User login
Management Challenges in Sarcoidosis
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
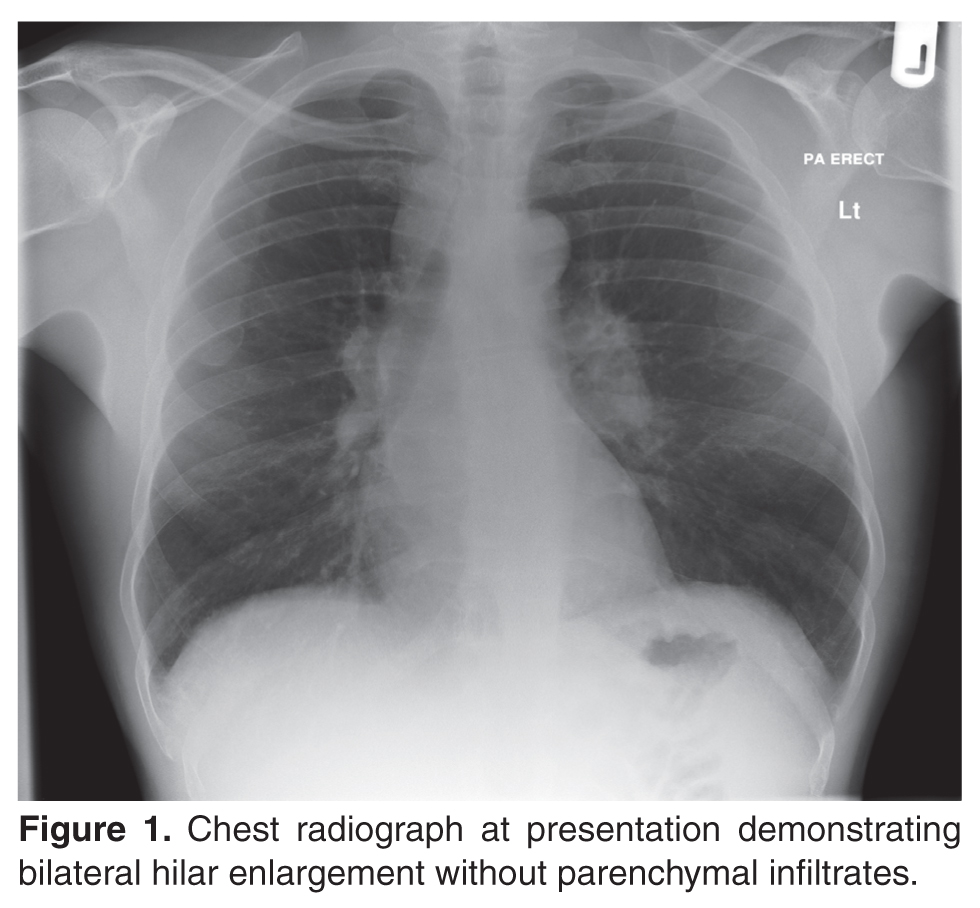
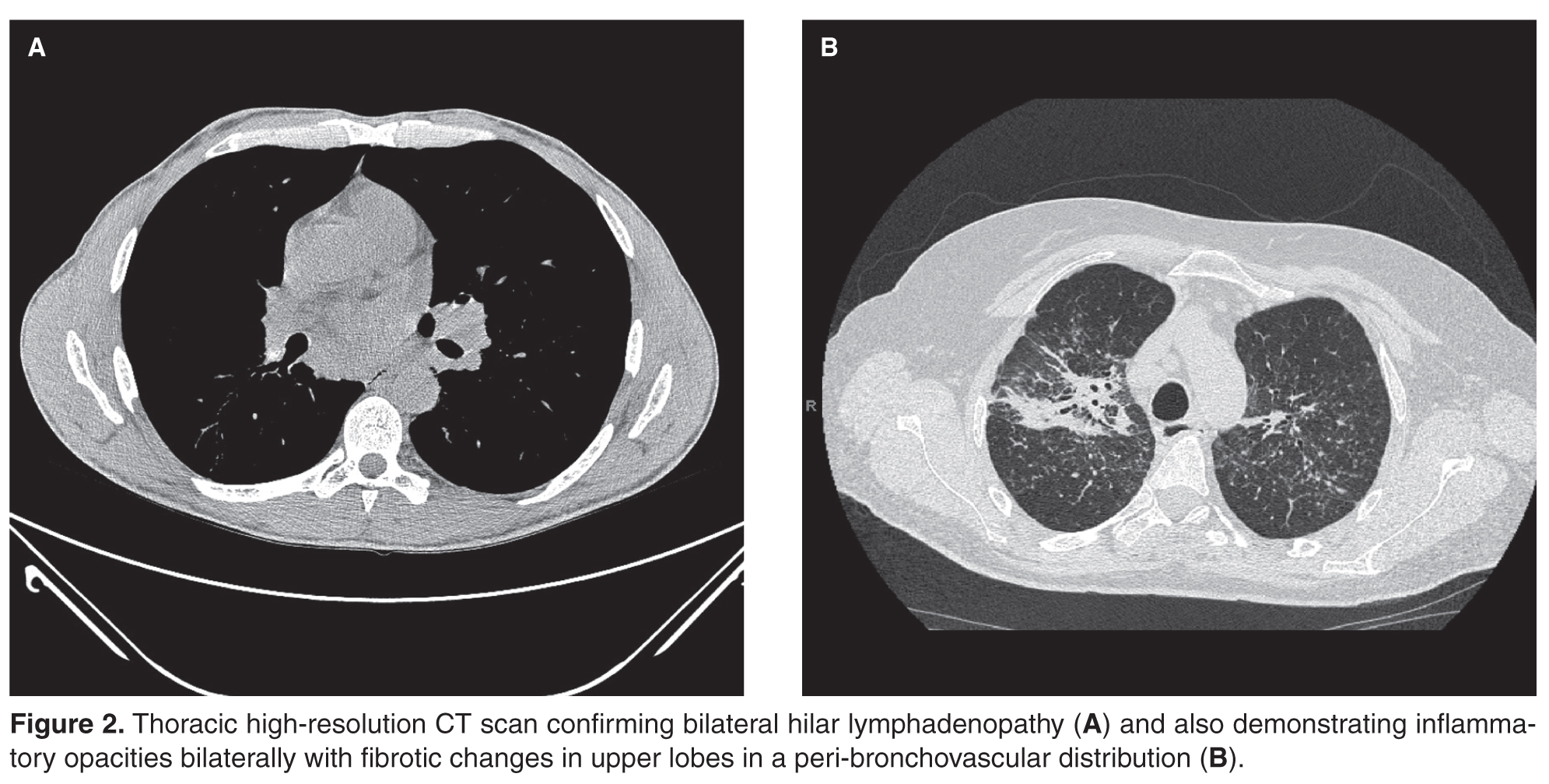
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
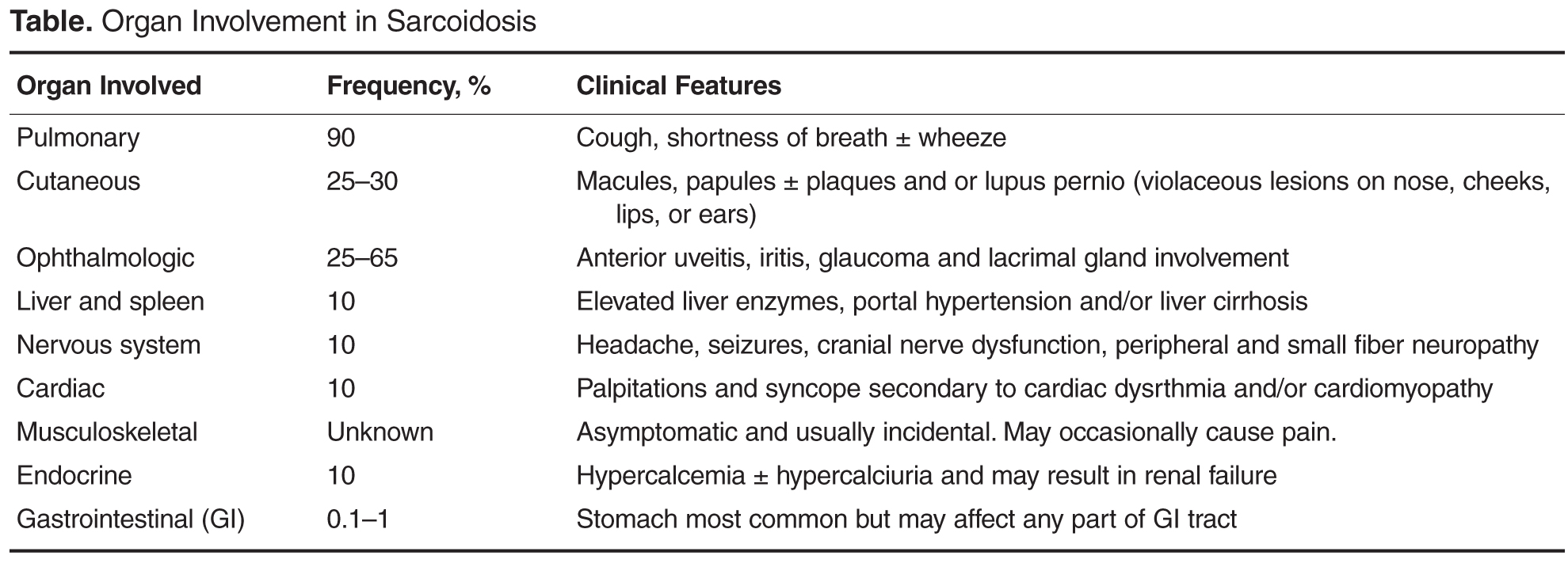
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.
Symptomatic Intracranial Atherosclerotic Disease
From the Emory University School of Medicine, Atlanta, GA.
Abstract
- Objective: To provide an overview of the diagnosis, clinical presentation, and management of symptomatic intracranial atherosclerotic disease (ICAD).
- Methods: Review of the current literature in the context of a clinical case.
- Results: ICAD is a common cause of ischemic strokes or transient ischemic attacks (TIAs), especially among Asian, black, and Hispanic patients. ICAD can be identified with noninvasive arterial imaging such as CT angiography, MR angiography, or transcranial Doppler ultrasound of the head when evaluating for the cause of an ischemic stroke or TIA. Aggressive medical management with dual antiplatelet therapy and lifestyle and risk factor modification has emerged as effective first-line therapy. In patients who have recurrent ischemic symptoms while on aggressive medical management, endovascular treatment can be considered.
- Conclusion: When symptomatic ICAD is identified early, aggressive medical management is effective in reducing the risk of recurrent ischemic events in this patient population.
Symptomatic intracranial atherosclerotic disease (ICAD) may represent the most common cause of ischemic stroke worldwide and the cause of approximately 8% to 10% of ischemic strokes in the United States [1–7]. It is a particularly important clinical entity due to the high recurrence rate of ischemic events in this population. The estimated recurrent stroke risk from symptomatic ICAD has been reported to be as high as 14.9% in the first year after an initial ischemic event [1].There are multiple risk factors for ICAD. Non-modifiable risk factors include race (particularly Asian, black, or Hispanic race), age, and family history of coronary artery disease or stroke; modifiable risk factors include diabetes, hypertension, and hyperlipidemia [2].
Case Study
Initial Presentation
A 78-year-old right-handed man presents to the outpatient clinic for follow-up evaluation after inpatient admission for acute ischemic stroke. The patient has an established medical history of hypertension, diabetes mellitus (diagnosed 10 years ago), dyslipidemia, and a 50 pack-year history of tobacco use.
Two weeks prior to the clinic visit, the patient presented to the emergency department (ED) via emergency medical services with right face and arm weakness and numbness and the inability to speak that had been ongoing for approximately 1 hour. The patient’s wife reported to ED providers that the patient had a similar episode 1 month prior that resolved completely in 30 to 45 minutes (and for which the patient never sought medical attention). Home medications at the time of admission included aspirin 81 mg and pravastatin 20 mg daily, both of which he takes intermittently.
The initial blood pressure was 185/76 mm Hg and blood glucose was 225 mg/dL. Initial exam was remarkable for the inability to answer orientation questions (but able to follow simple commands), forced gaze deviation to the left, right lower facial weakness, weakness in the right arm with no antigravity movement, moderately decreased sensation of the right face and arm, severe expressive aphasia, and severe dysarthria. A CT of the head without contrast showed no evidence of hemorrhage. Patient was suspected of having an acute ischemic stroke and was given intravenous tPA.
What are the possible mechanisms for this patient’s presentation with ischemic stroke?
This patient is presenting with right face and arm weakness with sensory loss, gaze deviation to the left, and expressive aphasia. Abnormalities of speech and gaze paresis that localize to the left frontal lobe with cortical involvement make a subcortical or brainstem lacunar ischemic event less likely. The syndrome is suggestive of a large artery occlusion of the left middle cerebral artery. This type of large artery occlusive disease in the anterior circulation is most often due to an embolus (artery-to-artery or cardiac origin) event and requires arterial imaging of the head and neck acutely as endovascular thrombectomy has been associated with reduced disability compared to intravenous tPA alone.
Further Evaluation
What imaging is recommended to identify patients with ICAD?
The Stroke Outcomes and Neuroimaging of Intracranial Atherosclerosis (SONIA) trial sought to evaluate the reliability of noninvasive imaging modalities to identify a 50%–99% stenosis of large proximal cerebral arteries as compared with the gold standard of conventional angiography [3]. Qualifying vessels included the M1 segment of the middle cerebral artery, the carotid siphon, and intracranial vertebral and basilar arteries. Imaging techniques included MRA head without contrast (time of flight) and TCD ultrasound. Both of these imaging modalities demonstrated a high negative predictive value (NPV) for the presence of a 50%–99% intracranial stenosis; however, the positive predictive value (PPV) for TCD and MRA was only 55% and 66%, respectively. Subsequent studies of contrast-enhanced MRA and CTA of the headhave shown high PPV (78%–94%) and NPV (95%–100%) when compared with cerebral angiography for the detection of 70%–99% ICAD [4,5]. Together these data suggest that TCD and MRA of the head without contrast can be used as screening tools to rule out ICAD when normal; however, subsequent contrast-enhanced MRA or CTA is required when abnormalities are identified.
When choosing the best noninvasive imaging modality for any given patient, concomitant medical issues including renal disease, age, presence of a pacemaker, implantable cardiac defibrillator or other metal, and claustrophobia are factors to consider.
What are the potential mechanisms of ischemic stroke in patients with symptomatic ICAD?
Stroke in the symptomatic ICAD group occurs as a result of either (1) thrombus formation at the site of a high-grade stenosis due to an unstable atherosclerotic plaque, (2) low flow through a narrow, stenotic artery (hypoperfusion) or (3) a combination of atheroembolism and hypoperfusion [6]. While a thrombus can occlude the parent vessel at the site of stenosis, more commonly the thrombus migrates distally as an artery-artery embolism to a smaller caliber artery.
What medical regimen is recommended for this patient with symptomatic ICAD?

Dual Antiplatelet Therapy
DAT with aspirin and clopidogrel is thought to reduce ischemic events related to regional thromboembolism. The best clinical evidence of the use of DAT in symptomatic ICAD is found in outcomes data for the SAMMPRIS trial where 30-day recurrent ischemic stroke rates in the aggressive medical management arm were significantly lower than patients treated with aspirin or warfarin monotherapy as part of the Warfarin and Aspirin for Symptomatic Intracranial Arterial Stenosis (WASID) trial. A subgroup of patients enrolled in WASID with similar clinical characteristics to those in SAMMPRIS were found to have a 30-day rate of stroke or death of 10.7% and a collective 1-year rate of ischemic stroke, brain hemorrhage, or vascular death of 25% [2]. The corresponding rates in the SAMMPRIS medical management arm were 5.8% at 30 days and 17.5% at 1 year. Given that the recurrent stroke rates were significantly lower in as little as 30 days, these benefits have been hypothesized to be more likely secondary to the DAT regimen than other risk factor modification [7]. There is some evidence that longer term DAT with aspirin and clopidogrel up to 1 year may be associated with further reduction in stroke, MI, and vascular death with similar bleeding risk, but this needs to be evaluated in prospective studies [8].
Additional evidence of the benefit and potential mechanism from DAT in ICAD comes from CLAIR (Clopidogrel plus Aspirin versus Aspirin alone for Reducing Embolization in Patients with Acute Symptomatic Cerebral or Carotid Artery Stenosis Trial), a multicenter, randomized trial with blinded outcome assessment, with patients recruited at sites in Hong Kong, Singapore, China, Thailand, and Malaysia [9]. Patients were enrolled with an extracranial or intracranial stenosis (greater than or equal to 50%, as diagnosed by carotid duplex, transcranial Doppler, or magnetic resonance angiography) if they had a clinical diagnosis of acute ischemic stroke or transient ischemic attack (TIA) during the 7 days prior to enrollment and were found to have microembolic signals (suggesting microemboli of atherosclerotic origin) detected at baseline assessment with transcranial Doppler ultrasound. Patients were randomized to DAT with aspirin and clopidogrel versus aspirin monotherapy. Patients on DAT had significantly reduced microembolic signals compared with patients on aspirin monotherapy. Asymptomatic embolization with dual antiplatelet therapy may also proffer a reduction in clinical events in these patients with symptomatic ICAD.
Statin Therapy
Statin therapy is an integral part in the prevention of recurrent ischemic events in the symptomatic ICAD cohort. Post-hoc analyses from the WASID trial found that total cholesterol greater than 200 mg/dL was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death.
In 2013, the American Heart Association released new guidelines regarding the treatment of cholesterol to reduce atherosclerotic cardiovascular risk in adults [10]. Clinical atherosclerotic cardiovascular disease (ASCVD) is the manifestation of systemic atherosclerotic disease and defined as acute coronary syndromes, a history of myocardial infarction, stable or unstable angina, a history of coronary or other arterial revascularization, stroke, transient ischemic attack, or peripheral arterial disease presumed to be of atherosclerotic origin. Recommendations for secondary prevention in patients with clinical ASCVD include the use of high-intensity statin therapy (atorvastatin 40 or 80 mg, rosuvastatin 20 or 40 mg) for patients less than 75 years of age or moderate-intensity statin therapy (atorvastatin 10 or 20 mg, rosuvastatin 5 or 10 mg, pravastatin 40 mg, or simvastatin 20 or 40 mg) for patients > 75 years of age. In the SAMMPRIS trial, an LDL cholesterol level less than 70 mg/dL was targeted [7].
Blood Pressure Management
Post-hoc analysis of data from WASID demonstrated a statistically significant increase in recurrent stroke risk with increasing mean systolic and diastolic blood pressure (BP) [11]. This was particularly true in patients with mean SBP > 160 mm Hg. This is contrary to the common perception that BP should be maintained higher in patients with intracranial stenosis. In multivariable analysis, systolic BP greater than 140 mm Hg was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death. In the SAMMPRIS trial, the recommended BP goals for patients with symptomatic ICAD were less than 140/90 mm Hg in non-diabetic patients and less than 130/80 mm Hg in diabetic patients [7]. The timing and pace of blood pressure normalization for a recently symptomatic patient with ICAD is still unclear and needs further study.
Lifestyle Modification and Secondary Risk Factors
The SAMMPRIS protocol incorporated a lifestyle coach for all patients enrolled in the study. Lifestyle modification to achieve smoking cessation, regular physical activity, weight reduction for overweight patients, and glucose control in diabetes (goal hemoglobin A1c < 7.0%) were complementary to the pharmacotherapy (aspirin, clopidogrel, statin, and antihypertensive regimen) prescribed [7].
Patients should be encouraged to participate in aerobic exercise for at least 30 minutes at least 3 times weekly. Dietary modifications modeled after the Mediterranean diet should be encouraged. These should be coupled with additional efforts to address excessive weight as needed.
Successful smoking cessation proves one of the most challenging lifestyle modifications for this group of patients and may require the employment of an extended support system with both family and medical providers. Nicotine supplementation is a common first-line aid for cessation, which may be provided in the form of gum or transdermal patches. Additional pharmacotherapy to address central mechanisms of addiction may be necessary. Many patients benefit from the addition of an antidepressant therapy such as bupropion or an adjunctive medication such as varenicline (a nicotine receptor partial agonist that helps with breaking nicotine addiction). It is important to establish a quit date and detailed, multistep plan for cessation [12].
