. Although Mrs. P was suffering from complex delusional constructs, auditory hallucinations, and grossly disorganized behavior, these symptoms occurred only within the context of her depressive episode. New-onset delusional guilt relating to her childhood sexual trauma and hypochondriacal preoccupations within the context of pain complaints make psychotic depression more likely.5
Table 1
Psychiatric diseases in which patients may present with psychotic symptoms
| Bipolar depression |
| Borderline personality disorder |
| Brief psychotic disorder |
| Delirium |
| Delusional disorder |
| Dementia |
| Major depressive disorder |
| Psychotic disorder due to a general medical condition |
| Schizoaffective disorder |
| Schizophrenia |
| Shared psychotic disorder |
| Substance-induced psychosis |
| Source: Reference 3 |
Table 2
DSM-IV-TR criteria for major depressive episode
|
| Source: Reference 4 |
Depression, psychosis, and pain
From the beginning of Mrs. P’s treatment, we considered psychotic depression worsened—if not completely explained—her pain. Her somatic complaints appeared to be subtly woven into her delusional constructs. For instance, she complained that a device had been implanted in her head and she had the scar to prove it, pointing to the scar from her microvascular decompression surgery. Research indicates that depressive illness and chronic pain syndromes are highly comorbid and depressive illness can worsen pain syndromes.6,7 In addition, Mrs. P failed several medical and 1 surgical interventions for her pain condition that had high success rates. Her husband notes that when her outpatient psychiatrist started olanzapine 3 months ago for emerging psychotic symptoms, her pain complaints initially decreased with her psychotic symptoms, and she used less opiate medication during that time. Several months later Mrs. P’s pain complaints increased as her psychotic symptoms worsened. Second-generation antipsychotics have been evaluated as treatment for chronic pain syndromes, and may exert a primary analgesic effect.8,9 However, because of the correlation between her fluctuating psychotic symptoms and pain complaints, the more plausible explanation for olanzapine’s initial efficacy in treating Mrs. P’s pain is a secondary analgesic effect from decreased psychotic somatic preoccupation.
TREATMENT: ECT
Mrs. P is admitted to the inpatient psychiatric unit and placed on suicide precautions. Oxycodone/acetaminophen and clonazepam are tapered and limited to twice daily as needed. Escitalopram is tapered and discontinued. Thiothixene is tapered and replaced by olanzapine, 5 mg/d. Mrs. P receives 3 bifrontal, brief pulse-width ECT treatments. These result in marked improvement in her depressive and psychotic symptoms. In addition, her pain complaints become minimal. She becomes less preoccupied with her sexual trauma and grows to trust many staff members whom she previously believed were part of her traumatic childhood events. Mrs. P is no longer suicidal and asks to continue ECT treatments as an outpatient. She is discharged on olanzapine, 5 mg/d, trazodone, 100 mg/d for insomnia, benztropine, 2 mg/d, clonazepam 0.5 mg twice daily as needed for panic attacks, and oxycodone/acetaminophen, 5 mg/325 mg twice daily as needed for pain.
The authors’ observations
According to the Harvard South Shore Algorithm, treatment strategies for psychotic depression include antidepressant and antipsychotic combinations, lithium augmentation, clozapine, and ECT.10 Several factors made ECT the best option for Mrs. P. She had failed multiple treatment strategies and was suicidal. ECT is an effective treatment for MDD with psychotic features, single or recurrent episode.11 ECT can be used as a primary treatment before psychotropic medications or secondarily when there has been lack of clinical response to medications, intolerable side effects, deterioration in psychiatric condition, or suicidality.11,12 In addition, when treated with ECT, psychotic depression has a significantly higher remission rate than major depression without psychosis.12 Delusional guilt, psychomotor retardation, hypochondriacal preoccupations, loss of insight, paranoia, and obsessive-compulsive symptoms predict a favorable response.12 ECT also has demonstrated efficacy for treating pain secondary to psychotic depression or melancholic depression.13 In addition, ECT has been shown to have analgesic properties beyond treating underlying depression.14 Our primary focus was not to treat Mrs. P’s pain syndrome with ECT; however, in treating her psychotic depression we had hoped that her pain tolerance would improve and she would rely less on opiates.
OUTCOME: Pain relief
As an outpatient, Mrs. P receives 11 bifrontal ECT treatments in her initial series, followed by 7 bifrontal maintenance treatments. Her speech is more spontaneous, her grooming and hygiene improve, and she exhibits a brighter and more reactive affect. Suicidal ideation has resolved. Pain improves from a “10 out of 10” to a “2 out of 10.” Mrs. P consistently requires less oxycodone/acetaminophen. She relates better to her family and begins exploring new hobbies such as pottery. In addition to monthly maintenance bifrontal ECT treatments, she is stable on citalopram, 60 mg/d, and trazodone, 50 mg/d as needed for insomnia.
The authors’ observations
The relationship between depressive illness and chronic pain is complex. Treating a primary depressive illness can lead to improved functional outcomes and decreased disability from chronic pain complaints.15 Patients with comorbid chronic pain and depressive illness are more likely to suffer from unremitting pain despite compliance with evidence-based treatment strategies.16 Mrs. P had 2 co-occurring disorders: psychotic depression and chronic pain disorder secondary to glossopharyngeal neuralgia. Our opinion is that Mrs. P’s psychotic depression worsened her experience of pain.
Treatment strategies that address both depressive symptoms and chronic pain are ideal.17 These treatment modalities include psychotherapeutic techniques such as cognitive-behavioral therapy, medications, and somatic treatments such as ECT.18 In Mrs. P’s case, ECT was an effective treatment that caused remission of psychotic depressive symptoms, which lead to improved pain control and restored social and occupational functioning.
Related Resources
- Schreiber S, Shmueli D, Grunhaus L, et al. The influence of electroconvulsive therapy on pain threshold and pain tolerance in major depression patients before, during and after treatment. Eur J Pain. 2003;7(5):419-424.
- Suzuki K, Ebina Y, Shindo T, et al. Repeated electroconvulsive therapy courses improved chronic regional pain with depression caused by failed back syndrome. Med Sci Monit. 2009;15(4):CS77-CS79.
- Giesecke T, Gracely RH, Williams DA, et al. The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum. 2005;52(5):1577-1584.
Drug Brand Names
- Amitriptyline • Elavil
- Benztropine • Cogentin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Gabapentin • Neurontin
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Oxycodone/ acetaminophen • Vicodin
- Phenytoin • Dilantin
- Thiothixene • Navane
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosures
Dr. Kugler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Magid receives NARSAD grant support.
1. Kawashima M, Matsushima T, Inoue T, et al. Microvascular decompression for glossopharyngeal neuralgia through the transcondylar fossa (supracondylar transjugular tubercle) approach. Neurosurgery. 2010;66(6 suppl operative):275-280.
2. Ferroli P, Fioravanti A, Schiariti M, et al. Microvascular decompression for glossopharyngeal neuralgia: a long-term retrospective review of the Milan-Bologna experience in 31 consecutive cases. Acta Neurochir (Wien). 2009;151(10):1245-1250.
3. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York NY: Cambridge University Press; 2008.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev.Washington DC: American Psychiatric Association; 2000.
5. Rothschild AJ. Diagnosis and assessment. In: Rothschild AJ. Clinical manual for diagnosis and treatment of psychotic depression. Arlington VA: American Psychiatric Publishing Inc.; 2009:57-71.
6. Tunks ER, Crook J, Weir R. Epidemiology of chronic pain with psychological comorbidity: prevalence risk, course and prognosis. Can J Psychiatry. 2008;53(4):235-242.
7. Hooten MW, Shi Y, Gazelka HM, et al. The effects of depression and smoking on pain severity and opioid use in patients with chronic pain. Pain. 2011;152(1):223-229.
8. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
9. Seidel S, Aigner M, Ossege M, et al. Antipsychotics for acute and chronic pain in adults. J Pain Symptom Manage. 2010;39(4):768-778.
10. Hamoda HM, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on psychotic depression. Harv Rev Psychiatry. 2008;16(4):235-247.
11. American Psychiatric Association. Committee on Electroconvulsive Therapy, Weiner RD, eds. The practice of electroconvulsive therapy: recommendations for treatment, training and privileging. 2nd ed. Washington DC: American Psychiatric Association; 2001.
12. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus nonpsychotic depressed patients: a report from CORE. J ECT. 2001;17(4):244-253.
13. Rasmussen KG, Rummans TA. Electroconvulsive therapy in the management of chronic pain. Curr Pain Headache Rep. 2002;6(1):17-22.
14. Wasan AD, Artin K, Clark MR. A case-matching study of the analgesic properties of electroconvulsive therapy. Pain Med. 2004;5(1):50-58.
15. Teh FC, Zaslavsky AM, Reynolds CF, 3rd, et al. Effect of depression treatment on chronic pain outcomes. Psychosom Med. 2010;72(1):61-67.
16. Sertel Berk HO. The biopsychosocial factors that serve as predictors of the outcome of surgical modalities for chronic pain. Agri. 2010;22(3):93-97.
17. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity. Arch Intern Med. 2003;163(20):2433-2445.
18. Veehof MM, Oskam MJ, Schreurs KM, et al. Acceptance-based interventions for the treatment of chronic pain: a systematic review and meta-analysis. Pain. 2011;152(3):533-542.
1. Kawashima M, Matsushima T, Inoue T, et al. Microvascular decompression for glossopharyngeal neuralgia through the transcondylar fossa (supracondylar transjugular tubercle) approach. Neurosurgery. 2010;66(6 suppl operative):275-280.
2. Ferroli P, Fioravanti A, Schiariti M, et al. Microvascular decompression for glossopharyngeal neuralgia: a long-term retrospective review of the Milan-Bologna experience in 31 consecutive cases. Acta Neurochir (Wien). 2009;151(10):1245-1250.
3. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications. 3rd ed. New York NY: Cambridge University Press; 2008.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev.Washington DC: American Psychiatric Association; 2000.
5. Rothschild AJ. Diagnosis and assessment. In: Rothschild AJ. Clinical manual for diagnosis and treatment of psychotic depression. Arlington VA: American Psychiatric Publishing Inc.; 2009:57-71.
6. Tunks ER, Crook J, Weir R. Epidemiology of chronic pain with psychological comorbidity: prevalence risk, course and prognosis. Can J Psychiatry. 2008;53(4):235-242.
7. Hooten MW, Shi Y, Gazelka HM, et al. The effects of depression and smoking on pain severity and opioid use in patients with chronic pain. Pain. 2011;152(1):223-229.
8. Rico-Villademoros F, Hidalgo J, Dominguez I, et al. Atypical antipsychotics in the treatment of fibromyalgia: a case series with olanzapine. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):161-164.
9. Seidel S, Aigner M, Ossege M, et al. Antipsychotics for acute and chronic pain in adults. J Pain Symptom Manage. 2010;39(4):768-778.
10. Hamoda HM, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on psychotic depression. Harv Rev Psychiatry. 2008;16(4):235-247.
11. American Psychiatric Association. Committee on Electroconvulsive Therapy, Weiner RD, eds. The practice of electroconvulsive therapy: recommendations for treatment, training and privileging. 2nd ed. Washington DC: American Psychiatric Association; 2001.
12. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus nonpsychotic depressed patients: a report from CORE. J ECT. 2001;17(4):244-253.
13. Rasmussen KG, Rummans TA. Electroconvulsive therapy in the management of chronic pain. Curr Pain Headache Rep. 2002;6(1):17-22.
14. Wasan AD, Artin K, Clark MR. A case-matching study of the analgesic properties of electroconvulsive therapy. Pain Med. 2004;5(1):50-58.
15. Teh FC, Zaslavsky AM, Reynolds CF, 3rd, et al. Effect of depression treatment on chronic pain outcomes. Psychosom Med. 2010;72(1):61-67.
16. Sertel Berk HO. The biopsychosocial factors that serve as predictors of the outcome of surgical modalities for chronic pain. Agri. 2010;22(3):93-97.
17. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity. Arch Intern Med. 2003;163(20):2433-2445.
18. Veehof MM, Oskam MJ, Schreurs KM, et al. Acceptance-based interventions for the treatment of chronic pain: a systematic review and meta-analysis. Pain. 2011;152(3):533-542.
Obsessive and inattentive
CASE: Perfect breath
Mr. C, a 20-year-old college student, is diagnosed with obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder (ADHD), and tic disorder (TD). His obsessions consist of a persistent sense that he is not breathing “correctly” or “perfectly.” He compulsively holds his breath to “rush blood to my head” until “the pressure feels just right.” Mr. C says that his OCD has had longstanding, significant negative impact on his academic performance and capacity to engage in other activities. Tics have been present for years and manifest as coughing and throat-clearing. After multiple syncopal epi-sodes from breath-holding with Valsalva maneuver—some of which caused falls and head injury—Mr. C is admitted to a residential psychiatric unit specializing in treating OCD. At the time of his admission, his Yale-Brown Obsessive Compulsive Scale (Y-BOCS) scores1,2 are 23 total, 12 on the obsessions subscale, and 11 on the compulsions subscale, indicating moderate to severe illness. Cognitive-behavioral therapy (CBT) is offered, along with a combination of escitalopram, 60 mg/d, and quetiapine, 50 mg/d. Quetiapine is over-sedating at subtherapeutic doses and Mr. C’s compulsions worsen. He reports that “[it] took longer and longer to get the ‘just right’ feeling.’” Quetiapine is discontinued and risperidone, 0.5 mg/d, is started, which decreases the frequency of his tics. When he is discharged after a 36-day stay, Mr. C’s Y-BOCS scores are greatly improved at 13 total, 7 on the obsessions subscale, and 3 on the compulsions subscale.
Mr. C’s psychologist refers him to our outpatient clinic for continued psychiatric evaluation and treatment of his OCD, ADHD, and TD. At this time, he is prescribed escitalopram, 60 mg/d, and risperidone, 0.5 mg/d, along with CBT with his psychologist. We do not readminister the Y-BOCS at this time, but Mr. C reports that his OCD is “60% improved.” However, he describes prominent obsessive thoughts regarding his breathing similar to those he experienced before residential treatment. These obsessive thoughts arise in the context of specific environmental “triggers,” such as other people coughing or his own tics. The obsessions lead to compulsive urges to engage in breath-holding rituals. Mr. C experiences the thoughts and compulsions as deeply troubling and they consume 5 to 6 hours each day. Mr. C reports impaired concentration in class and during studying: “I can focus for 5 minutes, then not for 2 minutes, then for 3 minutes… I can never stay focused for more than a couple minutes,” before becoming distracted “by my OCD” or other environmental stimuli. We note on exam prominent breath-holding occurring several times per minute. Mr. C says his OCD has not impaired his ability to socialize.
Mr. C notes that he has been exposed to an array of CBT techniques, but he has difficulty using these techniques because his “mind wanders” or he lacks “motivation.” He admits he occasionally has taken a classmate’s ADHD medication (mixed amphetamine salts [MAS], dose unspecified) and found it improved his ability to focus on his academic work.
The authors’ observations
Researchers have established a relationship among OCD, ADHD, and TD across all combinations of comorbidity (OCD and ADHD,3 ADHD and TD,4 OCD and TD,5,6 and all 3 entities7). Data suggests a poorer prognosis for OCD when comorbid with either or both of these conditions.8 Researchers have raised concerns that psychostimulants could exacerbate or potentiate tic behaviors in patients with ADHD,9,10 although safe and effective use of these medications has been documented in controlled trials of patients with comorbid ADHD and tics.11-13 Furthermore, tic suppression has been reported with psychostimulants,14 as well as a differential effect of stimulants on motor vs vocal tics.15 Despite these data (Table 1),9-15 the FDA regards using psychostimulants in patients with TD as a contraindication,16 although clinicians often recognize that this practice may be unavoidable in some circumstances because of high comorbidity rates. Psychostimulants could exacerbate obsessions or compulsions in some patients because of their dopaminergic properties or through mitigation of the purported anti-obsessional properties of dopamine antagonists.17
Although there is evidence that the prevalence of prescribed psychostimulant abuse is low among ADHD patients,18 diversion of prescribed medication is a risk inherent in the use of these agents, particularly among college-age patients.19,20
Table 1
Evidence of effect of psychostimulants on tics
| Study/disorder(s) | Medication and study design | Relevant findings |
|---|---|---|
| Lipkin et al, 19949; ADHD without TD | Chart review (N = 122) to determine the incidence of tics or dyskinesias in children treated with stimulants | Approximately 9% of children developed tics or dyskinesias, which predominantly were transient, with <1% developing chronic tics or Tourette’s syndrome. Personal or family tic history and medication selection or dosage were not related to onset of tics or dyskinesias |
| Gadow et al, 199515; ADHD with TD | Methylphenidate variable dose, placebo-controlled, 2-week trials (N = 24) | All children’s ADHD symptoms improved. At a 0.1 mg/kg dose, motor tics observed in the classroom increased, but there were fewer vocal tics observed in the lunchroom |
| Castellanos et al, 199710; ADHD with Tourette’s syndrome | Methylphenidate, dextroamphetamine, variable-dose, double-blind, placebo-controlled, 9-week crossover (N = 20) | 3 patients had consistent worsening of tics while taking stimulants. Stimulants reduced hyperactivity rates compared with placebo (P = .03). Stimulants improved ADHD symptoms and had acceptable effects on tics. Methylphenidate was better tolerated than dextroamphetamine |
| Gadow et al, 199911; ADHD with TD | 34 methylphenidate-treated children, followed at 6-month intervals for 2 years | No evidence that frequency or severity of motor or vocal tics changed during maintenance therapy |
| Tourette Syndrome Study Group, 200213; ADHD with TD | Clonidine alone, methylphenidate alone, clonidine plus methylphenidate, or placebo | Worsening of tics was not reported in any group at a rate significantly higher than placebo. Tic severity was more reduced in the 2 clonidine groups than in the methylphenidate group |
| Lyon et al, 201014; ADHD with Tourette’s syndrome | Dexmethylphenidate, single-dose challenge. Ten patients with or without TSP | Acute dexmethylphenidate administration resulted in tic suppression but did not augment TSP |
| Gadow et al, 200712; ADHD with TD | Double-blind, placebo-controlled, 2-week trials each of 3 doses of methylphenidate and placebo (N = 71) | MPH-IR did not alter the overall severity of TD or OCD behaviors. Teacher ratings indicated that MPH-IR therapy decreased tic frequency and severity |
| ADHD: attention-deficit/hyperactivity disorder; MPH-IR: methylphenidate immediate release; OCD: obsessive-compulsive disorder; TD: tic disorder; TSP: tic suppression protocol | ||
TREATMENT: Weighing options
To manage impaired attention and executive function difficulties secondary to ADHD, we offer Mr. C several options, including bupropion, modafinil, and memantine augmentation. Mr. C asks for a psychostimulant because exam week is approaching and he wants a treatment with quick therapeutic effect. We discuss with Mr. C the potential for dopaminergic agents, such as psychostimulants, to exacerbate tics or OCD symptoms. Ultimately, we prescribe immediate-release MAS, 20 mg/d.
Two days later, Mr. C says he has taken 3 MAS doses and describes a marked reduction in obsessions, significant decrease in frequency of “triggers,” and greater capacity to use CBT saying, “when I am [triggered], I am able to move past the urges without doing any compulsions.” Daily time spent “stuck on” obsessions or compulsions decreases from 5 to 6 hours per day to “about 2 and a half minutes.”
Mr. C reports a modest increase in the prevalence of tics, experienced as “little throat clears and quick stuttering of breath.” He notes that, although in the past such tics would be followed by urges for “perfecting the tic and making it feel just right,” he presently “had no desire to do so.”
OUTCOME: Sharper focus
Increasing MAS immediate release from 20 mg/d to 30 mg/d suppresses Mr. C’s obsessions and compulsions for 8 hours. On the 19th day of treatment, MAS immediate release was replaced with an extended release formulation, 30 mg/d, which preserves therapeutic effect and tolerability for 16 weeks. Repeat Y-BOCS yields 9 total, 3 on obsessions subscale, and 6 on compulsions subscale scores.
One month later, Mr. C reports that his symptoms have been “improving ever since” the previous appointment. He continues to be able to access skills for managing his OCD and is doing well in his 2 accelerated summer courses, saying “I focus really well” in 3-hour class sessions. On exam, tic behaviors are nearly absent. Mr. C describes occasional bouts of anxiety associated with urges to engage in tic behaviors, in turn arising from fear of symptomatic recurrence as he worked toward stopping smoking as advised by his primary care physician and psychiatrist.
The authors’ observations
The results of the repeat Y-BOCS are consistent with improvement in obsessions but possible worsening of compulsions since Mr. C was discharged from residential treatment. Alternatively, compulsions may have worsened immediately after discharge and declined again with introduction of MAS.
A substantial body of literature describes the challenges associated with treating ADHD with comorbid tics, including the relative degree of risk of tic exacerbation associated with treating ADHD with psychostimulants. The range of FDA-approved pharmacologic options for treatment of this comorbidity is limited (Table 2),21 particularly given the risk for tardive dyskinesia associated with the typical antipsychotics haloperidol and chlorpromazine. Data support using the α-2 agonist clonidine to treat hyperactivity associated with ADHD22 and TD23 and an extended-release preparation of this medication is FDA-approved for the former but not the latter indication (an α-2A receptor subtype agonist, guanfacine, also is FDA-approved for ADHD in pediatric patients). Mr. C’s experience of robust, sustained reduction in obsessions, if not compulsions, after treatment with MAS is consistent with the few studies of stimulant use in ADHD with comorbid OCD.24,25
Effective treatment of ADHD may help Mr. C better access CBT strategies and thereby potentiate treatment of comorbid OCD.
Table 2
FDA-approved medications for ADHD, OCD, and TD
| Disorder | Medications |
|---|---|
| ADHD | Amphetamine (racemic), atomoxetine, chlorpromazine (hyperactivity), clonidine extended release, dexmethylphenidate, dextroamphetamine, guanfacine extended release, haloperidol (hyperactivity, second-line), lisdexamfetamine, methylphenidate (racemic) |
| OCD | Clomipramine, fluoxetine, fluvoxamine, paroxetine, sertraline |
| TD/Tourette’s syndrome | Haloperidol (Tourette’s), pimozide (Tourette’s) |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder; TD: tic disorder Source: Reference 21 | |
Related Resources
- Pliszka SR. Treating ADHD and comorbid disorders: psychosocial and psychopharmacological interventions. New York, NY: The Guilford Press; 2011.
- Pollak Y, Benarroch F, Kanengisser L, et al. Tourette syndrome-associated psychopathology: roles of comorbid attention-deficit hyperactivity disorder and obsessive-compulsive disorder. J Dev Behav Pediatr. 2009;30(5):413-419.
Drug Brand Names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin, Zyban
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clonidine extended release • Kapvay
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guanfacine • Intuniv, Tenex
- Haloperidol • Haldol
- Lisdexamfetamine • Vyvanse
- Memantine • Namenda
- Methylphenidate • Methylin, Ritalin
- Modafinil • Provigil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: I. Development, use and reliability. Arch Gen Psych. 1989;46(11):1006-1011.
2. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: II. Validity. Arch Gen Psych. 1989;46(11):1012-1016.
3. Geller DA, Biederman J, Faraone S, et al. Re-examining comorbidity of obsessive compulsive and attention-deficit hyperactivity disorder using an empirically derived taxonomy. Eur Child Adolesc Psychiatry. 2004;13(2):83-91.
4. Freeman RD. Attention deficit hyperactivity disorder in the presence of Tourette syndrome. Neurol Clin. 1997;15(2):411-420.
5. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin North Am. 2006;29(2):353-370.
6. Eapen V, Fox-Hiley P, Banerjee S, et al. Clinical features and associated psychopathology in a Tourette syndrome cohort. Acta Neurol Scand. 2004;109(4):255-260.
7. Kano Y, Ohta M, Nagai Y, et al. Association between Tourette syndrome and comorbidities in Japan. Brain Dev. 2010;32(3):201-207.
8. Grados M, Riddle M. Do all obsessive-compulsive disorder subtypes respond to medication? Int Rev Psychiatry. 2008;20(2):189-193.
9. Lipkin PH, Goldstein IH, Adesman AR. Tics and dyskinesias associated with stimulant treatment in attention-deficit/hyperactivity disorder. Arch Pediatr Adolesc Med. 1994;148(8):859-861.
10. Castellanos FX, Giedd JN, Elia J, et al. Controlled stimulant treatment of ADHD and comorbid Tourette’s syndrome: effects of stimulant and dose. J Am Acad Child Adolesc Psychiatry. 1997;36(5):589-596.
11. Gadow K, Sverd J, Sprafkin J, et al. Long-term methylphenidate therapy in children with comorbid attention-deficit hyperactivity disorder and chronic multiple tic disorder. Arch Gen Psychiatry. 1999;56(4):330-333.
12. Gadow KD, Sverd J, Nolan EE, et al. Immediate-release methylphenidate for ADHD in children with comorbid chronic multiple tic disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):840-848.
13. Tourette’s Syndrome Study Group. Treatment of ADHD in children with tics: a randomized controlled trial. Neurology. 2002;58(4):527-536.
14. Lyon GJ, Samar SM, Conelea C, et al. Testing tic suppression: comparing the effects of dexmethylphenidate to no mediation in children and adolescents with attention-deficit/hyperactivity disorder and Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):283-289.
15. Gadow KD, Sverd J, Sprafkin J, et al. Efficacy of methylphenidate for attention-deficit hyperactivity disorder in children with tic disorder. Arch Gen Psychiatry. 1995;52(6):444-455.
16. Bloch MH, Panza KE, Landerso-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
17. McDougle CJ, Goodman WK, Price LH. Dopamine antagonists in tic-related and psychotic spectrum obsessive compulsive disorder. J Clin Psychiatry. 1994;55(suppl):24-31.
18. Wilens TE, Morrison NR. The intersection of attention-deficit/hyperactivity disorder and substance abuse. Curr Opin Psychiatry. 2011;24(4):280-285.
19. Kollins SH. A qualitative review of issues arising in the use of psycho-stimulant medications in patients with ADHD and co-morbid substance use disorders. Curr Med Res Opin. 2008;24(5):1345-1357.
20. Schubiner H. Substance abuse in patients with attention-deficit hyperactivity disorder: therapeutic implications. CNS Drugs. 2005;19(8):643-655.
21. Stahl SM. The prescriber’s guide. Stahl’s essential psychopharmacology. 3rd ed. New York NY: Cambridge University Press; 2009.
22. Jain R, Segal S, Kollins SH, et al. Clonidine extended-release tablets for pediatric patients with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(2):171-179.
23. Hedderick EF, Morris CM, Singer HS. Double-blind crossover study of clonidine and levetiracetam in Tourette syndrome. Pediatr Neurol. 2009;40(6):420-425.
24. Joffe RT, Swinson RP, Levitt AJ. Acute psychostimulant challenge in primary obsessive-compulsive disorder. J Clin Psychopharmacol. 1991;11(4):237-241.
25. Insel TR, Hamilton JA, Guttmacher LB, et al. D-amphetamine in obsessive-compulsive disorder. Psychopharmacology (Berl). 1983;80(3):231-235.
CASE: Perfect breath
Mr. C, a 20-year-old college student, is diagnosed with obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder (ADHD), and tic disorder (TD). His obsessions consist of a persistent sense that he is not breathing “correctly” or “perfectly.” He compulsively holds his breath to “rush blood to my head” until “the pressure feels just right.” Mr. C says that his OCD has had longstanding, significant negative impact on his academic performance and capacity to engage in other activities. Tics have been present for years and manifest as coughing and throat-clearing. After multiple syncopal epi-sodes from breath-holding with Valsalva maneuver—some of which caused falls and head injury—Mr. C is admitted to a residential psychiatric unit specializing in treating OCD. At the time of his admission, his Yale-Brown Obsessive Compulsive Scale (Y-BOCS) scores1,2 are 23 total, 12 on the obsessions subscale, and 11 on the compulsions subscale, indicating moderate to severe illness. Cognitive-behavioral therapy (CBT) is offered, along with a combination of escitalopram, 60 mg/d, and quetiapine, 50 mg/d. Quetiapine is over-sedating at subtherapeutic doses and Mr. C’s compulsions worsen. He reports that “[it] took longer and longer to get the ‘just right’ feeling.’” Quetiapine is discontinued and risperidone, 0.5 mg/d, is started, which decreases the frequency of his tics. When he is discharged after a 36-day stay, Mr. C’s Y-BOCS scores are greatly improved at 13 total, 7 on the obsessions subscale, and 3 on the compulsions subscale.
Mr. C’s psychologist refers him to our outpatient clinic for continued psychiatric evaluation and treatment of his OCD, ADHD, and TD. At this time, he is prescribed escitalopram, 60 mg/d, and risperidone, 0.5 mg/d, along with CBT with his psychologist. We do not readminister the Y-BOCS at this time, but Mr. C reports that his OCD is “60% improved.” However, he describes prominent obsessive thoughts regarding his breathing similar to those he experienced before residential treatment. These obsessive thoughts arise in the context of specific environmental “triggers,” such as other people coughing or his own tics. The obsessions lead to compulsive urges to engage in breath-holding rituals. Mr. C experiences the thoughts and compulsions as deeply troubling and they consume 5 to 6 hours each day. Mr. C reports impaired concentration in class and during studying: “I can focus for 5 minutes, then not for 2 minutes, then for 3 minutes… I can never stay focused for more than a couple minutes,” before becoming distracted “by my OCD” or other environmental stimuli. We note on exam prominent breath-holding occurring several times per minute. Mr. C says his OCD has not impaired his ability to socialize.
Mr. C notes that he has been exposed to an array of CBT techniques, but he has difficulty using these techniques because his “mind wanders” or he lacks “motivation.” He admits he occasionally has taken a classmate’s ADHD medication (mixed amphetamine salts [MAS], dose unspecified) and found it improved his ability to focus on his academic work.
The authors’ observations
Researchers have established a relationship among OCD, ADHD, and TD across all combinations of comorbidity (OCD and ADHD,3 ADHD and TD,4 OCD and TD,5,6 and all 3 entities7). Data suggests a poorer prognosis for OCD when comorbid with either or both of these conditions.8 Researchers have raised concerns that psychostimulants could exacerbate or potentiate tic behaviors in patients with ADHD,9,10 although safe and effective use of these medications has been documented in controlled trials of patients with comorbid ADHD and tics.11-13 Furthermore, tic suppression has been reported with psychostimulants,14 as well as a differential effect of stimulants on motor vs vocal tics.15 Despite these data (Table 1),9-15 the FDA regards using psychostimulants in patients with TD as a contraindication,16 although clinicians often recognize that this practice may be unavoidable in some circumstances because of high comorbidity rates. Psychostimulants could exacerbate obsessions or compulsions in some patients because of their dopaminergic properties or through mitigation of the purported anti-obsessional properties of dopamine antagonists.17
Although there is evidence that the prevalence of prescribed psychostimulant abuse is low among ADHD patients,18 diversion of prescribed medication is a risk inherent in the use of these agents, particularly among college-age patients.19,20
Table 1
Evidence of effect of psychostimulants on tics
| Study/disorder(s) | Medication and study design | Relevant findings |
|---|---|---|
| Lipkin et al, 19949; ADHD without TD | Chart review (N = 122) to determine the incidence of tics or dyskinesias in children treated with stimulants | Approximately 9% of children developed tics or dyskinesias, which predominantly were transient, with <1% developing chronic tics or Tourette’s syndrome. Personal or family tic history and medication selection or dosage were not related to onset of tics or dyskinesias |
| Gadow et al, 199515; ADHD with TD | Methylphenidate variable dose, placebo-controlled, 2-week trials (N = 24) | All children’s ADHD symptoms improved. At a 0.1 mg/kg dose, motor tics observed in the classroom increased, but there were fewer vocal tics observed in the lunchroom |
| Castellanos et al, 199710; ADHD with Tourette’s syndrome | Methylphenidate, dextroamphetamine, variable-dose, double-blind, placebo-controlled, 9-week crossover (N = 20) | 3 patients had consistent worsening of tics while taking stimulants. Stimulants reduced hyperactivity rates compared with placebo (P = .03). Stimulants improved ADHD symptoms and had acceptable effects on tics. Methylphenidate was better tolerated than dextroamphetamine |
| Gadow et al, 199911; ADHD with TD | 34 methylphenidate-treated children, followed at 6-month intervals for 2 years | No evidence that frequency or severity of motor or vocal tics changed during maintenance therapy |
| Tourette Syndrome Study Group, 200213; ADHD with TD | Clonidine alone, methylphenidate alone, clonidine plus methylphenidate, or placebo | Worsening of tics was not reported in any group at a rate significantly higher than placebo. Tic severity was more reduced in the 2 clonidine groups than in the methylphenidate group |
| Lyon et al, 201014; ADHD with Tourette’s syndrome | Dexmethylphenidate, single-dose challenge. Ten patients with or without TSP | Acute dexmethylphenidate administration resulted in tic suppression but did not augment TSP |
| Gadow et al, 200712; ADHD with TD | Double-blind, placebo-controlled, 2-week trials each of 3 doses of methylphenidate and placebo (N = 71) | MPH-IR did not alter the overall severity of TD or OCD behaviors. Teacher ratings indicated that MPH-IR therapy decreased tic frequency and severity |
| ADHD: attention-deficit/hyperactivity disorder; MPH-IR: methylphenidate immediate release; OCD: obsessive-compulsive disorder; TD: tic disorder; TSP: tic suppression protocol | ||
TREATMENT: Weighing options
To manage impaired attention and executive function difficulties secondary to ADHD, we offer Mr. C several options, including bupropion, modafinil, and memantine augmentation. Mr. C asks for a psychostimulant because exam week is approaching and he wants a treatment with quick therapeutic effect. We discuss with Mr. C the potential for dopaminergic agents, such as psychostimulants, to exacerbate tics or OCD symptoms. Ultimately, we prescribe immediate-release MAS, 20 mg/d.
Two days later, Mr. C says he has taken 3 MAS doses and describes a marked reduction in obsessions, significant decrease in frequency of “triggers,” and greater capacity to use CBT saying, “when I am [triggered], I am able to move past the urges without doing any compulsions.” Daily time spent “stuck on” obsessions or compulsions decreases from 5 to 6 hours per day to “about 2 and a half minutes.”
Mr. C reports a modest increase in the prevalence of tics, experienced as “little throat clears and quick stuttering of breath.” He notes that, although in the past such tics would be followed by urges for “perfecting the tic and making it feel just right,” he presently “had no desire to do so.”
OUTCOME: Sharper focus
Increasing MAS immediate release from 20 mg/d to 30 mg/d suppresses Mr. C’s obsessions and compulsions for 8 hours. On the 19th day of treatment, MAS immediate release was replaced with an extended release formulation, 30 mg/d, which preserves therapeutic effect and tolerability for 16 weeks. Repeat Y-BOCS yields 9 total, 3 on obsessions subscale, and 6 on compulsions subscale scores.
One month later, Mr. C reports that his symptoms have been “improving ever since” the previous appointment. He continues to be able to access skills for managing his OCD and is doing well in his 2 accelerated summer courses, saying “I focus really well” in 3-hour class sessions. On exam, tic behaviors are nearly absent. Mr. C describes occasional bouts of anxiety associated with urges to engage in tic behaviors, in turn arising from fear of symptomatic recurrence as he worked toward stopping smoking as advised by his primary care physician and psychiatrist.
The authors’ observations
The results of the repeat Y-BOCS are consistent with improvement in obsessions but possible worsening of compulsions since Mr. C was discharged from residential treatment. Alternatively, compulsions may have worsened immediately after discharge and declined again with introduction of MAS.
