User login
Precipitously and certainly psychotic—but what’s the cause?
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
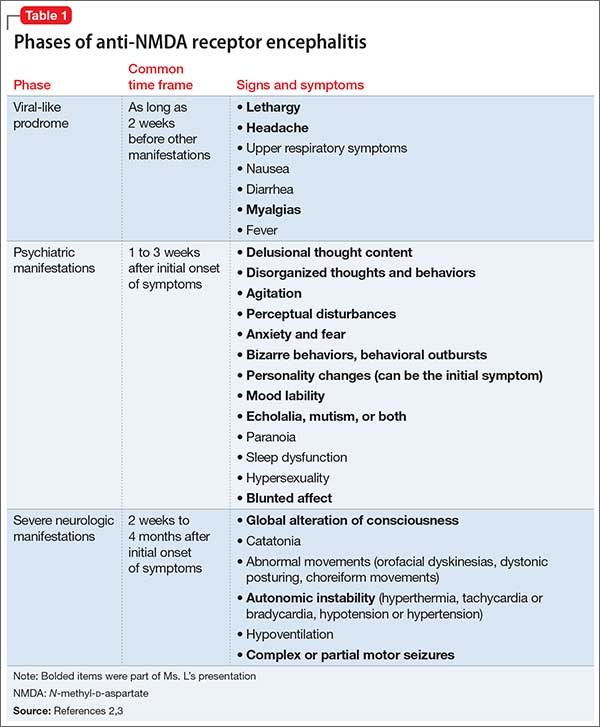
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).

This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).

What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).
This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).
What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).
This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).
What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
Manic after taking a vacation
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.

Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).

OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.
Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).
OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.
Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).
OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
Unresponsive and mute after he smoked ‘Spice’
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).

Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10

In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.

1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).
Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10
In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).
Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10
In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.
1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
Delusions, hypersexuality, and a steep cognitive decline
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2

Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.

TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2
Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.
TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2
Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.
TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
Manic and nonadherent, with a diagnosis of breast cancer
CASE Diagnosis, mood changes
Ms. A, age 58, is a white female with a history of chronic bipolar I disorder who is being evaluated as a new patient in an academic psychiatric clinic. Recently, she was diagnosed with ER+, PR+, and HER2+ ductal carcinoma. She does not take her prescribed mood stabilizers.
After her cancer diagnosis, Ms. A experiences new-onset agitation, including irritable mood, suicidal thoughts, tearfulness, decreased need for sleep, fast speech, excessive spending, and anorexia. She reports that she hears the voice of God telling her that she could cure her breast cancer through prayer and herbal remedies. Her treatment team, comprising her primary care provider and surgical oncologist, consider several medication adjustments, but are unsure of their effects on Ms. A’s mental health, progression of cancer, and cancer treatment.
What is the most likely cause of Ms. A’s psychiatric symptoms?
a) anxiety from having a diagnosis of cancer
b) stress reaction
c) panic attack
d) manic or mixed phase of bipolar I disorder
The authors’ observations
Treating breast cancer with concurrent severe mental illness is complex and challenging for the patient, family, and health care providers. Mental health and oncology clinicians must collaborate when treating these patients because of overlapping pathophysiology and medication interactions. A comprehensive evaluation is required to tease apart whether a patient is simply demoralized by her new diagnosis, or if a more serious mood disorder is present.
Worldwide, breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death among women.1 The mean age of women diagnosed with breast cancer is 61 years; 61% of these women are alive 15 years after diagnosis, representing the largest group of female cancer survivors.
The incidence of breast cancer is reported to be higher in women with bipolar disorder compared with the general population.2-4 This positive correlation might be associated with a high rate of smoking, poor health-related behaviors, and, possibly, medication side effects. A genome-wide association study found significant associations between bipolar disorder and the breast cancer-related genes BRCA2 and PALB2.5
Antipsychotics and prolactin
Antipsychotics play an important role in managing bipolar disorder; several, however, are known to raise the serum prolactin level 10- to 20-fold. A high prolactin level could be associated with progression of breast cancer. All antipsychotics have label warnings regarding their use in women with breast cancer.
The prolactin receptor is overexpressed in >95% of breast cancer cells, regardless of estrogen-receptor status. The role of prolactin in development of new breast cancer is open to debate. The effect of a high prolactin level in women with diagnosed breast cancer is unknown, although available preclinical data suggest that high levels should be avoided. Psychiatric clinicians should consider checking the serum prolactin level or switching to a treatment strategy that avoids iatrogenic prolactin elevation. This risk must be carefully weighed against the mood-stabilizing properties of antipsychotics.6
TREATMENT Consider comorbidities
Ms. A receives supportive psychotherapy in addition to quetiapine, 400 mg/d, and valproic acid, 1,500 mg/d. This regimen helps her successfully complete the initial phase of breast cancer treatment, which consists of a single mastectomy, adjuvant chemotherapy (doxorubicin and cyclophosphamide followed by paclitaxel and trastuzumab). She is now on endocrine therapy with tamoxifen.
Ms. A, calm, much improved mood symptoms, and euthymic, has questions regarding her mental health, cancer prognosis, and potential medication side effects with continued cancer treatment.
Which drug used to treat breast cancer might relieve Ms. A’s manic symptoms?
a) cyclophosphamide
b) tamoxifen
c) trastuzumab
d) pamidronate
The authors’ observations
Recent evidence suggests that tamoxifen reduces symptoms of bipolar mania more rapidly than many standard medications for bipolar disorder. Tamoxifen is the only available centrally active protein kinase C (PKC) inhibitor,7 although lithium and valproic acid also might inhibit PKC activity. PKC regulates presynaptic and postsynaptic neurotransmission, neuronal excitability, and neurotransmitter release. PKC is thought to be overactive during mania, possibly because of an increase in membrane-bound PKC and PKC translocation from the cytosol to membrane.7,8
Preliminary clinical trials suggest that tamoxifen significantly reduces manic symptoms in patients with bipolar disorder within 5 days of initiation.7 These findings have been confirmed in animal studies and in 1 single-blind and 4 double-blind placebo-controlled clinical studies over the past 15 years.9
Tamoxifen is a selective estrogen-receptor modulator used to prevent recurrence in receptor-positive breast cancer. Cytochrome P450 (CYP) 2D6 is the principal enzyme that converts tamoxifen to its active metabolite, endoxifen. Inhibition of tamoxifen conversion to endoxifen by CYP2D6 inhibitors could decrease the efficacy of tamoxifen therapy and might increase the risk of breast cancer recurrence. Although antidepressants generally are not recommended as a first-line agent for bipolar disorder, several selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors are potent, moderate, or mild inhibitors of CYP2D610 (Table 1). Approximately 7% of women have nonfunctional CYP2D6 alleles and have a lower endoxifen level.11
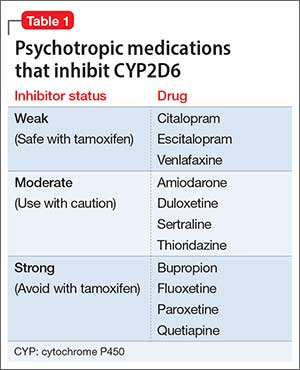
Treating breast cancer
The mainstays of breast cancer treatment are surgery, radiation therapy, chemotherapy, hormone therapy, and targeted monoclonal antibody therapy. The protocol of choice depends on the stage of cancer, estrogen receptor status, expression of human epidermal growth factor receptor 2 (HER-2), treatment history, and the patient’s menopausal status. Overexpression of HER-2 oncoprotein, found in 25% to 30% of breast cancers, has been shown to promote cell transformation. HER-2 overexpression is associated with aggressive tumor phenotypes, lymph node involvement, and resistance to chemotherapy and endocrine therapy. Therefore, the HER-2 oncoprotein is a key target for treatment. Often, several therapies are combined to prevent recurrence of disease.
Breast cancer treatment often can cause demoralization, menopausal symptoms, sleep disturbance, impaired sexual function, infertility, and disturbed body image. It also can trigger psychiatric symptoms in patients with, or without, a history of mental illness.
Trastuzumab is a recombinant humanized monoclonal antibody against HER-2, and is approved for treating HER-2 positive breast cancer. However, approximately 50% of patients with HER-2 overexpression do not respond to trastuzumab alone or combined with chemotherapy, and nearly all patients develop resistance to trastuzumab, leading to recurrence.12 This medication is still used in practice, and research regarding antiepileptic drugs working in synergy with this monoclonal antibody is underway.
OUTCOME Stability achieved
Quetiapine and valproic acid are first-line choices for Ms. A because (1) she would be on long-term tamoxifen to maintain cancer remission maintenance and (2) she is in a manic phase of bipolar disorder. Tamoxifen also could improve her manic symptoms. This medication regimen might enhance the action of cancer treatments and also could reduce adverse effects of cancer treatment, such as insomnia associated with tamoxifen.
After the team educates Ms. A about how her psychiatric medications could benefit her cancer treatment, she becomes more motivated to stay on her regimen. Ms. A does well on these medications and after 18 months has not experienced exacerbation of psychiatric symptoms or recurrence of cancer.
The authors’ observations
There are 3 major classes of mood stabilizers for treating bipolar disorder: lithium, antiepileptic drugs, and atypical antipsychotics.13 In a setting of cancer, mood stabilizers are prescribed for managing mania or drug-induced agitation or anxiety associated with steroid use, brain metastases, and other medical conditions. They also can be used to treat neuropathic pain and hot flashes and seizure prophylaxis.13
Valproic acid
Valproic acid can help treat mood lability, impulsivity, and disinhibition, whether these symptoms are due to primary psychiatric illness or secondary to cancer metastasis. It is a first-line agent for manic and mixed bipolar states, and can be titrated quickly to achieve optimal benefit. Valproic acid also has been described as a histone deacetylase (HDAC) inhibitor, known to attenuate apoptotic activity, making it of interest as a treatment for cancer.14 HDAC inhibitors have been shown to:
- induce differentiation and cell cycle arrest
- activate the extrinsic or intrinsic pathways of apoptosis
- inhibit invasion, migration, and angiogenesis in different cancer cell lines.15
In regard to breast cancer, valproic acid inhibits growth of cell lines independent of estrogen receptors, increases the action of such breast cancer treatments as tamoxifen, raloxifene, fulvestrant, and letrozole, and induces solid tumor regression.14 Valproic acid also reduces cancer cell viability and could act as a powerful antiproliferative agent in estrogen-sensitive breast cancer cells.16
Valproic acid reduces cell growth-inducing apoptosis and cell cycle arrest in ERα-positive breast cancer cells, although it has no significant apoptotic effect in ERα-negative cells.16 However, evidence does support the ability of valproic acid to restore an estrogen-sensitive phenotype in ERα-negative breast cancer cells, allowing successful treatment with the anti-estrogen tamoxifen in vitro.10
Antipsychotics
Antipsychotics act as dopamine D2 receptor antagonists within the hypothalamic-pituitary-adrenal axis, thus increasing the serum prolactin level. Among atypicals, risperidone and its active metabolite, paliperidone, produce the greatest increase in the prolactin level, whereas quetiapine, clozapine, and aripiprazole minimally elevate the prolactin level.
Hyperprolactinemia correlates with rapid breast cancer progression and inferior prognosis, regardless of breast cancer receptor typing. Therefore, prolactin-sparing antipsychotics are preferred when treating a patient with comorbid bipolar disorder and breast cancer. Checking the serum prolactin level might help guide treatment. The literature is mixed regarding antipsychotic use and new mammary tumorigenesis; current research does not support antipsychotic choice based on future risk of breast cancer.6
Other adverse effects from antipsychotic use for bipolar disorder could have an impact on patients with breast cancer. Several of these medications could ameliorate side effects of advanced cancer and chemotherapy. Quetiapine, for example, might improve tamoxifen-induced insomnia in women with breast cancer because of its high affinity for serotonergic receptors, thus enhancing central serotonergic neurotransmitters and decreasing excitatory glutamatergic transmission.17
In any type of advanced cancer, nausea and vomiting are common, independent of chemotherapy and medication regimens. Metabolic derangement, vestibular dysfunction, CNS disorders, and visceral metastasis all contribute to hyperemesis. Olanzapine has been shown to significantly reduce refractory nausea and can cause weight gain and improved appetite, which benefits cachectic patients.18
Last, clozapine is one of the more effective antipsychotic medications, but also carries a risk of neutropenia. In patients with neutropenia secondary to chemotherapy, clozapine could increase the risk of infection in an immunocompromised patient.19 Granulocyte colony stimulating factor might be useful as a rescue medication for treatment-emergent neutropenia.19
Treatment considerations
Cancer patients might be unable or unwilling to seek services for mental health during their cancer treatment, and many who have a diagnosis of psychiatric illness might stop following up with psychiatric care when cancer treatment takes priority. It is critical for clinicians to be aware of the current literature regarding the impact of mood-stabilizing medication on cancer treatment. Monitoring for drug interactions is essential, and electronic drug interaction tools, such as Lexicomp, may be useful for this purpose.13 Because of special vulnerabilities in this population, cautious and judicious prescribing practices are advised.
The risk-benefit profile for medications for bipolar disorder must be considered before they are initiated or changes are made to the regimen (Table 2). Changing an effective mood stabilizer to gain benefits in breast cancer prognosis is not recommended in most cases, because benefits have been shown to be only significant in preclinical research; currently, there are no clinical guidelines. However, medication adjustments should be made with these theoretical benefits in mind, as long as the treatment of bipolar disorder remains effective.
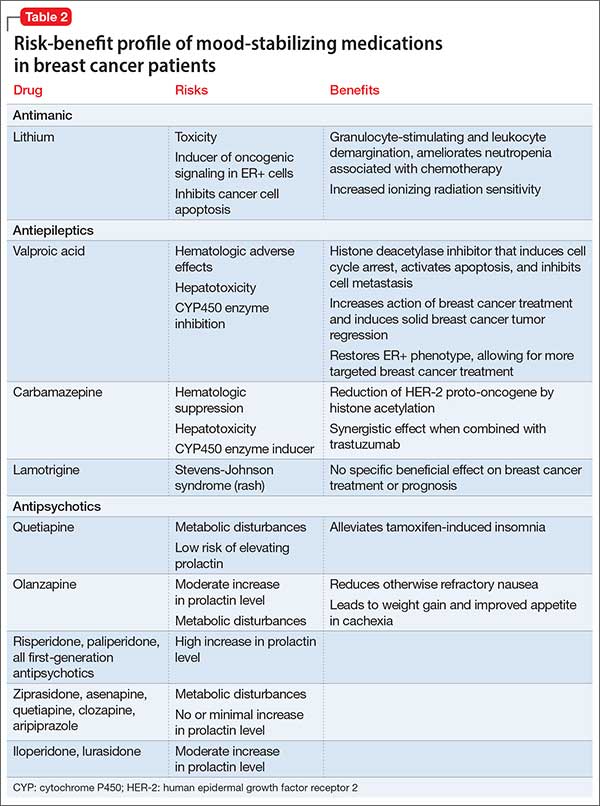
Regardless of what treatment regimen the health team decides on, several underlying issues that affect patient care must be considered in this population. Successfully treating breast cancer in a woman with severe mental illness only can be accomplished when her mental illness is under control. Once she is psychiatrically stable, it is important for her to have a basic understanding of how cancer can affect the body and know the reasons behind treatment.
It is imperative that physicians provide their patients with a general understanding of their comorbid disorders, and find ways to help patients remain adherent with treatment of both diseases. Many patients feel demoralized by a cancer diagnosis and adherence to a medication regimen might be a difficult task among those with bipolar disorder who also are socially isolated, lack education, or have poor recall of treatment recommendations.20
Bottom Line
Managing comorbid bipolar disorder and breast cancer might seem daunting,
but treatments for the 2 diseases can work in synergy. You have an opportunity to
educate patients and colleagues in treating bipolar disorder and comorbid breast
cancer. Optimizing care using known psychopharmacologic data can not only lead
to better outcomes, but might additionally offer some hope and reason to remain
treatment-adherent for patients suffering from this complex comorbidity.
Related Resources
• Agarwala P, Riba MB. Tailoring depression treatment for women with breast cancer. Current Psychiatry. 2010;9(11): 39-40,45-46,48-49.
• Cunningham R, Sarfati D, Stanley J, et al. Cancer survival in the context of mental illness: a national cohort study. Gen Hosp Psychiatry. 2015;37(6):501-506.
Drug Brand Names
Amiodarone • Cordarone
Aripiprazole • Abilify
Asenapine • Saphris
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Cyclophosphamide • Cytoxan, Neosar
Doxorubicin • Doxil, Adriamycin
Duloxetine • Cymbalta
Escitalopram • Lexapro
Fluoxetine • Prozac
Fulvestrant • Faslodex
Iloperidone • Fanapt
Lamotrigine • Lamictal
Letrozole • Femara
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paclitaxel • Onxol
Paliperidone • Invega
Pamidronate • Aredia
Paroxetine • Paxil
Quetiapine • Seroquel
Raloxifene • Evista
Risperidone • Risperdal
Sertraline • Zoloft
Tamoxifen • Nolvadex
Thioridazine • Mellaril
Trastuzumab • Herceptin
Valproic acid • Depakene
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69-90.
2. American Cancer Society. Cancer facts and figures 2014. Atlanta, GA: American Cancer Society; 2014.
3. BarChana M, Levav I, Lipshitz I, et al. Enhanced cancer risk among patients with bipolar disorder. J Affect Disord. 2008;108(1-2):43-48.
4. Hung YP, Liu CJ, Tsai CF, et al. Incidence and risk of mood disorders in patients with breast cancers in Taiwan: a nationwide population-based study. Psychooncology. 2013;22(10):2227-2234.
5. Tesli M, Athanasiu L, Mattingsdal M, et al. Association analysis of PALB2 and BRCA2 in bipolar disorder and schizophrenia in a scandinavian case–control sample. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(7):1276-1282.
6. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
7. Armani F, Andersen ML, Galduróz JC. Tamoxifen use for the management of mania: a review of current preclinical evidence. Psychopharmacology (Berl). 2014;231(4):639-649.
8. Zarate CA Jr, Singh JB, Carlson PJ, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9(6):561-570.
9. Zarate CA, Manji HK. Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs. 2009;23(7):569-582.
10. Fortunati N, Bertino S, Costantino L, et al. Valproic acid restores ER alpha and antiestrogen sensitivity to ER alpha-negative breast cancer cells. Mol Cell Endocrinol. 2010;314(1):17-22.
11. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in cancer. Curr Psychiatry Rep. 2014;17(1):529.
12. Meng Q, Chen X, Sun L, et al. Carbamazepine promotes Her-2 protein degradation in breast cancer cells by modulating HDAC6 activity and acetylation of Hsp90. Mol Cell Biochem. 2011;348(1-2):165-171.
13. Altamura AC, Lietti L, Dobrea C, et al. Mood stabilizers for patients with bipolar disorder: the state of the art. Expert Rev Neurother. 2011;11(1):85-99.
14. Chateauvieux S, Morceau F, Dicato M, et al. Molecular and therapeutic potential and toxicity of valproic acid [published online July 29, 2010]. J Biomed Biotechnol. doi: 10.1155/2010/479364.
15. Jafary H, Ahmadian S, Soleimani M. The enhanced apoptosis and antiproliferative response to combined treatment with valproate and nicotinamide in MCF-7 breast cancer cells. Tumour Biol. 2013;35(3):2701-2710.
16. Fortunati N, Bertino S, Costantino L, et al. Valproic acid is a selective antiproliferative agent in estrogen-sensitive breast cancer cells. Cancer Lett. 2008;259(2):156-164.
17. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
18. Srivastava M, Brito-Dellan N, Davis MP, et al. Olanzapine as an antiemetic in refractory nausea and vomiting in advanced cancer. J Pain Symptom Manage. 2003;25(6):578-582.
19. Sankaranarayanan A, Mulchandani M, Tirupati S. Clozapine, cancer chemotherapy and neutropenia - dilemmas in management. Psychiatr Danub. 2013;25(4):419-422.
20. Cole M, Padmanabhan A. Breast cancer treatment of women with schizophrenia and bipolar disorder from Philadelphia, PA: lessons learned and suggestions for improvement. J Cancer Educ. 2012;27(4):774-779.
CASE Diagnosis, mood changes
Ms. A, age 58, is a white female with a history of chronic bipolar I disorder who is being evaluated as a new patient in an academic psychiatric clinic. Recently, she was diagnosed with ER+, PR+, and HER2+ ductal carcinoma. She does not take her prescribed mood stabilizers.
After her cancer diagnosis, Ms. A experiences new-onset agitation, including irritable mood, suicidal thoughts, tearfulness, decreased need for sleep, fast speech, excessive spending, and anorexia. She reports that she hears the voice of God telling her that she could cure her breast cancer through prayer and herbal remedies. Her treatment team, comprising her primary care provider and surgical oncologist, consider several medication adjustments, but are unsure of their effects on Ms. A’s mental health, progression of cancer, and cancer treatment.
What is the most likely cause of Ms. A’s psychiatric symptoms?
a) anxiety from having a diagnosis of cancer
b) stress reaction
c) panic attack
d) manic or mixed phase of bipolar I disorder
The authors’ observations
Treating breast cancer with concurrent severe mental illness is complex and challenging for the patient, family, and health care providers. Mental health and oncology clinicians must collaborate when treating these patients because of overlapping pathophysiology and medication interactions. A comprehensive evaluation is required to tease apart whether a patient is simply demoralized by her new diagnosis, or if a more serious mood disorder is present.
Worldwide, breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death among women.1 The mean age of women diagnosed with breast cancer is 61 years; 61% of these women are alive 15 years after diagnosis, representing the largest group of female cancer survivors.
The incidence of breast cancer is reported to be higher in women with bipolar disorder compared with the general population.2-4 This positive correlation might be associated with a high rate of smoking, poor health-related behaviors, and, possibly, medication side effects. A genome-wide association study found significant associations between bipolar disorder and the breast cancer-related genes BRCA2 and PALB2.5
Antipsychotics and prolactin
Antipsychotics play an important role in managing bipolar disorder; several, however, are known to raise the serum prolactin level 10- to 20-fold. A high prolactin level could be associated with progression of breast cancer. All antipsychotics have label warnings regarding their use in women with breast cancer.
The prolactin receptor is overexpressed in >95% of breast cancer cells, regardless of estrogen-receptor status. The role of prolactin in development of new breast cancer is open to debate. The effect of a high prolactin level in women with diagnosed breast cancer is unknown, although available preclinical data suggest that high levels should be avoided. Psychiatric clinicians should consider checking the serum prolactin level or switching to a treatment strategy that avoids iatrogenic prolactin elevation. This risk must be carefully weighed against the mood-stabilizing properties of antipsychotics.6
TREATMENT Consider comorbidities
Ms. A receives supportive psychotherapy in addition to quetiapine, 400 mg/d, and valproic acid, 1,500 mg/d. This regimen helps her successfully complete the initial phase of breast cancer treatment, which consists of a single mastectomy, adjuvant chemotherapy (doxorubicin and cyclophosphamide followed by paclitaxel and trastuzumab). She is now on endocrine therapy with tamoxifen.
Ms. A, calm, much improved mood symptoms, and euthymic, has questions regarding her mental health, cancer prognosis, and potential medication side effects with continued cancer treatment.
Which drug used to treat breast cancer might relieve Ms. A’s manic symptoms?
a) cyclophosphamide
b) tamoxifen
c) trastuzumab
d) pamidronate
The authors’ observations
Recent evidence suggests that tamoxifen reduces symptoms of bipolar mania more rapidly than many standard medications for bipolar disorder. Tamoxifen is the only available centrally active protein kinase C (PKC) inhibitor,7 although lithium and valproic acid also might inhibit PKC activity. PKC regulates presynaptic and postsynaptic neurotransmission, neuronal excitability, and neurotransmitter release. PKC is thought to be overactive during mania, possibly because of an increase in membrane-bound PKC and PKC translocation from the cytosol to membrane.7,8
Preliminary clinical trials suggest that tamoxifen significantly reduces manic symptoms in patients with bipolar disorder within 5 days of initiation.7 These findings have been confirmed in animal studies and in 1 single-blind and 4 double-blind placebo-controlled clinical studies over the past 15 years.9
Tamoxifen is a selective estrogen-receptor modulator used to prevent recurrence in receptor-positive breast cancer. Cytochrome P450 (CYP) 2D6 is the principal enzyme that converts tamoxifen to its active metabolite, endoxifen. Inhibition of tamoxifen conversion to endoxifen by CYP2D6 inhibitors could decrease the efficacy of tamoxifen therapy and might increase the risk of breast cancer recurrence. Although antidepressants generally are not recommended as a first-line agent for bipolar disorder, several selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors are potent, moderate, or mild inhibitors of CYP2D610 (Table 1). Approximately 7% of women have nonfunctional CYP2D6 alleles and have a lower endoxifen level.11
Treating breast cancer
The mainstays of breast cancer treatment are surgery, radiation therapy, chemotherapy, hormone therapy, and targeted monoclonal antibody therapy. The protocol of choice depends on the stage of cancer, estrogen receptor status, expression of human epidermal growth factor receptor 2 (HER-2), treatment history, and the patient’s menopausal status. Overexpression of HER-2 oncoprotein, found in 25% to 30% of breast cancers, has been shown to promote cell transformation. HER-2 overexpression is associated with aggressive tumor phenotypes, lymph node involvement, and resistance to chemotherapy and endocrine therapy. Therefore, the HER-2 oncoprotein is a key target for treatment. Often, several therapies are combined to prevent recurrence of disease.
Breast cancer treatment often can cause demoralization, menopausal symptoms, sleep disturbance, impaired sexual function, infertility, and disturbed body image. It also can trigger psychiatric symptoms in patients with, or without, a history of mental illness.
Trastuzumab is a recombinant humanized monoclonal antibody against HER-2, and is approved for treating HER-2 positive breast cancer. However, approximately 50% of patients with HER-2 overexpression do not respond to trastuzumab alone or combined with chemotherapy, and nearly all patients develop resistance to trastuzumab, leading to recurrence.12 This medication is still used in practice, and research regarding antiepileptic drugs working in synergy with this monoclonal antibody is underway.
OUTCOME Stability achieved
Quetiapine and valproic acid are first-line choices for Ms. A because (1) she would be on long-term tamoxifen to maintain cancer remission maintenance and (2) she is in a manic phase of bipolar disorder. Tamoxifen also could improve her manic symptoms. This medication regimen might enhance the action of cancer treatments and also could reduce adverse effects of cancer treatment, such as insomnia associated with tamoxifen.
After the team educates Ms. A about how her psychiatric medications could benefit her cancer treatment, she becomes more motivated to stay on her regimen. Ms. A does well on these medications and after 18 months has not experienced exacerbation of psychiatric symptoms or recurrence of cancer.
The authors’ observations
There are 3 major classes of mood stabilizers for treating bipolar disorder: lithium, antiepileptic drugs, and atypical antipsychotics.13 In a setting of cancer, mood stabilizers are prescribed for managing mania or drug-induced agitation or anxiety associated with steroid use, brain metastases, and other medical conditions. They also can be used to treat neuropathic pain and hot flashes and seizure prophylaxis.13
Valproic acid
Valproic acid can help treat mood lability, impulsivity, and disinhibition, whether these symptoms are due to primary psychiatric illness or secondary to cancer metastasis. It is a first-line agent for manic and mixed bipolar states, and can be titrated quickly to achieve optimal benefit. Valproic acid also has been described as a histone deacetylase (HDAC) inhibitor, known to attenuate apoptotic activity, making it of interest as a treatment for cancer.14 HDAC inhibitors have been shown to:
- induce differentiation and cell cycle arrest
- activate the extrinsic or intrinsic pathways of apoptosis
- inhibit invasion, migration, and angiogenesis in different cancer cell lines.15
