User login
Phototoxic Contact Dermatitis From Over-the-counter 8-Methoxypsoralen
To the Editor:
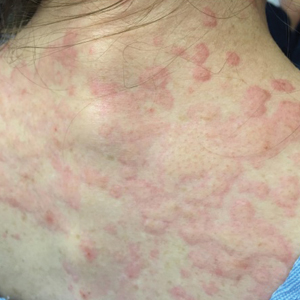
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.

The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
To the Editor:
A 71-year-old Hispanic man with a history of vitiligo presented with an acute-onset blistering rash on the face, arms, and hands. Physical examination demonstrated photodistributed erythematous plaques with overlying vesicles and erosions with hemorrhagic crust on the face, neck, dorsal aspects of the hands, and wrists (Figure). Further history revealed that the patient applied a new cream that was recommended to treat vitiligo the night before the rash onset; he obtained the cream from a Central American market without a prescription. He had gone running in the park without any form of sun protection and then developed the rash within several hours. He denied taking any other medications or supplements. The involvement of sun-protected areas (ie, upper eyelids, nasolabial folds, submental area) was explained when the patient further elaborated that he had performed supine exercises during his outdoor recreation. He brought his new cream into the clinic, which was found to contain prescription-strength methoxsalen (8-methoxypsoralen), confirming the diagnosis of acute phototoxic contact dermatitis. The acute reaction had subsided, and the patient already had discontinued the causative agent. He was counseled on further avoidance of the cream and sun-protective measures.
The photosensitizing properties of certain compounds have been harnessed for therapeutic purposes. For example, psoralen plus UVA therapy has been used for psoriasis and vitiligo and photodynamic therapy for actinic keratoses and superficial nonmelanoma skin cancers.1 However, these agents can induce severe phototoxicity if UV light exposure is not carefully monitored, as seen in our patient. This case is a classic example of phototoxic contact dermatitis and highlights the importance of obtaining a detailed patient history to allow for proper diagnosis and identification of the causative agent. Importantly, because prescription-strength topical medications are readily available over-the-counter, particularly in stores specializing in international goods, patients should be questioned about the use of all topical and systemic medications, both prescription and nonprescription.2
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
- Richard EG. The science and (lost) art of psoralen plus UVA phototherapy. Dermatol Clin. 2020;38:11-23. doi:10.1016/j.det.2019.08.002
- Kimyon RS, Schlarbaum JP, Liou YL, et al. Prescription-strengthtopical corticosteroids available over the counter: cross-sectional study of 80 stores in 13 United States cities. J Am Acad Dermatol. 2020;82:524-525. doi:10.1016/j.jaad.2019.10.035
Practice Points
- Phototoxic contact dermatitis is an irritant reaction resembling an exaggerated sunburn that occurs with the use of a photosensitizing agent and UV light exposure.
- A range of topical and systemic medications, plants, and natural products can elicit phototoxic reactions.
- With the wide availability of prescription-strength over-the-counter medications, a detailed history often is necessary to identify the causative agents of phototoxic contact dermatitis and ensure future avoidance.

Pencil-core Granuloma Forming 62 Years After Initial Injury
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.

An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
.")
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.
An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
To the Editor:
Trauma from a pencil tip can sometimes result in a fragment of lead being left embedded within the skin. Pencil lead is composed of 66% graphite carbon, 26% aluminum silicate, and 8% paraffin.1,2 While the toxicity of these individual elements is low, paraffin can cause nonallergic foreign-body reactions, aluminum silicate can induce epithelioid granulomatous reactions, and graphite has been reported to cause chronic granulomatous reactions in the lungs of those who work with graphite.2 Penetrating trauma with a pencil can result in the formation of a cutaneous granulomatous reaction that can sometimes occur years to decades after the initial injury.3,4 Several cases of pencil-core granulomas have been published, with lag times between the initial trauma and lesion growth as long as 58 years.1-10 The pencil-core granuloma may simulate malignant melanoma, as it presents clinically as a growing, darkly pigmented lesion, thus prompting biopsy. We present a case of a pencil-core granuloma that began to grow 62 years after the initial trauma.
A 72-year-old woman was referred to our clinic for evaluation of a dark nodule on the forehead. The lesion had been present since the age of 10 years, reportedly from an accidental stabbing with a pencil. The lesion had been flat, stable, and asymptomatic since the trauma occurred; however, the patient reported that approximately 9 months prior to presentation, it had started growing and became painful. Physical examination revealed a 1.0-cm, round, bluish-black nodule on the right superior forehead (Figure 1A). No satellite lesions or local lymphadenopathy were noted on general examination.
An elliptical excision of the lesion with 1-cm margins revealed a bluish-black mass extending through the dermis, through the frontalis muscle, and into the periosteum and frontal bone (Figure 1B). A No. 15 blade was then used to remove the remaining pigment from the outer table of the frontal bone. Histopathologic findings demonstrated a sarcoidal granulomatous dermatitis associated with abundant, nonpolarizable, black, granular pigment consistent with carbon tattoo. This foreign material was readily identifiable in large extracellular deposits and also within histiocytes, including numerous multinucleated giant cells (Figure 2). Immunostaining for MART-1 and SOX-10 antigens failed to demonstrate a melanocytic proliferation. These findings were consistent with a sarcoidal foreign-body granulomatous reaction to carbon tattoo following traumatic graphite implantation.
Granulomatous reactions to carbon tattoo may be sarcoidal (foreign-body granulomatous dermatitis), palisading, or rarely tuberculoid (caseating). Sarcoidal granulomatous tattoo reactions may occur in patients with sarcoidosis due to koebnerization, and histology alone is not discriminatory; however, in our patient, the absence of underlying sarcoidosis or clinical or histologic findings of sarcoidosis outside of the site of the pencil-core granuloma excluded that possibility.11 Pencil-core granulomas are characterized by a delayed foreign-body reaction to retained fragments of lead often years following a penetrating trauma with a pencil. Previous reports have described various lag times from injury to lesion growth of up to 58 years.1-10 Our patient claimed to have noticed the lesion growing and becoming painful only after a 62-year lag time following the initial trauma. To our knowledge, this is the longest lag time between the initial pencil injury and induction of the foreign-body reaction reported in the literature. Clinically, the lesion appeared and behaved very similar to a melanoma, prompting further treatment and evaluation.
It has been suggested that the lag period between the initial trauma and the rapid growth of the lesion may correspond to the amount of time required for the breakdown of the pencil lead to a critical size followed by the dispersal of those particles within the interstitium, where they can induce a granulomatous reaction.1,2,9 One case described a patient who reported that the growth and clinical change of the pencil-core granuloma only started when the patient accidentally hit the area where the trauma had occurred 31 years prior.1 This additional trauma may have caused further mechanical breakdown of the lead to set off the tissue reaction. In our case, the patient did not recall any additional trauma to the head prior to the onset of growth of the nodule on the forehead.
Our case indicates that carbon tattoo may be a possible sequela of a penetrating injury from a pencil with retained pencil lead fragments; however, many of these carbon tattoos may remain stable throughout the remainder of the patient’s life.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
- Gormley RH, Kovach SJ III, Zhang PJ. Role for trauma in inducing pencil “lead” granuloma in the skin. J Am Acad Dermatol. 2010;62:1074-1075.
- Terasawa N, Kishimoto S, Kibe Y, et al. Graphite foreign body granuloma. Br J Dermatol. 1999;141:774-776.
- Fukunaga Y, Hashimoto I, Nakanishi H, et al. Pencil-core granuloma of the face: report of two rare cases. J Plast Reconstr Aesthet Surg. 2011;64:1235-1237.
- Aswani VH, Kim SL. Fifty-three years after a pencil puncture wound. Case Rep Dermatol. 2015;7:303-305.
- Taylor B, Frumkin A, Pitha JV. Delayed reaction to “lead” pencil simulating melanoma. Cutis. 1988;42:199-201.
- Granick MS, Erickson ER, Solomon MP. Pencil-core granuloma. Plast Reconstr Surg. 1992;89:136-138.
- Andreano J. Stump the experts. foreign body granuloma. J Dermatol Surg Oncol. 1992;18:277, 343.
- Yoshitatsu S, Takagi T. A case of giant pencil-core granuloma. J Dermatol. 2000;27:329-332.
- Hatano Y, Asada Y, Komada S, et al. A case of pencil core granuloma with an unusual temporal profile. Dermatology. 2000;201:151-153.
- Seitz IA, Silva BA, Schechter LS. Unusual sequela from a pencil stab wound reveals a retained graphite foreign body. Pediatr Emerg Care. 2014;30:568-570.
- Motaparthi K. Tattoo ink. In: Cockerell CJ, Hall BJ, eds. Nonneoplastic Dermatopathology. 2nd ed. Amirsys; 2016: 270.
Practice Points
- Pencil-core granulomas can arise even decades after the lead is embedded in the skin.
- It is important to biopsy to confirm the diagnosis, as pencil-core granulomas can very closely mimic melanomas.

Lower Leg Hyperpigmentation in MYH9-Related Disorder
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.

Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.
Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
To the Editor:
MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.1 We describe a case of lower leg hyperpigmentation secondary to hemosiderin deposition from MYH9-related disorder.
A 31-year-old woman with a history of MYH9-related disorder and mixed connective tissue disease presented to the outpatient dermatology clinic with asymptomatic brown patches on the lower legs (Figure) of 10 years’ duration. She also had epistaxis, hearing loss, renal disease, and menorrhagia secondary to MYH9-related disorder. The patient had been started on hydroxychloroquine 2 years earlier by rheumatology for mixed connective tissue disorder. A biopsy was not performed, given the risk of bleeding from thrombocytopenia. Ammonium lactate lotion was recommended for the leg patches. No further interventions were undertaken. At 6-month follow-up, hyperpigmentation on the lower legs was stable. The patient expressed no desire for cosmetic intervention.
Prior to discovery of a common gene, MYH9-related disorder was classified as 4 overlapping syndromes: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.2 More than 30 MYH9 mutations have been identified, all of which encode for myosin-9, a subunit of myosin IIA,1,3 that is a nonmuscle myosin needed for cell movement, shape, and cytokinesis. Although most cells use myosin IIA to IIC, certain cells, such as platelets and neutrophils, use myosin IIA exclusively.
In neutrophils of patients with MYH9-related disorder, nonfunctional myosin-9 clumps to form hallmark inclusion bodies, which are seen on the peripheral blood smear. Macrothrombocytopenia, another hallmark of MYH9-related disorder, also can be seen on the peripheral smear of all affected patients. Approximately 30%of patients develop clinical manifestations of the disorder (eg, bleeding, renal failure, hearing loss, presenile cataracts). Bleeding tendency usually is mild; epistaxis and menorrhagia are the most common hematologic manifestations.4
We attribute the lower leg hyperpigmentation in our patient to a severe phenotype of MYH9-related disorder. In addition to hyperpigmentation, our patient had menorrhagia requiring treatment with tranexamic acid, renal failure, and hearing loss, further pointing to a more severe phenotype. Furthermore, it is likely that our patient’s hyperpigmentation was made worse by hydroxychloroquine and a coexisting diagnosis of mixed connective tissue disease, which led to a propensity for increased vessel fragility in the setting of thrombocytopenia.
The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder. The presence of small red blood cells (RBCs) in microcytic anemia and large platelets of MYH9-related disorder can lead to a situation in which platelets travel near the center of the lumen of blood vessels, while RBCs travel to the periphery. This decrease in the platelet-endothelium interaction increases the risk for bleeding. Our patient’s hemoglobin level was within reference range, without evidence of iron-deficiency anemia. Correction of iron-deficiency anemia, if applicable, can prevent bleeding brought on by the mechanism of decreased platelet-endothelium interaction and avoid unnecessary antiplatelet medication because of misdiagnosis based on an erroneous platelet count.
The workup of MYH9-related disorder also should include audiography, ophthalmologic examination, and renal function testing for hearing loss, cataracts, and renal disease, respectively. Referral to genetics also may be warranted.
It also is of clinical interest that automated cell counters may underestimate the count of abnormally large platelets in MYH9-related disorder, counting them as RBCs or white blood cells. The platelet count in MYH9-related disorder may be underestimated by 4-fold or greater.4-7
Treatment of leg hyperpigmentation can prove challenging, given the location of dermal hemosiderin. Topical therapy likely is ineffective. Lasers and intense pulsed light therapy are treatment modalities to consider for the hyperpigmentation of MYH9-related disorder. There have been reports of improved cosmesis in dermal hemosiderin depositional disorders, such as venous stasis.4 Our patient was given ammonium lactate lotion to thicken collagen, possibly preventing future bleeding episodes.
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
- Pecci A, Canobbio I, Balduini A, et al. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14:3169-3178. doi:10.1093/hmg/ddi344
- Seri M, Pecci A, Di Bari F, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore). 2003;82:203-215. doi:10.1097/01.md.0000076006.64510.5c
- Medline Plus. MYH9-related disorder. National Library of Medicine website. Updated August 18, 2020. Accessed January 21, 2022. https://ghr.nlm.nih.gov/condition/myh9-related-disorder#diagnosis
- Althaus K, Greinachar A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. doi:10.1055/s-0029-1220327
- Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell-specific regulation mechanisms in MYH9 disorders. Blood. 2008;111:3015-3023. doi:10.1182/blood-2007-10-116194
- Arrondel C, Vodovar N, Knebelmann B, et al. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65-74. doi:10.1681/ASN.V13165
- Selleng K, Lubenow LE, Greinacher A, et al. Perioperative management of MYH9 hereditary macrothrombocytopenia (Fechtner syndrome). Eur J Haematol. 2007;79:263-268. doi:10.1111/j.1600-0609.2007.00913.x
Practice Points
- MYH9-related disorder is an autosomal-dominant disorder characterized by macrothrombocytopenia and neutrophil inclusions secondary to defective myosin-9.
- Leg hyperpigmentation can occur secondary to hemosiderin deposition from MYH9-related disorder.
- The workup of suspected MYH9-related disorder includes exclusion of iron-deficiency anemia, which can increase bleeding in patients with the disorder.
- Lasers and intense pulsed light therapy are modalities to consider for the hyperpigmentation of MYH9- related disorder.