A substantial body of literature describes the challenges associated with treating ADHD with comorbid tics, including the relative degree of risk of tic exacerbation associated with treating ADHD with psychostimulants. The range of FDA-approved pharmacologic options for treatment of this comorbidity is limited (Table 2),21 particularly given the risk for tardive dyskinesia associated with the typical antipsychotics haloperidol and chlorpromazine. Data support using the α-2 agonist clonidine to treat hyperactivity associated with ADHD22 and TD23 and an extended-release preparation of this medication is FDA-approved for the former but not the latter indication (an α-2A receptor subtype agonist, guanfacine, also is FDA-approved for ADHD in pediatric patients). Mr. C’s experience of robust, sustained reduction in obsessions, if not compulsions, after treatment with MAS is consistent with the few studies of stimulant use in ADHD with comorbid OCD.24,25
Effective treatment of ADHD may help Mr. C better access CBT strategies and thereby potentiate treatment of comorbid OCD.
Table 2
FDA-approved medications for ADHD, OCD, and TD
| Disorder | Medications |
|---|---|
| ADHD | Amphetamine (racemic), atomoxetine, chlorpromazine (hyperactivity), clonidine extended release, dexmethylphenidate, dextroamphetamine, guanfacine extended release, haloperidol (hyperactivity, second-line), lisdexamfetamine, methylphenidate (racemic) |
| OCD | Clomipramine, fluoxetine, fluvoxamine, paroxetine, sertraline |
| TD/Tourette’s syndrome | Haloperidol (Tourette’s), pimozide (Tourette’s) |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder; TD: tic disorder Source: Reference 21 | |
Related Resources
- Pliszka SR. Treating ADHD and comorbid disorders: psychosocial and psychopharmacological interventions. New York, NY: The Guilford Press; 2011.
- Pollak Y, Benarroch F, Kanengisser L, et al. Tourette syndrome-associated psychopathology: roles of comorbid attention-deficit hyperactivity disorder and obsessive-compulsive disorder. J Dev Behav Pediatr. 2009;30(5):413-419.
Drug Brand Names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin, Zyban
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clonidine extended release • Kapvay
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guanfacine • Intuniv, Tenex
- Haloperidol • Haldol
- Lisdexamfetamine • Vyvanse
- Memantine • Namenda
- Methylphenidate • Methylin, Ritalin
- Modafinil • Provigil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Perfect breath
Mr. C, a 20-year-old college student, is diagnosed with obsessive-compulsive disorder (OCD), attention-deficit/hyperactivity disorder (ADHD), and tic disorder (TD). His obsessions consist of a persistent sense that he is not breathing “correctly” or “perfectly.” He compulsively holds his breath to “rush blood to my head” until “the pressure feels just right.” Mr. C says that his OCD has had longstanding, significant negative impact on his academic performance and capacity to engage in other activities. Tics have been present for years and manifest as coughing and throat-clearing. After multiple syncopal epi-sodes from breath-holding with Valsalva maneuver—some of which caused falls and head injury—Mr. C is admitted to a residential psychiatric unit specializing in treating OCD. At the time of his admission, his Yale-Brown Obsessive Compulsive Scale (Y-BOCS) scores1,2 are 23 total, 12 on the obsessions subscale, and 11 on the compulsions subscale, indicating moderate to severe illness. Cognitive-behavioral therapy (CBT) is offered, along with a combination of escitalopram, 60 mg/d, and quetiapine, 50 mg/d. Quetiapine is over-sedating at subtherapeutic doses and Mr. C’s compulsions worsen. He reports that “[it] took longer and longer to get the ‘just right’ feeling.’” Quetiapine is discontinued and risperidone, 0.5 mg/d, is started, which decreases the frequency of his tics. When he is discharged after a 36-day stay, Mr. C’s Y-BOCS scores are greatly improved at 13 total, 7 on the obsessions subscale, and 3 on the compulsions subscale.
Mr. C’s psychologist refers him to our outpatient clinic for continued psychiatric evaluation and treatment of his OCD, ADHD, and TD. At this time, he is prescribed escitalopram, 60 mg/d, and risperidone, 0.5 mg/d, along with CBT with his psychologist. We do not readminister the Y-BOCS at this time, but Mr. C reports that his OCD is “60% improved.” However, he describes prominent obsessive thoughts regarding his breathing similar to those he experienced before residential treatment. These obsessive thoughts arise in the context of specific environmental “triggers,” such as other people coughing or his own tics. The obsessions lead to compulsive urges to engage in breath-holding rituals. Mr. C experiences the thoughts and compulsions as deeply troubling and they consume 5 to 6 hours each day. Mr. C reports impaired concentration in class and during studying: “I can focus for 5 minutes, then not for 2 minutes, then for 3 minutes… I can never stay focused for more than a couple minutes,” before becoming distracted “by my OCD” or other environmental stimuli. We note on exam prominent breath-holding occurring several times per minute. Mr. C says his OCD has not impaired his ability to socialize.
Mr. C notes that he has been exposed to an array of CBT techniques, but he has difficulty using these techniques because his “mind wanders” or he lacks “motivation.” He admits he occasionally has taken a classmate’s ADHD medication (mixed amphetamine salts [MAS], dose unspecified) and found it improved his ability to focus on his academic work.
The authors’ observations
Researchers have established a relationship among OCD, ADHD, and TD across all combinations of comorbidity (OCD and ADHD,3 ADHD and TD,4 OCD and TD,5,6 and all 3 entities7). Data suggests a poorer prognosis for OCD when comorbid with either or both of these conditions.8 Researchers have raised concerns that psychostimulants could exacerbate or potentiate tic behaviors in patients with ADHD,9,10 although safe and effective use of these medications has been documented in controlled trials of patients with comorbid ADHD and tics.11-13 Furthermore, tic suppression has been reported with psychostimulants,14 as well as a differential effect of stimulants on motor vs vocal tics.15 Despite these data (Table 1),9-15 the FDA regards using psychostimulants in patients with TD as a contraindication,16 although clinicians often recognize that this practice may be unavoidable in some circumstances because of high comorbidity rates. Psychostimulants could exacerbate obsessions or compulsions in some patients because of their dopaminergic properties or through mitigation of the purported anti-obsessional properties of dopamine antagonists.17
Although there is evidence that the prevalence of prescribed psychostimulant abuse is low among ADHD patients,18 diversion of prescribed medication is a risk inherent in the use of these agents, particularly among college-age patients.19,20
Table 1
Evidence of effect of psychostimulants on tics
| Study/disorder(s) | Medication and study design | Relevant findings |
|---|---|---|
| Lipkin et al, 19949; ADHD without TD | Chart review (N = 122) to determine the incidence of tics or dyskinesias in children treated with stimulants | Approximately 9% of children developed tics or dyskinesias, which predominantly were transient, with <1% developing chronic tics or Tourette’s syndrome. Personal or family tic history and medication selection or dosage were not related to onset of tics or dyskinesias |
| Gadow et al, 199515; ADHD with TD | Methylphenidate variable dose, placebo-controlled, 2-week trials (N = 24) | All children’s ADHD symptoms improved. At a 0.1 mg/kg dose, motor tics observed in the classroom increased, but there were fewer vocal tics observed in the lunchroom |
| Castellanos et al, 199710; ADHD with Tourette’s syndrome | Methylphenidate, dextroamphetamine, variable-dose, double-blind, placebo-controlled, 9-week crossover (N = 20) | 3 patients had consistent worsening of tics while taking stimulants. Stimulants reduced hyperactivity rates compared with placebo (P = .03). Stimulants improved ADHD symptoms and had acceptable effects on tics. Methylphenidate was better tolerated than dextroamphetamine |
| Gadow et al, 199911; ADHD with TD | 34 methylphenidate-treated children, followed at 6-month intervals for 2 years | No evidence that frequency or severity of motor or vocal tics changed during maintenance therapy |
| Tourette Syndrome Study Group, 200213; ADHD with TD | Clonidine alone, methylphenidate alone, clonidine plus methylphenidate, or placebo | Worsening of tics was not reported in any group at a rate significantly higher than placebo. Tic severity was more reduced in the 2 clonidine groups than in the methylphenidate group |
| Lyon et al, 201014; ADHD with Tourette’s syndrome | Dexmethylphenidate, single-dose challenge. Ten patients with or without TSP | Acute dexmethylphenidate administration resulted in tic suppression but did not augment TSP |
| Gadow et al, 200712; ADHD with TD | Double-blind, placebo-controlled, 2-week trials each of 3 doses of methylphenidate and placebo (N = 71) | MPH-IR did not alter the overall severity of TD or OCD behaviors. Teacher ratings indicated that MPH-IR therapy decreased tic frequency and severity |
| ADHD: attention-deficit/hyperactivity disorder; MPH-IR: methylphenidate immediate release; OCD: obsessive-compulsive disorder; TD: tic disorder; TSP: tic suppression protocol | ||
TREATMENT: Weighing options
To manage impaired attention and executive function difficulties secondary to ADHD, we offer Mr. C several options, including bupropion, modafinil, and memantine augmentation. Mr. C asks for a psychostimulant because exam week is approaching and he wants a treatment with quick therapeutic effect. We discuss with Mr. C the potential for dopaminergic agents, such as psychostimulants, to exacerbate tics or OCD symptoms. Ultimately, we prescribe immediate-release MAS, 20 mg/d.
Two days later, Mr. C says he has taken 3 MAS doses and describes a marked reduction in obsessions, significant decrease in frequency of “triggers,” and greater capacity to use CBT saying, “when I am [triggered], I am able to move past the urges without doing any compulsions.” Daily time spent “stuck on” obsessions or compulsions decreases from 5 to 6 hours per day to “about 2 and a half minutes.”
Mr. C reports a modest increase in the prevalence of tics, experienced as “little throat clears and quick stuttering of breath.” He notes that, although in the past such tics would be followed by urges for “perfecting the tic and making it feel just right,” he presently “had no desire to do so.”
OUTCOME: Sharper focus
Increasing MAS immediate release from 20 mg/d to 30 mg/d suppresses Mr. C’s obsessions and compulsions for 8 hours. On the 19th day of treatment, MAS immediate release was replaced with an extended release formulation, 30 mg/d, which preserves therapeutic effect and tolerability for 16 weeks. Repeat Y-BOCS yields 9 total, 3 on obsessions subscale, and 6 on compulsions subscale scores.
One month later, Mr. C reports that his symptoms have been “improving ever since” the previous appointment. He continues to be able to access skills for managing his OCD and is doing well in his 2 accelerated summer courses, saying “I focus really well” in 3-hour class sessions. On exam, tic behaviors are nearly absent. Mr. C describes occasional bouts of anxiety associated with urges to engage in tic behaviors, in turn arising from fear of symptomatic recurrence as he worked toward stopping smoking as advised by his primary care physician and psychiatrist.
The authors’ observations
The results of the repeat Y-BOCS are consistent with improvement in obsessions but possible worsening of compulsions since Mr. C was discharged from residential treatment. Alternatively, compulsions may have worsened immediately after discharge and declined again with introduction of MAS.
A substantial body of literature describes the challenges associated with treating ADHD with comorbid tics, including the relative degree of risk of tic exacerbation associated with treating ADHD with psychostimulants. The range of FDA-approved pharmacologic options for treatment of this comorbidity is limited (Table 2),21 particularly given the risk for tardive dyskinesia associated with the typical antipsychotics haloperidol and chlorpromazine. Data support using the α-2 agonist clonidine to treat hyperactivity associated with ADHD22 and TD23 and an extended-release preparation of this medication is FDA-approved for the former but not the latter indication (an α-2A receptor subtype agonist, guanfacine, also is FDA-approved for ADHD in pediatric patients). Mr. C’s experience of robust, sustained reduction in obsessions, if not compulsions, after treatment with MAS is consistent with the few studies of stimulant use in ADHD with comorbid OCD.24,25
Effective treatment of ADHD may help Mr. C better access CBT strategies and thereby potentiate treatment of comorbid OCD.
Table 2
FDA-approved medications for ADHD, OCD, and TD
| Disorder | Medications |
|---|---|
| ADHD | Amphetamine (racemic), atomoxetine, chlorpromazine (hyperactivity), clonidine extended release, dexmethylphenidate, dextroamphetamine, guanfacine extended release, haloperidol (hyperactivity, second-line), lisdexamfetamine, methylphenidate (racemic) |
| OCD | Clomipramine, fluoxetine, fluvoxamine, paroxetine, sertraline |
| TD/Tourette’s syndrome | Haloperidol (Tourette’s), pimozide (Tourette’s) |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder; TD: tic disorder Source: Reference 21 | |
Related Resources
- Pliszka SR. Treating ADHD and comorbid disorders: psychosocial and psychopharmacological interventions. New York, NY: The Guilford Press; 2011.
- Pollak Y, Benarroch F, Kanengisser L, et al. Tourette syndrome-associated psychopathology: roles of comorbid attention-deficit hyperactivity disorder and obsessive-compulsive disorder. J Dev Behav Pediatr. 2009;30(5):413-419.
Drug Brand Names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin, Zyban
- Chlorpromazine • Thorazine
- Clomipramine • Anafranil
- Clonidine extended release • Kapvay
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guanfacine • Intuniv, Tenex
- Haloperidol • Haldol
- Lisdexamfetamine • Vyvanse
- Memantine • Namenda
- Methylphenidate • Methylin, Ritalin
- Modafinil • Provigil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: I. Development, use and reliability. Arch Gen Psych. 1989;46(11):1006-1011.
2. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: II. Validity. Arch Gen Psych. 1989;46(11):1012-1016.
3. Geller DA, Biederman J, Faraone S, et al. Re-examining comorbidity of obsessive compulsive and attention-deficit hyperactivity disorder using an empirically derived taxonomy. Eur Child Adolesc Psychiatry. 2004;13(2):83-91.
4. Freeman RD. Attention deficit hyperactivity disorder in the presence of Tourette syndrome. Neurol Clin. 1997;15(2):411-420.
5. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin North Am. 2006;29(2):353-370.
6. Eapen V, Fox-Hiley P, Banerjee S, et al. Clinical features and associated psychopathology in a Tourette syndrome cohort. Acta Neurol Scand. 2004;109(4):255-260.
7. Kano Y, Ohta M, Nagai Y, et al. Association between Tourette syndrome and comorbidities in Japan. Brain Dev. 2010;32(3):201-207.
8. Grados M, Riddle M. Do all obsessive-compulsive disorder subtypes respond to medication? Int Rev Psychiatry. 2008;20(2):189-193.
9. Lipkin PH, Goldstein IH, Adesman AR. Tics and dyskinesias associated with stimulant treatment in attention-deficit/hyperactivity disorder. Arch Pediatr Adolesc Med. 1994;148(8):859-861.
10. Castellanos FX, Giedd JN, Elia J, et al. Controlled stimulant treatment of ADHD and comorbid Tourette’s syndrome: effects of stimulant and dose. J Am Acad Child Adolesc Psychiatry. 1997;36(5):589-596.
11. Gadow K, Sverd J, Sprafkin J, et al. Long-term methylphenidate therapy in children with comorbid attention-deficit hyperactivity disorder and chronic multiple tic disorder. Arch Gen Psychiatry. 1999;56(4):330-333.
12. Gadow KD, Sverd J, Nolan EE, et al. Immediate-release methylphenidate for ADHD in children with comorbid chronic multiple tic disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):840-848.
13. Tourette’s Syndrome Study Group. Treatment of ADHD in children with tics: a randomized controlled trial. Neurology. 2002;58(4):527-536.
14. Lyon GJ, Samar SM, Conelea C, et al. Testing tic suppression: comparing the effects of dexmethylphenidate to no mediation in children and adolescents with attention-deficit/hyperactivity disorder and Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):283-289.
15. Gadow KD, Sverd J, Sprafkin J, et al. Efficacy of methylphenidate for attention-deficit hyperactivity disorder in children with tic disorder. Arch Gen Psychiatry. 1995;52(6):444-455.
16. Bloch MH, Panza KE, Landerso-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
17. McDougle CJ, Goodman WK, Price LH. Dopamine antagonists in tic-related and psychotic spectrum obsessive compulsive disorder. J Clin Psychiatry. 1994;55(suppl):24-31.
18. Wilens TE, Morrison NR. The intersection of attention-deficit/hyperactivity disorder and substance abuse. Curr Opin Psychiatry. 2011;24(4):280-285.
19. Kollins SH. A qualitative review of issues arising in the use of psycho-stimulant medications in patients with ADHD and co-morbid substance use disorders. Curr Med Res Opin. 2008;24(5):1345-1357.
20. Schubiner H. Substance abuse in patients with attention-deficit hyperactivity disorder: therapeutic implications. CNS Drugs. 2005;19(8):643-655.
21. Stahl SM. The prescriber’s guide. Stahl’s essential psychopharmacology. 3rd ed. New York NY: Cambridge University Press; 2009.
22. Jain R, Segal S, Kollins SH, et al. Clonidine extended-release tablets for pediatric patients with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(2):171-179.
23. Hedderick EF, Morris CM, Singer HS. Double-blind crossover study of clonidine and levetiracetam in Tourette syndrome. Pediatr Neurol. 2009;40(6):420-425.
24. Joffe RT, Swinson RP, Levitt AJ. Acute psychostimulant challenge in primary obsessive-compulsive disorder. J Clin Psychopharmacol. 1991;11(4):237-241.
25. Insel TR, Hamilton JA, Guttmacher LB, et al. D-amphetamine in obsessive-compulsive disorder. Psychopharmacology (Berl). 1983;80(3):231-235.
1. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: I. Development, use and reliability. Arch Gen Psych. 1989;46(11):1006-1011.
2. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale: II. Validity. Arch Gen Psych. 1989;46(11):1012-1016.
3. Geller DA, Biederman J, Faraone S, et al. Re-examining comorbidity of obsessive compulsive and attention-deficit hyperactivity disorder using an empirically derived taxonomy. Eur Child Adolesc Psychiatry. 2004;13(2):83-91.
4. Freeman RD. Attention deficit hyperactivity disorder in the presence of Tourette syndrome. Neurol Clin. 1997;15(2):411-420.
5. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin North Am. 2006;29(2):353-370.
6. Eapen V, Fox-Hiley P, Banerjee S, et al. Clinical features and associated psychopathology in a Tourette syndrome cohort. Acta Neurol Scand. 2004;109(4):255-260.
7. Kano Y, Ohta M, Nagai Y, et al. Association between Tourette syndrome and comorbidities in Japan. Brain Dev. 2010;32(3):201-207.
8. Grados M, Riddle M. Do all obsessive-compulsive disorder subtypes respond to medication? Int Rev Psychiatry. 2008;20(2):189-193.
9. Lipkin PH, Goldstein IH, Adesman AR. Tics and dyskinesias associated with stimulant treatment in attention-deficit/hyperactivity disorder. Arch Pediatr Adolesc Med. 1994;148(8):859-861.
10. Castellanos FX, Giedd JN, Elia J, et al. Controlled stimulant treatment of ADHD and comorbid Tourette’s syndrome: effects of stimulant and dose. J Am Acad Child Adolesc Psychiatry. 1997;36(5):589-596.
11. Gadow K, Sverd J, Sprafkin J, et al. Long-term methylphenidate therapy in children with comorbid attention-deficit hyperactivity disorder and chronic multiple tic disorder. Arch Gen Psychiatry. 1999;56(4):330-333.
12. Gadow KD, Sverd J, Nolan EE, et al. Immediate-release methylphenidate for ADHD in children with comorbid chronic multiple tic disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):840-848.
13. Tourette’s Syndrome Study Group. Treatment of ADHD in children with tics: a randomized controlled trial. Neurology. 2002;58(4):527-536.
14. Lyon GJ, Samar SM, Conelea C, et al. Testing tic suppression: comparing the effects of dexmethylphenidate to no mediation in children and adolescents with attention-deficit/hyperactivity disorder and Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):283-289.
15. Gadow KD, Sverd J, Sprafkin J, et al. Efficacy of methylphenidate for attention-deficit hyperactivity disorder in children with tic disorder. Arch Gen Psychiatry. 1995;52(6):444-455.
16. Bloch MH, Panza KE, Landerso-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
17. McDougle CJ, Goodman WK, Price LH. Dopamine antagonists in tic-related and psychotic spectrum obsessive compulsive disorder. J Clin Psychiatry. 1994;55(suppl):24-31.
18. Wilens TE, Morrison NR. The intersection of attention-deficit/hyperactivity disorder and substance abuse. Curr Opin Psychiatry. 2011;24(4):280-285.
19. Kollins SH. A qualitative review of issues arising in the use of psycho-stimulant medications in patients with ADHD and co-morbid substance use disorders. Curr Med Res Opin. 2008;24(5):1345-1357.
20. Schubiner H. Substance abuse in patients with attention-deficit hyperactivity disorder: therapeutic implications. CNS Drugs. 2005;19(8):643-655.
21. Stahl SM. The prescriber’s guide. Stahl’s essential psychopharmacology. 3rd ed. New York NY: Cambridge University Press; 2009.
22. Jain R, Segal S, Kollins SH, et al. Clonidine extended-release tablets for pediatric patients with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50(2):171-179.
23. Hedderick EF, Morris CM, Singer HS. Double-blind crossover study of clonidine and levetiracetam in Tourette syndrome. Pediatr Neurol. 2009;40(6):420-425.
24. Joffe RT, Swinson RP, Levitt AJ. Acute psychostimulant challenge in primary obsessive-compulsive disorder. J Clin Psychopharmacol. 1991;11(4):237-241.
25. Insel TR, Hamilton JA, Guttmacher LB, et al. D-amphetamine in obsessive-compulsive disorder. Psychopharmacology (Berl). 1983;80(3):231-235.
A paranoid, violent teenager
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Sleepless and paranoid
Ms. V, age 16, is referred to our psychiatric hospital from a juvenile detention center after she is charged with killing her sister with a hammer. She reports paranoid delusions, including believing that her sister was poisoning her food. Ms. V’s troubling behavior increased in the 6 months before the murder. She began to ask her mother to smell her food for possible poison. Her school grades dropped and she experienced decreased sleep and appetite. According to her mother, Ms. V’s insomnia worsened recently because of her paranoid thinking, which was evident when she noticed that her daughter slept with a hammer. Ms. V stopped socializing with her peers and no longer went to the gym.
Ms. V’s mother describes her daughter’s negative symptoms as consisting of social isolation and a flat affect. There was no evidence of auditory or visual hallucinations. After noticing the change in her daughter’s behavior, Ms. V’s mother attempted to schedule an appointment with a mental health professional, but there was a 2-month waiting list.
Ms. V cleaned her room before the murder, which was uncharacteristic of her routine behavior. On the day of the murder, Ms. V approached her sister while she was sleeping on the sofa and struck her on the head several times with a hammer. After the sister died, neighbors spotted Ms. V washing blood off her hands in their backyard with a sprinkler. Soaked in blood, she approached one of the neighbors and said that someone had been killed in the house. The neighbors called the police and Ms. V was arrested. She did not express remorse. She did not exhibit physical aggression toward others before the murder. Ms. V’s sense of entitlement and grandiosity persisted after the murder.
The authors’ observations
Paranoid delusions are fixed false beliefs with severe fears of others that may impair functioning at school or work, in personal relationships, and in other social dimensions. Paranoid thinking can have diverse presentations, ranging from social concerns such as fear of rejection to severe threat perceptions of people trying to cause substantial physical harm.1 Paranoid thoughts can be a result of misinterpretation of language, a personality disorder, anxiety, or psychosis.
Feelings of low self-esteem2 and anger1 may develop in a patient experiencing paranoid ideations. When anger begins to escalate, it may erupt into violent behavior. In Ms. V’s case, her paranoid ideations increased until she killed her younger sister. Ms. V’s case is similar to a mass shooting near Tucson, AZ on January 8, 2011 in that it possibly could have been prevented with earlier psychiatric intervention (Box).3-6
On January 8, 2011, a mass shooting occurred near Tucson, AZ that killed 6 and wounded 13. The suspect, 22-year-old Jared Lee Loughner, refused to cooperate with authorities by invoking his right to remain silent.3 Although the motives behind this crime remain undisclosed, mental illness appears to be a contributing factor.
Reports indicate that Mr. Loughner was abusing drugs and those close to him had noticed personality changes.4,5 The college he was attending advised Mr. Loughner to undergo a mental health evaluation, but he refused and dropped out of school.4,5 While in custody after the shooting, Mr. Loughner was diagnosed with paranoid schizophrenia, deemed incompetent to stand trial, and ordered to receive psychiatric treatment.6
This tragic mass shooting and similar incidents have led to questions regarding the adequacy of the mental health care infrastructure in United States. Experts suggest that this tragedy could have been prevented with more aggressive psychiatric prevention and intervention. Critical analysis of similar recent cases and expert opinions are needed to address this problem effectively.
EVALUATION: Remorseless
At admission, Ms. V’s affect is restricted and, at times, inappropriate. She is guarded about discussing the homicide but describes paranoid thoughts about her sister poisoning her. She is eager to learn if the police had found poison in her food. Her speech is soft with good articulation. Based on her presentation, her intelligence is average. She shows no evidence of remorse and is preoccupied with her sister poisoning her.
The Rorschach Inkblot Technique reveals positive evidence for a severe thought disorder. Ms. V’s thinking seems regressed. Ms. V’s medical workup, including MRI, electroencephalogram, and laboratory tests, are all within normal limits.
In the 5th grade, Ms. V’s primary care provider prescribed amphetamine and dextroamphetamine for attention-deficit/hyperactivity disorder, but she discontinued the drug after 1 year. Ms. V has never been hospitalized for psychiatric illness. She had no chronic medical conditions and no developmental delays.
Ms. V also has a history of periodic temper problems characterized by verbal aggression such as threatening the assistant principal at her school, and throwing her cellphone at her mother a few weeks before the murder, but no other aggressive episodes. Ms. V’s history does not suggest conduct disorder. She has no history of suicidal ideation or suicide attempts. Ms. V has used alcohol since age 15, but her mother reports that she was not a heavy or frequent user. Her last reported alcohol use was 10 days before the murder. A maternal uncle had been diagnosed with schizophrenia.
Before the murder, Ms. V lived with her sister and mother. Her parents were divorced. At age 9, Ms. V was sexually abused by a soccer coach; however, she denied symptoms of posttraumatic stress disorder related to the sexual abuse. She had no criminal history before the murder.
The authors’ observations
Based on Ms. V’s presentation and history, schizophrenia, paranoid type seems to be the most likely diagnosis because of her negative symptoms, including affective flattening, positive family history for schizophrenia, and paranoid delusions leading to dysfunction (Table).7 Delusional disorder seems less likely because Ms. V is young and has negative symptoms. Because she is generally healthy and her medical workup is negative, psychotic disorder due to a general medical condition is ruled out. She does not appear to be over-reporting, malingering, or exaggerating symptoms. In the context of psychosis, adolescent psychopathy does not seem likely even though there is evidence of grandiosity and a lack of remorse.
Table
DSM-IV-TR criteria for schizophrenia
A. Characteristic symptoms: ≥2 of the following, each present for a significant portion of time during a 1-month period:
|
| B. Social/occupational dysfunction: For a significant portion of the time since the onset of the disturbance, ≥1 major areas of functioning such as work, interpersonal relations, or self-care are markedly below the level achieved prior to the onset |
| C. Duration: Continuous signs of the disturbance persist for at least 6 months. This 6-month period must include at least 1 month of symptoms that meet Criterion A and may include periods of prodromal or residual symptoms |
| D. Schizoaffective and mood disorder exclusion: Schizoaffective disorder and mood disorder with psychotic features have been ruled out because either (1) no major depressive, manic, or mixed episodes have occurred concurrently with the active-phase symptoms; or (2) if mood episodes have occurred during active-phase symptoms, their total duration has been brief relative to the duration of the active and residual periods |
| E. Substance/general medical condition exclusion: The disturbance is not due to the direct physiological effects of a substance or a general medical condition |
| F. Relationship to a pervasive developmental disorder: If there is a history of autistic disorder or another pervasive developmental disorder, the additional diagnosis of schizophrenia is made only if prominent delusions or hallucinations are also present for at least 1 month |
| Diagnostic criteria for paranoid type: A type of schizophrenia in which the following criteria are met: A. Preoccupation with ≥1 delusions or frequent auditory hallucinations B. None of the following are prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect |
| Source: Reference 7 |
The authors’ observations
Various treatments can be used for paranoia with aggression, but the severity of the paranoia should be assessed before initiating treatment. Although categorizing paranoid ideations as mild, moderate, and severe is mainly a clinical judgment, Freeman et al1 have attempted to design a paranoia hierarchy from social concerns to severe threats. CBT8 and antipsychotic medication may help reduce mild to moderate paranoid delusions, particularly those associated with schizophrenia or mood disorders. For severe paranoia, hospitalization should carefully be considered.
When a patient exhibits moderate paranoia, the probability of progressing to severe symptoms or improving to mild symptoms depends on several variables. Pharmacologic treatment, family insight, and social support may be important variables in such circumstances. Psychoeducation for the family is vital.
In patients experiencing paranoia, violence may be prevented by proper assessment and treatment. The patient’s family should be educated about paranoid ideation and the need for treatment to improve symptoms and ensure safety. The long-term effects of untreated paranoia and types of treatment modalities available should be discussed with the family and the patient. During these teaching sessions, focus on improving the overall insight of the family and the patient about the psychotic illness to improve treatment adherence.9 This step may be challenging if the family is resistant to the patient receiving mental health treatment.
Gaining a detailed clinical history of a patient’s paranoia is important. A clinician should look for changes in behavior, such as the patient becoming quieter or more hostile, and impaired academic or social functioning. After gathering sufficient evidence contrary to the delusion, clinicians can help patients improve their reality testing.
Rule out medical and neurologic conditions that may be contributing to paranoia and aggression.
TREATMENT: Some improvement
Ms. V is started on risperidone, 1 mg/d, which leads to a partial response. She starts interacting more with staff and her peers on the unit, but her delusions of her sister poisioning her persist. Given the severity of the crime, Ms. V is sent to adult court, where she is found not guilty by reason of insanity and committed to a state hospital.
The authors’ observations
New-onset paranoia is a serious symptom that requires immediate evaluation and treatment. We recommend an approach presened in a flowchart (Figure) that highlights the importance of early intervention and aggressive treatment.
The MacArthur Violence Risk Assessment Study10 indicated that a “suspicious” attitude toward others can be a precipitating cause for increased violence in some cases. In light of ongoing controversy regarding the link between violence and mental illnesses such as schizophrenia,10-12 addressing an individual’s psychiatric illness early is preferable to prevent possible complications such as violent crimes. Because patients with paranoid ideations may have severely impaired ego control, they may be at risk for acting out aggressive and/or destructive urges. Therefore, new-onset paranoia should be thought of as a medical emergency similar to chest pain. Although accurately predicting and preventing violence may be impossible, in Ms. V’s case, earlier mental health treatment and intervention may have been able to prevent a murder.
Figure: Paranoia: A suggested approach to treatment
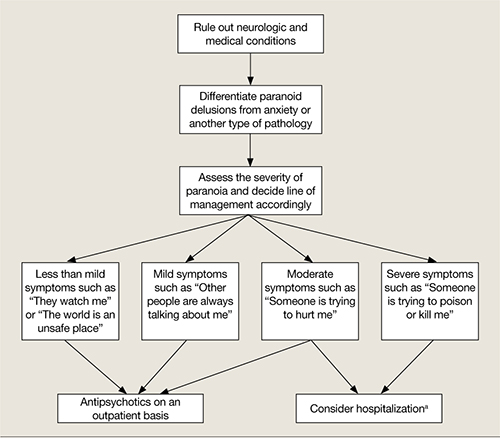
aBased on clinical judgment and extent of social support
Symptoms may become less severe or more severe (bidirectional). Strong social support has a positive effect on all levels and complements therapy. Regular counseling sessions and enhanced family insight about the patient’s paranoia helps strengthen social support
- Marneros A, Pillmann F, Wustmann T. Delusional disorders—are they simply paranoid schizophrenia? [published online ahead of print November 15, 2010]. Schizophr Bull. doi: 10.1093/schbul/sbq125.
Drug Brand Names
Amphetamine and dextroamphetamine • Adderall
Risperidone • Risperdal
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Personal and clinical details of this case were altered to maintain patient confidentiality.
1. Freeman D, Garety PA, Bebbington PE, et al. Psychological investigation of the structure of paranoia in a non-clinical population. Br J Psychiatry. 2005;186:427-435.
2. Kendler KS, Hays P. Paranoid psychosis (delusional disorder) and schizophrenia. A family history study. Arch Gen Psychiatry. 1981;38(5):547-551.
3. CNN Wire Staff. Police “actively pursuing” second person in Tucson shooting. CNN. http://us.cnn.com/2011/CRIME/01/08/arizona.shooting. Published January 9 2011. Accessed January 9, 2012.
4. Lipton E, Savage C, Shane S. Arizona suspect’s recent acts offer hints of alienation. The New York Times. January 8 2011:A8. http://www.nytimes.com/2011/01/09/us/politics/09shooter.html. Accessed January 10, 2012.
5. Berger J. Mental health warnings preceded rampage as Arizona gunman likely went untreated. http://www.foxnews.com/us/2011/01/10/mental-health-warnings-preceded-arizona-rampage-evidence-gunman-sought. Published January 10, 2011. Accessed January 11, 2012.
6. Lacey M. Suspect in shooting of Giffords ruled unfit for trial. The New York Times. May 25 2011:A1. http://www.nytimes.com/2011/05/26/us/26loughner.html. Accessed January 5, 2012.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
8. Turkington D, Kingdon D, Weiden PJ. Cognitive behavior therapy for schizophrenia. Am J Psychiatry. 2006;163(3):365-373.
9. Smith CM, Barzman DH, Pristach CA. Effect of patient and family insight on compliance of schizophrenic patients. J Clin Pharmacol. 1997;37(2):147-154.
10. Appelbaum PS, Robbins PC, Monahan J. Violence and delusions: data from the MacArthur Violence Risk Assessment Study. Am J Psychiatry. 2000;157(4):566-572.
11. Mullen PE. A reassessment of the link between mental disorder and violent behaviour and its implications for clinical practice. Aust N Z J Psychiatry. 1997;31(1):3-11.
12. Fazel S, Gulati G, Linsell L, et al. Schizophrenia and violence: systematic review and meta-analysis. PLoS Med. 2009;6(8):e1000120.-
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Sleepless and paranoid
Ms. V, age 16, is referred to our psychiatric hospital from a juvenile detention center after she is charged with killing her sister with a hammer. She reports paranoid delusions, including believing that her sister was poisoning her food. Ms. V’s troubling behavior increased in the 6 months before the murder. She began to ask her mother to smell her food for possible poison. Her school grades dropped and she experienced decreased sleep and appetite. According to her mother, Ms. V’s insomnia worsened recently because of her paranoid thinking, which was evident when she noticed that her daughter slept with a hammer. Ms. V stopped socializing with her peers and no longer went to the gym.
Ms. V’s mother describes her daughter’s negative symptoms as consisting of social isolation and a flat affect. There was no evidence of auditory or visual hallucinations. After noticing the change in her daughter’s behavior, Ms. V’s mother attempted to schedule an appointment with a mental health professional, but there was a 2-month waiting list.
Ms. V cleaned her room before the murder, which was uncharacteristic of her routine behavior. On the day of the murder, Ms. V approached her sister while she was sleeping on the sofa and struck her on the head several times with a hammer. After the sister died, neighbors spotted Ms. V washing blood off her hands in their backyard with a sprinkler. Soaked in blood, she approached one of the neighbors and said that someone had been killed in the house. The neighbors called the police and Ms. V was arrested. She did not express remorse. She did not exhibit physical aggression toward others before the murder. Ms. V’s sense of entitlement and grandiosity persisted after the murder.
The authors’ observations
Paranoid delusions are fixed false beliefs with severe fears of others that may impair functioning at school or work, in personal relationships, and in other social dimensions. Paranoid thinking can have diverse presentations, ranging from social concerns such as fear of rejection to severe threat perceptions of people trying to cause substantial physical harm.1 Paranoid thoughts can be a result of misinterpretation of language, a personality disorder, anxiety, or psychosis.