Exceptions and Other Considerations
These evidence-based recommendations are directed toward a specific group of patients with high-grade stenosis (70%–99%), a single symptomatic vessel, and relatively short segments of stenosis (< 14 mm) based on the inclusion and exclusion criteria for the SAMMPRIS trial [7]. Additionally, these patients were not enrolled during the immediate period following an acute ischemic event. There is less evidence on the management of patients who present with < 70% symptomatic ICAD but we can infer that therapy with DAT, statins, and risk factor modification are still of benefit for patients with 50%–69% stenosis or those requiring treatment during the acute evaluation [13]. Additionally, some of these SAMMPRIS exclusion criteria were included to select patients who could be considered intracranial stenting candidates (eg, short segment stenosis, single vessel disease, etc) and are not considered to impact on the benefits of the aggressive medical management regimen.
Further Evaluation
The patient was initiated on antithrombotic therapy on hospital day 2 (24 hours after IV tPA administration) with aspirin 325 mg daily and clopidogrel 300 mg oral load followed by 75 mg daily, ranitidine 150 mg twice a day to reduce gastroesophageal reflux and atorvastatin 80 mg daily. On hospital day 3, his neurological exam remained stable and he was initiated on losartan 25 mg daily with a plan to titrate the dose up as an outpatient to achieve target blood pressure. The patient and his wife were advised to maintain a blood pressure log and provide it to his physicians at follow-up and was provided smoking cessation counseling with a plan to remain off tobacco after discharge utilizing nicotine patches. The patient was discharged from the hospital 4 days after admission with a diagnosis of acute ischemic stroke secondary to symptomatic ICAD from regional thromboembolism. Early outpatient follow-up was scheduled with his primary physician within 1 week after discharge to evaluate BP and a vascular neurologist within 1 month after discharge.
What factors predict recurrent stroke in patients with symptomatic ICAD?
As evidenced in the WASID post-hoc analysis, female sex, prior ischemic stroke (versus TIA), time from qualifying event to enrollment (≤ 17 days after ischemic event), severity of stenosis (≥ 70% stenosis) of the symptomatic vessel, and history of diabetes were identified as independent risk factors for recurrent stroke in this population [14].
How do we manage patients who continue to have symptoms despite optimal medical therapy?
When evaluating symptomatic ICAD patients with recur-rent neurologic symptoms, several important factors should be considered:
- Do the recurrent neurologic symptoms localize to the original ICAD location?
Patients with symptomatic ICAD often have multiple risk factors that put them at risk for other subtypes of ischemic stroke including lacunar stroke. Repeat neuro-imaging with brain MRI is recommended to confirm the localization of a recurrent stroke is inside or outside of the territory of the ICAD.
- Were the recurrent neurologic symptoms provoked?
Patients with ICAD can be uniquely sensitive to fluctuations in cerebral blood flow and oxygenation. Diabetic patients with ICAD can develop autonomic neuropathy that results in orthostatic hypotension. If patients have recurrent transient ischemic events associated with postural changes, adequate treatment of orthostasis can reduce recurrent ischemic events. Other provoked circumstances include patients who develop anemia and patients with untreated severe obstructive sleep apnea who may awake with recurrent ischemic events due to hypoxemia.
- Is the patient adhering to the medication regimen?
Patients with symptomatic ICAD frequently have multiple medications to take for treatment of their comorbidities. Discussion of the number of missed doses of medications over the prior month is important to ascertain adherence. Patients should also be counseled to avoid concomitant medications that may cause drug interactions; given the known drug interactions between nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin, specific counseling on the avoidance of NSAIDs should be encouraged.
- Are the patient’s risk factors optimally managed?
Risk factor control should be assessed on a routine (eg every 3-6 months) basis and again if an ICAD patient has recurrent ischemic symptoms. Regular review of blood pressure logs, interval assessment of lipids, optimization of glucose control with serial hemoglobin A1c, tobacco cessation, and attention to weight management are paramount.
- Does the patient have an appropriate metabolic response to antiplatelet therapy?
If risk factors are optimized and the patient reports adherence to their medication regimen, aspirin and clopidogrel response should be evaluated. Aspirin resistance can be assessed by measuring the urinary level of 11-dehydrothromboxane B2 [15]. A urine level >1500 pg/mg creatinine should be expected in healthy, aspirin-free individuals; however, if this level is identified in a patient who is prescribed aspirin, aspirin resistance can be diagnosed. Various causes of aspirin resistance have been reported including inadequate adherence to aspirin therapy, concomitant use of a NSAID, genetic mutations in the COX-1 gene, non-platelet sources of thromboxane A2, and high platelet turnover [15]. Aspirin dosage adjustments should be made in consultation with a hematologist.
Resistance to clopidogrel has been less widely evaluated, but one meta-analysis estimated a mean prevalence of clopidogrel non-responsiveness at 21% [16]. While there is limited data on the optimal assessment of clopidogrel responsiveness in stroke patients, on-treatment platelet reactivity has been measured in parallel by means of light transmittance aggregometry, Verifynow P2Y12 and Platelet works assays, and the IMPACT-R and PFA-100 system in one study of patients undergoing coronary stent implantation [17]; of the platelet function assays assessed, only light transmittance aggregometry, Verifynow, and Platelet works assays were significantly associated with clinical outcomes in patients undergoing coronary stent implantation. Various causes of clopidogrel resistance have been reported including inadequate adherence to clopidogrel therapy, concomitant use of medications that interfere with clopidogrel prodrug conversion in the liver to its active metabolite, and genetic mutations in the cytochrome p450 3A4 gene [18,19]. Clopidogrel dosage adjustments should be made in consultation with a hematologist.
While the majority of patients will have no recurrent ischemic symptoms on aggressive medical therapy, some patients may continue to experience recurrent ischemic stroke or TIA secondary to ICAD despite optimal medical management. Patients who have hypoperfusion resulting in borderzone infarctions may be at higher risk for recurrent ischemic symptoms despite optimal medical therapy [20]. The risks of intracranial stenting, including stroke and death, must be weighed against the potential benefits. In the angioplasty plus stenting arm of the SAMMPRIS trial, the risk of stroke or death at 30 days was 14.7% [7]. In the angioplasty plus stenting arm of the VISSIT clinical trial evaluating symptomatic ICAD patients, the 30-day risk of stroke or death was 24% [21]. While intracranial angioplasty without stenting has been proposed as an alternative, there have been no randomized clinical trials to evaluate its efficacy beyond medical therapy alone in symptomatic ICAD patients. If endovascular treatment is considered, neuro-interventionists with high volume experience appear to have lower peri-procedural complications than those with low volume experience [22].
Another strategy that may be considered in symptomatic ICAD patients who have recurrent ischemic cerebral events due to symptomatic intracranial stenosis despite optimal medical therapy is indirect revascularization via encephaloduroarteriosynangiosis (EDAS). A single center retrospective study of 36 patients with ICAD and recent (< 30 days) TIAs or nondisabling strokes in the territory of the stenotic vessel (degree of stenosis not stated) and evidence of hypoperfusion and poor collateral flow on MR perfusion and/or conventional angiogram underwent EDAS [23]. Over a 2-year follow-up period, 5.6% of patients had ischemic strokes (1 stroke was periprocedural), compared with the estimated 17.2% risk of stroke in the SAMMPRIS cohort.
While endovascular and surgical techniques for revascularization of symptomatic ICAD are options for cases with medically refractory ischemic events due to hypoperfusion, further studies are needed to determine the safety of and optimal timing for these treatments.
Post-Discharge Follow-up Evaluation
At 1 month after discharge, the patient denied any new or recurrent signs or symptoms of stroke. Home blood pressure logs showed that BP was at target. Orthostatic BPs were assessed and no evidence of hypotension on standing was identified. Repeat laboratory evaluation was remarkable for hemoglobin A1c of 6.8% and LDL cholesterol of 64 mg/dL. The patient and his wife have been walking briskly 3 times per week for 45 minutes and continue to work on dietary modifications modeled after the Mediterranean diet. He is scheduled to follow up with his vascular neurologist 3 months after the stroke to discuss transition from DAT to aspirin monotherapy.
Conclusion
Intracranial atherosclerotic disease is a common cause of ischemic stroke, particularly amongst Asian, black, and Hispanic patients. Identification of ICAD can be performed with noninvasive arterial imaging including CT angiography or contrast-enhanced MR angiography as part of an ischemic stroke workup. Optimal medical management with early DAT with aspirin and clopidogrel along with aggressive risk factor and lifestyle modification has emerged as an effective first-line therapy. In patients with recurrent ischemic symptoms while optimally medically managed, endovascular therapy with angioplasty with or without stenting may be considered.
Corresponding author: Fadi Nahab, MD, 1635 Clifton Rd., Clinic B, Suite 2200, Atlanta, GA 30322, [email protected].
Financial disclosures: None.
1. Derdeyn C, Chimowitz M, Lynn MJ, et al. Aggressive medical treatment with or without stenting in high-risk patients with intracranial arterial stenosis (SAMMPRIS): the final results of a randomized trial. Lancet 2014;383:333–41.
2. Chimowitz, MI, Lynn MJ, Howlett-Smith H, et al. Comparision of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2004;351:1250–1.
3. Feldmann E, Wilterdink JL, Kosinski A, et al. The stroke outcomes and neuroimaging of intracranial atherosclerosis (SONIA) trial. Neurology 2007;68:2099–106.
4. Willinek WA, von Falkenhausen M, Born M, et al. Noninvasive detection of steno-occlusive disease of the supra-aortic arteries with three-dimensional contrast-enhanced magnetic resonance angiography: a prospective, intra-individual comparative analysis with digital subtraction angiography. Stroke 2005;36:38–43.
5. Bash S, Villablanca JP, Jahan R, et al. Intracranial vascular stenosis and occlusive disease: evaluation with CT angiography, MR angiography, and digital subtraction angiography. Am J Neuroradiol 2005;26:1012–21.
6. Qureshi A, Caplan L. Intracranial atherosclerosis. Lancet 2014;383:984–98.
7. Chimowitz MI, Lynn MJ, Derdeyn CP, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med 2011;365:993–1003.
8. Winningham M, Kasshout T, Bamford L, et al. Optimal duration of dual antiplatelet therapy for symptomatic intracranial stenosis. Stroke 2015;46:ATP100.
9. Wong KS, Chen C, Fu J, et al. Clopidogrel plus aspirin versus aspirin alone for reducing embolization in patients with acute symptomatic cerebral or carotid artery stenosis (CLAIR study): a randomized, open-label, blinded-endpoint trial. Lancet Neurol 2010;9:489–97.
10. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S1–45.
11. Turan TN, Cotsonis G, Lynn MJ, et al. Relationship between blood pressure and stroke recurrence in patients with intracranial arterial stenosis. Circulation 2007;115:2969–75.
12. Agency for Healthcare Research and Quality. Clinical guidelines for prescribing pharmacotherapy for smoking cessation. Accessed 8 May 2016 at www.ahrq.gov/professionals/clinicians-providers/guidelines-recommendations/tobacco/prescrib.html.
13. Nahab F, Kingston C, Frankel MR, et al. Early aggressive medical management for patients with symptomatic intracranial stenosis. J Stroke Cerebrovasc Dis 2013;22:87–91.
14. Kasner SE, Chimowitz MI, Lynn MJ, et al. Predictors of ischemic stroke in the territory of a symptomatic intracranial arterial stenosis. Circulation 2006;113:555–63.
15. Quest Diagnostics. Aspirin resistance. Accessed 23 Mar 2016 at www.questdiagnostics.com/testcenter/testguide.action?dc=TS_Aspirin_Resistance.
16. Alberts MJ. Platelet function testing for aspirin resistance is reasonable to do. Stroke 2010;41:2400–401.
17. Feher G, Feher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol 2010;26:171–86.
18. Taubert D, Kastrati A, Harlfinger S, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost 2004;92:311–6.
19. Lau WC, Gurbel PA, Watkins PB, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004;109:166–71.
20. Wabnitz AM, Derdeyn CP, Fiorella DJ, et al; for the SAMMPRIS Investigators. Infarct patterns in the anterior circulation as predictors of recurrent stroke in the medical arm of SAMMPRIS. Stroke 2016;47:A103.
21. Zaidat OO, Fitzsimmons BF, Woodward BK, et al. Effect of a balloon-expandable intracranial stent vs medical therapy on risk of stroke in patients with symptomatic intracranial stenosis: the VISSIT randomized clinical trial. JAMA 2015;313:1240–8.
22. Nahab F, Lynn MJ, Kasner SE, et al. Risk factors associated with major cerebrovascular complications after intracranial stenting. Neurology 2009;72:2014–9.
23. Gonzalez N, Dusick J, Connolly M, et al. Encephaloduroarteriosynangiosis for adult intracranial arterial steno-occlusive disease: long-term single-center experience with 107 operations. J Neurosurg 2015;123:654–61.
From the Emory University School of Medicine, Atlanta, GA.
Abstract
- Objective: To provide an overview of the diagnosis, clinical presentation, and management of symptomatic intracranial atherosclerotic disease (ICAD).
- Methods: Review of the current literature in the context of a clinical case.
- Results: ICAD is a common cause of ischemic strokes or transient ischemic attacks (TIAs), especially among Asian, black, and Hispanic patients. ICAD can be identified with noninvasive arterial imaging such as CT angiography, MR angiography, or transcranial Doppler ultrasound of the head when evaluating for the cause of an ischemic stroke or TIA. Aggressive medical management with dual antiplatelet therapy and lifestyle and risk factor modification has emerged as effective first-line therapy. In patients who have recurrent ischemic symptoms while on aggressive medical management, endovascular treatment can be considered.
- Conclusion: When symptomatic ICAD is identified early, aggressive medical management is effective in reducing the risk of recurrent ischemic events in this patient population.
Symptomatic intracranial atherosclerotic disease (ICAD) may represent the most common cause of ischemic stroke worldwide and the cause of approximately 8% to 10% of ischemic strokes in the United States [1–7]. It is a particularly important clinical entity due to the high recurrence rate of ischemic events in this population. The estimated recurrent stroke risk from symptomatic ICAD has been reported to be as high as 14.9% in the first year after an initial ischemic event [1].There are multiple risk factors for ICAD. Non-modifiable risk factors include race (particularly Asian, black, or Hispanic race), age, and family history of coronary artery disease or stroke; modifiable risk factors include diabetes, hypertension, and hyperlipidemia [2].
Case Study
Initial Presentation
A 78-year-old right-handed man presents to the outpatient clinic for follow-up evaluation after inpatient admission for acute ischemic stroke. The patient has an established medical history of hypertension, diabetes mellitus (diagnosed 10 years ago), dyslipidemia, and a 50 pack-year history of tobacco use.
Two weeks prior to the clinic visit, the patient presented to the emergency department (ED) via emergency medical services with right face and arm weakness and numbness and the inability to speak that had been ongoing for approximately 1 hour. The patient’s wife reported to ED providers that the patient had a similar episode 1 month prior that resolved completely in 30 to 45 minutes (and for which the patient never sought medical attention). Home medications at the time of admission included aspirin 81 mg and pravastatin 20 mg daily, both of which he takes intermittently.
The initial blood pressure was 185/76 mm Hg and blood glucose was 225 mg/dL. Initial exam was remarkable for the inability to answer orientation questions (but able to follow simple commands), forced gaze deviation to the left, right lower facial weakness, weakness in the right arm with no antigravity movement, moderately decreased sensation of the right face and arm, severe expressive aphasia, and severe dysarthria. A CT of the head without contrast showed no evidence of hemorrhage. Patient was suspected of having an acute ischemic stroke and was given intravenous tPA.
What are the possible mechanisms for this patient’s presentation with ischemic stroke?
This patient is presenting with right face and arm weakness with sensory loss, gaze deviation to the left, and expressive aphasia. Abnormalities of speech and gaze paresis that localize to the left frontal lobe with cortical involvement make a subcortical or brainstem lacunar ischemic event less likely. The syndrome is suggestive of a large artery occlusion of the left middle cerebral artery. This type of large artery occlusive disease in the anterior circulation is most often due to an embolus (artery-to-artery or cardiac origin) event and requires arterial imaging of the head and neck acutely as endovascular thrombectomy has been associated with reduced disability compared to intravenous tPA alone.
Further Evaluation
What imaging is recommended to identify patients with ICAD?
The Stroke Outcomes and Neuroimaging of Intracranial Atherosclerosis (SONIA) trial sought to evaluate the reliability of noninvasive imaging modalities to identify a 50%–99% stenosis of large proximal cerebral arteries as compared with the gold standard of conventional angiography [3]. Qualifying vessels included the M1 segment of the middle cerebral artery, the carotid siphon, and intracranial vertebral and basilar arteries. Imaging techniques included MRA head without contrast (time of flight) and TCD ultrasound. Both of these imaging modalities demonstrated a high negative predictive value (NPV) for the presence of a 50%–99% intracranial stenosis; however, the positive predictive value (PPV) for TCD and MRA was only 55% and 66%, respectively. Subsequent studies of contrast-enhanced MRA and CTA of the headhave shown high PPV (78%–94%) and NPV (95%–100%) when compared with cerebral angiography for the detection of 70%–99% ICAD [4,5]. Together these data suggest that TCD and MRA of the head without contrast can be used as screening tools to rule out ICAD when normal; however, subsequent contrast-enhanced MRA or CTA is required when abnormalities are identified.
When choosing the best noninvasive imaging modality for any given patient, concomitant medical issues including renal disease, age, presence of a pacemaker, implantable cardiac defibrillator or other metal, and claustrophobia are factors to consider.
What are the potential mechanisms of ischemic stroke in patients with symptomatic ICAD?
Stroke in the symptomatic ICAD group occurs as a result of either (1) thrombus formation at the site of a high-grade stenosis due to an unstable atherosclerotic plaque, (2) low flow through a narrow, stenotic artery (hypoperfusion) or (3) a combination of atheroembolism and hypoperfusion [6]. While a thrombus can occlude the parent vessel at the site of stenosis, more commonly the thrombus migrates distally as an artery-artery embolism to a smaller caliber artery.
What medical regimen is recommended for this patient with symptomatic ICAD?
Dual Antiplatelet Therapy
DAT with aspirin and clopidogrel is thought to reduce ischemic events related to regional thromboembolism. The best clinical evidence of the use of DAT in symptomatic ICAD is found in outcomes data for the SAMMPRIS trial where 30-day recurrent ischemic stroke rates in the aggressive medical management arm were significantly lower than patients treated with aspirin or warfarin monotherapy as part of the Warfarin and Aspirin for Symptomatic Intracranial Arterial Stenosis (WASID) trial. A subgroup of patients enrolled in WASID with similar clinical characteristics to those in SAMMPRIS were found to have a 30-day rate of stroke or death of 10.7% and a collective 1-year rate of ischemic stroke, brain hemorrhage, or vascular death of 25% [2]. The corresponding rates in the SAMMPRIS medical management arm were 5.8% at 30 days and 17.5% at 1 year. Given that the recurrent stroke rates were significantly lower in as little as 30 days, these benefits have been hypothesized to be more likely secondary to the DAT regimen than other risk factor modification [7]. There is some evidence that longer term DAT with aspirin and clopidogrel up to 1 year may be associated with further reduction in stroke, MI, and vascular death with similar bleeding risk, but this needs to be evaluated in prospective studies [8].
Additional evidence of the benefit and potential mechanism from DAT in ICAD comes from CLAIR (Clopidogrel plus Aspirin versus Aspirin alone for Reducing Embolization in Patients with Acute Symptomatic Cerebral or Carotid Artery Stenosis Trial), a multicenter, randomized trial with blinded outcome assessment, with patients recruited at sites in Hong Kong, Singapore, China, Thailand, and Malaysia [9]. Patients were enrolled with an extracranial or intracranial stenosis (greater than or equal to 50%, as diagnosed by carotid duplex, transcranial Doppler, or magnetic resonance angiography) if they had a clinical diagnosis of acute ischemic stroke or transient ischemic attack (TIA) during the 7 days prior to enrollment and were found to have microembolic signals (suggesting microemboli of atherosclerotic origin) detected at baseline assessment with transcranial Doppler ultrasound. Patients were randomized to DAT with aspirin and clopidogrel versus aspirin monotherapy. Patients on DAT had significantly reduced microembolic signals compared with patients on aspirin monotherapy. Asymptomatic embolization with dual antiplatelet therapy may also proffer a reduction in clinical events in these patients with symptomatic ICAD.
Statin Therapy
Statin therapy is an integral part in the prevention of recurrent ischemic events in the symptomatic ICAD cohort. Post-hoc analyses from the WASID trial found that total cholesterol greater than 200 mg/dL was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death.
In 2013, the American Heart Association released new guidelines regarding the treatment of cholesterol to reduce atherosclerotic cardiovascular risk in adults [10]. Clinical atherosclerotic cardiovascular disease (ASCVD) is the manifestation of systemic atherosclerotic disease and defined as acute coronary syndromes, a history of myocardial infarction, stable or unstable angina, a history of coronary or other arterial revascularization, stroke, transient ischemic attack, or peripheral arterial disease presumed to be of atherosclerotic origin. Recommendations for secondary prevention in patients with clinical ASCVD include the use of high-intensity statin therapy (atorvastatin 40 or 80 mg, rosuvastatin 20 or 40 mg) for patients less than 75 years of age or moderate-intensity statin therapy (atorvastatin 10 or 20 mg, rosuvastatin 5 or 10 mg, pravastatin 40 mg, or simvastatin 20 or 40 mg) for patients > 75 years of age. In the SAMMPRIS trial, an LDL cholesterol level less than 70 mg/dL was targeted [7].
