User login
Linear Scleroderma Associated With Neurofibromatosis Type I
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
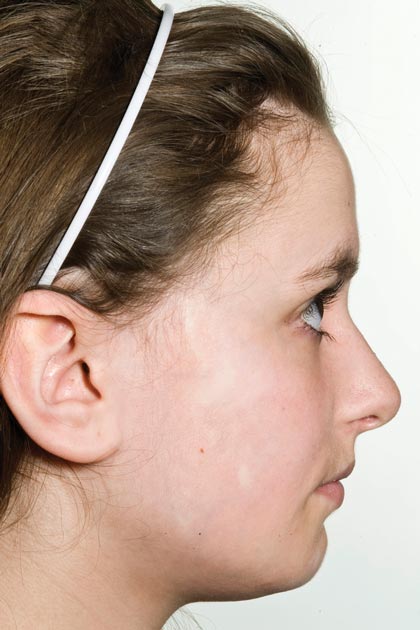
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
| 
|
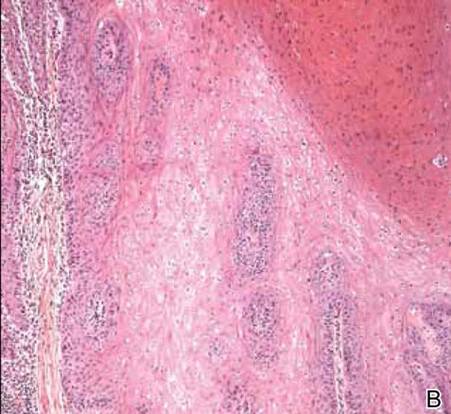
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
|
|
|
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
|
|
|
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
Verrucous Carcinoma on the Lower Extremities
To the Editor:
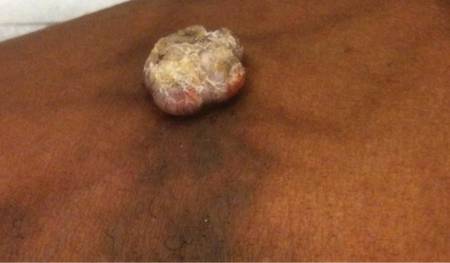
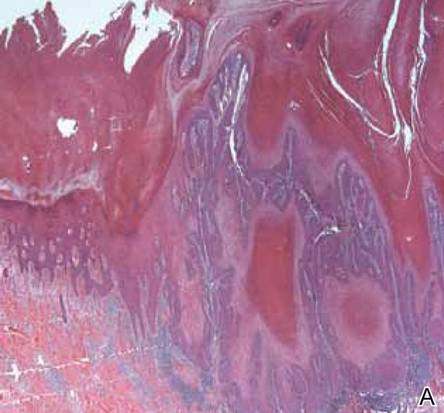
A 38-year-old black man presented with a slowly enlarging growth on the left thigh of 7 years’ duration. The lesion would occasionally scrape off but always recurred. He reported that the tumor developed in the area of a prior nevus. He reported no direct trauma to the area, chronic inflammation, or similar lesions elsewhere. His medical history included gastroesophageal reflux disease and inactive sarcoidosis. Physical examination revealed a 3×3×1-cm exophytic, hyperkeratotic, erythematous nodule with surrounding stellate and branching hyperpigmentation on the anterior aspect of the thigh (Figure 1). Pathologic examination demonstrated hyperkeratosis with an endophytic proliferation of mildly atypical keratinocytes with broad blunted rete ridges (Figure 2). Complete excision of the lesion was performed.
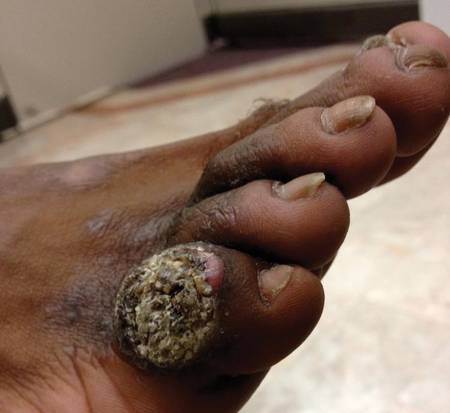
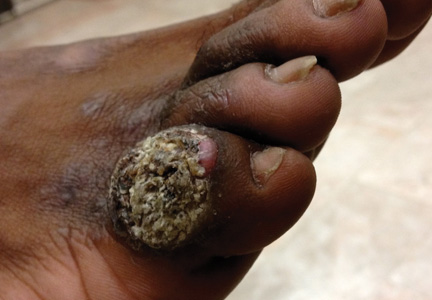
A 33-year-old black man presented with a rapidly growing lesion on the right fifth toe of 3 months’ duration. The patient originally believed the initial small papule was a corn, and after attempts to shave it down with a razor blade, the lesion grew rapidly into a large painful tumor. He reported no prior trauma to the area or history of a similar lesion. Physical examination revealed a 2×2×0.5-cm hyperkeratotic, papillated, hard nodule with a heaped-up border and no ulceration or drainage (Figure 3). A shave biopsy of the lesion was obtained. Microscopic examination revealed hyperkeratosis, parakeratosis, and papillomatosis with deep extension of mildly atypical keratinocytes into the dermis. Small toe amputation was performed by an orthopedic surgeon.
|
Verrucous carcinoma, first described by Ackerman1 in 1948, is an uncommon, low-grade, well-differentiated variant of squamous cell carcinoma. It presents as a slow-growing, bulky, exophytic tumor with a broad base. The tumor can ulcerate or present with surface sinus tracts that drain foul-smelling material. Typically, the tumor occurs in the fifth to sixth decades of life, with men outnumbering women by a ratio of 5.3 to 1.2 The prevalence of verrucous carcinoma in black individuals is unknown. A review of nonmelanoma skin cancers in skin of color identifies squamous cell carcinoma as the most common cutaneous carcinoma but does not report on the rare verrucous variant.3
Verrucous carcinoma is found in a variety of mucosal and skin surfaces. Verrucous carcinoma of the oral cavity, found most commonly on the buccal mucosa, is known as florid oral papillomatosis or Ackerman carcinoma. Cutaneous verrucous carcinoma is referred to as carcinoma cuniculatum or epithelioma cuniculatum and is predominantly located on the plantar surface of the foot. It is less commonly reported on the palm, scalp, face, extremities, and back. Verrucous carcinoma found in the anogenital area is referred to as the Buschke-Löwenstein tumor.4
Histologically, the lesion shows minimal cytologic atypia. Topped by an undulating keratinized mass, the deep margin of the tumor advances as a broad bulbous projection, compressing the underlying connective tissue in a bulldozing manner. Typically there also are keratin-filled sinuses and intraepidermal microabscesses.1
Human papillomavirus types 6, 11, 16, and 18 may be involved in the induction of the tumor. Human papillomavirus types 6 and 11 are frequently associated with the Buschke-Löwenstein tumor,2,4 while carcinoma cuniculatum is most commonly associated with human papillomavirus 16.5-7 In several cases of verrucous carcinoma, the tumor was reported to arise from preexisting lesions with chronic inflammation, such as a chronic ulcer, inflamed cyst, or burn scar.2 Ackerman carcinoma has been associated with the use of snuff, chewing tobacco, and betel nuts.
Morbidity and mortality from verrucous carcinoma arises from local invasion and infiltration into adjacent bone. The tumor rarely metastasizes, with regional lymph nodes being the only reported site of metastasis.8 The treatment of cutaneous verrucous carcinoma is complete surgical excision. Mohs micrographic surgery is preferred because it minimizes recurrence risk.4 Radiation therapy is contraindicated because it has been reported to cause the tumor to become more aggressive.9,10 Although local recurrence may occur, the prognosis is usually favorable.
1. Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
2. Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin). a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
3. Jackson BA. Nonmelanoma skin cancer in persons of color. Semin Cutan Med Surg. 2009;28:93-95.
4. Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21; quiz 22-24.
5. Assaf C, Steinhoff M, Petrov I, et al. Verrucous carcinoma of the axilla: case report and review. J Cutan Pathol. 2004;31:199-204.
6. Schell BJ, Rosen T, Rády P, et al. Verrucous carcinoma of the foot associated with human papillomavirus type 16. J Am Acad Dermatol. 2001;45:49-55.
7. Miyamoto T, Sasaoka R, Hagari Y, et al. Association of cutaneous verrucous carcinoma with human papillomavirus type 16. Br J Dermatol. 1999;140:168-169.
8. Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases [published online ahead of print July 11, 2008]. Oral Oncol. 2009;45:47-51.
9. Perez CA, Krans FT, Evans JC, et al. Anaplastic transformation in verrucous carcinoma of the oral cavity after radiation therapy. Radiology. 1966;86:108-115.
10. Proffett SD, Spooner TR, Kosek JC. Origin of undifferentiated neoplasm from verrucous epidermal carcinoma of oral cavity following irradiation. Cancer. 1970;26:389-393.
To the Editor:
A 38-year-old black man presented with a slowly enlarging growth on the left thigh of 7 years’ duration. The lesion would occasionally scrape off but always recurred. He reported that the tumor developed in the area of a prior nevus. He reported no direct trauma to the area, chronic inflammation, or similar lesions elsewhere. His medical history included gastroesophageal reflux disease and inactive sarcoidosis. Physical examination revealed a 3×3×1-cm exophytic, hyperkeratotic, erythematous nodule with surrounding stellate and branching hyperpigmentation on the anterior aspect of the thigh (Figure 1). Pathologic examination demonstrated hyperkeratosis with an endophytic proliferation of mildly atypical keratinocytes with broad blunted rete ridges (Figure 2). Complete excision of the lesion was performed.
A 33-year-old black man presented with a rapidly growing lesion on the right fifth toe of 3 months’ duration. The patient originally believed the initial small papule was a corn, and after attempts to shave it down with a razor blade, the lesion grew rapidly into a large painful tumor. He reported no prior trauma to the area or history of a similar lesion. Physical examination revealed a 2×2×0.5-cm hyperkeratotic, papillated, hard nodule with a heaped-up border and no ulceration or drainage (Figure 3). A shave biopsy of the lesion was obtained. Microscopic examination revealed hyperkeratosis, parakeratosis, and papillomatosis with deep extension of mildly atypical keratinocytes into the dermis. Small toe amputation was performed by an orthopedic surgeon.
|
|
Verrucous carcinoma, first described by Ackerman1 in 1948, is an uncommon, low-grade, well-differentiated variant of squamous cell carcinoma. It presents as a slow-growing, bulky, exophytic tumor with a broad base. The tumor can ulcerate or present with surface sinus tracts that drain foul-smelling material. Typically, the tumor occurs in the fifth to sixth decades of life, with men outnumbering women by a ratio of 5.3 to 1.2 The prevalence of verrucous carcinoma in black individuals is unknown. A review of nonmelanoma skin cancers in skin of color identifies squamous cell carcinoma as the most common cutaneous carcinoma but does not report on the rare verrucous variant.3
Verrucous carcinoma is found in a variety of mucosal and skin surfaces. Verrucous carcinoma of the oral cavity, found most commonly on the buccal mucosa, is known as florid oral papillomatosis or Ackerman carcinoma. Cutaneous verrucous carcinoma is referred to as carcinoma cuniculatum or epithelioma cuniculatum and is predominantly located on the plantar surface of the foot. It is less commonly reported on the palm, scalp, face, extremities, and back. Verrucous carcinoma found in the anogenital area is referred to as the Buschke-Löwenstein tumor.4
Histologically, the lesion shows minimal cytologic atypia. Topped by an undulating keratinized mass, the deep margin of the tumor advances as a broad bulbous projection, compressing the underlying connective tissue in a bulldozing manner. Typically there also are keratin-filled sinuses and intraepidermal microabscesses.1
Human papillomavirus types 6, 11, 16, and 18 may be involved in the induction of the tumor. Human papillomavirus types 6 and 11 are frequently associated with the Buschke-Löwenstein tumor,2,4 while carcinoma cuniculatum is most commonly associated with human papillomavirus 16.5-7 In several cases of verrucous carcinoma, the tumor was reported to arise from preexisting lesions with chronic inflammation, such as a chronic ulcer, inflamed cyst, or burn scar.2 Ackerman carcinoma has been associated with the use of snuff, chewing tobacco, and betel nuts.
Morbidity and mortality from verrucous carcinoma arises from local invasion and infiltration into adjacent bone. The tumor rarely metastasizes, with regional lymph nodes being the only reported site of metastasis.8 The treatment of cutaneous verrucous carcinoma is complete surgical excision. Mohs micrographic surgery is preferred because it minimizes recurrence risk.4 Radiation therapy is contraindicated because it has been reported to cause the tumor to become more aggressive.9,10 Although local recurrence may occur, the prognosis is usually favorable.
To the Editor:
A 38-year-old black man presented with a slowly enlarging growth on the left thigh of 7 years’ duration. The lesion would occasionally scrape off but always recurred. He reported that the tumor developed in the area of a prior nevus. He reported no direct trauma to the area, chronic inflammation, or similar lesions elsewhere. His medical history included gastroesophageal reflux disease and inactive sarcoidosis. Physical examination revealed a 3×3×1-cm exophytic, hyperkeratotic, erythematous nodule with surrounding stellate and branching hyperpigmentation on the anterior aspect of the thigh (Figure 1). Pathologic examination demonstrated hyperkeratosis with an endophytic proliferation of mildly atypical keratinocytes with broad blunted rete ridges (Figure 2). Complete excision of the lesion was performed.