In regard to breast cancer, valproic acid inhibits growth of cell lines independent of estrogen receptors, increases the action of such breast cancer treatments as tamoxifen, raloxifene, fulvestrant, and letrozole, and induces solid tumor regression.14 Valproic acid also reduces cancer cell viability and could act as a powerful antiproliferative agent in estrogen-sensitive breast cancer cells.16
Valproic acid reduces cell growth-inducing apoptosis and cell cycle arrest in ERα-positive breast cancer cells, although it has no significant apoptotic effect in ERα-negative cells.16 However, evidence does support the ability of valproic acid to restore an estrogen-sensitive phenotype in ERα-negative breast cancer cells, allowing successful treatment with the anti-estrogen tamoxifen in vitro.10
Antipsychotics
Antipsychotics act as dopamine D2 receptor antagonists within the hypothalamic-pituitary-adrenal axis, thus increasing the serum prolactin level. Among atypicals, risperidone and its active metabolite, paliperidone, produce the greatest increase in the prolactin level, whereas quetiapine, clozapine, and aripiprazole minimally elevate the prolactin level.
Hyperprolactinemia correlates with rapid breast cancer progression and inferior prognosis, regardless of breast cancer receptor typing. Therefore, prolactin-sparing antipsychotics are preferred when treating a patient with comorbid bipolar disorder and breast cancer. Checking the serum prolactin level might help guide treatment. The literature is mixed regarding antipsychotic use and new mammary tumorigenesis; current research does not support antipsychotic choice based on future risk of breast cancer.6
Other adverse effects from antipsychotic use for bipolar disorder could have an impact on patients with breast cancer. Several of these medications could ameliorate side effects of advanced cancer and chemotherapy. Quetiapine, for example, might improve tamoxifen-induced insomnia in women with breast cancer because of its high affinity for serotonergic receptors, thus enhancing central serotonergic neurotransmitters and decreasing excitatory glutamatergic transmission.17
In any type of advanced cancer, nausea and vomiting are common, independent of chemotherapy and medication regimens. Metabolic derangement, vestibular dysfunction, CNS disorders, and visceral metastasis all contribute to hyperemesis. Olanzapine has been shown to significantly reduce refractory nausea and can cause weight gain and improved appetite, which benefits cachectic patients.18
Last, clozapine is one of the more effective antipsychotic medications, but also carries a risk of neutropenia. In patients with neutropenia secondary to chemotherapy, clozapine could increase the risk of infection in an immunocompromised patient.19 Granulocyte colony stimulating factor might be useful as a rescue medication for treatment-emergent neutropenia.19
Treatment considerations
Cancer patients might be unable or unwilling to seek services for mental health during their cancer treatment, and many who have a diagnosis of psychiatric illness might stop following up with psychiatric care when cancer treatment takes priority. It is critical for clinicians to be aware of the current literature regarding the impact of mood-stabilizing medication on cancer treatment. Monitoring for drug interactions is essential, and electronic drug interaction tools, such as Lexicomp, may be useful for this purpose.13 Because of special vulnerabilities in this population, cautious and judicious prescribing practices are advised.
The risk-benefit profile for medications for bipolar disorder must be considered before they are initiated or changes are made to the regimen (Table 2). Changing an effective mood stabilizer to gain benefits in breast cancer prognosis is not recommended in most cases, because benefits have been shown to be only significant in preclinical research; currently, there are no clinical guidelines. However, medication adjustments should be made with these theoretical benefits in mind, as long as the treatment of bipolar disorder remains effective.
Regardless of what treatment regimen the health team decides on, several underlying issues that affect patient care must be considered in this population. Successfully treating breast cancer in a woman with severe mental illness only can be accomplished when her mental illness is under control. Once she is psychiatrically stable, it is important for her to have a basic understanding of how cancer can affect the body and know the reasons behind treatment.
It is imperative that physicians provide their patients with a general understanding of their comorbid disorders, and find ways to help patients remain adherent with treatment of both diseases. Many patients feel demoralized by a cancer diagnosis and adherence to a medication regimen might be a difficult task among those with bipolar disorder who also are socially isolated, lack education, or have poor recall of treatment recommendations.20
Bottom Line
Managing comorbid bipolar disorder and breast cancer might seem daunting,
but treatments for the 2 diseases can work in synergy. You have an opportunity to
educate patients and colleagues in treating bipolar disorder and comorbid breast
cancer. Optimizing care using known psychopharmacologic data can not only lead
to better outcomes, but might additionally offer some hope and reason to remain
treatment-adherent for patients suffering from this complex comorbidity.
Related Resources
• Agarwala P, Riba MB. Tailoring depression treatment for women with breast cancer. Current Psychiatry. 2010;9(11): 39-40,45-46,48-49.
• Cunningham R, Sarfati D, Stanley J, et al. Cancer survival in the context of mental illness: a national cohort study. Gen Hosp Psychiatry. 2015;37(6):501-506.
Drug Brand Names
Amiodarone • Cordarone
Aripiprazole • Abilify
Asenapine • Saphris
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Cyclophosphamide • Cytoxan, Neosar
Doxorubicin • Doxil, Adriamycin
Duloxetine • Cymbalta
Escitalopram • Lexapro
Fluoxetine • Prozac
Fulvestrant • Faslodex
Iloperidone • Fanapt
Lamotrigine • Lamictal
Letrozole • Femara
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paclitaxel • Onxol
Paliperidone • Invega
Pamidronate • Aredia
Paroxetine • Paxil
Quetiapine • Seroquel
Raloxifene • Evista
Risperidone • Risperdal
Sertraline • Zoloft
Tamoxifen • Nolvadex
Thioridazine • Mellaril
Trastuzumab • Herceptin
Valproic acid • Depakene
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Diagnosis, mood changes
Ms. A, age 58, is a white female with a history of chronic bipolar I disorder who is being evaluated as a new patient in an academic psychiatric clinic. Recently, she was diagnosed with ER+, PR+, and HER2+ ductal carcinoma. She does not take her prescribed mood stabilizers.
After her cancer diagnosis, Ms. A experiences new-onset agitation, including irritable mood, suicidal thoughts, tearfulness, decreased need for sleep, fast speech, excessive spending, and anorexia. She reports that she hears the voice of God telling her that she could cure her breast cancer through prayer and herbal remedies. Her treatment team, comprising her primary care provider and surgical oncologist, consider several medication adjustments, but are unsure of their effects on Ms. A’s mental health, progression of cancer, and cancer treatment.
What is the most likely cause of Ms. A’s psychiatric symptoms?
a) anxiety from having a diagnosis of cancer
b) stress reaction
c) panic attack
d) manic or mixed phase of bipolar I disorder
The authors’ observations
Treating breast cancer with concurrent severe mental illness is complex and challenging for the patient, family, and health care providers. Mental health and oncology clinicians must collaborate when treating these patients because of overlapping pathophysiology and medication interactions. A comprehensive evaluation is required to tease apart whether a patient is simply demoralized by her new diagnosis, or if a more serious mood disorder is present.
Worldwide, breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death among women.1 The mean age of women diagnosed with breast cancer is 61 years; 61% of these women are alive 15 years after diagnosis, representing the largest group of female cancer survivors.
The incidence of breast cancer is reported to be higher in women with bipolar disorder compared with the general population.2-4 This positive correlation might be associated with a high rate of smoking, poor health-related behaviors, and, possibly, medication side effects. A genome-wide association study found significant associations between bipolar disorder and the breast cancer-related genes BRCA2 and PALB2.5
Antipsychotics and prolactin
Antipsychotics play an important role in managing bipolar disorder; several, however, are known to raise the serum prolactin level 10- to 20-fold. A high prolactin level could be associated with progression of breast cancer. All antipsychotics have label warnings regarding their use in women with breast cancer.
The prolactin receptor is overexpressed in >95% of breast cancer cells, regardless of estrogen-receptor status. The role of prolactin in development of new breast cancer is open to debate. The effect of a high prolactin level in women with diagnosed breast cancer is unknown, although available preclinical data suggest that high levels should be avoided. Psychiatric clinicians should consider checking the serum prolactin level or switching to a treatment strategy that avoids iatrogenic prolactin elevation. This risk must be carefully weighed against the mood-stabilizing properties of antipsychotics.6
TREATMENT Consider comorbidities
Ms. A receives supportive psychotherapy in addition to quetiapine, 400 mg/d, and valproic acid, 1,500 mg/d. This regimen helps her successfully complete the initial phase of breast cancer treatment, which consists of a single mastectomy, adjuvant chemotherapy (doxorubicin and cyclophosphamide followed by paclitaxel and trastuzumab). She is now on endocrine therapy with tamoxifen.
Ms. A, calm, much improved mood symptoms, and euthymic, has questions regarding her mental health, cancer prognosis, and potential medication side effects with continued cancer treatment.
Which drug used to treat breast cancer might relieve Ms. A’s manic symptoms?
a) cyclophosphamide
b) tamoxifen
c) trastuzumab
d) pamidronate
The authors’ observations
Recent evidence suggests that tamoxifen reduces symptoms of bipolar mania more rapidly than many standard medications for bipolar disorder. Tamoxifen is the only available centrally active protein kinase C (PKC) inhibitor,7 although lithium and valproic acid also might inhibit PKC activity. PKC regulates presynaptic and postsynaptic neurotransmission, neuronal excitability, and neurotransmitter release. PKC is thought to be overactive during mania, possibly because of an increase in membrane-bound PKC and PKC translocation from the cytosol to membrane.7,8
Preliminary clinical trials suggest that tamoxifen significantly reduces manic symptoms in patients with bipolar disorder within 5 days of initiation.7 These findings have been confirmed in animal studies and in 1 single-blind and 4 double-blind placebo-controlled clinical studies over the past 15 years.9
Tamoxifen is a selective estrogen-receptor modulator used to prevent recurrence in receptor-positive breast cancer. Cytochrome P450 (CYP) 2D6 is the principal enzyme that converts tamoxifen to its active metabolite, endoxifen. Inhibition of tamoxifen conversion to endoxifen by CYP2D6 inhibitors could decrease the efficacy of tamoxifen therapy and might increase the risk of breast cancer recurrence. Although antidepressants generally are not recommended as a first-line agent for bipolar disorder, several selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors are potent, moderate, or mild inhibitors of CYP2D610 (Table 1). Approximately 7% of women have nonfunctional CYP2D6 alleles and have a lower endoxifen level.11
Treating breast cancer
The mainstays of breast cancer treatment are surgery, radiation therapy, chemotherapy, hormone therapy, and targeted monoclonal antibody therapy. The protocol of choice depends on the stage of cancer, estrogen receptor status, expression of human epidermal growth factor receptor 2 (HER-2), treatment history, and the patient’s menopausal status. Overexpression of HER-2 oncoprotein, found in 25% to 30% of breast cancers, has been shown to promote cell transformation. HER-2 overexpression is associated with aggressive tumor phenotypes, lymph node involvement, and resistance to chemotherapy and endocrine therapy. Therefore, the HER-2 oncoprotein is a key target for treatment. Often, several therapies are combined to prevent recurrence of disease.
Breast cancer treatment often can cause demoralization, menopausal symptoms, sleep disturbance, impaired sexual function, infertility, and disturbed body image. It also can trigger psychiatric symptoms in patients with, or without, a history of mental illness.
Trastuzumab is a recombinant humanized monoclonal antibody against HER-2, and is approved for treating HER-2 positive breast cancer. However, approximately 50% of patients with HER-2 overexpression do not respond to trastuzumab alone or combined with chemotherapy, and nearly all patients develop resistance to trastuzumab, leading to recurrence.12 This medication is still used in practice, and research regarding antiepileptic drugs working in synergy with this monoclonal antibody is underway.
OUTCOME Stability achieved
Quetiapine and valproic acid are first-line choices for Ms. A because (1) she would be on long-term tamoxifen to maintain cancer remission maintenance and (2) she is in a manic phase of bipolar disorder. Tamoxifen also could improve her manic symptoms. This medication regimen might enhance the action of cancer treatments and also could reduce adverse effects of cancer treatment, such as insomnia associated with tamoxifen.
After the team educates Ms. A about how her psychiatric medications could benefit her cancer treatment, she becomes more motivated to stay on her regimen. Ms. A does well on these medications and after 18 months has not experienced exacerbation of psychiatric symptoms or recurrence of cancer.
The authors’ observations
There are 3 major classes of mood stabilizers for treating bipolar disorder: lithium, antiepileptic drugs, and atypical antipsychotics.13 In a setting of cancer, mood stabilizers are prescribed for managing mania or drug-induced agitation or anxiety associated with steroid use, brain metastases, and other medical conditions. They also can be used to treat neuropathic pain and hot flashes and seizure prophylaxis.13
Valproic acid
Valproic acid can help treat mood lability, impulsivity, and disinhibition, whether these symptoms are due to primary psychiatric illness or secondary to cancer metastasis. It is a first-line agent for manic and mixed bipolar states, and can be titrated quickly to achieve optimal benefit. Valproic acid also has been described as a histone deacetylase (HDAC) inhibitor, known to attenuate apoptotic activity, making it of interest as a treatment for cancer.14 HDAC inhibitors have been shown to:
- induce differentiation and cell cycle arrest
- activate the extrinsic or intrinsic pathways of apoptosis
- inhibit invasion, migration, and angiogenesis in different cancer cell lines.15
In regard to breast cancer, valproic acid inhibits growth of cell lines independent of estrogen receptors, increases the action of such breast cancer treatments as tamoxifen, raloxifene, fulvestrant, and letrozole, and induces solid tumor regression.14 Valproic acid also reduces cancer cell viability and could act as a powerful antiproliferative agent in estrogen-sensitive breast cancer cells.16
Valproic acid reduces cell growth-inducing apoptosis and cell cycle arrest in ERα-positive breast cancer cells, although it has no significant apoptotic effect in ERα-negative cells.16 However, evidence does support the ability of valproic acid to restore an estrogen-sensitive phenotype in ERα-negative breast cancer cells, allowing successful treatment with the anti-estrogen tamoxifen in vitro.10
Antipsychotics
Antipsychotics act as dopamine D2 receptor antagonists within the hypothalamic-pituitary-adrenal axis, thus increasing the serum prolactin level. Among atypicals, risperidone and its active metabolite, paliperidone, produce the greatest increase in the prolactin level, whereas quetiapine, clozapine, and aripiprazole minimally elevate the prolactin level.
Hyperprolactinemia correlates with rapid breast cancer progression and inferior prognosis, regardless of breast cancer receptor typing. Therefore, prolactin-sparing antipsychotics are preferred when treating a patient with comorbid bipolar disorder and breast cancer. Checking the serum prolactin level might help guide treatment. The literature is mixed regarding antipsychotic use and new mammary tumorigenesis; current research does not support antipsychotic choice based on future risk of breast cancer.6
Other adverse effects from antipsychotic use for bipolar disorder could have an impact on patients with breast cancer. Several of these medications could ameliorate side effects of advanced cancer and chemotherapy. Quetiapine, for example, might improve tamoxifen-induced insomnia in women with breast cancer because of its high affinity for serotonergic receptors, thus enhancing central serotonergic neurotransmitters and decreasing excitatory glutamatergic transmission.17
In any type of advanced cancer, nausea and vomiting are common, independent of chemotherapy and medication regimens. Metabolic derangement, vestibular dysfunction, CNS disorders, and visceral metastasis all contribute to hyperemesis. Olanzapine has been shown to significantly reduce refractory nausea and can cause weight gain and improved appetite, which benefits cachectic patients.18
Last, clozapine is one of the more effective antipsychotic medications, but also carries a risk of neutropenia. In patients with neutropenia secondary to chemotherapy, clozapine could increase the risk of infection in an immunocompromised patient.19 Granulocyte colony stimulating factor might be useful as a rescue medication for treatment-emergent neutropenia.19
Treatment considerations
Cancer patients might be unable or unwilling to seek services for mental health during their cancer treatment, and many who have a diagnosis of psychiatric illness might stop following up with psychiatric care when cancer treatment takes priority. It is critical for clinicians to be aware of the current literature regarding the impact of mood-stabilizing medication on cancer treatment. Monitoring for drug interactions is essential, and electronic drug interaction tools, such as Lexicomp, may be useful for this purpose.13 Because of special vulnerabilities in this population, cautious and judicious prescribing practices are advised.
The risk-benefit profile for medications for bipolar disorder must be considered before they are initiated or changes are made to the regimen (Table 2). Changing an effective mood stabilizer to gain benefits in breast cancer prognosis is not recommended in most cases, because benefits have been shown to be only significant in preclinical research; currently, there are no clinical guidelines. However, medication adjustments should be made with these theoretical benefits in mind, as long as the treatment of bipolar disorder remains effective.
Regardless of what treatment regimen the health team decides on, several underlying issues that affect patient care must be considered in this population. Successfully treating breast cancer in a woman with severe mental illness only can be accomplished when her mental illness is under control. Once she is psychiatrically stable, it is important for her to have a basic understanding of how cancer can affect the body and know the reasons behind treatment.
It is imperative that physicians provide their patients with a general understanding of their comorbid disorders, and find ways to help patients remain adherent with treatment of both diseases. Many patients feel demoralized by a cancer diagnosis and adherence to a medication regimen might be a difficult task among those with bipolar disorder who also are socially isolated, lack education, or have poor recall of treatment recommendations.20
Bottom Line
Managing comorbid bipolar disorder and breast cancer might seem daunting,
but treatments for the 2 diseases can work in synergy. You have an opportunity to
educate patients and colleagues in treating bipolar disorder and comorbid breast
cancer. Optimizing care using known psychopharmacologic data can not only lead
to better outcomes, but might additionally offer some hope and reason to remain
treatment-adherent for patients suffering from this complex comorbidity.
Related Resources
• Agarwala P, Riba MB. Tailoring depression treatment for women with breast cancer. Current Psychiatry. 2010;9(11): 39-40,45-46,48-49.
• Cunningham R, Sarfati D, Stanley J, et al. Cancer survival in the context of mental illness: a national cohort study. Gen Hosp Psychiatry. 2015;37(6):501-506.
Drug Brand Names
Amiodarone • Cordarone
Aripiprazole • Abilify
Asenapine • Saphris
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Cyclophosphamide • Cytoxan, Neosar
Doxorubicin • Doxil, Adriamycin
Duloxetine • Cymbalta
Escitalopram • Lexapro
Fluoxetine • Prozac
Fulvestrant • Faslodex
Iloperidone • Fanapt
Lamotrigine • Lamictal
Letrozole • Femara
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paclitaxel • Onxol
Paliperidone • Invega
Pamidronate • Aredia
Paroxetine • Paxil
Quetiapine • Seroquel
Raloxifene • Evista
Risperidone • Risperdal
Sertraline • Zoloft
Tamoxifen • Nolvadex
Thioridazine • Mellaril
Trastuzumab • Herceptin
Valproic acid • Depakene
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69-90.
2. American Cancer Society. Cancer facts and figures 2014. Atlanta, GA: American Cancer Society; 2014.
3. BarChana M, Levav I, Lipshitz I, et al. Enhanced cancer risk among patients with bipolar disorder. J Affect Disord. 2008;108(1-2):43-48.
4. Hung YP, Liu CJ, Tsai CF, et al. Incidence and risk of mood disorders in patients with breast cancers in Taiwan: a nationwide population-based study. Psychooncology. 2013;22(10):2227-2234.
5. Tesli M, Athanasiu L, Mattingsdal M, et al. Association analysis of PALB2 and BRCA2 in bipolar disorder and schizophrenia in a scandinavian case–control sample. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(7):1276-1282.
6. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
7. Armani F, Andersen ML, Galduróz JC. Tamoxifen use for the management of mania: a review of current preclinical evidence. Psychopharmacology (Berl). 2014;231(4):639-649.
8. Zarate CA Jr, Singh JB, Carlson PJ, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9(6):561-570.
9. Zarate CA, Manji HK. Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs. 2009;23(7):569-582.
10. Fortunati N, Bertino S, Costantino L, et al. Valproic acid restores ER alpha and antiestrogen sensitivity to ER alpha-negative breast cancer cells. Mol Cell Endocrinol. 2010;314(1):17-22.
11. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in cancer. Curr Psychiatry Rep. 2014;17(1):529.
12. Meng Q, Chen X, Sun L, et al. Carbamazepine promotes Her-2 protein degradation in breast cancer cells by modulating HDAC6 activity and acetylation of Hsp90. Mol Cell Biochem. 2011;348(1-2):165-171.
13. Altamura AC, Lietti L, Dobrea C, et al. Mood stabilizers for patients with bipolar disorder: the state of the art. Expert Rev Neurother. 2011;11(1):85-99.
14. Chateauvieux S, Morceau F, Dicato M, et al. Molecular and therapeutic potential and toxicity of valproic acid [published online July 29, 2010]. J Biomed Biotechnol. doi: 10.1155/2010/479364.
15. Jafary H, Ahmadian S, Soleimani M. The enhanced apoptosis and antiproliferative response to combined treatment with valproate and nicotinamide in MCF-7 breast cancer cells. Tumour Biol. 2013;35(3):2701-2710.
16. Fortunati N, Bertino S, Costantino L, et al. Valproic acid is a selective antiproliferative agent in estrogen-sensitive breast cancer cells. Cancer Lett. 2008;259(2):156-164.
17. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
18. Srivastava M, Brito-Dellan N, Davis MP, et al. Olanzapine as an antiemetic in refractory nausea and vomiting in advanced cancer. J Pain Symptom Manage. 2003;25(6):578-582.
19. Sankaranarayanan A, Mulchandani M, Tirupati S. Clozapine, cancer chemotherapy and neutropenia - dilemmas in management. Psychiatr Danub. 2013;25(4):419-422.
20. Cole M, Padmanabhan A. Breast cancer treatment of women with schizophrenia and bipolar disorder from Philadelphia, PA: lessons learned and suggestions for improvement. J Cancer Educ. 2012;27(4):774-779.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69-90.
2. American Cancer Society. Cancer facts and figures 2014. Atlanta, GA: American Cancer Society; 2014.
3. BarChana M, Levav I, Lipshitz I, et al. Enhanced cancer risk among patients with bipolar disorder. J Affect Disord. 2008;108(1-2):43-48.
4. Hung YP, Liu CJ, Tsai CF, et al. Incidence and risk of mood disorders in patients with breast cancers in Taiwan: a nationwide population-based study. Psychooncology. 2013;22(10):2227-2234.
5. Tesli M, Athanasiu L, Mattingsdal M, et al. Association analysis of PALB2 and BRCA2 in bipolar disorder and schizophrenia in a scandinavian case–control sample. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(7):1276-1282.
6. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
7. Armani F, Andersen ML, Galduróz JC. Tamoxifen use for the management of mania: a review of current preclinical evidence. Psychopharmacology (Berl). 2014;231(4):639-649.
8. Zarate CA Jr, Singh JB, Carlson PJ, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9(6):561-570.
9. Zarate CA, Manji HK. Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs. 2009;23(7):569-582.
10. Fortunati N, Bertino S, Costantino L, et al. Valproic acid restores ER alpha and antiestrogen sensitivity to ER alpha-negative breast cancer cells. Mol Cell Endocrinol. 2010;314(1):17-22.
11. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in cancer. Curr Psychiatry Rep. 2014;17(1):529.
12. Meng Q, Chen X, Sun L, et al. Carbamazepine promotes Her-2 protein degradation in breast cancer cells by modulating HDAC6 activity and acetylation of Hsp90. Mol Cell Biochem. 2011;348(1-2):165-171.
13. Altamura AC, Lietti L, Dobrea C, et al. Mood stabilizers for patients with bipolar disorder: the state of the art. Expert Rev Neurother. 2011;11(1):85-99.
14. Chateauvieux S, Morceau F, Dicato M, et al. Molecular and therapeutic potential and toxicity of valproic acid [published online July 29, 2010]. J Biomed Biotechnol. doi: 10.1155/2010/479364.
15. Jafary H, Ahmadian S, Soleimani M. The enhanced apoptosis and antiproliferative response to combined treatment with valproate and nicotinamide in MCF-7 breast cancer cells. Tumour Biol. 2013;35(3):2701-2710.
16. Fortunati N, Bertino S, Costantino L, et al. Valproic acid is a selective antiproliferative agent in estrogen-sensitive breast cancer cells. Cancer Lett. 2008;259(2):156-164.
17. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
18. Srivastava M, Brito-Dellan N, Davis MP, et al. Olanzapine as an antiemetic in refractory nausea and vomiting in advanced cancer. J Pain Symptom Manage. 2003;25(6):578-582.
19. Sankaranarayanan A, Mulchandani M, Tirupati S. Clozapine, cancer chemotherapy and neutropenia - dilemmas in management. Psychiatr Danub. 2013;25(4):419-422.
20. Cole M, Padmanabhan A. Breast cancer treatment of women with schizophrenia and bipolar disorder from Philadelphia, PA: lessons learned and suggestions for improvement. J Cancer Educ. 2012;27(4):774-779.