Blood Pressure Management
Post-hoc analysis of data from WASID demonstrated a statistically significant increase in recurrent stroke risk with increasing mean systolic and diastolic blood pressure (BP) [11]. This was particularly true in patients with mean SBP > 160 mm Hg. This is contrary to the common perception that BP should be maintained higher in patients with intracranial stenosis. In multivariable analysis, systolic BP greater than 140 mm Hg was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death. In the SAMMPRIS trial, the recommended BP goals for patients with symptomatic ICAD were less than 140/90 mm Hg in non-diabetic patients and less than 130/80 mm Hg in diabetic patients [7]. The timing and pace of blood pressure normalization for a recently symptomatic patient with ICAD is still unclear and needs further study.
Lifestyle Modification and Secondary Risk Factors
The SAMMPRIS protocol incorporated a lifestyle coach for all patients enrolled in the study. Lifestyle modification to achieve smoking cessation, regular physical activity, weight reduction for overweight patients, and glucose control in diabetes (goal hemoglobin A1c < 7.0%) were complementary to the pharmacotherapy (aspirin, clopidogrel, statin, and antihypertensive regimen) prescribed [7].
Patients should be encouraged to participate in aerobic exercise for at least 30 minutes at least 3 times weekly. Dietary modifications modeled after the Mediterranean diet should be encouraged. These should be coupled with additional efforts to address excessive weight as needed.
Successful smoking cessation proves one of the most challenging lifestyle modifications for this group of patients and may require the employment of an extended support system with both family and medical providers. Nicotine supplementation is a common first-line aid for cessation, which may be provided in the form of gum or transdermal patches. Additional pharmacotherapy to address central mechanisms of addiction may be necessary. Many patients benefit from the addition of an antidepressant therapy such as bupropion or an adjunctive medication such as varenicline (a nicotine receptor partial agonist that helps with breaking nicotine addiction). It is important to establish a quit date and detailed, multistep plan for cessation [12].
Exceptions and Other Considerations
These evidence-based recommendations are directed toward a specific group of patients with high-grade stenosis (70%–99%), a single symptomatic vessel, and relatively short segments of stenosis (< 14 mm) based on the inclusion and exclusion criteria for the SAMMPRIS trial [7]. Additionally, these patients were not enrolled during the immediate period following an acute ischemic event. There is less evidence on the management of patients who present with < 70% symptomatic ICAD but we can infer that therapy with DAT, statins, and risk factor modification are still of benefit for patients with 50%–69% stenosis or those requiring treatment during the acute evaluation [13]. Additionally, some of these SAMMPRIS exclusion criteria were included to select patients who could be considered intracranial stenting candidates (eg, short segment stenosis, single vessel disease, etc) and are not considered to impact on the benefits of the aggressive medical management regimen.
Further Evaluation
The patient was initiated on antithrombotic therapy on hospital day 2 (24 hours after IV tPA administration) with aspirin 325 mg daily and clopidogrel 300 mg oral load followed by 75 mg daily, ranitidine 150 mg twice a day to reduce gastroesophageal reflux and atorvastatin 80 mg daily. On hospital day 3, his neurological exam remained stable and he was initiated on losartan 25 mg daily with a plan to titrate the dose up as an outpatient to achieve target blood pressure. The patient and his wife were advised to maintain a blood pressure log and provide it to his physicians at follow-up and was provided smoking cessation counseling with a plan to remain off tobacco after discharge utilizing nicotine patches. The patient was discharged from the hospital 4 days after admission with a diagnosis of acute ischemic stroke secondary to symptomatic ICAD from regional thromboembolism. Early outpatient follow-up was scheduled with his primary physician within 1 week after discharge to evaluate BP and a vascular neurologist within 1 month after discharge.
What factors predict recurrent stroke in patients with symptomatic ICAD?
As evidenced in the WASID post-hoc analysis, female sex, prior ischemic stroke (versus TIA), time from qualifying event to enrollment (≤ 17 days after ischemic event), severity of stenosis (≥ 70% stenosis) of the symptomatic vessel, and history of diabetes were identified as independent risk factors for recurrent stroke in this population [14].
How do we manage patients who continue to have symptoms despite optimal medical therapy?
When evaluating symptomatic ICAD patients with recur-rent neurologic symptoms, several important factors should be considered:
- Do the recurrent neurologic symptoms localize to the original ICAD location?
Patients with symptomatic ICAD often have multiple risk factors that put them at risk for other subtypes of ischemic stroke including lacunar stroke. Repeat neuro-imaging with brain MRI is recommended to confirm the localization of a recurrent stroke is inside or outside of the territory of the ICAD.
- Were the recurrent neurologic symptoms provoked?
Patients with ICAD can be uniquely sensitive to fluctuations in cerebral blood flow and oxygenation. Diabetic patients with ICAD can develop autonomic neuropathy that results in orthostatic hypotension. If patients have recurrent transient ischemic events associated with postural changes, adequate treatment of orthostasis can reduce recurrent ischemic events. Other provoked circumstances include patients who develop anemia and patients with untreated severe obstructive sleep apnea who may awake with recurrent ischemic events due to hypoxemia.
- Is the patient adhering to the medication regimen?
Patients with symptomatic ICAD frequently have multiple medications to take for treatment of their comorbidities. Discussion of the number of missed doses of medications over the prior month is important to ascertain adherence. Patients should also be counseled to avoid concomitant medications that may cause drug interactions; given the known drug interactions between nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin, specific counseling on the avoidance of NSAIDs should be encouraged.
- Are the patient’s risk factors optimally managed?
Risk factor control should be assessed on a routine (eg every 3-6 months) basis and again if an ICAD patient has recurrent ischemic symptoms. Regular review of blood pressure logs, interval assessment of lipids, optimization of glucose control with serial hemoglobin A1c, tobacco cessation, and attention to weight management are paramount.
- Does the patient have an appropriate metabolic response to antiplatelet therapy?
If risk factors are optimized and the patient reports adherence to their medication regimen, aspirin and clopidogrel response should be evaluated. Aspirin resistance can be assessed by measuring the urinary level of 11-dehydrothromboxane B2 [15]. A urine level >1500 pg/mg creatinine should be expected in healthy, aspirin-free individuals; however, if this level is identified in a patient who is prescribed aspirin, aspirin resistance can be diagnosed. Various causes of aspirin resistance have been reported including inadequate adherence to aspirin therapy, concomitant use of a NSAID, genetic mutations in the COX-1 gene, non-platelet sources of thromboxane A2, and high platelet turnover [15]. Aspirin dosage adjustments should be made in consultation with a hematologist.
Resistance to clopidogrel has been less widely evaluated, but one meta-analysis estimated a mean prevalence of clopidogrel non-responsiveness at 21% [16]. While there is limited data on the optimal assessment of clopidogrel responsiveness in stroke patients, on-treatment platelet reactivity has been measured in parallel by means of light transmittance aggregometry, Verifynow P2Y12 and Platelet works assays, and the IMPACT-R and PFA-100 system in one study of patients undergoing coronary stent implantation [17]; of the platelet function assays assessed, only light transmittance aggregometry, Verifynow, and Platelet works assays were significantly associated with clinical outcomes in patients undergoing coronary stent implantation. Various causes of clopidogrel resistance have been reported including inadequate adherence to clopidogrel therapy, concomitant use of medications that interfere with clopidogrel prodrug conversion in the liver to its active metabolite, and genetic mutations in the cytochrome p450 3A4 gene [18,19]. Clopidogrel dosage adjustments should be made in consultation with a hematologist.
While the majority of patients will have no recurrent ischemic symptoms on aggressive medical therapy, some patients may continue to experience recurrent ischemic stroke or TIA secondary to ICAD despite optimal medical management. Patients who have hypoperfusion resulting in borderzone infarctions may be at higher risk for recurrent ischemic symptoms despite optimal medical therapy [20]. The risks of intracranial stenting, including stroke and death, must be weighed against the potential benefits. In the angioplasty plus stenting arm of the SAMMPRIS trial, the risk of stroke or death at 30 days was 14.7% [7]. In the angioplasty plus stenting arm of the VISSIT clinical trial evaluating symptomatic ICAD patients, the 30-day risk of stroke or death was 24% [21]. While intracranial angioplasty without stenting has been proposed as an alternative, there have been no randomized clinical trials to evaluate its efficacy beyond medical therapy alone in symptomatic ICAD patients. If endovascular treatment is considered, neuro-interventionists with high volume experience appear to have lower peri-procedural complications than those with low volume experience [22].
Another strategy that may be considered in symptomatic ICAD patients who have recurrent ischemic cerebral events due to symptomatic intracranial stenosis despite optimal medical therapy is indirect revascularization via encephaloduroarteriosynangiosis (EDAS). A single center retrospective study of 36 patients with ICAD and recent (< 30 days) TIAs or nondisabling strokes in the territory of the stenotic vessel (degree of stenosis not stated) and evidence of hypoperfusion and poor collateral flow on MR perfusion and/or conventional angiogram underwent EDAS [23]. Over a 2-year follow-up period, 5.6% of patients had ischemic strokes (1 stroke was periprocedural), compared with the estimated 17.2% risk of stroke in the SAMMPRIS cohort.
While endovascular and surgical techniques for revascularization of symptomatic ICAD are options for cases with medically refractory ischemic events due to hypoperfusion, further studies are needed to determine the safety of and optimal timing for these treatments.
Post-Discharge Follow-up Evaluation
At 1 month after discharge, the patient denied any new or recurrent signs or symptoms of stroke. Home blood pressure logs showed that BP was at target. Orthostatic BPs were assessed and no evidence of hypotension on standing was identified. Repeat laboratory evaluation was remarkable for hemoglobin A1c of 6.8% and LDL cholesterol of 64 mg/dL. The patient and his wife have been walking briskly 3 times per week for 45 minutes and continue to work on dietary modifications modeled after the Mediterranean diet. He is scheduled to follow up with his vascular neurologist 3 months after the stroke to discuss transition from DAT to aspirin monotherapy.
Conclusion
Intracranial atherosclerotic disease is a common cause of ischemic stroke, particularly amongst Asian, black, and Hispanic patients. Identification of ICAD can be performed with noninvasive arterial imaging including CT angiography or contrast-enhanced MR angiography as part of an ischemic stroke workup. Optimal medical management with early DAT with aspirin and clopidogrel along with aggressive risk factor and lifestyle modification has emerged as an effective first-line therapy. In patients with recurrent ischemic symptoms while optimally medically managed, endovascular therapy with angioplasty with or without stenting may be considered.
Corresponding author: Fadi Nahab, MD, 1635 Clifton Rd., Clinic B, Suite 2200, Atlanta, GA 30322, [email protected].
Financial disclosures: None.
From the Emory University School of Medicine, Atlanta, GA.
Abstract
- Objective: To provide an overview of the diagnosis, clinical presentation, and management of symptomatic intracranial atherosclerotic disease (ICAD).
- Methods: Review of the current literature in the context of a clinical case.
- Results: ICAD is a common cause of ischemic strokes or transient ischemic attacks (TIAs), especially among Asian, black, and Hispanic patients. ICAD can be identified with noninvasive arterial imaging such as CT angiography, MR angiography, or transcranial Doppler ultrasound of the head when evaluating for the cause of an ischemic stroke or TIA. Aggressive medical management with dual antiplatelet therapy and lifestyle and risk factor modification has emerged as effective first-line therapy. In patients who have recurrent ischemic symptoms while on aggressive medical management, endovascular treatment can be considered.
- Conclusion: When symptomatic ICAD is identified early, aggressive medical management is effective in reducing the risk of recurrent ischemic events in this patient population.
Symptomatic intracranial atherosclerotic disease (ICAD) may represent the most common cause of ischemic stroke worldwide and the cause of approximately 8% to 10% of ischemic strokes in the United States [1–7]. It is a particularly important clinical entity due to the high recurrence rate of ischemic events in this population. The estimated recurrent stroke risk from symptomatic ICAD has been reported to be as high as 14.9% in the first year after an initial ischemic event [1].There are multiple risk factors for ICAD. Non-modifiable risk factors include race (particularly Asian, black, or Hispanic race), age, and family history of coronary artery disease or stroke; modifiable risk factors include diabetes, hypertension, and hyperlipidemia [2].
Case Study
Initial Presentation
A 78-year-old right-handed man presents to the outpatient clinic for follow-up evaluation after inpatient admission for acute ischemic stroke. The patient has an established medical history of hypertension, diabetes mellitus (diagnosed 10 years ago), dyslipidemia, and a 50 pack-year history of tobacco use.
Two weeks prior to the clinic visit, the patient presented to the emergency department (ED) via emergency medical services with right face and arm weakness and numbness and the inability to speak that had been ongoing for approximately 1 hour. The patient’s wife reported to ED providers that the patient had a similar episode 1 month prior that resolved completely in 30 to 45 minutes (and for which the patient never sought medical attention). Home medications at the time of admission included aspirin 81 mg and pravastatin 20 mg daily, both of which he takes intermittently.
The initial blood pressure was 185/76 mm Hg and blood glucose was 225 mg/dL. Initial exam was remarkable for the inability to answer orientation questions (but able to follow simple commands), forced gaze deviation to the left, right lower facial weakness, weakness in the right arm with no antigravity movement, moderately decreased sensation of the right face and arm, severe expressive aphasia, and severe dysarthria. A CT of the head without contrast showed no evidence of hemorrhage. Patient was suspected of having an acute ischemic stroke and was given intravenous tPA.
What are the possible mechanisms for this patient’s presentation with ischemic stroke?
This patient is presenting with right face and arm weakness with sensory loss, gaze deviation to the left, and expressive aphasia. Abnormalities of speech and gaze paresis that localize to the left frontal lobe with cortical involvement make a subcortical or brainstem lacunar ischemic event less likely. The syndrome is suggestive of a large artery occlusion of the left middle cerebral artery. This type of large artery occlusive disease in the anterior circulation is most often due to an embolus (artery-to-artery or cardiac origin) event and requires arterial imaging of the head and neck acutely as endovascular thrombectomy has been associated with reduced disability compared to intravenous tPA alone.
Further Evaluation
What imaging is recommended to identify patients with ICAD?
The Stroke Outcomes and Neuroimaging of Intracranial Atherosclerosis (SONIA) trial sought to evaluate the reliability of noninvasive imaging modalities to identify a 50%–99% stenosis of large proximal cerebral arteries as compared with the gold standard of conventional angiography [3]. Qualifying vessels included the M1 segment of the middle cerebral artery, the carotid siphon, and intracranial vertebral and basilar arteries. Imaging techniques included MRA head without contrast (time of flight) and TCD ultrasound. Both of these imaging modalities demonstrated a high negative predictive value (NPV) for the presence of a 50%–99% intracranial stenosis; however, the positive predictive value (PPV) for TCD and MRA was only 55% and 66%, respectively. Subsequent studies of contrast-enhanced MRA and CTA of the headhave shown high PPV (78%–94%) and NPV (95%–100%) when compared with cerebral angiography for the detection of 70%–99% ICAD [4,5]. Together these data suggest that TCD and MRA of the head without contrast can be used as screening tools to rule out ICAD when normal; however, subsequent contrast-enhanced MRA or CTA is required when abnormalities are identified.
When choosing the best noninvasive imaging modality for any given patient, concomitant medical issues including renal disease, age, presence of a pacemaker, implantable cardiac defibrillator or other metal, and claustrophobia are factors to consider.
What are the potential mechanisms of ischemic stroke in patients with symptomatic ICAD?
Stroke in the symptomatic ICAD group occurs as a result of either (1) thrombus formation at the site of a high-grade stenosis due to an unstable atherosclerotic plaque, (2) low flow through a narrow, stenotic artery (hypoperfusion) or (3) a combination of atheroembolism and hypoperfusion [6]. While a thrombus can occlude the parent vessel at the site of stenosis, more commonly the thrombus migrates distally as an artery-artery embolism to a smaller caliber artery.
What medical regimen is recommended for this patient with symptomatic ICAD?
Dual Antiplatelet Therapy
DAT with aspirin and clopidogrel is thought to reduce ischemic events related to regional thromboembolism. The best clinical evidence of the use of DAT in symptomatic ICAD is found in outcomes data for the SAMMPRIS trial where 30-day recurrent ischemic stroke rates in the aggressive medical management arm were significantly lower than patients treated with aspirin or warfarin monotherapy as part of the Warfarin and Aspirin for Symptomatic Intracranial Arterial Stenosis (WASID) trial. A subgroup of patients enrolled in WASID with similar clinical characteristics to those in SAMMPRIS were found to have a 30-day rate of stroke or death of 10.7% and a collective 1-year rate of ischemic stroke, brain hemorrhage, or vascular death of 25% [2]. The corresponding rates in the SAMMPRIS medical management arm were 5.8% at 30 days and 17.5% at 1 year. Given that the recurrent stroke rates were significantly lower in as little as 30 days, these benefits have been hypothesized to be more likely secondary to the DAT regimen than other risk factor modification [7]. There is some evidence that longer term DAT with aspirin and clopidogrel up to 1 year may be associated with further reduction in stroke, MI, and vascular death with similar bleeding risk, but this needs to be evaluated in prospective studies [8].
Additional evidence of the benefit and potential mechanism from DAT in ICAD comes from CLAIR (Clopidogrel plus Aspirin versus Aspirin alone for Reducing Embolization in Patients with Acute Symptomatic Cerebral or Carotid Artery Stenosis Trial), a multicenter, randomized trial with blinded outcome assessment, with patients recruited at sites in Hong Kong, Singapore, China, Thailand, and Malaysia [9]. Patients were enrolled with an extracranial or intracranial stenosis (greater than or equal to 50%, as diagnosed by carotid duplex, transcranial Doppler, or magnetic resonance angiography) if they had a clinical diagnosis of acute ischemic stroke or transient ischemic attack (TIA) during the 7 days prior to enrollment and were found to have microembolic signals (suggesting microemboli of atherosclerotic origin) detected at baseline assessment with transcranial Doppler ultrasound. Patients were randomized to DAT with aspirin and clopidogrel versus aspirin monotherapy. Patients on DAT had significantly reduced microembolic signals compared with patients on aspirin monotherapy. Asymptomatic embolization with dual antiplatelet therapy may also proffer a reduction in clinical events in these patients with symptomatic ICAD.
Statin Therapy
Statin therapy is an integral part in the prevention of recurrent ischemic events in the symptomatic ICAD cohort. Post-hoc analyses from the WASID trial found that total cholesterol greater than 200 mg/dL was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death.
In 2013, the American Heart Association released new guidelines regarding the treatment of cholesterol to reduce atherosclerotic cardiovascular risk in adults [10]. Clinical atherosclerotic cardiovascular disease (ASCVD) is the manifestation of systemic atherosclerotic disease and defined as acute coronary syndromes, a history of myocardial infarction, stable or unstable angina, a history of coronary or other arterial revascularization, stroke, transient ischemic attack, or peripheral arterial disease presumed to be of atherosclerotic origin. Recommendations for secondary prevention in patients with clinical ASCVD include the use of high-intensity statin therapy (atorvastatin 40 or 80 mg, rosuvastatin 20 or 40 mg) for patients less than 75 years of age or moderate-intensity statin therapy (atorvastatin 10 or 20 mg, rosuvastatin 5 or 10 mg, pravastatin 40 mg, or simvastatin 20 or 40 mg) for patients > 75 years of age. In the SAMMPRIS trial, an LDL cholesterol level less than 70 mg/dL was targeted [7].
Blood Pressure Management
Post-hoc analysis of data from WASID demonstrated a statistically significant increase in recurrent stroke risk with increasing mean systolic and diastolic blood pressure (BP) [11]. This was particularly true in patients with mean SBP > 160 mm Hg. This is contrary to the common perception that BP should be maintained higher in patients with intracranial stenosis. In multivariable analysis, systolic BP greater than 140 mm Hg was associated with an increased risk of ischemic stroke, myocardial infarction, or vascular death. In the SAMMPRIS trial, the recommended BP goals for patients with symptomatic ICAD were less than 140/90 mm Hg in non-diabetic patients and less than 130/80 mm Hg in diabetic patients [7]. The timing and pace of blood pressure normalization for a recently symptomatic patient with ICAD is still unclear and needs further study.