A 33-year-old black man presented with a rapidly growing lesion on the right fifth toe of 3 months’ duration. The patient originally believed the initial small papule was a corn, and after attempts to shave it down with a razor blade, the lesion grew rapidly into a large painful tumor. He reported no prior trauma to the area or history of a similar lesion. Physical examination revealed a 2×2×0.5-cm hyperkeratotic, papillated, hard nodule with a heaped-up border and no ulceration or drainage (Figure 3). A shave biopsy of the lesion was obtained. Microscopic examination revealed hyperkeratosis, parakeratosis, and papillomatosis with deep extension of mildly atypical keratinocytes into the dermis. Small toe amputation was performed by an orthopedic surgeon.
|
|
Verrucous carcinoma, first described by Ackerman1 in 1948, is an uncommon, low-grade, well-differentiated variant of squamous cell carcinoma. It presents as a slow-growing, bulky, exophytic tumor with a broad base. The tumor can ulcerate or present with surface sinus tracts that drain foul-smelling material. Typically, the tumor occurs in the fifth to sixth decades of life, with men outnumbering women by a ratio of 5.3 to 1.2 The prevalence of verrucous carcinoma in black individuals is unknown. A review of nonmelanoma skin cancers in skin of color identifies squamous cell carcinoma as the most common cutaneous carcinoma but does not report on the rare verrucous variant.3
Verrucous carcinoma is found in a variety of mucosal and skin surfaces. Verrucous carcinoma of the oral cavity, found most commonly on the buccal mucosa, is known as florid oral papillomatosis or Ackerman carcinoma. Cutaneous verrucous carcinoma is referred to as carcinoma cuniculatum or epithelioma cuniculatum and is predominantly located on the plantar surface of the foot. It is less commonly reported on the palm, scalp, face, extremities, and back. Verrucous carcinoma found in the anogenital area is referred to as the Buschke-Löwenstein tumor.4
Histologically, the lesion shows minimal cytologic atypia. Topped by an undulating keratinized mass, the deep margin of the tumor advances as a broad bulbous projection, compressing the underlying connective tissue in a bulldozing manner. Typically there also are keratin-filled sinuses and intraepidermal microabscesses.1
Human papillomavirus types 6, 11, 16, and 18 may be involved in the induction of the tumor. Human papillomavirus types 6 and 11 are frequently associated with the Buschke-Löwenstein tumor,2,4 while carcinoma cuniculatum is most commonly associated with human papillomavirus 16.5-7 In several cases of verrucous carcinoma, the tumor was reported to arise from preexisting lesions with chronic inflammation, such as a chronic ulcer, inflamed cyst, or burn scar.2 Ackerman carcinoma has been associated with the use of snuff, chewing tobacco, and betel nuts.
Morbidity and mortality from verrucous carcinoma arises from local invasion and infiltration into adjacent bone. The tumor rarely metastasizes, with regional lymph nodes being the only reported site of metastasis.8 The treatment of cutaneous verrucous carcinoma is complete surgical excision. Mohs micrographic surgery is preferred because it minimizes recurrence risk.4 Radiation therapy is contraindicated because it has been reported to cause the tumor to become more aggressive.9,10 Although local recurrence may occur, the prognosis is usually favorable.
1. Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
2. Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin). a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
3. Jackson BA. Nonmelanoma skin cancer in persons of color. Semin Cutan Med Surg. 2009;28:93-95.
4. Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21; quiz 22-24.
5. Assaf C, Steinhoff M, Petrov I, et al. Verrucous carcinoma of the axilla: case report and review. J Cutan Pathol. 2004;31:199-204.
6. Schell BJ, Rosen T, Rády P, et al. Verrucous carcinoma of the foot associated with human papillomavirus type 16. J Am Acad Dermatol. 2001;45:49-55.
7. Miyamoto T, Sasaoka R, Hagari Y, et al. Association of cutaneous verrucous carcinoma with human papillomavirus type 16. Br J Dermatol. 1999;140:168-169.
8. Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases [published online ahead of print July 11, 2008]. Oral Oncol. 2009;45:47-51.
9. Perez CA, Krans FT, Evans JC, et al. Anaplastic transformation in verrucous carcinoma of the oral cavity after radiation therapy. Radiology. 1966;86:108-115.
10. Proffett SD, Spooner TR, Kosek JC. Origin of undifferentiated neoplasm from verrucous epidermal carcinoma of oral cavity following irradiation. Cancer. 1970;26:389-393.
1. Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
2. Kao GF, Graham JH, Helwig EB. Carcinoma cuniculatum (verrucous carcinoma of the skin). a clinicopathologic study of 46 cases with ultrastructural observations. Cancer. 1982;49:2395-2403.
3. Jackson BA. Nonmelanoma skin cancer in persons of color. Semin Cutan Med Surg. 2009;28:93-95.
4. Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21; quiz 22-24.
5. Assaf C, Steinhoff M, Petrov I, et al. Verrucous carcinoma of the axilla: case report and review. J Cutan Pathol. 2004;31:199-204.
6. Schell BJ, Rosen T, Rády P, et al. Verrucous carcinoma of the foot associated with human papillomavirus type 16. J Am Acad Dermatol. 2001;45:49-55.
7. Miyamoto T, Sasaoka R, Hagari Y, et al. Association of cutaneous verrucous carcinoma with human papillomavirus type 16. Br J Dermatol. 1999;140:168-169.
8. Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases [published online ahead of print July 11, 2008]. Oral Oncol. 2009;45:47-51.
9. Perez CA, Krans FT, Evans JC, et al. Anaplastic transformation in verrucous carcinoma of the oral cavity after radiation therapy. Radiology. 1966;86:108-115.
10. Proffett SD, Spooner TR, Kosek JC. Origin of undifferentiated neoplasm from verrucous epidermal carcinoma of oral cavity following irradiation. Cancer. 1970;26:389-393.
Sweet Syndrome Presenting With an Unusual Morphology
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
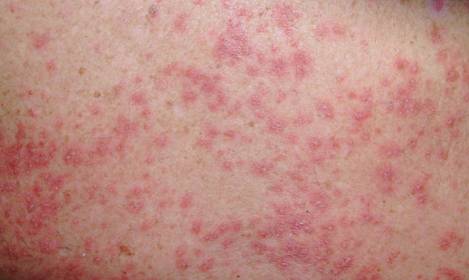
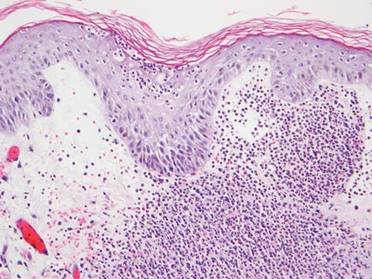
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
| 
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
|
|
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
|
|
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
Bilateral Auricular Swelling: Marginal Zone Lymphoma With Cutaneous Involvement
To the Editor:
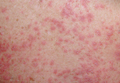
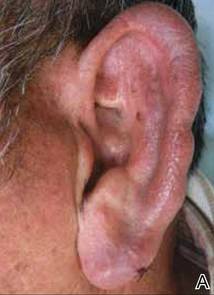
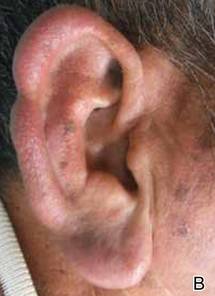
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
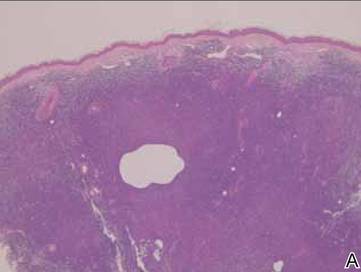
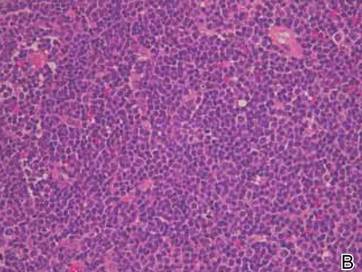
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.
To the Editor:
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
|
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
|
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
To the Editor:
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
|
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
|
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.

Nephrogenic Systemic Fibrosis Following Gadolinium Administration
To the Editor:
Nephrogenic systemic fibrosis (NSF) is an emerging medical entity in patients with renal disease, which results in progressive cutaneous and systemic fibrosis. It is a rare disorder that has been recognized in patients with renal impairment since 2000.1 Patients with NSF demonstrate symmetric dermal and subcutaneous fibrosis evidenced by increasing skin induration on clinical examination. Nephrogenic systemic fibrosis most commonly involves the lower extremities, and after extending to the upper extremities and trunk, it sporadically involves the head and neck.
The clinical manifestation of NSF begins with edema, followed by marked dermal induration, sclerotic plaques, and joint contractures that can lead to considerable disability. Pathogenesis remains to be elucidated; it has been hypothesized that it could be related to gadolinium (Gd). Currently, there is no treatment of this unremitting disease.1-3 We report the case of a patient affected by NSF after administration of Gd for magnetic resonance angiography.
A 56-year-old woman was referred to the department of dermatology at the University of Maryland (Baltimore, Maryland) with persistent swelling of the lower legs, forearms, and trunk of 5 months’ duration. She had end-stage renal disease of nonspecific origin. Five months prior to presentation, she had magnetic resonance angiography, during which 10 mmol of Gd was administered. After, she developed a persistent rash and swelling of the lower legs. On presentation, physical examination revealed symmetric, shiny, pigmented papules and plaques on the forearms, buttocks, thighs, and legs, with no facial involvement (Figure).
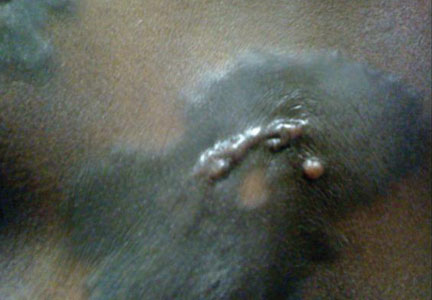
A skin biopsy of thigh lesions showed a diffuse dermal proliferation of bland spindle cells associated with dermal fibrosis. A CD4+ cellular infiltrate showed extension into the subcutaneous tissue. Deposition of Gd also was noted in the skin. She was treated with corticosteroid therapy, and after 2 months she reported softening of the affected skin. On 6-month follow-up, her skin lesions did not progress and there was no evidence of systemic involvement. Additionally, renal function had improved.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, was first described by Cowper et al1 in 2000. Since then, more than 215 cases have been reported in the literature. Clinically, it is characterized by acute onset of cutaneous hardening and thickening of the extremities and the trunk, often resulting in flexion contractures. There may be varying surface changes such as pigmentation, peau d’orange texture, and shiny sclerosis. Patients often experience unpleasant symptoms such as pain, pruritus, stiffness, and paresthesia. Systemic involvement has been documented in the heart, lung, tendons, muscle, testes, and lamina dura.1-4
Histologic findings of NSF are diffuse dermal fibrosis with increased cellular infiltrates comprised of bland spindled fibrocytes. These fibrocytes express CD34 and type I procollagen. Collagen bundles are thickened but retain clefting, and elastic fibers often are prominent. This fibrotic pattern typically extends to the subcutaneous fat septa, which are widened and collagenous. The epidermis generally is uninvolved. Other findings include dermal mucin deposition, calcification of collagen and vessels, increased CD68+ histiocytes, increased factor XIIIa and dendritic cells, and neoangiogenesis. Rarely, multinucleated giant cells and Miescher radial granulomas with lymphocytic aggregates mimicking erythema nodosum have been described.2-4
Dermatologic entities with similar clinical and histopathologic features, including scleroderma, scleromyxedema, lipodermatosclerosis, erythema nodosum, eosinophilic fasciitis, and spindle cell neoplasms, should be excluded.1-4 The exact pathogenetic mechanisms of NSF have yet to be determined, but there is strong evidence that Gd plays an important causative role.1 In fact, almost all patients with NSF have been exposed to Gd. Gadolinium has been documented in affected skin of patients with NSF and has been shown to induce NSF-like changes in rat models.
Other clinical factors that have been associated with NSF include erythropoietin, elevated serum calcium and phosphate levels, vascular injury or surgery, iron metabolic abnormalities, and metabolic acidosis. It is likely that many factors in the unique physiologic state of patients with renal failure contribute to the abnormal fibrotic reaction to Gd-containing contrast agents in NSF. Gadolinium is a member of a group of 15 elemental metals termed lanthanoids and has been used extensively worldwide in magnetic resonance imaging as a component of intravenously administered contrast agents. Currently, 6 such agents are approved for use in the United States: gadopentetate dimeglumine, gadoteridol, gadodiamide, gadoversetamide, gadobenate dimeglumine, and gadoxetate sodium. All are chelated Gd products, with the chelate serving to prevent toxicity from free Gd ions.
In patients with no renal function abnormalities, the biologic half-life of Gd-based magnetic resonance contrast agents (GBCAs) is 1.5 to 2.0 hours. However, in patients with abnormal kidney function, this half-life is inversely prolonged, proportional to the glomerular filtration rate.5-7 The link between GBCA administration and NSF is compelling, though other etiologic associations have been reported. Surgical or vascular procedures, history of a hypercoagulable state, erythropoietin administration, and immune suppression have been proposed as triggering factors in NSF. The proposed mechanisms responsible for fibrosis in NSF have centered on a collagen-producing cell in the peripheral blood termed the circulating fibrocyte. These cells express CD34 and CD45RO antigens and are capable of producing type I collagen.
Circulating fibrocytes traffic to areas of chronic antigenic stimulation promoting wound repair and fibrotic reactions. Some authors have proposed that materials deposited in the skin might serve as targets for circulating fibrocytes.8 Circulating fibrocytes also are known to produce inflammatory cytokines including IL-1 and chemokines such as platelet-derived growth factor, transforming growth factor b, and others capable of propagating fibrotic responses. Increased expression of transforming growth factor has been reported in dendritic cells in NSF lesions and Parsons et al9 postulated that transglutaminase-2 activation of this protein may be responsible for inciting fibrosis in NSF. Transglutaminases also are known to be directly activated by Gd.10,11
Transmetalation has been proposed as a possible operative phenomenon responsible for NSF. Several cations including zinc, copper, iron, and carbon are known to compete with Gd and may displace it from the ligand, with anions such as OHe, PO4 3e, and CO3 2e binding the resultant free Gd. Some GBCAs contain excess ligand to diminish potential free Gd concentrations. In fact, substantial elevations of serum calcium and phosphorus in patients with NSF have been noted in a large series of patients with NSF. Calciphylaxis, an often catastrophic condition arising in patients with renal failure, has been described in association with NSF, and sodium thiosulfate has been used with success in treating both conditions.10 In addition, Sanyal et al12 noted a substantially higher serum calcium in NSF cases compared with controls.