Scleral Plaques in Nephrogenic Systemic Fibrosis
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.

Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2

- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
Practice Points
- It is important to examine the eyes in a patient with sclerotic skin changes on physical examination.
- The presence of yellow scleral plaques strongly is associated with a diagnosis of nephrogenic systemic fibrosis.

Febrile Ulceronecrotic Mucha-Habermann Disease: A Rare Form of Pityriasis Lichenoides et Varioliformis Acuta
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.

A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.

The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.

At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.

Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
Practice Points
- Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare variant of pityriasis lichenoides et varioliformis acuta, characterized by ulceronecrotic lesions, fever, and systemic symptoms.
- A variety of treatments including immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been used in patients with FUMHD.

Blastomycosislike Pyoderma: Verrucous Hyperpigmented Plaques on the Pretibial Shins
To the Editor:
Blastomycosislike pyoderma (BLP), also commonly referred to as pyoderma vegetans, is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 It is characterized by a collection of neutrophilic abscesses with pseudoepitheliomatous hyperplasia that coalesce into crusted plaques.
A 15-year-old adolescent girl with a history of type 1 diabetes mellitus was admitted for diabetic ketoacidosis. The patient presented with bilateral pretibial lesions of 6 years’ duration that developed after swimming in a pool following reported trauma to the site. These pruritic plaques had grown slowly and were occasionally tender. Of note, with episodes of hyperglycemia, the lesions developed purulent drainage.
Upon admission to the hospital and subsequent dermatology consultation, physical examination revealed the right pretibial shin had a 15×5-cm, gray-brown, hyperpigmented, verrucous, tender plaque with purulent drainage and overlying crust (Figure 1). The left pretibial shin had a similar smaller lesion (Figure 2). Laboratory test results were notable for a white blood cell count of 41.84 cells/µL (reference range, 3.8–10.5 cells/µL), blood glucose level of 586 mg/dL (reference range, 70–99 mg/dL), and hemoglobin A1c of 11.7% (reference range, 4.0%–5.6%). A biopsy specimen from the right pretibial shin was stained with hematoxylin and eosin for dermatopathologic evaluation as well as sent for tissue culture. Tissue and wound cultures grew Staphylococcus aureus and group B Streptococcus with no fungal or acid-fast bacilli growth.
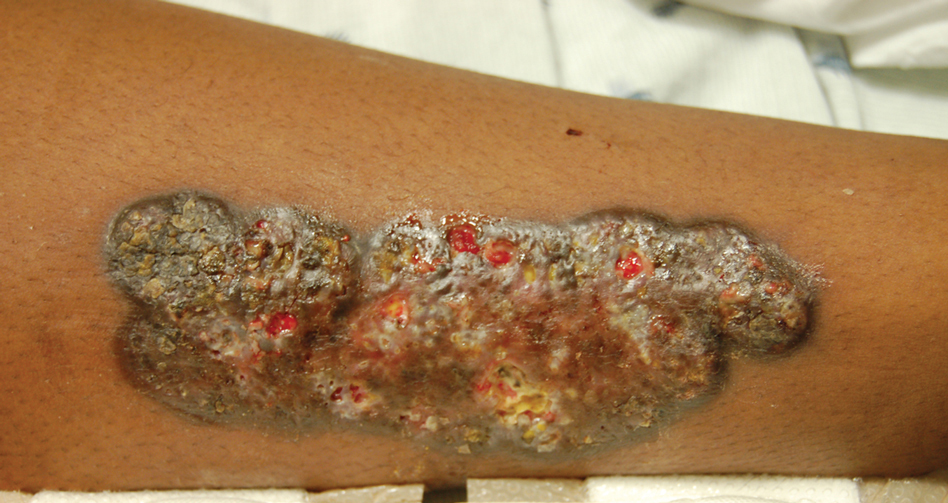
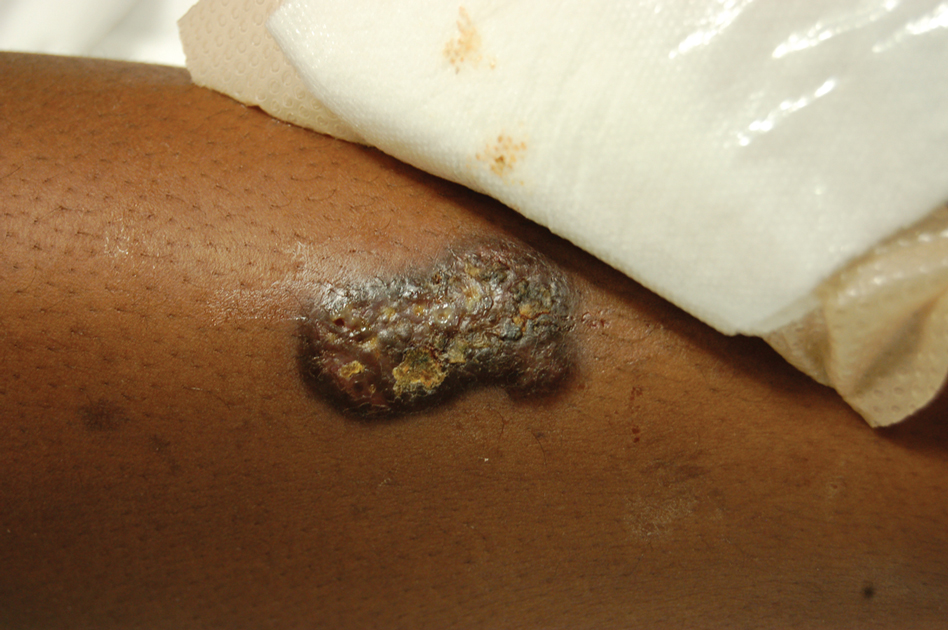
Blood cultures were negative for bacteria. Results of radiographic imaging were negative for osteomyelitis. Biopsy specimens from the right pretibial plaque showed a markedly inflamed, ruptured follicular unit with a dense dermal lympho-neutrophilic infiltrate and overlying pseudoepitheliomatous hyperplasia (Figure 3). Periodic acid–Schiff, Gomori methenamine-silver, acid-fast bacilli, and Giemsa stains were negative for organisms. No granules consistent with a Splendore-Hoeppli phenomenon were observed. These observations were consistent with a diagnosis of BLP.
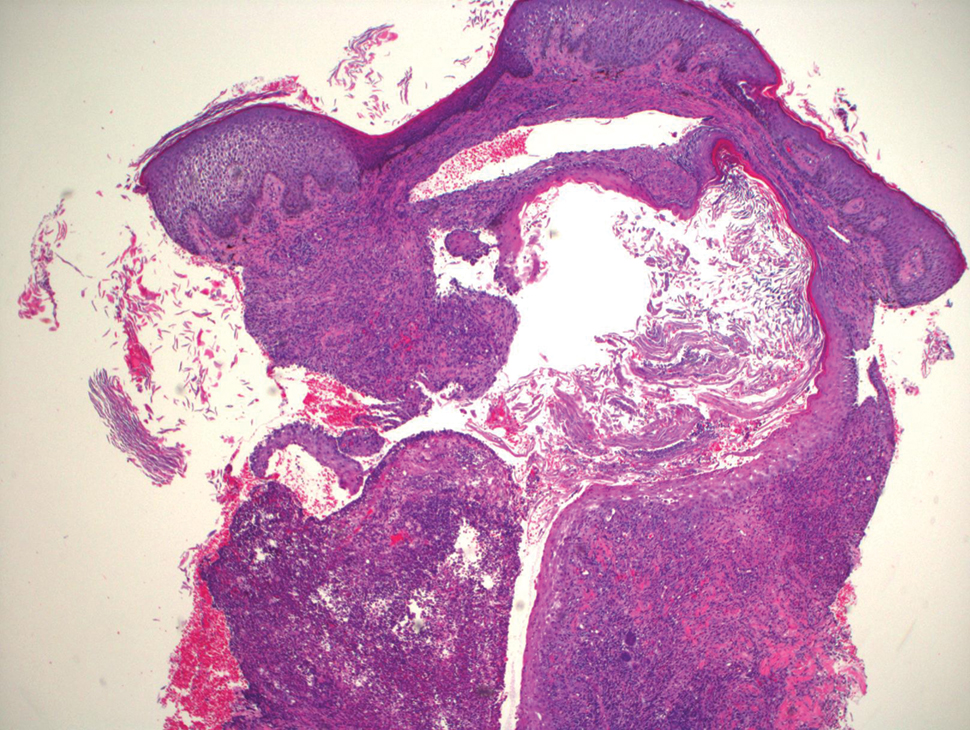
Blastomycosislike pyoderma is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 Pediatric cases also are uncommon. Blastomycosislike pyoderma most commonly is caused by infection with S aureus or group A streptococci, but several other organisms have been implicated.2 Clinically, BLP is similar to cutaneous botryomycosis, as both are caused by similar organisms.3 However, while BLP is limited to the skin, botryomycosis may involve visceral organs.
Blastomycosislike pyoderma typically presents as verrucous, hyperkeratotic, purulent plaques with raised borders. It most commonly occurs on the face, scalp, axillae, trunk, and distal extremities. Predisposing factors include immunosuppressed states such as poor nutrition, HIV, malignancy, alcoholism, and diabetes mellitus.3,4 Hyperglycemia is thought to suppress helper T cell (TH1)–dependent immunity, which may explain why our patient’s lesions worsened with hyperglycemic episodes.5Histopathology revealed pseudoepitheliomatous hyperplasia with neutrophilic abscesses.1 The distinguishing feature between botryomycosis and BLP is the development of grains known as the Splendore-Hoeppli phenomenon in botryomycosis.6 The grains are eosinophilic and contain the causative infectious agent. The presence of these grains is consistent with botryomycosis but is not pathognomonic, as it also can be found in several bacterial, fungal, and parasitic infections.3,6
The differential diagnosis of BLP includes atypical mycobacterial infection, pyoderma gangrenosum, fungal infection, and tuberculosis verrucosa cutis.7
Although BLP is caused by bacteria, response to systemic antibiotics is variable. Other treatment modalities include dapsone, systemic and intralesional corticosteroids, retinoids, debridement, CO2 laser, and excision.6,8 Lesions typically start out localized, but it is not uncommon for them to spread to distal or vulnerable tissue, such as sites of trauma or inflammation. Our patient was started on oral trimethoprim-sulfamethoxazole and showed improvement, but she worsened with subsequent hyperglycemic episodes when antibiotics were discontinued.
1. Adis¸en E, Tezel F, Gürer MA. Pyoderma vegetans: a case for discussion. Acta Derm Venereol. 2009;89:186-188.
2. Scuderi S, O’Brien B, Robertson I, et al. Heterogeneity of blastomycosis-like pyoderma: a selection of cases from the last 35 years. Australas J Dermatol. 2017;58:139-141.
3. Marschalko, M. Pyoderma vegetans: report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol Alp Pannonica Adriat. 1995;4:55-59.
4. Cerullo L, Zussman J, Young L. An unusual presentation of blastomycosislike pyoderma (pyoderma vegetans) and a review of the literature. Cutis. 2009;84:201-204.
5. Tanaka Y. Immunosuppressive mechanisms in diabetes mellitus [in Japanese]. Nihon Rinsho. 2008;66:2233-2237.
6. Hussein MR. Mucocutaneous Splendore-Hoeppli phenomenon. J Cutan Pathol. 2008;35:979-988.
7. Lee YS, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
8. Kobraei KB, Wesson SK. Blastomycosis-like pyoderma: response to systemic retinoid therapy. Int J Dermatol. 2010;49:1336-1338.
To the Editor:
Blastomycosislike pyoderma (BLP), also commonly referred to as pyoderma vegetans, is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 It is characterized by a collection of neutrophilic abscesses with pseudoepitheliomatous hyperplasia that coalesce into crusted plaques.
A 15-year-old adolescent girl with a history of type 1 diabetes mellitus was admitted for diabetic ketoacidosis. The patient presented with bilateral pretibial lesions of 6 years’ duration that developed after swimming in a pool following reported trauma to the site. These pruritic plaques had grown slowly and were occasionally tender. Of note, with episodes of hyperglycemia, the lesions developed purulent drainage.
Upon admission to the hospital and subsequent dermatology consultation, physical examination revealed the right pretibial shin had a 15×5-cm, gray-brown, hyperpigmented, verrucous, tender plaque with purulent drainage and overlying crust (Figure 1). The left pretibial shin had a similar smaller lesion (Figure 2). Laboratory test results were notable for a white blood cell count of 41.84 cells/µL (reference range, 3.8–10.5 cells/µL), blood glucose level of 586 mg/dL (reference range, 70–99 mg/dL), and hemoglobin A1c of 11.7% (reference range, 4.0%–5.6%). A biopsy specimen from the right pretibial shin was stained with hematoxylin and eosin for dermatopathologic evaluation as well as sent for tissue culture. Tissue and wound cultures grew Staphylococcus aureus and group B Streptococcus with no fungal or acid-fast bacilli growth.
Blood cultures were negative for bacteria. Results of radiographic imaging were negative for osteomyelitis. Biopsy specimens from the right pretibial plaque showed a markedly inflamed, ruptured follicular unit with a dense dermal lympho-neutrophilic infiltrate and overlying pseudoepitheliomatous hyperplasia (Figure 3). Periodic acid–Schiff, Gomori methenamine-silver, acid-fast bacilli, and Giemsa stains were negative for organisms. No granules consistent with a Splendore-Hoeppli phenomenon were observed. These observations were consistent with a diagnosis of BLP.
Blastomycosislike pyoderma is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 Pediatric cases also are uncommon. Blastomycosislike pyoderma most commonly is caused by infection with S aureus or group A streptococci, but several other organisms have been implicated.2 Clinically, BLP is similar to cutaneous botryomycosis, as both are caused by similar organisms.3 However, while BLP is limited to the skin, botryomycosis may involve visceral organs.