Feelings of low self-esteem2 and anger1 may develop in a patient experiencing paranoid ideations. When anger begins to escalate, it may erupt into violent behavior. In Ms. V’s case, her paranoid ideations increased until she killed her younger sister. Ms. V’s case is similar to a mass shooting near Tucson, AZ on January 8, 2011 in that it possibly could have been prevented with earlier psychiatric intervention (Box).3-6
On January 8, 2011, a mass shooting occurred near Tucson, AZ that killed 6 and wounded 13. The suspect, 22-year-old Jared Lee Loughner, refused to cooperate with authorities by invoking his right to remain silent.3 Although the motives behind this crime remain undisclosed, mental illness appears to be a contributing factor.
Reports indicate that Mr. Loughner was abusing drugs and those close to him had noticed personality changes.4,5 The college he was attending advised Mr. Loughner to undergo a mental health evaluation, but he refused and dropped out of school.4,5 While in custody after the shooting, Mr. Loughner was diagnosed with paranoid schizophrenia, deemed incompetent to stand trial, and ordered to receive psychiatric treatment.6
This tragic mass shooting and similar incidents have led to questions regarding the adequacy of the mental health care infrastructure in United States. Experts suggest that this tragedy could have been prevented with more aggressive psychiatric prevention and intervention. Critical analysis of similar recent cases and expert opinions are needed to address this problem effectively.
EVALUATION: Remorseless
At admission, Ms. V’s affect is restricted and, at times, inappropriate. She is guarded about discussing the homicide but describes paranoid thoughts about her sister poisoning her. She is eager to learn if the police had found poison in her food. Her speech is soft with good articulation. Based on her presentation, her intelligence is average. She shows no evidence of remorse and is preoccupied with her sister poisoning her.
The Rorschach Inkblot Technique reveals positive evidence for a severe thought disorder. Ms. V’s thinking seems regressed. Ms. V’s medical workup, including MRI, electroencephalogram, and laboratory tests, are all within normal limits.
In the 5th grade, Ms. V’s primary care provider prescribed amphetamine and dextroamphetamine for attention-deficit/hyperactivity disorder, but she discontinued the drug after 1 year. Ms. V has never been hospitalized for psychiatric illness. She had no chronic medical conditions and no developmental delays.
Ms. V also has a history of periodic temper problems characterized by verbal aggression such as threatening the assistant principal at her school, and throwing her cellphone at her mother a few weeks before the murder, but no other aggressive episodes. Ms. V’s history does not suggest conduct disorder. She has no history of suicidal ideation or suicide attempts. Ms. V has used alcohol since age 15, but her mother reports that she was not a heavy or frequent user. Her last reported alcohol use was 10 days before the murder. A maternal uncle had been diagnosed with schizophrenia.
Before the murder, Ms. V lived with her sister and mother. Her parents were divorced. At age 9, Ms. V was sexually abused by a soccer coach; however, she denied symptoms of posttraumatic stress disorder related to the sexual abuse. She had no criminal history before the murder.
The authors’ observations
Based on Ms. V’s presentation and history, schizophrenia, paranoid type seems to be the most likely diagnosis because of her negative symptoms, including affective flattening, positive family history for schizophrenia, and paranoid delusions leading to dysfunction (Table).7 Delusional disorder seems less likely because Ms. V is young and has negative symptoms. Because she is generally healthy and her medical workup is negative, psychotic disorder due to a general medical condition is ruled out. She does not appear to be over-reporting, malingering, or exaggerating symptoms. In the context of psychosis, adolescent psychopathy does not seem likely even though there is evidence of grandiosity and a lack of remorse.
Table
DSM-IV-TR criteria for schizophrenia
A. Characteristic symptoms: ≥2 of the following, each present for a significant portion of time during a 1-month period:
|
| B. Social/occupational dysfunction: For a significant portion of the time since the onset of the disturbance, ≥1 major areas of functioning such as work, interpersonal relations, or self-care are markedly below the level achieved prior to the onset |
| C. Duration: Continuous signs of the disturbance persist for at least 6 months. This 6-month period must include at least 1 month of symptoms that meet Criterion A and may include periods of prodromal or residual symptoms |
| D. Schizoaffective and mood disorder exclusion: Schizoaffective disorder and mood disorder with psychotic features have been ruled out because either (1) no major depressive, manic, or mixed episodes have occurred concurrently with the active-phase symptoms; or (2) if mood episodes have occurred during active-phase symptoms, their total duration has been brief relative to the duration of the active and residual periods |
| E. Substance/general medical condition exclusion: The disturbance is not due to the direct physiological effects of a substance or a general medical condition |
| F. Relationship to a pervasive developmental disorder: If there is a history of autistic disorder or another pervasive developmental disorder, the additional diagnosis of schizophrenia is made only if prominent delusions or hallucinations are also present for at least 1 month |
| Diagnostic criteria for paranoid type: A type of schizophrenia in which the following criteria are met: A. Preoccupation with ≥1 delusions or frequent auditory hallucinations B. None of the following are prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect |
| Source: Reference 7 |
The authors’ observations
Various treatments can be used for paranoia with aggression, but the severity of the paranoia should be assessed before initiating treatment. Although categorizing paranoid ideations as mild, moderate, and severe is mainly a clinical judgment, Freeman et al1 have attempted to design a paranoia hierarchy from social concerns to severe threats. CBT8 and antipsychotic medication may help reduce mild to moderate paranoid delusions, particularly those associated with schizophrenia or mood disorders. For severe paranoia, hospitalization should carefully be considered.
When a patient exhibits moderate paranoia, the probability of progressing to severe symptoms or improving to mild symptoms depends on several variables. Pharmacologic treatment, family insight, and social support may be important variables in such circumstances. Psychoeducation for the family is vital.
In patients experiencing paranoia, violence may be prevented by proper assessment and treatment. The patient’s family should be educated about paranoid ideation and the need for treatment to improve symptoms and ensure safety. The long-term effects of untreated paranoia and types of treatment modalities available should be discussed with the family and the patient. During these teaching sessions, focus on improving the overall insight of the family and the patient about the psychotic illness to improve treatment adherence.9 This step may be challenging if the family is resistant to the patient receiving mental health treatment.
Gaining a detailed clinical history of a patient’s paranoia is important. A clinician should look for changes in behavior, such as the patient becoming quieter or more hostile, and impaired academic or social functioning. After gathering sufficient evidence contrary to the delusion, clinicians can help patients improve their reality testing.
Rule out medical and neurologic conditions that may be contributing to paranoia and aggression.
TREATMENT: Some improvement
Ms. V is started on risperidone, 1 mg/d, which leads to a partial response. She starts interacting more with staff and her peers on the unit, but her delusions of her sister poisioning her persist. Given the severity of the crime, Ms. V is sent to adult court, where she is found not guilty by reason of insanity and committed to a state hospital.
The authors’ observations
New-onset paranoia is a serious symptom that requires immediate evaluation and treatment. We recommend an approach presened in a flowchart (Figure) that highlights the importance of early intervention and aggressive treatment.
The MacArthur Violence Risk Assessment Study10 indicated that a “suspicious” attitude toward others can be a precipitating cause for increased violence in some cases. In light of ongoing controversy regarding the link between violence and mental illnesses such as schizophrenia,10-12 addressing an individual’s psychiatric illness early is preferable to prevent possible complications such as violent crimes. Because patients with paranoid ideations may have severely impaired ego control, they may be at risk for acting out aggressive and/or destructive urges. Therefore, new-onset paranoia should be thought of as a medical emergency similar to chest pain. Although accurately predicting and preventing violence may be impossible, in Ms. V’s case, earlier mental health treatment and intervention may have been able to prevent a murder.
Figure: Paranoia: A suggested approach to treatment
aBased on clinical judgment and extent of social support
Symptoms may become less severe or more severe (bidirectional). Strong social support has a positive effect on all levels and complements therapy. Regular counseling sessions and enhanced family insight about the patient’s paranoia helps strengthen social support
- Marneros A, Pillmann F, Wustmann T. Delusional disorders—are they simply paranoid schizophrenia? [published online ahead of print November 15, 2010]. Schizophr Bull. doi: 10.1093/schbul/sbq125.
Drug Brand Names
Amphetamine and dextroamphetamine • Adderall
Risperidone • Risperdal
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Personal and clinical details of this case were altered to maintain patient confidentiality.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Sleepless and paranoid
Ms. V, age 16, is referred to our psychiatric hospital from a juvenile detention center after she is charged with killing her sister with a hammer. She reports paranoid delusions, including believing that her sister was poisoning her food. Ms. V’s troubling behavior increased in the 6 months before the murder. She began to ask her mother to smell her food for possible poison. Her school grades dropped and she experienced decreased sleep and appetite. According to her mother, Ms. V’s insomnia worsened recently because of her paranoid thinking, which was evident when she noticed that her daughter slept with a hammer. Ms. V stopped socializing with her peers and no longer went to the gym.
Ms. V’s mother describes her daughter’s negative symptoms as consisting of social isolation and a flat affect. There was no evidence of auditory or visual hallucinations. After noticing the change in her daughter’s behavior, Ms. V’s mother attempted to schedule an appointment with a mental health professional, but there was a 2-month waiting list.
Ms. V cleaned her room before the murder, which was uncharacteristic of her routine behavior. On the day of the murder, Ms. V approached her sister while she was sleeping on the sofa and struck her on the head several times with a hammer. After the sister died, neighbors spotted Ms. V washing blood off her hands in their backyard with a sprinkler. Soaked in blood, she approached one of the neighbors and said that someone had been killed in the house. The neighbors called the police and Ms. V was arrested. She did not express remorse. She did not exhibit physical aggression toward others before the murder. Ms. V’s sense of entitlement and grandiosity persisted after the murder.
The authors’ observations
Paranoid delusions are fixed false beliefs with severe fears of others that may impair functioning at school or work, in personal relationships, and in other social dimensions. Paranoid thinking can have diverse presentations, ranging from social concerns such as fear of rejection to severe threat perceptions of people trying to cause substantial physical harm.1 Paranoid thoughts can be a result of misinterpretation of language, a personality disorder, anxiety, or psychosis.
Feelings of low self-esteem2 and anger1 may develop in a patient experiencing paranoid ideations. When anger begins to escalate, it may erupt into violent behavior. In Ms. V’s case, her paranoid ideations increased until she killed her younger sister. Ms. V’s case is similar to a mass shooting near Tucson, AZ on January 8, 2011 in that it possibly could have been prevented with earlier psychiatric intervention (Box).3-6
On January 8, 2011, a mass shooting occurred near Tucson, AZ that killed 6 and wounded 13. The suspect, 22-year-old Jared Lee Loughner, refused to cooperate with authorities by invoking his right to remain silent.3 Although the motives behind this crime remain undisclosed, mental illness appears to be a contributing factor.
Reports indicate that Mr. Loughner was abusing drugs and those close to him had noticed personality changes.4,5 The college he was attending advised Mr. Loughner to undergo a mental health evaluation, but he refused and dropped out of school.4,5 While in custody after the shooting, Mr. Loughner was diagnosed with paranoid schizophrenia, deemed incompetent to stand trial, and ordered to receive psychiatric treatment.6
This tragic mass shooting and similar incidents have led to questions regarding the adequacy of the mental health care infrastructure in United States. Experts suggest that this tragedy could have been prevented with more aggressive psychiatric prevention and intervention. Critical analysis of similar recent cases and expert opinions are needed to address this problem effectively.
EVALUATION: Remorseless
At admission, Ms. V’s affect is restricted and, at times, inappropriate. She is guarded about discussing the homicide but describes paranoid thoughts about her sister poisoning her. She is eager to learn if the police had found poison in her food. Her speech is soft with good articulation. Based on her presentation, her intelligence is average. She shows no evidence of remorse and is preoccupied with her sister poisoning her.
The Rorschach Inkblot Technique reveals positive evidence for a severe thought disorder. Ms. V’s thinking seems regressed. Ms. V’s medical workup, including MRI, electroencephalogram, and laboratory tests, are all within normal limits.
In the 5th grade, Ms. V’s primary care provider prescribed amphetamine and dextroamphetamine for attention-deficit/hyperactivity disorder, but she discontinued the drug after 1 year. Ms. V has never been hospitalized for psychiatric illness. She had no chronic medical conditions and no developmental delays.
Ms. V also has a history of periodic temper problems characterized by verbal aggression such as threatening the assistant principal at her school, and throwing her cellphone at her mother a few weeks before the murder, but no other aggressive episodes. Ms. V’s history does not suggest conduct disorder. She has no history of suicidal ideation or suicide attempts. Ms. V has used alcohol since age 15, but her mother reports that she was not a heavy or frequent user. Her last reported alcohol use was 10 days before the murder. A maternal uncle had been diagnosed with schizophrenia.
Before the murder, Ms. V lived with her sister and mother. Her parents were divorced. At age 9, Ms. V was sexually abused by a soccer coach; however, she denied symptoms of posttraumatic stress disorder related to the sexual abuse. She had no criminal history before the murder.
The authors’ observations
Based on Ms. V’s presentation and history, schizophrenia, paranoid type seems to be the most likely diagnosis because of her negative symptoms, including affective flattening, positive family history for schizophrenia, and paranoid delusions leading to dysfunction (Table).7 Delusional disorder seems less likely because Ms. V is young and has negative symptoms. Because she is generally healthy and her medical workup is negative, psychotic disorder due to a general medical condition is ruled out. She does not appear to be over-reporting, malingering, or exaggerating symptoms. In the context of psychosis, adolescent psychopathy does not seem likely even though there is evidence of grandiosity and a lack of remorse.
Table
DSM-IV-TR criteria for schizophrenia
A. Characteristic symptoms: ≥2 of the following, each present for a significant portion of time during a 1-month period:
|
| B. Social/occupational dysfunction: For a significant portion of the time since the onset of the disturbance, ≥1 major areas of functioning such as work, interpersonal relations, or self-care are markedly below the level achieved prior to the onset |
| C. Duration: Continuous signs of the disturbance persist for at least 6 months. This 6-month period must include at least 1 month of symptoms that meet Criterion A and may include periods of prodromal or residual symptoms |
| D. Schizoaffective and mood disorder exclusion: Schizoaffective disorder and mood disorder with psychotic features have been ruled out because either (1) no major depressive, manic, or mixed episodes have occurred concurrently with the active-phase symptoms; or (2) if mood episodes have occurred during active-phase symptoms, their total duration has been brief relative to the duration of the active and residual periods |
| E. Substance/general medical condition exclusion: The disturbance is not due to the direct physiological effects of a substance or a general medical condition |
| F. Relationship to a pervasive developmental disorder: If there is a history of autistic disorder or another pervasive developmental disorder, the additional diagnosis of schizophrenia is made only if prominent delusions or hallucinations are also present for at least 1 month |
| Diagnostic criteria for paranoid type: A type of schizophrenia in which the following criteria are met: A. Preoccupation with ≥1 delusions or frequent auditory hallucinations B. None of the following are prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect |
| Source: Reference 7 |
The authors’ observations
Various treatments can be used for paranoia with aggression, but the severity of the paranoia should be assessed before initiating treatment. Although categorizing paranoid ideations as mild, moderate, and severe is mainly a clinical judgment, Freeman et al1 have attempted to design a paranoia hierarchy from social concerns to severe threats. CBT8 and antipsychotic medication may help reduce mild to moderate paranoid delusions, particularly those associated with schizophrenia or mood disorders. For severe paranoia, hospitalization should carefully be considered.
When a patient exhibits moderate paranoia, the probability of progressing to severe symptoms or improving to mild symptoms depends on several variables. Pharmacologic treatment, family insight, and social support may be important variables in such circumstances. Psychoeducation for the family is vital.
In patients experiencing paranoia, violence may be prevented by proper assessment and treatment. The patient’s family should be educated about paranoid ideation and the need for treatment to improve symptoms and ensure safety. The long-term effects of untreated paranoia and types of treatment modalities available should be discussed with the family and the patient. During these teaching sessions, focus on improving the overall insight of the family and the patient about the psychotic illness to improve treatment adherence.9 This step may be challenging if the family is resistant to the patient receiving mental health treatment.
Gaining a detailed clinical history of a patient’s paranoia is important. A clinician should look for changes in behavior, such as the patient becoming quieter or more hostile, and impaired academic or social functioning. After gathering sufficient evidence contrary to the delusion, clinicians can help patients improve their reality testing.
Rule out medical and neurologic conditions that may be contributing to paranoia and aggression.
TREATMENT: Some improvement
Ms. V is started on risperidone, 1 mg/d, which leads to a partial response. She starts interacting more with staff and her peers on the unit, but her delusions of her sister poisioning her persist. Given the severity of the crime, Ms. V is sent to adult court, where she is found not guilty by reason of insanity and committed to a state hospital.
The authors’ observations
New-onset paranoia is a serious symptom that requires immediate evaluation and treatment. We recommend an approach presened in a flowchart (Figure) that highlights the importance of early intervention and aggressive treatment.
The MacArthur Violence Risk Assessment Study10 indicated that a “suspicious” attitude toward others can be a precipitating cause for increased violence in some cases. In light of ongoing controversy regarding the link between violence and mental illnesses such as schizophrenia,10-12 addressing an individual’s psychiatric illness early is preferable to prevent possible complications such as violent crimes. Because patients with paranoid ideations may have severely impaired ego control, they may be at risk for acting out aggressive and/or destructive urges. Therefore, new-onset paranoia should be thought of as a medical emergency similar to chest pain. Although accurately predicting and preventing violence may be impossible, in Ms. V’s case, earlier mental health treatment and intervention may have been able to prevent a murder.
Figure: Paranoia: A suggested approach to treatment
aBased on clinical judgment and extent of social support
Symptoms may become less severe or more severe (bidirectional). Strong social support has a positive effect on all levels and complements therapy. Regular counseling sessions and enhanced family insight about the patient’s paranoia helps strengthen social support
- Marneros A, Pillmann F, Wustmann T. Delusional disorders—are they simply paranoid schizophrenia? [published online ahead of print November 15, 2010]. Schizophr Bull. doi: 10.1093/schbul/sbq125.
Drug Brand Names
Amphetamine and dextroamphetamine • Adderall
Risperidone • Risperdal
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Personal and clinical details of this case were altered to maintain patient confidentiality.
1. Freeman D, Garety PA, Bebbington PE, et al. Psychological investigation of the structure of paranoia in a non-clinical population. Br J Psychiatry. 2005;186:427-435.
2. Kendler KS, Hays P. Paranoid psychosis (delusional disorder) and schizophrenia. A family history study. Arch Gen Psychiatry. 1981;38(5):547-551.
3. CNN Wire Staff. Police “actively pursuing” second person in Tucson shooting. CNN. http://us.cnn.com/2011/CRIME/01/08/arizona.shooting. Published January 9 2011. Accessed January 9, 2012.
4. Lipton E, Savage C, Shane S. Arizona suspect’s recent acts offer hints of alienation. The New York Times. January 8 2011:A8. http://www.nytimes.com/2011/01/09/us/politics/09shooter.html. Accessed January 10, 2012.
5. Berger J. Mental health warnings preceded rampage as Arizona gunman likely went untreated. http://www.foxnews.com/us/2011/01/10/mental-health-warnings-preceded-arizona-rampage-evidence-gunman-sought. Published January 10, 2011. Accessed January 11, 2012.
6. Lacey M. Suspect in shooting of Giffords ruled unfit for trial. The New York Times. May 25 2011:A1. http://www.nytimes.com/2011/05/26/us/26loughner.html. Accessed January 5, 2012.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
8. Turkington D, Kingdon D, Weiden PJ. Cognitive behavior therapy for schizophrenia. Am J Psychiatry. 2006;163(3):365-373.
9. Smith CM, Barzman DH, Pristach CA. Effect of patient and family insight on compliance of schizophrenic patients. J Clin Pharmacol. 1997;37(2):147-154.
10. Appelbaum PS, Robbins PC, Monahan J. Violence and delusions: data from the MacArthur Violence Risk Assessment Study. Am J Psychiatry. 2000;157(4):566-572.
11. Mullen PE. A reassessment of the link between mental disorder and violent behaviour and its implications for clinical practice. Aust N Z J Psychiatry. 1997;31(1):3-11.
12. Fazel S, Gulati G, Linsell L, et al. Schizophrenia and violence: systematic review and meta-analysis. PLoS Med. 2009;6(8):e1000120.-
1. Freeman D, Garety PA, Bebbington PE, et al. Psychological investigation of the structure of paranoia in a non-clinical population. Br J Psychiatry. 2005;186:427-435.
2. Kendler KS, Hays P. Paranoid psychosis (delusional disorder) and schizophrenia. A family history study. Arch Gen Psychiatry. 1981;38(5):547-551.
3. CNN Wire Staff. Police “actively pursuing” second person in Tucson shooting. CNN. http://us.cnn.com/2011/CRIME/01/08/arizona.shooting. Published January 9 2011. Accessed January 9, 2012.
4. Lipton E, Savage C, Shane S. Arizona suspect’s recent acts offer hints of alienation. The New York Times. January 8 2011:A8. http://www.nytimes.com/2011/01/09/us/politics/09shooter.html. Accessed January 10, 2012.
5. Berger J. Mental health warnings preceded rampage as Arizona gunman likely went untreated. http://www.foxnews.com/us/2011/01/10/mental-health-warnings-preceded-arizona-rampage-evidence-gunman-sought. Published January 10, 2011. Accessed January 11, 2012.
6. Lacey M. Suspect in shooting of Giffords ruled unfit for trial. The New York Times. May 25 2011:A1. http://www.nytimes.com/2011/05/26/us/26loughner.html. Accessed January 5, 2012.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
8. Turkington D, Kingdon D, Weiden PJ. Cognitive behavior therapy for schizophrenia. Am J Psychiatry. 2006;163(3):365-373.
9. Smith CM, Barzman DH, Pristach CA. Effect of patient and family insight on compliance of schizophrenic patients. J Clin Pharmacol. 1997;37(2):147-154.
10. Appelbaum PS, Robbins PC, Monahan J. Violence and delusions: data from the MacArthur Violence Risk Assessment Study. Am J Psychiatry. 2000;157(4):566-572.
11. Mullen PE. A reassessment of the link between mental disorder and violent behaviour and its implications for clinical practice. Aust N Z J Psychiatry. 1997;31(1):3-11.
12. Fazel S, Gulati G, Linsell L, et al. Schizophrenia and violence: systematic review and meta-analysis. PLoS Med. 2009;6(8):e1000120.-
The delirious substance abuser
CASE: Hurt and confused
Emergency medical services (EMS) are called to Ms. K’s apartment after her roommate found her lying on the floor moaning. The roommate tells EMS that Ms. K, age 29, appeared confused and was slurring her words, and reports that this change in her awareness progressed rapidly over a few hours. EMS personnel find that Ms. K has multiple contusions on her arms and face, which they presume to be self-inflicted. A marijuana pipe is discovered at Ms. K’s apartment.
In the emergency room (ER), Ms. K is inattentive and has difficulty following simple commands. Her speech is mumbled and her thoughts are disorganized. She displays psychomotor restlessness in the form of combativeness. Ms. K cannot provide meaningful historical data and is disoriented to place and time. The ER staff requests a psychiatric consultation.
Family members reveal that Ms. K has no preexisting medical conditions, is not taking prescription medications, but has a history of substance abuse (sporadic cocaine and cannabis use). Her family is unaware of recent substance use.
Physical examination reveals tachycardia (heart rate 110 to 120 beats per minute), hypotension (blood pressure 78/49 mm Hg), hypothermia (temperature 88ºF), and peripheral pulse oximetry of 84%. Her pupils are dilated and reactive to light; no conjunctival injection is noted. Her lung fields are clear on auscultation, but she is noted to have a rapid, irregular heartbeat. The abdomen is positive for bowel sounds, soft on palpation, and without any repositioning or notable overt signs of tenderness. Ms. K’s toes show purple discoloration with poor capillary refill. The dorsalis pedis pulses are reported to be 1+ bilaterally; however, the remainder of the arterial pulse examination is normal.
Her sodium, potassium, and chloride values are normal, but she has an abnormal anion gap (28.1 mEq/L), blood urea nitrogen (53 mg/dL), creatinine (2.9 mg/dL), creatine kinase (10,857 U/L), creatine kinase MB (432.6 ng/mL), and hyperglycemia (glucose 425 mg/dL). Arterial blood gas reveals hypoxia (Po2 of 55 mm Hg), with metabolic acidosis (sodium bicarbonate 10 with compensatory Pco2 of 33 mm Hg). Her urine is cloudy, positive for protein, ketones, hemoglobin, and glucose. She is thought to have a high anion gap acidosis related to dehydration, lactic acidosis (lactic acid 20 mEq/L), and hyperglycemia. Urine toxicology is positive for cannabinoids; ethylene glycol and methanol screen negatively, which rules these out as potential contributors to her high anion gap acidosis.
Ms. K is intubated and IV fluids are initiated for rhabdomyolysis and acute renal failure. Dialysis is implemented on a short-term basis. Her mental state improves gradually over 3 days.
The authors’ observations
Based on the abrupt onset of inattention and confusion, disorganized speech, memory impairments, and psychomotor agitation, we made an initial diagnosis of delirium; however, the precise etiology remained unclear. DSM-IV-TR diagnostic criteria for delirium are described in Table 1. Although delirium due to multiple etiologies does not have a DSM-IV-TR coding designation, we speculated that multiple causes contributed to Ms. K’s presentation. Acute renal failure secondary to dehydration as well as rhabdomyolysis, hypoxia, and hyperglycemia were implicated as general medical conditions etiologically linked to delirium. Because Ms. K has no preexisting medical conditions and her roommate and family stated she had a history of substance abuse, we also considered a presumptive diagnosis of substance-induced delirium. The medical team speculated that, based on information provided by her family, Ms. K may have had a seizure or may have fallen, which would account for her multiple contusions, and could have led to muscle injury and breakdown and the resultant rhabdomyolysis.
The possibility of cannabinoid-induced delirium has been reported, albeit rarely.1-3 However, Ms. K’s presentation—hypothermia, variable heart rate, lack of dry mucous membranes—was not consistent with significant anticholinergic toxicity or cannabinoid intoxication (Table 2).
By contrast, cocaine-induced delirium has been reported and initially appeared to be a plausible cause of Ms. K’s symptoms (Table 2). Delirium related to excess ingestion of cocaine may be related to the drug’s secondary effects resulting in rhabdomyolysis and renal dysfunction.4-6 Although several mechanisms underlying this relationship have been proposed, no single specific mechanism has been identified. The basis for cocaine ingestion and the resultant metabolic and renal effects, as observed in Ms. K’s case, likely are multifactorial. Mechanisms of the rhabdomyolysis might include:
- blockade of synaptic catecholamine reuptake and induction of adrenergic agonism, resulting in vasoconstriction and ischemia and leading to muscle damage
- cocaine-induced seizures and/or prolonged unconsciousness, leading to muscle compression and breakdown of muscle tissue
- a period of exertion induced by cocaine, precipitating an excited delirium and associated rhabdomyolysis
- a surge in dopamine concentrations, similar to neuroleptic malignant syndrome, precipitates hyperthermia, muscle rigidity, and psychomotor agitation, disrupting neuromuscular homeostasis and leading to rhabdomyolysis.
We were uncertain about the plausibility that acute cocaine intoxication caused Ms. K’s medical sequelae, in light of her toxicology findings. If cocaine use was the inciting event, and because the delirium reportedly had developed over several hours, we would expect cocaine to be detected in the toxicology screen. However, it was not detected. Cocaine can remain detectable in urine for 2 to 4 days,7 which raised our speculation that remote cocaine abuse could account for Ms. K’s current presentation and the timeline the roommate initially relayed to EMS personnel was inaccurate. We needed to clarify the timeline and progression of Ms. K’s symptoms with the roommate. In addition, we suggested to the medical team that alternative substances of abuse could be causing Ms. K’s symptoms and the roommate might be the only person who could unveil this possibility.
Table 1
DSM-IV-TR criteria for delirium due to multiple etiologies
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbances) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the delirium has >1 etiology (eg, >1 etiological general medical condition, a general medical condition plus substance intoxication or medication side effect) |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
Table 2
Diagnostic criteria for cannabis and cocaine intoxication
| Diagnostic criteria | Cannabis intoxication | Cocaine intoxication |
|---|---|---|
| Recurrent use | + | + |
| Symptom onset | During or shortly after use | During or shortly after use |
| Behavioral changes | Impaired motor coordination | Hypervigilance, stereotyped behaviors |
| Psychological changes | Euphoria, anxiety, sensation of slowed time, social withdrawal, impaired judgment | Euphoria, anxiety, tension, anger, changes in sociability, interpersonal sensitivity, impaired social or occupational functioning |
| Associated criteria (≥2) | Conjunctival injection, increased appetite, dry mouth, tachycardia | Tachycardia or bradycardia, papillary dilation, elevated or lowered blood pressure, chills/perspiration, nausea/vomiting, evidence of weight loss, psychomotor changes, muscular weakness, chest pain, cardiac arrhythmias, seizure, dyskinesia, dystonia, delirium, coma |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | ||
HISTORY: Unknown substance
Ms. K’s roommate is contacted for supplemental history. The roommate reports that recently he observed Ms. K “snorting” a brown/tan-colored substance. He had not seen her use this substance previously, and when he asked her what it was, she reportedly said that it was “PeeVee” (also called “bath salts”) purchased over the Internet.
The authors’ observations
MDPV is a novel chemical compound that is used as a recreational drug (Table 3).8 It commonly is acquired from Internet sources and sold as “bath salts.” Its use first emerged in approximately 2004, and its popularity has been increasing because of its easy availability and relatively low cost.9 The American Association of Poison Control Centers received 302 calls related to MDPV toxicity in 2010 and 5,625 calls related to MDPV use between January 1 and October 31, 2011.10,11
MDPV has psychoactive properties, with stimulant effects acting as a norepinephrine-dopamine reuptake inhibitor.8,9,12 When snorted, ingested orally, or inserted rectally, the agent produces effects comparable to cocaine or psychostimulants such as methylphenidate or dextroamphetamine.
Acute effects of MDPV include heightened alertness, diminished need for sleep, hyperarousal, and euphoria.8,9 These symptoms often are accompanied by increases in heart rate and blood pressure, sweating, and peripheral vasoconstriction. Individuals may abuse MDPV to acquire sustained attention, reduce their need for sleep, or for aphrodisiac effects. In many cases, anxiety and irritability can accompany the desired euphoric effects. For some, the euphoric effects can be superseded by anxiety or agitation. Mood and attention effects are estimated to last 3 to 4 hours; however, tachycardia and hypertension can persist for 6 to 8 hours.
MDPV use can trigger cravings and lead to binging. Euphoric stimulation with MDPV can become dysphoric as the dose and duration of use increase. Extended use has been associated with agitation, irritability, aggression, panic and marked anxiety, psychosis, and delirium.8,9 Anxiety can range from mild dysphoric stimulation to extreme panic-like states. In moderate forms, a state of sympathetic discharge can occur, producing physiologic effects resembling panic attacks, including hypertension, tachycardia, sweating, and peripheral vasoconstriction. In more severe cases, users may experience a feeling of impending doom, marked distress, and frank psychosis. Patients may experience disorientation and unsystematized paranoid delusions. Case reports of intoxication have described self-injurious behaviors, such as cutting, which may account for the contusions observed on Ms. K’s face and arms. Increasingly, MDPV use has resulted in ER presentations with patients manifesting abrupt onset confusion, anxiety, and self-injurious behaviors.
The mechanisms underlying MDPV-induced delirium have not been definitively identified. Given the similarities in mechanism of action between MDPV and cocaine, causes for delirium related to MDPV are similarly presumed to be multifactorial. The course of delirium associated with MDPV intoxication is self-limited and requires supportive measures.8,9
Suspect MDPV abuse in patients who present with signs or symptoms of stimulant intoxication but have a negative toxicology screen for cocaine and other psychostimulants. MDPV is not detected on routine toxicology assessments; however, it can be identified through laboratories with gas chromatography/mass spectroscopy capabilities. However, the time needed to obtain the results may exceed the clinical course of the patient’s delirium. One of the limitations in Ms. K’s case was the lack of gas chromatography/mass spectroscopy to confirm MDPV ingestion. Ms. K’s roommate could not locate any unused brown powder within their apartment to bring in for laboratory investigations. Recently, screening assessments for MDPV have become commercially available (see Related Resources).
Table 3
Overview of MDPV features
| Chemical name | 3,4-methylenedioxypyrovalerone |
| Popular names | MDPV, PV, PeeVee, Super coke, Magic |
| Sources | Sold as “bath salts” by Internet sources, “head shops,” and gas stations |
| Mode of use | Oral, snorting, smoking, rectal insertion, intravenous |
| Acute effects | Increased energy, perception of heightened alertness/attention, aphrodisiac properties, increased sociability |
| Adverse psychological effects | Anxiety (panic attacks), irritability, agitation, confusion, suicidal ideations, visual distortions |
| Adverse physical effects | Insomnia/overstimulation, bruxism, muscle twitching, pupil dilation/blurred vision, anorexia, headache, nausea/vomiting, hyperthermia, irregular heart beat, tachycardia, dyspnea, fatigue |
| Effects of protracted use | Dysphoria, depression, anhedonia |
| LD50 | Unknown |
| LD50: lethal dose; MDPV: methylenedioxypyrovalerone Source: Reference 8 | |
OUTCOME: Referral to treatment
Dialysis is discontinued within 1 day of hospitalization. Ms. K’s peripheral arterial perfusion improves, as does her thermoregulatory status. Her mental status improvements coincide with improvements in her physical and metabolic status.
Ms. K is able to sustain attention when speaking with interviewers. She is aware of her surroundings and is no longer distracted by extraneous stimuli. Her speech is articulate and her thoughts are linear. There is no evidence of any residual thought disorganization, delusions, or hallucinations.
Initially, Ms. K is reluctant to acknowledge her substance use, but eventually, she concedes to acquiring a stimulant from an Internet source and abusing it in undetermined amounts. She had no experience with using MDPV and did not know how to avoid ingesting dangerous amounts. We educate Ms. K about the dangers she faced during this hospitalization and the potential life-threatening outcomes. She is amenable to pursuing outpatient substance abuse treatment. Her roommate is enlisted to facilitate her follow-up with this treatment.
The authors’ observations
Managing MDPV toxicity presents a diagnostic dilemma for medical personnel and psychiatrists when evaluating and managing acute delirium. MDPV ingestion may go unrecognized in clinical settings because toxicology assessments for it are not readily available and patients’ historical information may be unreliable.
Because of the seriousness of sequelae associated with MDPV use, state and federal agencies have intervened. Until recently, bath salts did not have a controlled substance designation. In October 2011, the US Drug Enforcement Administration (DEA) ruled to make MDPV a controlled substance for 1 year, with the possibility of a 6-month extension.13 Although this ruling is temporary, it makes possession, sale, or distribution of these chemicals, or the products that contain them, illegal in the United States. In the interim, the DEA and the US Department of Health and Human Services will determine whether MDPV should remain a controlled substance.
- American Screening Corp. (MDPV screening). www.americanscreeningcorp.com.
- U.S. Drug Enforcement Administration. 3, 4-Methylenedioxypyrovalerone (MDPV). www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf.
- Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones [published online ahead of print November 23, 2011]. J Med Toxicol. doi: 10.1007/s13181-011-0193-z.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. André C, Jaber-Filho JA, Bento RM, et al. Delirium following ingestion of marijuana in chocolate cookies. CNS Spectr. 2006;11(4):262-264.
2. Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38(1):1-20.
3. Meyer ME. Psychiatric consequences of marijuana use: the state of the evidence. In: Tinklenberg JR ed. Marijuana and health hazards: methodologic issues in current research. New York, NY: Academic Press; 1975:33–152.
4. Ruttenber AJ, Lawler-Heavner J, Yin M, et al. Fatal excited delirium following cocaine use: epidemiologic findings provide new evidence for mechanisms of cocaine toxicity. J Forensic Sci. 1997;42(1):25-31.
5. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20(2):120-127.
6. Singhal PC, Rubin RB, Peters A, et al. Rhabdomyolysis and acute renal failure associated with cocaine abuse. J Toxicol Clin Toxicol. 1990;28(3):321-330.
7. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83(1):66-76.