Gadolinium plays an important role in the pathology of NSF and is confirmed by the presence of Gd in skin biopsies.
1. Cowper SE, Robin HS, Steinberg SM, et al. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356:1000-1001.
2. Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathologiocal definition and workup recommendations [published online ahead of print July 2, 2011]. J Am Acad Dermatol. 2011;65:1095-1106.
3. Gupta A, Shamseddin MK, Khaira A. Pathomechanisms of nephrogenic systemic fibrosis: new insights [published online ahead of print July 25, 2011]. Clin Exp Dermatol. 2011;36:763-768.
4. Zou Z, Ma L. Nephrogenic systemic fibrosis: review of 408 biopsy-confirmed cases. Indian J Dermatol. 2011;56:65-73.
5. Pan D, Schmieder AH, Wickline SA, et al. Manganese-based MRI contrast agents: past, present and future. Tetrahedron. 2011;67:8431-8444.
6. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18:188-198.
7. Wang Y, Alkasab TK, Narin O, et al. Incidence of nephrogenic systemic fibrosis after adoption of restrictive gadolinium-based contrast agent guidelines [published online ahead of print May 17, 2011]. Radiology. 2011;260:105-111.
8. Ortonne N, Lipsker D, Chantrel F, et al. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150:1050-1052.
9. Parsons AC, Yosipovitch G, Sheehan DJ, et al. Transglutaminases: the missing link in nephrogenic systemic fibrosis. Am J Dermatopathol. 2007;29:433-436.
10. Wahba IM, Simpson EL, White K. Gadolinium is not the only trigger for nephrogenic systemic fibrosis: insights from two cases and review of the recent literature [published online ahead of print August 16, 2007]. Am J Transplant. 2007;7:2425-2432.
11. Goveia M, Chan BP, Patel PR. Evaluating the role of recombinant erythropoietin in nephrogenic systemic fibrosis [published online ahead of print August 8, 2007]. J Am Acad Dermatol. 2007;57:725-727.
12. Sanyal S, Marckmann P, Scherer S, et al. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis–an autopsy-based review [published online ahead of print March 25, 2011]. Nephrol Dial Transplant. 2011;26:3616-3626.
To the Editor:
Nephrogenic systemic fibrosis (NSF) is an emerging medical entity in patients with renal disease, which results in progressive cutaneous and systemic fibrosis. It is a rare disorder that has been recognized in patients with renal impairment since 2000.1 Patients with NSF demonstrate symmetric dermal and subcutaneous fibrosis evidenced by increasing skin induration on clinical examination. Nephrogenic systemic fibrosis most commonly involves the lower extremities, and after extending to the upper extremities and trunk, it sporadically involves the head and neck.
The clinical manifestation of NSF begins with edema, followed by marked dermal induration, sclerotic plaques, and joint contractures that can lead to considerable disability. Pathogenesis remains to be elucidated; it has been hypothesized that it could be related to gadolinium (Gd). Currently, there is no treatment of this unremitting disease.1-3 We report the case of a patient affected by NSF after administration of Gd for magnetic resonance angiography.
A 56-year-old woman was referred to the department of dermatology at the University of Maryland (Baltimore, Maryland) with persistent swelling of the lower legs, forearms, and trunk of 5 months’ duration. She had end-stage renal disease of nonspecific origin. Five months prior to presentation, she had magnetic resonance angiography, during which 10 mmol of Gd was administered. After, she developed a persistent rash and swelling of the lower legs. On presentation, physical examination revealed symmetric, shiny, pigmented papules and plaques on the forearms, buttocks, thighs, and legs, with no facial involvement (Figure).
A skin biopsy of thigh lesions showed a diffuse dermal proliferation of bland spindle cells associated with dermal fibrosis. A CD4+ cellular infiltrate showed extension into the subcutaneous tissue. Deposition of Gd also was noted in the skin. She was treated with corticosteroid therapy, and after 2 months she reported softening of the affected skin. On 6-month follow-up, her skin lesions did not progress and there was no evidence of systemic involvement. Additionally, renal function had improved.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, was first described by Cowper et al1 in 2000. Since then, more than 215 cases have been reported in the literature. Clinically, it is characterized by acute onset of cutaneous hardening and thickening of the extremities and the trunk, often resulting in flexion contractures. There may be varying surface changes such as pigmentation, peau d’orange texture, and shiny sclerosis. Patients often experience unpleasant symptoms such as pain, pruritus, stiffness, and paresthesia. Systemic involvement has been documented in the heart, lung, tendons, muscle, testes, and lamina dura.1-4
Histologic findings of NSF are diffuse dermal fibrosis with increased cellular infiltrates comprised of bland spindled fibrocytes. These fibrocytes express CD34 and type I procollagen. Collagen bundles are thickened but retain clefting, and elastic fibers often are prominent. This fibrotic pattern typically extends to the subcutaneous fat septa, which are widened and collagenous. The epidermis generally is uninvolved. Other findings include dermal mucin deposition, calcification of collagen and vessels, increased CD68+ histiocytes, increased factor XIIIa and dendritic cells, and neoangiogenesis. Rarely, multinucleated giant cells and Miescher radial granulomas with lymphocytic aggregates mimicking erythema nodosum have been described.2-4
Dermatologic entities with similar clinical and histopathologic features, including scleroderma, scleromyxedema, lipodermatosclerosis, erythema nodosum, eosinophilic fasciitis, and spindle cell neoplasms, should be excluded.1-4 The exact pathogenetic mechanisms of NSF have yet to be determined, but there is strong evidence that Gd plays an important causative role.1 In fact, almost all patients with NSF have been exposed to Gd. Gadolinium has been documented in affected skin of patients with NSF and has been shown to induce NSF-like changes in rat models.
Other clinical factors that have been associated with NSF include erythropoietin, elevated serum calcium and phosphate levels, vascular injury or surgery, iron metabolic abnormalities, and metabolic acidosis. It is likely that many factors in the unique physiologic state of patients with renal failure contribute to the abnormal fibrotic reaction to Gd-containing contrast agents in NSF. Gadolinium is a member of a group of 15 elemental metals termed lanthanoids and has been used extensively worldwide in magnetic resonance imaging as a component of intravenously administered contrast agents. Currently, 6 such agents are approved for use in the United States: gadopentetate dimeglumine, gadoteridol, gadodiamide, gadoversetamide, gadobenate dimeglumine, and gadoxetate sodium. All are chelated Gd products, with the chelate serving to prevent toxicity from free Gd ions.
In patients with no renal function abnormalities, the biologic half-life of Gd-based magnetic resonance contrast agents (GBCAs) is 1.5 to 2.0 hours. However, in patients with abnormal kidney function, this half-life is inversely prolonged, proportional to the glomerular filtration rate.5-7 The link between GBCA administration and NSF is compelling, though other etiologic associations have been reported. Surgical or vascular procedures, history of a hypercoagulable state, erythropoietin administration, and immune suppression have been proposed as triggering factors in NSF. The proposed mechanisms responsible for fibrosis in NSF have centered on a collagen-producing cell in the peripheral blood termed the circulating fibrocyte. These cells express CD34 and CD45RO antigens and are capable of producing type I collagen.
Circulating fibrocytes traffic to areas of chronic antigenic stimulation promoting wound repair and fibrotic reactions. Some authors have proposed that materials deposited in the skin might serve as targets for circulating fibrocytes.8 Circulating fibrocytes also are known to produce inflammatory cytokines including IL-1 and chemokines such as platelet-derived growth factor, transforming growth factor b, and others capable of propagating fibrotic responses. Increased expression of transforming growth factor has been reported in dendritic cells in NSF lesions and Parsons et al9 postulated that transglutaminase-2 activation of this protein may be responsible for inciting fibrosis in NSF. Transglutaminases also are known to be directly activated by Gd.10,11
Transmetalation has been proposed as a possible operative phenomenon responsible for NSF. Several cations including zinc, copper, iron, and carbon are known to compete with Gd and may displace it from the ligand, with anions such as OHe, PO4 3e, and CO3 2e binding the resultant free Gd. Some GBCAs contain excess ligand to diminish potential free Gd concentrations. In fact, substantial elevations of serum calcium and phosphorus in patients with NSF have been noted in a large series of patients with NSF. Calciphylaxis, an often catastrophic condition arising in patients with renal failure, has been described in association with NSF, and sodium thiosulfate has been used with success in treating both conditions.10 In addition, Sanyal et al12 noted a substantially higher serum calcium in NSF cases compared with controls.
Gadolinium plays an important role in the pathology of NSF and is confirmed by the presence of Gd in skin biopsies.
To the Editor:
Nephrogenic systemic fibrosis (NSF) is an emerging medical entity in patients with renal disease, which results in progressive cutaneous and systemic fibrosis. It is a rare disorder that has been recognized in patients with renal impairment since 2000.1 Patients with NSF demonstrate symmetric dermal and subcutaneous fibrosis evidenced by increasing skin induration on clinical examination. Nephrogenic systemic fibrosis most commonly involves the lower extremities, and after extending to the upper extremities and trunk, it sporadically involves the head and neck.
The clinical manifestation of NSF begins with edema, followed by marked dermal induration, sclerotic plaques, and joint contractures that can lead to considerable disability. Pathogenesis remains to be elucidated; it has been hypothesized that it could be related to gadolinium (Gd). Currently, there is no treatment of this unremitting disease.1-3 We report the case of a patient affected by NSF after administration of Gd for magnetic resonance angiography.
A 56-year-old woman was referred to the department of dermatology at the University of Maryland (Baltimore, Maryland) with persistent swelling of the lower legs, forearms, and trunk of 5 months’ duration. She had end-stage renal disease of nonspecific origin. Five months prior to presentation, she had magnetic resonance angiography, during which 10 mmol of Gd was administered. After, she developed a persistent rash and swelling of the lower legs. On presentation, physical examination revealed symmetric, shiny, pigmented papules and plaques on the forearms, buttocks, thighs, and legs, with no facial involvement (Figure).
A skin biopsy of thigh lesions showed a diffuse dermal proliferation of bland spindle cells associated with dermal fibrosis. A CD4+ cellular infiltrate showed extension into the subcutaneous tissue. Deposition of Gd also was noted in the skin. She was treated with corticosteroid therapy, and after 2 months she reported softening of the affected skin. On 6-month follow-up, her skin lesions did not progress and there was no evidence of systemic involvement. Additionally, renal function had improved.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, was first described by Cowper et al1 in 2000. Since then, more than 215 cases have been reported in the literature. Clinically, it is characterized by acute onset of cutaneous hardening and thickening of the extremities and the trunk, often resulting in flexion contractures. There may be varying surface changes such as pigmentation, peau d’orange texture, and shiny sclerosis. Patients often experience unpleasant symptoms such as pain, pruritus, stiffness, and paresthesia. Systemic involvement has been documented in the heart, lung, tendons, muscle, testes, and lamina dura.1-4
Histologic findings of NSF are diffuse dermal fibrosis with increased cellular infiltrates comprised of bland spindled fibrocytes. These fibrocytes express CD34 and type I procollagen. Collagen bundles are thickened but retain clefting, and elastic fibers often are prominent. This fibrotic pattern typically extends to the subcutaneous fat septa, which are widened and collagenous. The epidermis generally is uninvolved. Other findings include dermal mucin deposition, calcification of collagen and vessels, increased CD68+ histiocytes, increased factor XIIIa and dendritic cells, and neoangiogenesis. Rarely, multinucleated giant cells and Miescher radial granulomas with lymphocytic aggregates mimicking erythema nodosum have been described.2-4
Dermatologic entities with similar clinical and histopathologic features, including scleroderma, scleromyxedema, lipodermatosclerosis, erythema nodosum, eosinophilic fasciitis, and spindle cell neoplasms, should be excluded.1-4 The exact pathogenetic mechanisms of NSF have yet to be determined, but there is strong evidence that Gd plays an important causative role.1 In fact, almost all patients with NSF have been exposed to Gd. Gadolinium has been documented in affected skin of patients with NSF and has been shown to induce NSF-like changes in rat models.
Other clinical factors that have been associated with NSF include erythropoietin, elevated serum calcium and phosphate levels, vascular injury or surgery, iron metabolic abnormalities, and metabolic acidosis. It is likely that many factors in the unique physiologic state of patients with renal failure contribute to the abnormal fibrotic reaction to Gd-containing contrast agents in NSF. Gadolinium is a member of a group of 15 elemental metals termed lanthanoids and has been used extensively worldwide in magnetic resonance imaging as a component of intravenously administered contrast agents. Currently, 6 such agents are approved for use in the United States: gadopentetate dimeglumine, gadoteridol, gadodiamide, gadoversetamide, gadobenate dimeglumine, and gadoxetate sodium. All are chelated Gd products, with the chelate serving to prevent toxicity from free Gd ions.
In patients with no renal function abnormalities, the biologic half-life of Gd-based magnetic resonance contrast agents (GBCAs) is 1.5 to 2.0 hours. However, in patients with abnormal kidney function, this half-life is inversely prolonged, proportional to the glomerular filtration rate.5-7 The link between GBCA administration and NSF is compelling, though other etiologic associations have been reported. Surgical or vascular procedures, history of a hypercoagulable state, erythropoietin administration, and immune suppression have been proposed as triggering factors in NSF. The proposed mechanisms responsible for fibrosis in NSF have centered on a collagen-producing cell in the peripheral blood termed the circulating fibrocyte. These cells express CD34 and CD45RO antigens and are capable of producing type I collagen.