Blastomycosislike pyoderma typically presents as verrucous, hyperkeratotic, purulent plaques with raised borders. It most commonly occurs on the face, scalp, axillae, trunk, and distal extremities. Predisposing factors include immunosuppressed states such as poor nutrition, HIV, malignancy, alcoholism, and diabetes mellitus.3,4 Hyperglycemia is thought to suppress helper T cell (TH1)–dependent immunity, which may explain why our patient’s lesions worsened with hyperglycemic episodes.5Histopathology revealed pseudoepitheliomatous hyperplasia with neutrophilic abscesses.1 The distinguishing feature between botryomycosis and BLP is the development of grains known as the Splendore-Hoeppli phenomenon in botryomycosis.6 The grains are eosinophilic and contain the causative infectious agent. The presence of these grains is consistent with botryomycosis but is not pathognomonic, as it also can be found in several bacterial, fungal, and parasitic infections.3,6
The differential diagnosis of BLP includes atypical mycobacterial infection, pyoderma gangrenosum, fungal infection, and tuberculosis verrucosa cutis.7
Although BLP is caused by bacteria, response to systemic antibiotics is variable. Other treatment modalities include dapsone, systemic and intralesional corticosteroids, retinoids, debridement, CO2 laser, and excision.6,8 Lesions typically start out localized, but it is not uncommon for them to spread to distal or vulnerable tissue, such as sites of trauma or inflammation. Our patient was started on oral trimethoprim-sulfamethoxazole and showed improvement, but she worsened with subsequent hyperglycemic episodes when antibiotics were discontinued.
To the Editor:
Blastomycosislike pyoderma (BLP), also commonly referred to as pyoderma vegetans, is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 It is characterized by a collection of neutrophilic abscesses with pseudoepitheliomatous hyperplasia that coalesce into crusted plaques.
A 15-year-old adolescent girl with a history of type 1 diabetes mellitus was admitted for diabetic ketoacidosis. The patient presented with bilateral pretibial lesions of 6 years’ duration that developed after swimming in a pool following reported trauma to the site. These pruritic plaques had grown slowly and were occasionally tender. Of note, with episodes of hyperglycemia, the lesions developed purulent drainage.
Upon admission to the hospital and subsequent dermatology consultation, physical examination revealed the right pretibial shin had a 15×5-cm, gray-brown, hyperpigmented, verrucous, tender plaque with purulent drainage and overlying crust (Figure 1). The left pretibial shin had a similar smaller lesion (Figure 2). Laboratory test results were notable for a white blood cell count of 41.84 cells/µL (reference range, 3.8–10.5 cells/µL), blood glucose level of 586 mg/dL (reference range, 70–99 mg/dL), and hemoglobin A1c of 11.7% (reference range, 4.0%–5.6%). A biopsy specimen from the right pretibial shin was stained with hematoxylin and eosin for dermatopathologic evaluation as well as sent for tissue culture. Tissue and wound cultures grew Staphylococcus aureus and group B Streptococcus with no fungal or acid-fast bacilli growth.
Blood cultures were negative for bacteria. Results of radiographic imaging were negative for osteomyelitis. Biopsy specimens from the right pretibial plaque showed a markedly inflamed, ruptured follicular unit with a dense dermal lympho-neutrophilic infiltrate and overlying pseudoepitheliomatous hyperplasia (Figure 3). Periodic acid–Schiff, Gomori methenamine-silver, acid-fast bacilli, and Giemsa stains were negative for organisms. No granules consistent with a Splendore-Hoeppli phenomenon were observed. These observations were consistent with a diagnosis of BLP.
Blastomycosislike pyoderma is a rare cutaneous bacterial infection that often mimics other fungal, inflammatory, or neoplastic disorders.1 Pediatric cases also are uncommon. Blastomycosislike pyoderma most commonly is caused by infection with S aureus or group A streptococci, but several other organisms have been implicated.2 Clinically, BLP is similar to cutaneous botryomycosis, as both are caused by similar organisms.3 However, while BLP is limited to the skin, botryomycosis may involve visceral organs.
Blastomycosislike pyoderma typically presents as verrucous, hyperkeratotic, purulent plaques with raised borders. It most commonly occurs on the face, scalp, axillae, trunk, and distal extremities. Predisposing factors include immunosuppressed states such as poor nutrition, HIV, malignancy, alcoholism, and diabetes mellitus.3,4 Hyperglycemia is thought to suppress helper T cell (TH1)–dependent immunity, which may explain why our patient’s lesions worsened with hyperglycemic episodes.5Histopathology revealed pseudoepitheliomatous hyperplasia with neutrophilic abscesses.1 The distinguishing feature between botryomycosis and BLP is the development of grains known as the Splendore-Hoeppli phenomenon in botryomycosis.6 The grains are eosinophilic and contain the causative infectious agent. The presence of these grains is consistent with botryomycosis but is not pathognomonic, as it also can be found in several bacterial, fungal, and parasitic infections.3,6
The differential diagnosis of BLP includes atypical mycobacterial infection, pyoderma gangrenosum, fungal infection, and tuberculosis verrucosa cutis.7
Although BLP is caused by bacteria, response to systemic antibiotics is variable. Other treatment modalities include dapsone, systemic and intralesional corticosteroids, retinoids, debridement, CO2 laser, and excision.6,8 Lesions typically start out localized, but it is not uncommon for them to spread to distal or vulnerable tissue, such as sites of trauma or inflammation. Our patient was started on oral trimethoprim-sulfamethoxazole and showed improvement, but she worsened with subsequent hyperglycemic episodes when antibiotics were discontinued.
1. Adis¸en E, Tezel F, Gürer MA. Pyoderma vegetans: a case for discussion. Acta Derm Venereol. 2009;89:186-188.
2. Scuderi S, O’Brien B, Robertson I, et al. Heterogeneity of blastomycosis-like pyoderma: a selection of cases from the last 35 years. Australas J Dermatol. 2017;58:139-141.
3. Marschalko, M. Pyoderma vegetans: report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol Alp Pannonica Adriat. 1995;4:55-59.
4. Cerullo L, Zussman J, Young L. An unusual presentation of blastomycosislike pyoderma (pyoderma vegetans) and a review of the literature. Cutis. 2009;84:201-204.
5. Tanaka Y. Immunosuppressive mechanisms in diabetes mellitus [in Japanese]. Nihon Rinsho. 2008;66:2233-2237.
6. Hussein MR. Mucocutaneous Splendore-Hoeppli phenomenon. J Cutan Pathol. 2008;35:979-988.
7. Lee YS, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
8. Kobraei KB, Wesson SK. Blastomycosis-like pyoderma: response to systemic retinoid therapy. Int J Dermatol. 2010;49:1336-1338.
1. Adis¸en E, Tezel F, Gürer MA. Pyoderma vegetans: a case for discussion. Acta Derm Venereol. 2009;89:186-188.
2. Scuderi S, O’Brien B, Robertson I, et al. Heterogeneity of blastomycosis-like pyoderma: a selection of cases from the last 35 years. Australas J Dermatol. 2017;58:139-141.
3. Marschalko, M. Pyoderma vegetans: report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol Alp Pannonica Adriat. 1995;4:55-59.
4. Cerullo L, Zussman J, Young L. An unusual presentation of blastomycosislike pyoderma (pyoderma vegetans) and a review of the literature. Cutis. 2009;84:201-204.
5. Tanaka Y. Immunosuppressive mechanisms in diabetes mellitus [in Japanese]. Nihon Rinsho. 2008;66:2233-2237.
6. Hussein MR. Mucocutaneous Splendore-Hoeppli phenomenon. J Cutan Pathol. 2008;35:979-988.
7. Lee YS, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
8. Kobraei KB, Wesson SK. Blastomycosis-like pyoderma: response to systemic retinoid therapy. Int J Dermatol. 2010;49:1336-1338.
Practice Points
- Blastomycosislike pyoderma is a rare condition secondary to bacterial infection, but as the name suggests, it also can resemble cutaneous blastomycosis.
- Blastomycosislike pyoderma most commonly occurs in immunocompromised patients.
- The most common histologic findings include suppurative and neutrophilic inflammation with pseudoepitheliomatous hyperplasia.
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap in a Pregnant Patient
To the Editor:
A 34-year-old pregnant woman at 5 weeks’ gestation was transferred to dermatology from an outside hospital with a full-body rash. Three days after noting a fever and generalized body aches, she developed a painful rash on the legs that had gradually spread to the arms, trunk, and face. Symptoms of eyelid pruritus and edema initially were improved with intravenous (IV) steroids at an emergency department visit, but they started to flare soon thereafter with worsening mucosal involvement and dysphagia. After a second visit to the emergency department and repeat treatment with IV steroids, she was transferred to our institution for a higher level of care.