Lifestyle Modification and Secondary Risk Factors
The SAMMPRIS protocol incorporated a lifestyle coach for all patients enrolled in the study. Lifestyle modification to achieve smoking cessation, regular physical activity, weight reduction for overweight patients, and glucose control in diabetes (goal hemoglobin A1c < 7.0%) were complementary to the pharmacotherapy (aspirin, clopidogrel, statin, and antihypertensive regimen) prescribed [7].
Patients should be encouraged to participate in aerobic exercise for at least 30 minutes at least 3 times weekly. Dietary modifications modeled after the Mediterranean diet should be encouraged. These should be coupled with additional efforts to address excessive weight as needed.
Successful smoking cessation proves one of the most challenging lifestyle modifications for this group of patients and may require the employment of an extended support system with both family and medical providers. Nicotine supplementation is a common first-line aid for cessation, which may be provided in the form of gum or transdermal patches. Additional pharmacotherapy to address central mechanisms of addiction may be necessary. Many patients benefit from the addition of an antidepressant therapy such as bupropion or an adjunctive medication such as varenicline (a nicotine receptor partial agonist that helps with breaking nicotine addiction). It is important to establish a quit date and detailed, multistep plan for cessation [12].
Exceptions and Other Considerations
These evidence-based recommendations are directed toward a specific group of patients with high-grade stenosis (70%–99%), a single symptomatic vessel, and relatively short segments of stenosis (< 14 mm) based on the inclusion and exclusion criteria for the SAMMPRIS trial [7]. Additionally, these patients were not enrolled during the immediate period following an acute ischemic event. There is less evidence on the management of patients who present with < 70% symptomatic ICAD but we can infer that therapy with DAT, statins, and risk factor modification are still of benefit for patients with 50%–69% stenosis or those requiring treatment during the acute evaluation [13]. Additionally, some of these SAMMPRIS exclusion criteria were included to select patients who could be considered intracranial stenting candidates (eg, short segment stenosis, single vessel disease, etc) and are not considered to impact on the benefits of the aggressive medical management regimen.
Further Evaluation
The patient was initiated on antithrombotic therapy on hospital day 2 (24 hours after IV tPA administration) with aspirin 325 mg daily and clopidogrel 300 mg oral load followed by 75 mg daily, ranitidine 150 mg twice a day to reduce gastroesophageal reflux and atorvastatin 80 mg daily. On hospital day 3, his neurological exam remained stable and he was initiated on losartan 25 mg daily with a plan to titrate the dose up as an outpatient to achieve target blood pressure. The patient and his wife were advised to maintain a blood pressure log and provide it to his physicians at follow-up and was provided smoking cessation counseling with a plan to remain off tobacco after discharge utilizing nicotine patches. The patient was discharged from the hospital 4 days after admission with a diagnosis of acute ischemic stroke secondary to symptomatic ICAD from regional thromboembolism. Early outpatient follow-up was scheduled with his primary physician within 1 week after discharge to evaluate BP and a vascular neurologist within 1 month after discharge.
What factors predict recurrent stroke in patients with symptomatic ICAD?
As evidenced in the WASID post-hoc analysis, female sex, prior ischemic stroke (versus TIA), time from qualifying event to enrollment (≤ 17 days after ischemic event), severity of stenosis (≥ 70% stenosis) of the symptomatic vessel, and history of diabetes were identified as independent risk factors for recurrent stroke in this population [14].
How do we manage patients who continue to have symptoms despite optimal medical therapy?
When evaluating symptomatic ICAD patients with recur-rent neurologic symptoms, several important factors should be considered:
- Do the recurrent neurologic symptoms localize to the original ICAD location?
Patients with symptomatic ICAD often have multiple risk factors that put them at risk for other subtypes of ischemic stroke including lacunar stroke. Repeat neuro-imaging with brain MRI is recommended to confirm the localization of a recurrent stroke is inside or outside of the territory of the ICAD.
- Were the recurrent neurologic symptoms provoked?
Patients with ICAD can be uniquely sensitive to fluctuations in cerebral blood flow and oxygenation. Diabetic patients with ICAD can develop autonomic neuropathy that results in orthostatic hypotension. If patients have recurrent transient ischemic events associated with postural changes, adequate treatment of orthostasis can reduce recurrent ischemic events. Other provoked circumstances include patients who develop anemia and patients with untreated severe obstructive sleep apnea who may awake with recurrent ischemic events due to hypoxemia.
- Is the patient adhering to the medication regimen?
Patients with symptomatic ICAD frequently have multiple medications to take for treatment of their comorbidities. Discussion of the number of missed doses of medications over the prior month is important to ascertain adherence. Patients should also be counseled to avoid concomitant medications that may cause drug interactions; given the known drug interactions between nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin, specific counseling on the avoidance of NSAIDs should be encouraged.
- Are the patient’s risk factors optimally managed?
Risk factor control should be assessed on a routine (eg every 3-6 months) basis and again if an ICAD patient has recurrent ischemic symptoms. Regular review of blood pressure logs, interval assessment of lipids, optimization of glucose control with serial hemoglobin A1c, tobacco cessation, and attention to weight management are paramount.
- Does the patient have an appropriate metabolic response to antiplatelet therapy?
If risk factors are optimized and the patient reports adherence to their medication regimen, aspirin and clopidogrel response should be evaluated. Aspirin resistance can be assessed by measuring the urinary level of 11-dehydrothromboxane B2 [15]. A urine level >1500 pg/mg creatinine should be expected in healthy, aspirin-free individuals; however, if this level is identified in a patient who is prescribed aspirin, aspirin resistance can be diagnosed. Various causes of aspirin resistance have been reported including inadequate adherence to aspirin therapy, concomitant use of a NSAID, genetic mutations in the COX-1 gene, non-platelet sources of thromboxane A2, and high platelet turnover [15]. Aspirin dosage adjustments should be made in consultation with a hematologist.
Resistance to clopidogrel has been less widely evaluated, but one meta-analysis estimated a mean prevalence of clopidogrel non-responsiveness at 21% [16]. While there is limited data on the optimal assessment of clopidogrel responsiveness in stroke patients, on-treatment platelet reactivity has been measured in parallel by means of light transmittance aggregometry, Verifynow P2Y12 and Platelet works assays, and the IMPACT-R and PFA-100 system in one study of patients undergoing coronary stent implantation [17]; of the platelet function assays assessed, only light transmittance aggregometry, Verifynow, and Platelet works assays were significantly associated with clinical outcomes in patients undergoing coronary stent implantation. Various causes of clopidogrel resistance have been reported including inadequate adherence to clopidogrel therapy, concomitant use of medications that interfere with clopidogrel prodrug conversion in the liver to its active metabolite, and genetic mutations in the cytochrome p450 3A4 gene [18,19]. Clopidogrel dosage adjustments should be made in consultation with a hematologist.
While the majority of patients will have no recurrent ischemic symptoms on aggressive medical therapy, some patients may continue to experience recurrent ischemic stroke or TIA secondary to ICAD despite optimal medical management. Patients who have hypoperfusion resulting in borderzone infarctions may be at higher risk for recurrent ischemic symptoms despite optimal medical therapy [20]. The risks of intracranial stenting, including stroke and death, must be weighed against the potential benefits. In the angioplasty plus stenting arm of the SAMMPRIS trial, the risk of stroke or death at 30 days was 14.7% [7]. In the angioplasty plus stenting arm of the VISSIT clinical trial evaluating symptomatic ICAD patients, the 30-day risk of stroke or death was 24% [21]. While intracranial angioplasty without stenting has been proposed as an alternative, there have been no randomized clinical trials to evaluate its efficacy beyond medical therapy alone in symptomatic ICAD patients. If endovascular treatment is considered, neuro-interventionists with high volume experience appear to have lower peri-procedural complications than those with low volume experience [22].
Another strategy that may be considered in symptomatic ICAD patients who have recurrent ischemic cerebral events due to symptomatic intracranial stenosis despite optimal medical therapy is indirect revascularization via encephaloduroarteriosynangiosis (EDAS). A single center retrospective study of 36 patients with ICAD and recent (< 30 days) TIAs or nondisabling strokes in the territory of the stenotic vessel (degree of stenosis not stated) and evidence of hypoperfusion and poor collateral flow on MR perfusion and/or conventional angiogram underwent EDAS [23]. Over a 2-year follow-up period, 5.6% of patients had ischemic strokes (1 stroke was periprocedural), compared with the estimated 17.2% risk of stroke in the SAMMPRIS cohort.
While endovascular and surgical techniques for revascularization of symptomatic ICAD are options for cases with medically refractory ischemic events due to hypoperfusion, further studies are needed to determine the safety of and optimal timing for these treatments.
Post-Discharge Follow-up Evaluation
At 1 month after discharge, the patient denied any new or recurrent signs or symptoms of stroke. Home blood pressure logs showed that BP was at target. Orthostatic BPs were assessed and no evidence of hypotension on standing was identified. Repeat laboratory evaluation was remarkable for hemoglobin A1c of 6.8% and LDL cholesterol of 64 mg/dL. The patient and his wife have been walking briskly 3 times per week for 45 minutes and continue to work on dietary modifications modeled after the Mediterranean diet. He is scheduled to follow up with his vascular neurologist 3 months after the stroke to discuss transition from DAT to aspirin monotherapy.
Conclusion
Intracranial atherosclerotic disease is a common cause of ischemic stroke, particularly amongst Asian, black, and Hispanic patients. Identification of ICAD can be performed with noninvasive arterial imaging including CT angiography or contrast-enhanced MR angiography as part of an ischemic stroke workup. Optimal medical management with early DAT with aspirin and clopidogrel along with aggressive risk factor and lifestyle modification has emerged as an effective first-line therapy. In patients with recurrent ischemic symptoms while optimally medically managed, endovascular therapy with angioplasty with or without stenting may be considered.
Corresponding author: Fadi Nahab, MD, 1635 Clifton Rd., Clinic B, Suite 2200, Atlanta, GA 30322, [email protected].
Financial disclosures: None.
1. Derdeyn C, Chimowitz M, Lynn MJ, et al. Aggressive medical treatment with or without stenting in high-risk patients with intracranial arterial stenosis (SAMMPRIS): the final results of a randomized trial. Lancet 2014;383:333–41.
2. Chimowitz, MI, Lynn MJ, Howlett-Smith H, et al. Comparision of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2004;351:1250–1.
3. Feldmann E, Wilterdink JL, Kosinski A, et al. The stroke outcomes and neuroimaging of intracranial atherosclerosis (SONIA) trial. Neurology 2007;68:2099–106.
4. Willinek WA, von Falkenhausen M, Born M, et al. Noninvasive detection of steno-occlusive disease of the supra-aortic arteries with three-dimensional contrast-enhanced magnetic resonance angiography: a prospective, intra-individual comparative analysis with digital subtraction angiography. Stroke 2005;36:38–43.
5. Bash S, Villablanca JP, Jahan R, et al. Intracranial vascular stenosis and occlusive disease: evaluation with CT angiography, MR angiography, and digital subtraction angiography. Am J Neuroradiol 2005;26:1012–21.
6. Qureshi A, Caplan L. Intracranial atherosclerosis. Lancet 2014;383:984–98.
7. Chimowitz MI, Lynn MJ, Derdeyn CP, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med 2011;365:993–1003.
8. Winningham M, Kasshout T, Bamford L, et al. Optimal duration of dual antiplatelet therapy for symptomatic intracranial stenosis. Stroke 2015;46:ATP100.
9. Wong KS, Chen C, Fu J, et al. Clopidogrel plus aspirin versus aspirin alone for reducing embolization in patients with acute symptomatic cerebral or carotid artery stenosis (CLAIR study): a randomized, open-label, blinded-endpoint trial. Lancet Neurol 2010;9:489–97.
10. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S1–45.
11. Turan TN, Cotsonis G, Lynn MJ, et al. Relationship between blood pressure and stroke recurrence in patients with intracranial arterial stenosis. Circulation 2007;115:2969–75.
12. Agency for Healthcare Research and Quality. Clinical guidelines for prescribing pharmacotherapy for smoking cessation. Accessed 8 May 2016 at www.ahrq.gov/professionals/clinicians-providers/guidelines-recommendations/tobacco/prescrib.html.
13. Nahab F, Kingston C, Frankel MR, et al. Early aggressive medical management for patients with symptomatic intracranial stenosis. J Stroke Cerebrovasc Dis 2013;22:87–91.
14. Kasner SE, Chimowitz MI, Lynn MJ, et al. Predictors of ischemic stroke in the territory of a symptomatic intracranial arterial stenosis. Circulation 2006;113:555–63.
15. Quest Diagnostics. Aspirin resistance. Accessed 23 Mar 2016 at www.questdiagnostics.com/testcenter/testguide.action?dc=TS_Aspirin_Resistance.
16. Alberts MJ. Platelet function testing for aspirin resistance is reasonable to do. Stroke 2010;41:2400–401.
17. Feher G, Feher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol 2010;26:171–86.
18. Taubert D, Kastrati A, Harlfinger S, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost 2004;92:311–6.
19. Lau WC, Gurbel PA, Watkins PB, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004;109:166–71.
20. Wabnitz AM, Derdeyn CP, Fiorella DJ, et al; for the SAMMPRIS Investigators. Infarct patterns in the anterior circulation as predictors of recurrent stroke in the medical arm of SAMMPRIS. Stroke 2016;47:A103.
21. Zaidat OO, Fitzsimmons BF, Woodward BK, et al. Effect of a balloon-expandable intracranial stent vs medical therapy on risk of stroke in patients with symptomatic intracranial stenosis: the VISSIT randomized clinical trial. JAMA 2015;313:1240–8.
22. Nahab F, Lynn MJ, Kasner SE, et al. Risk factors associated with major cerebrovascular complications after intracranial stenting. Neurology 2009;72:2014–9.
23. Gonzalez N, Dusick J, Connolly M, et al. Encephaloduroarteriosynangiosis for adult intracranial arterial steno-occlusive disease: long-term single-center experience with 107 operations. J Neurosurg 2015;123:654–61.
1. Derdeyn C, Chimowitz M, Lynn MJ, et al. Aggressive medical treatment with or without stenting in high-risk patients with intracranial arterial stenosis (SAMMPRIS): the final results of a randomized trial. Lancet 2014;383:333–41.
2. Chimowitz, MI, Lynn MJ, Howlett-Smith H, et al. Comparision of warfarin and aspirin for symptomatic intracranial arterial stenosis. N Engl J Med 2004;351:1250–1.
3. Feldmann E, Wilterdink JL, Kosinski A, et al. The stroke outcomes and neuroimaging of intracranial atherosclerosis (SONIA) trial. Neurology 2007;68:2099–106.
4. Willinek WA, von Falkenhausen M, Born M, et al. Noninvasive detection of steno-occlusive disease of the supra-aortic arteries with three-dimensional contrast-enhanced magnetic resonance angiography: a prospective, intra-individual comparative analysis with digital subtraction angiography. Stroke 2005;36:38–43.
5. Bash S, Villablanca JP, Jahan R, et al. Intracranial vascular stenosis and occlusive disease: evaluation with CT angiography, MR angiography, and digital subtraction angiography. Am J Neuroradiol 2005;26:1012–21.
6. Qureshi A, Caplan L. Intracranial atherosclerosis. Lancet 2014;383:984–98.
7. Chimowitz MI, Lynn MJ, Derdeyn CP, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med 2011;365:993–1003.
8. Winningham M, Kasshout T, Bamford L, et al. Optimal duration of dual antiplatelet therapy for symptomatic intracranial stenosis. Stroke 2015;46:ATP100.
9. Wong KS, Chen C, Fu J, et al. Clopidogrel plus aspirin versus aspirin alone for reducing embolization in patients with acute symptomatic cerebral or carotid artery stenosis (CLAIR study): a randomized, open-label, blinded-endpoint trial. Lancet Neurol 2010;9:489–97.
10. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S1–45.
11. Turan TN, Cotsonis G, Lynn MJ, et al. Relationship between blood pressure and stroke recurrence in patients with intracranial arterial stenosis. Circulation 2007;115:2969–75.
12. Agency for Healthcare Research and Quality. Clinical guidelines for prescribing pharmacotherapy for smoking cessation. Accessed 8 May 2016 at www.ahrq.gov/professionals/clinicians-providers/guidelines-recommendations/tobacco/prescrib.html.
13. Nahab F, Kingston C, Frankel MR, et al. Early aggressive medical management for patients with symptomatic intracranial stenosis. J Stroke Cerebrovasc Dis 2013;22:87–91.
14. Kasner SE, Chimowitz MI, Lynn MJ, et al. Predictors of ischemic stroke in the territory of a symptomatic intracranial arterial stenosis. Circulation 2006;113:555–63.
15. Quest Diagnostics. Aspirin resistance. Accessed 23 Mar 2016 at www.questdiagnostics.com/testcenter/testguide.action?dc=TS_Aspirin_Resistance.
16. Alberts MJ. Platelet function testing for aspirin resistance is reasonable to do. Stroke 2010;41:2400–401.
17. Feher G, Feher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol 2010;26:171–86.
18. Taubert D, Kastrati A, Harlfinger S, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost 2004;92:311–6.
19. Lau WC, Gurbel PA, Watkins PB, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004;109:166–71.
20. Wabnitz AM, Derdeyn CP, Fiorella DJ, et al; for the SAMMPRIS Investigators. Infarct patterns in the anterior circulation as predictors of recurrent stroke in the medical arm of SAMMPRIS. Stroke 2016;47:A103.
21. Zaidat OO, Fitzsimmons BF, Woodward BK, et al. Effect of a balloon-expandable intracranial stent vs medical therapy on risk of stroke in patients with symptomatic intracranial stenosis: the VISSIT randomized clinical trial. JAMA 2015;313:1240–8.
22. Nahab F, Lynn MJ, Kasner SE, et al. Risk factors associated with major cerebrovascular complications after intracranial stenting. Neurology 2009;72:2014–9.
23. Gonzalez N, Dusick J, Connolly M, et al. Encephaloduroarteriosynangiosis for adult intracranial arterial steno-occlusive disease: long-term single-center experience with 107 operations. J Neurosurg 2015;123:654–61.
Treated with a mood stabilizer, he becomes incontinent and walks oddly
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.

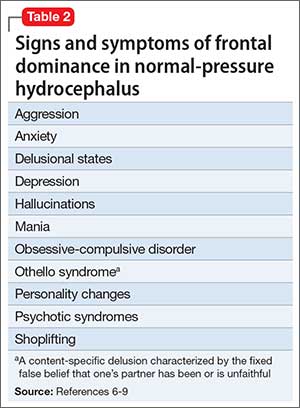
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
CASE Rapid decline
Mr. X, age 67, is a businessman who had a diagnosis of bipolar depression 8 years ago, and who is being evaluated now for new-onset cognitive impairment, gait disturbance that resembles child-like steps, dyskinesia, and urinary incontinence of approximately 2 months’ duration. He has been treated for bipolar depression with valproic acid, 1,000 mg/d, and venlafaxine, 150 mg/d, without complaint until now, since the diagnosis was made 8 years ago. The serum valproic acid level, tested every month, is within the therapeutic range; liver function tests, ordered every 6 months, also are within the normal range.
Mr. X has become confined to his bedroom and needs assistance to walk. He has to be lifted to a standing position by 2 attendants, who bear his weight and instruct him to take one step at a time. He wears a diaper and needs assistance shaving, showering, and getting dressed. When the treatment team asks him about his condition, Mr. X turns to his wife to respond on his behalf. He is slow to speak and struggles to remember the details about his condition or the duration of his disability.
Mr. X is referred to a neurologist, based on cognitive impairment and gait disturbance, who orders an MRI scan of the brain that shows enlarged ventricles and some cortical atrophy (Figure 1). A neurosurgeon removes approximately 25 mL of CSF as a diagnostic and therapeutic intervention.
Videography of his ambulation, recorded before and after the CSF tap, shows slight improvement in gait. Mr. X is seen by a neurosurgery team, who recommends that he receive a ventriculoperitoneal shunt for hydrocephalus.
While awaiting surgical treatment, Mr. X’s psychotropic medications are withheld, and he is closely monitored for reemergence of psychiatric symptoms. Mr. X shows gradual but significant improvement in his gait within 8 to 10 weeks. His dyskinesia improves significantly, as does his cognitive function.
What additional testing is recommended beyond MRI?
a) complete blood count with differential
b) blood ammonia level
c) neuropsychological evaluation
d) APOE-e4 genetic testing
e) all the above
The authors’ observations
Normal pressure hydrocephalus (NPH) is characterized by gait disturbance, dementia, or urinary incontinence that is associated with dilation of the brain’s ventricular system with normal opening CSF pressure (Table 1). Several studies have reported that patients with NPH might exhibit neuropsychiatric symptoms,1-4 possibly related to alterations in central neurotransmitter activity.5 NPH patients could present with symptoms reflecting frontal dominance (Table 2,6-9). In a study of 35 patients with idiopathic NPH in a tertiary hospital in Brazil,10 psychiatric symptoms were established by formal psychiatric evaluation in 71%, notably anxiety, depression, and psychotic syndromes.