8. Psychonaut WebMapping Research Group. MDPV report. London United Kingdom: Institute of Psychiatry, King’s College. http://www.psychonautproject.eu/documents/reports/MDPV.pdf. Accessed November 23, 2011.
9. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
10. American Association of Poison Control Centers. Bath salts data. http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%2011.03.2011.pdf. Updated November 3 2011. Accessed November 23, 2011.
11. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan November 13, 2010-March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(19):624-627.
12. Westphal F, Junge T, Rösner P, et al. Mass and NMR spectroscopic characterization of 3, 4-methylenedioxypyrovalerone: a designer drug with α-pyrrolidinophenone structure. Forensic Sci Int. 2009;190(1-3):1-8.
13. U.S. Drug Enforcement Administration. Chemicals used in “bath salts” now under federal control and regulation. http://www.justice.gov/dea/pubs/pressrel/pr102111.html. Accessed November 23, 2011.
CASE: Hurt and confused
Emergency medical services (EMS) are called to Ms. K’s apartment after her roommate found her lying on the floor moaning. The roommate tells EMS that Ms. K, age 29, appeared confused and was slurring her words, and reports that this change in her awareness progressed rapidly over a few hours. EMS personnel find that Ms. K has multiple contusions on her arms and face, which they presume to be self-inflicted. A marijuana pipe is discovered at Ms. K’s apartment.
In the emergency room (ER), Ms. K is inattentive and has difficulty following simple commands. Her speech is mumbled and her thoughts are disorganized. She displays psychomotor restlessness in the form of combativeness. Ms. K cannot provide meaningful historical data and is disoriented to place and time. The ER staff requests a psychiatric consultation.
Family members reveal that Ms. K has no preexisting medical conditions, is not taking prescription medications, but has a history of substance abuse (sporadic cocaine and cannabis use). Her family is unaware of recent substance use.
Physical examination reveals tachycardia (heart rate 110 to 120 beats per minute), hypotension (blood pressure 78/49 mm Hg), hypothermia (temperature 88ºF), and peripheral pulse oximetry of 84%. Her pupils are dilated and reactive to light; no conjunctival injection is noted. Her lung fields are clear on auscultation, but she is noted to have a rapid, irregular heartbeat. The abdomen is positive for bowel sounds, soft on palpation, and without any repositioning or notable overt signs of tenderness. Ms. K’s toes show purple discoloration with poor capillary refill. The dorsalis pedis pulses are reported to be 1+ bilaterally; however, the remainder of the arterial pulse examination is normal.
Her sodium, potassium, and chloride values are normal, but she has an abnormal anion gap (28.1 mEq/L), blood urea nitrogen (53 mg/dL), creatinine (2.9 mg/dL), creatine kinase (10,857 U/L), creatine kinase MB (432.6 ng/mL), and hyperglycemia (glucose 425 mg/dL). Arterial blood gas reveals hypoxia (Po2 of 55 mm Hg), with metabolic acidosis (sodium bicarbonate 10 with compensatory Pco2 of 33 mm Hg). Her urine is cloudy, positive for protein, ketones, hemoglobin, and glucose. She is thought to have a high anion gap acidosis related to dehydration, lactic acidosis (lactic acid 20 mEq/L), and hyperglycemia. Urine toxicology is positive for cannabinoids; ethylene glycol and methanol screen negatively, which rules these out as potential contributors to her high anion gap acidosis.
Ms. K is intubated and IV fluids are initiated for rhabdomyolysis and acute renal failure. Dialysis is implemented on a short-term basis. Her mental state improves gradually over 3 days.
The authors’ observations
Based on the abrupt onset of inattention and confusion, disorganized speech, memory impairments, and psychomotor agitation, we made an initial diagnosis of delirium; however, the precise etiology remained unclear. DSM-IV-TR diagnostic criteria for delirium are described in Table 1. Although delirium due to multiple etiologies does not have a DSM-IV-TR coding designation, we speculated that multiple causes contributed to Ms. K’s presentation. Acute renal failure secondary to dehydration as well as rhabdomyolysis, hypoxia, and hyperglycemia were implicated as general medical conditions etiologically linked to delirium. Because Ms. K has no preexisting medical conditions and her roommate and family stated she had a history of substance abuse, we also considered a presumptive diagnosis of substance-induced delirium. The medical team speculated that, based on information provided by her family, Ms. K may have had a seizure or may have fallen, which would account for her multiple contusions, and could have led to muscle injury and breakdown and the resultant rhabdomyolysis.
The possibility of cannabinoid-induced delirium has been reported, albeit rarely.1-3 However, Ms. K’s presentation—hypothermia, variable heart rate, lack of dry mucous membranes—was not consistent with significant anticholinergic toxicity or cannabinoid intoxication (Table 2).
By contrast, cocaine-induced delirium has been reported and initially appeared to be a plausible cause of Ms. K’s symptoms (Table 2). Delirium related to excess ingestion of cocaine may be related to the drug’s secondary effects resulting in rhabdomyolysis and renal dysfunction.4-6 Although several mechanisms underlying this relationship have been proposed, no single specific mechanism has been identified. The basis for cocaine ingestion and the resultant metabolic and renal effects, as observed in Ms. K’s case, likely are multifactorial. Mechanisms of the rhabdomyolysis might include:
- blockade of synaptic catecholamine reuptake and induction of adrenergic agonism, resulting in vasoconstriction and ischemia and leading to muscle damage
- cocaine-induced seizures and/or prolonged unconsciousness, leading to muscle compression and breakdown of muscle tissue
- a period of exertion induced by cocaine, precipitating an excited delirium and associated rhabdomyolysis
- a surge in dopamine concentrations, similar to neuroleptic malignant syndrome, precipitates hyperthermia, muscle rigidity, and psychomotor agitation, disrupting neuromuscular homeostasis and leading to rhabdomyolysis.
We were uncertain about the plausibility that acute cocaine intoxication caused Ms. K’s medical sequelae, in light of her toxicology findings. If cocaine use was the inciting event, and because the delirium reportedly had developed over several hours, we would expect cocaine to be detected in the toxicology screen. However, it was not detected. Cocaine can remain detectable in urine for 2 to 4 days,7 which raised our speculation that remote cocaine abuse could account for Ms. K’s current presentation and the timeline the roommate initially relayed to EMS personnel was inaccurate. We needed to clarify the timeline and progression of Ms. K’s symptoms with the roommate. In addition, we suggested to the medical team that alternative substances of abuse could be causing Ms. K’s symptoms and the roommate might be the only person who could unveil this possibility.
Table 1
DSM-IV-TR criteria for delirium due to multiple etiologies
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbances) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the delirium has >1 etiology (eg, >1 etiological general medical condition, a general medical condition plus substance intoxication or medication side effect) |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
Table 2
Diagnostic criteria for cannabis and cocaine intoxication
| Diagnostic criteria | Cannabis intoxication | Cocaine intoxication |
|---|---|---|
| Recurrent use | + | + |
| Symptom onset | During or shortly after use | During or shortly after use |
| Behavioral changes | Impaired motor coordination | Hypervigilance, stereotyped behaviors |
| Psychological changes | Euphoria, anxiety, sensation of slowed time, social withdrawal, impaired judgment | Euphoria, anxiety, tension, anger, changes in sociability, interpersonal sensitivity, impaired social or occupational functioning |
| Associated criteria (≥2) | Conjunctival injection, increased appetite, dry mouth, tachycardia | Tachycardia or bradycardia, papillary dilation, elevated or lowered blood pressure, chills/perspiration, nausea/vomiting, evidence of weight loss, psychomotor changes, muscular weakness, chest pain, cardiac arrhythmias, seizure, dyskinesia, dystonia, delirium, coma |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | ||
HISTORY: Unknown substance
Ms. K’s roommate is contacted for supplemental history. The roommate reports that recently he observed Ms. K “snorting” a brown/tan-colored substance. He had not seen her use this substance previously, and when he asked her what it was, she reportedly said that it was “PeeVee” (also called “bath salts”) purchased over the Internet.
The authors’ observations
MDPV is a novel chemical compound that is used as a recreational drug (Table 3).8 It commonly is acquired from Internet sources and sold as “bath salts.” Its use first emerged in approximately 2004, and its popularity has been increasing because of its easy availability and relatively low cost.9 The American Association of Poison Control Centers received 302 calls related to MDPV toxicity in 2010 and 5,625 calls related to MDPV use between January 1 and October 31, 2011.10,11
MDPV has psychoactive properties, with stimulant effects acting as a norepinephrine-dopamine reuptake inhibitor.8,9,12 When snorted, ingested orally, or inserted rectally, the agent produces effects comparable to cocaine or psychostimulants such as methylphenidate or dextroamphetamine.
Acute effects of MDPV include heightened alertness, diminished need for sleep, hyperarousal, and euphoria.8,9 These symptoms often are accompanied by increases in heart rate and blood pressure, sweating, and peripheral vasoconstriction. Individuals may abuse MDPV to acquire sustained attention, reduce their need for sleep, or for aphrodisiac effects. In many cases, anxiety and irritability can accompany the desired euphoric effects. For some, the euphoric effects can be superseded by anxiety or agitation. Mood and attention effects are estimated to last 3 to 4 hours; however, tachycardia and hypertension can persist for 6 to 8 hours.
MDPV use can trigger cravings and lead to binging. Euphoric stimulation with MDPV can become dysphoric as the dose and duration of use increase. Extended use has been associated with agitation, irritability, aggression, panic and marked anxiety, psychosis, and delirium.8,9 Anxiety can range from mild dysphoric stimulation to extreme panic-like states. In moderate forms, a state of sympathetic discharge can occur, producing physiologic effects resembling panic attacks, including hypertension, tachycardia, sweating, and peripheral vasoconstriction. In more severe cases, users may experience a feeling of impending doom, marked distress, and frank psychosis. Patients may experience disorientation and unsystematized paranoid delusions. Case reports of intoxication have described self-injurious behaviors, such as cutting, which may account for the contusions observed on Ms. K’s face and arms. Increasingly, MDPV use has resulted in ER presentations with patients manifesting abrupt onset confusion, anxiety, and self-injurious behaviors.
The mechanisms underlying MDPV-induced delirium have not been definitively identified. Given the similarities in mechanism of action between MDPV and cocaine, causes for delirium related to MDPV are similarly presumed to be multifactorial. The course of delirium associated with MDPV intoxication is self-limited and requires supportive measures.8,9
Suspect MDPV abuse in patients who present with signs or symptoms of stimulant intoxication but have a negative toxicology screen for cocaine and other psychostimulants. MDPV is not detected on routine toxicology assessments; however, it can be identified through laboratories with gas chromatography/mass spectroscopy capabilities. However, the time needed to obtain the results may exceed the clinical course of the patient’s delirium. One of the limitations in Ms. K’s case was the lack of gas chromatography/mass spectroscopy to confirm MDPV ingestion. Ms. K’s roommate could not locate any unused brown powder within their apartment to bring in for laboratory investigations. Recently, screening assessments for MDPV have become commercially available (see Related Resources).
Table 3
Overview of MDPV features
| Chemical name | 3,4-methylenedioxypyrovalerone |
| Popular names | MDPV, PV, PeeVee, Super coke, Magic |
| Sources | Sold as “bath salts” by Internet sources, “head shops,” and gas stations |
| Mode of use | Oral, snorting, smoking, rectal insertion, intravenous |
| Acute effects | Increased energy, perception of heightened alertness/attention, aphrodisiac properties, increased sociability |
| Adverse psychological effects | Anxiety (panic attacks), irritability, agitation, confusion, suicidal ideations, visual distortions |
| Adverse physical effects | Insomnia/overstimulation, bruxism, muscle twitching, pupil dilation/blurred vision, anorexia, headache, nausea/vomiting, hyperthermia, irregular heart beat, tachycardia, dyspnea, fatigue |
| Effects of protracted use | Dysphoria, depression, anhedonia |
| LD50 | Unknown |
| LD50: lethal dose; MDPV: methylenedioxypyrovalerone Source: Reference 8 | |
OUTCOME: Referral to treatment
Dialysis is discontinued within 1 day of hospitalization. Ms. K’s peripheral arterial perfusion improves, as does her thermoregulatory status. Her mental status improvements coincide with improvements in her physical and metabolic status.
Ms. K is able to sustain attention when speaking with interviewers. She is aware of her surroundings and is no longer distracted by extraneous stimuli. Her speech is articulate and her thoughts are linear. There is no evidence of any residual thought disorganization, delusions, or hallucinations.
Initially, Ms. K is reluctant to acknowledge her substance use, but eventually, she concedes to acquiring a stimulant from an Internet source and abusing it in undetermined amounts. She had no experience with using MDPV and did not know how to avoid ingesting dangerous amounts. We educate Ms. K about the dangers she faced during this hospitalization and the potential life-threatening outcomes. She is amenable to pursuing outpatient substance abuse treatment. Her roommate is enlisted to facilitate her follow-up with this treatment.
The authors’ observations
Managing MDPV toxicity presents a diagnostic dilemma for medical personnel and psychiatrists when evaluating and managing acute delirium. MDPV ingestion may go unrecognized in clinical settings because toxicology assessments for it are not readily available and patients’ historical information may be unreliable.
Because of the seriousness of sequelae associated with MDPV use, state and federal agencies have intervened. Until recently, bath salts did not have a controlled substance designation. In October 2011, the US Drug Enforcement Administration (DEA) ruled to make MDPV a controlled substance for 1 year, with the possibility of a 6-month extension.13 Although this ruling is temporary, it makes possession, sale, or distribution of these chemicals, or the products that contain them, illegal in the United States. In the interim, the DEA and the US Department of Health and Human Services will determine whether MDPV should remain a controlled substance.
- American Screening Corp. (MDPV screening). www.americanscreeningcorp.com.
- U.S. Drug Enforcement Administration. 3, 4-Methylenedioxypyrovalerone (MDPV). www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf.
- Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones [published online ahead of print November 23, 2011]. J Med Toxicol. doi: 10.1007/s13181-011-0193-z.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Hurt and confused
Emergency medical services (EMS) are called to Ms. K’s apartment after her roommate found her lying on the floor moaning. The roommate tells EMS that Ms. K, age 29, appeared confused and was slurring her words, and reports that this change in her awareness progressed rapidly over a few hours. EMS personnel find that Ms. K has multiple contusions on her arms and face, which they presume to be self-inflicted. A marijuana pipe is discovered at Ms. K’s apartment.
In the emergency room (ER), Ms. K is inattentive and has difficulty following simple commands. Her speech is mumbled and her thoughts are disorganized. She displays psychomotor restlessness in the form of combativeness. Ms. K cannot provide meaningful historical data and is disoriented to place and time. The ER staff requests a psychiatric consultation.
Family members reveal that Ms. K has no preexisting medical conditions, is not taking prescription medications, but has a history of substance abuse (sporadic cocaine and cannabis use). Her family is unaware of recent substance use.
Physical examination reveals tachycardia (heart rate 110 to 120 beats per minute), hypotension (blood pressure 78/49 mm Hg), hypothermia (temperature 88ºF), and peripheral pulse oximetry of 84%. Her pupils are dilated and reactive to light; no conjunctival injection is noted. Her lung fields are clear on auscultation, but she is noted to have a rapid, irregular heartbeat. The abdomen is positive for bowel sounds, soft on palpation, and without any repositioning or notable overt signs of tenderness. Ms. K’s toes show purple discoloration with poor capillary refill. The dorsalis pedis pulses are reported to be 1+ bilaterally; however, the remainder of the arterial pulse examination is normal.
Her sodium, potassium, and chloride values are normal, but she has an abnormal anion gap (28.1 mEq/L), blood urea nitrogen (53 mg/dL), creatinine (2.9 mg/dL), creatine kinase (10,857 U/L), creatine kinase MB (432.6 ng/mL), and hyperglycemia (glucose 425 mg/dL). Arterial blood gas reveals hypoxia (Po2 of 55 mm Hg), with metabolic acidosis (sodium bicarbonate 10 with compensatory Pco2 of 33 mm Hg). Her urine is cloudy, positive for protein, ketones, hemoglobin, and glucose. She is thought to have a high anion gap acidosis related to dehydration, lactic acidosis (lactic acid 20 mEq/L), and hyperglycemia. Urine toxicology is positive for cannabinoids; ethylene glycol and methanol screen negatively, which rules these out as potential contributors to her high anion gap acidosis.
Ms. K is intubated and IV fluids are initiated for rhabdomyolysis and acute renal failure. Dialysis is implemented on a short-term basis. Her mental state improves gradually over 3 days.
The authors’ observations
Based on the abrupt onset of inattention and confusion, disorganized speech, memory impairments, and psychomotor agitation, we made an initial diagnosis of delirium; however, the precise etiology remained unclear. DSM-IV-TR diagnostic criteria for delirium are described in Table 1. Although delirium due to multiple etiologies does not have a DSM-IV-TR coding designation, we speculated that multiple causes contributed to Ms. K’s presentation. Acute renal failure secondary to dehydration as well as rhabdomyolysis, hypoxia, and hyperglycemia were implicated as general medical conditions etiologically linked to delirium. Because Ms. K has no preexisting medical conditions and her roommate and family stated she had a history of substance abuse, we also considered a presumptive diagnosis of substance-induced delirium. The medical team speculated that, based on information provided by her family, Ms. K may have had a seizure or may have fallen, which would account for her multiple contusions, and could have led to muscle injury and breakdown and the resultant rhabdomyolysis.
The possibility of cannabinoid-induced delirium has been reported, albeit rarely.1-3 However, Ms. K’s presentation—hypothermia, variable heart rate, lack of dry mucous membranes—was not consistent with significant anticholinergic toxicity or cannabinoid intoxication (Table 2).
By contrast, cocaine-induced delirium has been reported and initially appeared to be a plausible cause of Ms. K’s symptoms (Table 2). Delirium related to excess ingestion of cocaine may be related to the drug’s secondary effects resulting in rhabdomyolysis and renal dysfunction.4-6 Although several mechanisms underlying this relationship have been proposed, no single specific mechanism has been identified. The basis for cocaine ingestion and the resultant metabolic and renal effects, as observed in Ms. K’s case, likely are multifactorial. Mechanisms of the rhabdomyolysis might include:
- blockade of synaptic catecholamine reuptake and induction of adrenergic agonism, resulting in vasoconstriction and ischemia and leading to muscle damage
- cocaine-induced seizures and/or prolonged unconsciousness, leading to muscle compression and breakdown of muscle tissue
- a period of exertion induced by cocaine, precipitating an excited delirium and associated rhabdomyolysis
- a surge in dopamine concentrations, similar to neuroleptic malignant syndrome, precipitates hyperthermia, muscle rigidity, and psychomotor agitation, disrupting neuromuscular homeostasis and leading to rhabdomyolysis.
We were uncertain about the plausibility that acute cocaine intoxication caused Ms. K’s medical sequelae, in light of her toxicology findings. If cocaine use was the inciting event, and because the delirium reportedly had developed over several hours, we would expect cocaine to be detected in the toxicology screen. However, it was not detected. Cocaine can remain detectable in urine for 2 to 4 days,7 which raised our speculation that remote cocaine abuse could account for Ms. K’s current presentation and the timeline the roommate initially relayed to EMS personnel was inaccurate. We needed to clarify the timeline and progression of Ms. K’s symptoms with the roommate. In addition, we suggested to the medical team that alternative substances of abuse could be causing Ms. K’s symptoms and the roommate might be the only person who could unveil this possibility.
Table 1
DSM-IV-TR criteria for delirium due to multiple etiologies
| A. Disturbance of consciousness (ie, reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention |
| B. A change in cognition (such as memory deficit, disorientation, language disturbances) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia |
| C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day |
| D. There is evidence from the history, physical examination, or laboratory findings that the delirium has >1 etiology (eg, >1 etiological general medical condition, a general medical condition plus substance intoxication or medication side effect) |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
Table 2
Diagnostic criteria for cannabis and cocaine intoxication
| Diagnostic criteria | Cannabis intoxication | Cocaine intoxication |
|---|---|---|
| Recurrent use | + | + |
| Symptom onset | During or shortly after use | During or shortly after use |
| Behavioral changes | Impaired motor coordination | Hypervigilance, stereotyped behaviors |
| Psychological changes | Euphoria, anxiety, sensation of slowed time, social withdrawal, impaired judgment | Euphoria, anxiety, tension, anger, changes in sociability, interpersonal sensitivity, impaired social or occupational functioning |
| Associated criteria (≥2) | Conjunctival injection, increased appetite, dry mouth, tachycardia | Tachycardia or bradycardia, papillary dilation, elevated or lowered blood pressure, chills/perspiration, nausea/vomiting, evidence of weight loss, psychomotor changes, muscular weakness, chest pain, cardiac arrhythmias, seizure, dyskinesia, dystonia, delirium, coma |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | ||
HISTORY: Unknown substance
Ms. K’s roommate is contacted for supplemental history. The roommate reports that recently he observed Ms. K “snorting” a brown/tan-colored substance. He had not seen her use this substance previously, and when he asked her what it was, she reportedly said that it was “PeeVee” (also called “bath salts”) purchased over the Internet.
The authors’ observations
MDPV is a novel chemical compound that is used as a recreational drug (Table 3).8 It commonly is acquired from Internet sources and sold as “bath salts.” Its use first emerged in approximately 2004, and its popularity has been increasing because of its easy availability and relatively low cost.9 The American Association of Poison Control Centers received 302 calls related to MDPV toxicity in 2010 and 5,625 calls related to MDPV use between January 1 and October 31, 2011.10,11
MDPV has psychoactive properties, with stimulant effects acting as a norepinephrine-dopamine reuptake inhibitor.8,9,12 When snorted, ingested orally, or inserted rectally, the agent produces effects comparable to cocaine or psychostimulants such as methylphenidate or dextroamphetamine.
Acute effects of MDPV include heightened alertness, diminished need for sleep, hyperarousal, and euphoria.8,9 These symptoms often are accompanied by increases in heart rate and blood pressure, sweating, and peripheral vasoconstriction. Individuals may abuse MDPV to acquire sustained attention, reduce their need for sleep, or for aphrodisiac effects. In many cases, anxiety and irritability can accompany the desired euphoric effects. For some, the euphoric effects can be superseded by anxiety or agitation. Mood and attention effects are estimated to last 3 to 4 hours; however, tachycardia and hypertension can persist for 6 to 8 hours.
MDPV use can trigger cravings and lead to binging. Euphoric stimulation with MDPV can become dysphoric as the dose and duration of use increase. Extended use has been associated with agitation, irritability, aggression, panic and marked anxiety, psychosis, and delirium.8,9 Anxiety can range from mild dysphoric stimulation to extreme panic-like states. In moderate forms, a state of sympathetic discharge can occur, producing physiologic effects resembling panic attacks, including hypertension, tachycardia, sweating, and peripheral vasoconstriction. In more severe cases, users may experience a feeling of impending doom, marked distress, and frank psychosis. Patients may experience disorientation and unsystematized paranoid delusions. Case reports of intoxication have described self-injurious behaviors, such as cutting, which may account for the contusions observed on Ms. K’s face and arms. Increasingly, MDPV use has resulted in ER presentations with patients manifesting abrupt onset confusion, anxiety, and self-injurious behaviors.
The mechanisms underlying MDPV-induced delirium have not been definitively identified. Given the similarities in mechanism of action between MDPV and cocaine, causes for delirium related to MDPV are similarly presumed to be multifactorial. The course of delirium associated with MDPV intoxication is self-limited and requires supportive measures.8,9
Suspect MDPV abuse in patients who present with signs or symptoms of stimulant intoxication but have a negative toxicology screen for cocaine and other psychostimulants. MDPV is not detected on routine toxicology assessments; however, it can be identified through laboratories with gas chromatography/mass spectroscopy capabilities. However, the time needed to obtain the results may exceed the clinical course of the patient’s delirium. One of the limitations in Ms. K’s case was the lack of gas chromatography/mass spectroscopy to confirm MDPV ingestion. Ms. K’s roommate could not locate any unused brown powder within their apartment to bring in for laboratory investigations. Recently, screening assessments for MDPV have become commercially available (see Related Resources).
Table 3
Overview of MDPV features
| Chemical name | 3,4-methylenedioxypyrovalerone |
| Popular names | MDPV, PV, PeeVee, Super coke, Magic |
| Sources | Sold as “bath salts” by Internet sources, “head shops,” and gas stations |
| Mode of use | Oral, snorting, smoking, rectal insertion, intravenous |
| Acute effects | Increased energy, perception of heightened alertness/attention, aphrodisiac properties, increased sociability |
| Adverse psychological effects | Anxiety (panic attacks), irritability, agitation, confusion, suicidal ideations, visual distortions |
| Adverse physical effects | Insomnia/overstimulation, bruxism, muscle twitching, pupil dilation/blurred vision, anorexia, headache, nausea/vomiting, hyperthermia, irregular heart beat, tachycardia, dyspnea, fatigue |
| Effects of protracted use | Dysphoria, depression, anhedonia |
| LD50 | Unknown |
| LD50: lethal dose; MDPV: methylenedioxypyrovalerone Source: Reference 8 | |
OUTCOME: Referral to treatment
Dialysis is discontinued within 1 day of hospitalization. Ms. K’s peripheral arterial perfusion improves, as does her thermoregulatory status. Her mental status improvements coincide with improvements in her physical and metabolic status.
Ms. K is able to sustain attention when speaking with interviewers. She is aware of her surroundings and is no longer distracted by extraneous stimuli. Her speech is articulate and her thoughts are linear. There is no evidence of any residual thought disorganization, delusions, or hallucinations.
Initially, Ms. K is reluctant to acknowledge her substance use, but eventually, she concedes to acquiring a stimulant from an Internet source and abusing it in undetermined amounts. She had no experience with using MDPV and did not know how to avoid ingesting dangerous amounts. We educate Ms. K about the dangers she faced during this hospitalization and the potential life-threatening outcomes. She is amenable to pursuing outpatient substance abuse treatment. Her roommate is enlisted to facilitate her follow-up with this treatment.
The authors’ observations
Managing MDPV toxicity presents a diagnostic dilemma for medical personnel and psychiatrists when evaluating and managing acute delirium. MDPV ingestion may go unrecognized in clinical settings because toxicology assessments for it are not readily available and patients’ historical information may be unreliable.
Because of the seriousness of sequelae associated with MDPV use, state and federal agencies have intervened. Until recently, bath salts did not have a controlled substance designation. In October 2011, the US Drug Enforcement Administration (DEA) ruled to make MDPV a controlled substance for 1 year, with the possibility of a 6-month extension.13 Although this ruling is temporary, it makes possession, sale, or distribution of these chemicals, or the products that contain them, illegal in the United States. In the interim, the DEA and the US Department of Health and Human Services will determine whether MDPV should remain a controlled substance.
- American Screening Corp. (MDPV screening). www.americanscreeningcorp.com.
- U.S. Drug Enforcement Administration. 3, 4-Methylenedioxypyrovalerone (MDPV). www.deadiversion.usdoj.gov/drugs_concern/mdpv.pdf.
- Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones [published online ahead of print November 23, 2011]. J Med Toxicol. doi: 10.1007/s13181-011-0193-z.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. André C, Jaber-Filho JA, Bento RM, et al. Delirium following ingestion of marijuana in chocolate cookies. CNS Spectr. 2006;11(4):262-264.
2. Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38(1):1-20.
3. Meyer ME. Psychiatric consequences of marijuana use: the state of the evidence. In: Tinklenberg JR ed. Marijuana and health hazards: methodologic issues in current research. New York, NY: Academic Press; 1975:33–152.
4. Ruttenber AJ, Lawler-Heavner J, Yin M, et al. Fatal excited delirium following cocaine use: epidemiologic findings provide new evidence for mechanisms of cocaine toxicity. J Forensic Sci. 1997;42(1):25-31.
5. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20(2):120-127.
6. Singhal PC, Rubin RB, Peters A, et al. Rhabdomyolysis and acute renal failure associated with cocaine abuse. J Toxicol Clin Toxicol. 1990;28(3):321-330.
7. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83(1):66-76.
8. Psychonaut WebMapping Research Group. MDPV report. London United Kingdom: Institute of Psychiatry, King’s College. http://www.psychonautproject.eu/documents/reports/MDPV.pdf. Accessed November 23, 2011.
9. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
10. American Association of Poison Control Centers. Bath salts data. http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%2011.03.2011.pdf. Updated November 3 2011. Accessed November 23, 2011.
11. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan November 13, 2010-March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(19):624-627.
12. Westphal F, Junge T, Rösner P, et al. Mass and NMR spectroscopic characterization of 3, 4-methylenedioxypyrovalerone: a designer drug with α-pyrrolidinophenone structure. Forensic Sci Int. 2009;190(1-3):1-8.
13. U.S. Drug Enforcement Administration. Chemicals used in “bath salts” now under federal control and regulation. http://www.justice.gov/dea/pubs/pressrel/pr102111.html. Accessed November 23, 2011.
1. André C, Jaber-Filho JA, Bento RM, et al. Delirium following ingestion of marijuana in chocolate cookies. CNS Spectr. 2006;11(4):262-264.
2. Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38(1):1-20.
3. Meyer ME. Psychiatric consequences of marijuana use: the state of the evidence. In: Tinklenberg JR ed. Marijuana and health hazards: methodologic issues in current research. New York, NY: Academic Press; 1975:33–152.
4. Ruttenber AJ, Lawler-Heavner J, Yin M, et al. Fatal excited delirium following cocaine use: epidemiologic findings provide new evidence for mechanisms of cocaine toxicity. J Forensic Sci. 1997;42(1):25-31.
5. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20(2):120-127.
6. Singhal PC, Rubin RB, Peters A, et al. Rhabdomyolysis and acute renal failure associated with cocaine abuse. J Toxicol Clin Toxicol. 1990;28(3):321-330.
7. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83(1):66-76.
8. Psychonaut WebMapping Research Group. MDPV report. London United Kingdom: Institute of Psychiatry, King’s College. http://www.psychonautproject.eu/documents/reports/MDPV.pdf. Accessed November 23, 2011.
9. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365(10):967-968.
10. American Association of Poison Control Centers. Bath salts data. http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%2011.03.2011.pdf. Updated November 3 2011. Accessed November 23, 2011.
11. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan November 13, 2010-March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(19):624-627.
12. Westphal F, Junge T, Rösner P, et al. Mass and NMR spectroscopic characterization of 3, 4-methylenedioxypyrovalerone: a designer drug with α-pyrrolidinophenone structure. Forensic Sci Int. 2009;190(1-3):1-8.
13. U.S. Drug Enforcement Administration. Chemicals used in “bath salts” now under federal control and regulation. http://www.justice.gov/dea/pubs/pressrel/pr102111.html. Accessed November 23, 2011.
Disoriented and forgetful
CASE: Disoriented and delusional
Ms. P, a 53-year-old registered nurse, is admitted to the inpatient psychiatric unit with confusion, markedly disorganized thought processes, delayed verbal responsiveness, mood lability, and persecutory delusions. Shortly before hospitalization, Ms. P traveled approximately 360 miles from her daughter’s home with a male companion. Noting changes in her mental status, the man brought Ms. P to the local hospital. She was then transferred to our facility.
At admission, Ms. P is not oriented to time. She denies auditory or visual hallucinations and does not display psychomotor agitation or retardation. She reports her mood as sad and her affect is mildly labile. Insight and judgment are considered poor.
Five years ago, Ms. P and her mother were diagnosed with Fabry’s disease (FD) based on genetic analysis. Both women are carriers for the mutations and Ms. P’s mother was found to have almost absent galactosidase activity.
The authors’ observations
FD is an X-linked recessive glycolipid storage disease caused by deficient activity of the lysosomal storage enzyme α-galactosidase A. The disorder affects both men and women and leads to progressive intracellular accumulation of globotriaosylceramide and other related glycosphingolipids.1,2 The earliest FD symptoms—burning pain and acroparesthesias—typically appear in childhood (Table 1).2 FD often is misdiagnosed in women because women tend to display neurologic symptoms later than men, with typical symptom onset in the teenage years.3,4 Often, these symptoms are confused with psychiatric disorders or vague neurologic or pain syndromes.5 In patients with no family history of FD, accurate diagnosis may not be made until adulthood.
Laboratory, dermatologic, and genetic tests can accurately determine the presence of FD.1 However, because multiple organ systems are involved, initially attributing symptoms to FD is challenging, particularly in women.1,3,5 For men, diagnosis can be established by measuring plasma or urinary globotriaosylceramide or plasma α-galactosidase A in addition to genetic analysis. In women, genetic analysis is a better diagnosis strategy because elevations in globotriaosylceramide or α-galactosidase A may not be prominent. An algorithm for diagnosing and assessing patients with FD has been proposed.2
Table 1
Typical signs and symptoms of Fabry’s disease
| Typical time at onset | Signs/symptoms |
|---|---|
| Childhood and adolescence (age ≤16) | Neuropathic pain Ophthalmologic abnormalities (cornea verticillata and tortuous retinal blood vessels) Hearing impairment Dyshidrosis (hypohidrosis and hyperhidrosis) Hypersensitivity to heat and cold Gastrointestinal disturbances and abdominal pain Lethargy and tiredness Angiokeratomas Onset of renal and cardiac signs (eg, microalbuminuria, proteinuria, abnormal heart rate variability) |
| Early adulthood (age 17 to 30) | Extension of any of the above Proteinuria and progressive renal failure Cardiomyopathy Transient ischemic attacks, strokes Facial dysmorphism |
| Later adulthood (age >30) | Worsening of any of the above Heart disease (eg, left ventricular hypertrophy, angina, arrhythmia, and dyspnea) Transient ischemic attacks, strokes Osteopenia and osteoporosis |
| Source: Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659, by permission of Oxford University Press | |
HISTORY: Cognitive deterioration
Ms. P has had psychiatric symptoms such as depression and anxiety since childhood. However, 3 years ago she started to experience psychological and cognitive deterioration. Medical records indicate that Ms. P described memory and concentration problems over the previous few years. She also reported pain, weakness, and numbness in her left leg after surgery for a work-related back injury, for which she received a financial settlement through workers’ compensation. Shortly thereafter, Ms. P separated from her third husband, moved in with her parents, and found work as a psychiatric nurse. She was dismissed after 6 weeks because she could not learn the electronic medical record system and had difficulty with memory and attention. Her performance on the Mini-Mental State Exam6 at that time was 28 out of 30, which was within normal limits.
After her parents died 3 years ago, Ms. P lived with her daughter, who became her primary caregiver and legal guardian. Ms. P’s daughter notes that her mother’s impulsive and risky behaviors grew more pronounced. Ms. P went on shopping sprees and became sexually promiscuous.
Ms. P’s psychiatric history includes childhood sexual abuse, hospitalization for a suicide attempt at age 19, and courses of psychotherapy and pharmacotherapy. In addition to FD, Ms. P’s medical history consists of coronary artery disease, type 2 diabetes mellitus, hypercholesterolemia, obesity, arthritis, back pain, fibromyalgia, and gastroesophageal reflux disease. Her family history is notable for alcohol abuse (both parents and a brother), lung cancer (mother), myocardial infarction (father), and Alzheimer’s disease (father).
The authors’ observations
Because α-galactosidase A is ubiquitous throughout the body, in addition to neurologic symptoms, FD involves multiple organ systems, with possible dermatologic, renal, gastrointestinal, cardiac, and cerebrovascular dysfunction. Despite growth in FD research, including the Fabry Outcomes Survey,3 the psychosocial and neuropsychiatric implications of the disease remain unclear.7 Behavioral presentations are idiosyncratic and unstable over time, depending on the structures impacted by progressive glycosphingolipid accumulation. Premature cardiovascular events (onset between age 30 and 40 for women), greater incidence of ischemic stroke or transient ischemic attack (7% to 30%), and frequent evidence of white matter lesions put FD patients at greater risk for developing presenile vascular dementia.1,3 Nearly all male FD patients with dementia show some evidence of stroke or transient ischemic attack; cognitive functioning has not been well explored in female patients.4 In a heterogeneous sample of 15 FD patients age 7 to 61, Segal et al8 noted deficits in attention, processing speed, and executive function .75 to 1.95 standard deviations below normative means. No patients in this study had a history of stroke or transient ischemic attack; neuroimaging studies were not reported. Kolodny and Pastores9 suggested multiple mechanisms for cognitive disruption, suggesting that mild dementia late in the disease course could be secondary to diffuse leukomalacia, multiple strokes, or possibly to lipid storage in hippocampal and frontal lobe neurons.