The patient denied taking any new medications in the 2 months prior to the onset of the rash. Her medication history only consisted of over-the-counter prenatal vitamins, a single use of over-the-counter migraine medication (containing acetaminophen, aspirin, and caffeine as active ingredients), and a possible use of ibuprofen or acetaminophen separately. She reported ocular discomfort and blurriness, dysphagia, dysuria, and vaginal discomfort. Physical examination revealed dusky red to violaceous macules and patches that involved approximately 65% of the body surface area (BSA), with bullae involving approximately 10% BSA. The face was diffusely red and edematous with crusted erosions and scattered bullae on the cheeks. Mucosal involvement was notable for injected conjunctivae and erosions present on the upper hard palate of the mouth and lips (Figure, A). Erythematous macules with dusky centers coalescing into patches with overlying vesicles and bullae were scattered on the arms (Figure, B), hands, trunk (Figure, C), and legs. The Nikolsky sign was positive. The vulva was swollen and covered with erythematous macules with dusky centers.
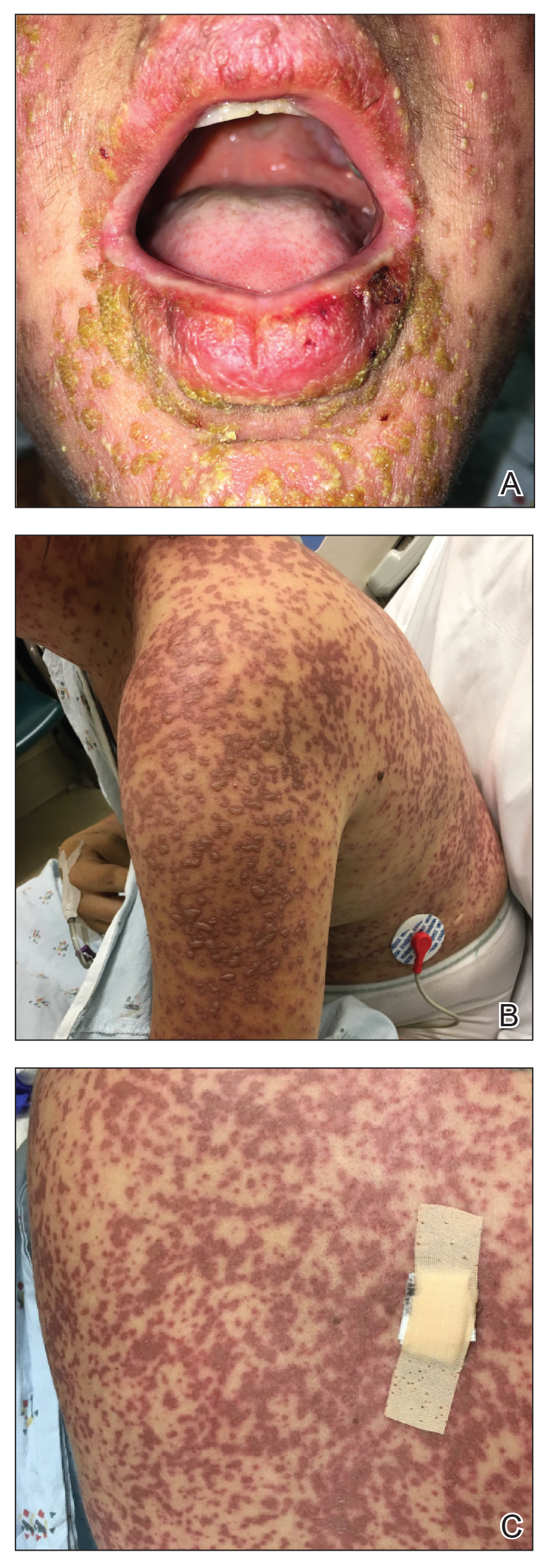
A biopsy from the upper back revealed a vacuolar interface with subepidermal bullae and confluent keratinocyte necrosis with many CD8+ cells and scattered granzyme B. Given these results in conjunction with the clinical findings, a diagnosis of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) overlap was made. In addition to providing supportive care, the patient was started on a 4-day course of IV immunoglobulin (IVIG)(3g/kg total) and prednisone 60 mg daily, tapered over several weeks with a good clinical response. At outpatient follow-up she was found to have postinflammatory hypopigmentation on the face, trunk, and extremities, as well as tear duct scarring, but she had no vulvovaginal scarring or stenosis. She was progressing well in her pregnancy with no serious complications for 4 months after admission, at which point she was lost to follow-up.
Stevens-Johnson syndrome and TEN represent a spectrum of severe mucocutaneous reactions with high morbidity and mortality. Medications are the leading trigger, followed by infection. The most common inciting medications include antibacterial sulfonamides, antiepileptics such as carbamazepine and lamotrigine, nonsteroidal anti-inflammatory drugs, nevirapine, and allopurinol. The onset of symptoms from 1 to 4 weeks combined with characteristic morphologic features helps distinguish SJS/TEN from other drug eruptions. The initial presentation classically consists of a flulike prodrome followed by mucocutaneous eruption. Skin lesions often present as a diffuse erythema or ill-defined, coalescing, erythematous macules with purpuric centers that may evolve into vesicles and bullae with sloughing of the skin. Histopathology reveals full-thickness epidermal necrosis with detachment.1
Erythema multiforme and Mycoplasma-induced rash and mucositis (MIRM) are high on the differential diagnosis. Distinguishing features of erythema multiforme include the morphology of targetoid lesions and a common distribution on the extremities, in addition to the limited bullae and epidermal detachment in comparison with SJS/TEN. In MIRM, mucositis often is more severe and extensive, with multiple mucosal surfaces affected. It typically has less cutaneous involvement than SJS/TEN, though clinical variants can include diffuse rash and affect fewer than 2 mucosal sites.2 Depending on the timing of rash onset, Mycoplasma IgM/IgG titers may be drawn to further support the diagnosis. A diagnosis of MIRM was not favored in our patient due to lack of respiratory symptoms, normal chest radiography, and negative Mycoplasma IgM and IgG titers.
Stevens-Johnson syndrome/toxic epidermal necrolysis overlap has been reported in pregnant patients, typically in association with HIV infection or new medication exposure.3 A combination of genetic susceptibility and an altered immune system during pregnancy may contribute to the pathogenesis, involving a cytotoxic T-cell mediated reaction with release of inflammatory cytokines.1 Interestingly, these factors that may predispose a patient to developing SJS/TEN may not pass on to the neonate, evidenced by a few cases that showed no reaction in the newborn when given the same offending drug.4
Stevens-Johnson syndrome/toxic epidermal necrolysis more frequently presents in the second or third trimester, with no increase in maternal mortality and an equally high survival rate of the fetus.1,5 Unique sequelae in pregnant patients may include vaginal stenosis, vulvar swelling, and postpartum sepsis. Fetal complications can include low birth weight, preterm delivery, and respiratory distress. The fetus rarely exhibits cutaneous manifestations of the disease.6
A multidisciplinary approach to the diagnosis and management of SJS/TEN overlap in special patient populations such as pregnant women is vital. Supportive measures consisting of wound care, fluid and electrolyte management, infection monitoring, and nutritional support have sufficed in treating SJS/TEN in pregnant patients.3 Although adjunctive therapy with systemic corticosteroids, IVIG, cyclosporine, and tumor necrosis factor inhibitors commonly are used in clinical practice, the safety of these treatments in pregnant patients affected by SJS/TEN has not been established. However, use of these medications for other indications, primarily rheumatologic diseases, has been reported to be safe in the pregnant population.7 If necessary, glucocorticoids should be used in the lowest effective dose to avoid complications such as premature rupture of membranes; intrauterine growth restriction; and increased risk for pregnancy-induced hypertension, gestational diabetes, osteoporosis, and infection. Little is known about IVIG use in pregnancy. While it has not been associated with increased risk of fetal malformations, it may cross the placenta in a notable amount when administered after 30 weeks’ gestation.7
Unlike most cases of SJS/TEN in pregnancy that largely were associated with HIV infection or drug exposure, primarily antiretrovirals such as nevirapine or antiepileptics, our case is a rare incidence of SJS/TEN in a pregnant patient with no clear medication or infectious trigger. Although the causative drug was unclear, we suspected it was secondary to nonsteroidal anti-inflammatory drug use. The patient had a SCORTEN (SCORe of Toxic Epidermal Necrosis) of 0, which portends a relatively good prognosis with an estimated mortality rate of approximately 3% (Table).8 However, the large BSA involvement of the morbilliform rash warranted aggressive management to prevent the involved skin from fully detaching.
1. Struck MF, Illert T, Liss Y, et al. Toxic epidermal necrolysis in pregnancy: case report and review of the literature. J Burn Care Res. 2010;31:816-821. doi:10.1097/BCR.0b013e3181eed441
2. Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245.e4. doi:10.1016/j.jaad.2014.06.026
3. Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes in twenty-two consecutive pregnant HIV infected women. PLoS One. 2015;10:1-11. doi:10.1371/journal.pone.0135501
4. Velter C, Hotz C, Ingen-Housz-Oro S. Stevens-Johnson syndrome during pregnancy: case report of a newborn treated with the culprit drug. JAMA Dermatol. 2018;154:224-225. doi:10.1001/jamadermatol.2017.4607
5. El Daief SG, Das S, Ekekwe G, et al. A successful pregnancy outcome after Stevens-Johnson syndrome. J Obstet Gynaecol (Lahore). 2014;34:445-446. doi:10.3109/01443615.2014.914897
6. Rodriguez G, Trent JT, Mirzabeigi M. Toxic epidermal necrolysis in a mother and fetus. J Am Acad Dermatol. 2006;55(5 suppl):96-98. doi:10.1016/j.jaad.2005.09.023
7. Bermas BL. Safety of rheumatic disease medication use during pregnancy and lactation. UptoDate website. Updated March 24, 2021. Accessed December 16, 2021. https://www.uptodate.com/contents/safety-of-rheumatic-disease-medication-use-during-pregnancy-and-lactation#H11
8. Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153. doi:10.1046/j.1523-1747.2000.00061.x
To the Editor:
A 34-year-old pregnant woman at 5 weeks’ gestation was transferred to dermatology from an outside hospital with a full-body rash. Three days after noting a fever and generalized body aches, she developed a painful rash on the legs that had gradually spread to the arms, trunk, and face. Symptoms of eyelid pruritus and edema initially were improved with intravenous (IV) steroids at an emergency department visit, but they started to flare soon thereafter with worsening mucosal involvement and dysphagia. After a second visit to the emergency department and repeat treatment with IV steroids, she was transferred to our institution for a higher level of care.