Circulating fibrocytes traffic to areas of chronic antigenic stimulation promoting wound repair and fibrotic reactions. Some authors have proposed that materials deposited in the skin might serve as targets for circulating fibrocytes.8 Circulating fibrocytes also are known to produce inflammatory cytokines including IL-1 and chemokines such as platelet-derived growth factor, transforming growth factor b, and others capable of propagating fibrotic responses. Increased expression of transforming growth factor has been reported in dendritic cells in NSF lesions and Parsons et al9 postulated that transglutaminase-2 activation of this protein may be responsible for inciting fibrosis in NSF. Transglutaminases also are known to be directly activated by Gd.10,11
Transmetalation has been proposed as a possible operative phenomenon responsible for NSF. Several cations including zinc, copper, iron, and carbon are known to compete with Gd and may displace it from the ligand, with anions such as OHe, PO4 3e, and CO3 2e binding the resultant free Gd. Some GBCAs contain excess ligand to diminish potential free Gd concentrations. In fact, substantial elevations of serum calcium and phosphorus in patients with NSF have been noted in a large series of patients with NSF. Calciphylaxis, an often catastrophic condition arising in patients with renal failure, has been described in association with NSF, and sodium thiosulfate has been used with success in treating both conditions.10 In addition, Sanyal et al12 noted a substantially higher serum calcium in NSF cases compared with controls.
Gadolinium plays an important role in the pathology of NSF and is confirmed by the presence of Gd in skin biopsies.
1. Cowper SE, Robin HS, Steinberg SM, et al. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356:1000-1001.
2. Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathologiocal definition and workup recommendations [published online ahead of print July 2, 2011]. J Am Acad Dermatol. 2011;65:1095-1106.
3. Gupta A, Shamseddin MK, Khaira A. Pathomechanisms of nephrogenic systemic fibrosis: new insights [published online ahead of print July 25, 2011]. Clin Exp Dermatol. 2011;36:763-768.
4. Zou Z, Ma L. Nephrogenic systemic fibrosis: review of 408 biopsy-confirmed cases. Indian J Dermatol. 2011;56:65-73.
5. Pan D, Schmieder AH, Wickline SA, et al. Manganese-based MRI contrast agents: past, present and future. Tetrahedron. 2011;67:8431-8444.
6. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18:188-198.
7. Wang Y, Alkasab TK, Narin O, et al. Incidence of nephrogenic systemic fibrosis after adoption of restrictive gadolinium-based contrast agent guidelines [published online ahead of print May 17, 2011]. Radiology. 2011;260:105-111.
8. Ortonne N, Lipsker D, Chantrel F, et al. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150:1050-1052.
9. Parsons AC, Yosipovitch G, Sheehan DJ, et al. Transglutaminases: the missing link in nephrogenic systemic fibrosis. Am J Dermatopathol. 2007;29:433-436.
10. Wahba IM, Simpson EL, White K. Gadolinium is not the only trigger for nephrogenic systemic fibrosis: insights from two cases and review of the recent literature [published online ahead of print August 16, 2007]. Am J Transplant. 2007;7:2425-2432.
11. Goveia M, Chan BP, Patel PR. Evaluating the role of recombinant erythropoietin in nephrogenic systemic fibrosis [published online ahead of print August 8, 2007]. J Am Acad Dermatol. 2007;57:725-727.
12. Sanyal S, Marckmann P, Scherer S, et al. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis–an autopsy-based review [published online ahead of print March 25, 2011]. Nephrol Dial Transplant. 2011;26:3616-3626.
1. Cowper SE, Robin HS, Steinberg SM, et al. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356:1000-1001.
2. Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathologiocal definition and workup recommendations [published online ahead of print July 2, 2011]. J Am Acad Dermatol. 2011;65:1095-1106.
3. Gupta A, Shamseddin MK, Khaira A. Pathomechanisms of nephrogenic systemic fibrosis: new insights [published online ahead of print July 25, 2011]. Clin Exp Dermatol. 2011;36:763-768.
4. Zou Z, Ma L. Nephrogenic systemic fibrosis: review of 408 biopsy-confirmed cases. Indian J Dermatol. 2011;56:65-73.
5. Pan D, Schmieder AH, Wickline SA, et al. Manganese-based MRI contrast agents: past, present and future. Tetrahedron. 2011;67:8431-8444.
6. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18:188-198.
7. Wang Y, Alkasab TK, Narin O, et al. Incidence of nephrogenic systemic fibrosis after adoption of restrictive gadolinium-based contrast agent guidelines [published online ahead of print May 17, 2011]. Radiology. 2011;260:105-111.
8. Ortonne N, Lipsker D, Chantrel F, et al. Presence of CD45RO+ CD34+ cells with collagen synthesis activity in nephrogenic fibrosing dermopathy: a new pathogenic hypothesis. Br J Dermatol. 2004;150:1050-1052.
9. Parsons AC, Yosipovitch G, Sheehan DJ, et al. Transglutaminases: the missing link in nephrogenic systemic fibrosis. Am J Dermatopathol. 2007;29:433-436.
10. Wahba IM, Simpson EL, White K. Gadolinium is not the only trigger for nephrogenic systemic fibrosis: insights from two cases and review of the recent literature [published online ahead of print August 16, 2007]. Am J Transplant. 2007;7:2425-2432.
11. Goveia M, Chan BP, Patel PR. Evaluating the role of recombinant erythropoietin in nephrogenic systemic fibrosis [published online ahead of print August 8, 2007]. J Am Acad Dermatol. 2007;57:725-727.
12. Sanyal S, Marckmann P, Scherer S, et al. Multiorgan gadolinium (Gd) deposition and fibrosis in a patient with nephrogenic systemic fibrosis–an autopsy-based review [published online ahead of print March 25, 2011]. Nephrol Dial Transplant. 2011;26:3616-3626.
Cutaneous Infection With Mycobacterium kansasii in a Patient With Myelodysplastic Syndrome and Sweet Syndrome
To the Editor:
A 68-year-old man with a history of myelodysplastic syndrome and recurrent Sweet syndrome presented with left leg lesions of 3 months’ duration. The lesions originated as a solitary nodule on the left calf and subsequently developed into multiple nonpainful, nonpruritic, erythematous plaques of varying sizes with violaceous coloration and overlying necrotic eschar, occupying the entire anterior aspect of the left lower leg and left popliteal fossa (Figure). The patient denied any trauma or associated symptoms but had a history of Sweet syndrome that manifested as lesions on the arms and legs for which he took 6 mg of prednisone daily to prevent recurrence.
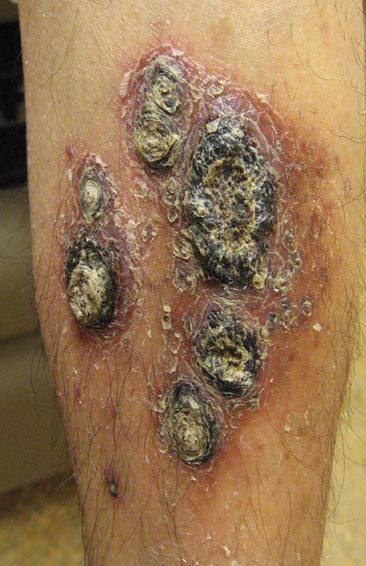
Histologic examination revealed nodular and diffuse chronic granulomatous and acute inflammatory infiltrate. Stains for bacteria, fungi, and acid-fast bacilli were negative. Cultures subsequently grew Mycobacterium kansasii, and the patient was started on isoniazid 300 mg daily, rifampin 600 mg daily, ethambutol 800 mg daily, and pyridoxine 50 mg daily. Chest radiograph and computed tomography showed no evidence of pulmonary disease and 2 blood cultures were negative for growth. The patient subsequently developed weakness that he attributed to the antibiotics and he decided to discontinue all treatment.
At 11 months the lesions showed no change; however, magnetic resonance imaging of the leg was suggestive of osteomyelitis. The patient was started on clarithromycin 500 mg twice daily with planned addition of isoniazid. The patient refused any additional antibiotics but agreed to continue the clarithromycin treatment for one year. He was subsequently lost to dermatology follow-up.
Nontuberculous mycobacteria (NTM) infection is a rare sequela of hematologic malignancy, seen in only 1.5% of patients.1 The NTM most commonly seen in hematologic malignancy are generally the fast-growing species Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium fortuitum, or Mycobacterium phlei, rather than slow growers Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium kansasii, Mycobacterium marinum, and Mycobacterium xenopi. Mycobacterium kansasii infection, such as seen in our patient, accounts for only 18% of cases.1 This case is further distinguished by the fact that cutaneous infections with NTM also are generally caused by fast-growing organisms such as Mycobacterium abscessus-chelonae complex and M fortuitum, rather than the slow-growing M kansasii.2,3
Mycobacterium kansasii is a slow-growing, acid-fast bacillus found in local water reservoirs, swimming pools, sewers, and tap water where it can live for up to 12 months.2,4,5Mycobacterium kansasii is traditionally considered the most virulent NTM.3,6 It most frequently causes a pulmonary infection in the immunosuppressed and patients with chronic bronchopulmonary disease.6,7 Disseminated disease is less common and is primarily seen in immunocompromised patients, particularly in human immunodeficiency virus–positive patients, transplant recipients, and patients with hematologic malignancies.1,6,8 Disseminated disease rarely has been seen in patients with normal immune function.2,3
Cutaneous M kansasii infection has only infrequently been described. Most patients tend to be middle-aged men, with a median affected age of 43 years.2,7,9,10 One review of cutaneous cases found that 72% had some form of altered immunity and more than 50% of those patients were on chronic steroids. The same review found that of the cases of cutaneous M kansasii in patients with altered immunity, only 30% had disseminated disease.10 Our patient was immunocompromised but showed no evidence of disseminated disease, as displayed by negative chest radiograph and computed tomography, lack of pulmonary symptoms, and negative blood cultures. As a 68-year-old man with myelodysplastic syndrome on chronic steroids with no disseminated disease, our patient fits well into these demographics, aside from his advanced age.
Cutaneous M kansasii infection has a variable presentation, manifesting as solitary lesions, nodules, pustules, seromas, erythematous plaques, verrucous lesions, ulcers, and as cellulitis.5,7,9-12 Immune competent individuals were more likely to present with raised lesions or ulcers, whereas immune compromised individuals had a more diffuse presentation of cellulitis or seromas with variable histology.6,8 Our patient, though immune compromised, presented with multiple erythematous plaques with eschars, which further endorses having a high clinical suspicion, as the lesions display marked heterogeneity.
Treatment of M kansasii infection consists of at least 1 year of isoniazid 300 mg daily, rifampin 600 mg daily, and ethambutol 15 mg/kg daily, with possible addition of streptomycin.8,13Mycobacterium kansasii infection necessitates multidrug treatment due to the broad range of resistance exhibited by different isolated strains.14
Response to treatment in cutaneous M kansasii greatly depends on the underlying disease state of the individual. Generally, immune competent individuals do very well, while the course in immune compromised patients depends on their degree of illness. Patients with disseminated disease generally do poorly.4,7,10 In at least one case of cutaneous disease, dissemination developed as a sequela, thus suggesting treatment is needed even in stable lesions.2 Dissemination was a concern with our patient given the magnetic resonance imaging findings suggestive of osteomyelitis. Although treatment generally consists of triple therapy with isoniazid, rifampin, and ethambutol, given the high frequency of adverse effects due to isoniazid or rifampin, as was seen in our patient, the drug regimen might have to be altered to suit the patient. Susceptibility testing is desirable to aid in tailoring the treatment.8,13 Furthermore, as the duration of treatment is at least 1 year, diligent follow-up must be maintained to avoid incomplete treatment.
The unpredictable presentation of cutaneous M kansasii infection coupled with the variable history necessitates a high level of clinical suspicion and a low threshold for culturing lesions. Furthermore, the long duration and complexity of the antibiotic regimen and the high incidence of adverse reactions demands strict follow-up, especially given the risk for progression to disseminated disease.
1. Chen CY, Sheng WH, Lai CC. Mycobacterial infections in adult patients with hematological malignancy. Eur J Clin Microbiol Infect Dis. 2012;31:1059-1066.
2. Han SH, Kim KM, Chin BS, et al. Disseminated Mycobacterium kansasii infection associated with skin lesions: a case report and comprehensive review of the literature. J Korean Med Sci. 2010;25:304-308.
3. Razavi B, Cleveland MG. Cutaneous infection due to Mycobacterium kansasii. Diagn Microbiol Infect Dis. 2000;38:173-175.
4. Portaels F. Epidemiology of mycobacterial diseases. Clin Dermatol. 1995;13:207-222.
5. Nomura Y, Nishie W, Shibaki A, et al. Disseminated cutaneous Mycobacterium kansasii infection in a patient infected with the human immunodeficiency virus. Clin Exp Dermatol. 2009;34:625-626.
6. Bloch KC, Zwerling L, Pletcher MJ, et al. Incidence and clinical implications of isolation of Mycobacterium kansasii: results of a 5-year, population-based study. Ann Intern Med. 1998;129:698-704.
7. Breathnach A, Levell N, Munro C, et al. Cutaneous Mycobacterium kansasii infection: case report and review. Clin Infect Dis. 1995;20:812-817.
8. Pintado V, Gómez-Mampaso E, Martín-Dávila P. Mycobacterium kansasii infection in patients infected with the human immunodeficiency virus. Eur J Clin Microbiol Infect Dis. 1999;18:582-586.
9. Stengem J, Grande KK, Hsu S. Localized primary cutaneous Mycobacterium kansasii infection in an immunocompromised patient. J Am Acad Dermatol. 1999;41(5, pt 2):854-856.