Agitated and hallucinating, with a throbbing headache
CASE Psychotic, and nonadherent
Mr. K, a 42-year-old Fijian man, is brought to the emergency department by his older brother for evaluation of behavioral agitation. Mr. K is belligerent and threatens to kill his family members. Three years earlier, he was given a diagnosis of schizophrenia and treated at an inpatient psychiatric unit.
At that time, Mr. K was stabilized on risperidone, 4 mg/d. However, he did not follow-up with treatment after discharge and has not taken any psychotropic medications since that time.
His brother reports that Mr. K has been slowly deteriorating, talking to himself, staying up at night, and getting into arguments with his family over his delusional beliefs. Although Mr. K once worked as a security guard, he has not worked in 8 years. He has been living with his family, who are no longer willing to accept him into their home because they fear he might harm them.
In the emergency department, Mr. K reports that he has a throbbing headache. Blood pressure is 177/101 mm Hg; heart rate is 103 beats per minute; respiratory rate is 18 breaths per minute; weight is 185 lb; and body mass index (BMI) is 31.8. Physical examination is unremarkable.
Laboratory values show that sodium is 131 mEq/L; potassium, 3.7 mEq/L; bicarbonate, 26 mEq/L; glucose, 420 mg/dL; hemoglobin A1c,12.7; and urine glucose, 3+. Mr. K denies being told he has diabetes.
What are Mr. K’s risk factors for diabetes?
a) schizophrenia
b) physical inactivity
c) obesity
d) Fijian ethnicity
e) all of the above
The authors’ observations
The prevalence of type 2 diabetes mellitus (T2DM) in persons with schizophrenia or a schizoaffective disorder is twice that of the general population.1-4 Multiple variables contribute to the increased prevalence of diabetes in this population, including genetic predisposition, environmental and cultural factors related to diet and physical activity, a high rate of smoking,5,6 iatrogenic causes (metabolic dysregulation and weight gain from antipsychotic treatment), and socioeconomic factors (poverty, lack of access to health care). In addition, symptoms of psychosis such as thought disorder, delusions, hallucinations, and cognitive decline in persons with chronic schizophrenia and schizoaffective disorder can make basic health maintenance difficult.
In Mr. K’s case, premorbid risk of diabetes was elevated because of his Fijian ethnicity.7 With a BMI of 31.8, obesity further increased that risk.5,6,8 In addition, his untreated chronic mental illness, lack of access to health care, low socioeconomic status, long-standing smoking habit, and previous exposure to antipsychotics also increased his risk of T2DM.
The interaction between diabetes and psychosis contributes to a vicious cycle that makes both conditions worse if either, or both, are untreated. In general, medical comorbidities are associated with depression and neurocognitive impairment in persons with schizophrenia.9 Specifically, diabetes is associated with lower global cognitive functioning among persons with schizophrenia.10 Poor cognitive functioning can, in turn, decrease the patient’s ability to manage his medical illness. Also, persons with schizophrenia are less likely to be treated for diabetes, dyslipidemia, and hypertension, as in Mr. K’s case.11
How would you treat Mr. K’s newly diagnosed diabetes?
a) refer him to a primary care physician
b) start an oral agent
c) start sliding-scale insulin
d) start long-acting insulin
e) recommend a carbohydrate-controlled diet=
TREATMENT Stabilization
Mr. K is admitted to the medical unit for treatment of hyperglycemia. The team starts him on amlodipine, 5 mg/d, for hypertension; aripiprazole, 10 mg/d, for psychosis; and sliding-scale insulin (lispro) and 20 units of insulin (glargine) nightly for diabetes. Mr. K’s blood glucose level is well regulated on this regimen; after being medically cleared, he is transferred to the inpatient psychiatric unit.
EVALUATION Denies symptoms
Mr. K appears older than his stated age, is poorly groomed, and is dressed in a hospital gown. He is isolated and appears to be internally preoccupied. He repeatedly denies hearing auditory hallucinations, but often is overheard responding to internal stimuli and mumbling indecipherably in a low tone. His speech is decreased and his affect is flat and guarded. He states that he is not “mental” but that he came to the hospital for “tooth pain.” Every day he asks when he can return home and he asks the staff to call his family to take him home. When informed that his family is not able to care for him, Mr. K states that he would live in a house he owns in Fiji, which his family members state is untrue.
How would you treat Mr. K’s psychosis?
a) continue aripiprazole
b) switch to risperidone, an agent to which he previously responded
c) switch to olanzapine because he has not been sleeping well
d) switch to haloperidol because of diabetes
The authors’ observations
Pharmacotherapy for patients with comorbid schizophrenia and diabetes requires consideration of clinical and psychosocial factors unique to this population.
Antipsychotic choice
Selection of an antipsychotic agent to address psychosis requires considering its efficacy, side-effect profile, and adherence rates, as well as its potential effects on metabolic regulation and weight (Table 1). Typical antipsychotics are less likely than atypical antipsychotics to cause metabolic dysregulation.12 When treatment with atypical antipsychotics cannot be avoided—such as when side effects or an allergic reaction develop—consider selecting a relatively weight-neutral drug with a lower potential for metabolic dysregulation, such as aripiprazole. However, many times, using an agent with significant effects on metabolic regulation cannot be avoided, making treating and monitoring the resulting metabolic effects even more significant.