Psychiatric comorbidity
Psychiatric illness, such as depression or a personality disorder, may be comorbid with FD, although pathologic mechanisms remain unclear.7,10,11 Hypothesized mechanisms include:
- psychosocial stress from chronic disease
- white matter changes
- disruption of impaired L-arginine-nitric oxide pathways.7,12
Crosbie et al13 noted that FD patients presented with greater psychological distress as measured by the Minnesota Multiphasic Personality Inventory-2 than patients with Gaucher disease or chronic heart disease. However, no significant differences were found between patients with FD and those diagnosed with a pain disorder. In the Segal et al study, out of 11 adult FD patients, 4 were diagnosed with major depressive disorder, 1 with schizophrenia, 2 with schizotypal personality disorder, and 1 with borderline personality disorder.8
EVALUATION: Brain abnormalities
Head CT scans (conducted 2 years ago and 6 months ago) revealed prominent cortical sulci likely caused by underlying volume loss, especially in bifrontal areas. A brain MRI performed 2 months ago indicated a moderate degree of subcortical atrophy in bilateral frontal and parietal regions. These radiology findings suggest mild to moderate frontal atrophy, mild degree of white matter changes, and slightly enlarged ventricles. An EEG showed background slowing and lack of an alpha rhythm, indicative of cerebral cortical dysfunction.
Ms. P’s α-galactosidase A level was within normal limits; however, normal enzyme levels frequently are reported in symptomatic and asymptomatic female FD patients.14 A dermatology consult confirmed the presence of skin findings characteristic of FD (ie, multiple cherry red papules extensively distributed throughout Ms. P’s chest, abdomen, and back, as well as upper and lower extremities).
Ms. P completed 2 neuropsychological assessments separated by 5 months. For a summary of the results of these tests, see the table titled “Ms. P’s neuropsychological assessment results”. Both assessments revealed grossly impaired intellectual capacity, memory, processing speed, and motor functioning. During the assessment, Ms. P could understand all directions with minimal changes from standardized protocols. Ms. P became insistent that she would not be able to complete memory tasks successfully. She gave up prematurely on tasks, saying they were too difficult. She admitted to guessing on several items because she did not want to continue the task.
Ms. P’s performance on tasks measuring effort and validity of a person’s neuropsychological presentation was consistent with someone exaggerating neurologic symptoms. A person with true dementia may perform as poorly as Ms. P did. However, Ms. P’s scores likely underestimated her level of functioning, even if she was experiencing dementia. Ms. P could not complete tasks individuals with severe dementia complete successfully, such as simple addition and subtraction and digit repetition. Ms. P recalled several recent and remote events, such as her breakfast menu and location of her first assessment, but could not recall words practiced multiple times. Although Ms. P’s scores on a complex card-sorting task were in the impaired range, a detailed review of her pattern indicated that although Ms. P could not generate any correct sorting categories, she made few repetitive responses and errors. This pattern is consistent with someone who understands task requirements, but deliberately avoids answering correctly. This suggests that she retained some ability for hypotheses generation and problem solving; however, because she exaggerated her symptoms, specific deficits could not be determined.
The authors’ observations
Ms. P presented with an interesting manifestation of neuropsychiatric symptoms in the context of FD; however, common cardiac and cerebrovascular features of the disease were not fully developed. Ms. P experienced progressive cognitive and behavioral changes for 2 years before her admission (Table 2), which may represent a prodromal period leading up to what appeared to be a frontally mediated dementia syndrome. Müller et al15 described a patient with FD who displayed a behavioral profile similar to Ms. P’s that included increasingly unstable mood for at least 3 years, borderline personality disorder features, and rapidly fluctuating mood. A case study reported that risperidone, 1 mg/d, used to treat psychosis in a male FD patient caused extrapyramidal symptoms.16
Ms. P presented with no evidence of stroke or transient ischemic attacks, which is atypical for FD patients with cognitive impairment. However, neuroimaging did reveal frontal atrophy that may be associated with her impulse control deficits, risk-taking behavior, emotional instability, and poor judgment. Her cognitive testing was notable for impairment and exaggeration of symptoms consistent with personality disorder symptoms. Possible reasons for exaggeration include a desire to maintain the sick role or secondary gain related to obtaining disability income.
Ms. P’s behavior pattern could be caused by dementia with frontal features, possibly secondary to FD, in combination with personality and psychiatric pathology.
The mainstay of FD treatment is enzyme replacement therapy (ERT), which addresses the underlying enzyme deficiency. Available research indicates that ERT may reduce symptom severity and slow disease progression; however, further studies are needed to determine if it will reduce outcomes such as stroke, ischemic heart disease, or renal disease.2
Table 2
Symptoms that preceded Ms. P’s admission
| Time frame | Symptoms |
|---|---|
| 24 months before admission | Depressed mood Decreased ability to manage independent activities of daily living (eg, finances, cooking) Minimal objective cognitive impairment |
| 12 months before admission | Increased depression Mild to moderate decline in cognitive functioning Visual and auditory hallucinations Impulsivity/poor impulse control Irrational decision-making Increased risky behavior |
| 6 months before admission | Severe cognitive decline with cognitive symptom exaggeration Psychiatric symptom exaggeration Disorganized thinking Continued risky behavior and poor decision-making |
TREATMENT: Persistent deficits
Ms. P is started on risperidone rapidly titrated to 4 mg/d for delusional thinking and behavioral disturbance. After initially improving, she develops delirium when risperidone is increased to 4 mg/d. She has visual hallucinations, marked confusion with disorientation, worsened short-term memory, and an unsteady, shuffling gait. Risperidone is tapered and discontinued and Ms. P’s motor symptoms resolve within 2 days; however, she remains confused and delusional. We start her on quetiapine, 25 mg/d titrated to 50 mg/d, and her agitation and delusional thinking progressively decline. Memantine, titrated to 20 mg/d, and rivastigmine, started at 3 mg/d titrated to 9 mg/d, are added to address her cognitive symptoms.
Over several weeks, Ms. P’s mental status slowly improves and her drug-induced delirium completely resolves. However, she has persistent cognitive impairment characterized by compromised short-term memory and poor insight into her medical and psychological condition. She maintains unrealistic expectations about her ability to live independently and return to the workforce. The treatment team recommends that Ms. P’s daughter pursue guardianship and that she receive around-the-clock supervision after discharge from the hospital.
Table
Ms. P’s neuropsychological assessment results
| June | November | |
|---|---|---|
| Intellectual functioning | ||
| Wechsler Adult Intelligence Scale-III | ||
| FSIQ | 60 | |
| VIQ | 68 | |
| PIQ | 56 | |
| Ravens Colored Progressive Matrices | 70 | |
| Premorbid intellectual functioning estimates | ||
| Peabody Picture Vocabulary Test-2 | 89 | |
| Barona Demographic Estimate | 104 | 104 |
| North American Adult Reading Test | 99 | |
| Memory functioning | ||
| Wechsler Memory Scale-III | ||
| Immediate memory | 45 | |
| General delay memory | 47 | |
| Auditory recognition delay | 55 | |
| California Verbal Learning Test-II | ||
| Trial 1 (immediate recall) | <60 (raw = 3) | |
| Trial 5 | <60 (raw = 3) | |
| Total Words Learned | <60 (raw = 15) | |
| Short Delay Free Recall | <60 (raw = 2) | |
| Long Delay Free Recall | <60 (raw = 4) | |
| Executive functioning | ||
| Trail Making Test A | 88 | 88 |
| Trail Making Test B | failed to understand | failed to understand |
| Wisconsin Card Sort-64 | ||
| Number of categories | <60 (raw = 0) | |
| Errors | 81 | |
| Percent conceptual level responses | 74 | |
| Perseverative responses | 107 | |
| Perseverative errors | 108 | |
| COWAT FAS | 65 | 69 |
| Category exemplar | 69 | 80 |
| Motor functioning | ||
| Finger Tapping Dominant Hand | 68 | |
| Finger Tapping Non-Dominant Hand | 62 | |
| Invalidity/effort | ||
| TOMM | ||
| Trial 1 | raw = 34 | raw = 37 |
| Trial 2 | raw = 42 | raw = 45 |
| Recognition | raw = 44 | |
| MSVT verbal | fail | |
| MSVT nonverbal | fail | |
| Scores provided are standardized (mean = 100; SD = 15). Raw scores are also provided when indicated. COWAT: Controlled oral word association test; FSIQ: Full Scale IQ; MSVT: Medical Symptom Validity Test; PIQ: Performance IQ; TOMM: Test of Memory Malingering; VIQ: Verbal IQ | ||
Related Resources
- National Institute of Neurological Disorders and Stroke. Fabry disease information page. www.ninds.nih.gov/disorders/fabrys/fabrys.htm.
- National Fabry Disease Foundation. www.thenfdf.org.
- Rozenfeld P, Neumann PM. Treatment of Fabry disease: current and emerging strategies. Curr Pharm Biotechnol. 2011;12(6):916-922.
Drug Brand Names
- Donepezil • Aricept
- Memantine • Namenda
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Rivastigmine • Exelon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539-548.
2. Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659.
3. Deegan PB, Baehner AF, Barba Romero MA, et al. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43(4):347-352.
4. Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurol. 2006;5(9):791-795.
5. Møller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol. 2007;3(2):95-106.
6. Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
7. Müller MJ. Neuropsychiatric and psychosocial aspects of Fabry disease. In: Mehta A Beck M, Sunder-Plassman G, eds. Fabry disease: perspectives from 5 years of FOS. Oxford, United Kingdom: Oxford PharmaGenesis Ltd; 2006. http://www.ncbi.nlm.nih.gov/books/nbk11618. Accessed October 31, 2011.
8. Segal P, Kohn Y, Pollak Y, et al. Psychiatric and cognitive profile in Anderson-Fabry patients: a preliminary study. J Inherit Metab Dis. 2010;33(4):429-436.
9. Kolodny EH, Pastores GM. Anderson-Fabry disease: Extrarenal neurologic manifestations. J Am Soc Nephrol. 2002;13(suppl 2):S150-153.
10. Grewal RP. Psychiatric disorders in patients with Fabry disease. Int J Psychiatry Med. 1993;23(3):307-312.
11. Müller MJ, Müller KM, Dascalescu A, et al. Psychiatric and neuropsychological signs and symptoms in patients with Fabry disease: literature review [in German]. Fortschr Neurol Psychiatr. 2005;73(11):687-693.
12. Segal P, Raas-Rothschild A. Neuropsychiatric manifestations of AFD. In: Elstein D Altarescu G, Beck M, eds. Fabry disease. New York, NY: Springer; 2010:321–324.
13. Crosbie TW, Packman W, Packman S. Psychological aspects of patients with Fabry disease. J Inherit Metab Dis. 2009;32(6):745-753.
14. Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J Am Coll Cardiol. 2008;51(21):2082.-
15. Müller MJ, Fellgiebel A, Scheurich A, et al. Recurrent brief depression in female patient with Fabry disease. Bipolar Disord. 2006;8(4):418-419.
16. Shen YC, Haw-Ming L, Lin CC, et al. Psychosis in a patient with Fabry’s disease and treatment with aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):779-780.
CASE: Disoriented and delusional
Ms. P, a 53-year-old registered nurse, is admitted to the inpatient psychiatric unit with confusion, markedly disorganized thought processes, delayed verbal responsiveness, mood lability, and persecutory delusions. Shortly before hospitalization, Ms. P traveled approximately 360 miles from her daughter’s home with a male companion. Noting changes in her mental status, the man brought Ms. P to the local hospital. She was then transferred to our facility.
At admission, Ms. P is not oriented to time. She denies auditory or visual hallucinations and does not display psychomotor agitation or retardation. She reports her mood as sad and her affect is mildly labile. Insight and judgment are considered poor.
Five years ago, Ms. P and her mother were diagnosed with Fabry’s disease (FD) based on genetic analysis. Both women are carriers for the mutations and Ms. P’s mother was found to have almost absent galactosidase activity.
The authors’ observations
FD is an X-linked recessive glycolipid storage disease caused by deficient activity of the lysosomal storage enzyme α-galactosidase A. The disorder affects both men and women and leads to progressive intracellular accumulation of globotriaosylceramide and other related glycosphingolipids.1,2 The earliest FD symptoms—burning pain and acroparesthesias—typically appear in childhood (Table 1).2 FD often is misdiagnosed in women because women tend to display neurologic symptoms later than men, with typical symptom onset in the teenage years.3,4 Often, these symptoms are confused with psychiatric disorders or vague neurologic or pain syndromes.5 In patients with no family history of FD, accurate diagnosis may not be made until adulthood.
Laboratory, dermatologic, and genetic tests can accurately determine the presence of FD.1 However, because multiple organ systems are involved, initially attributing symptoms to FD is challenging, particularly in women.1,3,5 For men, diagnosis can be established by measuring plasma or urinary globotriaosylceramide or plasma α-galactosidase A in addition to genetic analysis. In women, genetic analysis is a better diagnosis strategy because elevations in globotriaosylceramide or α-galactosidase A may not be prominent. An algorithm for diagnosing and assessing patients with FD has been proposed.2
Table 1
Typical signs and symptoms of Fabry’s disease
| Typical time at onset | Signs/symptoms |
|---|---|
| Childhood and adolescence (age ≤16) | Neuropathic pain Ophthalmologic abnormalities (cornea verticillata and tortuous retinal blood vessels) Hearing impairment Dyshidrosis (hypohidrosis and hyperhidrosis) Hypersensitivity to heat and cold Gastrointestinal disturbances and abdominal pain Lethargy and tiredness Angiokeratomas Onset of renal and cardiac signs (eg, microalbuminuria, proteinuria, abnormal heart rate variability) |
| Early adulthood (age 17 to 30) | Extension of any of the above Proteinuria and progressive renal failure Cardiomyopathy Transient ischemic attacks, strokes Facial dysmorphism |
| Later adulthood (age >30) | Worsening of any of the above Heart disease (eg, left ventricular hypertrophy, angina, arrhythmia, and dyspnea) Transient ischemic attacks, strokes Osteopenia and osteoporosis |
| Source: Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659, by permission of Oxford University Press | |
HISTORY: Cognitive deterioration
Ms. P has had psychiatric symptoms such as depression and anxiety since childhood. However, 3 years ago she started to experience psychological and cognitive deterioration. Medical records indicate that Ms. P described memory and concentration problems over the previous few years. She also reported pain, weakness, and numbness in her left leg after surgery for a work-related back injury, for which she received a financial settlement through workers’ compensation. Shortly thereafter, Ms. P separated from her third husband, moved in with her parents, and found work as a psychiatric nurse. She was dismissed after 6 weeks because she could not learn the electronic medical record system and had difficulty with memory and attention. Her performance on the Mini-Mental State Exam6 at that time was 28 out of 30, which was within normal limits.
After her parents died 3 years ago, Ms. P lived with her daughter, who became her primary caregiver and legal guardian. Ms. P’s daughter notes that her mother’s impulsive and risky behaviors grew more pronounced. Ms. P went on shopping sprees and became sexually promiscuous.
Ms. P’s psychiatric history includes childhood sexual abuse, hospitalization for a suicide attempt at age 19, and courses of psychotherapy and pharmacotherapy. In addition to FD, Ms. P’s medical history consists of coronary artery disease, type 2 diabetes mellitus, hypercholesterolemia, obesity, arthritis, back pain, fibromyalgia, and gastroesophageal reflux disease. Her family history is notable for alcohol abuse (both parents and a brother), lung cancer (mother), myocardial infarction (father), and Alzheimer’s disease (father).
The authors’ observations
Because α-galactosidase A is ubiquitous throughout the body, in addition to neurologic symptoms, FD involves multiple organ systems, with possible dermatologic, renal, gastrointestinal, cardiac, and cerebrovascular dysfunction. Despite growth in FD research, including the Fabry Outcomes Survey,3 the psychosocial and neuropsychiatric implications of the disease remain unclear.7 Behavioral presentations are idiosyncratic and unstable over time, depending on the structures impacted by progressive glycosphingolipid accumulation. Premature cardiovascular events (onset between age 30 and 40 for women), greater incidence of ischemic stroke or transient ischemic attack (7% to 30%), and frequent evidence of white matter lesions put FD patients at greater risk for developing presenile vascular dementia.1,3 Nearly all male FD patients with dementia show some evidence of stroke or transient ischemic attack; cognitive functioning has not been well explored in female patients.4 In a heterogeneous sample of 15 FD patients age 7 to 61, Segal et al8 noted deficits in attention, processing speed, and executive function .75 to 1.95 standard deviations below normative means. No patients in this study had a history of stroke or transient ischemic attack; neuroimaging studies were not reported. Kolodny and Pastores9 suggested multiple mechanisms for cognitive disruption, suggesting that mild dementia late in the disease course could be secondary to diffuse leukomalacia, multiple strokes, or possibly to lipid storage in hippocampal and frontal lobe neurons.
Psychiatric comorbidity
Psychiatric illness, such as depression or a personality disorder, may be comorbid with FD, although pathologic mechanisms remain unclear.7,10,11 Hypothesized mechanisms include:
- psychosocial stress from chronic disease
- white matter changes
- disruption of impaired L-arginine-nitric oxide pathways.7,12
Crosbie et al13 noted that FD patients presented with greater psychological distress as measured by the Minnesota Multiphasic Personality Inventory-2 than patients with Gaucher disease or chronic heart disease. However, no significant differences were found between patients with FD and those diagnosed with a pain disorder. In the Segal et al study, out of 11 adult FD patients, 4 were diagnosed with major depressive disorder, 1 with schizophrenia, 2 with schizotypal personality disorder, and 1 with borderline personality disorder.8
EVALUATION: Brain abnormalities
Head CT scans (conducted 2 years ago and 6 months ago) revealed prominent cortical sulci likely caused by underlying volume loss, especially in bifrontal areas. A brain MRI performed 2 months ago indicated a moderate degree of subcortical atrophy in bilateral frontal and parietal regions. These radiology findings suggest mild to moderate frontal atrophy, mild degree of white matter changes, and slightly enlarged ventricles. An EEG showed background slowing and lack of an alpha rhythm, indicative of cerebral cortical dysfunction.
Ms. P’s α-galactosidase A level was within normal limits; however, normal enzyme levels frequently are reported in symptomatic and asymptomatic female FD patients.14 A dermatology consult confirmed the presence of skin findings characteristic of FD (ie, multiple cherry red papules extensively distributed throughout Ms. P’s chest, abdomen, and back, as well as upper and lower extremities).
Ms. P completed 2 neuropsychological assessments separated by 5 months. For a summary of the results of these tests, see the table titled “Ms. P’s neuropsychological assessment results”. Both assessments revealed grossly impaired intellectual capacity, memory, processing speed, and motor functioning. During the assessment, Ms. P could understand all directions with minimal changes from standardized protocols. Ms. P became insistent that she would not be able to complete memory tasks successfully. She gave up prematurely on tasks, saying they were too difficult. She admitted to guessing on several items because she did not want to continue the task.
Ms. P’s performance on tasks measuring effort and validity of a person’s neuropsychological presentation was consistent with someone exaggerating neurologic symptoms. A person with true dementia may perform as poorly as Ms. P did. However, Ms. P’s scores likely underestimated her level of functioning, even if she was experiencing dementia. Ms. P could not complete tasks individuals with severe dementia complete successfully, such as simple addition and subtraction and digit repetition. Ms. P recalled several recent and remote events, such as her breakfast menu and location of her first assessment, but could not recall words practiced multiple times. Although Ms. P’s scores on a complex card-sorting task were in the impaired range, a detailed review of her pattern indicated that although Ms. P could not generate any correct sorting categories, she made few repetitive responses and errors. This pattern is consistent with someone who understands task requirements, but deliberately avoids answering correctly. This suggests that she retained some ability for hypotheses generation and problem solving; however, because she exaggerated her symptoms, specific deficits could not be determined.
The authors’ observations
Ms. P presented with an interesting manifestation of neuropsychiatric symptoms in the context of FD; however, common cardiac and cerebrovascular features of the disease were not fully developed. Ms. P experienced progressive cognitive and behavioral changes for 2 years before her admission (Table 2), which may represent a prodromal period leading up to what appeared to be a frontally mediated dementia syndrome. Müller et al15 described a patient with FD who displayed a behavioral profile similar to Ms. P’s that included increasingly unstable mood for at least 3 years, borderline personality disorder features, and rapidly fluctuating mood. A case study reported that risperidone, 1 mg/d, used to treat psychosis in a male FD patient caused extrapyramidal symptoms.16
Ms. P presented with no evidence of stroke or transient ischemic attacks, which is atypical for FD patients with cognitive impairment. However, neuroimaging did reveal frontal atrophy that may be associated with her impulse control deficits, risk-taking behavior, emotional instability, and poor judgment. Her cognitive testing was notable for impairment and exaggeration of symptoms consistent with personality disorder symptoms. Possible reasons for exaggeration include a desire to maintain the sick role or secondary gain related to obtaining disability income.
Ms. P’s behavior pattern could be caused by dementia with frontal features, possibly secondary to FD, in combination with personality and psychiatric pathology.
The mainstay of FD treatment is enzyme replacement therapy (ERT), which addresses the underlying enzyme deficiency. Available research indicates that ERT may reduce symptom severity and slow disease progression; however, further studies are needed to determine if it will reduce outcomes such as stroke, ischemic heart disease, or renal disease.2
Table 2
Symptoms that preceded Ms. P’s admission
| Time frame | Symptoms |
|---|---|
| 24 months before admission | Depressed mood Decreased ability to manage independent activities of daily living (eg, finances, cooking) Minimal objective cognitive impairment |
| 12 months before admission | Increased depression Mild to moderate decline in cognitive functioning Visual and auditory hallucinations Impulsivity/poor impulse control Irrational decision-making Increased risky behavior |
| 6 months before admission | Severe cognitive decline with cognitive symptom exaggeration Psychiatric symptom exaggeration Disorganized thinking Continued risky behavior and poor decision-making |
TREATMENT: Persistent deficits
Ms. P is started on risperidone rapidly titrated to 4 mg/d for delusional thinking and behavioral disturbance. After initially improving, she develops delirium when risperidone is increased to 4 mg/d. She has visual hallucinations, marked confusion with disorientation, worsened short-term memory, and an unsteady, shuffling gait. Risperidone is tapered and discontinued and Ms. P’s motor symptoms resolve within 2 days; however, she remains confused and delusional. We start her on quetiapine, 25 mg/d titrated to 50 mg/d, and her agitation and delusional thinking progressively decline. Memantine, titrated to 20 mg/d, and rivastigmine, started at 3 mg/d titrated to 9 mg/d, are added to address her cognitive symptoms.
Over several weeks, Ms. P’s mental status slowly improves and her drug-induced delirium completely resolves. However, she has persistent cognitive impairment characterized by compromised short-term memory and poor insight into her medical and psychological condition. She maintains unrealistic expectations about her ability to live independently and return to the workforce. The treatment team recommends that Ms. P’s daughter pursue guardianship and that she receive around-the-clock supervision after discharge from the hospital.
Table
Ms. P’s neuropsychological assessment results
| June | November | |
|---|---|---|
| Intellectual functioning | ||
| Wechsler Adult Intelligence Scale-III | ||
| FSIQ | 60 | |
| VIQ | 68 | |
| PIQ | 56 | |
| Ravens Colored Progressive Matrices | 70 | |
| Premorbid intellectual functioning estimates | ||
| Peabody Picture Vocabulary Test-2 | 89 | |
| Barona Demographic Estimate | 104 | 104 |
| North American Adult Reading Test | 99 | |
| Memory functioning | ||
| Wechsler Memory Scale-III | ||
| Immediate memory | 45 | |
| General delay memory | 47 | |
| Auditory recognition delay | 55 | |
| California Verbal Learning Test-II | ||
| Trial 1 (immediate recall) | <60 (raw = 3) | |
| Trial 5 | <60 (raw = 3) | |
| Total Words Learned | <60 (raw = 15) | |
| Short Delay Free Recall | <60 (raw = 2) | |
| Long Delay Free Recall | <60 (raw = 4) | |
| Executive functioning | ||
| Trail Making Test A | 88 | 88 |
| Trail Making Test B | failed to understand | failed to understand |
| Wisconsin Card Sort-64 | ||
| Number of categories | <60 (raw = 0) | |
| Errors | 81 | |
| Percent conceptual level responses | 74 | |
| Perseverative responses | 107 | |
| Perseverative errors | 108 | |
| COWAT FAS | 65 | 69 |
| Category exemplar | 69 | 80 |
| Motor functioning | ||
| Finger Tapping Dominant Hand | 68 | |
| Finger Tapping Non-Dominant Hand | 62 | |
| Invalidity/effort | ||
| TOMM | ||
| Trial 1 | raw = 34 | raw = 37 |
| Trial 2 | raw = 42 | raw = 45 |
| Recognition | raw = 44 | |
| MSVT verbal | fail | |
| MSVT nonverbal | fail | |
| Scores provided are standardized (mean = 100; SD = 15). Raw scores are also provided when indicated. COWAT: Controlled oral word association test; FSIQ: Full Scale IQ; MSVT: Medical Symptom Validity Test; PIQ: Performance IQ; TOMM: Test of Memory Malingering; VIQ: Verbal IQ | ||
Related Resources
- National Institute of Neurological Disorders and Stroke. Fabry disease information page. www.ninds.nih.gov/disorders/fabrys/fabrys.htm.
- National Fabry Disease Foundation. www.thenfdf.org.
- Rozenfeld P, Neumann PM. Treatment of Fabry disease: current and emerging strategies. Curr Pharm Biotechnol. 2011;12(6):916-922.
Drug Brand Names
- Donepezil • Aricept
- Memantine • Namenda
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Rivastigmine • Exelon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Disoriented and delusional
Ms. P, a 53-year-old registered nurse, is admitted to the inpatient psychiatric unit with confusion, markedly disorganized thought processes, delayed verbal responsiveness, mood lability, and persecutory delusions. Shortly before hospitalization, Ms. P traveled approximately 360 miles from her daughter’s home with a male companion. Noting changes in her mental status, the man brought Ms. P to the local hospital. She was then transferred to our facility.
At admission, Ms. P is not oriented to time. She denies auditory or visual hallucinations and does not display psychomotor agitation or retardation. She reports her mood as sad and her affect is mildly labile. Insight and judgment are considered poor.
Five years ago, Ms. P and her mother were diagnosed with Fabry’s disease (FD) based on genetic analysis. Both women are carriers for the mutations and Ms. P’s mother was found to have almost absent galactosidase activity.
The authors’ observations
FD is an X-linked recessive glycolipid storage disease caused by deficient activity of the lysosomal storage enzyme α-galactosidase A. The disorder affects both men and women and leads to progressive intracellular accumulation of globotriaosylceramide and other related glycosphingolipids.1,2 The earliest FD symptoms—burning pain and acroparesthesias—typically appear in childhood (Table 1).2 FD often is misdiagnosed in women because women tend to display neurologic symptoms later than men, with typical symptom onset in the teenage years.3,4 Often, these symptoms are confused with psychiatric disorders or vague neurologic or pain syndromes.5 In patients with no family history of FD, accurate diagnosis may not be made until adulthood.
Laboratory, dermatologic, and genetic tests can accurately determine the presence of FD.1 However, because multiple organ systems are involved, initially attributing symptoms to FD is challenging, particularly in women.1,3,5 For men, diagnosis can be established by measuring plasma or urinary globotriaosylceramide or plasma α-galactosidase A in addition to genetic analysis. In women, genetic analysis is a better diagnosis strategy because elevations in globotriaosylceramide or α-galactosidase A may not be prominent. An algorithm for diagnosing and assessing patients with FD has been proposed.2
Table 1
Typical signs and symptoms of Fabry’s disease
| Typical time at onset | Signs/symptoms |
|---|---|
| Childhood and adolescence (age ≤16) | Neuropathic pain Ophthalmologic abnormalities (cornea verticillata and tortuous retinal blood vessels) Hearing impairment Dyshidrosis (hypohidrosis and hyperhidrosis) Hypersensitivity to heat and cold Gastrointestinal disturbances and abdominal pain Lethargy and tiredness Angiokeratomas Onset of renal and cardiac signs (eg, microalbuminuria, proteinuria, abnormal heart rate variability) |
| Early adulthood (age 17 to 30) | Extension of any of the above Proteinuria and progressive renal failure Cardiomyopathy Transient ischemic attacks, strokes Facial dysmorphism |
| Later adulthood (age >30) | Worsening of any of the above Heart disease (eg, left ventricular hypertrophy, angina, arrhythmia, and dyspnea) Transient ischemic attacks, strokes Osteopenia and osteoporosis |
| Source: Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659, by permission of Oxford University Press | |
HISTORY: Cognitive deterioration
Ms. P has had psychiatric symptoms such as depression and anxiety since childhood. However, 3 years ago she started to experience psychological and cognitive deterioration. Medical records indicate that Ms. P described memory and concentration problems over the previous few years. She also reported pain, weakness, and numbness in her left leg after surgery for a work-related back injury, for which she received a financial settlement through workers’ compensation. Shortly thereafter, Ms. P separated from her third husband, moved in with her parents, and found work as a psychiatric nurse. She was dismissed after 6 weeks because she could not learn the electronic medical record system and had difficulty with memory and attention. Her performance on the Mini-Mental State Exam6 at that time was 28 out of 30, which was within normal limits.
After her parents died 3 years ago, Ms. P lived with her daughter, who became her primary caregiver and legal guardian. Ms. P’s daughter notes that her mother’s impulsive and risky behaviors grew more pronounced. Ms. P went on shopping sprees and became sexually promiscuous.
Ms. P’s psychiatric history includes childhood sexual abuse, hospitalization for a suicide attempt at age 19, and courses of psychotherapy and pharmacotherapy. In addition to FD, Ms. P’s medical history consists of coronary artery disease, type 2 diabetes mellitus, hypercholesterolemia, obesity, arthritis, back pain, fibromyalgia, and gastroesophageal reflux disease. Her family history is notable for alcohol abuse (both parents and a brother), lung cancer (mother), myocardial infarction (father), and Alzheimer’s disease (father).
The authors’ observations
Because α-galactosidase A is ubiquitous throughout the body, in addition to neurologic symptoms, FD involves multiple organ systems, with possible dermatologic, renal, gastrointestinal, cardiac, and cerebrovascular dysfunction. Despite growth in FD research, including the Fabry Outcomes Survey,3 the psychosocial and neuropsychiatric implications of the disease remain unclear.7 Behavioral presentations are idiosyncratic and unstable over time, depending on the structures impacted by progressive glycosphingolipid accumulation. Premature cardiovascular events (onset between age 30 and 40 for women), greater incidence of ischemic stroke or transient ischemic attack (7% to 30%), and frequent evidence of white matter lesions put FD patients at greater risk for developing presenile vascular dementia.1,3 Nearly all male FD patients with dementia show some evidence of stroke or transient ischemic attack; cognitive functioning has not been well explored in female patients.4 In a heterogeneous sample of 15 FD patients age 7 to 61, Segal et al8 noted deficits in attention, processing speed, and executive function .75 to 1.95 standard deviations below normative means. No patients in this study had a history of stroke or transient ischemic attack; neuroimaging studies were not reported. Kolodny and Pastores9 suggested multiple mechanisms for cognitive disruption, suggesting that mild dementia late in the disease course could be secondary to diffuse leukomalacia, multiple strokes, or possibly to lipid storage in hippocampal and frontal lobe neurons.
Psychiatric comorbidity
Psychiatric illness, such as depression or a personality disorder, may be comorbid with FD, although pathologic mechanisms remain unclear.7,10,11 Hypothesized mechanisms include:
- psychosocial stress from chronic disease
- white matter changes
- disruption of impaired L-arginine-nitric oxide pathways.7,12
Crosbie et al13 noted that FD patients presented with greater psychological distress as measured by the Minnesota Multiphasic Personality Inventory-2 than patients with Gaucher disease or chronic heart disease. However, no significant differences were found between patients with FD and those diagnosed with a pain disorder. In the Segal et al study, out of 11 adult FD patients, 4 were diagnosed with major depressive disorder, 1 with schizophrenia, 2 with schizotypal personality disorder, and 1 with borderline personality disorder.8
EVALUATION: Brain abnormalities
Head CT scans (conducted 2 years ago and 6 months ago) revealed prominent cortical sulci likely caused by underlying volume loss, especially in bifrontal areas. A brain MRI performed 2 months ago indicated a moderate degree of subcortical atrophy in bilateral frontal and parietal regions. These radiology findings suggest mild to moderate frontal atrophy, mild degree of white matter changes, and slightly enlarged ventricles. An EEG showed background slowing and lack of an alpha rhythm, indicative of cerebral cortical dysfunction.
Ms. P’s α-galactosidase A level was within normal limits; however, normal enzyme levels frequently are reported in symptomatic and asymptomatic female FD patients.14 A dermatology consult confirmed the presence of skin findings characteristic of FD (ie, multiple cherry red papules extensively distributed throughout Ms. P’s chest, abdomen, and back, as well as upper and lower extremities).
Ms. P completed 2 neuropsychological assessments separated by 5 months. For a summary of the results of these tests, see the table titled “Ms. P’s neuropsychological assessment results”. Both assessments revealed grossly impaired intellectual capacity, memory, processing speed, and motor functioning. During the assessment, Ms. P could understand all directions with minimal changes from standardized protocols. Ms. P became insistent that she would not be able to complete memory tasks successfully. She gave up prematurely on tasks, saying they were too difficult. She admitted to guessing on several items because she did not want to continue the task.
Ms. P’s performance on tasks measuring effort and validity of a person’s neuropsychological presentation was consistent with someone exaggerating neurologic symptoms. A person with true dementia may perform as poorly as Ms. P did. However, Ms. P’s scores likely underestimated her level of functioning, even if she was experiencing dementia. Ms. P could not complete tasks individuals with severe dementia complete successfully, such as simple addition and subtraction and digit repetition. Ms. P recalled several recent and remote events, such as her breakfast menu and location of her first assessment, but could not recall words practiced multiple times. Although Ms. P’s scores on a complex card-sorting task were in the impaired range, a detailed review of her pattern indicated that although Ms. P could not generate any correct sorting categories, she made few repetitive responses and errors. This pattern is consistent with someone who understands task requirements, but deliberately avoids answering correctly. This suggests that she retained some ability for hypotheses generation and problem solving; however, because she exaggerated her symptoms, specific deficits could not be determined.
The authors’ observations
Ms. P presented with an interesting manifestation of neuropsychiatric symptoms in the context of FD; however, common cardiac and cerebrovascular features of the disease were not fully developed. Ms. P experienced progressive cognitive and behavioral changes for 2 years before her admission (Table 2), which may represent a prodromal period leading up to what appeared to be a frontally mediated dementia syndrome. Müller et al15 described a patient with FD who displayed a behavioral profile similar to Ms. P’s that included increasingly unstable mood for at least 3 years, borderline personality disorder features, and rapidly fluctuating mood. A case study reported that risperidone, 1 mg/d, used to treat psychosis in a male FD patient caused extrapyramidal symptoms.16
Ms. P presented with no evidence of stroke or transient ischemic attacks, which is atypical for FD patients with cognitive impairment. However, neuroimaging did reveal frontal atrophy that may be associated with her impulse control deficits, risk-taking behavior, emotional instability, and poor judgment. Her cognitive testing was notable for impairment and exaggeration of symptoms consistent with personality disorder symptoms. Possible reasons for exaggeration include a desire to maintain the sick role or secondary gain related to obtaining disability income.
Ms. P’s behavior pattern could be caused by dementia with frontal features, possibly secondary to FD, in combination with personality and psychiatric pathology.