Mechanism responsible for gait disturbance
Gait disturbance typically is the first and most prominent symptom of the NPH triad. Gait disturbance in NPH can be progressive because of expansion of the ventricular system, mainly the lateral ventricles, leading to pressure on the corticospinal motor fibers descending to the lumbosacral spinal cord. Although there is no one type of gait disturbance indicative of NPH, it often is described as shuffling, magnetic, and wide-based.11 Slowness of gait and gait imbalance or disequilibrium are common and more likely to respond to shunting.12
Drug-induced gait disturbance is likely to result in parkinsonian symptoms.13 A possible mechanism involves inhibition of neurite outgrowth. Qian et al14 found that therapeutic plasma levels of valproic acid reduced cell proliferation and neurite outgrowth, using SY5Y neuroblastoma cells as a neuronal model. Researchers also reported that valproic acid reduced mRNA and protein levels of neurofilament 160; a possible mechanistic explanation involves inhibition of neurite outgrowth that leads to gait disturbance. These effects reversed 2 days after stopping valproic acid.
Another possible mechanism is related to γ-aminobutyric acid (GABA) pathway disturbance leading to dopamine inhibition. This postulates that valproic acid or a metabolite of valproic acid, such as Δ-2-valproate, which may be a more potent inhibitor of the GABA-degrading enzyme than valproic acid, could cause a transient inhibitory effect on dopaminergic pathways.15
Mechanism of mood stabilizer action
Valproic acid is incorporated into neuronal membranes in a saturable manner and appears to displace naturally occurring branched-chain phospholipids.16 Chronic valproic acid use reduces protein kinase C (PKC) activity in patients with mania.17 Elevated PKC activity has been observed in patients with mania and in animal models of mania.18 Valproic acid has antioxidant effects and has reversed early DNA damage caused by amphetamine in an animal model of mania.19 Valproic acid and lithium both reduce inositol biosynthesis; the mechanism of action for valproic acid is unique, however, resulting from decreased myo-inositol-1-phosphate synthase inhibition.20
There is not a strong correlation between serum valproic acid levels and antimanic effects, but levels in the range of 50 to 150 μg/mL generally are required for therapeutic effect.
Neuropsychiatric adverse effects of valproic acid
With most antiepileptic drugs, adverse effects mainly are dose-related and include sedation, drowsiness, incoordination, nausea, and fatigue. Careful dose titration can reduce the risk of these adverse effects. Research on mothers with epilepsy has shown an association between valproic acid exposure in utero and lower IQ and a higher prevalence of autism spectrum disorder in children.21
Adverse effects on cognitive functioning are infrequent; valproic acid improves cognition in select patients.22 In a 20-week randomized, observer-blinded, parallel-group trial, adding valproic acid to carbamazepine resulted in improvement in short-term verbal memory.23 In a group of geriatric patients (mean age 77 years), no adverse cognitive effects were observed with valproic acid use.24
Masmoudi et al25 evaluated dementia and extrapyramidal symptoms associated with long-term valproic acid use. Among the side effects attributed to valproic acid, parkinsonian syndromes and cognitive impairment were not commonly reported. In a prospective study, Armon et al26 found several abnormal symptoms and signs related to motor and cognitive function impairment in patients on long-term valproic acid therapy. These side effects might be related to a disturbance in the GABAergic pathways in the basal ganglia system. Note that Δ2-valproic acid, a metabolite of valproic acid, preferentially accumulates in select areas of the brain: the substantia nigra, superior and inferior colliculus, hippocampus, and medulla.
What is the next best step in management?
a) surgically implant a shunt
b) adjust the dosage of valproic acid
c) switch to monotherapy
d) switch to an alternative psychotropic medication
e) provide observation and follow-up
The authors’ observations
Unusual appearances of NPH symptoms could hinder early diagnosis and proper treatment. Mr. X was taking valproic acid and venlafaxine for bipolar depression, without any complaints, and was asymptomatic for 8 years—until he developed symptoms of NPH.
In patients who have what can be considered classic symptoms of NPH and are taking valproic acid, consider discontinuing the drug on a trial basis before resorting to a more invasive procedure. This strategy could significantly reduce the cost of health care and contribute to the overall well-being of the patient.
NPH associated with chronic valproic acid use is rare, supported by only 1 case report13 in our literature review. Based on the severity of symptoms and chance for misdiagnosis, it is essential to identify such cases and differentiate them from others with underlying neuropathology or a secondary cause, such as age-related dementia or Parkinson’s disease, to avoid the burden of unnecessary diagnostic testing on the patient and physician.
Family history also is important in cases presenting with sensorineural hearing loss,13 which follows a pattern of maternal inheritance. Consider genetic testing in such cases.
Earlier diagnosis of valproic acid-induced NPH enables specific interventions and treatment. Treatment of NPH includes one of several forms of shunting and appropriate neuroleptic therapy for behavioral symptoms. Although there is a significant risk (40% to 50%) of psychiatric and behavioral symptoms as a shunt-related complication, as many as 60% of operated patients showed objective improvement. This makes the diagnosis of NPH, and referral for appropriate surgical treatment of NPH, an important challenge to the psychiatrist.27
OUTCOME No reemergence
Findings on a repeat MRI 2.5 months after the CSF tap remain unchanged. Surgery is cancelled and medications are discontinued. Mr. X is advised to continue outpatient follow-up for monitoring of re-emerging symptoms of bipolar depression.
At a follow-up visit, Mr. X’s condition has returned to baseline. He ambulates spontaneously and responds to questions without evidence of cognitive deficit. He no longer is incontinent.
Follow-up MRI is performed and indicated normal results.
Neuropsychological testing is deemed unnecessary because Mr. X has fully recovered from cognitive clouding (and there would be no baseline results against which to compare current findings). Based on the medication history, the team concludes that prolonged use of valproic acid may have led to development of signs and symptoms of an NPH-like syndrome.
The authors’ observations
Awareness of an association of NPH with neuropsychiatric changes is important for clinical psychiatrists because early assessment and appropriate intervention can prevent associated long-term complications. Valproic acid is considered a relatively safe medication with few neurologic side effects, but the association of an NPH-like syndrome with chronic valproic acid use, documented in this case report, emphasizes the importance of studying long-term consequences of using valproic acid in geriatric patients. More such case reports need to be evaluated to study the association of neuropsychiatric complications with chronic valproic use in the geriatric population.
Mr. X apparently had cerebral atrophy with enlarged ventricles that was consistently evident for 10 years (Figure 2), although he has been maintained on valproic acid for 8 years. What is intriguing in this case is that discontinuing valproic acid relieved the triad of incontinence, imbalance, and memory deficits indicative of NPH. Mr. X remains free of these symptoms.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
1. Pinner G, Johnson H, Bouman WP, et al. Psychiatric manifestations of normal-pressure hydrocephalus: a short review and unusual case. Int Psychogeriatr. 1997;9(4):465-470.
2. Alao AO, Naprawa SA. Psychiatric complications of hydrocephalus. Int J Psychiatry Med. 2001;31(3):337-340.
3. Lindqvist G, Andersson H, Bilting M, et al. Normal pressure hydrocephalus: psychiatric findings before and after shunt operation classified in a new diagnostic system for organic psychiatry. Acta Psychiatr Scand Suppl. 1993;373:18-32.
4. Kito Y, Kazui H, Kubo Y, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol. 2009;21(3):165-174.
5. Markianos M, Lafazanos S, Koutsis G, et al. CSF neurotransmitter metabolites and neuropsychiatric symptomatology in patients with normal pressure hydrocephalus. Clin Neurol Neurosurg. 2009;111(3):231-234.
6. McIntyre AW, Emsley RA. Shoplifting associated with normal-pressure hydrocephalus: report of a case. J Geriatr Psychiatry Neurol. 1990;3(4):229-230.
7. Kwentus JA, Hart RP. Normal pressure hydrocephalus presenting as mania. J Nerv Ment Dis. 1987;175(8):500-502.
8. Bloom KK, Kraft WA. Paranoia—an unusual presentation of hydrocephalus. Am J Phys Med Rehabil. 1998;77(2):157-159.
9. Yusim A, Anbarasan D, Bernstein C, et al. Normal pressure hydrocephalus presenting as Othello syndrome: case presentation and review of the literature. Am J Psychiatry. 2008;165(9):1119-1125.
10. Oliveira MF, Oliveira JR, Rotta JM, et al. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr. 2014;72(6):435-438.
11. Marmarou A, Young HF, Aygok GA, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102(6):987-997.
12. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord. 2007;13(7):434-437.
13. Evans MD, Shinar R, Yaari R. Reversible dementia and gait disturbance after prolonged use of valproic acid. Seizure. 2011;20(6):509-511.
14. Qian Y, Zheng Y, Tiffany-Castiglioni E. Valproate reversibly reduces neurite outgrowth by human SY5Y neuroblastoma cells. Brain Res. 2009;1302:21-33.
15. Löscher W. Pharmacological, toxicological and neurochemical effects of delta 2(E)-valproate in animals. Pharm Weekbl Sci. 1992;14(3A):139-143.
16. Siafaka-Kapadai A, Patiris M, Bowden C, et al. Incorporation of [3H]-valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol. 1998;56(2):207-212.
17. Hahn CG, Umapathy, Wagn HY, et al. Lithium and valproic acid treatments reduce PKC activation and receptor-G-protein coupling in platelets of bipolar manic patients. J Psychiatr Res. 2005;39(4):35-63.
18. Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160-1171.
19. Andreazza AC, Frey BN, Stertz L, et al. Effects of lithium and valproate on DNA damage and oxidative stress markers in an animal model of mania [abstract P10]. Bipolar Disord. 2007;9(suppl 1):16.
20. Galit S, Shirley M, Ora K, et al. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol Rep. 2007;59(4):402-407.
21. Kennedy GM, Lhatoo SD. CNS adverse events associated with antiepileptic drugs. CNS Drugs. 2008;22(9):739-760.
22. Prevey ML, Delaney RC, Cramer JA, et al. Effect of valproate on cognitive functioning. Comparison with carbamazepine. The Department of Veteran Affairs Epilepsy Cooperative Study 264 Group. Arch Neurol. 1996;53(10):1008-1016.
23. Aldenkamp AP, Baker G, Mulder OG, et al. A multicenter randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41(9):1167-1178.
24. Craig I, Tallis R. Impact of valproate and phenytoin on cognitive function in elderly patients: results of a single-blind randomized comparative study. Epilepsia. 1994;35(2):381-390.
25. Masmoudi K, Gras-Champel V, Bonnet I, et al. Dementia and extrapyramidal problems caused by long-term valproic acid [in French]. Therapie. 2000;55(5):629-634.
26. Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 1996;47(3):626-635.
27. Price TR, Tucker GJ. Psychiatric and behavioral manifestations of normal pressure hydrocephalus. A case report and brief review. J Nerv Ment Dis. 1977;164(1):51-55.
Precipitously and certainly psychotic—but what’s the cause?
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
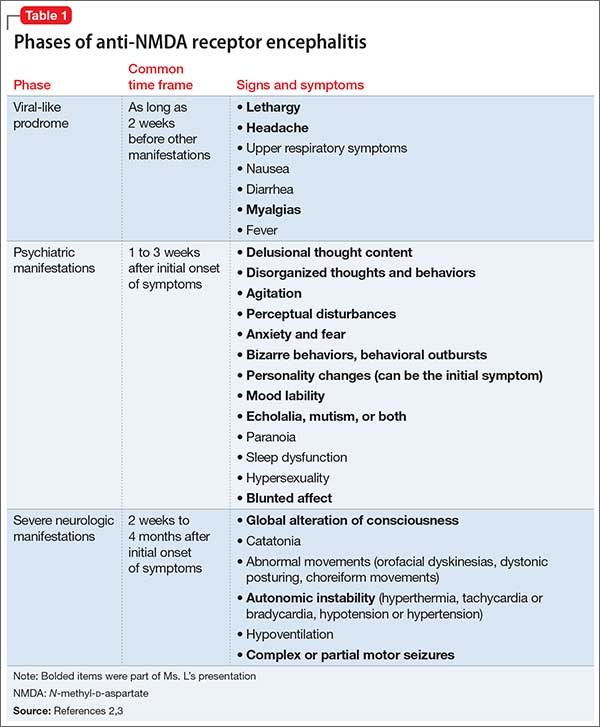
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).

This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).

What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).
This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).
What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
CASE Sudden personality change
Ms. L, age 38, is brought to the university hospital’s emergency department (ED) under police escort after she awoke in the middle of the night screaming, “I found it out! I’m a lie! Life is a lie!” and began threatening suicide. This prompted her spouse to call emergency services because of concerns about her safety.
Over the preceding 9 days—and, most precipitously, over the last 24 hours—Ms. L has experienced a dramatic “change in her personality,” according to her spouse. In the ED, she is oriented to person, place, and time. Her vital signs are within normal limits, other than a mild tachycardia. Complete blood count and complete metabolic profile are unremarkable and a urine drug screen is positive only for benzodiazepines (she recently was prescribed alprazolam). Ms. L smiles inappropriately at the ED physicians and confides that she is hearing music by The Lumineers, despite silence in her room.
The psychiatry service is consulted after she is seen making threats of harm to her family members.
EVALUATION Confusion
Over past several weeks, Ms. L has experienced rapid onset of neurovegetative symptoms, with poor oral intake, increased somnolence, neglect of hygiene, excessive time spent in bed, and weight loss of 15 to 20 lb, according to her spouse. She also has been complaining of foggy mentation, weakening handgrip, and tinnitus. She has no previous psychiatric history.
She recently established care with an outpatient neurologist and infectious disease specialist to address these symptoms. Outpatient EEG and sexually transmitted infection (STI) tests were scheduled but not yet obtained. Ms. L’s spouse observes that her drastic “personality change” over the preceding 24 hours coincided with her feeling upset and offended by a physician’s recommendation to obtain STI tests (it is unclear why the physician recommended these tests).
Ms. L had presented to another local ED 4 times over several weeks for various complaints, and had been prescribed alprazolam, 0.5 mg, 3 times a day as needed, and buspirone, 15 mg/d, for anxiety. She also had received a short course of doxycycline, 200 mg/d, which she did not finish, for treatment of presumed Lyme disease. According to her spouse, Ms. L had completed a course of doxycycline for Lyme disease 1 year earlier, but the medical records are not available for review.
During the interview, Ms. L is fairly well groomed but appears confused; she asks her spouse if she is “real” and states that she feels “crazy.” She seems uncomfortable and is guarded, with a minimally reactive, anxious affect. She has general psychomotor slowing and her speech is soft and monotonous, with prominent latency. She reports passive suicidal ideations as well as active auditory hallucinations of a musical quality.
The Mini-Mental State Examination (MMSE) score is 19/30, indicating moderate cognitive impairment, and she is unable to complete attention, executive function, 3-stage command, and delayed word recall tasks. She reports fatigue, diarrhea, and decreased appetite. Her physical examination is notable for an overweight white woman without focal neurologic deficits. Her family psychiatric history reveals bipolar disorder in 2 distant relatives.
In the ED, Ms. L is given 3 provisional diagnoses:
- adjustment disorder, because of her reaction to the proposed STI testing
- psychotic disorder not otherwise specified, because of her obvious psychosis of unknown cause
- rule out delirium due to a general medical condition, because of her sudden onset of attention, perception, and memory difficulties.
As Ms. L sits in her room, her abnormal behaviors become more apparent. She starts to endorse active suicidal ideations and becomes aggressive, trying to choke her spouse, shouting, jumping on her bed, and attempting to strike herself. For her safety, she is physically restrained and given IM haloperidol, 10 mg, and IM lorazepam, 2 mg.
What would you do next to treat Ms. L?
a) Admit her to the psychiatric unit for monitoring and treatment of psychosis and consider additional antipsychotics for agitation
b) Perform a bedside lumbar puncture to assess for findings suggestive of a CNS infection or anomaly
c) Sedate her with IM ketamine, intubate her, and admit her to the intensive care unit (ICU) for further medical workup
d) Begin IV antibiotic therapy with ceftriaxone for early-disseminated Lyme disease with CNS involvement
The authors’ observations
Clearly, Ms. L was psychotic. However, psychosis is a nonspecific term used to describe a heterogeneous group of phenomena in which one experiences an impaired sense of reality. Although commonly caused by psychiatric disorders, psychosis can arise from a variety of causes.1 Ms. L’s initial physical examination and laboratory studies were within the normal range, but her mental status exam and MMSE were abnormal. At this point, selecting the appropriate setting for further observation, workup, and treatment became important.
TREATMENT The right setting
Given the abrupt onset of Ms. L’s symptoms, the treatment team is concerned about active neurologic or infectious disease. However, no acute laboratory or physical examination findings support this hypothesis, and the ED physicians conclude that no further emergent workup is indicated. Because Ms. L is threatening harm to herself and others, she cannot be safely discharged. The treatment team decides the safest option is to admit Ms. L to the inpatient psychiatric unit for observation, further non-emergent workup, and consultation with the neurology service.
At admission. Ms. L is cooperative and calm, lying in bed comfortably. She obeys simple commands; a brief neurologic examination is remarkable for a sedated female without focal motor or sensory deficits. Although her answers to questions are brief, they are appropriate. She sleeps without incident for approximately 10 hours.
The next morning. Ms. L does not awaken to verbal or gentle physical stimuli. Upon sternal rub, she awakens and forcefully squeezes the examiner’s arm, after which she closes her eyes and does not answer further questions (but does resist passive eye opening). After several minutes, she begins exhibiting verbigeration, shouting repeated phrases such as “The birds are in my ears” and “No, I am not okay.”
An emergent EEG is ordered because the team is concerned about nonconvulsive status epilepticus and the neurology service is consulted about the need for an urgent lumbar puncture. Without any obvious abnormal physical examination findings, however, the neurology team’s initial assessment attributes Ms. L’s presentation to a primary psychiatric illness and does not recommend a lumbar puncture or EEG.
That day and night, Ms. L has several episodes of agitation with a disorganized thought process and perseverative speech. She appears distraught and exhibits menacing behaviors. She is poorly redirectable and physically hostile toward staff, requiring several emergent doses of IM haloperidol and IM lorazepam, to which she responds minimally. Ms. L is placed on constant observation, requiring frequent redirection from the rooms of other patients and intermittent seclusion because of her violent, destructive behavior.
The next day. Ms. L remains grossly agitated and psychotic. Although an EEG is ordered, it is not performed because the technicians are concerned about their safety. With her unclear history of Lyme disease and concern for an infectious encephalopathy, Ms. L’s history and symptoms are discussed with the infectious disease service. Given her abrupt onset of symptoms, including auditory hallucinations, they express concern for herpes simplex encephalitis and recommend emergent treatment with IV acyclovir and ceftriaxone.
This recommendation, however, causes a practical conundrum. Because of state laws and differences in staff training, the treatment team believes that the inpatient psychiatric unit is not the appropriate setting to administer these IV treatments. At the same time, hospital security, nursing staff, and the receiving medical team are concerned about transporting Ms. L to the general medical floor.
In the ICU. After discussion, the teams decide that the safest and least traumatic option is to transport Ms. L to the ICU after she is sedated and intubated. In the ICU, she undergoes empirical treatment for herpes simplex encephalitis and further medical workup.
An EEG reveals findings suggestive of severe encephalopathy. A lumbar puncture shows lymphocytic pleocytosis with an opening pressure of 28 cm H2O and normal protein and glucose levels. Her serum C-reactive protein is slightly elevated at 1.4 mg/dL. She also is found to have an elevated herpes simplex virus (HSV)-2 IgG antibody.
Subsequent hospital stay. Ms. L has 2 episodes of seizure-like activity, for which she is treated with levetiracetam, 2,000 mg/d, increased to 3,000 mg/d. She is sedated for several days to allow broad treatment with antiviral and antibiotic medications. Although she experiences intermittent fevers and tachycardia, cultures of blood, urine, and cerebrospinal fluid (CSF) show no growth. Similarly, a test of serum HSV IgM antibodies is negative.
CT of the chest, abdomen, and pelvis reveals no findings suggestive of malignancy but does show a solid-appearing 6-mm nodule in her right lung. Magnetic resonance angiography of the head and neck shows no evidence of abnormalities other than atrophy of the superior cerebellar vermis and a subtle focus of T2/FLAIR signal abnormality in the medial portion of the left occipital lobe.