Glycemic control
Initiating an agent for glycemic control in persons with newly diagnosed diabetes requires weighing many factors, including mode of delivery (oral or subcutaneous), level of glycemic control required, comorbid medical illness (such as renal impairment and heart failure), cost, and potential long-term side effects of the medication (Table 2).13 Oral agents have a slower effect on lowering the blood glucose level, and each agent carries contraindications, but generally they are more affordable. Many present a low risk of hypoglycemia but sulfonylureas are an exception. In addition, metformin could lead to weight loss over the long term, which further lowers the risk of diabetes.

If, however, tight and immediate glycemic control is needed, subcutaneous insulin injections might be required, although this method is more costly, carries an increased risk of hypoglycemic episodes acutely, and leads to weight gain in the long run because of induced hunger and increased appetite.
Psychosocial factors
In addition to the clinical variables above, treating diabetes in patients with comorbid schizophrenia requires considering other psychosocial factors that might not be readily apparent (Table 3). For example, patients with schizophrenia might have decreased motivation, self-efficacy, and capacity for self-care when it comes to health maintenance and medication adherence.14 Chronically mentally ill persons might have reduced cognitive functioning that could affect their ability to maintain complicated medication regimens, such as administering insulin and monitoring blood glucose.
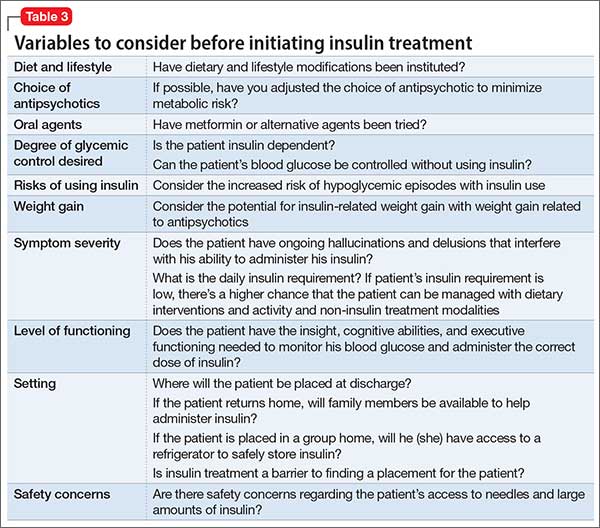
In addition, easy access to hypodermic needles and large amounts of insulin could become a safety concern in patients with ongoing hallucinations, delusions, and thought disorder, despite antipsychotic treatment. For example, a patient with schizophrenia and diabetes who has been maintained on insulin might begin hearing voices that tell her to inject her eyeballs with insulin. Similarly, although the risk of hypoglycemic episodes in patients treated with insulin is well known, patients with schizophrenia have a higher risk of acute complications of diabetes than other patients with diabetes.15Psychosocial factors, such as placement, support system, and follow-up care need to be considered. In some instances, the need to administer daily subcutaneous insulin could be a barrier to placement if the facility does not have the staff or expertise to monitor blood glucose and administer insulin.
If the patient is returning home, then the patient or a caretaker will need to be trained to monitor blood glucose and administer insulin. This might be difficult for persons with chronic mental illness who have lost the support and care of their family. Also, consider the issue of storing insulin, which requires refrigeration. Because of the potential complications involved in using insulin in patients with schizophrenia, practitioners should consider managing non-insulin dependent diabetes with an oral hypoglycemic agent, when possible, along with lifestyle modifications.
OUTCOME Weight loss, discharge
Mr. K is transitioned from aripiprazole to higher-potency oral risperidone, titrated to 6 mg/d. At this dosage, he continues to maintain a delusion about owning a house in Fiji, but was seen responding to internal stimuli less often. The treatment team places him on a diabetic and low-sodium diet of 1,800 kcal/d. His fasting blood glucose levels range in the 70s to 110s mg/dL during his first week of hospitalization.
The treatment team starts Mr. K on oral metformin, titrated to 1,000 mg twice daily, while tapering him off insulin lispro and glargine over the course of hospitalization. The transition from insulin to metformin also is considered because Mr. K’s daily insulin requirement is rather low (<0.5 units/kg).
Mr. K’s course is prolonged because his treatment team initiates the process of conservatorship and placement in the community. Approximately 6 months after his admission, Mr. K is discharged to an unlocked facility with support from an intensive outpatient mental health program. At 6 months follow-up with his outpatient provider, Mr. K’s hemoglobin A1c is 7.0 and he weighs 155 lb with a BMI of 26.5.
The authors’ observations
Despite Mr. K’s initial elevated hemoglobin A1c of 12.7 and weight of 185 lb, over approximately 6 months he experiences a 5.7-unit drop in hemoglobin A1c and weight loss of 30 lb with dietary management and metformin—without the need for other agents. Other options for weight-neutral treatment of T2DM include exenatide, which also is available as a once weekly injectable formulation, canagliflozin, and the gliptins (sitagliptin, saxagliptin, and linagliptin) (Table 4).
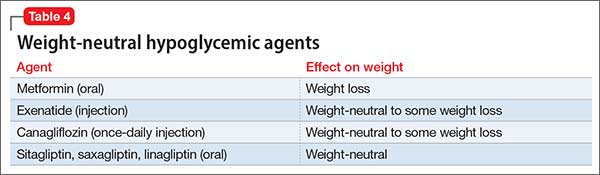
Mr. K’s improved control of his diabetes occurred despite initiation of an atypical antipsychotic, which would have been expected to cause additional weight gain and make his diabetes worse secondary to metabolic effects.12 Treatment with metformin in particular has been associated with weight loss in patients with16 and without17 comorbid schizophrenia, including those with antipsychotic-induced weight gain.18-20
Bottom Line
Persons with schizophrenia are at higher risk of type 2 diabetes mellitus for many reasons, including metabolic effects of antipsychotics, reduced cognitive functioning, poor self-care, and limited access to health care. Typical antipsychotics are less likely to cause metabolic dysregulation but, if an atypical is needed, one that is relatively weight-neutral should be selected. When choosing an agent for glycemic control, consider a patient’s or a caretaker’s ability to administer medications, monitor blood glucose, and follow dietary recommendations.
Related Resources
• Cohn T. The link between schizophrenia and diabetes. Current Psychiatry. 2012;11(10):28-34,46.
• Annamalai A, Tek C. An overview of diabetes management in schizophrenia patients: office based strategies for primary care practitioners and endocrinologists [published online March 23, 2015]. Int J Endocrinol. 2015;2015:969182. doi: 10.1155/2015/969182.
Drug Brand Names
Amlodipine • Norvasc
Aripiprazole • Abilify
Asenapine • Saphris
Canagliflozin • Invokana
Exenatide • Bydureon, Byetta
Glargine • Lantus
Linagliptin • Tradjenta
Lispro • Humalog
Lurasidone • Latuda
Metformin • Glucophage
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Saxagliptin • Onglyza
Sitagliptin • Januvia
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. Heald A. Physical health in schizophrenia: a challenge for antipsychotic therapy. Eur Psychiatry. 2010;25(suppl 2):S6-S11.
3. El-Mallakh P. Doing my best: poverty and self-care among individuals with schizophrenia and diabetes mellitus. Arch Psychiatr Nurs. 2007;21(1):49-60; discussion 61-63.
4. Rouillon F, Sorbara F. Schizophrenia and diabetes: epidemiological data. Eur Psychiatry. 2005;20(suppl 4):S345-S348.
5. Willi C, Bodenmann P, Ghali WA, et al. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2007;298(22):2654-2664.
6. Dome P, Lazary J, Kalapos MP, et al. Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev. 2010;34(3):295-342.
7. National Institute of Diabetes and Digestive and Kidney Diseases. For people of African, Mediterranean, or Southeast Asian heritage: important information about diabetes blood tests. http://www.diabetes.niddk.nih.gov/dm/ pubs/asianamerican/index.htm. Published October 2011. Accessed November 18, 2015.
8. Shaten BJ, Smith GD, Kuller LH, et al. Risk factors for the development of type 2 diabetes among men enrolled in the usual care group of the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16(10):1331-1339.
9. Chwastiak LA, Rosenheck RA, McEvoy JP, et al. Interrelationships of psychiatric symptom severity, medical comorbidity, and functioning in schizophrenia. Psychiatr Serv. 2006;57(8):1102-1109.
10. Takayanagi Y, Cascella NG, Sawa A, et al. Diabetes is associated with lower global cognitive function in schizophrenia. Schizophr Res. 2012;142(1-3):183-187.
11. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
12. Meyer JM, Davis VG, Goff DC, et al. Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE Schizophrenia Trial: prospective data from phase 1. Schizophr Res. 2008;101(1-3):273-286.
13. Inzucchi SE. Clinical practice. Management of hyperglycemia in the hospital setting. N Engl J Med. 2006; 355(18):1903-1911.
14. Chen SR, Chien YP, Kang CM, et al. Comparing self-efficacy and self-care behaviours between outpatients with comorbid schizophrenia and type 2 diabetes and outpatients with only type 2 diabetes. J Psychiatr Ment Health Nurs. 2014;21(5):414-422.
15. Becker T, Hux J. Risk of acute complications of diabetes among people with schizophrenia in Ontario, Canada. Diabetes Care. 2011;34(2):398-402.
16. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care. 2012;35(4):731-737.
17. Jarskog LF, Hamer RM, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
18. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-430. doi: 10.4088/JCP.12m08186.
19. Wang M, Tong JH, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
20. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6): 1385-1403.
CASE Psychotic, and nonadherent
Mr. K, a 42-year-old Fijian man, is brought to the emergency department by his older brother for evaluation of behavioral agitation. Mr. K is belligerent and threatens to kill his family members. Three years earlier, he was given a diagnosis of schizophrenia and treated at an inpatient psychiatric unit.
At that time, Mr. K was stabilized on risperidone, 4 mg/d. However, he did not follow-up with treatment after discharge and has not taken any psychotropic medications since that time.
His brother reports that Mr. K has been slowly deteriorating, talking to himself, staying up at night, and getting into arguments with his family over his delusional beliefs. Although Mr. K once worked as a security guard, he has not worked in 8 years. He has been living with his family, who are no longer willing to accept him into their home because they fear he might harm them.
In the emergency department, Mr. K reports that he has a throbbing headache. Blood pressure is 177/101 mm Hg; heart rate is 103 beats per minute; respiratory rate is 18 breaths per minute; weight is 185 lb; and body mass index (BMI) is 31.8. Physical examination is unremarkable.
Laboratory values show that sodium is 131 mEq/L; potassium, 3.7 mEq/L; bicarbonate, 26 mEq/L; glucose, 420 mg/dL; hemoglobin A1c,12.7; and urine glucose, 3+. Mr. K denies being told he has diabetes.
What are Mr. K’s risk factors for diabetes?
a) schizophrenia
b) physical inactivity
c) obesity
d) Fijian ethnicity
e) all of the above
The authors’ observations
The prevalence of type 2 diabetes mellitus (T2DM) in persons with schizophrenia or a schizoaffective disorder is twice that of the general population.1-4 Multiple variables contribute to the increased prevalence of diabetes in this population, including genetic predisposition, environmental and cultural factors related to diet and physical activity, a high rate of smoking,5,6 iatrogenic causes (metabolic dysregulation and weight gain from antipsychotic treatment), and socioeconomic factors (poverty, lack of access to health care). In addition, symptoms of psychosis such as thought disorder, delusions, hallucinations, and cognitive decline in persons with chronic schizophrenia and schizoaffective disorder can make basic health maintenance difficult.
In Mr. K’s case, premorbid risk of diabetes was elevated because of his Fijian ethnicity.7 With a BMI of 31.8, obesity further increased that risk.5,6,8 In addition, his untreated chronic mental illness, lack of access to health care, low socioeconomic status, long-standing smoking habit, and previous exposure to antipsychotics also increased his risk of T2DM.
The interaction between diabetes and psychosis contributes to a vicious cycle that makes both conditions worse if either, or both, are untreated. In general, medical comorbidities are associated with depression and neurocognitive impairment in persons with schizophrenia.9 Specifically, diabetes is associated with lower global cognitive functioning among persons with schizophrenia.10 Poor cognitive functioning can, in turn, decrease the patient’s ability to manage his medical illness. Also, persons with schizophrenia are less likely to be treated for diabetes, dyslipidemia, and hypertension, as in Mr. K’s case.11
How would you treat Mr. K’s newly diagnosed diabetes?
a) refer him to a primary care physician
b) start an oral agent
c) start sliding-scale insulin
d) start long-acting insulin
e) recommend a carbohydrate-controlled diet=
TREATMENT Stabilization
Mr. K is admitted to the medical unit for treatment of hyperglycemia. The team starts him on amlodipine, 5 mg/d, for hypertension; aripiprazole, 10 mg/d, for psychosis; and sliding-scale insulin (lispro) and 20 units of insulin (glargine) nightly for diabetes. Mr. K’s blood glucose level is well regulated on this regimen; after being medically cleared, he is transferred to the inpatient psychiatric unit.
EVALUATION Denies symptoms
Mr. K appears older than his stated age, is poorly groomed, and is dressed in a hospital gown. He is isolated and appears to be internally preoccupied. He repeatedly denies hearing auditory hallucinations, but often is overheard responding to internal stimuli and mumbling indecipherably in a low tone. His speech is decreased and his affect is flat and guarded. He states that he is not “mental” but that he came to the hospital for “tooth pain.” Every day he asks when he can return home and he asks the staff to call his family to take him home. When informed that his family is not able to care for him, Mr. K states that he would live in a house he owns in Fiji, which his family members state is untrue.
How would you treat Mr. K’s psychosis?
a) continue aripiprazole
b) switch to risperidone, an agent to which he previously responded
c) switch to olanzapine because he has not been sleeping well
d) switch to haloperidol because of diabetes
The authors’ observations
Pharmacotherapy for patients with comorbid schizophrenia and diabetes requires consideration of clinical and psychosocial factors unique to this population.
Antipsychotic choice
Selection of an antipsychotic agent to address psychosis requires considering its efficacy, side-effect profile, and adherence rates, as well as its potential effects on metabolic regulation and weight (Table 1). Typical antipsychotics are less likely than atypical antipsychotics to cause metabolic dysregulation.12 When treatment with atypical antipsychotics cannot be avoided—such as when side effects or an allergic reaction develop—consider selecting a relatively weight-neutral drug with a lower potential for metabolic dysregulation, such as aripiprazole. However, many times, using an agent with significant effects on metabolic regulation cannot be avoided, making treating and monitoring the resulting metabolic effects even more significant.
Glycemic control
Initiating an agent for glycemic control in persons with newly diagnosed diabetes requires weighing many factors, including mode of delivery (oral or subcutaneous), level of glycemic control required, comorbid medical illness (such as renal impairment and heart failure), cost, and potential long-term side effects of the medication (Table 2).13 Oral agents have a slower effect on lowering the blood glucose level, and each agent carries contraindications, but generally they are more affordable. Many present a low risk of hypoglycemia but sulfonylureas are an exception. In addition, metformin could lead to weight loss over the long term, which further lowers the risk of diabetes.
If, however, tight and immediate glycemic control is needed, subcutaneous insulin injections might be required, although this method is more costly, carries an increased risk of hypoglycemic episodes acutely, and leads to weight gain in the long run because of induced hunger and increased appetite.
Psychosocial factors
In addition to the clinical variables above, treating diabetes in patients with comorbid schizophrenia requires considering other psychosocial factors that might not be readily apparent (Table 3). For example, patients with schizophrenia might have decreased motivation, self-efficacy, and capacity for self-care when it comes to health maintenance and medication adherence.14 Chronically mentally ill persons might have reduced cognitive functioning that could affect their ability to maintain complicated medication regimens, such as administering insulin and monitoring blood glucose.
In addition, easy access to hypodermic needles and large amounts of insulin could become a safety concern in patients with ongoing hallucinations, delusions, and thought disorder, despite antipsychotic treatment. For example, a patient with schizophrenia and diabetes who has been maintained on insulin might begin hearing voices that tell her to inject her eyeballs with insulin. Similarly, although the risk of hypoglycemic episodes in patients treated with insulin is well known, patients with schizophrenia have a higher risk of acute complications of diabetes than other patients with diabetes.15Psychosocial factors, such as placement, support system, and follow-up care need to be considered. In some instances, the need to administer daily subcutaneous insulin could be a barrier to placement if the facility does not have the staff or expertise to monitor blood glucose and administer insulin.
If the patient is returning home, then the patient or a caretaker will need to be trained to monitor blood glucose and administer insulin. This might be difficult for persons with chronic mental illness who have lost the support and care of their family. Also, consider the issue of storing insulin, which requires refrigeration. Because of the potential complications involved in using insulin in patients with schizophrenia, practitioners should consider managing non-insulin dependent diabetes with an oral hypoglycemic agent, when possible, along with lifestyle modifications.
OUTCOME Weight loss, discharge
Mr. K is transitioned from aripiprazole to higher-potency oral risperidone, titrated to 6 mg/d. At this dosage, he continues to maintain a delusion about owning a house in Fiji, but was seen responding to internal stimuli less often. The treatment team places him on a diabetic and low-sodium diet of 1,800 kcal/d. His fasting blood glucose levels range in the 70s to 110s mg/dL during his first week of hospitalization.
The treatment team starts Mr. K on oral metformin, titrated to 1,000 mg twice daily, while tapering him off insulin lispro and glargine over the course of hospitalization. The transition from insulin to metformin also is considered because Mr. K’s daily insulin requirement is rather low (<0.5 units/kg).
Mr. K’s course is prolonged because his treatment team initiates the process of conservatorship and placement in the community. Approximately 6 months after his admission, Mr. K is discharged to an unlocked facility with support from an intensive outpatient mental health program. At 6 months follow-up with his outpatient provider, Mr. K’s hemoglobin A1c is 7.0 and he weighs 155 lb with a BMI of 26.5.
The authors’ observations
Despite Mr. K’s initial elevated hemoglobin A1c of 12.7 and weight of 185 lb, over approximately 6 months he experiences a 5.7-unit drop in hemoglobin A1c and weight loss of 30 lb with dietary management and metformin—without the need for other agents. Other options for weight-neutral treatment of T2DM include exenatide, which also is available as a once weekly injectable formulation, canagliflozin, and the gliptins (sitagliptin, saxagliptin, and linagliptin) (Table 4).