The mainstay of FD treatment is enzyme replacement therapy (ERT), which addresses the underlying enzyme deficiency. Available research indicates that ERT may reduce symptom severity and slow disease progression; however, further studies are needed to determine if it will reduce outcomes such as stroke, ischemic heart disease, or renal disease.2
Table 2
Symptoms that preceded Ms. P’s admission
| Time frame | Symptoms |
|---|---|
| 24 months before admission | Depressed mood Decreased ability to manage independent activities of daily living (eg, finances, cooking) Minimal objective cognitive impairment |
| 12 months before admission | Increased depression Mild to moderate decline in cognitive functioning Visual and auditory hallucinations Impulsivity/poor impulse control Irrational decision-making Increased risky behavior |
| 6 months before admission | Severe cognitive decline with cognitive symptom exaggeration Psychiatric symptom exaggeration Disorganized thinking Continued risky behavior and poor decision-making |
TREATMENT: Persistent deficits
Ms. P is started on risperidone rapidly titrated to 4 mg/d for delusional thinking and behavioral disturbance. After initially improving, she develops delirium when risperidone is increased to 4 mg/d. She has visual hallucinations, marked confusion with disorientation, worsened short-term memory, and an unsteady, shuffling gait. Risperidone is tapered and discontinued and Ms. P’s motor symptoms resolve within 2 days; however, she remains confused and delusional. We start her on quetiapine, 25 mg/d titrated to 50 mg/d, and her agitation and delusional thinking progressively decline. Memantine, titrated to 20 mg/d, and rivastigmine, started at 3 mg/d titrated to 9 mg/d, are added to address her cognitive symptoms.
Over several weeks, Ms. P’s mental status slowly improves and her drug-induced delirium completely resolves. However, she has persistent cognitive impairment characterized by compromised short-term memory and poor insight into her medical and psychological condition. She maintains unrealistic expectations about her ability to live independently and return to the workforce. The treatment team recommends that Ms. P’s daughter pursue guardianship and that she receive around-the-clock supervision after discharge from the hospital.
Table
Ms. P’s neuropsychological assessment results
| June | November | |
|---|---|---|
| Intellectual functioning | ||
| Wechsler Adult Intelligence Scale-III | ||
| FSIQ | 60 | |
| VIQ | 68 | |
| PIQ | 56 | |
| Ravens Colored Progressive Matrices | 70 | |
| Premorbid intellectual functioning estimates | ||
| Peabody Picture Vocabulary Test-2 | 89 | |
| Barona Demographic Estimate | 104 | 104 |
| North American Adult Reading Test | 99 | |
| Memory functioning | ||
| Wechsler Memory Scale-III | ||
| Immediate memory | 45 | |
| General delay memory | 47 | |
| Auditory recognition delay | 55 | |
| California Verbal Learning Test-II | ||
| Trial 1 (immediate recall) | <60 (raw = 3) | |
| Trial 5 | <60 (raw = 3) | |
| Total Words Learned | <60 (raw = 15) | |
| Short Delay Free Recall | <60 (raw = 2) | |
| Long Delay Free Recall | <60 (raw = 4) | |
| Executive functioning | ||
| Trail Making Test A | 88 | 88 |
| Trail Making Test B | failed to understand | failed to understand |
| Wisconsin Card Sort-64 | ||
| Number of categories | <60 (raw = 0) | |
| Errors | 81 | |
| Percent conceptual level responses | 74 | |
| Perseverative responses | 107 | |
| Perseverative errors | 108 | |
| COWAT FAS | 65 | 69 |
| Category exemplar | 69 | 80 |
| Motor functioning | ||
| Finger Tapping Dominant Hand | 68 | |
| Finger Tapping Non-Dominant Hand | 62 | |
| Invalidity/effort | ||
| TOMM | ||
| Trial 1 | raw = 34 | raw = 37 |
| Trial 2 | raw = 42 | raw = 45 |
| Recognition | raw = 44 | |
| MSVT verbal | fail | |
| MSVT nonverbal | fail | |
| Scores provided are standardized (mean = 100; SD = 15). Raw scores are also provided when indicated. COWAT: Controlled oral word association test; FSIQ: Full Scale IQ; MSVT: Medical Symptom Validity Test; PIQ: Performance IQ; TOMM: Test of Memory Malingering; VIQ: Verbal IQ | ||
Related Resources
- National Institute of Neurological Disorders and Stroke. Fabry disease information page. www.ninds.nih.gov/disorders/fabrys/fabrys.htm.
- National Fabry Disease Foundation. www.thenfdf.org.
- Rozenfeld P, Neumann PM. Treatment of Fabry disease: current and emerging strategies. Curr Pharm Biotechnol. 2011;12(6):916-922.
Drug Brand Names
- Donepezil • Aricept
- Memantine • Namenda
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Rivastigmine • Exelon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539-548.
2. Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659.
3. Deegan PB, Baehner AF, Barba Romero MA, et al. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43(4):347-352.
4. Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurol. 2006;5(9):791-795.
5. Møller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol. 2007;3(2):95-106.
6. Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
7. Müller MJ. Neuropsychiatric and psychosocial aspects of Fabry disease. In: Mehta A Beck M, Sunder-Plassman G, eds. Fabry disease: perspectives from 5 years of FOS. Oxford, United Kingdom: Oxford PharmaGenesis Ltd; 2006. http://www.ncbi.nlm.nih.gov/books/nbk11618. Accessed October 31, 2011.
8. Segal P, Kohn Y, Pollak Y, et al. Psychiatric and cognitive profile in Anderson-Fabry patients: a preliminary study. J Inherit Metab Dis. 2010;33(4):429-436.
9. Kolodny EH, Pastores GM. Anderson-Fabry disease: Extrarenal neurologic manifestations. J Am Soc Nephrol. 2002;13(suppl 2):S150-153.
10. Grewal RP. Psychiatric disorders in patients with Fabry disease. Int J Psychiatry Med. 1993;23(3):307-312.
11. Müller MJ, Müller KM, Dascalescu A, et al. Psychiatric and neuropsychological signs and symptoms in patients with Fabry disease: literature review [in German]. Fortschr Neurol Psychiatr. 2005;73(11):687-693.
12. Segal P, Raas-Rothschild A. Neuropsychiatric manifestations of AFD. In: Elstein D Altarescu G, Beck M, eds. Fabry disease. New York, NY: Springer; 2010:321–324.
13. Crosbie TW, Packman W, Packman S. Psychological aspects of patients with Fabry disease. J Inherit Metab Dis. 2009;32(6):745-753.
14. Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J Am Coll Cardiol. 2008;51(21):2082.-
15. Müller MJ, Fellgiebel A, Scheurich A, et al. Recurrent brief depression in female patient with Fabry disease. Bipolar Disord. 2006;8(4):418-419.
16. Shen YC, Haw-Ming L, Lin CC, et al. Psychosis in a patient with Fabry’s disease and treatment with aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):779-780.
1. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539-548.
2. Mehta A, Beck M, Eyskens F, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641-659.
3. Deegan PB, Baehner AF, Barba Romero MA, et al. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43(4):347-352.
4. Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurol. 2006;5(9):791-795.
5. Møller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol. 2007;3(2):95-106.
6. Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
7. Müller MJ. Neuropsychiatric and psychosocial aspects of Fabry disease. In: Mehta A Beck M, Sunder-Plassman G, eds. Fabry disease: perspectives from 5 years of FOS. Oxford, United Kingdom: Oxford PharmaGenesis Ltd; 2006. http://www.ncbi.nlm.nih.gov/books/nbk11618. Accessed October 31, 2011.
8. Segal P, Kohn Y, Pollak Y, et al. Psychiatric and cognitive profile in Anderson-Fabry patients: a preliminary study. J Inherit Metab Dis. 2010;33(4):429-436.
9. Kolodny EH, Pastores GM. Anderson-Fabry disease: Extrarenal neurologic manifestations. J Am Soc Nephrol. 2002;13(suppl 2):S150-153.
10. Grewal RP. Psychiatric disorders in patients with Fabry disease. Int J Psychiatry Med. 1993;23(3):307-312.
11. Müller MJ, Müller KM, Dascalescu A, et al. Psychiatric and neuropsychological signs and symptoms in patients with Fabry disease: literature review [in German]. Fortschr Neurol Psychiatr. 2005;73(11):687-693.
12. Segal P, Raas-Rothschild A. Neuropsychiatric manifestations of AFD. In: Elstein D Altarescu G, Beck M, eds. Fabry disease. New York, NY: Springer; 2010:321–324.
13. Crosbie TW, Packman W, Packman S. Psychological aspects of patients with Fabry disease. J Inherit Metab Dis. 2009;32(6):745-753.
14. Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J Am Coll Cardiol. 2008;51(21):2082.-
15. Müller MJ, Fellgiebel A, Scheurich A, et al. Recurrent brief depression in female patient with Fabry disease. Bipolar Disord. 2006;8(4):418-419.
16. Shen YC, Haw-Ming L, Lin CC, et al. Psychosis in a patient with Fabry’s disease and treatment with aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):779-780.
A questionable diagnosis
CASE: Space traveler
Mr. O, age 69, is a patient at a long-term psychiatric hospital. He has a 56-year psychiatric history, a current diagnosis of schizoaffective disorder, and suffered a torn rotator cuff approximately 5 years ago. His medication regimen is haloperidol decanoate, 100 mg IM every month, duloxetine, 60 mg/d, and naproxen, as needed for chronic pain.
He frequently lies on the floor. Attendants urge him to get up and join groups or sit with other patients but he complains of pain and soon finds another spot on the floor to use as a bed.
Eight months earlier, a homeless shelter sent Mr. O to the emergency room (ER) because he tried to eat a dollar bill and a sock. In the ER he was inattentive, with loose associations and bizarre delusions; he believed he was on a spaceship. Mr. O was admitted to the hospital, where clinicians noted that his behavior remained bizarre and he complained of insomnia. They also noted a history of setting fires, which complicated discharge planning and contributed to their decision to transfer him to our psychiatric facility for longer-term care.
During our initial interview, Mr. O readily picks himself off the floor. His responses are logical and direct but abrupt and unelaborated. His first and most vehement complaint is pain. Zolpidem, he says, is the only treatment that helps.
He says he began using zolpidem approximately 5 years ago because pain from a shoulder injury kept him awake at night. When he could not obtain the drug by prescription, he bought it on the street. One day when living in the homeless shelter, he took 30 or 40 mg of zolpidem, then “blacked out” and awoke in the ER.
His first experience with psychiatric treatment was the result of problems getting along with his single mother because of “petty things” such as shooting off a BB gun in their apartment, he says. As a teenager he was sent to a boarding school; as a young adult, to a psychiatric hospital. After his release he returned to his mother’s apartment. He worked steadily for 20 years before he obtained Social Security benefits, and then worked intermittently “off the books” until approximately 15 years ago. Mr. O lived with his mother until her death 17 years earlier, and then in her apartment alone until a fire, which he set accidentally by smoking in bed after taking zolpidem, forced him to leave 3 years ago. He says, “My whole life was in that place.” He was admitted to a psychiatric hospital for an unknown reason, which was his first psychiatric admission in 40 years. After he was released from the hospital, Mr. O lived in various homeless shelters and adult homes until his current hospitalization.
The author’s observations
An effective and well-tolerated drug with a reputation for rarely being abused, zolpidem is widely prescribed as a hypnotic. Zolpidem and benzodiazepines have different chemical structures but both act at the GABAA receptor and have comparable behavioral effects.1 The reported incidence of zolpidem abuse is much lower than the reported rate of benzodiazepine abuse when used for sleep2; however, abuse, dependence, and withdrawal have been reported.2-4 Zolpidem abuse seems to be more common among patients with a history of abusing other substances or a history of psychiatric illness.2 A French study4 found that abusers fell into 2 groups. The younger group (median age 35) used higher doses—a median of 300 mg/d—and took zolpidem in the daytime to achieve euphoria. A second, older group (median age 42) used lower doses—a median of 200 mg/d—at nighttime to sleep.
There are few reports of delirium and symptoms such as visual hallucinations and distortions associated with zolpidem use.5,6 These reactions have occurred in persons without a history of psychosis. They usually are associated with doses ≥10 mg.
In the ER Mr. O showed a disturbance in consciousness with inability to focus attention and a perceptual disturbance (he believed he was in a spaceship) that developed over hours to days. He met criteria for delirium, possibly caused by zolpidem, but his presentation also could have been attributable to an underlying psychiatric disorder.
ER and inpatient psychiatrists noted Mr. O was intoxicated with zolpidem when the shelter brought him to the ER, but both groups diagnosed schizoaffective disorder and treated him with antipsychotics. They saw his >50-year psychiatric history as evidence of an underlying, long-standing condition such as schizoaffective illness.
However, features of Mr. O’s illness are not typical of a chronic psychotic illness. He recalls psychiatric hospitalizations in his youth and recently, but not for the 40 years in between. Mr. O says he has never experienced auditory hallucinations. For these reasons, our treatment team obtains old medical records to investigate his early history (Table).
Table
Mr. O’s clinical course
| Age | Symptoms/behaviors | Diagnosis |
|---|---|---|
| 17 | Temper tantrums and destructive behaviors. No delusions or hallucinations but a flat affect and hostile attitude | Primary behavior disorder, simple adult maladjustment |
| 22 | Returned to the psychiatric hospital when his welfare payments stopped; “psychopathic” symptoms; described as defiant and resented authority and regular work | Primary behavior disorder |
| 24 | His mother complained that he stole from her and carried a weapon; while hospitalized, described as manageable and without overt psychotic symptoms | Primary behavior disorder |
| 26 | Arrested for causing property damage while intoxicated on alcohol; silly laugh, loose associations, irrelevant and incoherent speech, and believed hospital staff were against him | Psychosis with psychopathic personality |
| 66 | A fire that he set accidentally while smoking in bed after taking zolpidem destroyed his home | Diagnosis unknown |
| 68 | Transferred from a homeless shelter to the ER after he took 30 to 40 mg of zolpidem and exhibited bizarre behaviors | Schizoaffective disorder |
| 69 | More spontaneous, remains logical and relevant after haloperidol is discontinued; no delusions or hallucinations, still complains of pain | Substance use disorder and personality disorder |
| ER: emergency room | ||
HISTORY: Destructive and defiant
Mr. O’s mother reported that he had been a nervous, restless child who would scream and yell at the slightest provocation. At age 10 he became wantonly destructive. His mother bought him an expensive toy that he destroyed after a short time; he asked for another toy, which he also destroyed. When such behavior became more frequent, she took him to a city hospital, where he was treated for 6 weeks and released at age 13. He was sent to a boarding school but soon was expelled for drinking and selling beer.
Mr. O was admitted to long-term psychiatric facilities 6 times in the next 10 years, from the late 1950s to the late 1960s. He was first admitted at age 17 for temper tantrums during which he fired an air rifle and smashed windows in the home he shared with his mother. During examination he had no delusions or hallucinations but did have flat affect and a hostile attitude. Doctors documented that almost all his tantrums were as a result of interactions with his mother.
Records from this psychiatric admission state that Mr. O showed no unusual distractibility, “psychotic trends,” or paranoid thinking. After approximately 6 months in the hospital he was discharged home with the diagnosis of primary behavior disorder, simple adult maladjustment. Mr. O, who was age 18 at the time, and his mother were eager for him to complete high school and learn auto mechanics.
Nine months later, he returned to the psychiatric facility because of excessive drinking and inability to secure employment, according to his records. In the hospital, he was productive and reliable. When he was discharged home 3 months later, doctors wrote that his determination to stop drinking was firmly fixed. They encouraged Mr. O to complete high school as a night student and find employment during the day. His mother was delighted with his improvement.
A third admission, less than 2 months later, occurred after he broke a window during an argument with his mother. He had a job but quit. After 5 months he was discharged with the same diagnosis of primary behavior disorder, but his mother would not let him back in her home. He was referred to the social service department to be placed on welfare.
A year later, Mr. O had trouble managing his welfare allotment and moved repeatedly. He said he returned to the psychiatric hospital because his welfare payments had been discontinued. During this admission, doctors noted “psychopathic” symptoms; Mr. O was defiant and resented authority and regular work. Mr. O eloped from the hospital several times and brought beer into the building. After 18 months he was discharged with the same diagnosis, with plans to apply for welfare. He was not prescribed medication.
Mr. O’s fifth admission came nearly 2 years later after his mother complained that he stole from her home and carried a weapon. In the hospital he was described as manageable and without overt psychotic symptoms. When he was discharged a little more than a year after being admitted, doctors wrote that he was a psychopath who had a history of drinking, stealing, and delinquent tendencies as a teenager. His diagnosis remained primary behavior disorder.
A year after this discharge, Mr. O was arrested for causing serious property damage when he was intoxicated on alcohol. Subsequently he was readmitted.
After a few months in the hospital, Mr. O changed. He developed a silly laugh, loose associations, irrelevant and incoherent speech, and a belief that hospital staff were against him. Although Mr. O denied auditory hallucinations, a psychiatrist wrote that he seemed to be experiencing hallucinations and prescribed chlorpromazine. The next day Mr. O slashed his arms and legs in several places, requiring many sutures. His diagnosis was changed to psychosis with psychopathic personality. However, within a few months, psychiatrists determined that Mr. O had recovered, so they stopped chlorpromazine. Months later, clinicians wrote that Mr. O was idle most of the time, neat, clean, and not involved in arguments with other patients. He was discharged after 1 month in the hospital.
Over the years, psychiatrists had differing opinions about Mr. O’s diagnosis. One noted that his mental illness was characterized by emotional instability and poor judgment. He had impulsive reactions without regard for others, rapid mood swings, irritability, and depression with transient paranoia. Another clinician detected evidence of schizoid personality disorder because Mr. O did not experience hallucinations or a gross thought disorder, but did have rambling, circumstantial, autistic (unrealistic), and ambivalent thought content. Another psychiatrist wrote Mr. O best fit in the category of psychosis with psychopathic personality, which was his diagnosis at discharge from his sixth hospitalization.
The author’s observations
Mr. O’s old medical records revealed the diagnostic thinking and treatment practices of a past era. They did not demonstrate that Mr. O met current criteria for schizophrenia or schizoaffective disorder, although he may have had a brief psychotic episode. Because there was little support for a diagnosis of schizoaffective illness and haloperidol use, we stopped the drug but continued duloxetine for chronic pain. It was clear that he has a substance use disorder and perhaps met criteria for antisocial personality disorder.
OUTCOME: Further explanations
Approximately 2 months after stopping haloperidol, Mr. O is more spontaneous, logical, and relevant. He does not have delusions or hallucinations. Despite further attempts at pain management with physical therapy and increased doses of duloxetine, he still complains of pain. We do not prescribe zolpidem.
Mr. O is unwilling to discuss the incident more than 40 years ago when he cut his arms and legs except to say, “That’s the past. My life wasn’t so good at that time.” When we ask why he had been a client of Adult Protective Services 5 years before he was burned out of his apartment, he admitted that he was 21 months in arrears in his rent. “I used to do this thing called crack,” he explains. He was discharged to an adult home with a prescription for duloxetine after he promised to never smoke in his room again.
Related Resource
- Aggarwal A, Sharma DD. Zolpidem withdrawal delirium: a case report. J Neuropsychiatry Clin Neurosci. 2010;22(4):451.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Duloxetine • Cymbalta
- Haloperidol decanoate • Haloperidol decanoate
- Haloperidol • Haldol
- Naproxen • Naproxyn, Aleve, others
- Zolpidem • Ambien
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rush CR. Behavioral pharmacology of zolpidem relative to benzodiazepines: a review. Pharmacol Biochem Behav. 1998;61(3):253-269.
2. Hajak G, Müller WE, Wittchen HU, et al. Abuse and dependence potential for the non-benzodiazepine hypnotics zolpidem and zopiclone: a review of case reports and epidemiological data. Addiction. 2003;98(10):1371-1378.
3. Madrak LN, Rosenberg M. Zolpidem abuse. Am J Psychiatry. 2001;158(8):1330-1331.
4. Victorri-Vigneau C, Dailly E, Veyrac G, et al. Evidence of zolpidem abuse and dependence: results of the French Centre for Evaluation and Information on Pharmacodepencence (CEIP) network survey. Br J Clin Pharmacol. 2007;64(2):198-209.
5. Markowitz JS, Brewerton TD. Zolpidem-induced psychosis. Ann Clin Psychiatry. 1996;8(2):89-91.
6. Tsai MJ, Huang YB, Wu PC. A novel clinical pattern of visual hallucination after zolpidem use. J Toxicol Clin Toxicol. 2003;41(6):869-872.
CASE: Space traveler
Mr. O, age 69, is a patient at a long-term psychiatric hospital. He has a 56-year psychiatric history, a current diagnosis of schizoaffective disorder, and suffered a torn rotator cuff approximately 5 years ago. His medication regimen is haloperidol decanoate, 100 mg IM every month, duloxetine, 60 mg/d, and naproxen, as needed for chronic pain.
He frequently lies on the floor. Attendants urge him to get up and join groups or sit with other patients but he complains of pain and soon finds another spot on the floor to use as a bed.
Eight months earlier, a homeless shelter sent Mr. O to the emergency room (ER) because he tried to eat a dollar bill and a sock. In the ER he was inattentive, with loose associations and bizarre delusions; he believed he was on a spaceship. Mr. O was admitted to the hospital, where clinicians noted that his behavior remained bizarre and he complained of insomnia. They also noted a history of setting fires, which complicated discharge planning and contributed to their decision to transfer him to our psychiatric facility for longer-term care.
During our initial interview, Mr. O readily picks himself off the floor. His responses are logical and direct but abrupt and unelaborated. His first and most vehement complaint is pain. Zolpidem, he says, is the only treatment that helps.
He says he began using zolpidem approximately 5 years ago because pain from a shoulder injury kept him awake at night. When he could not obtain the drug by prescription, he bought it on the street. One day when living in the homeless shelter, he took 30 or 40 mg of zolpidem, then “blacked out” and awoke in the ER.
His first experience with psychiatric treatment was the result of problems getting along with his single mother because of “petty things” such as shooting off a BB gun in their apartment, he says. As a teenager he was sent to a boarding school; as a young adult, to a psychiatric hospital. After his release he returned to his mother’s apartment. He worked steadily for 20 years before he obtained Social Security benefits, and then worked intermittently “off the books” until approximately 15 years ago. Mr. O lived with his mother until her death 17 years earlier, and then in her apartment alone until a fire, which he set accidentally by smoking in bed after taking zolpidem, forced him to leave 3 years ago. He says, “My whole life was in that place.” He was admitted to a psychiatric hospital for an unknown reason, which was his first psychiatric admission in 40 years. After he was released from the hospital, Mr. O lived in various homeless shelters and adult homes until his current hospitalization.
The author’s observations
An effective and well-tolerated drug with a reputation for rarely being abused, zolpidem is widely prescribed as a hypnotic. Zolpidem and benzodiazepines have different chemical structures but both act at the GABAA receptor and have comparable behavioral effects.1 The reported incidence of zolpidem abuse is much lower than the reported rate of benzodiazepine abuse when used for sleep2; however, abuse, dependence, and withdrawal have been reported.2-4 Zolpidem abuse seems to be more common among patients with a history of abusing other substances or a history of psychiatric illness.2 A French study4 found that abusers fell into 2 groups. The younger group (median age 35) used higher doses—a median of 300 mg/d—and took zolpidem in the daytime to achieve euphoria. A second, older group (median age 42) used lower doses—a median of 200 mg/d—at nighttime to sleep.
There are few reports of delirium and symptoms such as visual hallucinations and distortions associated with zolpidem use.5,6 These reactions have occurred in persons without a history of psychosis. They usually are associated with doses ≥10 mg.
In the ER Mr. O showed a disturbance in consciousness with inability to focus attention and a perceptual disturbance (he believed he was in a spaceship) that developed over hours to days. He met criteria for delirium, possibly caused by zolpidem, but his presentation also could have been attributable to an underlying psychiatric disorder.
ER and inpatient psychiatrists noted Mr. O was intoxicated with zolpidem when the shelter brought him to the ER, but both groups diagnosed schizoaffective disorder and treated him with antipsychotics. They saw his >50-year psychiatric history as evidence of an underlying, long-standing condition such as schizoaffective illness.
However, features of Mr. O’s illness are not typical of a chronic psychotic illness. He recalls psychiatric hospitalizations in his youth and recently, but not for the 40 years in between. Mr. O says he has never experienced auditory hallucinations. For these reasons, our treatment team obtains old medical records to investigate his early history (Table).
Table
Mr. O’s clinical course
| Age | Symptoms/behaviors | Diagnosis |
|---|---|---|
| 17 | Temper tantrums and destructive behaviors. No delusions or hallucinations but a flat affect and hostile attitude | Primary behavior disorder, simple adult maladjustment |
| 22 | Returned to the psychiatric hospital when his welfare payments stopped; “psychopathic” symptoms; described as defiant and resented authority and regular work | Primary behavior disorder |
| 24 | His mother complained that he stole from her and carried a weapon; while hospitalized, described as manageable and without overt psychotic symptoms | Primary behavior disorder |
| 26 | Arrested for causing property damage while intoxicated on alcohol; silly laugh, loose associations, irrelevant and incoherent speech, and believed hospital staff were against him | Psychosis with psychopathic personality |
| 66 | A fire that he set accidentally while smoking in bed after taking zolpidem destroyed his home | Diagnosis unknown |
| 68 | Transferred from a homeless shelter to the ER after he took 30 to 40 mg of zolpidem and exhibited bizarre behaviors | Schizoaffective disorder |
| 69 | More spontaneous, remains logical and relevant after haloperidol is discontinued; no delusions or hallucinations, still complains of pain | Substance use disorder and personality disorder |
| ER: emergency room | ||
HISTORY: Destructive and defiant
Mr. O’s mother reported that he had been a nervous, restless child who would scream and yell at the slightest provocation. At age 10 he became wantonly destructive. His mother bought him an expensive toy that he destroyed after a short time; he asked for another toy, which he also destroyed. When such behavior became more frequent, she took him to a city hospital, where he was treated for 6 weeks and released at age 13. He was sent to a boarding school but soon was expelled for drinking and selling beer.
Mr. O was admitted to long-term psychiatric facilities 6 times in the next 10 years, from the late 1950s to the late 1960s. He was first admitted at age 17 for temper tantrums during which he fired an air rifle and smashed windows in the home he shared with his mother. During examination he had no delusions or hallucinations but did have flat affect and a hostile attitude. Doctors documented that almost all his tantrums were as a result of interactions with his mother.
Records from this psychiatric admission state that Mr. O showed no unusual distractibility, “psychotic trends,” or paranoid thinking. After approximately 6 months in the hospital he was discharged home with the diagnosis of primary behavior disorder, simple adult maladjustment. Mr. O, who was age 18 at the time, and his mother were eager for him to complete high school and learn auto mechanics.
Nine months later, he returned to the psychiatric facility because of excessive drinking and inability to secure employment, according to his records. In the hospital, he was productive and reliable. When he was discharged home 3 months later, doctors wrote that his determination to stop drinking was firmly fixed. They encouraged Mr. O to complete high school as a night student and find employment during the day. His mother was delighted with his improvement.
A third admission, less than 2 months later, occurred after he broke a window during an argument with his mother. He had a job but quit. After 5 months he was discharged with the same diagnosis of primary behavior disorder, but his mother would not let him back in her home. He was referred to the social service department to be placed on welfare.
A year later, Mr. O had trouble managing his welfare allotment and moved repeatedly. He said he returned to the psychiatric hospital because his welfare payments had been discontinued. During this admission, doctors noted “psychopathic” symptoms; Mr. O was defiant and resented authority and regular work. Mr. O eloped from the hospital several times and brought beer into the building. After 18 months he was discharged with the same diagnosis, with plans to apply for welfare. He was not prescribed medication.
Mr. O’s fifth admission came nearly 2 years later after his mother complained that he stole from her home and carried a weapon. In the hospital he was described as manageable and without overt psychotic symptoms. When he was discharged a little more than a year after being admitted, doctors wrote that he was a psychopath who had a history of drinking, stealing, and delinquent tendencies as a teenager. His diagnosis remained primary behavior disorder.
A year after this discharge, Mr. O was arrested for causing serious property damage when he was intoxicated on alcohol. Subsequently he was readmitted.
After a few months in the hospital, Mr. O changed. He developed a silly laugh, loose associations, irrelevant and incoherent speech, and a belief that hospital staff were against him. Although Mr. O denied auditory hallucinations, a psychiatrist wrote that he seemed to be experiencing hallucinations and prescribed chlorpromazine. The next day Mr. O slashed his arms and legs in several places, requiring many sutures. His diagnosis was changed to psychosis with psychopathic personality. However, within a few months, psychiatrists determined that Mr. O had recovered, so they stopped chlorpromazine. Months later, clinicians wrote that Mr. O was idle most of the time, neat, clean, and not involved in arguments with other patients. He was discharged after 1 month in the hospital.
Over the years, psychiatrists had differing opinions about Mr. O’s diagnosis. One noted that his mental illness was characterized by emotional instability and poor judgment. He had impulsive reactions without regard for others, rapid mood swings, irritability, and depression with transient paranoia. Another clinician detected evidence of schizoid personality disorder because Mr. O did not experience hallucinations or a gross thought disorder, but did have rambling, circumstantial, autistic (unrealistic), and ambivalent thought content. Another psychiatrist wrote Mr. O best fit in the category of psychosis with psychopathic personality, which was his diagnosis at discharge from his sixth hospitalization.
The author’s observations
Mr. O’s old medical records revealed the diagnostic thinking and treatment practices of a past era. They did not demonstrate that Mr. O met current criteria for schizophrenia or schizoaffective disorder, although he may have had a brief psychotic episode. Because there was little support for a diagnosis of schizoaffective illness and haloperidol use, we stopped the drug but continued duloxetine for chronic pain. It was clear that he has a substance use disorder and perhaps met criteria for antisocial personality disorder.
OUTCOME: Further explanations
Approximately 2 months after stopping haloperidol, Mr. O is more spontaneous, logical, and relevant. He does not have delusions or hallucinations. Despite further attempts at pain management with physical therapy and increased doses of duloxetine, he still complains of pain. We do not prescribe zolpidem.
Mr. O is unwilling to discuss the incident more than 40 years ago when he cut his arms and legs except to say, “That’s the past. My life wasn’t so good at that time.” When we ask why he had been a client of Adult Protective Services 5 years before he was burned out of his apartment, he admitted that he was 21 months in arrears in his rent. “I used to do this thing called crack,” he explains. He was discharged to an adult home with a prescription for duloxetine after he promised to never smoke in his room again.
Related Resource
- Aggarwal A, Sharma DD. Zolpidem withdrawal delirium: a case report. J Neuropsychiatry Clin Neurosci. 2010;22(4):451.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Duloxetine • Cymbalta
- Haloperidol decanoate • Haloperidol decanoate
- Haloperidol • Haldol
- Naproxen • Naproxyn, Aleve, others
- Zolpidem • Ambien
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Space traveler
Mr. O, age 69, is a patient at a long-term psychiatric hospital. He has a 56-year psychiatric history, a current diagnosis of schizoaffective disorder, and suffered a torn rotator cuff approximately 5 years ago. His medication regimen is haloperidol decanoate, 100 mg IM every month, duloxetine, 60 mg/d, and naproxen, as needed for chronic pain.
He frequently lies on the floor. Attendants urge him to get up and join groups or sit with other patients but he complains of pain and soon finds another spot on the floor to use as a bed.
Eight months earlier, a homeless shelter sent Mr. O to the emergency room (ER) because he tried to eat a dollar bill and a sock. In the ER he was inattentive, with loose associations and bizarre delusions; he believed he was on a spaceship. Mr. O was admitted to the hospital, where clinicians noted that his behavior remained bizarre and he complained of insomnia. They also noted a history of setting fires, which complicated discharge planning and contributed to their decision to transfer him to our psychiatric facility for longer-term care.
During our initial interview, Mr. O readily picks himself off the floor. His responses are logical and direct but abrupt and unelaborated. His first and most vehement complaint is pain. Zolpidem, he says, is the only treatment that helps.
He says he began using zolpidem approximately 5 years ago because pain from a shoulder injury kept him awake at night. When he could not obtain the drug by prescription, he bought it on the street. One day when living in the homeless shelter, he took 30 or 40 mg of zolpidem, then “blacked out” and awoke in the ER.
His first experience with psychiatric treatment was the result of problems getting along with his single mother because of “petty things” such as shooting off a BB gun in their apartment, he says. As a teenager he was sent to a boarding school; as a young adult, to a psychiatric hospital. After his release he returned to his mother’s apartment. He worked steadily for 20 years before he obtained Social Security benefits, and then worked intermittently “off the books” until approximately 15 years ago. Mr. O lived with his mother until her death 17 years earlier, and then in her apartment alone until a fire, which he set accidentally by smoking in bed after taking zolpidem, forced him to leave 3 years ago. He says, “My whole life was in that place.” He was admitted to a psychiatric hospital for an unknown reason, which was his first psychiatric admission in 40 years. After he was released from the hospital, Mr. O lived in various homeless shelters and adult homes until his current hospitalization.
The author’s observations
An effective and well-tolerated drug with a reputation for rarely being abused, zolpidem is widely prescribed as a hypnotic. Zolpidem and benzodiazepines have different chemical structures but both act at the GABAA receptor and have comparable behavioral effects.1 The reported incidence of zolpidem abuse is much lower than the reported rate of benzodiazepine abuse when used for sleep2; however, abuse, dependence, and withdrawal have been reported.2-4 Zolpidem abuse seems to be more common among patients with a history of abusing other substances or a history of psychiatric illness.2 A French study4 found that abusers fell into 2 groups. The younger group (median age 35) used higher doses—a median of 300 mg/d—and took zolpidem in the daytime to achieve euphoria. A second, older group (median age 42) used lower doses—a median of 200 mg/d—at nighttime to sleep.
There are few reports of delirium and symptoms such as visual hallucinations and distortions associated with zolpidem use.5,6 These reactions have occurred in persons without a history of psychosis. They usually are associated with doses ≥10 mg.
In the ER Mr. O showed a disturbance in consciousness with inability to focus attention and a perceptual disturbance (he believed he was in a spaceship) that developed over hours to days. He met criteria for delirium, possibly caused by zolpidem, but his presentation also could have been attributable to an underlying psychiatric disorder.
ER and inpatient psychiatrists noted Mr. O was intoxicated with zolpidem when the shelter brought him to the ER, but both groups diagnosed schizoaffective disorder and treated him with antipsychotics. They saw his >50-year psychiatric history as evidence of an underlying, long-standing condition such as schizoaffective illness.
However, features of Mr. O’s illness are not typical of a chronic psychotic illness. He recalls psychiatric hospitalizations in his youth and recently, but not for the 40 years in between. Mr. O says he has never experienced auditory hallucinations. For these reasons, our treatment team obtains old medical records to investigate his early history (Table).
Table
Mr. O’s clinical course
| Age | Symptoms/behaviors | Diagnosis |
|---|---|---|
| 17 | Temper tantrums and destructive behaviors. No delusions or hallucinations but a flat affect and hostile attitude | Primary behavior disorder, simple adult maladjustment |
| 22 | Returned to the psychiatric hospital when his welfare payments stopped; “psychopathic” symptoms; described as defiant and resented authority and regular work | Primary behavior disorder |
| 24 | His mother complained that he stole from her and carried a weapon; while hospitalized, described as manageable and without overt psychotic symptoms | Primary behavior disorder |
| 26 | Arrested for causing property damage while intoxicated on alcohol; silly laugh, loose associations, irrelevant and incoherent speech, and believed hospital staff were against him | Psychosis with psychopathic personality |
| 66 | A fire that he set accidentally while smoking in bed after taking zolpidem destroyed his home | Diagnosis unknown |
| 68 | Transferred from a homeless shelter to the ER after he took 30 to 40 mg of zolpidem and exhibited bizarre behaviors | Schizoaffective disorder |
| 69 | More spontaneous, remains logical and relevant after haloperidol is discontinued; no delusions or hallucinations, still complains of pain | Substance use disorder and personality disorder |
| ER: emergency room | ||
HISTORY: Destructive and defiant
Mr. O’s mother reported that he had been a nervous, restless child who would scream and yell at the slightest provocation. At age 10 he became wantonly destructive. His mother bought him an expensive toy that he destroyed after a short time; he asked for another toy, which he also destroyed. When such behavior became more frequent, she took him to a city hospital, where he was treated for 6 weeks and released at age 13. He was sent to a boarding school but soon was expelled for drinking and selling beer.