10. Czelusta A, Moore AY. Cutaneous Mycobacterium kansasii infection in a patient with systemic lupus erythematosus: case report and review. J Am Acad Dermatol. 1999;40(2, pt 2):359-363.
11. Curcó N, Pagerols X, Gómez L, et al. Mycobacterium kansasii infection limited to the skin in a patient with AIDS. Br J Dermatol. 1996;135:324-326.
12. Hanke CW, Temofeew RK, Slama SL. Mycobacterium kansasii infection with multiple cutaneous lesions. J Am Acad Dermatol. 1987;16(5, pt 2):1122-1128.
13. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculousmycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
14. da Silva Telles MA, Chimara E, Ferrazoli L, Riley LW. Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J Med Microbiol. 2005;54:975-979.
To the Editor:
A 68-year-old man with a history of myelodysplastic syndrome and recurrent Sweet syndrome presented with left leg lesions of 3 months’ duration. The lesions originated as a solitary nodule on the left calf and subsequently developed into multiple nonpainful, nonpruritic, erythematous plaques of varying sizes with violaceous coloration and overlying necrotic eschar, occupying the entire anterior aspect of the left lower leg and left popliteal fossa (Figure). The patient denied any trauma or associated symptoms but had a history of Sweet syndrome that manifested as lesions on the arms and legs for which he took 6 mg of prednisone daily to prevent recurrence.
Histologic examination revealed nodular and diffuse chronic granulomatous and acute inflammatory infiltrate. Stains for bacteria, fungi, and acid-fast bacilli were negative. Cultures subsequently grew Mycobacterium kansasii, and the patient was started on isoniazid 300 mg daily, rifampin 600 mg daily, ethambutol 800 mg daily, and pyridoxine 50 mg daily. Chest radiograph and computed tomography showed no evidence of pulmonary disease and 2 blood cultures were negative for growth. The patient subsequently developed weakness that he attributed to the antibiotics and he decided to discontinue all treatment.
At 11 months the lesions showed no change; however, magnetic resonance imaging of the leg was suggestive of osteomyelitis. The patient was started on clarithromycin 500 mg twice daily with planned addition of isoniazid. The patient refused any additional antibiotics but agreed to continue the clarithromycin treatment for one year. He was subsequently lost to dermatology follow-up.
Nontuberculous mycobacteria (NTM) infection is a rare sequela of hematologic malignancy, seen in only 1.5% of patients.1 The NTM most commonly seen in hematologic malignancy are generally the fast-growing species Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium fortuitum, or Mycobacterium phlei, rather than slow growers Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium kansasii, Mycobacterium marinum, and Mycobacterium xenopi. Mycobacterium kansasii infection, such as seen in our patient, accounts for only 18% of cases.1 This case is further distinguished by the fact that cutaneous infections with NTM also are generally caused by fast-growing organisms such as Mycobacterium abscessus-chelonae complex and M fortuitum, rather than the slow-growing M kansasii.2,3
Mycobacterium kansasii is a slow-growing, acid-fast bacillus found in local water reservoirs, swimming pools, sewers, and tap water where it can live for up to 12 months.2,4,5Mycobacterium kansasii is traditionally considered the most virulent NTM.3,6 It most frequently causes a pulmonary infection in the immunosuppressed and patients with chronic bronchopulmonary disease.6,7 Disseminated disease is less common and is primarily seen in immunocompromised patients, particularly in human immunodeficiency virus–positive patients, transplant recipients, and patients with hematologic malignancies.1,6,8 Disseminated disease rarely has been seen in patients with normal immune function.2,3
Cutaneous M kansasii infection has only infrequently been described. Most patients tend to be middle-aged men, with a median affected age of 43 years.2,7,9,10 One review of cutaneous cases found that 72% had some form of altered immunity and more than 50% of those patients were on chronic steroids. The same review found that of the cases of cutaneous M kansasii in patients with altered immunity, only 30% had disseminated disease.10 Our patient was immunocompromised but showed no evidence of disseminated disease, as displayed by negative chest radiograph and computed tomography, lack of pulmonary symptoms, and negative blood cultures. As a 68-year-old man with myelodysplastic syndrome on chronic steroids with no disseminated disease, our patient fits well into these demographics, aside from his advanced age.
Cutaneous M kansasii infection has a variable presentation, manifesting as solitary lesions, nodules, pustules, seromas, erythematous plaques, verrucous lesions, ulcers, and as cellulitis.5,7,9-12 Immune competent individuals were more likely to present with raised lesions or ulcers, whereas immune compromised individuals had a more diffuse presentation of cellulitis or seromas with variable histology.6,8 Our patient, though immune compromised, presented with multiple erythematous plaques with eschars, which further endorses having a high clinical suspicion, as the lesions display marked heterogeneity.
Treatment of M kansasii infection consists of at least 1 year of isoniazid 300 mg daily, rifampin 600 mg daily, and ethambutol 15 mg/kg daily, with possible addition of streptomycin.8,13Mycobacterium kansasii infection necessitates multidrug treatment due to the broad range of resistance exhibited by different isolated strains.14
Response to treatment in cutaneous M kansasii greatly depends on the underlying disease state of the individual. Generally, immune competent individuals do very well, while the course in immune compromised patients depends on their degree of illness. Patients with disseminated disease generally do poorly.4,7,10 In at least one case of cutaneous disease, dissemination developed as a sequela, thus suggesting treatment is needed even in stable lesions.2 Dissemination was a concern with our patient given the magnetic resonance imaging findings suggestive of osteomyelitis. Although treatment generally consists of triple therapy with isoniazid, rifampin, and ethambutol, given the high frequency of adverse effects due to isoniazid or rifampin, as was seen in our patient, the drug regimen might have to be altered to suit the patient. Susceptibility testing is desirable to aid in tailoring the treatment.8,13 Furthermore, as the duration of treatment is at least 1 year, diligent follow-up must be maintained to avoid incomplete treatment.
The unpredictable presentation of cutaneous M kansasii infection coupled with the variable history necessitates a high level of clinical suspicion and a low threshold for culturing lesions. Furthermore, the long duration and complexity of the antibiotic regimen and the high incidence of adverse reactions demands strict follow-up, especially given the risk for progression to disseminated disease.
To the Editor:
A 68-year-old man with a history of myelodysplastic syndrome and recurrent Sweet syndrome presented with left leg lesions of 3 months’ duration. The lesions originated as a solitary nodule on the left calf and subsequently developed into multiple nonpainful, nonpruritic, erythematous plaques of varying sizes with violaceous coloration and overlying necrotic eschar, occupying the entire anterior aspect of the left lower leg and left popliteal fossa (Figure). The patient denied any trauma or associated symptoms but had a history of Sweet syndrome that manifested as lesions on the arms and legs for which he took 6 mg of prednisone daily to prevent recurrence.
Histologic examination revealed nodular and diffuse chronic granulomatous and acute inflammatory infiltrate. Stains for bacteria, fungi, and acid-fast bacilli were negative. Cultures subsequently grew Mycobacterium kansasii, and the patient was started on isoniazid 300 mg daily, rifampin 600 mg daily, ethambutol 800 mg daily, and pyridoxine 50 mg daily. Chest radiograph and computed tomography showed no evidence of pulmonary disease and 2 blood cultures were negative for growth. The patient subsequently developed weakness that he attributed to the antibiotics and he decided to discontinue all treatment.
At 11 months the lesions showed no change; however, magnetic resonance imaging of the leg was suggestive of osteomyelitis. The patient was started on clarithromycin 500 mg twice daily with planned addition of isoniazid. The patient refused any additional antibiotics but agreed to continue the clarithromycin treatment for one year. He was subsequently lost to dermatology follow-up.
Nontuberculous mycobacteria (NTM) infection is a rare sequela of hematologic malignancy, seen in only 1.5% of patients.1 The NTM most commonly seen in hematologic malignancy are generally the fast-growing species Mycobacterium abscessus, Mycobacterium chelonae, Mycobacterium fortuitum, or Mycobacterium phlei, rather than slow growers Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium kansasii, Mycobacterium marinum, and Mycobacterium xenopi. Mycobacterium kansasii infection, such as seen in our patient, accounts for only 18% of cases.1 This case is further distinguished by the fact that cutaneous infections with NTM also are generally caused by fast-growing organisms such as Mycobacterium abscessus-chelonae complex and M fortuitum, rather than the slow-growing M kansasii.2,3
Mycobacterium kansasii is a slow-growing, acid-fast bacillus found in local water reservoirs, swimming pools, sewers, and tap water where it can live for up to 12 months.2,4,5Mycobacterium kansasii is traditionally considered the most virulent NTM.3,6 It most frequently causes a pulmonary infection in the immunosuppressed and patients with chronic bronchopulmonary disease.6,7 Disseminated disease is less common and is primarily seen in immunocompromised patients, particularly in human immunodeficiency virus–positive patients, transplant recipients, and patients with hematologic malignancies.1,6,8 Disseminated disease rarely has been seen in patients with normal immune function.2,3
Cutaneous M kansasii infection has only infrequently been described. Most patients tend to be middle-aged men, with a median affected age of 43 years.2,7,9,10 One review of cutaneous cases found that 72% had some form of altered immunity and more than 50% of those patients were on chronic steroids. The same review found that of the cases of cutaneous M kansasii in patients with altered immunity, only 30% had disseminated disease.10 Our patient was immunocompromised but showed no evidence of disseminated disease, as displayed by negative chest radiograph and computed tomography, lack of pulmonary symptoms, and negative blood cultures. As a 68-year-old man with myelodysplastic syndrome on chronic steroids with no disseminated disease, our patient fits well into these demographics, aside from his advanced age.
Cutaneous M kansasii infection has a variable presentation, manifesting as solitary lesions, nodules, pustules, seromas, erythematous plaques, verrucous lesions, ulcers, and as cellulitis.5,7,9-12 Immune competent individuals were more likely to present with raised lesions or ulcers, whereas immune compromised individuals had a more diffuse presentation of cellulitis or seromas with variable histology.6,8 Our patient, though immune compromised, presented with multiple erythematous plaques with eschars, which further endorses having a high clinical suspicion, as the lesions display marked heterogeneity.
Treatment of M kansasii infection consists of at least 1 year of isoniazid 300 mg daily, rifampin 600 mg daily, and ethambutol 15 mg/kg daily, with possible addition of streptomycin.8,13Mycobacterium kansasii infection necessitates multidrug treatment due to the broad range of resistance exhibited by different isolated strains.14
Response to treatment in cutaneous M kansasii greatly depends on the underlying disease state of the individual. Generally, immune competent individuals do very well, while the course in immune compromised patients depends on their degree of illness. Patients with disseminated disease generally do poorly.4,7,10 In at least one case of cutaneous disease, dissemination developed as a sequela, thus suggesting treatment is needed even in stable lesions.2 Dissemination was a concern with our patient given the magnetic resonance imaging findings suggestive of osteomyelitis. Although treatment generally consists of triple therapy with isoniazid, rifampin, and ethambutol, given the high frequency of adverse effects due to isoniazid or rifampin, as was seen in our patient, the drug regimen might have to be altered to suit the patient. Susceptibility testing is desirable to aid in tailoring the treatment.8,13 Furthermore, as the duration of treatment is at least 1 year, diligent follow-up must be maintained to avoid incomplete treatment.
The unpredictable presentation of cutaneous M kansasii infection coupled with the variable history necessitates a high level of clinical suspicion and a low threshold for culturing lesions. Furthermore, the long duration and complexity of the antibiotic regimen and the high incidence of adverse reactions demands strict follow-up, especially given the risk for progression to disseminated disease.
1. Chen CY, Sheng WH, Lai CC. Mycobacterial infections in adult patients with hematological malignancy. Eur J Clin Microbiol Infect Dis. 2012;31:1059-1066.
2. Han SH, Kim KM, Chin BS, et al. Disseminated Mycobacterium kansasii infection associated with skin lesions: a case report and comprehensive review of the literature. J Korean Med Sci. 2010;25:304-308.
3. Razavi B, Cleveland MG. Cutaneous infection due to Mycobacterium kansasii. Diagn Microbiol Infect Dis. 2000;38:173-175.
4. Portaels F. Epidemiology of mycobacterial diseases. Clin Dermatol. 1995;13:207-222.
5. Nomura Y, Nishie W, Shibaki A, et al. Disseminated cutaneous Mycobacterium kansasii infection in a patient infected with the human immunodeficiency virus. Clin Exp Dermatol. 2009;34:625-626.
6. Bloch KC, Zwerling L, Pletcher MJ, et al. Incidence and clinical implications of isolation of Mycobacterium kansasii: results of a 5-year, population-based study. Ann Intern Med. 1998;129:698-704.
7. Breathnach A, Levell N, Munro C, et al. Cutaneous Mycobacterium kansasii infection: case report and review. Clin Infect Dis. 1995;20:812-817.
8. Pintado V, Gómez-Mampaso E, Martín-Dávila P. Mycobacterium kansasii infection in patients infected with the human immunodeficiency virus. Eur J Clin Microbiol Infect Dis. 1999;18:582-586.
9. Stengem J, Grande KK, Hsu S. Localized primary cutaneous Mycobacterium kansasii infection in an immunocompromised patient. J Am Acad Dermatol. 1999;41(5, pt 2):854-856.
10. Czelusta A, Moore AY. Cutaneous Mycobacterium kansasii infection in a patient with systemic lupus erythematosus: case report and review. J Am Acad Dermatol. 1999;40(2, pt 2):359-363.
11. Curcó N, Pagerols X, Gómez L, et al. Mycobacterium kansasii infection limited to the skin in a patient with AIDS. Br J Dermatol. 1996;135:324-326.
12. Hanke CW, Temofeew RK, Slama SL. Mycobacterium kansasii infection with multiple cutaneous lesions. J Am Acad Dermatol. 1987;16(5, pt 2):1122-1128.
13. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculousmycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
14. da Silva Telles MA, Chimara E, Ferrazoli L, Riley LW. Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J Med Microbiol. 2005;54:975-979.
1. Chen CY, Sheng WH, Lai CC. Mycobacterial infections in adult patients with hematological malignancy. Eur J Clin Microbiol Infect Dis. 2012;31:1059-1066.
2. Han SH, Kim KM, Chin BS, et al. Disseminated Mycobacterium kansasii infection associated with skin lesions: a case report and comprehensive review of the literature. J Korean Med Sci. 2010;25:304-308.
3. Razavi B, Cleveland MG. Cutaneous infection due to Mycobacterium kansasii. Diagn Microbiol Infect Dis. 2000;38:173-175.
4. Portaels F. Epidemiology of mycobacterial diseases. Clin Dermatol. 1995;13:207-222.
5. Nomura Y, Nishie W, Shibaki A, et al. Disseminated cutaneous Mycobacterium kansasii infection in a patient infected with the human immunodeficiency virus. Clin Exp Dermatol. 2009;34:625-626.
6. Bloch KC, Zwerling L, Pletcher MJ, et al. Incidence and clinical implications of isolation of Mycobacterium kansasii: results of a 5-year, population-based study. Ann Intern Med. 1998;129:698-704.
7. Breathnach A, Levell N, Munro C, et al. Cutaneous Mycobacterium kansasii infection: case report and review. Clin Infect Dis. 1995;20:812-817.
8. Pintado V, Gómez-Mampaso E, Martín-Dávila P. Mycobacterium kansasii infection in patients infected with the human immunodeficiency virus. Eur J Clin Microbiol Infect Dis. 1999;18:582-586.
9. Stengem J, Grande KK, Hsu S. Localized primary cutaneous Mycobacterium kansasii infection in an immunocompromised patient. J Am Acad Dermatol. 1999;41(5, pt 2):854-856.
10. Czelusta A, Moore AY. Cutaneous Mycobacterium kansasii infection in a patient with systemic lupus erythematosus: case report and review. J Am Acad Dermatol. 1999;40(2, pt 2):359-363.
11. Curcó N, Pagerols X, Gómez L, et al. Mycobacterium kansasii infection limited to the skin in a patient with AIDS. Br J Dermatol. 1996;135:324-326.
12. Hanke CW, Temofeew RK, Slama SL. Mycobacterium kansasii infection with multiple cutaneous lesions. J Am Acad Dermatol. 1987;16(5, pt 2):1122-1128.
13. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculousmycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
14. da Silva Telles MA, Chimara E, Ferrazoli L, Riley LW. Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J Med Microbiol. 2005;54:975-979.
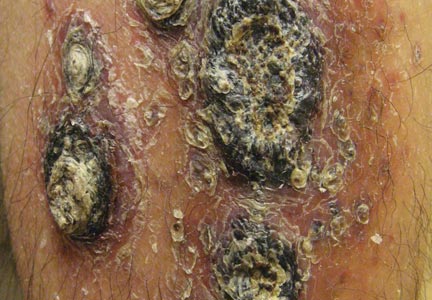
Progressive Cribriform and Zosteriform Hyperpigmentation
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
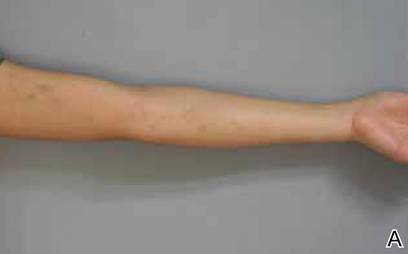
|
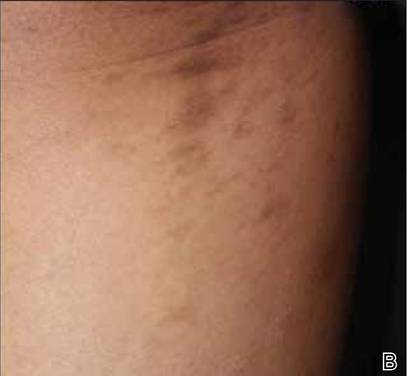
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
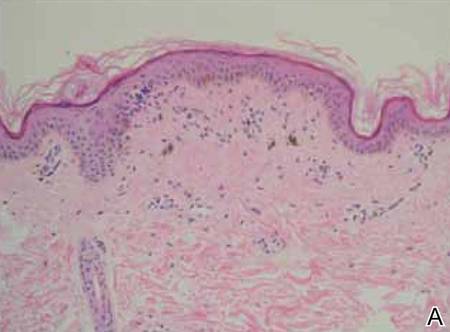
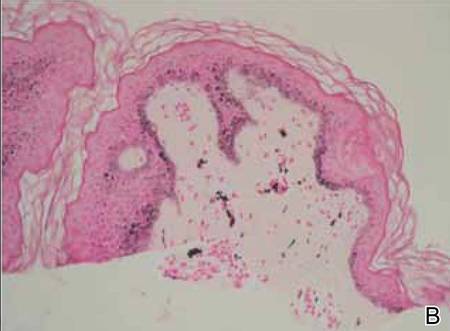
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
|
|
|
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
|
|
|
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
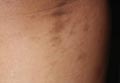
Levonorgestrel-Releasing Intrauterine System Causes a Lichenoid Drug Eruption
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
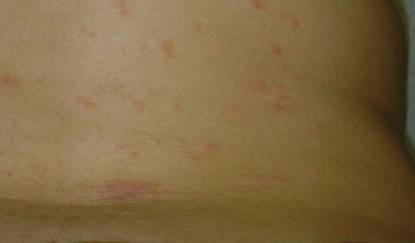
|
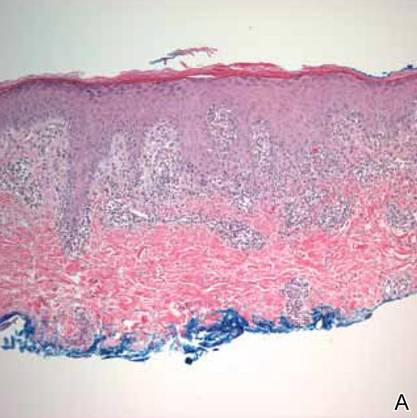

A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
|
|
A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
|
|
A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.

Leukemia Cutis Presenting as Scrotal Ulcerations in a Patient With Acute Myelogenous Leukemia
To the Editor:
Cutaneous manifestations of leukemia can be defined as specific or nonspecific skin lesions. Specific skin lesions include papules, nodules, tumors, or ulcerating plaques caused by the infiltration of leukemic cells (eg, leukemia cutis), while nonspecific skin lesions include petechiae, purpura, ecchymosis, or infection. Leukemia cutis is a rare entity that denotes a poor prognosis. We present a case of leukemia cutis with scrotal ulcerations, which heralded the onset of acute myelogenous leukemia (AML). Other causes of painful cutaneous ulcers to consider in a patient with a hematologic malignancy include a variety of infectious and inflammatory etiologies.
A 63-year-old man with long-standing essential thrombocytosis was admitted to the hospital with increasing fatigue and fever. A complete blood cell count revealed anemia, thrombocytopenia, neutropenia, and leukocytosis with 49% blasts. He was immediately placed on broad-spectrum antifungal, antiviral, and antibiotic agents after blood and urine cultures were obtained and while subsequent diagnostic tests were being performed. The patient developed antibiotic-associated Clostridium difficile diarrhea after several days in the hospital. The nursing staff noted “genital irritation from feces” while cleaning the patient and reported the findings to the primary team. Dermatology was consulted for treatment recommendations and wound care.
A full-body skin examination revealed discrete and coalescing painful scrotal and perineal ulcers with fecal contamination (Figure 1), along with numerous petechial macules on the anteromedial thighs. The patient was unsure of the onset of the ulcers, but he claimed to have scrotal and perineal pain prior to admission, which suggested their long-standing presence. No crepitus or rapid contiguous spread was noted on serial physical examination over the next 12 to 24 hours. Punch biopsies for tissue culture and histopathologic examination were obtained from the scrotum.
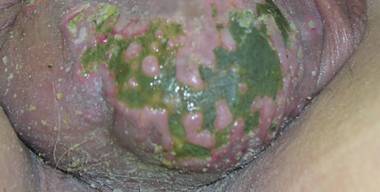
Subsequent diagnostic studies, including a bone marrow biopsy touch preparation that showed 44.7% blasts, along with cytogenetics analysis yielded the diagnosis of AML with an unfavorable karyotype: 5q deletion, 17p deletion, and monosomy 16. Induction chemotherapy with multiple cycles of a combination of cytarabine and daunorubicin was initiated, albeit with an unfavorable response. Blood, urine, bone marrow, and stool cultures remained negative. Serologic testing for herpes simplex virus (HSV), cytomegalovirus, and Epstein-Barr virus was negative.
Histopathology showed an ulcerated epidermis along with a dense dermal perivascular and interstitial infiltrate of myeloid blasts (Figure 2). Lesional cells were diffusely CD68+ (Figure 3), myeloperoxidase positive (Figure 4), and CD34-, correlating with findings on the bone marrow biopsy. Gomori methenamine-silver, periodic acid–Schiff, and Gram stains highlighted several bacterial cocci, yeast forms, and pseudohyphae on the ulcer surface with no organisms seen in the dermis. Tissue culture did not yield any organisms. There was no overt evidence of HSV infection. The histopathologic features were consistent with a diagnosis of leukemia cutis in the setting of AML. After failed chemotherapy and a complicated hospital course with a poor long-term prognosis, the patient and his family opted for home hospice care.
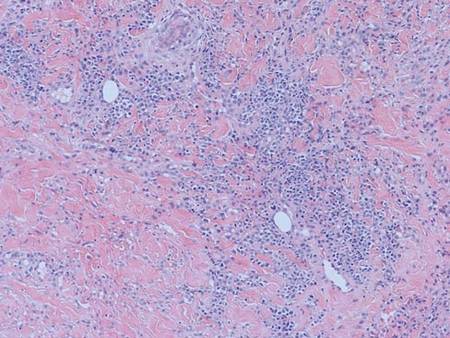
| 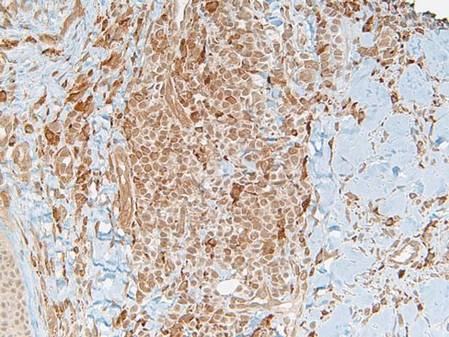
|
Cases of leukemia cutis manifesting as scrotal ulcerations are rare.1 Typically, most dermatologic lesions seen in leukemia patients are of a nonleukemic origin.2 Leukemia cutis is a rare entity that is associated with a poor prognosis.3 It was accordingly an ominous finding in our patient, as his AML had an unfavorable karyotype and was unresponsive to chemotherapy.
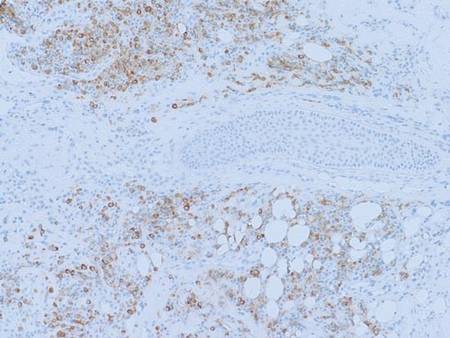
In hematologic malignancies, the morphology and distribution of specific cutaneous lesions tends to vary by the type of leukemia. For example, chronic lymphocytic leukemia presents with papules, nodules, and ulcerating plaques more often than acute lymphocytic leukemia.4 Acute myelocytic papules, nodules, or tumors tend to appear on the face, while chronic lesions commonly are seen on the face and extremities.5 Generally, for all forms of leukemia, lesions on the genitalia are uncommon.
This case of ulcerative scrotal AML lesions is unique, as myelogenous leukemia cutis typically appears as a tumor that infrequently ulcerates and rarely is located on the genitalia.6 Tissue culture, blood culture, viral serology, and biopsy failed to reveal an infectious etiology of the genital ulcerations in our patient. Therefore, the leukemic infiltrate likely preceded the ulceration and an antecedent infection did not elicit an inflammatory response, resulting in the attraction of a leukemic infiltrate to the area. This latter phenomenon also has been demonstrated in the setting of recent surgical scars, trauma, burns, herpes zoster, HSV, and intramuscular injection sites.1,7
The differential diagnosis of painful cutaneous ulcers in the setting of hematologic malignancy includes a variety of infectious and inflammatory etiologies such as ulcerative HSV, cytomegalovirus, Epstein-Barr virus, amoebic skin ulcerations, opportunistic fungal infections, bacterial infections, pyoderma gangrenosum, erythema nodosum, acute febrile neutrophilic dermatosis (Sweet syndrome), histoplasmosis,1 Fournier gangrene, and vasculitis, all of which were ruled out in our case.
We report a case of a patient with a history of essential thrombocytosis who developed leukemia cutis presenting as scrotal ulcerations, which preceded the diagnosis of AML. Ulcerative leukemic lesions on the genitalia are atypical and extremely rare. However, the lesions are notable because they may be suggestive of a poor prognostic outcome, as in our patient whose induction therapy with cytarabine and daunorubicin failed. We propose that leukemia cutis be included in the differential diagnosis of chronic skin ulcerations of the genitalia.
1. Zax RH, Kulp-Shorten CL, Callen JP. Leukemia cutis presenting as a scrotal ulcer. J Am Acad Dermatol. 1989;21(2, pt 2):410-413.