Mr. K’s improved control of his diabetes occurred despite initiation of an atypical antipsychotic, which would have been expected to cause additional weight gain and make his diabetes worse secondary to metabolic effects.12 Treatment with metformin in particular has been associated with weight loss in patients with16 and without17 comorbid schizophrenia, including those with antipsychotic-induced weight gain.18-20
Bottom Line
Persons with schizophrenia are at higher risk of type 2 diabetes mellitus for many reasons, including metabolic effects of antipsychotics, reduced cognitive functioning, poor self-care, and limited access to health care. Typical antipsychotics are less likely to cause metabolic dysregulation but, if an atypical is needed, one that is relatively weight-neutral should be selected. When choosing an agent for glycemic control, consider a patient’s or a caretaker’s ability to administer medications, monitor blood glucose, and follow dietary recommendations.
Related Resources
• Cohn T. The link between schizophrenia and diabetes. Current Psychiatry. 2012;11(10):28-34,46.
• Annamalai A, Tek C. An overview of diabetes management in schizophrenia patients: office based strategies for primary care practitioners and endocrinologists [published online March 23, 2015]. Int J Endocrinol. 2015;2015:969182. doi: 10.1155/2015/969182.
Drug Brand Names
Amlodipine • Norvasc
Aripiprazole • Abilify
Asenapine • Saphris
Canagliflozin • Invokana
Exenatide • Bydureon, Byetta
Glargine • Lantus
Linagliptin • Tradjenta
Lispro • Humalog
Lurasidone • Latuda
Metformin • Glucophage
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Saxagliptin • Onglyza
Sitagliptin • Januvia
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Psychotic, and nonadherent
Mr. K, a 42-year-old Fijian man, is brought to the emergency department by his older brother for evaluation of behavioral agitation. Mr. K is belligerent and threatens to kill his family members. Three years earlier, he was given a diagnosis of schizophrenia and treated at an inpatient psychiatric unit.
At that time, Mr. K was stabilized on risperidone, 4 mg/d. However, he did not follow-up with treatment after discharge and has not taken any psychotropic medications since that time.
His brother reports that Mr. K has been slowly deteriorating, talking to himself, staying up at night, and getting into arguments with his family over his delusional beliefs. Although Mr. K once worked as a security guard, he has not worked in 8 years. He has been living with his family, who are no longer willing to accept him into their home because they fear he might harm them.
In the emergency department, Mr. K reports that he has a throbbing headache. Blood pressure is 177/101 mm Hg; heart rate is 103 beats per minute; respiratory rate is 18 breaths per minute; weight is 185 lb; and body mass index (BMI) is 31.8. Physical examination is unremarkable.
Laboratory values show that sodium is 131 mEq/L; potassium, 3.7 mEq/L; bicarbonate, 26 mEq/L; glucose, 420 mg/dL; hemoglobin A1c,12.7; and urine glucose, 3+. Mr. K denies being told he has diabetes.
What are Mr. K’s risk factors for diabetes?
a) schizophrenia
b) physical inactivity
c) obesity
d) Fijian ethnicity
e) all of the above
The authors’ observations
The prevalence of type 2 diabetes mellitus (T2DM) in persons with schizophrenia or a schizoaffective disorder is twice that of the general population.1-4 Multiple variables contribute to the increased prevalence of diabetes in this population, including genetic predisposition, environmental and cultural factors related to diet and physical activity, a high rate of smoking,5,6 iatrogenic causes (metabolic dysregulation and weight gain from antipsychotic treatment), and socioeconomic factors (poverty, lack of access to health care). In addition, symptoms of psychosis such as thought disorder, delusions, hallucinations, and cognitive decline in persons with chronic schizophrenia and schizoaffective disorder can make basic health maintenance difficult.
In Mr. K’s case, premorbid risk of diabetes was elevated because of his Fijian ethnicity.7 With a BMI of 31.8, obesity further increased that risk.5,6,8 In addition, his untreated chronic mental illness, lack of access to health care, low socioeconomic status, long-standing smoking habit, and previous exposure to antipsychotics also increased his risk of T2DM.
The interaction between diabetes and psychosis contributes to a vicious cycle that makes both conditions worse if either, or both, are untreated. In general, medical comorbidities are associated with depression and neurocognitive impairment in persons with schizophrenia.9 Specifically, diabetes is associated with lower global cognitive functioning among persons with schizophrenia.10 Poor cognitive functioning can, in turn, decrease the patient’s ability to manage his medical illness. Also, persons with schizophrenia are less likely to be treated for diabetes, dyslipidemia, and hypertension, as in Mr. K’s case.11
How would you treat Mr. K’s newly diagnosed diabetes?
a) refer him to a primary care physician
b) start an oral agent
c) start sliding-scale insulin
d) start long-acting insulin
e) recommend a carbohydrate-controlled diet=
TREATMENT Stabilization
Mr. K is admitted to the medical unit for treatment of hyperglycemia. The team starts him on amlodipine, 5 mg/d, for hypertension; aripiprazole, 10 mg/d, for psychosis; and sliding-scale insulin (lispro) and 20 units of insulin (glargine) nightly for diabetes. Mr. K’s blood glucose level is well regulated on this regimen; after being medically cleared, he is transferred to the inpatient psychiatric unit.
EVALUATION Denies symptoms
Mr. K appears older than his stated age, is poorly groomed, and is dressed in a hospital gown. He is isolated and appears to be internally preoccupied. He repeatedly denies hearing auditory hallucinations, but often is overheard responding to internal stimuli and mumbling indecipherably in a low tone. His speech is decreased and his affect is flat and guarded. He states that he is not “mental” but that he came to the hospital for “tooth pain.” Every day he asks when he can return home and he asks the staff to call his family to take him home. When informed that his family is not able to care for him, Mr. K states that he would live in a house he owns in Fiji, which his family members state is untrue.
How would you treat Mr. K’s psychosis?
a) continue aripiprazole
b) switch to risperidone, an agent to which he previously responded
c) switch to olanzapine because he has not been sleeping well
d) switch to haloperidol because of diabetes
The authors’ observations
Pharmacotherapy for patients with comorbid schizophrenia and diabetes requires consideration of clinical and psychosocial factors unique to this population.
Antipsychotic choice
Selection of an antipsychotic agent to address psychosis requires considering its efficacy, side-effect profile, and adherence rates, as well as its potential effects on metabolic regulation and weight (Table 1). Typical antipsychotics are less likely than atypical antipsychotics to cause metabolic dysregulation.12 When treatment with atypical antipsychotics cannot be avoided—such as when side effects or an allergic reaction develop—consider selecting a relatively weight-neutral drug with a lower potential for metabolic dysregulation, such as aripiprazole. However, many times, using an agent with significant effects on metabolic regulation cannot be avoided, making treating and monitoring the resulting metabolic effects even more significant.
Glycemic control
Initiating an agent for glycemic control in persons with newly diagnosed diabetes requires weighing many factors, including mode of delivery (oral or subcutaneous), level of glycemic control required, comorbid medical illness (such as renal impairment and heart failure), cost, and potential long-term side effects of the medication (Table 2).13 Oral agents have a slower effect on lowering the blood glucose level, and each agent carries contraindications, but generally they are more affordable. Many present a low risk of hypoglycemia but sulfonylureas are an exception. In addition, metformin could lead to weight loss over the long term, which further lowers the risk of diabetes.
If, however, tight and immediate glycemic control is needed, subcutaneous insulin injections might be required, although this method is more costly, carries an increased risk of hypoglycemic episodes acutely, and leads to weight gain in the long run because of induced hunger and increased appetite.
Psychosocial factors
In addition to the clinical variables above, treating diabetes in patients with comorbid schizophrenia requires considering other psychosocial factors that might not be readily apparent (Table 3). For example, patients with schizophrenia might have decreased motivation, self-efficacy, and capacity for self-care when it comes to health maintenance and medication adherence.14 Chronically mentally ill persons might have reduced cognitive functioning that could affect their ability to maintain complicated medication regimens, such as administering insulin and monitoring blood glucose.
In addition, easy access to hypodermic needles and large amounts of insulin could become a safety concern in patients with ongoing hallucinations, delusions, and thought disorder, despite antipsychotic treatment. For example, a patient with schizophrenia and diabetes who has been maintained on insulin might begin hearing voices that tell her to inject her eyeballs with insulin. Similarly, although the risk of hypoglycemic episodes in patients treated with insulin is well known, patients with schizophrenia have a higher risk of acute complications of diabetes than other patients with diabetes.15Psychosocial factors, such as placement, support system, and follow-up care need to be considered. In some instances, the need to administer daily subcutaneous insulin could be a barrier to placement if the facility does not have the staff or expertise to monitor blood glucose and administer insulin.
If the patient is returning home, then the patient or a caretaker will need to be trained to monitor blood glucose and administer insulin. This might be difficult for persons with chronic mental illness who have lost the support and care of their family. Also, consider the issue of storing insulin, which requires refrigeration. Because of the potential complications involved in using insulin in patients with schizophrenia, practitioners should consider managing non-insulin dependent diabetes with an oral hypoglycemic agent, when possible, along with lifestyle modifications.
OUTCOME Weight loss, discharge
Mr. K is transitioned from aripiprazole to higher-potency oral risperidone, titrated to 6 mg/d. At this dosage, he continues to maintain a delusion about owning a house in Fiji, but was seen responding to internal stimuli less often. The treatment team places him on a diabetic and low-sodium diet of 1,800 kcal/d. His fasting blood glucose levels range in the 70s to 110s mg/dL during his first week of hospitalization.
The treatment team starts Mr. K on oral metformin, titrated to 1,000 mg twice daily, while tapering him off insulin lispro and glargine over the course of hospitalization. The transition from insulin to metformin also is considered because Mr. K’s daily insulin requirement is rather low (<0.5 units/kg).
Mr. K’s course is prolonged because his treatment team initiates the process of conservatorship and placement in the community. Approximately 6 months after his admission, Mr. K is discharged to an unlocked facility with support from an intensive outpatient mental health program. At 6 months follow-up with his outpatient provider, Mr. K’s hemoglobin A1c is 7.0 and he weighs 155 lb with a BMI of 26.5.
The authors’ observations
Despite Mr. K’s initial elevated hemoglobin A1c of 12.7 and weight of 185 lb, over approximately 6 months he experiences a 5.7-unit drop in hemoglobin A1c and weight loss of 30 lb with dietary management and metformin—without the need for other agents. Other options for weight-neutral treatment of T2DM include exenatide, which also is available as a once weekly injectable formulation, canagliflozin, and the gliptins (sitagliptin, saxagliptin, and linagliptin) (Table 4).
Mr. K’s improved control of his diabetes occurred despite initiation of an atypical antipsychotic, which would have been expected to cause additional weight gain and make his diabetes worse secondary to metabolic effects.12 Treatment with metformin in particular has been associated with weight loss in patients with16 and without17 comorbid schizophrenia, including those with antipsychotic-induced weight gain.18-20
Bottom Line
Persons with schizophrenia are at higher risk of type 2 diabetes mellitus for many reasons, including metabolic effects of antipsychotics, reduced cognitive functioning, poor self-care, and limited access to health care. Typical antipsychotics are less likely to cause metabolic dysregulation but, if an atypical is needed, one that is relatively weight-neutral should be selected. When choosing an agent for glycemic control, consider a patient’s or a caretaker’s ability to administer medications, monitor blood glucose, and follow dietary recommendations.
Related Resources
• Cohn T. The link between schizophrenia and diabetes. Current Psychiatry. 2012;11(10):28-34,46.
• Annamalai A, Tek C. An overview of diabetes management in schizophrenia patients: office based strategies for primary care practitioners and endocrinologists [published online March 23, 2015]. Int J Endocrinol. 2015;2015:969182. doi: 10.1155/2015/969182.
Drug Brand Names
Amlodipine • Norvasc
Aripiprazole • Abilify
Asenapine • Saphris
Canagliflozin • Invokana
Exenatide • Bydureon, Byetta
Glargine • Lantus
Linagliptin • Tradjenta
Lispro • Humalog
Lurasidone • Latuda
Metformin • Glucophage
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Saxagliptin • Onglyza
Sitagliptin • Januvia
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. Heald A. Physical health in schizophrenia: a challenge for antipsychotic therapy. Eur Psychiatry. 2010;25(suppl 2):S6-S11.
3. El-Mallakh P. Doing my best: poverty and self-care among individuals with schizophrenia and diabetes mellitus. Arch Psychiatr Nurs. 2007;21(1):49-60; discussion 61-63.
4. Rouillon F, Sorbara F. Schizophrenia and diabetes: epidemiological data. Eur Psychiatry. 2005;20(suppl 4):S345-S348.
5. Willi C, Bodenmann P, Ghali WA, et al. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2007;298(22):2654-2664.
6. Dome P, Lazary J, Kalapos MP, et al. Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev. 2010;34(3):295-342.
7. National Institute of Diabetes and Digestive and Kidney Diseases. For people of African, Mediterranean, or Southeast Asian heritage: important information about diabetes blood tests. http://www.diabetes.niddk.nih.gov/dm/ pubs/asianamerican/index.htm. Published October 2011. Accessed November 18, 2015.
8. Shaten BJ, Smith GD, Kuller LH, et al. Risk factors for the development of type 2 diabetes among men enrolled in the usual care group of the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16(10):1331-1339.
9. Chwastiak LA, Rosenheck RA, McEvoy JP, et al. Interrelationships of psychiatric symptom severity, medical comorbidity, and functioning in schizophrenia. Psychiatr Serv. 2006;57(8):1102-1109.
10. Takayanagi Y, Cascella NG, Sawa A, et al. Diabetes is associated with lower global cognitive function in schizophrenia. Schizophr Res. 2012;142(1-3):183-187.
11. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
12. Meyer JM, Davis VG, Goff DC, et al. Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE Schizophrenia Trial: prospective data from phase 1. Schizophr Res. 2008;101(1-3):273-286.
13. Inzucchi SE. Clinical practice. Management of hyperglycemia in the hospital setting. N Engl J Med. 2006; 355(18):1903-1911.
14. Chen SR, Chien YP, Kang CM, et al. Comparing self-efficacy and self-care behaviours between outpatients with comorbid schizophrenia and type 2 diabetes and outpatients with only type 2 diabetes. J Psychiatr Ment Health Nurs. 2014;21(5):414-422.
15. Becker T, Hux J. Risk of acute complications of diabetes among people with schizophrenia in Ontario, Canada. Diabetes Care. 2011;34(2):398-402.
16. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care. 2012;35(4):731-737.
17. Jarskog LF, Hamer RM, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
18. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-430. doi: 10.4088/JCP.12m08186.
19. Wang M, Tong JH, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
20. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6): 1385-1403.
1. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. Heald A. Physical health in schizophrenia: a challenge for antipsychotic therapy. Eur Psychiatry. 2010;25(suppl 2):S6-S11.
3. El-Mallakh P. Doing my best: poverty and self-care among individuals with schizophrenia and diabetes mellitus. Arch Psychiatr Nurs. 2007;21(1):49-60; discussion 61-63.
4. Rouillon F, Sorbara F. Schizophrenia and diabetes: epidemiological data. Eur Psychiatry. 2005;20(suppl 4):S345-S348.
5. Willi C, Bodenmann P, Ghali WA, et al. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2007;298(22):2654-2664.
6. Dome P, Lazary J, Kalapos MP, et al. Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev. 2010;34(3):295-342.
7. National Institute of Diabetes and Digestive and Kidney Diseases. For people of African, Mediterranean, or Southeast Asian heritage: important information about diabetes blood tests. http://www.diabetes.niddk.nih.gov/dm/ pubs/asianamerican/index.htm. Published October 2011. Accessed November 18, 2015.
8. Shaten BJ, Smith GD, Kuller LH, et al. Risk factors for the development of type 2 diabetes among men enrolled in the usual care group of the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16(10):1331-1339.
9. Chwastiak LA, Rosenheck RA, McEvoy JP, et al. Interrelationships of psychiatric symptom severity, medical comorbidity, and functioning in schizophrenia. Psychiatr Serv. 2006;57(8):1102-1109.
10. Takayanagi Y, Cascella NG, Sawa A, et al. Diabetes is associated with lower global cognitive function in schizophrenia. Schizophr Res. 2012;142(1-3):183-187.
11. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
12. Meyer JM, Davis VG, Goff DC, et al. Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE Schizophrenia Trial: prospective data from phase 1. Schizophr Res. 2008;101(1-3):273-286.
13. Inzucchi SE. Clinical practice. Management of hyperglycemia in the hospital setting. N Engl J Med. 2006; 355(18):1903-1911.
14. Chen SR, Chien YP, Kang CM, et al. Comparing self-efficacy and self-care behaviours between outpatients with comorbid schizophrenia and type 2 diabetes and outpatients with only type 2 diabetes. J Psychiatr Ment Health Nurs. 2014;21(5):414-422.
15. Becker T, Hux J. Risk of acute complications of diabetes among people with schizophrenia in Ontario, Canada. Diabetes Care. 2011;34(2):398-402.
16. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care. 2012;35(4):731-737.
17. Jarskog LF, Hamer RM, Catellier DJ, et al; METS Investigators. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032-1040.
18. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-430. doi: 10.4088/JCP.12m08186.
19. Wang M, Tong JH, Zhu G, et al. Metformin for treatment of antipsychotic-induced weight gain: a randomized, placebo-controlled study. Schizophr Res. 2012;138(1):54-57.
20. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6): 1385-1403.