The following weeks. Ms. L’s cognitive status improves markedly. Extensive studies—including serum ammonia, thyroid-stimulating hormone, Lyme disease antibody, vitamin B12, folate, beta-hCG, HIV, hepatitis B and C, Varicella zoster, syphilis, Lyme disease serology, CSF Eastern equine encephalitis, St. Louis encephalitis virus, West Nile virus, Ehrlichia chaffeensis, Babesia microti, Rocky Mountain spotted fever, John Cunningham virus, typhus fever, cryptococcal antigen, rabies, 2 serum tests for anti-N-methyl-D-aspartate (NMDA) receptor antibodies, and serum ceruloplasmin—are normal.
At discharge, Ms. L’s clinical presentation is thought to be most consistent with viral encephalitis, because of her CSF lymphocytic pleocytosis, fever, and improvement with supportive care. Because she improves, the team does not find it necessary to wait for results of pending studies, including a paraneoplastic autoantibody panel and a CSF anti-NMDA receptor antibody, before discharging her.
Readmission. Although the results of the paraneoplastic autoantibody panel are unremarkable, several weeks after discharge Ms. L’s CSF anti-NMDA receptor antibodies return positive, despite 2 earlier negative serum studies. She is readmitted to the neurology service for treatment with immunomodulators.
A positron-emission tomography scan is negative for malignancy. She is treated on an ongoing basis with immunomodulators; cognition improves such that she is able to start working again with good overall functioning. Despite this improvement, she experiences residual sequelae, including noise sensitivity, amnesia of the events surrounding her hospitalization, mild short-term memory deficits, and persistent affective blunting.
The authors’ observations
Psychosis is not exclusive to psychiatric syndromes and frequently is a symptom of an underlying neurologic, immunologic, metabolic, infectious, or oncologic abnormality.1 Anti-NMDA receptor encephalitis is an autoimmune disease in which antibodies attack NMDA-type glutamate receptors at central neuronal synapses and can produce psychosis, as seen with Ms. L2 (Table 12,3). The etiology of the disease is not fully understood. Determining the appropriate setting to perform a complete medical workup in a severely agitated patient after an initial negative medical workup can be challenging.
What’s the most appropriate treatment setting?
This case illustrates the importance, with any new-onset psychosis, of weighing heavily a carefully obtained psychiatric history, even in the absence of focal physical examination and initial laboratory abnormalities. It also highlights the challenge of determining the most appropriate initial setting for performing the important task of a complete medical workup for first-episode psychosis.
Ms. L initially was treated in the inpatient psychiatric unit because of safety concerns and practical limitations, but was later found to have a disease that could not be managed in that setting. She proved to be too agitated to obtain a full medical workup on the inpatient psychiatric or general medical floors and required transfer to the ICU. Despite her normal basic laboratory tests, her EEG and CSF studies did demonstrate abnormalities, suggesting these can be useful to the basic workup for psychosis of unknown cause (Table 21,2).
This case also demonstrates that negative serum anti-NMDA receptor antibody tests do not rule out the disease; one study found that only 85% of patients with CSF anti-NMDA receptor antibodies also had detectable antibodies in their serum and that detectability changed during the course of the disease.4 This supports the utility of a lumbar puncture as part of a basic initial workup for some cases of new-onset psychosis. Because clinical outcomes often correlate with early treatment, as with anti-NMDA receptor encephalitis, a timely diagnostic workup of psychosis often can be important.3,5 The ICU can be considered an appropriate setting for working up some patients who develop new, rapid-onset psychosis and severe agitation, even in the absence of initial laboratory or physical examination findings.
Ms. L’s case also illustrates the importance of completing a thorough medical workup for patients with new-onset psychosis before transferring them to an independent psychiatric hospital. Initially, the university’s psychiatric unit was at capacity and a bed was sought at outside psychiatric hospitals while Ms. L waited in the ED. Had Ms. L not been admitted to a large academic medical center, she may not have had access to the multidisciplinary collaboration that proved necessary for the appropriate diagnosis and treatment of her anti-NMDA receptor encephalitis (Table 35,6).
What prodromal symptoms occur as long as 2 weeks as an initial presentation in many patients with anti-NMDA receptor encephalitis?
a) Flu-like symptoms of lethargy, headache, gastrointestinal symptoms, myalgias, fevers, and upper respiratory symptoms
b) Delusions, hallucinations, disorganized behaviors and thoughts, behavioral outbursts, hypersexuality, mood lability, personality change, paranoia, echolalia, mutism, anxiety, agitation, aggression, hyperactivity, sleep dysfunction, and blunted affect
c) Dyskinesias, autonomic instability, central hypoventilation, and seizures
The authors’ observations
Lab results, vital signs, and physical examination should not supplant a careful history when determining an appropriate clinical course of action. As experts in the cognitive sciences, psychiatrists may be the most qualified in determining whether a patient with new-onset psychosis should undergo further medical testing before a condition is deemed to be solely of a psychiatric cause. As a neurologic disease of immunologic origin with psychiatric manifestations, anti-NMDA receptor encephalitis is a complex condition requiring collaboration among several specialists for appropriate management.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
1. Freudenreich O. Differential diagnosis of psychotic symptoms: medical “mimics.” Psychiatric Times. http://www.psychiatrictimes.com/forensic-psychiatry/differential-diagnosis-psychotic-symptoms-medical-%E2%80%9Cmimics%E2%80%9D. Published December 3, 2012. Accessed March 31, 2016.
2. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193.
3. Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74.
4. Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177.
5. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
6. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
Is the evidence compelling for using ketamine to treat resistant depression?
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.

as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
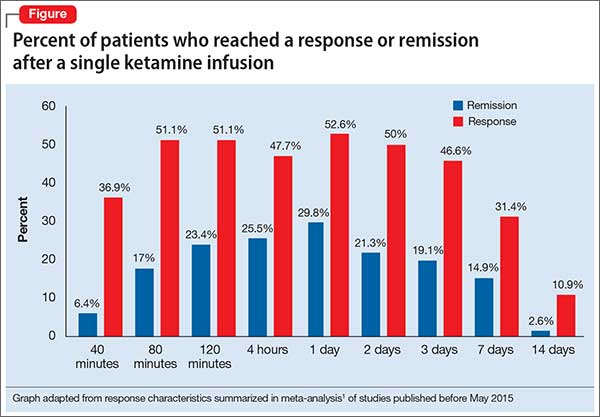
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.
as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.
as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
Manic after taking a vacation
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.

Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).

OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.
Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).
OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
CASE From soft-spoken to manic
Mr. K, age 36, an Asian male with no psychiatric history, arrives at the outpatient psychiatry clinic accompanied by his wife, after being referred from the emergency room the night before. He reports racing thoughts, euphoric mood, increased speech, hypergraphia, elevated self-esteem, decreased need for sleep, distractibility, and increased goal-directed activity. Notably, Mr. K states that he likes how he is feeling.
Mr. K’s wife says that his condition is a clear change from his baseline demeanor: soft-spoken and mild-mannered.
Mr. K reports that his symptoms started approximately 10 days earlier, after he returned from a cruise with his wife. During the cruise, he used a scopolamine patch to prevent motion sickness. Mr. K and his wife say that they believe that the scopolamine patch caused his symptoms.
Can scopolamine cause mania?
a) No
b) Yes; this is well-documented in the literature
c) It is theoretically possible because of scopolamine’s antidepressant and central anticholinergic effects
TREATMENT Lithium, close follow up
Mr. K has no history of psychiatric illness or substance use and no recent use of psychoactive substances—other than scopolamine—that could trigger a manic episode. His family history is significant for a younger brother who had a single manic episode at age 12 and a suicide attempt as a young adult.
Mr. K works full-time on rotating shifts—including some overnight shifts—as a manufacturing supervisor at a biotechnology company. He has been unable to work since returning from the cruise because of his psychiatric symptoms.
Mr. K is started on sustained-release (SR) lithium, 900 mg/d. In addition, the psychiatrist advises Mr. K to continue taking clonazepam, 0.5 to 1 mg as needed, which the emergency medicine physician prescribed, for insomnia. Mr. K is referred to a psychiatric intensive outpatient program (IOP), 3 days a week for 2 weeks, and is advised to stay home from work until symptoms stabilize.
Mr. K follows up closely with the psychiatrist in the clinic, every 1 to 2 weeks for the first month, as well as by several telephone and e-mail contacts. Lithium SR is titrated to 1,200 mg/d, to a therapeutic serum level of 1.1 mEq/L. Clonazepam is switched to quetiapine, 25 to 50 mg as needed, to address ongoing insomnia and to reduce the risk of dependency on clonazepam.
Mr. K’s mania gradually abates. He finishes the IOP and returns to work 3 weeks after his initial presentation. At an office visit, Mr. K’s wife gives the psychiatrist 2 scientific articles documenting the antidepressant effect of scopolamine.1,2 Mr. K and his wife both continue to believe that Mr. K’s manic episode was triggered by the scopolamine patch he used while on the cruise. They think this is important because Mr. K believes he would not have developed mania otherwise, and he does not want to take a mood stabilizer for the rest of his life.
The author’s observations
There are several proposed mechanisms for scopolamine’s antidepressant effect (Table 1).3-9 Scopolamine blocks central muscarinic cholinergic receptors, which reduces production of glutamate receptors and leads to reduced glutamate transmission and neurotoxicity.3,4 Scopolamine—similar to ketamine—could enhance synaptogenesis and synaptic signaling.5,6 Also, by blocking muscarinic autoreceptors, scopolamine results in an acute upregulation of acetylcholine release, which, in turn, influences the nicotinic, dopamine, serotonin, and neuropeptide Y systems. This action could contribute to anti-inflammatory effects, all of which can benefit mood.7-9 These antidepressant mechanisms also could explain why, theoretically, scopolamine could precipitate mania in a person predisposed to mental illness.
Proposed by Janowsky et al10 in 1972, the cholinergic−adrenergic balance hypothesis of affective disorders suggests that depression represents an excess of central cholinergic tone over adrenergic tone, and that mania represents the opposite imbalance. Several lines of evidence in the literature support this theory. For example, depressed patients have been found to have hypersensitive central cholinergic receptors.11,12 Also, central cholinesterase inhibition has been shown to affect pituitary hormone and epinephrine levels via central muscarinic receptors.13 In addition, scopolamine has been shown to attenuate these effects via the central anti-muscarinic mechanism.14
Rapid antidepressant therapy. Scopolamine is being studied as a rapid antidepressant treatment, although it usually is administered via IV infusion, rather than patch form, in trials.15-17 IV ketamine is another therapy being investigated for rapid treatment of depression, which might have downstream mechanisms of action related to scopolamine.5,18 Electroconvulsive therapy is a well-known for its quick antidepressant effect, which could involve synaptogenesis or effects on the neuroendocrine system.19 Sleep deprivation also can produce a rapid antidepressant effect20 (Table 21,2,5,6,15,16,18-20).
OUTCOME Prone to motion sickness
Approximately 3.5 months after his initial presentation, Mr. K continues to do well with treatment. He is euthymic and functioning well at work. He and his wife are preparing for the birth of their first child.
Mr. K is prone to motion sickness, and asks if he can take over-the-counter dimenhydrinate tablets for long car rides. He reports that dimenhydrinate has worked well for him in the past without triggering manic episodes, and he did not anticipate needing to take it very often.
What would you tell Mr. K about dimenhydrinate for motion sickness during car rides?
a) Mr. K should not take dimenhydrinate to prevent motion sickness because he experienced a manic episode triggered by a scopolamine patch
b) Mr. K can use dimenhydrinate as much as he wants to prevent motion sickness because it poses no risk of mania
c) Mr. K can use dimenhydrinate with caution and sparingly on a trial basis, as long as he is taking his mood stabilizer
FOLLOW UP Cautious use
The psychiatrist advised Mr. K to take dimenhydrinate cautiously when needed for long car rides. The psychiatrist feels this is safe because Mr. K is taking a mood stabilizer (lithium). Also, although dimenhydrinate has anticholinergic properties, occasional use is thought to pose less risk of triggering mania than the constant anticholinergic exposure over several days with a scopolamine patch. (The scopolamine patch contains 1.5 mg of the drug delivered over 3 days [ie, 0.5 mg/d]. In trials of IV scopolamine for depression, the dosage was 0.4 mcg/kg/d administered over 3 consecutive days.15-17 For an adult weighing 70 kg, this would be equivalent to 0.24 mg/d. Therefore, using a scopolamine patch over 3 days would appear to deliver a robust antidepressant-level dosage, even taking into account possible lower bioavailability with transdermal administration compared with IV infusion.)
The psychiatrist concludes that sporadic use of dimenhydrinate tablets for motion sickness during occasional long car rides poses less of a risk for Mr. K of triggering mania than repeat use of a scopolamine patch.
The author’s observations
Mr. K’s case is notable for several reasons:
- Novelty. This might be the first report of scopolamine-induced mania in the literature. In clinical trials by Furey and Drevets,15 Drevets and Furey,16 and Ellis et al,17 no study participants who received scopolamine infusion developed mania or hypomania. Although it is possible that Mr. K’s manic episode could have occurred spontaneously and was coincidental to his scopolamine use, there are valid reasons why scopolamine could trigger mania in a vulnerable person.
- Biochemical insight. The case underscores the role of the muscarinic cholinergic system in regulating mood.10
- Rational medical care. Sensible clinical decision-making was needed when Mr. K asked about using dimenhydrinate for motion sickness during car rides. Although there might not be definitively correct answers for questions that arose during Mr. K’s care (in the absence of research literature), theoretical understanding of the antidepressant effects of anticholinergic medications helped inform the psychiatrist’s responses to Mr. K and his wife.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
1. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
2. Jaffe RJ, Novakovic V, Peselow ED. Scopolamine as an antidepressant: a systematic review. Clin Neuropharmacol. 2013;36(1):24-26.
3. Rami A, Ausmeir F, Winckler J, et al. Differential effects of scopolamine on neuronal survival in ischemia and glutamate neurotoxicity: relationships to the excessive vulnerability of the dorsoseptal hippocampus. J Chem Neuroanat. 1997;13(3):201-208.
4. Benveniste M, Wilhelm J, Dingledine RJ, et al. Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res. 2010;1352:61-69.
5. Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
6. Voleti B, Navarria A, Liu R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-749.
7. Overstreet DH, Friedman E, Mathé AA, et al. The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739-759.
8. Tizabi Y, Getachew B, Rezvani AH, et al. Antidepressant-like effects of nicotine and reduced nicotinic receptor binding in the Fawn-Hooded rat, an animal model of co-morbid depression and alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(3):398-402.
9. Wang DW, Zhou RB, Yao YM. Role of cholinergic anti-inflammatory pathway in regulating host response and its interventional strategy for inflammatory diseases. Chin J Traumatol. 2009;12(6):355-364.
10. Janowsky DS, el-Yousef MK, Davis JM, et al. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2(7778):632-635.
11. Risch SC, Kalin NH, Janowsky DS. Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol. 1981;1(4):186-192.
12. Risch SC, Janowsky DS, Gillin JC. Muscarinic supersensitivity of anterior pituitary ACTH and β-endorphin release in major depressive illness. Peptides. 1983;4(5):789-792.
13. Risch SC, Janowsky DS, Mott MA, et al. Central and peripheral cholinesterase inhibition: effects on anterior pituitary and sympathomimetic function. Psychoneuroendocrinology. 1986;11(2):221-230.
14. Janowsky DS, Risch SC, Kennedy B, et al. Central muscarinic effects of physostigmine on mood, cardiovascular function, pituitary and adrenal neuroendocrine release. Psychopharmacology (Berl). 1986;89(2):150-154.
15. Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2006;63(10):1121-1129.
16. Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432-438.
17. Ellis JS, Zarate CA Jr, Luckenbaugh DA, et al. Antidepressant treatment history as a predictor of response to scopolamine: clinical implications. J Affect Disord. 2014;162:39-42.
18. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
19. Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Can J Psychiatry. 2011;56(1):13-18.
20. Wu JC, Kelsoe JR, Schachat C, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66(3):298-301.
When and why to initiate antipsychotic polypharmacy, and with which agents
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,

and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4

Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
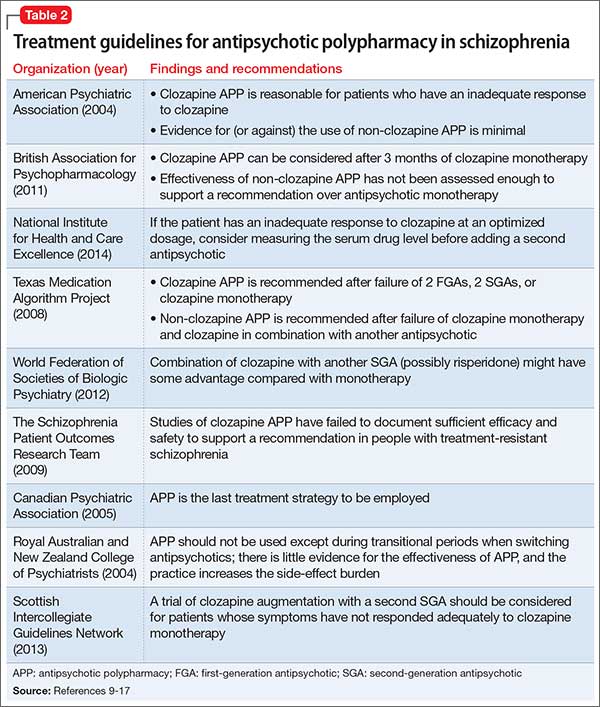

What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
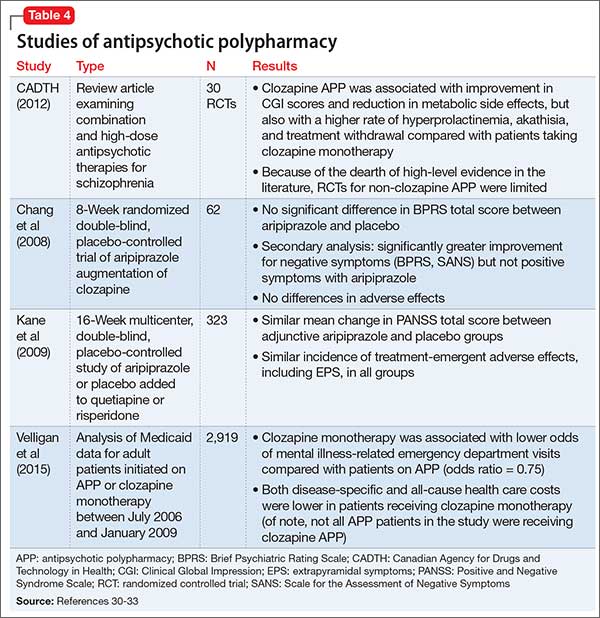
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
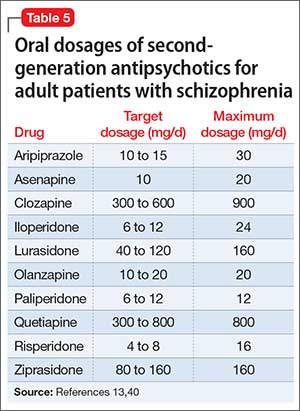
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,
and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4
Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,
and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4
Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
Recognition and Management of Children with Nonalcoholic Fatty Liver Disease
From the Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, Bronx, NY.
Abstract
- Objective: To review diagnostic challenges and management strategies in children with nonalcoholic fatty liver disease (NAFLD).
- Methods: Review of the literaure.
- Results: NAFLD is common in the United States and should be suspected in overweight or obese children with an elevated serum alanine aminotransferase level. The differential diagnosis for these patients is broad, however, and liver biopsy—the gold standard test—should be undertaken selectively after an appropriate workup. Patients should be counseled on lifestyle modifications, whereas vitamin E therapy can be initiated for those with biopsy-proven disease.
- Conclusion: Providers should have a high degree of suspicion for NAFLD, approaching the workup and diagnosis in an incremental, step-wise fashion. Further research is needed to standardize the diagnostic approach, identify reliable, noninvasive diagnostic measures, and develop novel treatment modalities.
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the Western world, affecting approximately 10% of children and a third of all adults in the United States [1–3]. It is a significant public health challenge and is estimated to soon be the number one indication for liver transplantation in adults.
NAFLD is a generic term encompassing 2 distinct conditions defined by their histopathology: simple steatosis and nonalcoholic steatohepatitis (NASH). Simple steatosis is characterized by predominantly macrovesicular—meaning large droplet—cytoplasmic lipid inclusions found in ≥ 5% of hepatocytes. NASH is defined as hepatic steatosis plus the additional features of inflammation, hepatocyte ballooning, and/or fibrosis. There are some adult data [4-6] and 1 retrospective pediatric study [7] demonstrating that over time, NAFLD may progress. That is, steatosis may progress to NASH and some patients with fibrosis will ultimately develop cirrhosis. If intervention is provided early in the histologic spectrum, NAFLD can be reversed [4,8] and late complications—such as cirrhosis, hepatocellular carcinoma, or liver transplantation—may be prevented.