The patient denied taking any new medications in the 2 months prior to the onset of the rash. Her medication history only consisted of over-the-counter prenatal vitamins, a single use of over-the-counter migraine medication (containing acetaminophen, aspirin, and caffeine as active ingredients), and a possible use of ibuprofen or acetaminophen separately. She reported ocular discomfort and blurriness, dysphagia, dysuria, and vaginal discomfort. Physical examination revealed dusky red to violaceous macules and patches that involved approximately 65% of the body surface area (BSA), with bullae involving approximately 10% BSA. The face was diffusely red and edematous with crusted erosions and scattered bullae on the cheeks. Mucosal involvement was notable for injected conjunctivae and erosions present on the upper hard palate of the mouth and lips (Figure, A). Erythematous macules with dusky centers coalescing into patches with overlying vesicles and bullae were scattered on the arms (Figure, B), hands, trunk (Figure, C), and legs. The Nikolsky sign was positive. The vulva was swollen and covered with erythematous macules with dusky centers.
A biopsy from the upper back revealed a vacuolar interface with subepidermal bullae and confluent keratinocyte necrosis with many CD8+ cells and scattered granzyme B. Given these results in conjunction with the clinical findings, a diagnosis of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) overlap was made. In addition to providing supportive care, the patient was started on a 4-day course of IV immunoglobulin (IVIG)(3g/kg total) and prednisone 60 mg daily, tapered over several weeks with a good clinical response. At outpatient follow-up she was found to have postinflammatory hypopigmentation on the face, trunk, and extremities, as well as tear duct scarring, but she had no vulvovaginal scarring or stenosis. She was progressing well in her pregnancy with no serious complications for 4 months after admission, at which point she was lost to follow-up.
Stevens-Johnson syndrome and TEN represent a spectrum of severe mucocutaneous reactions with high morbidity and mortality. Medications are the leading trigger, followed by infection. The most common inciting medications include antibacterial sulfonamides, antiepileptics such as carbamazepine and lamotrigine, nonsteroidal anti-inflammatory drugs, nevirapine, and allopurinol. The onset of symptoms from 1 to 4 weeks combined with characteristic morphologic features helps distinguish SJS/TEN from other drug eruptions. The initial presentation classically consists of a flulike prodrome followed by mucocutaneous eruption. Skin lesions often present as a diffuse erythema or ill-defined, coalescing, erythematous macules with purpuric centers that may evolve into vesicles and bullae with sloughing of the skin. Histopathology reveals full-thickness epidermal necrosis with detachment.1
Erythema multiforme and Mycoplasma-induced rash and mucositis (MIRM) are high on the differential diagnosis. Distinguishing features of erythema multiforme include the morphology of targetoid lesions and a common distribution on the extremities, in addition to the limited bullae and epidermal detachment in comparison with SJS/TEN. In MIRM, mucositis often is more severe and extensive, with multiple mucosal surfaces affected. It typically has less cutaneous involvement than SJS/TEN, though clinical variants can include diffuse rash and affect fewer than 2 mucosal sites.2 Depending on the timing of rash onset, Mycoplasma IgM/IgG titers may be drawn to further support the diagnosis. A diagnosis of MIRM was not favored in our patient due to lack of respiratory symptoms, normal chest radiography, and negative Mycoplasma IgM and IgG titers.
Stevens-Johnson syndrome/toxic epidermal necrolysis overlap has been reported in pregnant patients, typically in association with HIV infection or new medication exposure.3 A combination of genetic susceptibility and an altered immune system during pregnancy may contribute to the pathogenesis, involving a cytotoxic T-cell mediated reaction with release of inflammatory cytokines.1 Interestingly, these factors that may predispose a patient to developing SJS/TEN may not pass on to the neonate, evidenced by a few cases that showed no reaction in the newborn when given the same offending drug.4
Stevens-Johnson syndrome/toxic epidermal necrolysis more frequently presents in the second or third trimester, with no increase in maternal mortality and an equally high survival rate of the fetus.1,5 Unique sequelae in pregnant patients may include vaginal stenosis, vulvar swelling, and postpartum sepsis. Fetal complications can include low birth weight, preterm delivery, and respiratory distress. The fetus rarely exhibits cutaneous manifestations of the disease.6
A multidisciplinary approach to the diagnosis and management of SJS/TEN overlap in special patient populations such as pregnant women is vital. Supportive measures consisting of wound care, fluid and electrolyte management, infection monitoring, and nutritional support have sufficed in treating SJS/TEN in pregnant patients.3 Although adjunctive therapy with systemic corticosteroids, IVIG, cyclosporine, and tumor necrosis factor inhibitors commonly are used in clinical practice, the safety of these treatments in pregnant patients affected by SJS/TEN has not been established. However, use of these medications for other indications, primarily rheumatologic diseases, has been reported to be safe in the pregnant population.7 If necessary, glucocorticoids should be used in the lowest effective dose to avoid complications such as premature rupture of membranes; intrauterine growth restriction; and increased risk for pregnancy-induced hypertension, gestational diabetes, osteoporosis, and infection. Little is known about IVIG use in pregnancy. While it has not been associated with increased risk of fetal malformations, it may cross the placenta in a notable amount when administered after 30 weeks’ gestation.7
Unlike most cases of SJS/TEN in pregnancy that largely were associated with HIV infection or drug exposure, primarily antiretrovirals such as nevirapine or antiepileptics, our case is a rare incidence of SJS/TEN in a pregnant patient with no clear medication or infectious trigger. Although the causative drug was unclear, we suspected it was secondary to nonsteroidal anti-inflammatory drug use. The patient had a SCORTEN (SCORe of Toxic Epidermal Necrosis) of 0, which portends a relatively good prognosis with an estimated mortality rate of approximately 3% (Table).8 However, the large BSA involvement of the morbilliform rash warranted aggressive management to prevent the involved skin from fully detaching.
To the Editor:
A 34-year-old pregnant woman at 5 weeks’ gestation was transferred to dermatology from an outside hospital with a full-body rash. Three days after noting a fever and generalized body aches, she developed a painful rash on the legs that had gradually spread to the arms, trunk, and face. Symptoms of eyelid pruritus and edema initially were improved with intravenous (IV) steroids at an emergency department visit, but they started to flare soon thereafter with worsening mucosal involvement and dysphagia. After a second visit to the emergency department and repeat treatment with IV steroids, she was transferred to our institution for a higher level of care.
The patient denied taking any new medications in the 2 months prior to the onset of the rash. Her medication history only consisted of over-the-counter prenatal vitamins, a single use of over-the-counter migraine medication (containing acetaminophen, aspirin, and caffeine as active ingredients), and a possible use of ibuprofen or acetaminophen separately. She reported ocular discomfort and blurriness, dysphagia, dysuria, and vaginal discomfort. Physical examination revealed dusky red to violaceous macules and patches that involved approximately 65% of the body surface area (BSA), with bullae involving approximately 10% BSA. The face was diffusely red and edematous with crusted erosions and scattered bullae on the cheeks. Mucosal involvement was notable for injected conjunctivae and erosions present on the upper hard palate of the mouth and lips (Figure, A). Erythematous macules with dusky centers coalescing into patches with overlying vesicles and bullae were scattered on the arms (Figure, B), hands, trunk (Figure, C), and legs. The Nikolsky sign was positive. The vulva was swollen and covered with erythematous macules with dusky centers.