A girl refuses to eat solid food because she is afraid of choking
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6

Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
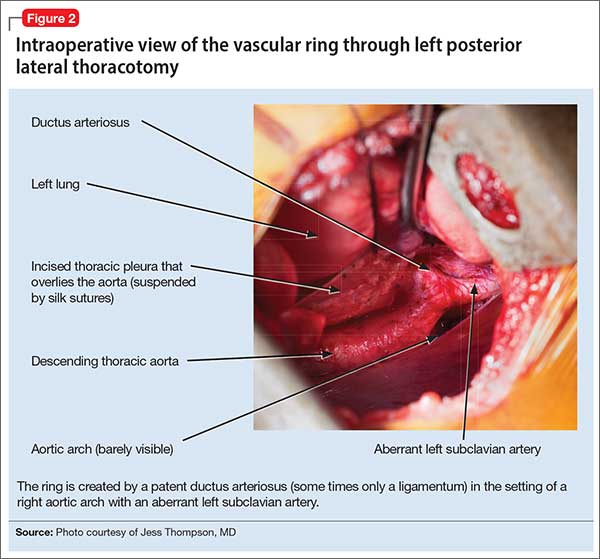
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6
Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6
Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.

Malignant catatonia and aphasia follow multiple-drug overdose
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
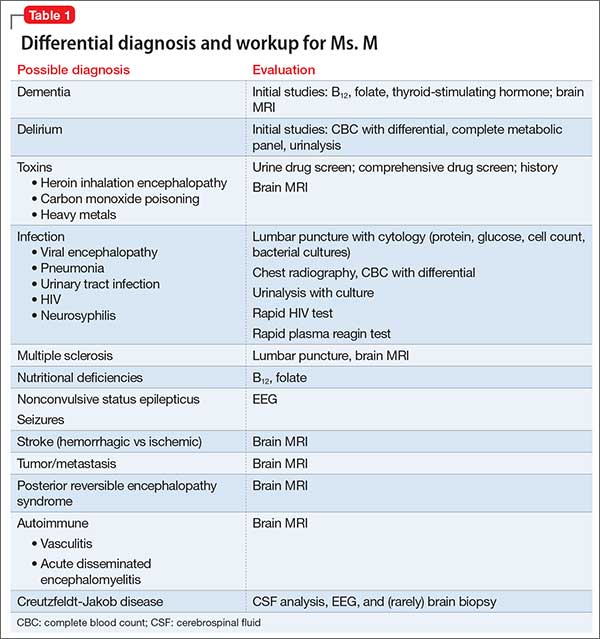
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.

Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
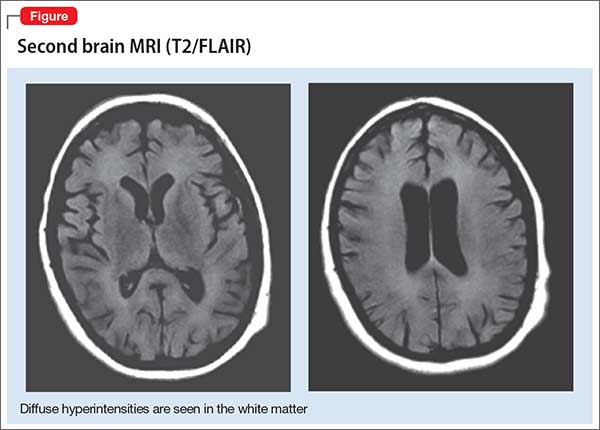
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
CASE Improvement, then decline
Ms. M, age 37, is brought to the hospital after her husband found her at home, after an unknown duration of impaired consciousness. Her husband reports that Ms. M had normal cognitive functioning before this event, with no difficulty completing activities of daily living. Ms. M’s medical and psychiatric histories are notable for type 2 diabetes mellitus, unspecified bipolar disorder, and opioid, cocaine, and alcohol use disorders. Her medications include paroxetine, 40 mg/d, and gabapentin, 1,200 mg/d.
First admission. Poor inspiratory effort and oxygen saturation of 70% leads to emergent intubation. Serum laboratory studies reveal a white blood cell (WBC) count at 10,900/μL and creatinine phosphokinase level of 25,000 U/L. Urine drug screen is positive for tetrahydrocannabinol, cocaine, and opioids.
Ms. M is admitted to the ICU for management of rhabdomyolysis and multi-organ system failure, including acute hypoxic kidney injury.
By hospital Day 7, the tube is extubated with no recorded physical neurologic deficits. Mental status exam is normal, except for impaired memory of events surrounding the admission. Ms. M is discharged home with a recommendation for outpatient follow-up.
2 Weeks later. Ms. M is brought to the emergency department after a progressive decrease in social interaction, limited oral intake, decline in activities of daily living, and urinary incontinence. Results from laboratory studies are within normal limits; brain MRI is negative; EEG shows generalized moderate slowing.
During psychiatric evaluation, Ms. M is mute and staring continuously. Examination reveals oppositional paratonia (gegenhalten), catalepsy, prominent negativism, and waxy flexibility, all suggestive of catatonia. IV lorazepam is initiated at 1 mg every 8 hours, titrated to 2 mg, 3 times a day.
Ms. M is transferred to a psychiatric hospital for further treatment of catatonia.
Second admission. Evaluation with the Bush-Francis Catatonia Rating Scale supported a diagnosis of catatonia, with the presence of >3 features from the 14-item screen and a score of 16 on the 23-item rating scale.1 After titrating lorazepam to 9 mg/d with minimal therapeutic impact, the psychiatry team consults the electroconvulsive therapy (ECT) service, who deems Ms. M to be an appropriate candidate and petitions for court-ordered ECT.
On hospital Day 8, Ms. M has a fever of 104°F, tachycardia at 180 beats per minute, increased rigidity, and a WBC count of 17,800/μL. She is transferred to the ICU, with a presumptive diagnosis of malignant catatonia.
The medical evaluation, including general laboratory studies, EEG, and spinal fluid analysis, is unremarkable. Because of vital sign instability, 2 ECT treatments are completed in the general hospital before Ms. M resumes psychiatric inpatient care.
By the tenth ECT treatment, Ms. M is no longer febrile and experiences no further autonomic instability or psychomotor features of catatonia. Despite these improvements, she is noted to have persistent word-finding difficulty.
Which test would you order as the next step in your work up?
a) EEG
b) lumbar puncture
c) MRI
d) CT
The authors’ observations
In approximately 25% of cases, catatonia is caused by a general medical condition2; as such, a comprehensive medical workup is vital for assessment and management of catatonic patients. In Ms. M’s case, we considered several medical causes, including nutritional deficiency, infection, a toxin, renal or hepatic impairment, hypothyroidism, seizure, and stroke. Evaluation included measurement of thyroid-stimulating hormone, vitamin B12, and folic acid levels; urinalysis and urine drug screen; chest radiography; lumbar puncture; neuroimaging; and EEG (Table 1).
Several conditions in the differential diagnosis were noteworthy. Ms. M’s severe and sudden neurologic decline, along with a positive urine drug screen for substances of abuse, raised concern about overdose leading to toxic encephalopathy or hypoxic brain injury. Ms. M’s oxygen saturation when she was found was moderately hypoxic at 70%, which is not a level associated with hypoxic brain damage.
We also considered posterior reversible encephalopathy syndrome (PRES), which presents variably with nausea, visual impairment, disturbance in consciousness, seizures, and focal neurologic signs.3 Although 67% to 80% of patients with PRES also have acute hypertension, blood pressure elevation is not necessary for the diagnosis.4 Similar to toxic leukoencephalopathy, PRES is diagnosed by brain MRI, with classic signs of posterior white-matter edema.
Case reports also describe an uncommon demyelinating syndrome, delayed post-hypoxic leukoencephalopathy (DPHL), which develops several weeks or months after a cerebral anoxic insult.5 In Ms. M’s case, brain MRI performed during her second medical hospitalization, 7 days after the initial neuropsychiatric decline, was unremarkable. Using this result to rule out DPHL would have been premature because pathognomonic abnormalities can appear as long as 40 days after the anoxic insult. Given our differential diagnosis, we ordered a repeat MRI.
Etiology and pathophysiology
First described in 1979, DPHL is rare, posing diagnostic challenges for clinical providers.6 Although the exact incidence of DPHL is unknown, the precipitating event typically involves cerebral anoxia, which can occur through carbon monoxide (CO) poisoning, strangulation, cardiac arrest, respiratory failure, and overdose from sedatives and narcotics (Table 2).7 DPHL was first observed in a small percentage (2.75%) of patients suffering from CO poisoning.8,9 Progression of the disease generally includes a period of unconsciousness, then a lucid interval that can last 2 to 40 days, followed by the abrupt onset of neuropsychiatric symptoms.10 The specific pathophysiologic mechanism is unknown, but has been hypothesized to involve inferior compensatory response to decreased oxygenation in the white matter.
Diagnosis and clinical features
DPHL can be divided into 2 clinical variations: parkinsonism and akinetic mutism. The former consists of conventional parkinsonian features along with agitation, apathy, hallucinations, dystonic posturing, and odd behaviors. The latter variant presents with apathy, minimal response to pain, functional bowel and bladder incontinence, mutism, and, at times, inappropriate laughter or tearfulness.5 Both variants share similar features with hypokinetic forms of catatonia.
DPHL is a diagnosis of exclusion. A careful history is critical to establish the possibility of a recent anoxic event. MRI findings, including hyperintensities in the cerebral white matter on T2-based sequencing, are suggestive of the disease. A choline peak on magnetic resonance spectroscopy also might be present in patients with DPHL, although it is not specific to the diagnosis.
Early reports of DPHL suggested an associated deficiency of arylsulfatase A, an enzyme required in the modulation of myelin; however, more recent case reports are conflicting.11 Familial mutations in the gene for arylsulfatase A also result in metachromatic leukodystrophy, and adult onset can present with psychiatric symptoms, including delusions and hallucinations.12
Treatment and prognosis
The treatment of DPHL consists primarily of supportive care and rehabilitation with physical, occupational, and speech therapy.11 With these measures, most patients improve after 3 to 6 months; however, a large percentage sustain some long-term cognitive deficit, the most prevalent symptom being frontal executive dysfunction.5
OUTCOME Supportive care
A second MRI shows diffuse hyperintensities in the white matter that spare the cerebellum and brainstem (Figure). This finding is pathognomonic for DPHL.
ECT is discontinued because there is no evidence to support ECT-associated improvement in DPHL. Moreover, ECT might worsen the clinical course through increased stress and metabolic demand on the brain.13
Because the primary treatment of DPHL is early rehabilitation, we consider that Ms. M would benefit most from increased supportive care and therapy. She is discharged to a brain injury rehabilitation facility, where metoprolol is prescribed for mild tachycardia, along with thiamine and vitamins B12 and D. Physical, occupational, and speech therapy are continued.
Approximately 3 weeks after admission to the rehabilitation program, Ms. M is discharged home. Although she improves in overall activities of daily living, she continues to experience moderate communication deficits and occasional external distractibility.
Bottom Line
Although delayed post-hypoxic leukoencephalopathy is considered rare, consider it in the differential diagnosis when a patient has a recent history of an anoxic event followed by the abrupt onset of neuropsychiatric symptoms. Keep in mind that the condition can be missed if an MRI is obtained too early, and the clinical signs can mimic hypokinetic catatonia.
Related Resources
• Meyer MA. Delayed post-hypoxic leukoencephalopathy: case report with a review of disease pathophysiology. Neurol Int. 2013;5(3):e13. doi: 10.4081/ni.2013.e13.
• Aljarallah S, Al-Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69.
Drug Brand Names
Gabapentin • Neurontin
Lorazepam • Ativan
Metoprolol • Lopressor
Paroxetine • Paxil
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of com
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
1. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
2. Azzam PN, Gopalan P. Prototypes of catatonia: diagnostic and therapeutic challenges in the general hospital. Psychosomatics. 2013;54(1):88-93.
3. Tormoehlen LM. Toxic leukoencephalopathies. Neurol Clin. 2011;29(3):591-605
4. Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In: Vincent JL, ed. Annual update in intensive care and emergency medicine. Heidelberg, Germany: Springer Berlin Heidelberg; 2011:631-653.
5. Schprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65-72.
6. Wallace IR, Dynan C, Esmonde T. One confused patient, many confused physicians: a case of delayed post-hypoxic leucoencephalopathy. QJM. 2010;103(3):193-194.
7. Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277(1-2):147-149.
8. Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40(7):433-435.
9. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27(8):1763-1765.
10. Shprecher DR, Flanigan KM, Smith AG, et al. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473-477.
11. Lee BH, Lyketsos CG. Delayed post-hypoxic leukoencephalopathy. Psychosomatics. 2001;42(6):530-533.
12. Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Insights into the neurobiology of psychosis. Arch Neurol. 1992;49(4):401-406.
13. Quinn DK, Abbott CC. Catatonia after cerebral hypoxia: do the usual treatments apply? Psychosomatics. 2014;55(6):525-535.
Psychosis and catatonia after dancing with a dangerous partner
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.