It is important to highlight that the above definitions are based on histology and that a liver biopsy cannot be reasonably obtained in such a large percentage of the U.S. population. This case-based review will therefore focus primarily on the current diagnostic challenges facing health care providers as well as management strategies in children with presumed NAFLD.
Case Study
Initial Presentation
As you finish your charts at the end of a busy clinic day, you identify 3 patients who may have NAFLD:

History
All 3 patients presented to your office for a routine annual physical before the start of the school year and are asymptomatic. None of the patients has a family history of liver disease and their previously diagnosed comorbidities are listed in the table above. No patient is taking medications other than patient C, who is on metformin.
All 3 children have a smooth, velvety rash on their necks consistent with acanthosis nigricans with an otherwise normal physical exam. The liver and spleen are difficult to palpate but are seemingly normal.
What is the typical presentation for a child with NAFLD?
Most children with NAFLD are asymptomatic, though some may present with vague right upper quadrant abdominal pain. It is unclear, however, if the pain is caused by NAFLD or is rather an unrelated symptom that brings the child to the attention of a physician. In addition, hepatomegaly can be found in over 30% to 40% of patients [9]. For children without abdominal pain or hepatomegaly, most are recognized by an elevated serum alanine aminotransferase (ALT) or findings of increased liver echogenicity on ultrasonography.
Serum Alanine Aminotransferase
Serum aminotransferases are one of the more common screening tests for NAFLD. However, ALT is highly insensitive at commonly used thresholds and is also nonspecific. As documented in the SAFETY study, the upper limit of normal for ALT in healthy children should be set around 25 U/L in boys and 22 U/L in girls [10]. Yet even at these thresholds, the sensitivity of ALT to diagnose NAFLD is 80% in boys and 92% in girls, whereas specificity is 79% and 85%, respectively [10]. These findings are largely consistent with adult studies [11–14]. Furthermore, ALT does not correlate well with disease severity and children may still have NASH or significant fibrosis with normal values. In a well-characterized cohort of 91 children with biopsy-proven NAFLD, for example, early fibrosis was identified in 12% of children with a normal ALT (≤ 22 U/L for girls and ≤ 25 U/L in boys) [15]. Advanced fibrosis or cirrhosis was seen in 9% of children with an ALT up to 2 times this upper limit [15]. Thus, reliance on the serum ALT may significantly underestimate the prevalence and severity of liver injury.
Ultrasonography
Children with NAFLD typically have findings of increased hepatic echogenicity on abdominal ultrasonography. However, there are multiple limitations to sonography. First, ultrasound is insensitive for identifying mild steatosis if less than 30% of hepatocytes are affected [16,17]. Second, increased hepatic echogenicity is nonspecific and may be caused by inflammation, fibrosis, or intrahepatic accumulation of iron, copper, or glycogen. Third, there can be considerable inter- and intra-operator variability. And lastly, there is some evidence that ultrasounds do not add benefit to diagnosing children with NAFLD [18].
Which patients are at risk for developing hepatic steatosis and NASH?
Weight, Age, and Gender
There is a strong, direct correlation between body mass index (BMI) and NAFLD. The Study of Child and Adolescent Liver Epidemiology (SCALE)—a sentinel pediatric autopsy study of 742 children—found that 5% of normal weight children, 16% of overweight children, and 38% of obese children had NAFLD. The SCALE study also demonstrated an increasing prevalence with age, such that NAFLD was present in 17.3% of 15- to 19-year-olds but only in 0.2% of 2- to 4-year-olds [1]. With regards to gender, NAFLD is roughly twice as prevalent in males [18–20]. While the exact etiology of this difference is unclear, hormonal differences are a leading hypothesis.
Ethnicity
NAFLD is most common in Hispanics, followed by Asians, Caucasians, and African Americans. Research suggests that genetics may be largely responsible for these ethnic disparities. For example, the I148M allele of PNPLA3 (a single nucleotide polymorphism) is strongly associated with steatosis, NASH, and fibrosis [21] and is most common in Hispanics, with a 50% carrier frequency in some cohorts [22]. Conversely, African Americans are more likely to carry the S453I allele of PNPLA3, which is associated with decreased hepatic steatosis [22]. There is also considerable variability within ethnic groups. For example, Mexican-American children appear to be at the highest risk for steatosis or NASH among Hispanics, whereas Filipino-American children are believed to have higher disease prevalence than Cambodian or Vietnamese Americans [1].
Comorbidities
NAFLD is associated with obesity, insulin resistance and diabetes, cardiovascular disease, the metabolic syndrome [23], decreased quality of life [24,25], and obstructive sleep apnea (OSA). These associations generally hold even after controlling for the other confounders listed. It is important to note that these data come largely from cross-sectional studies and direct causation has yet to be determined.
Insulin resistance in particular is strongly associated with NAFLD—so much so, in fact, that some consider it to be the hepatic manifestation of the metabolic syndrome. Additionally, children with features of the metabolic syndrome are more likely to have advanced histologic features of NAFLD [23]. There are also intriguing data from small pediatric studies to suggest that OSA may contribute to the development of hepatic fibrosis. In one study of 25 children with biopsy-proven NAFLD, for example, the presence of OSA and hypoxemia correlated with the degree of hepatic fibrosis [26]. In a slightly larger study of 65 children, OSA was also strongly associated with significant hepatic fibrosis (odds ratio, 5.91; 95% confidence interval, 3.23–7.42; P < 0.001). The duration of hypoxemia also correlated with histologic findings of inflammation and circulating biomarkers of apoptosis and fibrogenesis [27].
Other Laboratory Tests
Several studies have documented an association between elevated gamma-glutamyl transferase (GGT) and hepatic fibrosis [28,29], though others have been conflicting [30,31]. Pediatric studies have also demonstrated an inverse correlation between NASH and total bilirubin [32], serum potassium [33], and serum ceruloplasmin [34]. In addition, there are a number of serum biomarkers or biomarker panels commercially available for use in adults. Because similar efficacy data are unavailable in children, however, serum biomarkers should be primarily used for research purposes only.
Who should be screened for NAFLD? And how?
Published professional society recommendations differ significantly with regards to screening. In 2007, the American Academy of Pediatrics suggested screening obese children over 10 years of age or overweight children with additional risk factors with biannual liver tests [35]. There were no management recommendations made for elevated aminotransferase levels other than for subspecialty referral. In 2012, the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) recommended obtaining an ultrasound and liver tests in every obese child [36]. One month later, however, the American Gastroenterological Association, American Association for the Study of Liver Disease, and the American College of Gastroenterology published joint guidelines without screening recommendations “due to a paucity of evidence” [37].
Because these statements conflict and are based heavily on expert opinion, one should consider the risks, benefits, and costs to screening large numbers of patients. Until additional research clarifies this controversy, we suggest that providers individualize their screening practices to their population and the risks of each individual patient. For example, we would consider screening children who are obese; Hispanic or Asian; have multiple features of the metabolic syndrome; and/or those who have a family history of NAFLD. Further, we recommend screening children for NAFLD with serum liver enzymes only and not with ultrasonography.
Case Continued: Laboratory Results
ALT and GGT tests are ordered and the results are as follows:
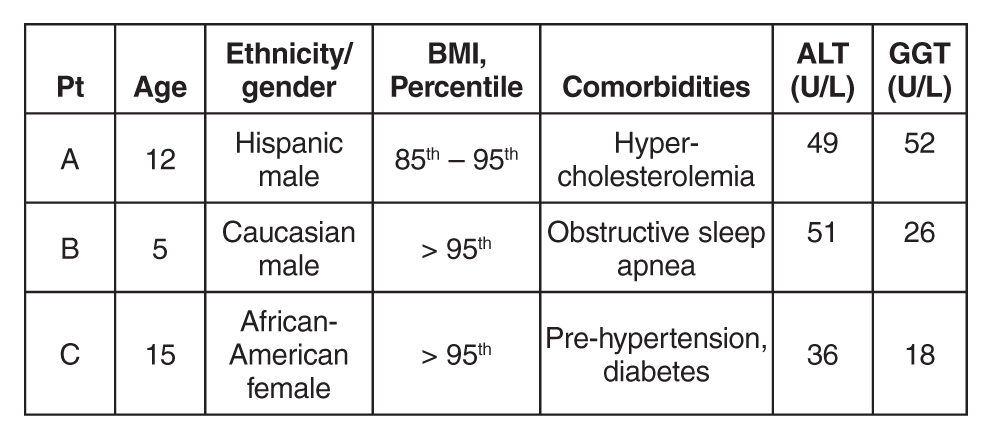
What is the differential for children with suspected NAFLD?

Autoimmune Hepatitis (AIH)
AIH is a progressive necro-inflammatory disorder of the liver characterized by elevated aminotransferases, positive autoantibodies, and distinctive histologic features. AIH is believed to occur in genetically predisposed patients in response to an environmental trigger. There is a female predominance and it can present in any age or ethnic group.
AIH is divided in 2 subtypes. Type 1 disease is characterized by a positive antinuclear (ANA) antibody and anti-smooth muscle antibody. More commonly, it presents in adolescence with an indolent course—many patients are asymptomatic until they develop features of cirrhosis and portal hypertension. Conversely, type 2 AIH is characterized by a positive liver kidney microsomal (LKM) antibody and tends to present acutely in young children. It is important to note that antibody titers can be falsely positive in a significant percentage of patients and, in such cases, are often mildly elevated [38]. We strongly suggest children with positive autoantibody titers be evaluated by a specialist.
Treatment should be started promptly to avoid progression to cirrhosis and should also done so in consultation with a pediatric gastroenterologist or hepatologist. The prognosis of AIH with immunosuppression is favorable, with long-term remission rates of approximately 80%. Transplantation is typically required in the remaining 10% to 20% [39].
Celiac Disease
Celiac disease is an autoimmune, inflammatory enteropathy caused by exposure to gluten in genetically susceptible individuals. Up to a third of all children presenting with celiac will have an elevated serum ALT [40]. Additional symptoms/features are both variable and nonspecific: abdominal pain, poor growth, diarrhea, or constipation, among others. Celiac is diagnosed by duodenal biopsy or a sufficiently elevated tissue transglutaminase antibody level [41]. Treatment with a strict gluten-free diet will resolve the enteropathy and normalize the serum aminotransferases.
Wilson’s Disease
Wilson’s disease is a metabolic disorder leading to copper deposition in the liver, brain, cornea, and kidneys. It is caused by an ATP7B gene mutation and inherited in an autosomal recessive fashion. Patients may present with asymptomatic liver disease, chronic hepatitis, acute liver failure, or with symptoms of portal hypertension. Neuropsychiatric symptoms may also be prominent. Screening tests include a serum ceruloplasmin and 24-hour urinary copper quantification. Because diagnosing Wilson’s disease can be challenging, however, further testing should occur in consultation with a pediatric gastroenterologist or hepatologist.
Viral Hepatitis
Chronic viral infections such as hepatitis B and C are still common etiologies of liver disease in the United States. However, universal vaccination and blood donor screening have reduced the risk of transmission; new antiviral agents will likely further decrease the prevalence and transmission risk over time. Acute viral hepatitis—cytomegalovirus, Epstein-Barr virus, hepatitis A, or hepatitis E—should also be considered in children who present with appropriate symptoms and an elevated ALT.
Drug-Induced
Drug-induced liver injury (DILI) can present with elevated serum aminotransferases (hepatocellular pattern), an elevated bilirubin (cholestatic pattern), or a mixed picture. Idiosyncratic DILI in children is commonly caused by antimicrobial or central nervous system agents and usually presents with a hepatocellular injury pattern. Substance abuse, including alcohol, is common and should also be investigated as the source of underlying liver disease.
Muscle Disease
Aspartate aminotransferase (AST) and ALT are present in hepatocytes, myocytes, and red blood cells, among other tissues. Thus, children with congenital myopathies or myositis can have elevated aminotransferases, typically with the AST higher than the ALT. In these patients, checking a creatine phosphokinase (CPK) level may lead to the correct diagnosis and limit unnecessary testing.
Other Metabolic Disorders
Myriad metabolic disorders present with liver disease and/or elevated serum aminotransferase levels. Individually, these conditions are rare but, collectively, are relatively common. Two of the more occult conditions—lysosomal acid lipase deficiency (LAL-D) and alpha-1 antitrypsin (A1A) deficiency—are discussed in further detail below.
LAL-D is an autosomal recessive disease resulting in the accumulation of cholesterol esters and triglycerides in lysosomes. Patients typically present with hepatomegaly and mildly elevated aminotransferases, an elevated LDL, low HDL cholesterol, and increased hepatic echogenicity on ultrasound. If a biopsy is obtained, microvesicular steatosis is predominant as opposed to macrovesicular steatosis found in NAFLD. The diagnosis of LAL-D can be made on a commercially available dry blood spot enzymatic assay or genetic testing and treatment has recently been FDA approved.
A1A deficiency is an autosomal recessive disease diagnosable by an alpha-1-antitrypsin phenotype. The clinical presentation is characterized by neonatal cholestasis in the infantile form and by hepatitis, cirrhosis and portal hypertension in older children. Classic symptoms of emphysema and chronic lung disease present in adulthood.
What further testing should be performed in children with suspected NAFLD?
For obese children with an elevated ALT or evidence of increased hepatic echogenicity, ESPGHAN recommends targeting the workup according to the child’s age [36]. According to their consensus statement, they recommend an upfront, thorough laboratory evaluation in children less than 10 years of age and consideration of a liver biopsy upon completion. For children over 10 years of age at low risk for NASH or fibrosis, additional laboratory evaluation is suggested 3 to 6 months after failed lifestyle interventions. In general, the recommended workup includes testing for conditions discussed in the section above such as viral hepatitis, AIH, Wilson’s disease, and others. If negative, ESPGHAN states that a liver biopsy should be “considered.”
The question of whether or not to obtain a liver biopsy is controversial, though there are several clear advantages to doing so. First, biopsy is the gold standard test for diagnosing NAFLD and there are no highly accurate, noninvasive tests currently approved for use in children. Second, biopsy is a more definitive means of ruling out competing diagnoses such as AIH. Third, biopsy may provide prognostic data. In a retrospective adult study of 136 patients, for example, those who presented with simple steatosis had a roughly 3% chance of progressing to cirrhosis within 10 years. If a patient within this cohort presented with NASH, however, the progression risk was approximately 30% within 5 years [42,43]. Fourth, due to potential side effects of medications, position papers recommend obtaining a liver biopsy prior to the initiation of pharmacotherapy [37]. Lastly, the risk for serious morbidity from a liver biopsy is low [44,45]. Alternatively, one must acknowledge the risks of liver biopsy: morbidity, sampling bias, invasiveness, cost, and sedation risks in children.
Case Continued: Biopsy Results
You refer your patients to a gastroenterologist. Tests for viral hepatitis, A1A deficiency, celiac disease, muscle disorders, Wilson’s disease, and AIH are negative. Ultimately, a liver biopsy is performed on all 3 children without complications. The results are presented below.
What is the treatment of NAFLD?
Lifestyle Modification
Lifestyle modifications are the mainstay of treatment for NAFLD. In adult studies, weight loss of more than 5% reduces hepatic steatosis whereas weight loss of more than 9% improves or eliminates NASH [47]. We recommend that children engage in age-appropriate, enjoyable, moderate- or vigorous-intensity aerobic activity for 60 minutes a day [48]. In addition, there should be a focus on reducing sedentary behavior by limiting screen time and a concerted effort to engage the family in lifestyle modifications.
Dietary interventions to treat NAFLD are less concrete but there is a growing body of literature to suggest that dietary fructose is particularly harmful. In adults, for example, fructose consumption is associated with the development of NAFLD [49] and hepatic fibrosis [50]. Recent data in adolescents has similarly documented an association between NAFLD incidence and energy-adjusted fructose intake [51]. It is worth highlighting that these clinical findings are also biologically plausible, as fructose is primarily metabolized within hepatocytes and has recently been shown to increase de novo lipogenesis [52,53]. In general, we suggest a well-balanced diet of unprocessed foods—that is, with limited added sugars—sufficient to induce gradual weight loss in older children or body weight maintenance in younger children.
Medications
Vitamin E is the only medication with proven efficacy in children, as demonstrated in the TONIC trial [20]. TONIC was a double-blind, multicenter, placebo-controlled study with 3 treatment arms: 800 IU of vitamin E daily, 1000 mg of metformin daily, or placebo. Metformin did not reduce the serum ALT or significantly improve liver histology and should therefore not be used for these indications. However, patients treated with vitamin E had a statistically significant improvement in the NAFLD activity score (a histologic grading system comprising steatosis, inflammation, and hepatocyte ballooning) and resolution of NASH when compared to placebo. For these reasons—as well as a paucity of other viable treatment options—vitamin E is routinely prescribed for children with biopsy-proven NASH. However, the long- term risks of high-dose vitamin E therapy in children are largely unknown.
Polyunsaturated fats such as docosahexaenoic acid (DHA) [54] and probiotics such as VSL #3 [55] have showed efficacy reducing hepatic steatosis in small, randomized clinical trials. Both medications need to be further validated before they can be recommended for use in children. Conversely, ursodeoxycholic acid has not been found to be efficacious in children with NAFLD [56], whereas phase IIb data on cysteamine is expected soon. There are currently insufficient data to recommend bariatric surgery as treatment for NAFLD in adolescence.
Case Continued: Follow-up
After their biopsies, both patients with NASH (patients A and B) are started on vitamin E therapy. All 3 patients continue to report for follow-up visits without short-term complications, though they have still been unable to significantly reduce their body mass index and have a persistently elevated serum ALT.
Summary
NAFLD is a common condition in the United States with serious personal and public health ramifications. This case-based review highlights the diagnostic and management challenges in children with NAFLD and the unique role primary care providers play in caring for these patients.
Corresponding author: Bryan Rudolph, MD, Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, 3415 Bainbridge Ave., Bronx, NY 10467, [email protected].
Financial disclosures: None.
1. Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388–93.
2. Welsh JA, Karpen S, Vos MB.Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr 2013;162:496–500.
3. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274–85.
4. McPherson S, Hardy T, Henderson E, et al. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 2015;62:1148–55.
5. Singh S, Allen AM, Wang Z, et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 2015;13:643–54.
6. Pais R, Charlotte F, Fedchuk L, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol 2013;59:550–6.
7. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, et al. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009;58:1538–44.
8. Mummadi RR, Kasturi KS, Chennareddygari S, et al. Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2008;6:1396–402.
9. Rashid M, Roberts EA. Nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2000;30:48–53.
10. Schwimmer JB, Dunn W, Norman GJ, et al. SAFETY study: alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010;138:1357–64.
11. Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med 2002;137:1–10.
12. Lee JK, Shim JH, Lee HC, et al. Estimation of the healthy upper limits for serum alanine aminotransferase in Asian populations with normal liver histology. Hepatology 2010;51:1577–83.
13. Kang HS, Um SH, Seo YS, et al. Healthy range for serum ALT and the clinical significance of "unhealthy" normal ALT levels in the Korean population. J Gastroenterol Hepatol 2011;26:292–9.
14. Zheng MH, Shi KQ, Fan YC, et al. Upper limits of normal for serum alanine aminotransferase levels in Chinese Han population. PLoS One 2012;7:e43736.
15. Molleston JP, Schwimmer JB, Yates KP, et al. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. J Pediatr 2014;164:707–13.
16. Dasarathy S, Dasarathy J, Khiyami A, et al. Validity of real time ultrasound in the diagnosis of hepatic steatosis: a prospective study. J Hepatol 2009;51:1061–7.
17. Nobili V, M. Pinzani M. Paediatric non-alcoholic fatty liver disease. Gut 2010;59:561–4.
18. Rudolph B, Rivas Y, Kulak S, et al. Yield of diagnostic tests in obese children with an elevated alanine aminotransferase. Acta Paediatr 2015;104:e557–63.
19. Nobili V, Manco M, Ciampalini P, et al. Metformin use in children with nonalcoholic fatty liver disease: an open-label, 24-month, observational pilot study. Clin Ther 2008;30:1168–76.
20. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659–68.
21. Krawczyk MP, Portincasa P, Lammert F. PNPLA3-associated steatohepatitis: toward a gene-based classification of fatty liver disease. Semin Liver Dis 2013;33:369–79.
22. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–5.
23. Patton HM, Yates K, Unalp-Arida A, et al. Association between metabolic syndrome and liver histology among children with nonalcoholic fatty liver disease. Am J Gastroenterol 2010;105:2093–102.
24. Kistler KD, Molleston J, Unalp A, et al., Symptoms and quality of life in obese children and adolescents with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2010;31:396–406.