A biopsy from the upper back revealed a vacuolar interface with subepidermal bullae and confluent keratinocyte necrosis with many CD8+ cells and scattered granzyme B. Given these results in conjunction with the clinical findings, a diagnosis of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) overlap was made. In addition to providing supportive care, the patient was started on a 4-day course of IV immunoglobulin (IVIG)(3g/kg total) and prednisone 60 mg daily, tapered over several weeks with a good clinical response. At outpatient follow-up she was found to have postinflammatory hypopigmentation on the face, trunk, and extremities, as well as tear duct scarring, but she had no vulvovaginal scarring or stenosis. She was progressing well in her pregnancy with no serious complications for 4 months after admission, at which point she was lost to follow-up.
Stevens-Johnson syndrome and TEN represent a spectrum of severe mucocutaneous reactions with high morbidity and mortality. Medications are the leading trigger, followed by infection. The most common inciting medications include antibacterial sulfonamides, antiepileptics such as carbamazepine and lamotrigine, nonsteroidal anti-inflammatory drugs, nevirapine, and allopurinol. The onset of symptoms from 1 to 4 weeks combined with characteristic morphologic features helps distinguish SJS/TEN from other drug eruptions. The initial presentation classically consists of a flulike prodrome followed by mucocutaneous eruption. Skin lesions often present as a diffuse erythema or ill-defined, coalescing, erythematous macules with purpuric centers that may evolve into vesicles and bullae with sloughing of the skin. Histopathology reveals full-thickness epidermal necrosis with detachment.1
Erythema multiforme and Mycoplasma-induced rash and mucositis (MIRM) are high on the differential diagnosis. Distinguishing features of erythema multiforme include the morphology of targetoid lesions and a common distribution on the extremities, in addition to the limited bullae and epidermal detachment in comparison with SJS/TEN. In MIRM, mucositis often is more severe and extensive, with multiple mucosal surfaces affected. It typically has less cutaneous involvement than SJS/TEN, though clinical variants can include diffuse rash and affect fewer than 2 mucosal sites.2 Depending on the timing of rash onset, Mycoplasma IgM/IgG titers may be drawn to further support the diagnosis. A diagnosis of MIRM was not favored in our patient due to lack of respiratory symptoms, normal chest radiography, and negative Mycoplasma IgM and IgG titers.
Stevens-Johnson syndrome/toxic epidermal necrolysis overlap has been reported in pregnant patients, typically in association with HIV infection or new medication exposure.3 A combination of genetic susceptibility and an altered immune system during pregnancy may contribute to the pathogenesis, involving a cytotoxic T-cell mediated reaction with release of inflammatory cytokines.1 Interestingly, these factors that may predispose a patient to developing SJS/TEN may not pass on to the neonate, evidenced by a few cases that showed no reaction in the newborn when given the same offending drug.4
Stevens-Johnson syndrome/toxic epidermal necrolysis more frequently presents in the second or third trimester, with no increase in maternal mortality and an equally high survival rate of the fetus.1,5 Unique sequelae in pregnant patients may include vaginal stenosis, vulvar swelling, and postpartum sepsis. Fetal complications can include low birth weight, preterm delivery, and respiratory distress. The fetus rarely exhibits cutaneous manifestations of the disease.6
A multidisciplinary approach to the diagnosis and management of SJS/TEN overlap in special patient populations such as pregnant women is vital. Supportive measures consisting of wound care, fluid and electrolyte management, infection monitoring, and nutritional support have sufficed in treating SJS/TEN in pregnant patients.3 Although adjunctive therapy with systemic corticosteroids, IVIG, cyclosporine, and tumor necrosis factor inhibitors commonly are used in clinical practice, the safety of these treatments in pregnant patients affected by SJS/TEN has not been established. However, use of these medications for other indications, primarily rheumatologic diseases, has been reported to be safe in the pregnant population.7 If necessary, glucocorticoids should be used in the lowest effective dose to avoid complications such as premature rupture of membranes; intrauterine growth restriction; and increased risk for pregnancy-induced hypertension, gestational diabetes, osteoporosis, and infection. Little is known about IVIG use in pregnancy. While it has not been associated with increased risk of fetal malformations, it may cross the placenta in a notable amount when administered after 30 weeks’ gestation.7
Unlike most cases of SJS/TEN in pregnancy that largely were associated with HIV infection or drug exposure, primarily antiretrovirals such as nevirapine or antiepileptics, our case is a rare incidence of SJS/TEN in a pregnant patient with no clear medication or infectious trigger. Although the causative drug was unclear, we suspected it was secondary to nonsteroidal anti-inflammatory drug use. The patient had a SCORTEN (SCORe of Toxic Epidermal Necrosis) of 0, which portends a relatively good prognosis with an estimated mortality rate of approximately 3% (Table).8 However, the large BSA involvement of the morbilliform rash warranted aggressive management to prevent the involved skin from fully detaching.
1. Struck MF, Illert T, Liss Y, et al. Toxic epidermal necrolysis in pregnancy: case report and review of the literature. J Burn Care Res. 2010;31:816-821. doi:10.1097/BCR.0b013e3181eed441
2. Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245.e4. doi:10.1016/j.jaad.2014.06.026
3. Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes in twenty-two consecutive pregnant HIV infected women. PLoS One. 2015;10:1-11. doi:10.1371/journal.pone.0135501
4. Velter C, Hotz C, Ingen-Housz-Oro S. Stevens-Johnson syndrome during pregnancy: case report of a newborn treated with the culprit drug. JAMA Dermatol. 2018;154:224-225. doi:10.1001/jamadermatol.2017.4607
5. El Daief SG, Das S, Ekekwe G, et al. A successful pregnancy outcome after Stevens-Johnson syndrome. J Obstet Gynaecol (Lahore). 2014;34:445-446. doi:10.3109/01443615.2014.914897
6. Rodriguez G, Trent JT, Mirzabeigi M. Toxic epidermal necrolysis in a mother and fetus. J Am Acad Dermatol. 2006;55(5 suppl):96-98. doi:10.1016/j.jaad.2005.09.023
7. Bermas BL. Safety of rheumatic disease medication use during pregnancy and lactation. UptoDate website. Updated March 24, 2021. Accessed December 16, 2021. https://www.uptodate.com/contents/safety-of-rheumatic-disease-medication-use-during-pregnancy-and-lactation#H11
8. Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153. doi:10.1046/j.1523-1747.2000.00061.x
1. Struck MF, Illert T, Liss Y, et al. Toxic epidermal necrolysis in pregnancy: case report and review of the literature. J Burn Care Res. 2010;31:816-821. doi:10.1097/BCR.0b013e3181eed441
2. Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245.e4. doi:10.1016/j.jaad.2014.06.026
3. Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes in twenty-two consecutive pregnant HIV infected women. PLoS One. 2015;10:1-11. doi:10.1371/journal.pone.0135501
4. Velter C, Hotz C, Ingen-Housz-Oro S. Stevens-Johnson syndrome during pregnancy: case report of a newborn treated with the culprit drug. JAMA Dermatol. 2018;154:224-225. doi:10.1001/jamadermatol.2017.4607
5. El Daief SG, Das S, Ekekwe G, et al. A successful pregnancy outcome after Stevens-Johnson syndrome. J Obstet Gynaecol (Lahore). 2014;34:445-446. doi:10.3109/01443615.2014.914897
6. Rodriguez G, Trent JT, Mirzabeigi M. Toxic epidermal necrolysis in a mother and fetus. J Am Acad Dermatol. 2006;55(5 suppl):96-98. doi:10.1016/j.jaad.2005.09.023
7. Bermas BL. Safety of rheumatic disease medication use during pregnancy and lactation. UptoDate website. Updated March 24, 2021. Accessed December 16, 2021. https://www.uptodate.com/contents/safety-of-rheumatic-disease-medication-use-during-pregnancy-and-lactation#H11
8. Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153. doi:10.1046/j.1523-1747.2000.00061.x
Practice Points
- Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of severe mucocutaneous reactions commonly presenting as drug eruptions.
- Pregnant patients affected by SJS/TEN represent a special patient population that requires a multidisciplinary approach for management and treatment.
- The rates of adverse outcomes for pregnant patients with SJS/TEN are low with timely diagnosis, removal of the offending agent, and supportive care as mainstays of treatment.
A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
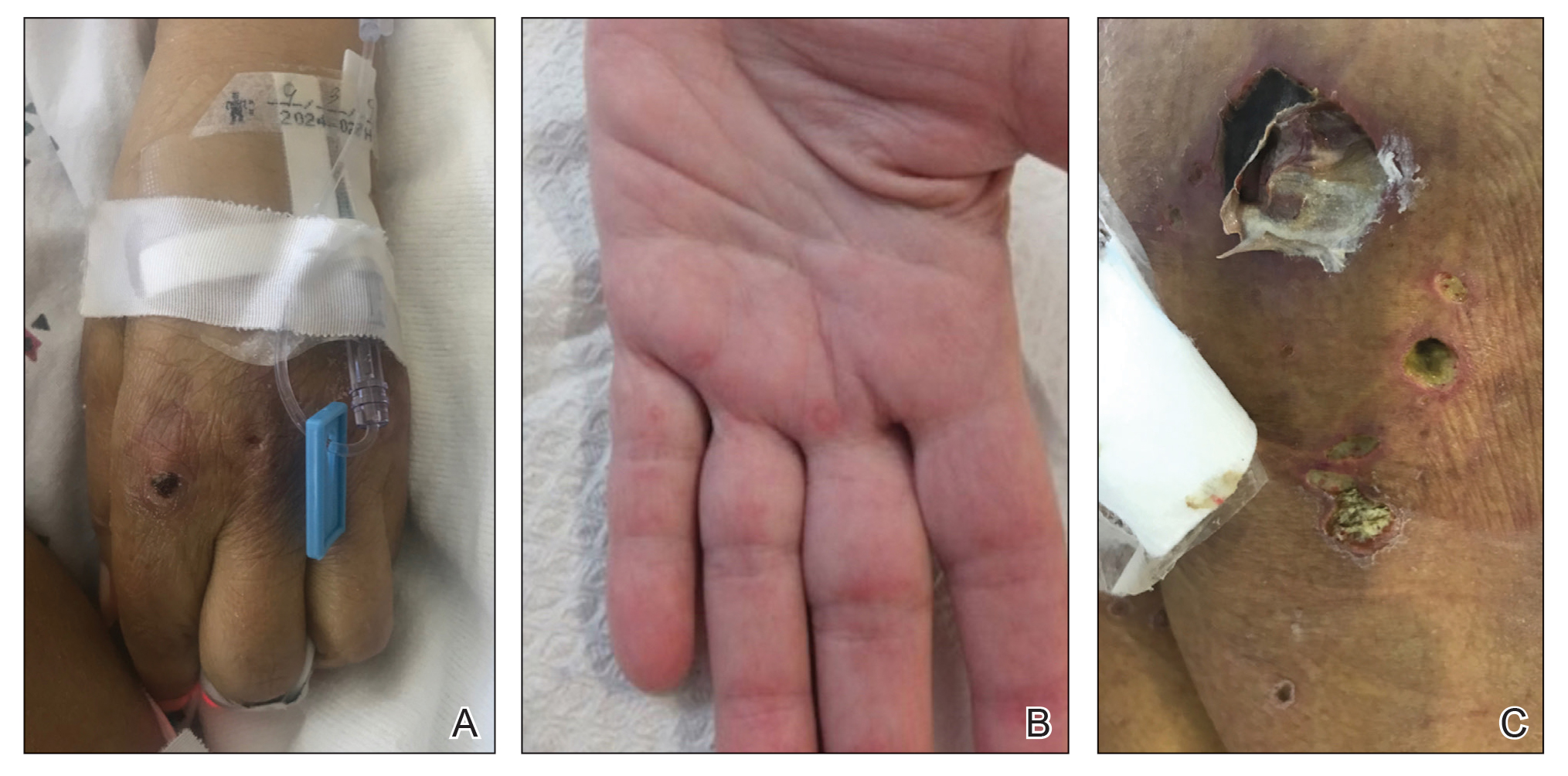
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
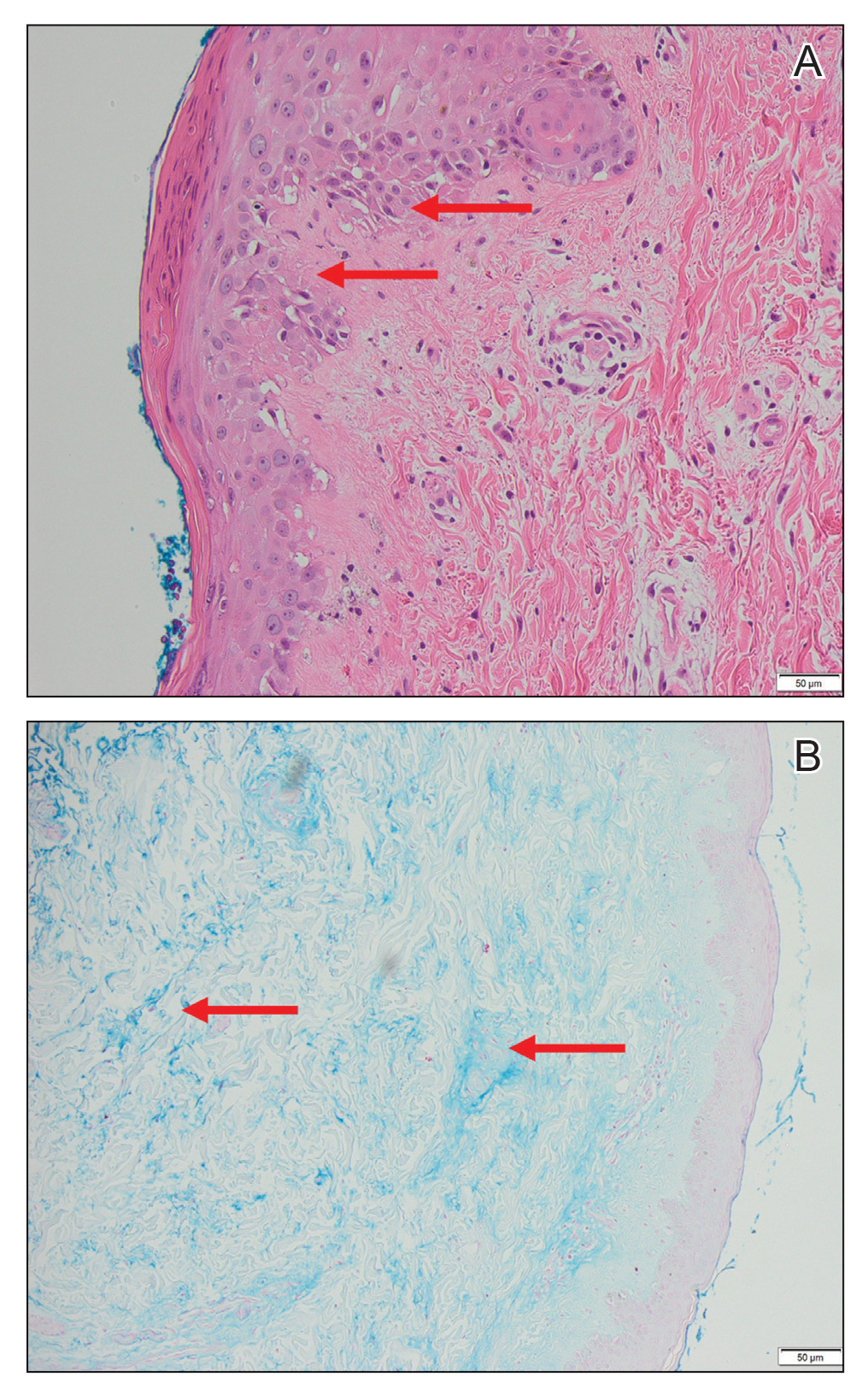
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.
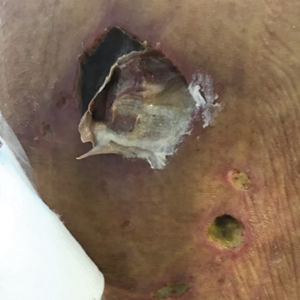
DRESS Syndrome Due to Cefdinir Mimicking Superinfected Eczema in a Pediatric Patient
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.