2. Wadhwa N, Batra M, Singh B. Nonhealing ulcer—a rare initial presentation of chronic myeloid leukemia diagnosed on aspiration cytology. Diagn Cytopathol. 2011;39:135-137.
3. Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
4. Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
5. Stawiski MA. Skin manifestations of leukemias and lymphomas. Cutis. 1978;21:814-818.
6. Costello MJ, Canizares O, Montague M, et al. Cutaneous manifestations of myelogenous leukemia. AMA Arch Derm. 1955;71:605-614.
7. Koizumi H, Kumakiri M, Ishizuka M, et al. Leukemia cutis in acute myelomonocytic leukemia: infiltration to minor traumas and scars. J Dermatol. 1991;18:281-285.
To the Editor:
Cutaneous manifestations of leukemia can be defined as specific or nonspecific skin lesions. Specific skin lesions include papules, nodules, tumors, or ulcerating plaques caused by the infiltration of leukemic cells (eg, leukemia cutis), while nonspecific skin lesions include petechiae, purpura, ecchymosis, or infection. Leukemia cutis is a rare entity that denotes a poor prognosis. We present a case of leukemia cutis with scrotal ulcerations, which heralded the onset of acute myelogenous leukemia (AML). Other causes of painful cutaneous ulcers to consider in a patient with a hematologic malignancy include a variety of infectious and inflammatory etiologies.
A 63-year-old man with long-standing essential thrombocytosis was admitted to the hospital with increasing fatigue and fever. A complete blood cell count revealed anemia, thrombocytopenia, neutropenia, and leukocytosis with 49% blasts. He was immediately placed on broad-spectrum antifungal, antiviral, and antibiotic agents after blood and urine cultures were obtained and while subsequent diagnostic tests were being performed. The patient developed antibiotic-associated Clostridium difficile diarrhea after several days in the hospital. The nursing staff noted “genital irritation from feces” while cleaning the patient and reported the findings to the primary team. Dermatology was consulted for treatment recommendations and wound care.
A full-body skin examination revealed discrete and coalescing painful scrotal and perineal ulcers with fecal contamination (Figure 1), along with numerous petechial macules on the anteromedial thighs. The patient was unsure of the onset of the ulcers, but he claimed to have scrotal and perineal pain prior to admission, which suggested their long-standing presence. No crepitus or rapid contiguous spread was noted on serial physical examination over the next 12 to 24 hours. Punch biopsies for tissue culture and histopathologic examination were obtained from the scrotum.
Subsequent diagnostic studies, including a bone marrow biopsy touch preparation that showed 44.7% blasts, along with cytogenetics analysis yielded the diagnosis of AML with an unfavorable karyotype: 5q deletion, 17p deletion, and monosomy 16. Induction chemotherapy with multiple cycles of a combination of cytarabine and daunorubicin was initiated, albeit with an unfavorable response. Blood, urine, bone marrow, and stool cultures remained negative. Serologic testing for herpes simplex virus (HSV), cytomegalovirus, and Epstein-Barr virus was negative.
Histopathology showed an ulcerated epidermis along with a dense dermal perivascular and interstitial infiltrate of myeloid blasts (Figure 2). Lesional cells were diffusely CD68+ (Figure 3), myeloperoxidase positive (Figure 4), and CD34-, correlating with findings on the bone marrow biopsy. Gomori methenamine-silver, periodic acid–Schiff, and Gram stains highlighted several bacterial cocci, yeast forms, and pseudohyphae on the ulcer surface with no organisms seen in the dermis. Tissue culture did not yield any organisms. There was no overt evidence of HSV infection. The histopathologic features were consistent with a diagnosis of leukemia cutis in the setting of AML. After failed chemotherapy and a complicated hospital course with a poor long-term prognosis, the patient and his family opted for home hospice care.
|
|
|
Cases of leukemia cutis manifesting as scrotal ulcerations are rare.1 Typically, most dermatologic lesions seen in leukemia patients are of a nonleukemic origin.2 Leukemia cutis is a rare entity that is associated with a poor prognosis.3 It was accordingly an ominous finding in our patient, as his AML had an unfavorable karyotype and was unresponsive to chemotherapy.
In hematologic malignancies, the morphology and distribution of specific cutaneous lesions tends to vary by the type of leukemia. For example, chronic lymphocytic leukemia presents with papules, nodules, and ulcerating plaques more often than acute lymphocytic leukemia.4 Acute myelocytic papules, nodules, or tumors tend to appear on the face, while chronic lesions commonly are seen on the face and extremities.5 Generally, for all forms of leukemia, lesions on the genitalia are uncommon.
This case of ulcerative scrotal AML lesions is unique, as myelogenous leukemia cutis typically appears as a tumor that infrequently ulcerates and rarely is located on the genitalia.6 Tissue culture, blood culture, viral serology, and biopsy failed to reveal an infectious etiology of the genital ulcerations in our patient. Therefore, the leukemic infiltrate likely preceded the ulceration and an antecedent infection did not elicit an inflammatory response, resulting in the attraction of a leukemic infiltrate to the area. This latter phenomenon also has been demonstrated in the setting of recent surgical scars, trauma, burns, herpes zoster, HSV, and intramuscular injection sites.1,7
The differential diagnosis of painful cutaneous ulcers in the setting of hematologic malignancy includes a variety of infectious and inflammatory etiologies such as ulcerative HSV, cytomegalovirus, Epstein-Barr virus, amoebic skin ulcerations, opportunistic fungal infections, bacterial infections, pyoderma gangrenosum, erythema nodosum, acute febrile neutrophilic dermatosis (Sweet syndrome), histoplasmosis,1 Fournier gangrene, and vasculitis, all of which were ruled out in our case.
We report a case of a patient with a history of essential thrombocytosis who developed leukemia cutis presenting as scrotal ulcerations, which preceded the diagnosis of AML. Ulcerative leukemic lesions on the genitalia are atypical and extremely rare. However, the lesions are notable because they may be suggestive of a poor prognostic outcome, as in our patient whose induction therapy with cytarabine and daunorubicin failed. We propose that leukemia cutis be included in the differential diagnosis of chronic skin ulcerations of the genitalia.
To the Editor:
Cutaneous manifestations of leukemia can be defined as specific or nonspecific skin lesions. Specific skin lesions include papules, nodules, tumors, or ulcerating plaques caused by the infiltration of leukemic cells (eg, leukemia cutis), while nonspecific skin lesions include petechiae, purpura, ecchymosis, or infection. Leukemia cutis is a rare entity that denotes a poor prognosis. We present a case of leukemia cutis with scrotal ulcerations, which heralded the onset of acute myelogenous leukemia (AML). Other causes of painful cutaneous ulcers to consider in a patient with a hematologic malignancy include a variety of infectious and inflammatory etiologies.
A 63-year-old man with long-standing essential thrombocytosis was admitted to the hospital with increasing fatigue and fever. A complete blood cell count revealed anemia, thrombocytopenia, neutropenia, and leukocytosis with 49% blasts. He was immediately placed on broad-spectrum antifungal, antiviral, and antibiotic agents after blood and urine cultures were obtained and while subsequent diagnostic tests were being performed. The patient developed antibiotic-associated Clostridium difficile diarrhea after several days in the hospital. The nursing staff noted “genital irritation from feces” while cleaning the patient and reported the findings to the primary team. Dermatology was consulted for treatment recommendations and wound care.
A full-body skin examination revealed discrete and coalescing painful scrotal and perineal ulcers with fecal contamination (Figure 1), along with numerous petechial macules on the anteromedial thighs. The patient was unsure of the onset of the ulcers, but he claimed to have scrotal and perineal pain prior to admission, which suggested their long-standing presence. No crepitus or rapid contiguous spread was noted on serial physical examination over the next 12 to 24 hours. Punch biopsies for tissue culture and histopathologic examination were obtained from the scrotum.
Subsequent diagnostic studies, including a bone marrow biopsy touch preparation that showed 44.7% blasts, along with cytogenetics analysis yielded the diagnosis of AML with an unfavorable karyotype: 5q deletion, 17p deletion, and monosomy 16. Induction chemotherapy with multiple cycles of a combination of cytarabine and daunorubicin was initiated, albeit with an unfavorable response. Blood, urine, bone marrow, and stool cultures remained negative. Serologic testing for herpes simplex virus (HSV), cytomegalovirus, and Epstein-Barr virus was negative.
Histopathology showed an ulcerated epidermis along with a dense dermal perivascular and interstitial infiltrate of myeloid blasts (Figure 2). Lesional cells were diffusely CD68+ (Figure 3), myeloperoxidase positive (Figure 4), and CD34-, correlating with findings on the bone marrow biopsy. Gomori methenamine-silver, periodic acid–Schiff, and Gram stains highlighted several bacterial cocci, yeast forms, and pseudohyphae on the ulcer surface with no organisms seen in the dermis. Tissue culture did not yield any organisms. There was no overt evidence of HSV infection. The histopathologic features were consistent with a diagnosis of leukemia cutis in the setting of AML. After failed chemotherapy and a complicated hospital course with a poor long-term prognosis, the patient and his family opted for home hospice care.
|
|
|
Cases of leukemia cutis manifesting as scrotal ulcerations are rare.1 Typically, most dermatologic lesions seen in leukemia patients are of a nonleukemic origin.2 Leukemia cutis is a rare entity that is associated with a poor prognosis.3 It was accordingly an ominous finding in our patient, as his AML had an unfavorable karyotype and was unresponsive to chemotherapy.
In hematologic malignancies, the morphology and distribution of specific cutaneous lesions tends to vary by the type of leukemia. For example, chronic lymphocytic leukemia presents with papules, nodules, and ulcerating plaques more often than acute lymphocytic leukemia.4 Acute myelocytic papules, nodules, or tumors tend to appear on the face, while chronic lesions commonly are seen on the face and extremities.5 Generally, for all forms of leukemia, lesions on the genitalia are uncommon.
This case of ulcerative scrotal AML lesions is unique, as myelogenous leukemia cutis typically appears as a tumor that infrequently ulcerates and rarely is located on the genitalia.6 Tissue culture, blood culture, viral serology, and biopsy failed to reveal an infectious etiology of the genital ulcerations in our patient. Therefore, the leukemic infiltrate likely preceded the ulceration and an antecedent infection did not elicit an inflammatory response, resulting in the attraction of a leukemic infiltrate to the area. This latter phenomenon also has been demonstrated in the setting of recent surgical scars, trauma, burns, herpes zoster, HSV, and intramuscular injection sites.1,7
The differential diagnosis of painful cutaneous ulcers in the setting of hematologic malignancy includes a variety of infectious and inflammatory etiologies such as ulcerative HSV, cytomegalovirus, Epstein-Barr virus, amoebic skin ulcerations, opportunistic fungal infections, bacterial infections, pyoderma gangrenosum, erythema nodosum, acute febrile neutrophilic dermatosis (Sweet syndrome), histoplasmosis,1 Fournier gangrene, and vasculitis, all of which were ruled out in our case.
We report a case of a patient with a history of essential thrombocytosis who developed leukemia cutis presenting as scrotal ulcerations, which preceded the diagnosis of AML. Ulcerative leukemic lesions on the genitalia are atypical and extremely rare. However, the lesions are notable because they may be suggestive of a poor prognostic outcome, as in our patient whose induction therapy with cytarabine and daunorubicin failed. We propose that leukemia cutis be included in the differential diagnosis of chronic skin ulcerations of the genitalia.
1. Zax RH, Kulp-Shorten CL, Callen JP. Leukemia cutis presenting as a scrotal ulcer. J Am Acad Dermatol. 1989;21(2, pt 2):410-413.
2. Wadhwa N, Batra M, Singh B. Nonhealing ulcer—a rare initial presentation of chronic myeloid leukemia diagnosed on aspiration cytology. Diagn Cytopathol. 2011;39:135-137.
3. Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
4. Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
5. Stawiski MA. Skin manifestations of leukemias and lymphomas. Cutis. 1978;21:814-818.
6. Costello MJ, Canizares O, Montague M, et al. Cutaneous manifestations of myelogenous leukemia. AMA Arch Derm. 1955;71:605-614.
7. Koizumi H, Kumakiri M, Ishizuka M, et al. Leukemia cutis in acute myelomonocytic leukemia: infiltration to minor traumas and scars. J Dermatol. 1991;18:281-285.
1. Zax RH, Kulp-Shorten CL, Callen JP. Leukemia cutis presenting as a scrotal ulcer. J Am Acad Dermatol. 1989;21(2, pt 2):410-413.
2. Wadhwa N, Batra M, Singh B. Nonhealing ulcer—a rare initial presentation of chronic myeloid leukemia diagnosed on aspiration cytology. Diagn Cytopathol. 2011;39:135-137.
3. Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
4. Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
5. Stawiski MA. Skin manifestations of leukemias and lymphomas. Cutis. 1978;21:814-818.
6. Costello MJ, Canizares O, Montague M, et al. Cutaneous manifestations of myelogenous leukemia. AMA Arch Derm. 1955;71:605-614.
7. Koizumi H, Kumakiri M, Ishizuka M, et al. Leukemia cutis in acute myelomonocytic leukemia: infiltration to minor traumas and scars. J Dermatol. 1991;18:281-285.
Remission of Psoriasis 13 Years After Autologous Stem Cell Transplant
To the Editor:
Remission of psoriasis after bone marrow transplant has been reported1; however, follow-up typically has been limited. We describe a case of a remote recurrence of psoriasis 13 years after autologous stem cell transplant.
In September 1997, a 48-year-old woman with a 20-year history of moderate to severe plaque psoriasis, previously managed with oral methotrexate (up to 30–40 mg weekly), presented to her primary care physician with a persistent cough and newly pervasive arthralgia and myalgia. A complete blood cell count showed a white blood cell count of 6.9 mg/dL (reference range, 3.8–10.8 mg/dL), hemoglobin level of 11.6 mg/dL (reference range, 11.7–15.5 mg/dL), and hematocrit level of 34% (reference range, 35.0%–45.0%). Bone survey demonstrated a 4×2-cm lytic expansile lesion in the occipital bone, and a bone marrow aspirate and biopsy revealed 40% plasma cells. She was subsequently diagnosed with stage IIIA IgA λ and free λ light chain multiple myeloma with an IgA level of 62.1 g/L (reference range, 1.24–2.17 g/L).