Mr. O was admitted to long-term psychiatric facilities 6 times in the next 10 years, from the late 1950s to the late 1960s. He was first admitted at age 17 for temper tantrums during which he fired an air rifle and smashed windows in the home he shared with his mother. During examination he had no delusions or hallucinations but did have flat affect and a hostile attitude. Doctors documented that almost all his tantrums were as a result of interactions with his mother.
Records from this psychiatric admission state that Mr. O showed no unusual distractibility, “psychotic trends,” or paranoid thinking. After approximately 6 months in the hospital he was discharged home with the diagnosis of primary behavior disorder, simple adult maladjustment. Mr. O, who was age 18 at the time, and his mother were eager for him to complete high school and learn auto mechanics.
Nine months later, he returned to the psychiatric facility because of excessive drinking and inability to secure employment, according to his records. In the hospital, he was productive and reliable. When he was discharged home 3 months later, doctors wrote that his determination to stop drinking was firmly fixed. They encouraged Mr. O to complete high school as a night student and find employment during the day. His mother was delighted with his improvement.
A third admission, less than 2 months later, occurred after he broke a window during an argument with his mother. He had a job but quit. After 5 months he was discharged with the same diagnosis of primary behavior disorder, but his mother would not let him back in her home. He was referred to the social service department to be placed on welfare.
A year later, Mr. O had trouble managing his welfare allotment and moved repeatedly. He said he returned to the psychiatric hospital because his welfare payments had been discontinued. During this admission, doctors noted “psychopathic” symptoms; Mr. O was defiant and resented authority and regular work. Mr. O eloped from the hospital several times and brought beer into the building. After 18 months he was discharged with the same diagnosis, with plans to apply for welfare. He was not prescribed medication.
Mr. O’s fifth admission came nearly 2 years later after his mother complained that he stole from her home and carried a weapon. In the hospital he was described as manageable and without overt psychotic symptoms. When he was discharged a little more than a year after being admitted, doctors wrote that he was a psychopath who had a history of drinking, stealing, and delinquent tendencies as a teenager. His diagnosis remained primary behavior disorder.
A year after this discharge, Mr. O was arrested for causing serious property damage when he was intoxicated on alcohol. Subsequently he was readmitted.
After a few months in the hospital, Mr. O changed. He developed a silly laugh, loose associations, irrelevant and incoherent speech, and a belief that hospital staff were against him. Although Mr. O denied auditory hallucinations, a psychiatrist wrote that he seemed to be experiencing hallucinations and prescribed chlorpromazine. The next day Mr. O slashed his arms and legs in several places, requiring many sutures. His diagnosis was changed to psychosis with psychopathic personality. However, within a few months, psychiatrists determined that Mr. O had recovered, so they stopped chlorpromazine. Months later, clinicians wrote that Mr. O was idle most of the time, neat, clean, and not involved in arguments with other patients. He was discharged after 1 month in the hospital.
Over the years, psychiatrists had differing opinions about Mr. O’s diagnosis. One noted that his mental illness was characterized by emotional instability and poor judgment. He had impulsive reactions without regard for others, rapid mood swings, irritability, and depression with transient paranoia. Another clinician detected evidence of schizoid personality disorder because Mr. O did not experience hallucinations or a gross thought disorder, but did have rambling, circumstantial, autistic (unrealistic), and ambivalent thought content. Another psychiatrist wrote Mr. O best fit in the category of psychosis with psychopathic personality, which was his diagnosis at discharge from his sixth hospitalization.
The author’s observations
Mr. O’s old medical records revealed the diagnostic thinking and treatment practices of a past era. They did not demonstrate that Mr. O met current criteria for schizophrenia or schizoaffective disorder, although he may have had a brief psychotic episode. Because there was little support for a diagnosis of schizoaffective illness and haloperidol use, we stopped the drug but continued duloxetine for chronic pain. It was clear that he has a substance use disorder and perhaps met criteria for antisocial personality disorder.
OUTCOME: Further explanations
Approximately 2 months after stopping haloperidol, Mr. O is more spontaneous, logical, and relevant. He does not have delusions or hallucinations. Despite further attempts at pain management with physical therapy and increased doses of duloxetine, he still complains of pain. We do not prescribe zolpidem.
Mr. O is unwilling to discuss the incident more than 40 years ago when he cut his arms and legs except to say, “That’s the past. My life wasn’t so good at that time.” When we ask why he had been a client of Adult Protective Services 5 years before he was burned out of his apartment, he admitted that he was 21 months in arrears in his rent. “I used to do this thing called crack,” he explains. He was discharged to an adult home with a prescription for duloxetine after he promised to never smoke in his room again.
Related Resource
- Aggarwal A, Sharma DD. Zolpidem withdrawal delirium: a case report. J Neuropsychiatry Clin Neurosci. 2010;22(4):451.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Duloxetine • Cymbalta
- Haloperidol decanoate • Haloperidol decanoate
- Haloperidol • Haldol
- Naproxen • Naproxyn, Aleve, others
- Zolpidem • Ambien
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rush CR. Behavioral pharmacology of zolpidem relative to benzodiazepines: a review. Pharmacol Biochem Behav. 1998;61(3):253-269.
2. Hajak G, Müller WE, Wittchen HU, et al. Abuse and dependence potential for the non-benzodiazepine hypnotics zolpidem and zopiclone: a review of case reports and epidemiological data. Addiction. 2003;98(10):1371-1378.
3. Madrak LN, Rosenberg M. Zolpidem abuse. Am J Psychiatry. 2001;158(8):1330-1331.
4. Victorri-Vigneau C, Dailly E, Veyrac G, et al. Evidence of zolpidem abuse and dependence: results of the French Centre for Evaluation and Information on Pharmacodepencence (CEIP) network survey. Br J Clin Pharmacol. 2007;64(2):198-209.
5. Markowitz JS, Brewerton TD. Zolpidem-induced psychosis. Ann Clin Psychiatry. 1996;8(2):89-91.
6. Tsai MJ, Huang YB, Wu PC. A novel clinical pattern of visual hallucination after zolpidem use. J Toxicol Clin Toxicol. 2003;41(6):869-872.
1. Rush CR. Behavioral pharmacology of zolpidem relative to benzodiazepines: a review. Pharmacol Biochem Behav. 1998;61(3):253-269.
2. Hajak G, Müller WE, Wittchen HU, et al. Abuse and dependence potential for the non-benzodiazepine hypnotics zolpidem and zopiclone: a review of case reports and epidemiological data. Addiction. 2003;98(10):1371-1378.
3. Madrak LN, Rosenberg M. Zolpidem abuse. Am J Psychiatry. 2001;158(8):1330-1331.
4. Victorri-Vigneau C, Dailly E, Veyrac G, et al. Evidence of zolpidem abuse and dependence: results of the French Centre for Evaluation and Information on Pharmacodepencence (CEIP) network survey. Br J Clin Pharmacol. 2007;64(2):198-209.
5. Markowitz JS, Brewerton TD. Zolpidem-induced psychosis. Ann Clin Psychiatry. 1996;8(2):89-91.
6. Tsai MJ, Huang YB, Wu PC. A novel clinical pattern of visual hallucination after zolpidem use. J Toxicol Clin Toxicol. 2003;41(6):869-872.
A curious case of depression
Discuss this article at www.facebook.com/CurrentPsychiatry
Mr. Z, age 61, is referred by his primary care clinician to the hospital’s medical service with increasing depressive symptoms and non-pruritic rash. He has a history of bipolar I disorder for >30 years. When the primary care physician evaluated Mr. Z, his vitals were normal, but blood work revealed mild anemia and thrombocytopenia of 34 x103/μL, which prompted referral to the hospital. During admission, the psychiatric consultation service is called to evaluate Mr. Z’s depressive symptoms.
Mr. Z reports having chronic sleep problems and feeling cold and tired, shivering at times, but has no pain. He says he’s worried because he feels severely depressed, worthless, and hopeless, but denies suicidal ideation and psychosis. Mr. Z says he started experiencing increasingly depressed mood, anhedonia, insomnia, fatigue, poor appetite, and concentration 2 months ago. At that time his outpatient psychiatrist started Mr. Z on risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime because of emerging mood symptoms, after he was off medication for 7 months. Mr. Z attributed his worsened mood symptoms to being overwhelmed by several psychosocial stressors, including going through a complicated divorce, financial problems, and homelessness after being evicted from his apartment.
A review of Mr. Z’s psychiatric history reveals several remote hospitalizations—the last was 7 years ago—for escalated manic symptoms after he stopped taking his medication. He denies past suicide attempts. Mr. Z says he is compliant with his current medication regimen—risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime. He denies illicit drug use and says he drinks “a couple of beers, mostly on weekends.” Family history is positive for depression and bipolar II disorder.
His medical history is significant for hypothyroidism after goiter removal 6 years ago, for which he takes levothyroxine, 150 mcg/d, and a sports injury-related splenectomy in childhood. He reports no allergies. Vital signs at the time of admission are temperature, 99.1°F; pulse, 98 beats per minute; respiration, 16 breaths per minute; blood pressure, 123/73 mm/Hg; and oxygen saturation, 97%.
During the interview, Mr. Z presents with tired facies and exhibits psychomotor retardation. He has to force himself to stay engaged in the evaluation and maintain eye contact. His speech is clear, regular, and soft. Mr. Z says he is “very depressed”; his affect is constricted, almost flat, stable, and consistent with depressed mood. His thought process is linear and somewhat concrete and his thought content is notable for hopelessness, although Mr. Z continues to deny suicidal or homicidal ideations. No hallucinations or apparent delusions are noted. Insight and judgment are fair. Mr. Z understands his current mental state; however, he displays some lack of knowledge regarding his current hospitalization. Cognition is intact.
The authors’ observations
The differential diagnosis in patients presenting with mood changes is extensive (Table 1)1 and in Mr. Z’s case includes several precipitating and perpetuating factors. Mr. Z presents with severe depressive symptoms and meets DSM-IV-TR criteria for a major depressive episode (MDE). This presentation is not typical of his bipolar I disorder because Mr. Z has never experienced an MDE and usually presents with escalating hypomanic/manic symptoms in the context of medication nonadherence. Nevertheless, Mr. Z has several risk factors for severe depression, including a family psychiatric history, multiple enduring social stressors and life crises, and medical conditions.
In the general population, the lifetime risk for developing depression is 8% to 17%.2 The risk of developing a mood disorder increases significantly if a first-degree relative is diagnosed with a mood disorder; the relative risk is 10.3 for bipolar disorder and 3.2 for depression.3 Additionally, Mr. Z is going through a complicated divorce, has financial problems, and is homeless, all of which could trigger an MDE. Furthermore, hypothyroidism shares many symptoms of depression, including fatigue, lethargy, anhedonia, cold intolerance, and low mood; mental status changes frequently are the initial presentation of thyroid problems.4 Physicians started Mr. Z on a new medication regimen (risperidone and divalproex) to control mood instability, which coincided with symptom onset. Atypical antipsychotics have been reported to precipitate depressive symptoms; their side effect profile includes extrapyramidal effects, such as flat affect, which can be mistaken for depression.5 Rapid valproate titration can mimic neurovegetative symptoms of depression and cause dose-dependent thrombocytopenia and rash, which could explain his initial presentation.6 Finally, Mr. Z’s history of traumatic splenectomy, change in mental status, and thrombocytopenia suggest an infectious etiology.
Table 1
Differential diagnosis in patients presenting with mood changes
| Cerebrovascular disease |
| Degenerative disorders (Parkinson’s disease, Huntington’s disease, Wilson’s disease) |
| Demyelinating disorders (multiple sclerosis, amyotrophic lateral sclerosis, lipid storage disease) |
| Endocrine disorders (Addison’s disease, Cushing’s disease, hyperthyroidism, hypothyroidism, hyperparathyroidism, pituitary dysfunction) |
| Epilepsy |
| Infectious diseases |
| Immune diseases |
| Metabolic encephalopathy |
| Neoplasm |
| Nutritional deficits (thiamine, niacin, vitamin B12) |
| Primary psychiatric disorders (mood disorders, dementia, sleep disorders) |
| Substance use |
| Toxins/medications |
| Traumatic brain injury |
| Source: Reference 1 |
Possible infectious causes
The increased prevalence of immune suppression due to human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS) or from therapeutic modalities such as cancer therapy or splenectomy has led to an increased number of chronic CNS infections, manifesting with an array of neuropsychiatric symptoms and nonspecific physiological reactions.1,7 Mr. Z complains of a 2-month period of worsening depression that could suggest an infectious process with an insidious onset. Some infectious agents that can cause chronic CNS infection and encephalopathy are presented in Table 2.8 HIV, tuberculosis, syphilis, Lyme disease, and herpes simplex virus are becoming more prevalent and can present with neuropsychiatric symptoms.1 For example, in addition to thrombocytopenia and low-grade fever, patients with HIV may exhibit a broad range of neuropsychiatric symptoms such as cognitive problems, impaired executive and motor functioning, sleep disturbance, and anxiety. These patients frequently present with low mood and neurovegetative symptoms of depression.7 Similarly, the same tick responsible for Lyme disease infection can transmit other infectious agents that can cause thrombocytopenia, including Babesia, Ehrlichia chaffeensis, Anaplasma phagocytophilum, and human Ewingii ehrlichiosis.
The authors’ observations
Diagnosis of a mood change, particularly an MDE, is clinical, based on careful psychiatric evaluation using standardized criteria rather than a specific lab test. However, some laboratory studies (Table 3)1 are useful in differentiating medical illnesses that may present with depression. Mr. Z’s presentation warrants investigating these tests. His history of traumatic splenectomy and night sweats suggests an infection. The team’s initial recommendations include laboratory tests, discontinuing divalproex because it may be causing thrombocytopenia, and decreasing risperidone to 2 mg/d to improve his fatigue and possibly developed extrapyramidal symptoms.
Table 2
Potential infectious causes of chronic encephalopathy
| Type of infection | Organism/disease |
|---|---|
| Mycobacterial | Mycobacterium tuberculosis |
| Spirochetal | Treponema pallidum (syphilis), Borrelia burgdorferi (Lyme disease), Leptospira |
| Bacterial | Brucella, Listeria, Nocardia, Actinomyces israelii, Whipple’s disease |
| Viral | HIV/AIDS, cytomegalovirus, varicella zoster virus, herpes simplex virus, enterovirus |
| Fungal | Histoplasmosis, coccidiosis, sporothrix, Blastomyces, Cryptococcus |
| Parasitic | Toxoplasmosis, taenia solium (cysticercosis), Schistosoma, Acanthamoeba |
| AIDS: acquired immunodeficiency syndrome; HIV: human immunodeficiency virus Source: Reference 8 | |
Table 3
Differentiating medical illnesses that may mimic depression
| Laboratory tests |
|
| Imaging studies |
|
| Other tests |
|
| Procedures |
|
| ABG: arterial blood gases; CBC: complete blood count; EEG: electroencephalogram; ELISA: enzyme-linked immunosorbent assay; HIV: human immunodeficiency virus; VDRL: venereal disease research laboratory Source: Reference 1 |
TREATMENT: Cause revealed
Mr. Z develops a persistent fever of 102°F with continuous profuse sweating and a hypotensive episode. Blood work reveals mild anemia, thrombocytopenia, and increased coagulation parameters with high D-dimer and low fibrinogen, consistent with diagnosis of disseminated intravascular coagulation (DIC) secondary to infectious etiology. Thyroid and HIV tests are negative. After further evaluation, Mr. Z remembers that 4 months earlier he removed several ticks from his legs after hunting; he also remembers experiencing shivering and night sweats several weeks before he was hospitalized. His blood smear is positive for babesiosis and further testing confirms positive Lyme antibodies. Mr. Z is started on aggressive hemodynamic stabilization and a pathogen-tailored course of antibiotics for several weeks. This results in improvement and discharge home in a stable condition. His depression and fatigue improve but do not fully remit by the time he is discharged.
The authors’ observations
Lyme disease is one of the fastest-growing infectious diseases in the United States.9 The prevalence of positive Lyme antibodies is 30% higher in psychiatric populations than the general population.10 Lyme disease is transmitted by deer tick bite, often undetected, that is infected with spirochete Borrelia burgdorferi. To be infectious, ticks need to be attached to the skin for 24 to 48 hours,11,12 although individual cases have reported transmission in <24 hours. The clinical manifestations of Lyme disease can be divided into 3 phases:
- early localized phase, characterized by the distinctive skin lesion erythema migrans with or without constitutional symptoms
- early disseminated phase, characterized by multiple erythema migrans lesions and neurologic and/or cardiac findings
- late or chronic disease associated with intermittent/persistent arthritis and/or neurologic problems.11,13
The clinical features of each stage frequently overlap and some patients in a later stage of Lyme disease do not have prior signs or symptoms of the disease. Because it is a multisystem disease, Lyme disease can attack the CNS in the form of neuroborreliosis, a clinical diagnosis, without involving other systems, and its neuropsychiatric manifestations can resemble neurosyphilis because both organisms are spirochetes.11,13,14 CNS disorders have been found in up to 40% of Lyme disease cases.11 In neuroborreliosis, cognitive problems usually predominate; however, neuroborreliosis can mimic multiple brain diseases presenting with various neurologic and psychiatric symptoms (Table 4)14,15 and can present at any time after the tick bite. Furthermore neuroborreliosis is difficult to diagnose because symptoms may remain dormant and emerge after several years.11,13,14Borrelia burgdorferi is challenging to isolate and grow in the lab, and enzyme-linked immunosorbent assay (ELISA) testing for antibodies is highly specific but not very sensitive,16 frequently giving false negative results. Western blot confirms the diagnosis.
Table 4
Late-stage neuropsychiatric symptoms of Lyme disease
| Cognitive problems, memory problems, forgetfulness, slowing of thought processing, dysfunction in visuospatial orientation, dyslexia |
| Depression |
| Mood swings |
| Psychosis |
| Violent behavior/irritability |
| OCD |
| Anxiety |
| Panic attacks |
| Sleep disorders |
| Seizures |
| ADHD-like symptoms |
| Autism-like behavior |
| Chronic fatigue syndrome |
| Fibromyalgia |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder Source: References 14,15 |
Mr. Z’s presentation also reflects co-infection with babesiosis. Babesia is malaria-like protozoa diagnosed by blood smear that can cause a fatal illness in immuno-compromised patients. The clinical picture varies from mild symptoms such as night sweats, chills, arthralgias, and anorexia with thrombocytopenia to severe and potentially fatal outcomes in immunocompromised patients, including DIC, acute renal failure, sepsis, congestive heart failure, and myocardial infarction.11 Risk factors for the severest forms of babesiosis are age >50, co-infection with Lyme disease, and splenectomy,11 all of which were present in Mr. Z. Co-infected patients experience fatigue, headache, anorexia, and emotional lability more frequently than those with Lyme disease alone.12
Treatment options
Treatment of Lyme disease/neuroborreliosis is complex. The mainstay approach is antibiotics. Despite adequate treatment, many patients experience continued impairment, including chronic pain, fatigue, and cognitive and psychiatric symptoms.11,14 There is some evidence Borrelia burgdorferi can persist and re-emerge after adequate treatment.14,17 The National Institute of Health sponsored several clinical trials of prolonged antibiotic treatment for chronic Lyme disease. Some results suggested improvement in fatigue and cognitive function, although these results were not sustained.18
There is a strong link between mental illness and increased prevalence of positive Lyme disease antibodies.10 Several studies report increased risk of infection during psychological stress that may be related to an altered immune system response.19 Evidence suggests that Borrelia burgdorferi can alter immune system response, making T cells more reactive not only to Borrelia burgdorferi antigens but also to host antigens,20 creating autoimmune inflammatory reactions that could explain chronic neuropsychiatric symptoms. It appears Lyme disease antigens can mimic certain autoantigens (for example, in the thyroid gland).21 Whether there is a role for autoimmune therapy in treating chronic symptoms needs to be investigated.
Once Lyme disease is diagnosed, educating patients and families becomes an important part of treatment because many patients report feeling stigmatized by the diagnosis. Referral to a Lyme disease support group may be beneficial. Patients with neuropsychiatric symptoms that persist after antibiotic treatment should be offered symptom-based treatment, including medications and therapy.
Related Resource
- Centers for Disease Control and Prevention. Lyme disease: Resources for clinicians. www.cdc.gov/lyme/healthcare/clinicians.html.
Drug Brand Names
- Divalproex sodium • Depakote
- Levothyroxine • Levoxyl, Synthroid
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stern TA, Rosenbaum JF, Fava M, et al. eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby; 2008;257-277.
2. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
3. Merikangas KR, Low NC. The epidemiology of mood disorders. Curr Psychiatry Rep. 2004;6:411-421.
4. Brown GM. Psychiatric and neurologic aspects of endocrine disease. In Krieger DT Hughes JC, eds. Neuroendocrinology. Sunderland, MA: Sinauer Associates; 1980;185-194.
5. Maguire GA. Comprehensive understanding of schizophrenia and its treatment. Am J Health Syst Pharm. 2002;59(17 suppl 5):S4-S11.
6. Dreifuss FE, Langer DH. Side effects of valproate. Am J Med. 1988;84(1A):34-41.
7. Perry S, Jacobsen P. Neuropsychiatric manifestations of AIDS-spectrum disorders. Hosp Community Psychiatry. 1986;37:135-142.
8. Hildebrand J, Aoun M. Chronic meningitis: still a diagnostic challenge. J Neurol. 2003;250(6):653-660.
9. Bacon RM, Kugeler KJ, Mead PS. Centers for Disease Control and Prevention (CDC). Surveillance for Lyme disease—United States 1992-2006. MMWR Surveill Summ. 2008;57(10):1-9.
10. Hájek T, Pasková B, Janovská D, et al. Higher prevalence of antibodies to Borrelia burgdorferi in psychiatric patients than in healthy subjects. Am J Psychiatry. 2002;159:297-301.
11. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134.
12. Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. Evidence for increased severity and duration of illness. JAMA. 1996;275(21):1657-1660.
13. Hildenbrand P, Craven DE, Jones R, et al. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am J Neuroradiol. 2009;30:1079-1087.
14. Fallon BA, Nields JA. Lyme disease: a neuropsychiatric illness. Am J Psychiatry. 1994;151:1571-1583.
15. Fallon BA, Nields JA, Burrascano JJ, et al. The neuropsychiatric manifestations of Lyme borreliosis. Psychiatr Q. 1992;63(1):95-117.
16. Stricker RB, Johnson L. Lyme wars: let’s tackle the testing. BMJ. 2007;335:1008.-
17. Hodzic E, Feng S, Holden K, et al. Persistence of Borrelia burgdorferi following antibiotic treatment in mice. Antimicrob Agents Chemother. 2008;52(5):1728-1736.
18. Fallon BA, Keilp JG, Corbera KM, et al. A randomized, placebo-controlled trial of repeated IV antibiotic therapy for Lyme encephalopathy. Neurology. 2008;70(13):992-1003.
19. Sheridan JF, Dobbs C, Brown D, et al. Psychoneuroimmunology: stress effects on pathogenesis and immunity during infection. Clin Microbiol Rev. 1994;7(2):200-212.
20. Yssel H, Shanafelt MC, Soderberg C, et al. Borrelia burgdorferi activates a T helper 1-like T cell subset in Lyme arthritis. J Exp Med. 1991;174:593-601.
21. Benvenga S, Guarneri F, Vaccaro M, et al. Homologies between proteins of Borrelia burgdorferi and thyroid autoantigens. Thyroid. 2004;14(11):964-966.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mr. Z, age 61, is referred by his primary care clinician to the hospital’s medical service with increasing depressive symptoms and non-pruritic rash. He has a history of bipolar I disorder for >30 years. When the primary care physician evaluated Mr. Z, his vitals were normal, but blood work revealed mild anemia and thrombocytopenia of 34 x103/μL, which prompted referral to the hospital. During admission, the psychiatric consultation service is called to evaluate Mr. Z’s depressive symptoms.
Mr. Z reports having chronic sleep problems and feeling cold and tired, shivering at times, but has no pain. He says he’s worried because he feels severely depressed, worthless, and hopeless, but denies suicidal ideation and psychosis. Mr. Z says he started experiencing increasingly depressed mood, anhedonia, insomnia, fatigue, poor appetite, and concentration 2 months ago. At that time his outpatient psychiatrist started Mr. Z on risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime because of emerging mood symptoms, after he was off medication for 7 months. Mr. Z attributed his worsened mood symptoms to being overwhelmed by several psychosocial stressors, including going through a complicated divorce, financial problems, and homelessness after being evicted from his apartment.
A review of Mr. Z’s psychiatric history reveals several remote hospitalizations—the last was 7 years ago—for escalated manic symptoms after he stopped taking his medication. He denies past suicide attempts. Mr. Z says he is compliant with his current medication regimen—risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime. He denies illicit drug use and says he drinks “a couple of beers, mostly on weekends.” Family history is positive for depression and bipolar II disorder.
His medical history is significant for hypothyroidism after goiter removal 6 years ago, for which he takes levothyroxine, 150 mcg/d, and a sports injury-related splenectomy in childhood. He reports no allergies. Vital signs at the time of admission are temperature, 99.1°F; pulse, 98 beats per minute; respiration, 16 breaths per minute; blood pressure, 123/73 mm/Hg; and oxygen saturation, 97%.
During the interview, Mr. Z presents with tired facies and exhibits psychomotor retardation. He has to force himself to stay engaged in the evaluation and maintain eye contact. His speech is clear, regular, and soft. Mr. Z says he is “very depressed”; his affect is constricted, almost flat, stable, and consistent with depressed mood. His thought process is linear and somewhat concrete and his thought content is notable for hopelessness, although Mr. Z continues to deny suicidal or homicidal ideations. No hallucinations or apparent delusions are noted. Insight and judgment are fair. Mr. Z understands his current mental state; however, he displays some lack of knowledge regarding his current hospitalization. Cognition is intact.
The authors’ observations
The differential diagnosis in patients presenting with mood changes is extensive (Table 1)1 and in Mr. Z’s case includes several precipitating and perpetuating factors. Mr. Z presents with severe depressive symptoms and meets DSM-IV-TR criteria for a major depressive episode (MDE). This presentation is not typical of his bipolar I disorder because Mr. Z has never experienced an MDE and usually presents with escalating hypomanic/manic symptoms in the context of medication nonadherence. Nevertheless, Mr. Z has several risk factors for severe depression, including a family psychiatric history, multiple enduring social stressors and life crises, and medical conditions.
In the general population, the lifetime risk for developing depression is 8% to 17%.2 The risk of developing a mood disorder increases significantly if a first-degree relative is diagnosed with a mood disorder; the relative risk is 10.3 for bipolar disorder and 3.2 for depression.3 Additionally, Mr. Z is going through a complicated divorce, has financial problems, and is homeless, all of which could trigger an MDE. Furthermore, hypothyroidism shares many symptoms of depression, including fatigue, lethargy, anhedonia, cold intolerance, and low mood; mental status changes frequently are the initial presentation of thyroid problems.4 Physicians started Mr. Z on a new medication regimen (risperidone and divalproex) to control mood instability, which coincided with symptom onset. Atypical antipsychotics have been reported to precipitate depressive symptoms; their side effect profile includes extrapyramidal effects, such as flat affect, which can be mistaken for depression.5 Rapid valproate titration can mimic neurovegetative symptoms of depression and cause dose-dependent thrombocytopenia and rash, which could explain his initial presentation.6 Finally, Mr. Z’s history of traumatic splenectomy, change in mental status, and thrombocytopenia suggest an infectious etiology.
Table 1
Differential diagnosis in patients presenting with mood changes
| Cerebrovascular disease |
| Degenerative disorders (Parkinson’s disease, Huntington’s disease, Wilson’s disease) |
| Demyelinating disorders (multiple sclerosis, amyotrophic lateral sclerosis, lipid storage disease) |
| Endocrine disorders (Addison’s disease, Cushing’s disease, hyperthyroidism, hypothyroidism, hyperparathyroidism, pituitary dysfunction) |
| Epilepsy |
| Infectious diseases |
| Immune diseases |
| Metabolic encephalopathy |
| Neoplasm |
| Nutritional deficits (thiamine, niacin, vitamin B12) |
| Primary psychiatric disorders (mood disorders, dementia, sleep disorders) |
| Substance use |
| Toxins/medications |
| Traumatic brain injury |
| Source: Reference 1 |
Possible infectious causes
The increased prevalence of immune suppression due to human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS) or from therapeutic modalities such as cancer therapy or splenectomy has led to an increased number of chronic CNS infections, manifesting with an array of neuropsychiatric symptoms and nonspecific physiological reactions.1,7 Mr. Z complains of a 2-month period of worsening depression that could suggest an infectious process with an insidious onset. Some infectious agents that can cause chronic CNS infection and encephalopathy are presented in Table 2.8 HIV, tuberculosis, syphilis, Lyme disease, and herpes simplex virus are becoming more prevalent and can present with neuropsychiatric symptoms.1 For example, in addition to thrombocytopenia and low-grade fever, patients with HIV may exhibit a broad range of neuropsychiatric symptoms such as cognitive problems, impaired executive and motor functioning, sleep disturbance, and anxiety. These patients frequently present with low mood and neurovegetative symptoms of depression.7 Similarly, the same tick responsible for Lyme disease infection can transmit other infectious agents that can cause thrombocytopenia, including Babesia, Ehrlichia chaffeensis, Anaplasma phagocytophilum, and human Ewingii ehrlichiosis.
The authors’ observations
Diagnosis of a mood change, particularly an MDE, is clinical, based on careful psychiatric evaluation using standardized criteria rather than a specific lab test. However, some laboratory studies (Table 3)1 are useful in differentiating medical illnesses that may present with depression. Mr. Z’s presentation warrants investigating these tests. His history of traumatic splenectomy and night sweats suggests an infection. The team’s initial recommendations include laboratory tests, discontinuing divalproex because it may be causing thrombocytopenia, and decreasing risperidone to 2 mg/d to improve his fatigue and possibly developed extrapyramidal symptoms.
Table 2
Potential infectious causes of chronic encephalopathy
| Type of infection | Organism/disease |
|---|---|
| Mycobacterial | Mycobacterium tuberculosis |
| Spirochetal | Treponema pallidum (syphilis), Borrelia burgdorferi (Lyme disease), Leptospira |
| Bacterial | Brucella, Listeria, Nocardia, Actinomyces israelii, Whipple’s disease |
| Viral | HIV/AIDS, cytomegalovirus, varicella zoster virus, herpes simplex virus, enterovirus |
| Fungal | Histoplasmosis, coccidiosis, sporothrix, Blastomyces, Cryptococcus |
| Parasitic | Toxoplasmosis, taenia solium (cysticercosis), Schistosoma, Acanthamoeba |
| AIDS: acquired immunodeficiency syndrome; HIV: human immunodeficiency virus Source: Reference 8 | |
Table 3
Differentiating medical illnesses that may mimic depression
| Laboratory tests |
|
| Imaging studies |
|
| Other tests |
|
| Procedures |
|
| ABG: arterial blood gases; CBC: complete blood count; EEG: electroencephalogram; ELISA: enzyme-linked immunosorbent assay; HIV: human immunodeficiency virus; VDRL: venereal disease research laboratory Source: Reference 1 |
TREATMENT: Cause revealed
Mr. Z develops a persistent fever of 102°F with continuous profuse sweating and a hypotensive episode. Blood work reveals mild anemia, thrombocytopenia, and increased coagulation parameters with high D-dimer and low fibrinogen, consistent with diagnosis of disseminated intravascular coagulation (DIC) secondary to infectious etiology. Thyroid and HIV tests are negative. After further evaluation, Mr. Z remembers that 4 months earlier he removed several ticks from his legs after hunting; he also remembers experiencing shivering and night sweats several weeks before he was hospitalized. His blood smear is positive for babesiosis and further testing confirms positive Lyme antibodies. Mr. Z is started on aggressive hemodynamic stabilization and a pathogen-tailored course of antibiotics for several weeks. This results in improvement and discharge home in a stable condition. His depression and fatigue improve but do not fully remit by the time he is discharged.
The authors’ observations
Lyme disease is one of the fastest-growing infectious diseases in the United States.9 The prevalence of positive Lyme antibodies is 30% higher in psychiatric populations than the general population.10 Lyme disease is transmitted by deer tick bite, often undetected, that is infected with spirochete Borrelia burgdorferi. To be infectious, ticks need to be attached to the skin for 24 to 48 hours,11,12 although individual cases have reported transmission in <24 hours. The clinical manifestations of Lyme disease can be divided into 3 phases:
- early localized phase, characterized by the distinctive skin lesion erythema migrans with or without constitutional symptoms
- early disseminated phase, characterized by multiple erythema migrans lesions and neurologic and/or cardiac findings
- late or chronic disease associated with intermittent/persistent arthritis and/or neurologic problems.11,13
The clinical features of each stage frequently overlap and some patients in a later stage of Lyme disease do not have prior signs or symptoms of the disease. Because it is a multisystem disease, Lyme disease can attack the CNS in the form of neuroborreliosis, a clinical diagnosis, without involving other systems, and its neuropsychiatric manifestations can resemble neurosyphilis because both organisms are spirochetes.11,13,14 CNS disorders have been found in up to 40% of Lyme disease cases.11 In neuroborreliosis, cognitive problems usually predominate; however, neuroborreliosis can mimic multiple brain diseases presenting with various neurologic and psychiatric symptoms (Table 4)14,15 and can present at any time after the tick bite. Furthermore neuroborreliosis is difficult to diagnose because symptoms may remain dormant and emerge after several years.11,13,14Borrelia burgdorferi is challenging to isolate and grow in the lab, and enzyme-linked immunosorbent assay (ELISA) testing for antibodies is highly specific but not very sensitive,16 frequently giving false negative results. Western blot confirms the diagnosis.
Table 4
Late-stage neuropsychiatric symptoms of Lyme disease
| Cognitive problems, memory problems, forgetfulness, slowing of thought processing, dysfunction in visuospatial orientation, dyslexia |
| Depression |
| Mood swings |
| Psychosis |
| Violent behavior/irritability |
| OCD |
| Anxiety |
| Panic attacks |
| Sleep disorders |
| Seizures |
| ADHD-like symptoms |
| Autism-like behavior |
| Chronic fatigue syndrome |
| Fibromyalgia |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder Source: References 14,15 |
Mr. Z’s presentation also reflects co-infection with babesiosis. Babesia is malaria-like protozoa diagnosed by blood smear that can cause a fatal illness in immuno-compromised patients. The clinical picture varies from mild symptoms such as night sweats, chills, arthralgias, and anorexia with thrombocytopenia to severe and potentially fatal outcomes in immunocompromised patients, including DIC, acute renal failure, sepsis, congestive heart failure, and myocardial infarction.11 Risk factors for the severest forms of babesiosis are age >50, co-infection with Lyme disease, and splenectomy,11 all of which were present in Mr. Z. Co-infected patients experience fatigue, headache, anorexia, and emotional lability more frequently than those with Lyme disease alone.12
Treatment options
Treatment of Lyme disease/neuroborreliosis is complex. The mainstay approach is antibiotics. Despite adequate treatment, many patients experience continued impairment, including chronic pain, fatigue, and cognitive and psychiatric symptoms.11,14 There is some evidence Borrelia burgdorferi can persist and re-emerge after adequate treatment.14,17 The National Institute of Health sponsored several clinical trials of prolonged antibiotic treatment for chronic Lyme disease. Some results suggested improvement in fatigue and cognitive function, although these results were not sustained.18
There is a strong link between mental illness and increased prevalence of positive Lyme disease antibodies.10 Several studies report increased risk of infection during psychological stress that may be related to an altered immune system response.19 Evidence suggests that Borrelia burgdorferi can alter immune system response, making T cells more reactive not only to Borrelia burgdorferi antigens but also to host antigens,20 creating autoimmune inflammatory reactions that could explain chronic neuropsychiatric symptoms. It appears Lyme disease antigens can mimic certain autoantigens (for example, in the thyroid gland).21 Whether there is a role for autoimmune therapy in treating chronic symptoms needs to be investigated.