The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.
The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, or drug-induced hypersensitivity syndrome, is a serious and potentially fatal multiorgan drug hypersensitivity reaction. Drug reaction with eosinophilia and systemic symptoms syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis (AD) in which children may have baseline eosinophilia from an atopic diathesis. The cutaneous exanthema also may be variable in presentation, further complicating diagnosis.1,2
A 3-year-old boy with AD since infancy and a history of anaphylaxis to peanuts presented to the emergency department with reported fever, rash, sore throat, and decreased oral intake. Ten days prior, the patient was treated for cellulitis of the left foot with a 7-day course of cefdinir with complete resolution of symptoms. Four days prior to admission, the patient started developing “bumps” on the face and fevers. He was seen at an outside facility, where a rapid test for Streptococcus was negative, and the patient was treated with ibuprofen and fluids for a presumed viral exanthem. The rash subsequently spread to involve the trunk and extremities. On the day of admission, the patient had a positive rapid test for Streptococcus and was referred to the emergency department with concern for superinfected eczema and eczema herpeticum. The patient recently traveled to Puerto Rico, where he had contact with an aunt with active herpes zoster but no other sick contacts. The patient’s immunizations were reported to be up-to-date.
Physical examination revealed the patient was afebrile but irritable and had erythematous crusted papules and patches on the face, arms, and legs, as well as erythematous dry patches on the chest, abdomen, and back (Figure). There were no conjunctival erythematous or oral erosions. The patient was admitted to the hospital for presumed superinfected AD and possible eczema herpeticum. He was started on intravenous clindamycin and acyclovir.
The following day, the patient had new facial edema and fever (temperature, 102.8 °F [39.36 °C]) in addition to palpable mobile cervical, axillary, and inguinal lymphadenopathy. He also was noted to have notably worsening eosinophilia from 1288 (14%) to 2570 (29.2%) cells/µL (reference range, 0%–5%) and new-onset transaminitis. Herpes and varicella-zoster direct fluorescent antibody tests, culture, and serum polymerase chain reaction were all negative, and acyclovir was discontinued. Repeat laboratory tests 12 hours later showed a continued uptrend in transaminitis. Serologies for acute and chronic cytomegalovirus; Epstein-Barr virus; and hepatitis A, B, and C were all nonreactive. The patient was started on intravenous methylprednisolone 1 mg/kg daily for suspected DRESS syndrome likely due to cefdinir.
The patient’s eosinophilia completely resolved (from approximately 2600 to 100 cells/µL) after 1 dose of steroids, and his transaminitis trended down over the next few days. He remained afebrile for the remainder of his admission, and his facial swelling and rash continued to improve. Bacterial culture from the skin grew oxacillin-susceptible Staphylococcus aureus and group A Streptococcus pyogenes. A blood culture was negative. The patient was discharged home to complete a 10-day course of clindamycin and was given topical steroids for the eczema. He continued on oral prednisolone 1 mg/kg daily for 10 days, after which the dose was tapered down for a total 1-month course of systemic corticosteroids. At 1-month follow-up after completing the course of steroids, he was doing well with normal hepatic enzyme levels and no recurrence of fever, facial edema, or rash. He continues to be followed for management of the AD.
Drug reaction with eosinophilia and systemic symptoms syndrome is a serious systemic adverse drug reaction, with high morbidity and even mortality, estimated at 10% in the adult population, though more specific pediatric mortality data are not available.1,2 The exact pathogenesis of DRESS syndrome has not been elucidated. Certain human leukocyte antigen class I alleles are predisposed to the development of DRESS syndrome, but there has not been a human leukocyte antigen subtype identified with beta-lactam–associated DRESS syndrome. Some studies have demonstrated a reactivation of human herpesvirus 6, human herpesvirus 7, and Epstein-Barr virus.3 One study involving 40 patients with DRESS syndrome identified viremia in 76% (29/38) of patients and identified CD8+ T-cell populations directed toward viral epitopes.3 Finally, DRESS syndrome may be related to the slow detoxification and elimination of intermediary products of offending medications that serve as an immunogenic stimulus for the inflammatory cascade.2
In adults, DRESS syndrome was first identified in association with phenytoin, but more recently other drugs have been identified, including other aromatic anticonvulsants (ie, lamotrigine, phenobarbital, carbamazepine), allopurinol, sulfonamides, antiretrovirals (particularly abacavir), and minocycline.2 In a 3-year pediatric prospective study, 11 cases of DRESS syndrome were identified: 4 cases due to lamotrigine, and 3 caused by penicillins.4 The trigger in our patient’s case was the beta-lactam, third-generation cephalosporin cefdinir, and his symptoms developed within 6 days of starting the medication. Many articles report that beta-lactams are a rare cause of DRESS syndrome, with only a handful of cases reported.1,5,6
The diagnosis of DRESS syndrome often can be delayed, as children present acutely febrile and toxic appearing. Unlike many adverse drug reactions, DRESS syndrome does not show rapid resolution with withdrawal of the causative agent, further complicating the diagnosis. The typical onset of DRESS syndrome generally ranges from 2 to 6 weeks after the initiation of the offending drug; however, faster onset of symptoms, similar to our case, has been noted in antibiotic-triggered cases. In the prospective pediatric series by Sasidharanpillai et al,4 the average time to onset among 3 antibiotic-triggered DRESS cases was 5.8 days vs 23.9 days among the 4 cases of lamotrigine-associated DRESS syndrome.
Our patient demonstrated the classic features of DRESS syndrome, including fever, rash, lymphadenopathy, facial edema, peripheral eosinophilia, atypical lymphocytosis, and hepatitis. Based on the proposed RegiSCAR scoring system, our patient was classified as a “definite” case of DRESS syndrome.1,7 Other hematologic findings in DRESS syndrome may include thrombocytopenia and anemia. The liver is the most commonly affected internal organ in DRESS syndrome, with pneumonitis, carditis, and nephritis reported less frequently.1 The pattern of liver injury in our patient was mixed (hepatocellular and cholestatic), the second most common pattern in patients with DRESS syndrome (the cholestatic pattern is most common).8
The exanthem of DRESS syndrome can vary in morphology, with up to 7% of patients reported to have eczemalike lesions in the multinational prospective RegiSCAR study.1 Other entities in the differential diagnosis for our patient included Kawasaki disease, where conjunctivitis and strawberry tongue are classically present, as well as erythrodermic AD, where internal organ involvement is not common.2 Our patient’s exanthem initially was considered to be a flare of AD with superimposed bacterial infection and possible eczema herpeticum. Although bacterial cultures did grow Staphylococcus and Streptococcus, viral studies were all negative, and this alone would not have explained the facial edema, rapidly rising eosinophil count, and transaminitis. The dramatic drop in his eosinophil count and decrease in hepatic enzymes after 1 dose of intravenous methylprednisolone also supported the diagnosis of DRESS syndrome.
Treatment recommendations remain largely anecdotal. Early systemic steroids generally are accepted as the first line of therapy, with a slow taper. Although the average required duration of systemic steroids in 1 series of adults was reported at 50.1 days,9 the duration was shorter (21–35 days) in a series of pediatric patients.4 Our patient’s clinical symptoms and laboratory values normalized after completing a 1-month steroid taper. Other therapies have been tried for recalcitrant cases, including intravenous immunoglobulin, plasmapheresis, rituximab, and valganciclovir.2
Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality. Although DRESS syndrome in pediatric patients presents with many similar clinical features as in adults, it may be a greater diagnostic challenge. As in adult cases, timely administration of systemic corticosteroids and tapering based on clinical signs and symptoms can lead to resolution of the hypersensitivity syndrome.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Picard D, Janela B, Descamps V, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Aouam K, Chaabane A, Toumi A, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) probably induced by cefotaxime: a report of two cases. Clin Med Res. 2012;10:32-35.
- Guleria VS, Dhillon M, Gill S, et al. Ceftriaxone induced drug rash with eosinophilia and systemic symptoms. J Res Pharm Pract. 2014;3:72-74.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Ang CC, Wang YS, Yoosuff EL, et al. Retrospective analysis of drug-induced hypersensitivity syndrome: a study of 27 patients. J Am Acad Dermatol. 2010;63:219-227.
Practice Points
- Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome shares many clinical features with viral exanthems and may be difficult to diagnose in the setting of atopic dermatitis in which children may have baseline eosinophilia from an atopic diathesis.
- Early clinical recognition of the signs and symptoms of DRESS syndrome in the setting of a new medication can decrease morbidity and mortality.
Acute Severe Urticaria From Minocycline
To the Editor:
Minocycline is a commonly prescribed semisynthetic tetracycline derivative used for long-term treatment of acne vulgaris.1 Given the continued popularity of minocycline and other tetracyclines in treating acne, more adverse side effects are being reported. We report a patient who experienced acute severe urticaria with angioedema from minocycline.
A 35-year-old woman with a history of acne vulgaris presented to the emergency department with urticaria and associated angioedema. Fifteen days after starting minocycline, she awoke with diffuse hives sparing only the abdomen that resolved with diphenhydramine. Later that day, she developed generalized pruritus, hives, and lip swelling. She received intravenous methylprednisolone, diphenhydramine, and famotidine in the emergency department. She returned to the emergency department the next day due to facial and lip swelling, diffuse urticaria that was most pronounced on the arms, and throat irritation. Intramuscular epinephrine was administered first followed by methylprednisolone, famotidine, and cetirizine. She was discharged and advised to start daily prednisone 50 mg and cetirizine 20 mg every evening.
She returned to the emergency department the following morning due to worsening generalized urticaria and angioedema of the lips. She denied any associated respiratory, joint, or gastrointestinal tract symptoms. She had several urticarial plaques on the scalp, face, and body (Figure), only sparing the abdomen. Her hives were erythematous, raised, pruritic, and blanching. There was no residual purpura, ecchymosis, or hyperpigmentation associated with the urticaria, and each lesion was present for less than 24 hours. There was no swelling on examination. Additionally, she was afebrile. The C4 level was 18 mg/dL (reference range, 15–45 mg/dL). She did not develop eosinophilia (absolute eosinophil count, 0/µL [reference range, 50–500/µL]), lymphocytosis (absolute lymphocyte count, 1300/µL [reference range, 1000–4800/µL]), or abnormal liver or renal function. She was hospitalized for 3 days with severe urticaria and required 7 days of prednisone 40 to 50 mg, fexofenadine 360 mg, and cetirizine 20 mg. A viral infection was considered as a possible etiology; however, she had no supporting signs or symptoms of an upper respiratory illness or other viral illness.
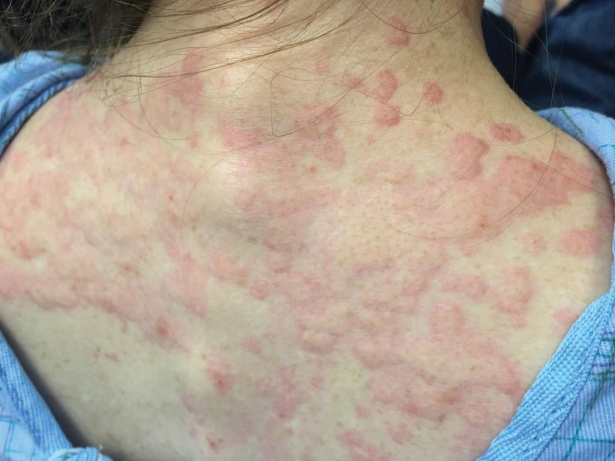
The patient’s minocycline use was considered the most likely etiology, as an oral contraceptive was the only other medication. She was labelled allergic to minocycline and discharged with intramuscular epinephrine. She was evaluated in the outpatient allergy immunology clinic 9 days later, and all her symptoms had resolved. Due to the severity of our patient’s reaction and the possibility of further severe reactions, an oral challenge was not carried out. Our patient was not interested in pursuing any further minocycline or other tetracycline-based therapy for her acne. She also was not interested in pursuing any minocycline skin-prick testing or oral challenge. One limitation to this case is our patient declining a confirmatory drug challenge; however, given the severity of the symptoms, the physicians involved agreed the patient's safety outweighed the benefits of confirmatory testing.
A PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms minocycline, drug hypersensitivity, urticaria, anaphylaxis, minocycline allergy, and angioedema yielded only 16 articles and correspondences. Reported adverse effects of minocycline included drug-induced lupus erythematosus, vasculitis, nausea, photosensitivity, and DRESS-like (drug reaction with eosinophilia and systemic symptoms syndrome) conditions. Three case reports of anaphylaxis/anaphylactoid reactions have been published,2-4 but cases of urticaria attributable to minocycline appear to be exceedingly rare.2,3 Reports of serum sickness in patients aged 15 to 62 years were rare. Women were noted to experience a higher frequency of adverse effects compared to men.5 Symptoms typically presented 3 to 28 days after initiation of minocycline. Data currently suggest that the pathogenesis of hypersensitivity reactions to minocycline remains unknown6; however, one hypothesis is that minocycline or its metabolites act as a superantigen, resulting in lymphocyte overactivation and massive cytokine release.7
Minocycline generally is well tolerated by patients. Physicians should be aware that minocycline is a possible causative agent of allergic drug reactions. Our patient’s presentation of severe acute urticaria with angioedema of the face and lips is a rarity.
- Levenson T, Masood D, Patterson R. Minocycline-induced serum sickness. Allergy Asthma Proc. 1996;17:79-81.
- Okano M, Imai S. Anaphylactoid symptoms due to oral minocycline. Acta Derm Venereol. 1996;76:164.
- Jang JW, Bae Y-J, Kim YG, et al. A case of anaphylaxis to oral minocycline. J Korean Med Sci. 2010;25:1233.
- Nakamura R, Tanaka A, Kinoshita H, et al. Minocycline-induced anaphylaxis mediated by antigen-specific immunoglobulin E [published online November 9, 2021]. J Dermatol. doi:10.1111/1346-8138.16228
- MacNeil M, Haase DA, Tremaine R, et al. Fever, lymphadenopathy, eosinophilia, lymphocytosis, hepatitis, and dermatitis: a severe adverse reaction to minocycline. J Am Acad Dermatol. 1997;36:347-350.
- DePaz S, Perez A, Gomez M, et al. Severe hypersensitivity reaction to minocycline. J Invest Allergol Clin Immunol. 1999;9:403-404.
- Somech R, Arav-Boger R, Assia A, et al. Complications of minocycline therapy for acne vulgaris: case reports and review of the literature. Pediatr Dermatol. 1999;16:469-472.
To the Editor:
Minocycline is a commonly prescribed semisynthetic tetracycline derivative used for long-term treatment of acne vulgaris.1 Given the continued popularity of minocycline and other tetracyclines in treating acne, more adverse side effects are being reported. We report a patient who experienced acute severe urticaria with angioedema from minocycline.
A 35-year-old woman with a history of acne vulgaris presented to the emergency department with urticaria and associated angioedema. Fifteen days after starting minocycline, she awoke with diffuse hives sparing only the abdomen that resolved with diphenhydramine. Later that day, she developed generalized pruritus, hives, and lip swelling. She received intravenous methylprednisolone, diphenhydramine, and famotidine in the emergency department. She returned to the emergency department the next day due to facial and lip swelling, diffuse urticaria that was most pronounced on the arms, and throat irritation. Intramuscular epinephrine was administered first followed by methylprednisolone, famotidine, and cetirizine. She was discharged and advised to start daily prednisone 50 mg and cetirizine 20 mg every evening.
She returned to the emergency department the following morning due to worsening generalized urticaria and angioedema of the lips. She denied any associated respiratory, joint, or gastrointestinal tract symptoms. She had several urticarial plaques on the scalp, face, and body (Figure), only sparing the abdomen. Her hives were erythematous, raised, pruritic, and blanching. There was no residual purpura, ecchymosis, or hyperpigmentation associated with the urticaria, and each lesion was present for less than 24 hours. There was no swelling on examination. Additionally, she was afebrile. The C4 level was 18 mg/dL (reference range, 15–45 mg/dL). She did not develop eosinophilia (absolute eosinophil count, 0/µL [reference range, 50–500/µL]), lymphocytosis (absolute lymphocyte count, 1300/µL [reference range, 1000–4800/µL]), or abnormal liver or renal function. She was hospitalized for 3 days with severe urticaria and required 7 days of prednisone 40 to 50 mg, fexofenadine 360 mg, and cetirizine 20 mg. A viral infection was considered as a possible etiology; however, she had no supporting signs or symptoms of an upper respiratory illness or other viral illness.
The patient’s minocycline use was considered the most likely etiology, as an oral contraceptive was the only other medication. She was labelled allergic to minocycline and discharged with intramuscular epinephrine. She was evaluated in the outpatient allergy immunology clinic 9 days later, and all her symptoms had resolved. Due to the severity of our patient’s reaction and the possibility of further severe reactions, an oral challenge was not carried out. Our patient was not interested in pursuing any further minocycline or other tetracycline-based therapy for her acne. She also was not interested in pursuing any minocycline skin-prick testing or oral challenge. One limitation to this case is our patient declining a confirmatory drug challenge; however, given the severity of the symptoms, the physicians involved agreed the patient's safety outweighed the benefits of confirmatory testing.
A PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms minocycline, drug hypersensitivity, urticaria, anaphylaxis, minocycline allergy, and angioedema yielded only 16 articles and correspondences. Reported adverse effects of minocycline included drug-induced lupus erythematosus, vasculitis, nausea, photosensitivity, and DRESS-like (drug reaction with eosinophilia and systemic symptoms syndrome) conditions. Three case reports of anaphylaxis/anaphylactoid reactions have been published,2-4 but cases of urticaria attributable to minocycline appear to be exceedingly rare.2,3 Reports of serum sickness in patients aged 15 to 62 years were rare. Women were noted to experience a higher frequency of adverse effects compared to men.5 Symptoms typically presented 3 to 28 days after initiation of minocycline. Data currently suggest that the pathogenesis of hypersensitivity reactions to minocycline remains unknown6; however, one hypothesis is that minocycline or its metabolites act as a superantigen, resulting in lymphocyte overactivation and massive cytokine release.7
Minocycline generally is well tolerated by patients. Physicians should be aware that minocycline is a possible causative agent of allergic drug reactions. Our patient’s presentation of severe acute urticaria with angioedema of the face and lips is a rarity.
To the Editor:
Minocycline is a commonly prescribed semisynthetic tetracycline derivative used for long-term treatment of acne vulgaris.1 Given the continued popularity of minocycline and other tetracyclines in treating acne, more adverse side effects are being reported. We report a patient who experienced acute severe urticaria with angioedema from minocycline.
A 35-year-old woman with a history of acne vulgaris presented to the emergency department with urticaria and associated angioedema. Fifteen days after starting minocycline, she awoke with diffuse hives sparing only the abdomen that resolved with diphenhydramine. Later that day, she developed generalized pruritus, hives, and lip swelling. She received intravenous methylprednisolone, diphenhydramine, and famotidine in the emergency department. She returned to the emergency department the next day due to facial and lip swelling, diffuse urticaria that was most pronounced on the arms, and throat irritation. Intramuscular epinephrine was administered first followed by methylprednisolone, famotidine, and cetirizine. She was discharged and advised to start daily prednisone 50 mg and cetirizine 20 mg every evening.
She returned to the emergency department the following morning due to worsening generalized urticaria and angioedema of the lips. She denied any associated respiratory, joint, or gastrointestinal tract symptoms. She had several urticarial plaques on the scalp, face, and body (Figure), only sparing the abdomen. Her hives were erythematous, raised, pruritic, and blanching. There was no residual purpura, ecchymosis, or hyperpigmentation associated with the urticaria, and each lesion was present for less than 24 hours. There was no swelling on examination. Additionally, she was afebrile. The C4 level was 18 mg/dL (reference range, 15–45 mg/dL). She did not develop eosinophilia (absolute eosinophil count, 0/µL [reference range, 50–500/µL]), lymphocytosis (absolute lymphocyte count, 1300/µL [reference range, 1000–4800/µL]), or abnormal liver or renal function. She was hospitalized for 3 days with severe urticaria and required 7 days of prednisone 40 to 50 mg, fexofenadine 360 mg, and cetirizine 20 mg. A viral infection was considered as a possible etiology; however, she had no supporting signs or symptoms of an upper respiratory illness or other viral illness.
The patient’s minocycline use was considered the most likely etiology, as an oral contraceptive was the only other medication. She was labelled allergic to minocycline and discharged with intramuscular epinephrine. She was evaluated in the outpatient allergy immunology clinic 9 days later, and all her symptoms had resolved. Due to the severity of our patient’s reaction and the possibility of further severe reactions, an oral challenge was not carried out. Our patient was not interested in pursuing any further minocycline or other tetracycline-based therapy for her acne. She also was not interested in pursuing any minocycline skin-prick testing or oral challenge. One limitation to this case is our patient declining a confirmatory drug challenge; however, given the severity of the symptoms, the physicians involved agreed the patient's safety outweighed the benefits of confirmatory testing.
A PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms minocycline, drug hypersensitivity, urticaria, anaphylaxis, minocycline allergy, and angioedema yielded only 16 articles and correspondences. Reported adverse effects of minocycline included drug-induced lupus erythematosus, vasculitis, nausea, photosensitivity, and DRESS-like (drug reaction with eosinophilia and systemic symptoms syndrome) conditions. Three case reports of anaphylaxis/anaphylactoid reactions have been published,2-4 but cases of urticaria attributable to minocycline appear to be exceedingly rare.2,3 Reports of serum sickness in patients aged 15 to 62 years were rare. Women were noted to experience a higher frequency of adverse effects compared to men.5 Symptoms typically presented 3 to 28 days after initiation of minocycline. Data currently suggest that the pathogenesis of hypersensitivity reactions to minocycline remains unknown6; however, one hypothesis is that minocycline or its metabolites act as a superantigen, resulting in lymphocyte overactivation and massive cytokine release.7
Minocycline generally is well tolerated by patients. Physicians should be aware that minocycline is a possible causative agent of allergic drug reactions. Our patient’s presentation of severe acute urticaria with angioedema of the face and lips is a rarity.
- Levenson T, Masood D, Patterson R. Minocycline-induced serum sickness. Allergy Asthma Proc. 1996;17:79-81.
- Okano M, Imai S. Anaphylactoid symptoms due to oral minocycline. Acta Derm Venereol. 1996;76:164.
- Jang JW, Bae Y-J, Kim YG, et al. A case of anaphylaxis to oral minocycline. J Korean Med Sci. 2010;25:1233.
- Nakamura R, Tanaka A, Kinoshita H, et al. Minocycline-induced anaphylaxis mediated by antigen-specific immunoglobulin E [published online November 9, 2021]. J Dermatol. doi:10.1111/1346-8138.16228
- MacNeil M, Haase DA, Tremaine R, et al. Fever, lymphadenopathy, eosinophilia, lymphocytosis, hepatitis, and dermatitis: a severe adverse reaction to minocycline. J Am Acad Dermatol. 1997;36:347-350.
- DePaz S, Perez A, Gomez M, et al. Severe hypersensitivity reaction to minocycline. J Invest Allergol Clin Immunol. 1999;9:403-404.
- Somech R, Arav-Boger R, Assia A, et al. Complications of minocycline therapy for acne vulgaris: case reports and review of the literature. Pediatr Dermatol. 1999;16:469-472.
- Levenson T, Masood D, Patterson R. Minocycline-induced serum sickness. Allergy Asthma Proc. 1996;17:79-81.
- Okano M, Imai S. Anaphylactoid symptoms due to oral minocycline. Acta Derm Venereol. 1996;76:164.
- Jang JW, Bae Y-J, Kim YG, et al. A case of anaphylaxis to oral minocycline. J Korean Med Sci. 2010;25:1233.
- Nakamura R, Tanaka A, Kinoshita H, et al. Minocycline-induced anaphylaxis mediated by antigen-specific immunoglobulin E [published online November 9, 2021]. J Dermatol. doi:10.1111/1346-8138.16228
- MacNeil M, Haase DA, Tremaine R, et al. Fever, lymphadenopathy, eosinophilia, lymphocytosis, hepatitis, and dermatitis: a severe adverse reaction to minocycline. J Am Acad Dermatol. 1997;36:347-350.
- DePaz S, Perez A, Gomez M, et al. Severe hypersensitivity reaction to minocycline. J Invest Allergol Clin Immunol. 1999;9:403-404.
- Somech R, Arav-Boger R, Assia A, et al. Complications of minocycline therapy for acne vulgaris: case reports and review of the literature. Pediatr Dermatol. 1999;16:469-472.
Practice Points
- Minocycline is a commonly prescribed long-term treatment for acne vulgaris.
- Minocycline-induced acute urticaria and anaphylaxis are rare adverse events.