Therapy was initiated with dexamethasone in combination with pamidronate and she was transitioned to cyclophosphamide chemotherapy. A peripheral autologous blood stem cell transplant followed. Throughout her initial treatment, the psoriasis continued to flare and she remained on methotrexate to help maintain control of the skin disease.
In November 1998, the patient underwent an autologous CD34+ stem cell transplant. There were no complications following the transplant and the multiple myeloma has remained in complete remission to date. Following the stem cell transplant, the patient remained free of psoriatic disease for just over 13 years without any intervention. Recurrent erythematous plaques with thick scale on the bilateral lower legs prompted her return to dermatology in August 2011. To date, she continues to note intermittent though mild flares of her psoriasis. She has responded well to methotrexate 7.5 mg weekly and clobetasol propionate ointment 0.05% as needed with satisfactory control of her disease.
The development of autoimmune disease following an allogenic bone marrow transplant such as psoriasis, thyroiditis, myasthenia gravis, insulin-dependent diabetes mellitus, and vitiligo has been well described.2-4 The causal relationship between allogenic hematopoetic stem cell transplant (HSCT) and subsequent autoimmune disease has been attributed to T cells and passive transfer of autoimmune disease from the donor to the recipient.1
In the case of autologous HSCT, the remission of autoimmune disease thereafter has been attributed to myeloablative conditioning, whereby high-dose chemotherapy eliminates self-reactive lymphocytes, resulting in ablation of peripheral reactive and alloreactive T cells with depletion of thymus and reactive B cells.1
In 1997, Cooley et al5 described 4 patients with autoimmune disease who experienced complete but temporary (up to 26 months) remission following autologous bone marrow or stem cell transplant. A comprehensive review of psoriasis cases that resolved after HSCT published by Kaffenberger et al6 in 2013 cited 6 cases of autologous and 13 cases of allogenic transplant that resulted in resolution of psoriatic disease. Recurrence of disease was witnessed in 3 of 13 allogenic HSCT patients and 5 of 6 autologous HSCT patients. Of note, all 5 of these patients developed recurrent disease within 24 months following autologous HSCT, albeit with more benign disease.6
Similar to those cases presented by Kaffenberger et al,6 our patient’s psoriatic disease was notably mild compared to her disease prior to HSCT. Although an unintended but welcome outcome of multiple myeloma treatment, the nearly quiescent nature of her skin disease following autologous HSCT has led to a dramatic improvement in her quality of life.
We highlight a unique case in which psoriatic skin disease recurred more than 1 decade after autologous HSCT.
1. Braiteh F, Hymes SR, Giralt SA, et al. Complete remission of psoriasis after autologous hematopoietic stem-cell transplantation for multiple myeloma. J Clin Oncol. 2008;26:4511-4513.
2. Kishimoto Y, Yamamoto Y, Matsumoto N, et al. Transfer of autoimmune thyroiditis and resolution of palmoplantar pustular psoriasis following allogeneic bone marrow transplantation. Bone Marrow Transpl. 1997;19:1041-1043.
3. Wahie S, Alexandroff A, Reynolds NY, et al. Psoriasis occurring after myeloablative therapy and autologous stem cell transplantation. Br J Dermatol. 2006;154:177-204.
4. Snowden JA, Heaton DC. Development of psoriasis after syngenic bone marrow transplant from psoriatic donor: further evidence for adoptive autoimmunity. Br J Dermatol. 1997;137:130-132.
5. Cooley HM, Snowden JA, Grigg AP, et al. Outcome of rheumatoid arthritis and psoriasis following autologous stem cell transplantation for hematologic malignancy. Arthritis Rheum. 1997;40:1712-1715.
6. Kaffenberger BH, Wong HK, Jarjour W, et al. Remission of psoriasis after allogenic but not autologous, hematopoietic stem-cell transplantation. J Am Acad Dermatol. 2013;68:489-492.
To the Editor:
Remission of psoriasis after bone marrow transplant has been reported1; however, follow-up typically has been limited. We describe a case of a remote recurrence of psoriasis 13 years after autologous stem cell transplant.
In September 1997, a 48-year-old woman with a 20-year history of moderate to severe plaque psoriasis, previously managed with oral methotrexate (up to 30–40 mg weekly), presented to her primary care physician with a persistent cough and newly pervasive arthralgia and myalgia. A complete blood cell count showed a white blood cell count of 6.9 mg/dL (reference range, 3.8–10.8 mg/dL), hemoglobin level of 11.6 mg/dL (reference range, 11.7–15.5 mg/dL), and hematocrit level of 34% (reference range, 35.0%–45.0%). Bone survey demonstrated a 4×2-cm lytic expansile lesion in the occipital bone, and a bone marrow aspirate and biopsy revealed 40% plasma cells. She was subsequently diagnosed with stage IIIA IgA λ and free λ light chain multiple myeloma with an IgA level of 62.1 g/L (reference range, 1.24–2.17 g/L).
Therapy was initiated with dexamethasone in combination with pamidronate and she was transitioned to cyclophosphamide chemotherapy. A peripheral autologous blood stem cell transplant followed. Throughout her initial treatment, the psoriasis continued to flare and she remained on methotrexate to help maintain control of the skin disease.
In November 1998, the patient underwent an autologous CD34+ stem cell transplant. There were no complications following the transplant and the multiple myeloma has remained in complete remission to date. Following the stem cell transplant, the patient remained free of psoriatic disease for just over 13 years without any intervention. Recurrent erythematous plaques with thick scale on the bilateral lower legs prompted her return to dermatology in August 2011. To date, she continues to note intermittent though mild flares of her psoriasis. She has responded well to methotrexate 7.5 mg weekly and clobetasol propionate ointment 0.05% as needed with satisfactory control of her disease.
The development of autoimmune disease following an allogenic bone marrow transplant such as psoriasis, thyroiditis, myasthenia gravis, insulin-dependent diabetes mellitus, and vitiligo has been well described.2-4 The causal relationship between allogenic hematopoetic stem cell transplant (HSCT) and subsequent autoimmune disease has been attributed to T cells and passive transfer of autoimmune disease from the donor to the recipient.1
In the case of autologous HSCT, the remission of autoimmune disease thereafter has been attributed to myeloablative conditioning, whereby high-dose chemotherapy eliminates self-reactive lymphocytes, resulting in ablation of peripheral reactive and alloreactive T cells with depletion of thymus and reactive B cells.1
In 1997, Cooley et al5 described 4 patients with autoimmune disease who experienced complete but temporary (up to 26 months) remission following autologous bone marrow or stem cell transplant. A comprehensive review of psoriasis cases that resolved after HSCT published by Kaffenberger et al6 in 2013 cited 6 cases of autologous and 13 cases of allogenic transplant that resulted in resolution of psoriatic disease. Recurrence of disease was witnessed in 3 of 13 allogenic HSCT patients and 5 of 6 autologous HSCT patients. Of note, all 5 of these patients developed recurrent disease within 24 months following autologous HSCT, albeit with more benign disease.6
Similar to those cases presented by Kaffenberger et al,6 our patient’s psoriatic disease was notably mild compared to her disease prior to HSCT. Although an unintended but welcome outcome of multiple myeloma treatment, the nearly quiescent nature of her skin disease following autologous HSCT has led to a dramatic improvement in her quality of life.
We highlight a unique case in which psoriatic skin disease recurred more than 1 decade after autologous HSCT.
To the Editor:
Remission of psoriasis after bone marrow transplant has been reported1; however, follow-up typically has been limited. We describe a case of a remote recurrence of psoriasis 13 years after autologous stem cell transplant.
In September 1997, a 48-year-old woman with a 20-year history of moderate to severe plaque psoriasis, previously managed with oral methotrexate (up to 30–40 mg weekly), presented to her primary care physician with a persistent cough and newly pervasive arthralgia and myalgia. A complete blood cell count showed a white blood cell count of 6.9 mg/dL (reference range, 3.8–10.8 mg/dL), hemoglobin level of 11.6 mg/dL (reference range, 11.7–15.5 mg/dL), and hematocrit level of 34% (reference range, 35.0%–45.0%). Bone survey demonstrated a 4×2-cm lytic expansile lesion in the occipital bone, and a bone marrow aspirate and biopsy revealed 40% plasma cells. She was subsequently diagnosed with stage IIIA IgA λ and free λ light chain multiple myeloma with an IgA level of 62.1 g/L (reference range, 1.24–2.17 g/L).
Therapy was initiated with dexamethasone in combination with pamidronate and she was transitioned to cyclophosphamide chemotherapy. A peripheral autologous blood stem cell transplant followed. Throughout her initial treatment, the psoriasis continued to flare and she remained on methotrexate to help maintain control of the skin disease.
In November 1998, the patient underwent an autologous CD34+ stem cell transplant. There were no complications following the transplant and the multiple myeloma has remained in complete remission to date. Following the stem cell transplant, the patient remained free of psoriatic disease for just over 13 years without any intervention. Recurrent erythematous plaques with thick scale on the bilateral lower legs prompted her return to dermatology in August 2011. To date, she continues to note intermittent though mild flares of her psoriasis. She has responded well to methotrexate 7.5 mg weekly and clobetasol propionate ointment 0.05% as needed with satisfactory control of her disease.
The development of autoimmune disease following an allogenic bone marrow transplant such as psoriasis, thyroiditis, myasthenia gravis, insulin-dependent diabetes mellitus, and vitiligo has been well described.2-4 The causal relationship between allogenic hematopoetic stem cell transplant (HSCT) and subsequent autoimmune disease has been attributed to T cells and passive transfer of autoimmune disease from the donor to the recipient.1
In the case of autologous HSCT, the remission of autoimmune disease thereafter has been attributed to myeloablative conditioning, whereby high-dose chemotherapy eliminates self-reactive lymphocytes, resulting in ablation of peripheral reactive and alloreactive T cells with depletion of thymus and reactive B cells.1
In 1997, Cooley et al5 described 4 patients with autoimmune disease who experienced complete but temporary (up to 26 months) remission following autologous bone marrow or stem cell transplant. A comprehensive review of psoriasis cases that resolved after HSCT published by Kaffenberger et al6 in 2013 cited 6 cases of autologous and 13 cases of allogenic transplant that resulted in resolution of psoriatic disease. Recurrence of disease was witnessed in 3 of 13 allogenic HSCT patients and 5 of 6 autologous HSCT patients. Of note, all 5 of these patients developed recurrent disease within 24 months following autologous HSCT, albeit with more benign disease.6
Similar to those cases presented by Kaffenberger et al,6 our patient’s psoriatic disease was notably mild compared to her disease prior to HSCT. Although an unintended but welcome outcome of multiple myeloma treatment, the nearly quiescent nature of her skin disease following autologous HSCT has led to a dramatic improvement in her quality of life.
We highlight a unique case in which psoriatic skin disease recurred more than 1 decade after autologous HSCT.
1. Braiteh F, Hymes SR, Giralt SA, et al. Complete remission of psoriasis after autologous hematopoietic stem-cell transplantation for multiple myeloma. J Clin Oncol. 2008;26:4511-4513.
2. Kishimoto Y, Yamamoto Y, Matsumoto N, et al. Transfer of autoimmune thyroiditis and resolution of palmoplantar pustular psoriasis following allogeneic bone marrow transplantation. Bone Marrow Transpl. 1997;19:1041-1043.
3. Wahie S, Alexandroff A, Reynolds NY, et al. Psoriasis occurring after myeloablative therapy and autologous stem cell transplantation. Br J Dermatol. 2006;154:177-204.
4. Snowden JA, Heaton DC. Development of psoriasis after syngenic bone marrow transplant from psoriatic donor: further evidence for adoptive autoimmunity. Br J Dermatol. 1997;137:130-132.
5. Cooley HM, Snowden JA, Grigg AP, et al. Outcome of rheumatoid arthritis and psoriasis following autologous stem cell transplantation for hematologic malignancy. Arthritis Rheum. 1997;40:1712-1715.
6. Kaffenberger BH, Wong HK, Jarjour W, et al. Remission of psoriasis after allogenic but not autologous, hematopoietic stem-cell transplantation. J Am Acad Dermatol. 2013;68:489-492.
1. Braiteh F, Hymes SR, Giralt SA, et al. Complete remission of psoriasis after autologous hematopoietic stem-cell transplantation for multiple myeloma. J Clin Oncol. 2008;26:4511-4513.
2. Kishimoto Y, Yamamoto Y, Matsumoto N, et al. Transfer of autoimmune thyroiditis and resolution of palmoplantar pustular psoriasis following allogeneic bone marrow transplantation. Bone Marrow Transpl. 1997;19:1041-1043.
3. Wahie S, Alexandroff A, Reynolds NY, et al. Psoriasis occurring after myeloablative therapy and autologous stem cell transplantation. Br J Dermatol. 2006;154:177-204.
4. Snowden JA, Heaton DC. Development of psoriasis after syngenic bone marrow transplant from psoriatic donor: further evidence for adoptive autoimmunity. Br J Dermatol. 1997;137:130-132.
5. Cooley HM, Snowden JA, Grigg AP, et al. Outcome of rheumatoid arthritis and psoriasis following autologous stem cell transplantation for hematologic malignancy. Arthritis Rheum. 1997;40:1712-1715.
6. Kaffenberger BH, Wong HK, Jarjour W, et al. Remission of psoriasis after allogenic but not autologous, hematopoietic stem-cell transplantation. J Am Acad Dermatol. 2013;68:489-492.