Once Lyme disease is diagnosed, educating patients and families becomes an important part of treatment because many patients report feeling stigmatized by the diagnosis. Referral to a Lyme disease support group may be beneficial. Patients with neuropsychiatric symptoms that persist after antibiotic treatment should be offered symptom-based treatment, including medications and therapy.
Related Resource
- Centers for Disease Control and Prevention. Lyme disease: Resources for clinicians. www.cdc.gov/lyme/healthcare/clinicians.html.
Drug Brand Names
- Divalproex sodium • Depakote
- Levothyroxine • Levoxyl, Synthroid
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mr. Z, age 61, is referred by his primary care clinician to the hospital’s medical service with increasing depressive symptoms and non-pruritic rash. He has a history of bipolar I disorder for >30 years. When the primary care physician evaluated Mr. Z, his vitals were normal, but blood work revealed mild anemia and thrombocytopenia of 34 x103/μL, which prompted referral to the hospital. During admission, the psychiatric consultation service is called to evaluate Mr. Z’s depressive symptoms.
Mr. Z reports having chronic sleep problems and feeling cold and tired, shivering at times, but has no pain. He says he’s worried because he feels severely depressed, worthless, and hopeless, but denies suicidal ideation and psychosis. Mr. Z says he started experiencing increasingly depressed mood, anhedonia, insomnia, fatigue, poor appetite, and concentration 2 months ago. At that time his outpatient psychiatrist started Mr. Z on risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime because of emerging mood symptoms, after he was off medication for 7 months. Mr. Z attributed his worsened mood symptoms to being overwhelmed by several psychosocial stressors, including going through a complicated divorce, financial problems, and homelessness after being evicted from his apartment.
A review of Mr. Z’s psychiatric history reveals several remote hospitalizations—the last was 7 years ago—for escalated manic symptoms after he stopped taking his medication. He denies past suicide attempts. Mr. Z says he is compliant with his current medication regimen—risperidone, 6 mg/d, and divalproex, 1,500 mg at bedtime. He denies illicit drug use and says he drinks “a couple of beers, mostly on weekends.” Family history is positive for depression and bipolar II disorder.
His medical history is significant for hypothyroidism after goiter removal 6 years ago, for which he takes levothyroxine, 150 mcg/d, and a sports injury-related splenectomy in childhood. He reports no allergies. Vital signs at the time of admission are temperature, 99.1°F; pulse, 98 beats per minute; respiration, 16 breaths per minute; blood pressure, 123/73 mm/Hg; and oxygen saturation, 97%.
During the interview, Mr. Z presents with tired facies and exhibits psychomotor retardation. He has to force himself to stay engaged in the evaluation and maintain eye contact. His speech is clear, regular, and soft. Mr. Z says he is “very depressed”; his affect is constricted, almost flat, stable, and consistent with depressed mood. His thought process is linear and somewhat concrete and his thought content is notable for hopelessness, although Mr. Z continues to deny suicidal or homicidal ideations. No hallucinations or apparent delusions are noted. Insight and judgment are fair. Mr. Z understands his current mental state; however, he displays some lack of knowledge regarding his current hospitalization. Cognition is intact.
The authors’ observations
The differential diagnosis in patients presenting with mood changes is extensive (Table 1)1 and in Mr. Z’s case includes several precipitating and perpetuating factors. Mr. Z presents with severe depressive symptoms and meets DSM-IV-TR criteria for a major depressive episode (MDE). This presentation is not typical of his bipolar I disorder because Mr. Z has never experienced an MDE and usually presents with escalating hypomanic/manic symptoms in the context of medication nonadherence. Nevertheless, Mr. Z has several risk factors for severe depression, including a family psychiatric history, multiple enduring social stressors and life crises, and medical conditions.
In the general population, the lifetime risk for developing depression is 8% to 17%.2 The risk of developing a mood disorder increases significantly if a first-degree relative is diagnosed with a mood disorder; the relative risk is 10.3 for bipolar disorder and 3.2 for depression.3 Additionally, Mr. Z is going through a complicated divorce, has financial problems, and is homeless, all of which could trigger an MDE. Furthermore, hypothyroidism shares many symptoms of depression, including fatigue, lethargy, anhedonia, cold intolerance, and low mood; mental status changes frequently are the initial presentation of thyroid problems.4 Physicians started Mr. Z on a new medication regimen (risperidone and divalproex) to control mood instability, which coincided with symptom onset. Atypical antipsychotics have been reported to precipitate depressive symptoms; their side effect profile includes extrapyramidal effects, such as flat affect, which can be mistaken for depression.5 Rapid valproate titration can mimic neurovegetative symptoms of depression and cause dose-dependent thrombocytopenia and rash, which could explain his initial presentation.6 Finally, Mr. Z’s history of traumatic splenectomy, change in mental status, and thrombocytopenia suggest an infectious etiology.
Table 1
Differential diagnosis in patients presenting with mood changes
| Cerebrovascular disease |
| Degenerative disorders (Parkinson’s disease, Huntington’s disease, Wilson’s disease) |
| Demyelinating disorders (multiple sclerosis, amyotrophic lateral sclerosis, lipid storage disease) |
| Endocrine disorders (Addison’s disease, Cushing’s disease, hyperthyroidism, hypothyroidism, hyperparathyroidism, pituitary dysfunction) |
| Epilepsy |
| Infectious diseases |
| Immune diseases |
| Metabolic encephalopathy |
| Neoplasm |
| Nutritional deficits (thiamine, niacin, vitamin B12) |
| Primary psychiatric disorders (mood disorders, dementia, sleep disorders) |
| Substance use |
| Toxins/medications |
| Traumatic brain injury |
| Source: Reference 1 |
Possible infectious causes
The increased prevalence of immune suppression due to human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS) or from therapeutic modalities such as cancer therapy or splenectomy has led to an increased number of chronic CNS infections, manifesting with an array of neuropsychiatric symptoms and nonspecific physiological reactions.1,7 Mr. Z complains of a 2-month period of worsening depression that could suggest an infectious process with an insidious onset. Some infectious agents that can cause chronic CNS infection and encephalopathy are presented in Table 2.8 HIV, tuberculosis, syphilis, Lyme disease, and herpes simplex virus are becoming more prevalent and can present with neuropsychiatric symptoms.1 For example, in addition to thrombocytopenia and low-grade fever, patients with HIV may exhibit a broad range of neuropsychiatric symptoms such as cognitive problems, impaired executive and motor functioning, sleep disturbance, and anxiety. These patients frequently present with low mood and neurovegetative symptoms of depression.7 Similarly, the same tick responsible for Lyme disease infection can transmit other infectious agents that can cause thrombocytopenia, including Babesia, Ehrlichia chaffeensis, Anaplasma phagocytophilum, and human Ewingii ehrlichiosis.
The authors’ observations
Diagnosis of a mood change, particularly an MDE, is clinical, based on careful psychiatric evaluation using standardized criteria rather than a specific lab test. However, some laboratory studies (Table 3)1 are useful in differentiating medical illnesses that may present with depression. Mr. Z’s presentation warrants investigating these tests. His history of traumatic splenectomy and night sweats suggests an infection. The team’s initial recommendations include laboratory tests, discontinuing divalproex because it may be causing thrombocytopenia, and decreasing risperidone to 2 mg/d to improve his fatigue and possibly developed extrapyramidal symptoms.
Table 2
Potential infectious causes of chronic encephalopathy
| Type of infection | Organism/disease |
|---|---|
| Mycobacterial | Mycobacterium tuberculosis |
| Spirochetal | Treponema pallidum (syphilis), Borrelia burgdorferi (Lyme disease), Leptospira |
| Bacterial | Brucella, Listeria, Nocardia, Actinomyces israelii, Whipple’s disease |
| Viral | HIV/AIDS, cytomegalovirus, varicella zoster virus, herpes simplex virus, enterovirus |
| Fungal | Histoplasmosis, coccidiosis, sporothrix, Blastomyces, Cryptococcus |
| Parasitic | Toxoplasmosis, taenia solium (cysticercosis), Schistosoma, Acanthamoeba |
| AIDS: acquired immunodeficiency syndrome; HIV: human immunodeficiency virus Source: Reference 8 | |
Table 3
Differentiating medical illnesses that may mimic depression
| Laboratory tests |
|
| Imaging studies |
|
| Other tests |
|
| Procedures |
|
| ABG: arterial blood gases; CBC: complete blood count; EEG: electroencephalogram; ELISA: enzyme-linked immunosorbent assay; HIV: human immunodeficiency virus; VDRL: venereal disease research laboratory Source: Reference 1 |
TREATMENT: Cause revealed
Mr. Z develops a persistent fever of 102°F with continuous profuse sweating and a hypotensive episode. Blood work reveals mild anemia, thrombocytopenia, and increased coagulation parameters with high D-dimer and low fibrinogen, consistent with diagnosis of disseminated intravascular coagulation (DIC) secondary to infectious etiology. Thyroid and HIV tests are negative. After further evaluation, Mr. Z remembers that 4 months earlier he removed several ticks from his legs after hunting; he also remembers experiencing shivering and night sweats several weeks before he was hospitalized. His blood smear is positive for babesiosis and further testing confirms positive Lyme antibodies. Mr. Z is started on aggressive hemodynamic stabilization and a pathogen-tailored course of antibiotics for several weeks. This results in improvement and discharge home in a stable condition. His depression and fatigue improve but do not fully remit by the time he is discharged.
The authors’ observations
Lyme disease is one of the fastest-growing infectious diseases in the United States.9 The prevalence of positive Lyme antibodies is 30% higher in psychiatric populations than the general population.10 Lyme disease is transmitted by deer tick bite, often undetected, that is infected with spirochete Borrelia burgdorferi. To be infectious, ticks need to be attached to the skin for 24 to 48 hours,11,12 although individual cases have reported transmission in <24 hours. The clinical manifestations of Lyme disease can be divided into 3 phases:
- early localized phase, characterized by the distinctive skin lesion erythema migrans with or without constitutional symptoms
- early disseminated phase, characterized by multiple erythema migrans lesions and neurologic and/or cardiac findings
- late or chronic disease associated with intermittent/persistent arthritis and/or neurologic problems.11,13
The clinical features of each stage frequently overlap and some patients in a later stage of Lyme disease do not have prior signs or symptoms of the disease. Because it is a multisystem disease, Lyme disease can attack the CNS in the form of neuroborreliosis, a clinical diagnosis, without involving other systems, and its neuropsychiatric manifestations can resemble neurosyphilis because both organisms are spirochetes.11,13,14 CNS disorders have been found in up to 40% of Lyme disease cases.11 In neuroborreliosis, cognitive problems usually predominate; however, neuroborreliosis can mimic multiple brain diseases presenting with various neurologic and psychiatric symptoms (Table 4)14,15 and can present at any time after the tick bite. Furthermore neuroborreliosis is difficult to diagnose because symptoms may remain dormant and emerge after several years.11,13,14Borrelia burgdorferi is challenging to isolate and grow in the lab, and enzyme-linked immunosorbent assay (ELISA) testing for antibodies is highly specific but not very sensitive,16 frequently giving false negative results. Western blot confirms the diagnosis.
Table 4
Late-stage neuropsychiatric symptoms of Lyme disease
| Cognitive problems, memory problems, forgetfulness, slowing of thought processing, dysfunction in visuospatial orientation, dyslexia |
| Depression |
| Mood swings |
| Psychosis |
| Violent behavior/irritability |
| OCD |
| Anxiety |
| Panic attacks |
| Sleep disorders |
| Seizures |
| ADHD-like symptoms |
| Autism-like behavior |
| Chronic fatigue syndrome |
| Fibromyalgia |
| ADHD: attention-deficit/hyperactivity disorder; OCD: obsessive-compulsive disorder Source: References 14,15 |
Mr. Z’s presentation also reflects co-infection with babesiosis. Babesia is malaria-like protozoa diagnosed by blood smear that can cause a fatal illness in immuno-compromised patients. The clinical picture varies from mild symptoms such as night sweats, chills, arthralgias, and anorexia with thrombocytopenia to severe and potentially fatal outcomes in immunocompromised patients, including DIC, acute renal failure, sepsis, congestive heart failure, and myocardial infarction.11 Risk factors for the severest forms of babesiosis are age >50, co-infection with Lyme disease, and splenectomy,11 all of which were present in Mr. Z. Co-infected patients experience fatigue, headache, anorexia, and emotional lability more frequently than those with Lyme disease alone.12
Treatment options
Treatment of Lyme disease/neuroborreliosis is complex. The mainstay approach is antibiotics. Despite adequate treatment, many patients experience continued impairment, including chronic pain, fatigue, and cognitive and psychiatric symptoms.11,14 There is some evidence Borrelia burgdorferi can persist and re-emerge after adequate treatment.14,17 The National Institute of Health sponsored several clinical trials of prolonged antibiotic treatment for chronic Lyme disease. Some results suggested improvement in fatigue and cognitive function, although these results were not sustained.18
There is a strong link between mental illness and increased prevalence of positive Lyme disease antibodies.10 Several studies report increased risk of infection during psychological stress that may be related to an altered immune system response.19 Evidence suggests that Borrelia burgdorferi can alter immune system response, making T cells more reactive not only to Borrelia burgdorferi antigens but also to host antigens,20 creating autoimmune inflammatory reactions that could explain chronic neuropsychiatric symptoms. It appears Lyme disease antigens can mimic certain autoantigens (for example, in the thyroid gland).21 Whether there is a role for autoimmune therapy in treating chronic symptoms needs to be investigated.
Once Lyme disease is diagnosed, educating patients and families becomes an important part of treatment because many patients report feeling stigmatized by the diagnosis. Referral to a Lyme disease support group may be beneficial. Patients with neuropsychiatric symptoms that persist after antibiotic treatment should be offered symptom-based treatment, including medications and therapy.
Related Resource
- Centers for Disease Control and Prevention. Lyme disease: Resources for clinicians. www.cdc.gov/lyme/healthcare/clinicians.html.
Drug Brand Names
- Divalproex sodium • Depakote
- Levothyroxine • Levoxyl, Synthroid
- Risperidone • Risperdal
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stern TA, Rosenbaum JF, Fava M, et al. eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby; 2008;257-277.
2. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
3. Merikangas KR, Low NC. The epidemiology of mood disorders. Curr Psychiatry Rep. 2004;6:411-421.
4. Brown GM. Psychiatric and neurologic aspects of endocrine disease. In Krieger DT Hughes JC, eds. Neuroendocrinology. Sunderland, MA: Sinauer Associates; 1980;185-194.
5. Maguire GA. Comprehensive understanding of schizophrenia and its treatment. Am J Health Syst Pharm. 2002;59(17 suppl 5):S4-S11.
6. Dreifuss FE, Langer DH. Side effects of valproate. Am J Med. 1988;84(1A):34-41.
7. Perry S, Jacobsen P. Neuropsychiatric manifestations of AIDS-spectrum disorders. Hosp Community Psychiatry. 1986;37:135-142.
8. Hildebrand J, Aoun M. Chronic meningitis: still a diagnostic challenge. J Neurol. 2003;250(6):653-660.
9. Bacon RM, Kugeler KJ, Mead PS. Centers for Disease Control and Prevention (CDC). Surveillance for Lyme disease—United States 1992-2006. MMWR Surveill Summ. 2008;57(10):1-9.
10. Hájek T, Pasková B, Janovská D, et al. Higher prevalence of antibodies to Borrelia burgdorferi in psychiatric patients than in healthy subjects. Am J Psychiatry. 2002;159:297-301.
11. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134.
12. Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. Evidence for increased severity and duration of illness. JAMA. 1996;275(21):1657-1660.
13. Hildenbrand P, Craven DE, Jones R, et al. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am J Neuroradiol. 2009;30:1079-1087.
14. Fallon BA, Nields JA. Lyme disease: a neuropsychiatric illness. Am J Psychiatry. 1994;151:1571-1583.
15. Fallon BA, Nields JA, Burrascano JJ, et al. The neuropsychiatric manifestations of Lyme borreliosis. Psychiatr Q. 1992;63(1):95-117.
16. Stricker RB, Johnson L. Lyme wars: let’s tackle the testing. BMJ. 2007;335:1008.-
17. Hodzic E, Feng S, Holden K, et al. Persistence of Borrelia burgdorferi following antibiotic treatment in mice. Antimicrob Agents Chemother. 2008;52(5):1728-1736.
18. Fallon BA, Keilp JG, Corbera KM, et al. A randomized, placebo-controlled trial of repeated IV antibiotic therapy for Lyme encephalopathy. Neurology. 2008;70(13):992-1003.
19. Sheridan JF, Dobbs C, Brown D, et al. Psychoneuroimmunology: stress effects on pathogenesis and immunity during infection. Clin Microbiol Rev. 1994;7(2):200-212.
20. Yssel H, Shanafelt MC, Soderberg C, et al. Borrelia burgdorferi activates a T helper 1-like T cell subset in Lyme arthritis. J Exp Med. 1991;174:593-601.
21. Benvenga S, Guarneri F, Vaccaro M, et al. Homologies between proteins of Borrelia burgdorferi and thyroid autoantigens. Thyroid. 2004;14(11):964-966.
1. Stern TA, Rosenbaum JF, Fava M, et al. eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby; 2008;257-277.
2. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
3. Merikangas KR, Low NC. The epidemiology of mood disorders. Curr Psychiatry Rep. 2004;6:411-421.
4. Brown GM. Psychiatric and neurologic aspects of endocrine disease. In Krieger DT Hughes JC, eds. Neuroendocrinology. Sunderland, MA: Sinauer Associates; 1980;185-194.
5. Maguire GA. Comprehensive understanding of schizophrenia and its treatment. Am J Health Syst Pharm. 2002;59(17 suppl 5):S4-S11.
6. Dreifuss FE, Langer DH. Side effects of valproate. Am J Med. 1988;84(1A):34-41.
7. Perry S, Jacobsen P. Neuropsychiatric manifestations of AIDS-spectrum disorders. Hosp Community Psychiatry. 1986;37:135-142.
8. Hildebrand J, Aoun M. Chronic meningitis: still a diagnostic challenge. J Neurol. 2003;250(6):653-660.
9. Bacon RM, Kugeler KJ, Mead PS. Centers for Disease Control and Prevention (CDC). Surveillance for Lyme disease—United States 1992-2006. MMWR Surveill Summ. 2008;57(10):1-9.
10. Hájek T, Pasková B, Janovská D, et al. Higher prevalence of antibodies to Borrelia burgdorferi in psychiatric patients than in healthy subjects. Am J Psychiatry. 2002;159:297-301.
11. Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089-1134.
12. Krause PJ, Telford SR 3rd, Spielman A, et al. Concurrent Lyme disease and babesiosis. Evidence for increased severity and duration of illness. JAMA. 1996;275(21):1657-1660.
13. Hildenbrand P, Craven DE, Jones R, et al. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am J Neuroradiol. 2009;30:1079-1087.
14. Fallon BA, Nields JA. Lyme disease: a neuropsychiatric illness. Am J Psychiatry. 1994;151:1571-1583.
15. Fallon BA, Nields JA, Burrascano JJ, et al. The neuropsychiatric manifestations of Lyme borreliosis. Psychiatr Q. 1992;63(1):95-117.
16. Stricker RB, Johnson L. Lyme wars: let’s tackle the testing. BMJ. 2007;335:1008.-
17. Hodzic E, Feng S, Holden K, et al. Persistence of Borrelia burgdorferi following antibiotic treatment in mice. Antimicrob Agents Chemother. 2008;52(5):1728-1736.
18. Fallon BA, Keilp JG, Corbera KM, et al. A randomized, placebo-controlled trial of repeated IV antibiotic therapy for Lyme encephalopathy. Neurology. 2008;70(13):992-1003.
19. Sheridan JF, Dobbs C, Brown D, et al. Psychoneuroimmunology: stress effects on pathogenesis and immunity during infection. Clin Microbiol Rev. 1994;7(2):200-212.
20. Yssel H, Shanafelt MC, Soderberg C, et al. Borrelia burgdorferi activates a T helper 1-like T cell subset in Lyme arthritis. J Exp Med. 1991;174:593-601.
21. Benvenga S, Guarneri F, Vaccaro M, et al. Homologies between proteins of Borrelia burgdorferi and thyroid autoantigens. Thyroid. 2004;14(11):964-966.
‘Scared’ and short of breath
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
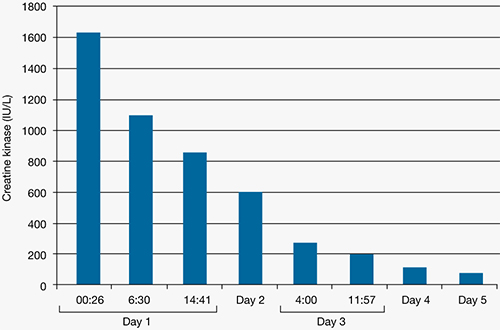
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
Unexpected improvement
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Relapsing psychosis
Ms. U, age 53, was diagnosed with paranoid schizophrenia at age 21 and has a continuous pattern of frequent relapses and inpatient admissions. She has received therapeutic doses of trifluoperazine, sertindole, haloperidol, loxapine, thioridazine, olanzapine, risperidone, clozapine, and several other antipsychotics not available in the United States. Clozapine had been prescribed at 600 mg/d (average blood level was 350 ng/mL), at times in combination with other antipsychotics or lithium.
Despite treatment, Ms. U has never achieved clinical stability. She has fluctuating yet persistent auditory hallucinations (eg, voices threatening to “announce disasters” or songs of a religious nature), associated disorganized behavior (eg, covering her ears or asking third parties “to turn off the radio”), severe hyponatremia secondary to potomania, paranoid ideation (eg, being followed by a “hidden camera”), and a strong tendency toward negativism, mutism, and emotional lability secondary to her psychotic symptoms. Her affect is predominantly poor and flattened, with very poor insight. Her symptoms are associated with progressive social isolation and poor grooming. Because of her worsening status, Ms. U was admitted to a residential facility 3 years ago.
Ms. U is single and the eldest of 2 siblings. Her parents are deceased; one parent may have committed suicide. She reports a family history of psychosis in her first cousins, but no history of hereditary neurologic disorders. Ms. U is a heavy smoker, did not complete college, and has a job in a family business.
The authors’ observations
Historically, the prevailing theory to explain the pathophysiology of schizophrenia has been the dopamine hypothesis, which links a hyperdopaminergic state in the mesolimbic system with acute psychosis. This theory could explain positive symptoms of schizophrenia but not other core domains, such as negative symptoms and cognitive dysfunction.1-3 The glutamate hypothesis postulates a hypoglutamatergic state can be the cause, at least in part, of various symptoms of psychosis, similar to those induced by phencyclidine and ketamine. Antagonists at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor are being studied as a way to influence this pathway,1 which is believed to be influenced by genetic factors.4
Glutamate, an amino acid, is the primary excitatory neurotransmitter in the brain. Its action is exerted in 2 types of receptors on the postsynaptic neuron: ionotropic and metabotropic.
The activation of NMDA receptors generated by glutamate and glycine coagonist can stimulate an uncontrolled release of calcium and subsequent cell death known as excitotoxicity. This phenomenon has been described in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, and Huntington’s disease. Although overstimulation of NMDA receptors induces neurodegeneration, NMDA hypoactivity has been observed in psychotic states.5
EVALUATION: Neurologic symptoms
A few months after arriving at the residential facility, Ms. U develops dysarthria and drooling, which the treatment team initially interprets as secondary to high doses of clozapine. In the absence of clinical response after clozapine dose reduction and with the subsequent appearance of dysphagia with solid foods and liquids, Ms. U is evaluated by a ear, nose, and throat physician, and later by a neurologist. Both clinicians describe frontal release signs, anarthria, facial hypomimia, bilateral mild central paresis, absence of soft palate elevation with symmetrical phonation, decreased gag reflex and palatal atrophy, fasciculations, and bilateral lingual mandibular reflex and diagnose Ms. U with progressive bulbar palsy, a variant of ALS.
The authors’ observations
ALS is a progressive, degenerative neuromuscular condition of unknown etiology affecting the corticospinal tracts and the anterior horn of the spinal cord, leading to dysfunction of the upper and lower motor neurons.6 It is more common in men, persons with diets rich in glutamate, and smokers.7,8
Riluzole is the only FDA-approved medication for ALS.9 It interferes with the responses mediated by the NMDA receptor, stabilizes inactive sodium voltage-dependent channels, inhibits glutamate release from synaptic endings, and activates extracellular reuptake of glutamate, all of which are thought to confer a neuroprotective effect.10
TREATMENT: Psychosis improves
As suggested by the neurology team, we begin riluzole, 50 mg every 12 hours. At this time Ms. U also is taking clozapine, 600 mg/d; lithium, 1200 mg/d; and haloperidol, 6 mg/d; her psychiatric symptoms have not changed since the initial evaluation at the residential facility.
Seven months after initiating riluzole Ms. U is more receptive, less querulant, and no longer experiences delusions or hallucinations. At the same time, she develops an interest in her clinical status regarding her ALS diagnosis, which reflects improved insight. One year after starting riluzole, she is more cooperative and adherent with treatment. Ms. U is able to reestablish relationships with her family. Clozapine and haloperidol are tapered and discontinued. Ms. U’s medication regimen includes risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; and lithium, 600 mg/d, in addition to riluzole, 50 mg every 12 hours.
An assessment 18 months after starting riluzole describes a Positive and Negative Syndrome Scale (PANSS) score of 9 for positive symptoms, 11 for negative, 35 for the general psychopathology, and -2 for the composite (Table 1). Laboratory tests are normal except for a mild normocytic, normochromic anemia. MRI shows no detectable lesions or changes in comparison with previous images.
Table 1
Ms. U’s clinical course
| PANSS score | Treatment | Mental status |
|---|---|---|
| Before starting riluzole | ||
| No PANSS reported | Clozapine, 600 mg/d; lithium, 1200 mg/d; haloperidol, 6 mg/d | Persistent auditory hallucinations. Persistent hallucinatory behavior. Paranoid delirious ideas. Negativism, mutism, and liability reactive to her psychosis state. Poor and flattened affect. Lack of disease awareness. Progressive social isolation. Loss of self care |
| After starting riluzole | ||
| Positive subscale: 9 (below 5th percentile) Negative subscale: 11 (between 5th-25th percentile) General psychopathology subscale: 35 (between 5th-25th percentile) Composite score: -2 (between 25th-50th percentiles) | Riluzole, 50 mg every 12 hours; risperidone, 1 mg/d; methotrimeprazine, 10 mg/d; venlafaxine, 75 mg/d; trazodone, 100 mg/d; lithium, 600 mg/d | Re-establishes relationships with family because she no longer experiences paranoid delusions. Behavioral improvement. Allows physical proximity to nursing and medical personnel. Attention to physical appearance. Participates in social and recreational activities outside the hospital. Absence of auditory hallucinations. Affective improvement with appropriate responses. Realistic anxiety and fear about ALS diagnosis |
| ALS: amyotrophic lateral sclerosis; PANSS: Positive and Negative Syndrome Scale | ||
The authors’ observations
We present a patient with schizophrenia and a continuous pattern of relapses, functional and social impairment, and partial remission of her psychosis despite the use of multiple typical and atypical antipsychotics at therapeutic doses. Ms. U received treatment with clozapine at therapeutic doses for >6 months without sustained improvement. After beginning riluzole, a glutamate pathway antagonist, and with no other changes to her medication regimen, Ms. U experienced substantial improvement in her mental status. This was evidenced by a significant decline in her paranoid delusions, disappearance of auditory hallucinations, and substantial improvement on her social performance.
This fact is consistent with previous observations where modulation of the glutamate pathway has been associated with improvement in depression and anxiety levels in different populations. This case report provides further evidence to the possibility that blocking this receptor is a promising approach to psychotic disorders.
Riluzole for psychiatric illness
Currently, there are 11 clinical trials investigating riluzole for psychiatric disorders, including OCD, depression, bipolar disorder, schizophrenia, and Tourette’s syndrome.11 Consistent with the altered glutamatergic neurotransmission implicated in mood and anxiety disorders, preliminary evidence suggests riluzole can effectively treat OCD, bipolar depression, unipolar depression, and comorbid OCD and depression (Table 2). Some investigators consider the glutamatergic pathway an essential target for future antidepressants and mood-stabilizing agents.12
Other drugs such as memantine, acamprosate, and lamotrigine act on this same pathway and therefore have a role in treating psychiatric and neurologic conditions. In the case of lamotrigine, the drug inhibits glutamate release through inhibition of voltage-dependent sodium and calcium channels13 and postsynaptic AMPA receptors14 and has been shown to effectively treat generalized epilepsies,15 bipolar depression,13,16 and depression and mood swings associated with Huntington’s disease.17
Acamprosate’s attenuation of hyperglutamatergic states through NMDA antagonism and metabotropic glutamate receptors and reduction of intracellular calcium release—therefore balancing the glutamatergic and GABAergic systems and conferring neuroprotective properties—has been effective in patients with alcohol use disorders.18,19
Memantine and amantadine act through NMDA antagonism and by modulating dopaminergic transmission and may have clinical roles beyond dementia treatment.
Table 2
Evidence of efficacy of riluzole for OCD and depression
| Study | Disorder | Findings |
|---|---|---|
| Pittenger et al, 2006a | OCD | Brain imaging reveals elevated glutamate levels in OCD patients; agents that reduce glutamate hyperactivity may be effective |
| Coric et al, 2005b | OCD | Among 13 patients with OCD who received riluzole, 54% demonstrated >35% reduction in Y-BOCS scores and 39% were considered treatment responders |
| Zarate et al, 2005c | Bipolar depression | In an 8-week add-on study of riluzole in combination with lithium of 14 patients with bipolar depression, riluzole showed efficacy as measured by MADRS score and was well tolerated |
| Singh et al, 2004d | Bipolar depression | Case report of a patient with bipolar II disorder and depression who had a good response to riluzole when lamotrigine was discontinued because of a maculopapular erythematic rash |
| Zarate et al, 2004e | Unipolar depression | In a 6-week, open-label trial, 19 treatment-resistant depressed patients received riluzole; significant improvement measured by MADRS, CGI-S, and HAM-A were noted at weeks 3 through 6 |
| Coric et al, 2003f | Comorbid OCD and major depressive disorder | Case report of a patient with symptomatic OCD and depression who did not respond to appropriate pharmacotherapy, including augmentation strategies; adding riluzole significantly attenuated both obsessions and depressive symptoms |
| CGI-S: Clinical Global Impressions-Severity; HAM-A: Hamilton Anxiety Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; OCD: obsessive-compulsive disorder; Y-BOCS: Yale-Brown Obsessive Compulsive Scale Source: a. Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. Neurotherapeutics. 2006;3(1):69-81. b. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424-428. c. Zarate CA Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432. d. Singh J, Zarate CA, Krystal AD. Case report: successful riluzole augmentation therapy in treatment-resistant bipolar depression following the development of rash with lamotrigine. Psychopharmacology. 2004;173(1-2):227-228. e. Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171-174. f. Coric V, Milanovic S, Wasylink S, et al. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology. 2003;167(2):219-220. | ||
Schizophrenia-ALS comorbidity
Some investigators have suggested20 the relative rarity of ALS in patients with schizophrenia is attributable to the neuroprotective effects of antipsychotics and antidepressants.21 If this is true, it is possible resistance to antipsychotics among some schizophrenia patients may be underpinned by the degree of cell injury and therefore of neurodegeneration, which may be the case with Ms. U.
Controlled, randomized, double-blind studies are needed to confirm our team’s assumptions. Our observation is limited by the lack of standardized scale measurements to assess all schizophrenia domains before starting riluzole and Ms. U’s clinical improvement could be associated with other factors such as passage of time or schizophrenia “burning out.” However, clinical observation and description from family members and hospital staff are important to consider in this case.
The improvement in schizophrenia symptoms observed from a drug with no action on dopamine blockade—a quality observed in all antipsychotics22—reinforces the possibility that targeting different pathways involved in the genesis of schizophrenia is a reasonable topic for future research. The possible use of riluzole and other glutamate-modulating drugs might influence positive, negative, and cognitive symptoms of schizophrenia.
Related Resources
- Kantrowitz JT, Javitt DC. Glutamate: new hope for schizophrenia treatment. Current Psychiatry. 2011;10(4):68-74.
- Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2011. Epub ahead of print.
Drug Brand Names
- Acamprosate • Campral
- Amantadine • Symmetrel
- Clozapine • Clozaril
- Haloperidol • Haldol
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Loxapine • Loxitane
- Methotrimeprazine • Nozinan
- Memantine • Namenda
- Olanzapine • Zyprexa
- Riluzole • Rilutek
- Risperidone • Risperdal
- Sertindole • Serdolect
- Thioridazine • Mellaril
- Trazodone • Desyrel, Oleptro
- Trifluoperazine • Stelazine
- Venlafaxine • Effexor
Disclosures
Dr. Millán-González is a consultant to AstraZeneca CAMCAR. Drs. Loizaga-Arniaz and Zúñiga-Montes report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.
1. Freudenreich O, Weiss AP, Goff DC. Psychosis and schizophrenia. In: Stern T Rosenbaum, JF, Fava M, et al, eds. Massachusetts general hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:371–389.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. Bowie CR, Harvey PD. Cognition in schizophrenia: impairments determinants, and functional importance. Psychiatr Clin North Am. 2005;28(3):613-633.
4. Waddington JL, Corvin AP, Donohoe G, et al. Functional genomics and schizophrenia: endophenotypes and mutant models. Psychiatr Clin North Am. 2007;30(3):365-399.
5. Morrow EM, Roffman JL, Wolf DH, et al. Psychiatric neuroscience: incorporating pathophysiology into clinical case formulation. In: Stern T, Rosenbaum, JF, Fava M, et al, eds. Massachusetts General Hospital comprehensive clinical psychiatry. Philadelphia, PA: Mosby, an Imprint of Elsevier; 2008:543–564.
6. Harrison T. Amyotrophic lateral sclerosis. In: Ferri’s clinical advisor 2010. Philadelphia PA. Mosby, an Imprint of Elsevier; 2011:57.
7. Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
8. Chancellor AM, Warlow CP. Adult onset motor neuron disease: worldwide mortality incidence and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
10. Distad BJ, Meekins GD, Liou LL, et al. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651.
11. ClinicalTrials.gov. U.S. National Institutes of Health. Available at: http://clinicaltrials.gov/ct2/results?intr=%22Riluzole%22. Accessed June 27, 2011.
12. Krystal JH, Sanacora G, Blumberg H, et al. Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry. 2002;7(suppl 1):S71-80.
13. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
14. Lee CY, Fu WM, Chen CC, et al. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia. 2008;49(5):888-897.
15. Patsalos PN. Properties of antiepileptic drugs in the treatment of idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):140-148.
16. Yatham LN, Kennedy SH, Schaffer A, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2009. Bipolar Disord. 2009;11(3):225-255.
17. Shen YC. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J Biol Psychiatry. 2008;9(2):147-149.
18. Scott LJ, Figgitt DP, Keam SJ, et al. Acamprosate: a review of its use in the maintenance of abstinence in patients with alcohol dependence. CNS Drugs. 2005;19(5):445-464.
19. De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19(6):517-537.
20. Stommel EW, Graber D, Montanye J, et al. Does treating schizophrenia reduce the chances of developing amyotrophic lateral sclerosis? Med Hypotheses. 2007;69(5):1021-1028.
21. Howland RH. Schizophrenia and amyotrophic lateral sclerosis. Compr Psychiatry. 1990;31(4):327-336.
22. Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27-38.