The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.

By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
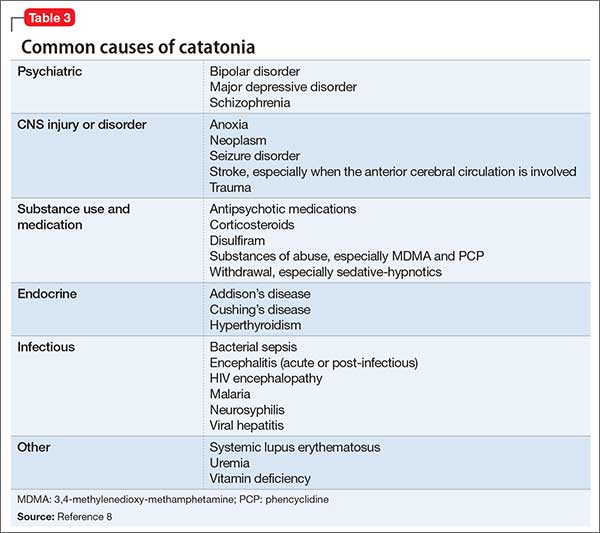
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.
The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.
By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Rigid, frightened, and mute
Mr. D, age 23, presents for evaluation immediately after discharge from another hospital, where he had been treated for altered mental status.
Ten days earlier, Mr. D’s friends obtained 2C-B (2,5-dimethoxy-4-bromophenethylamine), from the “Darknet,” an underground niche of the Internet. He ingested 20 mg of 2C-B in powder form. Although his friends recovered from a “safe trip,” Mr. D decompensated rapidly over the next few days with persistent psychosis, experiencing both auditory and visual hallucinations. He is “acting strange“ at work, and trying to find “hidden codes” in data. Mr. D also has persistent thought disorganization. He speaks of “connections” between people and things, and says that he is an alien in a spaceship. His friends and family report that he is talking rapidly and is sleeping only 2 or 3 hours each night. Mr. D abruptly quit his job as an analyst a few days after taking the drug.
Mr. D is a single, Ivy League-educated man and is described as hardworking and analytical. His family denies any recent mood changes or life stressors. They report that 1 month ago, Mr. D began smoking marijuana daily. He has no significant medical or psychiatric history, and no family history of psychiatric disorders.
What is your most likely diagnosis for Mr. D?
a) delirium due to a general medical condition
b) substance-induced psychotic disorder
c) catatonia due to a general medical condition
d) schizophrenia
e) bipolar I disorder, currently manic, with psychosis
The authors’ observations
Ring-substituted phenethylamines, commonly known as 2Cs, are designer drugs that are emerging as new substances of abuse.1 2C-B belongs to the phenethylamine subclass of monoamine alkaloids that includes more familiar drugs such as amphetamines, methamphetamines, and 3,4-methylenedioxy-methamphetamine (MDMA).2 It was first synthesized in 1974 by Alexander Shulgin, later described in his book Phenethylamines I Have Known and Loved: A Chemical Love Story, and its hallucinogenic activity is reported to be similar to LSD, mescaline, and psilocybin.3 The literature is scant on the acute effects of 2C intoxication or long-term sequelae of 2C ingestion.1 Most available information regarding the pharmacology of 2C-B comes from users who have reported their drug experiences on blogs, Web sites and forums, and in the media.4
2C-B usually is taken orally in powder or tablet form, in a dose of 10 to 50 mg.4 After an onset period of 20 to 90 minutes, the drug’s effect reaches maximum effect in 15 to 30 minutes, then plateaus for 2 to 7 hours, and comes down within 1 to 2 hours.4 2C-B is known to be orally active, and its hallucinogenic effects are mediated by its actions as a partial serotonin 5HT-2A and 5HT-2C receptor agonist.5 Entactogenic-stimulating effects have been reported at low doses (4 to 10 mg), whereas visual hallucinations with intense colors and object distortion have been reported at moderate doses (10 to 20 mg).4
2C-B, which users often take at parties or raves, appeared on the drug market in the mid 1980s and early 1990s under the names Nexus, Erox, Performax, Toonies, Bromo, Spectrum, and Venus and marketed as a replacement for MDMA after it became a Schedule I drug in the United States.4,6 Some users consume 2C-B in combination with other illicit drugs, including MDMA (called a “party pack”) or LSD (referred to as a “banana split”).6
According to the U.S. Drug Enforcement Agency, law enforcement authorities first seized 2C-B laboratories in California in 1986 and Arizona in 1992.6 Distribution of the drug has been sporadic since it became Schedule I in 1995, and it has been seized from several states, including Virginia, Nevada, Maine, Illinois, Missouri, South Dakota, and Kansas.6
EXAMINATION Passive and mute
On examination, Mr. D is lying in bed with eyes closed and extremities extended in an odd, rigid posture. He is resistant to attempts at passive movement, is nonresponsive to verbal commands, and is mute. A review of vital signs shows tachycardia, 110 beats per minute, but the physical exam is otherwise unremarkable. His Bush-Francis Catatonia Rating Scale (BFCRS) score is 17, indicating a diagnosis of catatonia. Mini-Mental Status Examination cannot be completed because Mr. D is unable to participate.
Laboratory studies reveal an elevated creatinine kinase (CK) level of 356 U/L. Results of a complete blood count, comprehensive metabolic panel, urinalysis, and thyroid-stimulating hormone are normal. Blood alcohol level is <10 mg/dL. Acetaminophen and salicylate levels are normal (<5 mg/dL). Records from his recent hospitalization reveal normal head CT, chest radiography, EEG, and urinalysis, and a negative urine drug screen.
What is the next step in managing Mr. D’s catatonic symptoms?
a) IV normal saline
b) IV lorazepam
c) emergent electroconvulsive therapy (ECT)
d) IM haloperidol
e) IM olanzapine
TREATMENT Saline and psychotropics
While in the emergency room, Mr. D receives 2 L of IV saline. His CK level falls to 137 U/L. A challenge with IV lorazepam, 2 mg, also is performed. Mr. D becomes talkative and follows commands with fluid movements, but his disorganized, delusional thoughts persist. BFCRS score has improved to 9 (Table 1). He is admitted to the psychiatric unit and started on oral lorazepam, 2 mg, 3 times daily, for catatonia, and olanzapine, 10 mg/d, for psychosis.
The differential diagnosis for Mr. D’s psychosis includes substance-induced psychotic disorder, schizophrenia, bipolar disorder, and psychosis with another organic cause (Table 2).7 Further medical workup is completed, including a urine drug screen, testing for HIV, hepatitis B, syphilis, lead and heavy metals, ceruloplasmin, vitamin B12, folate, antinuclear antibody, sedimentation rate, and brain MRI. Cannabinoids are detected in his urine drug screen. Another urine sample is sent to an outside lab to test for several synthetic drugs, including MDMA, 3,4-methylenedioxy- N-ethyl-amphetamine, 2C-B, 2C-C, 2C-I, and 2C-P, results of which also are negative.
By the second day of hospitalization, Mr. D appears less disorganized but continues to complain of “scrambled thoughts” and appears guarded. Despite initial response to IV lorazepam and its continuation in oral form, over the next day Mr. D appears more psychomotor-slowed, with motor stiffness. His score on the BFCRS increases, with significant posturing; vital signs remain stable, however.
What is your next step in managing his catatonic symptoms?
a) increase olanzapine
b) decrease olanzapine
c) decrease lorazepam
d) emergent ECT
e) switch to haloperidol
The authors’ observations
Although catatonia can be associated with a mood or psychotic disorder, it also can be induced by a medication or general medical condition (Table 3).8 It is thought that catatonia is associated with decreased γ-aminobutyric acid (GABA) and dopamine D2 receptor activity, and increased N-methyl-d-aspartate (NMDA) receptor activity.9 Antipsychotics could worsen catatonia through D2 blockade. Benzodiazepines, however, improve catatonia by increasing GABA and decreasing NMDA receptor activity. In this case, Mr. D was naïve to antipsychotics and seemed to be sensitive to them, as evidenced by his worsening symptoms.
Which condition should be considered in the differential diagnosis?
a) parkinsonian-hyperpyrexia syndrome
b) neuroleptic malignant syndrome (NMS)
c) stiff person syndrome
d) serotonin syndrome
e) CNS infection
The authors’ observations
NMS, catatonia, and parkinsonian-hyperpyrexia syndrome are all related to diminished action of dopamine at the D2 receptor. Although the mechanism of catatonia is not completely understood, NMS is thought to be caused by blockade at the D2 receptors by antipsychotics, whereas parkinsonian-hyperpyrexia syndrome is related to withdrawal of dopamine agonists. Because of the similarity in symptoms and proposed mechanisms, some experts hypothesize that NMS is a drug-induced malignant catatonia.10,11 Interestingly, NMS and catatonia respond to withdrawal of antipsychotics, and addition of benzodiazepines and ECT.
Mr. D showed posturing and other behavioral abnormalities, which are less common in NMS. Furthermore, although he had episodes of mild tachycardia, autonomic dysregulation—a hallmark of NMS—was not found. Given the common shared deficiency of activity at the D2 receptor in both NMS and catatonia, antipsychotics could cause or worsen either condition.
TREATMENT ECT
Mr. D’s olanzapine dosage is decreased to 2.5 mg/d. His catatonic symptoms improve with each dosage of oral lorazepam; however, effects seem to lessen and last for shorter periods over the following day. Additionally, Mr. D again becomes more disorganized, stiff, and unable to feed or bathe himself, and develops episodes of mild tachycardia.
Given Mr. D’s partial and poorly sustained response to lorazepam, a trial of ECT is pursued. On the third day of hospitalization, he receives ECT with bi-frontal lead placement at 25% energy. Concurrently, olanzapine is discontinued because of worsening muscle stiffness and concern about neuroleptic sensitivity. His BFCRS score after ECT is 2, and he is noted to be more interactive on the inpatient unit. He continues to receive ECT 3 times a week, with notable improvement, but ongoing psychotic symptoms and catatonic symptoms partially reemerge between ECT treatments. Lead placement is changed to bi-temporal by the third treatment, and the energy setting is increased from 25% to 50%, and to 75% by the sixth treatment. An additional nighttime dose of oral lorazepam, 2 mg, is added after the sixth treatment, in an attempt to reduce “wearing off” by morning.
After the seventh treatment, Mr. D is able to maintain logical conversation without re-emergence of catatonic symptoms over 2 days, signifying a turning point in the treatment course. The ECT energy setting is decreased to 50% to minimize potential memory deficits. His insight into his illness and treatment dramatically improve over the next few days. ECT is discontinued after the tenth treatment and Mr. D is discharged home to the care of his family.
The authors’ observations
Randomized clinical trials studying the effectiveness of ECT for catatonia are limited. Much of what we know about ECT comes from case reports that describe excellent outcomes for a variety of treatment-resistant illnesses, including catatonia in mood disorders, schizophrenia, autism, and other organic brain disease.12
Although benzodiazepines often are the first-line treatment for catatonia caused by any underlying illness, one study showed only 1 of 41 patients achieved remission with benzodiazepines, compared with 100% of those treated with ECT13; another study supported these results with 8 of 9 lorazepam non-responders responding to ECT.14 There are few case reports of substance-induced catatonia in the absence of other chronic mental illness, although none report use of ECT. However, a study showed no significant difference in the effectiveness of ECT for catatonia caused by an affective disorder or schizophrenia.15
Mr. D’s case exemplifies complete remission of catatonia induced by a psychoactive substance.
OUTCOME Steady improvement
Mr. D is followed in the outpatient clinic for 1 month after discharge; lorazepam is tapered successfully. During this time frame, psychotic and catatonic symptoms do not re-emerge. He reports some initial working memory deficits that improve steadily. There is no evidence of any significant psychiatric signs or symptoms, including neurovegetative symptoms of depression, mania or hypomania, perceptual disturbances, or disorganized thoughts or behaviors. He remains abstinent from alcohol, tobacco, and all psychoactive substances.
Bottom Line
Persistent psychosis and catatonia after the use of newer designer drugs such as 2C-B are rare, but these drugs carry serious potential complications that clinicians should be aware of. Benzodiazepines and electroconvulsive therapy have been proved effective for catatonia that is related to a number of psychiatric illnesses, often resulting in good outcomes. However, current evidence on their use is limited, particularly regarding treatment of substance-induced psychosis and catatonia.
Related Resources
• Meyer MR, Maurer HH. Metabolism of designer drugs of abuse: an updated review. Curr Drug Metab. 2010;11(5):468-482.
• Rickli A, Luethi D, Reinisch J, et al. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology. 2015;99:546-553.
Drug Brand Names
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
1. Dean BV, Stellpflug SJ, Burnett AM, et al. 2C or not 2C: phenethylamine designer drug review. J Med Toxicol. 2013;9(2):172-178.
2. Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
3. Shulgin A, Shulgin A. PiHKAL: a chemical love story. Berkley, CA: Transform Press; 1991.
4. Papoutsis I, Nikolaou P, Stefanidou M, et al. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicology. 2015;3(1):1-11.
5. Caudevilla-Gálligo F, Riba J, Ventura M, et al. 4-Bromo-2, 5-dimethoxyphenethylamine (2C-B): presence in the recreational drug market in Spain, pattern of use and subjective effects. J Psychopharmacol. 2012;26(7):1026-1035.
6. National Drug Intelligence Center. Information bulletin: 2C-B (Nexus) reappears on the club drug scene. http:// www.Justice.gov/archive/ndic/pubs0/665. Published May 2001. Accessed June 12, 2015.
7. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
8. Masand PS, Levenson JL, et al. Mania, catatonia, and psychosis. In: Levenson JL, ed. The American Psychiatric Publishing textbook of psychosomatic medicine. Washington, DC: American Psychiatric Publishing; 2005: 239-241.
9. Carroll BT. The universal field of hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr. 2000;5(7):26-33.
10. Lee JW. Neuroleptic-induced catatonia: clinical presentation, response to benzodiazepines, and relationship to neuroleptic malignant syndrome. J Clin Psychopharmacol. 2010;30(1):3-10.
11. Vancaester E, Santens P. Catatonia and neuroleptic malignant syndrome: two sides of a coin? Acta Neurol Belg. 2007;107(2):47-50.
12. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
13. Hatta K, Miyakawa K, Ota T, et al. Maximal response to electroconvulsive therapy for the treatment of catatonic symptoms. J ECT. 2007;23(4):233-235.
14. Payee H, Chandrasekaran R, Raju GV. Catatonic syndrome: treatment response to Lorazepam. Indian J Psychiatry. 1999;41(1):49-53.
15. Rohland BM, Carroll BT, Jacoby RG. ECT in the treatment of the catatonic syndrome. J Affect Disord. 1993;29(4):255-261.
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
3,4-methylenedioxy-methamphetamine, MDMA, Phenethylamines I Have Known and Loved: A Chemical Love Story, LSD, substance abuse, substance use
Malnourished and psychotic, and found incompetent to stand trial
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
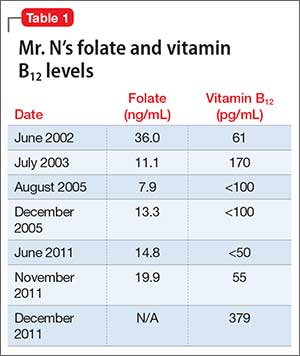
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
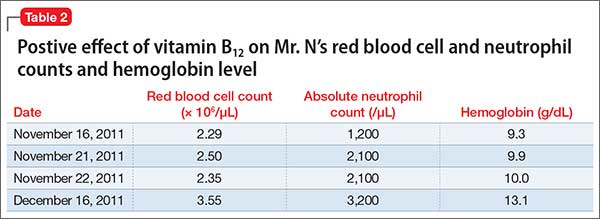
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. N, age 48, has chronic mental illness and has been in and out of psychiatric hospitals for 30 years, with diagnoses of bipolar disorder, not otherwise specified, without psychotic features and schizophrenia. He often is delusional and disorganized and does not adhere to treatment. Since age 18, his psychiatric care has been sporadic; during his last admission 3 years ago, he refused treatment and left the hospital against medical advice. Mr. N is homeless and often eats out of a dumpster.
Recently, Mr. N was arrested for cocaine possession, for which he was held in custody. His mental status continued to deteriorate while in jail, where he was evaluated by a forensics examiner.
Mr. N was declared incompetent to stand trial and was transferred to a state psychiatric hospital.
In the hospital, the treatment team finds that Mr. N is disorganized and preoccupied with thoughts of not wanting to “lose control” to the physicians. He shows no evidence of suicidal or homicidal ideation or perceptual disturbance. Mr. N has difficulty grasping concepts, making plans, and following through with them. He has poor insight and impulse control and impaired judgment.
Mr. N’s past and present diagnoses include bipolar disorder without psychotic features, schizophrenia, obsessive-compulsive personality disorder, paranoid personality traits, borderline intelligence, cellulitis of both legs, and chronic venous stasis. Although he was arrested for cocaine possession, we are not able to obtain much information about his history of substance abuse because of his poor mental status.
What could be causing Mr. N’s deteriorating mental status?
a) substance withdrawal
b) malnutrition
c) worsening schizophrenia
d) untreated infection due to cellulitis
HISTORY Sporadic care
Mr. N can provide few details of his early life. He was adopted as a child. He spent time in juvenile detention center. He completed 10th grade but did not graduate from high school. Symptoms of mental illness emerged at age 18. His employment history is consistent with chronic mental illness: His longest job, at a grocery store, lasted only 6 months. He has had multiple admissions to psychiatric hospitals. Over the years his treatment has included divalproex sodium, risperidone, paroxetine, chlorpromazine, thioridazine, amitriptyline, methylphenidate, and a multivitamin; however, he often is noncompliant with treatment and was not taking any medications when he arrived at the hospital.
EVALUATION Possible deficiency
The treatment team discusses guardianship, but the public administrator’s office provides little support because of Mr. N’s refusal to stay in one place. He was evicted from his last apartment because of hoarding behavior, which created a fire hazard. He has been homeless most of his adult life, which might have significantly restricted his diet.
A routine laboratory workup—complete blood count, basic metabolic panel, liver function test, thyroid-stimulating hormone, and lipids—is ordered, revealing an absolute neutrophil count (ANC) in the low range at 1,200/μL (normal range, 1,500 to 8,000/μL). Mr. N is offered treatment with a long-acting IM injection of risperidone because of his history of noncompliance, but he refuses the medication. Instead, he is started on oral risperidone, 2 mg/d.
The cellulitis of both lower limbs and chronic venous stasis are of concern; the medical team is consulted. Review of Mr. N’s medical records from an affiliated hospital reveals a history of vitamin B12 deficiency. Further tests show that the vitamin B12 level is low at <50 pg/mL (normal range, 160 to 950 pg/mL). Pernicious anemia had been ruled out after Mr. N tested negative for antibodies to intrinsic factor (a glycoprotein secreted in the stomach that is necessary for absorption of vitamin B12). Suspicion is that vitamin B12 deficiency is caused by Mr. N’s restricted diet in the context of chronic homelessness.
The authors’ observations
A review of the literature on vitamin B12 deficiency describes tingling or numbness, ataxia, and dementia; however, in rare cases, vitamin B12 deficiency presents with psychiatric symptoms, such as depression, mania, psychosis, dementia, and catatonia.1-13
We suspected that Mr. N’s vitamin B12 deficiency could have been affecting his mental status; consequently, we ordered routine laboratory work-up that included a complete blood count with differential and peripheral smear, which showed macrocytic anemia and ovalocytes. We also tested his vitamin B12 level, which was very low at 55 pg/mL. These results, combined with his previously recorded vitamin B12 level (Table 1), suggested deficiency.
TREATMENT Oral medication
Two months after starting risperidone, the medical team recommends IM vitamin B12 as first-line treatment, but Mr. N refuses. We considered guardianship ex parte for involuntary administration of IM B12 injection to prevent life-threatening consequences of a non-healing ulcer on his leg that was related to his cellulitis. Meanwhile, we reviewed the literature on vitamin B12 therapy, including route, dosage, and outcome.14-23 Mr. N agrees to oral vitamin B12, 1,000 μg/d,21 and we no longer consider guardianship ex parte. Mr. N’s vitamin B12 level and clinical picture improve 1 month after oral vitamin B12 is added to oral risperidone. His thought process is more organized, he is no longer paranoid, and he shows improved insight and judgement. ANC and neutrophil count improve as well (Table 2). Mr. N’s ulcer begins to heal despite his noncompliance with wound care.
The forensic examiner sees Mr. N after 3 months of continued therapy. His thought pattern is more organized and he is able to comprehend the criminal charges against him and to work with his attorney. He is determined competent by the forensic examiner; in a court hearing, the judge finds Mr. N competent to stand trial.
The authors’ observations
Based on our experience treating Mr. N, we think that it is important to establish an association between vitamin B12 deficiency and psychosis. Vitamin B12 deficiency is uncommon; however, serum levels do not need to be significantly low to produce severe neuropsychiatric morbidity, which has been reported with serum levels ≤457 pg/mL.2-5,24,25 It is more frequent than the other organic causes of psychosis5,10,24 and Mr. N’s improvement further strengthened the correlation.
Parenteral vitamin B12 therapy is the first-line treatment for a deficiency, but oral or sublingual vitamin B12 can be given to patients who are disabled, geriatric, or refuse parenteral administration.21 Only approximately 1% of oral vitamin B12 is absorbed in patients who do not have intrinsic factor. The daily requirement of vitamin B12 is 1.0 to 2.5 μg/d; large oral dosages of 1,000 to 5,000 μg/d therefore seem to be effective in correcting deficiency, even in the presence of intrinsic factor deficiency.15,20,21 Large oral dosages also benefit other hematological abnormalities, such as a low white blood cell count and neutropenia.
How vitamin B12 deficiency affects neuropsychiatric illness
Vitamin B12 is essential for methylation, a process crucial for the formation of neurotransmitters such as serotonin, dopamine, and epinephrine. A low level of vitamin B12 can interrupt methylation and cause accumulation of homocysteine and impaired metabolism of serotonin, dopamine, and epinephrine. Hyperhomocysteinemia can contribute to cerebral dysfunction by causing vascular injury.26
Vitamin B12 also is involved in tetrahydrobiopterin synthesis in the brain, which is pivotal for synthesis of monoamine neurotransmitters. Vitamin B12 deficiency can lead to accumulation of methyltetrahydrofolate, an excitatory neurotoxin. All of these can contribute to development of psychosis. Therefore, a defect in the methylation process could be responsible for the neuropsychiatric manifestations of vitamin B12 deficiency.
What did we learn from Mr. N?
In most people, vitamin B12 levels are normal, however, we recommend that clinicians consider vitamin B12 deficiency when a patient has new-onset or unresponsive psychosis,27 particularly in a homeless person or one who has a restricted diet.28 It is important to rule out vitamin B12 deficiency in a patient with a low serum folate level because folic acid therapy could exacerbate neurologic manifestations of underlying vitamin B12 deficiency and increase the risk of permanent nerve damage and cognitive decline.
We were intrigued to see improvement in Mr. N after we added vitamin B12 to his ongoing treatment with an antipsychotic. We did not believe that vitamin B12 supplementation was the sole reason his mental status improved enough to be found competent to stand trial, although we believe that initiating oral vitamin B12 was beneficial for Mr. N.
Last, this case supports the need for research to further explore the role of vitamin B12 in refractory psychosis, depression, and mania.
Bottom Line
Vitamin B12 deficiency can contribute to psychosis and other psychiatric disorders, especially in patients with a restricted diet, such as those who are homeless. Parenteral vitamin B12 therapy is the first-line treatment, but oral supplementation can be used if the patient refuses therapy. Large oral dosages of 1,000 to 5,000 μg/d seem to be effective in correcting vitamin B12 deficiency.
Related Resources
• Ramsey D, Muskin PR. Vitamin deficiencies and mental health: How are they linked? Current Psychiatry. 2013;12(1):37-43.
• Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
Drug Brand Names
Amitriptyline • Elavil
Chlorpromazine • Thorazine
Divalproex sodium • Depakote
Methylphenidate • Ritalin
Paroxetine • Paxil
Risperidone • Risperdal
Thioridazine • Mellaril
Acknowledgements
The authors thank Jan Jill-Jordan, PhD, for her help preparing the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.
1. Dogan M, Ozdemir O, Sal EA, et al. Psychotic disorder and extrapyramidal symptoms associated with vitamin B12 and folate deficiency. J Trop Pediatr. 2009;55(3):205-207.
2. Levine J, Stahl Z, Sela BA, et al. Elevated homocysteine levels in young male patients with schizophrenia. Am J Psychiatry. 2002;159(10):1790-1792.
3. Jauhar S, Blackett A, Srireddy P, et al. Pernicious anaemia presenting as catatonia without signs of anaemia or macrocytosis. Br J Psychiatry. 2010;197(3):244-245.
4. de Carvalho Abi-Abib R, Milech A, Ramalho FV, et al. Psychosis as the initial manifestation of pernicious anemia in a type 1 diabetes mellitus patient. Endocrinologist. 2010;20(5):224-225.
5. Berry N, Sagar R, Tripathi BM. Catatonia and other psychiatric symptoms with vitamin B12 deficiency. Acta Psychiatr Scand. 2003;108(2):156-159.
6. Zucker DK, Livingston RL, Nakra R, et al. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16(2):197-205.
7. Stanger O, Fowler B, Piertzik K, et al. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev Neurother. 2009;9(9):1393-1412.
8. Roze E, Gervais D, Demeret S, et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol. 2003;60(10):1457-1462.
9. Lewis AL, Pelic C, Kahn DA. Malignant catatonia in a patient with bipolar disorder, B12 deficiency, and neuroleptic malignant syndrome: one cause or three? J Psychiatr Pract. 2009;15(5):415-422.
10. Rajkumar AP, Jebaraj P. Chronic psychosis associated with vitamin B12 deficiency. J Assoc Physicians India. 2008;56:115-116.
11. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J. 2001;3(9):701-703.
12. Smith R, Oliver RA. Sudden onset of psychosis in association with vitamin-B12 deficiency. Br Med J. 1967;3(5556):34.
13. Russell RM, Baik HW. Clinical implications of vitamin B12 deficiency in the elderly. Nutrition in Clinical Care. 2001;4(4):214-220.
14. Sharabi A, Cohen E, Sulkes J, et al. Replacement therapy for vitamin B12 deficiency: comparison between the sublingual and oral route. Br J Clin Pharmacol. 2003; 56(6):635-638.
15. Chalmers RA, Bain MD, Costello I. Oral cobalamin therapy. Lancet. 2000;355(9198):148.
16. Borchardt J, Malnick S. Sublingual cobalamin for pernicious anaemia. Lancet. 1999;354(9195):2081.
17. Seal EC, Metz J, Flicker L, et al. A randomized, double-blind, placebo-controlled study of oral vitamin B12 supplementation in older patients with subnormal or borderline serum vitamin B12 concentrations. J Am Geriatr Soc. 2002;50(1):146-151.
18. Erkurt MA, Aydogdu I, Dikilitas M, et al. Effects of cyanocobalamin on immunity in patients with pernicious anemia. Med Princ Pract. 2008;17(2):131-135.
19. Andrès E, Kaltenbach G, Noel E, et al. Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol. 2003;25(3):161-166.
20. Wellmer J, Sturm KU, Herrmann W, et al. Oral treatment of vitamin B12 deficiency in subacute combined degeneration [in German]. Nervenarzt. 2006;77(10):1228-1231.
21. Lederle FA. Oral cobalamin for pernicious anemia. Medicine‘s best kept secret? JAMA. 1991;265(1):94-95.
22. Chalouhi C, Faesch S, Anthoine-Milhomme MC, et al. Neurological consequences of vitamin B12 deficiency and its treatment. Pediatr Emerg Care. 2008;24(8):538-541.
23. Andrès E, Federici L, Affenberger S, et al. B12 deficiency: a look beyond pernicious anemia. J Fam Pract. 2007;56(7):537-542.
24. Aaron S, Kumar S, Vijayan J, et al. Clinical and laboratory features and response to treatment in patients presenting with vitamin B12 deficiency related neurological syndromes. Neurol India. 2005;53(1):55-58.
25. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60(9):1296-1301.
26. Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A. 2007;143A(20):2430-2434.
27. Brett AS, Roberts MS. Screening for vitamin B12 deficiency in psychiatric patients. J Gen Intern Med. 1994;9(9):522-524.
28. Kaltenbach G, Noblet-Dick M, Barnier-Figue G, et al. Early normalization of low vitamin B12 levels by oral cobalamin therapy in three older patients with pernicious anemia. J Am Geriatr Soc. 2002;50(11):1914-1915.