25. Kerkar N, D'Urso C, Van Nostrand K, et al. Psychosocial outcomes for children with nonalcoholic fatty liver disease over time and compared with obese controls. J Pediatr Gastroenterol Nutr 2013;56:77–82.
26. Sundaram SS, Sokol RJ, Capocelli KE, et al. Obstructive sleep apnea and hypoxemia are associated with advanced liver histology in pediatric nonalcoholic fatty liver disease. J Pediatr 2014;164:699–706.
27. Nobili V, Cutrera R, Liccardo D, et al. Obstructive sleep apnea syndrome affects liver histology and inflammatory cell activation in pediatric nonalcoholic fatty liver disease, regardless of obesity/insulin resistance. Am J Respir Crit Care Med 2014;189:66–76.
28. Patton HM, Lavine JE, Van Natta ML, et al., Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008;135:1961–71.
29. Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641–9.
30. Nobili V, Parkes J, Bottazzo G, et al. Performance of ELF serum markers in predicting fibrosis stage in pediatric non-alcoholic fatty liver disease. Gastroenterology 2009;136:160–7.
31. Yang HR, Kim HR, Kim MJ, et al. Noninvasive parameters and hepatic fibrosis scores in children with nonalcoholic fatty liver disease. World J Gastroenterol 2012;18:1525–30.
32. Puri K, Nobili V, Melville K, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2013;57:114–8.
33. Tabbaa A, Shaker M, Lopez R, et al. Low serum potassium levels associated with disease severity in children with nonalcoholic fatty liver disease. Pediatr Gastroenterol Hepatol Nutr 2015;18:168–74.
34. Nobili V, Siotto M, Bedogni G, et al. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2013;56:370–5.
35. Barlow SE; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120 Suppl 4:S164–92.
36. Vajro P, Lenta S, Socha P, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2012;54:700–13.
37. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012;142:1592–609.
38. Vuppalanchi R, Gould RJ, Wilson LA, et al. Clinical significance of serum autoantibodies in patients with NAFLD: results from the nonalcoholic steatohepatitis clinical research network. Hepatol Int 2012;6:379–85.
39. Floreani A, Liberal R, Vergani D, et al. Autoimmune hepatitis: contrasts and comparisons in children and adults - a comprehensive review. J Autoimmun 2013;46:7–16.
40. Vajro P, Paolella G, Maggiore G, et al. Pediatric celiac disease, cryptogenic hypertransaminasemia, and autoimmune hepatitis. J Pediatr Gastroenterol Nutr 2013;56:663–70.
41. Husby S, Koletzko S, Korponay-Szabó IR, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54:136–60.
42. Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999;116:1413–9.
43. McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:521–33.
44. Ovchinsky N, Moreira RK, Lefkowitch JH, Lavine JE. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012;19:250–62.
45. Dezsőfi A, Baumann U, Dhawan A, et al. Liver biopsy in children: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2015;60:408–20.
46. Fusillo S, Rudolph B. Nonalcoholic fatty liver disease. Pediatr Rev 2015;36:198–205.
47. Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009;49:80–6.
48. School health guidelines to promote healthy eating and physical activity. MMWR Recomm Rep 2011;60(Rr-5):1–76.
49. Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–9.
50. Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1961–71.
51. O’Sullivan TA, Oddy WH, Bremner AP, et al. Lower fructose intake may help protect against development of nonalcoholic fatty liver in adolescents with obesity. J Pediatr Gastroenterol Nutr 2014;58:624–31.
52. Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 2008;138:1039–46.
53. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–34.
54. Nobili V, Alisi A, Della Corte C, et al., Docosahexaenoic acid for the treatment of fatty liver: randomised controlled trial in children. Nutr Metab Cardiovasc Dis 2013;23:1066–70.
55. Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2014;39:1276–85.
56. Vajro P, Franzese A, Valerio G, et al. Lack of efficacy of ursodeoxycholic acid for the treatment of liver abnormalities in obese children. J Pediatr 2000;136:739–43.
From the Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, Bronx, NY.
Abstract
- Objective: To review diagnostic challenges and management strategies in children with nonalcoholic fatty liver disease (NAFLD).
- Methods: Review of the literaure.
- Results: NAFLD is common in the United States and should be suspected in overweight or obese children with an elevated serum alanine aminotransferase level. The differential diagnosis for these patients is broad, however, and liver biopsy—the gold standard test—should be undertaken selectively after an appropriate workup. Patients should be counseled on lifestyle modifications, whereas vitamin E therapy can be initiated for those with biopsy-proven disease.
- Conclusion: Providers should have a high degree of suspicion for NAFLD, approaching the workup and diagnosis in an incremental, step-wise fashion. Further research is needed to standardize the diagnostic approach, identify reliable, noninvasive diagnostic measures, and develop novel treatment modalities.
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the Western world, affecting approximately 10% of children and a third of all adults in the United States [1–3]. It is a significant public health challenge and is estimated to soon be the number one indication for liver transplantation in adults.
NAFLD is a generic term encompassing 2 distinct conditions defined by their histopathology: simple steatosis and nonalcoholic steatohepatitis (NASH). Simple steatosis is characterized by predominantly macrovesicular—meaning large droplet—cytoplasmic lipid inclusions found in ≥ 5% of hepatocytes. NASH is defined as hepatic steatosis plus the additional features of inflammation, hepatocyte ballooning, and/or fibrosis. There are some adult data [4-6] and 1 retrospective pediatric study [7] demonstrating that over time, NAFLD may progress. That is, steatosis may progress to NASH and some patients with fibrosis will ultimately develop cirrhosis. If intervention is provided early in the histologic spectrum, NAFLD can be reversed [4,8] and late complications—such as cirrhosis, hepatocellular carcinoma, or liver transplantation—may be prevented.
It is important to highlight that the above definitions are based on histology and that a liver biopsy cannot be reasonably obtained in such a large percentage of the U.S. population. This case-based review will therefore focus primarily on the current diagnostic challenges facing health care providers as well as management strategies in children with presumed NAFLD.
Case Study
Initial Presentation
As you finish your charts at the end of a busy clinic day, you identify 3 patients who may have NAFLD:
History
All 3 patients presented to your office for a routine annual physical before the start of the school year and are asymptomatic. None of the patients has a family history of liver disease and their previously diagnosed comorbidities are listed in the table above. No patient is taking medications other than patient C, who is on metformin.
All 3 children have a smooth, velvety rash on their necks consistent with acanthosis nigricans with an otherwise normal physical exam. The liver and spleen are difficult to palpate but are seemingly normal.
What is the typical presentation for a child with NAFLD?
Most children with NAFLD are asymptomatic, though some may present with vague right upper quadrant abdominal pain. It is unclear, however, if the pain is caused by NAFLD or is rather an unrelated symptom that brings the child to the attention of a physician. In addition, hepatomegaly can be found in over 30% to 40% of patients [9]. For children without abdominal pain or hepatomegaly, most are recognized by an elevated serum alanine aminotransferase (ALT) or findings of increased liver echogenicity on ultrasonography.
Serum Alanine Aminotransferase
Serum aminotransferases are one of the more common screening tests for NAFLD. However, ALT is highly insensitive at commonly used thresholds and is also nonspecific. As documented in the SAFETY study, the upper limit of normal for ALT in healthy children should be set around 25 U/L in boys and 22 U/L in girls [10]. Yet even at these thresholds, the sensitivity of ALT to diagnose NAFLD is 80% in boys and 92% in girls, whereas specificity is 79% and 85%, respectively [10]. These findings are largely consistent with adult studies [11–14]. Furthermore, ALT does not correlate well with disease severity and children may still have NASH or significant fibrosis with normal values. In a well-characterized cohort of 91 children with biopsy-proven NAFLD, for example, early fibrosis was identified in 12% of children with a normal ALT (≤ 22 U/L for girls and ≤ 25 U/L in boys) [15]. Advanced fibrosis or cirrhosis was seen in 9% of children with an ALT up to 2 times this upper limit [15]. Thus, reliance on the serum ALT may significantly underestimate the prevalence and severity of liver injury.
Ultrasonography
Children with NAFLD typically have findings of increased hepatic echogenicity on abdominal ultrasonography. However, there are multiple limitations to sonography. First, ultrasound is insensitive for identifying mild steatosis if less than 30% of hepatocytes are affected [16,17]. Second, increased hepatic echogenicity is nonspecific and may be caused by inflammation, fibrosis, or intrahepatic accumulation of iron, copper, or glycogen. Third, there can be considerable inter- and intra-operator variability. And lastly, there is some evidence that ultrasounds do not add benefit to diagnosing children with NAFLD [18].
Which patients are at risk for developing hepatic steatosis and NASH?
Weight, Age, and Gender
There is a strong, direct correlation between body mass index (BMI) and NAFLD. The Study of Child and Adolescent Liver Epidemiology (SCALE)—a sentinel pediatric autopsy study of 742 children—found that 5% of normal weight children, 16% of overweight children, and 38% of obese children had NAFLD. The SCALE study also demonstrated an increasing prevalence with age, such that NAFLD was present in 17.3% of 15- to 19-year-olds but only in 0.2% of 2- to 4-year-olds [1]. With regards to gender, NAFLD is roughly twice as prevalent in males [18–20]. While the exact etiology of this difference is unclear, hormonal differences are a leading hypothesis.
Ethnicity
NAFLD is most common in Hispanics, followed by Asians, Caucasians, and African Americans. Research suggests that genetics may be largely responsible for these ethnic disparities. For example, the I148M allele of PNPLA3 (a single nucleotide polymorphism) is strongly associated with steatosis, NASH, and fibrosis [21] and is most common in Hispanics, with a 50% carrier frequency in some cohorts [22]. Conversely, African Americans are more likely to carry the S453I allele of PNPLA3, which is associated with decreased hepatic steatosis [22]. There is also considerable variability within ethnic groups. For example, Mexican-American children appear to be at the highest risk for steatosis or NASH among Hispanics, whereas Filipino-American children are believed to have higher disease prevalence than Cambodian or Vietnamese Americans [1].
Comorbidities
NAFLD is associated with obesity, insulin resistance and diabetes, cardiovascular disease, the metabolic syndrome [23], decreased quality of life [24,25], and obstructive sleep apnea (OSA). These associations generally hold even after controlling for the other confounders listed. It is important to note that these data come largely from cross-sectional studies and direct causation has yet to be determined.
Insulin resistance in particular is strongly associated with NAFLD—so much so, in fact, that some consider it to be the hepatic manifestation of the metabolic syndrome. Additionally, children with features of the metabolic syndrome are more likely to have advanced histologic features of NAFLD [23]. There are also intriguing data from small pediatric studies to suggest that OSA may contribute to the development of hepatic fibrosis. In one study of 25 children with biopsy-proven NAFLD, for example, the presence of OSA and hypoxemia correlated with the degree of hepatic fibrosis [26]. In a slightly larger study of 65 children, OSA was also strongly associated with significant hepatic fibrosis (odds ratio, 5.91; 95% confidence interval, 3.23–7.42; P < 0.001). The duration of hypoxemia also correlated with histologic findings of inflammation and circulating biomarkers of apoptosis and fibrogenesis [27].
Other Laboratory Tests
Several studies have documented an association between elevated gamma-glutamyl transferase (GGT) and hepatic fibrosis [28,29], though others have been conflicting [30,31]. Pediatric studies have also demonstrated an inverse correlation between NASH and total bilirubin [32], serum potassium [33], and serum ceruloplasmin [34]. In addition, there are a number of serum biomarkers or biomarker panels commercially available for use in adults. Because similar efficacy data are unavailable in children, however, serum biomarkers should be primarily used for research purposes only.
Who should be screened for NAFLD? And how?
Published professional society recommendations differ significantly with regards to screening. In 2007, the American Academy of Pediatrics suggested screening obese children over 10 years of age or overweight children with additional risk factors with biannual liver tests [35]. There were no management recommendations made for elevated aminotransferase levels other than for subspecialty referral. In 2012, the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) recommended obtaining an ultrasound and liver tests in every obese child [36]. One month later, however, the American Gastroenterological Association, American Association for the Study of Liver Disease, and the American College of Gastroenterology published joint guidelines without screening recommendations “due to a paucity of evidence” [37].
Because these statements conflict and are based heavily on expert opinion, one should consider the risks, benefits, and costs to screening large numbers of patients. Until additional research clarifies this controversy, we suggest that providers individualize their screening practices to their population and the risks of each individual patient. For example, we would consider screening children who are obese; Hispanic or Asian; have multiple features of the metabolic syndrome; and/or those who have a family history of NAFLD. Further, we recommend screening children for NAFLD with serum liver enzymes only and not with ultrasonography.
Case Continued: Laboratory Results
ALT and GGT tests are ordered and the results are as follows:
What is the differential for children with suspected NAFLD?
Autoimmune Hepatitis (AIH)
AIH is a progressive necro-inflammatory disorder of the liver characterized by elevated aminotransferases, positive autoantibodies, and distinctive histologic features. AIH is believed to occur in genetically predisposed patients in response to an environmental trigger. There is a female predominance and it can present in any age or ethnic group.
AIH is divided in 2 subtypes. Type 1 disease is characterized by a positive antinuclear (ANA) antibody and anti-smooth muscle antibody. More commonly, it presents in adolescence with an indolent course—many patients are asymptomatic until they develop features of cirrhosis and portal hypertension. Conversely, type 2 AIH is characterized by a positive liver kidney microsomal (LKM) antibody and tends to present acutely in young children. It is important to note that antibody titers can be falsely positive in a significant percentage of patients and, in such cases, are often mildly elevated [38]. We strongly suggest children with positive autoantibody titers be evaluated by a specialist.
Treatment should be started promptly to avoid progression to cirrhosis and should also done so in consultation with a pediatric gastroenterologist or hepatologist. The prognosis of AIH with immunosuppression is favorable, with long-term remission rates of approximately 80%. Transplantation is typically required in the remaining 10% to 20% [39].
Celiac Disease
Celiac disease is an autoimmune, inflammatory enteropathy caused by exposure to gluten in genetically susceptible individuals. Up to a third of all children presenting with celiac will have an elevated serum ALT [40]. Additional symptoms/features are both variable and nonspecific: abdominal pain, poor growth, diarrhea, or constipation, among others. Celiac is diagnosed by duodenal biopsy or a sufficiently elevated tissue transglutaminase antibody level [41]. Treatment with a strict gluten-free diet will resolve the enteropathy and normalize the serum aminotransferases.
Wilson’s Disease
Wilson’s disease is a metabolic disorder leading to copper deposition in the liver, brain, cornea, and kidneys. It is caused by an ATP7B gene mutation and inherited in an autosomal recessive fashion. Patients may present with asymptomatic liver disease, chronic hepatitis, acute liver failure, or with symptoms of portal hypertension. Neuropsychiatric symptoms may also be prominent. Screening tests include a serum ceruloplasmin and 24-hour urinary copper quantification. Because diagnosing Wilson’s disease can be challenging, however, further testing should occur in consultation with a pediatric gastroenterologist or hepatologist.
Viral Hepatitis
Chronic viral infections such as hepatitis B and C are still common etiologies of liver disease in the United States. However, universal vaccination and blood donor screening have reduced the risk of transmission; new antiviral agents will likely further decrease the prevalence and transmission risk over time. Acute viral hepatitis—cytomegalovirus, Epstein-Barr virus, hepatitis A, or hepatitis E—should also be considered in children who present with appropriate symptoms and an elevated ALT.
Drug-Induced
Drug-induced liver injury (DILI) can present with elevated serum aminotransferases (hepatocellular pattern), an elevated bilirubin (cholestatic pattern), or a mixed picture. Idiosyncratic DILI in children is commonly caused by antimicrobial or central nervous system agents and usually presents with a hepatocellular injury pattern. Substance abuse, including alcohol, is common and should also be investigated as the source of underlying liver disease.
Muscle Disease
Aspartate aminotransferase (AST) and ALT are present in hepatocytes, myocytes, and red blood cells, among other tissues. Thus, children with congenital myopathies or myositis can have elevated aminotransferases, typically with the AST higher than the ALT. In these patients, checking a creatine phosphokinase (CPK) level may lead to the correct diagnosis and limit unnecessary testing.
Other Metabolic Disorders
Myriad metabolic disorders present with liver disease and/or elevated serum aminotransferase levels. Individually, these conditions are rare but, collectively, are relatively common. Two of the more occult conditions—lysosomal acid lipase deficiency (LAL-D) and alpha-1 antitrypsin (A1A) deficiency—are discussed in further detail below.
LAL-D is an autosomal recessive disease resulting in the accumulation of cholesterol esters and triglycerides in lysosomes. Patients typically present with hepatomegaly and mildly elevated aminotransferases, an elevated LDL, low HDL cholesterol, and increased hepatic echogenicity on ultrasound. If a biopsy is obtained, microvesicular steatosis is predominant as opposed to macrovesicular steatosis found in NAFLD. The diagnosis of LAL-D can be made on a commercially available dry blood spot enzymatic assay or genetic testing and treatment has recently been FDA approved.
A1A deficiency is an autosomal recessive disease diagnosable by an alpha-1-antitrypsin phenotype. The clinical presentation is characterized by neonatal cholestasis in the infantile form and by hepatitis, cirrhosis and portal hypertension in older children. Classic symptoms of emphysema and chronic lung disease present in adulthood.
What further testing should be performed in children with suspected NAFLD?
For obese children with an elevated ALT or evidence of increased hepatic echogenicity, ESPGHAN recommends targeting the workup according to the child’s age [36]. According to their consensus statement, they recommend an upfront, thorough laboratory evaluation in children less than 10 years of age and consideration of a liver biopsy upon completion. For children over 10 years of age at low risk for NASH or fibrosis, additional laboratory evaluation is suggested 3 to 6 months after failed lifestyle interventions. In general, the recommended workup includes testing for conditions discussed in the section above such as viral hepatitis, AIH, Wilson’s disease, and others. If negative, ESPGHAN states that a liver biopsy should be “considered.”
The question of whether or not to obtain a liver biopsy is controversial, though there are several clear advantages to doing so. First, biopsy is the gold standard test for diagnosing NAFLD and there are no highly accurate, noninvasive tests currently approved for use in children. Second, biopsy is a more definitive means of ruling out competing diagnoses such as AIH. Third, biopsy may provide prognostic data. In a retrospective adult study of 136 patients, for example, those who presented with simple steatosis had a roughly 3% chance of progressing to cirrhosis within 10 years. If a patient within this cohort presented with NASH, however, the progression risk was approximately 30% within 5 years [42,43]. Fourth, due to potential side effects of medications, position papers recommend obtaining a liver biopsy prior to the initiation of pharmacotherapy [37]. Lastly, the risk for serious morbidity from a liver biopsy is low [44,45]. Alternatively, one must acknowledge the risks of liver biopsy: morbidity, sampling bias, invasiveness, cost, and sedation risks in children.
Case Continued: Biopsy Results
You refer your patients to a gastroenterologist. Tests for viral hepatitis, A1A deficiency, celiac disease, muscle disorders, Wilson’s disease, and AIH are negative. Ultimately, a liver biopsy is performed on all 3 children without complications. The results are presented below.
What is the treatment of NAFLD?
Lifestyle Modification
Lifestyle modifications are the mainstay of treatment for NAFLD. In adult studies, weight loss of more than 5% reduces hepatic steatosis whereas weight loss of more than 9% improves or eliminates NASH [47]. We recommend that children engage in age-appropriate, enjoyable, moderate- or vigorous-intensity aerobic activity for 60 minutes a day [48]. In addition, there should be a focus on reducing sedentary behavior by limiting screen time and a concerted effort to engage the family in lifestyle modifications.
Dietary interventions to treat NAFLD are less concrete but there is a growing body of literature to suggest that dietary fructose is particularly harmful. In adults, for example, fructose consumption is associated with the development of NAFLD [49] and hepatic fibrosis [50]. Recent data in adolescents has similarly documented an association between NAFLD incidence and energy-adjusted fructose intake [51]. It is worth highlighting that these clinical findings are also biologically plausible, as fructose is primarily metabolized within hepatocytes and has recently been shown to increase de novo lipogenesis [52,53]. In general, we suggest a well-balanced diet of unprocessed foods—that is, with limited added sugars—sufficient to induce gradual weight loss in older children or body weight maintenance in younger children.
Medications
Vitamin E is the only medication with proven efficacy in children, as demonstrated in the TONIC trial [20]. TONIC was a double-blind, multicenter, placebo-controlled study with 3 treatment arms: 800 IU of vitamin E daily, 1000 mg of metformin daily, or placebo. Metformin did not reduce the serum ALT or significantly improve liver histology and should therefore not be used for these indications. However, patients treated with vitamin E had a statistically significant improvement in the NAFLD activity score (a histologic grading system comprising steatosis, inflammation, and hepatocyte ballooning) and resolution of NASH when compared to placebo. For these reasons—as well as a paucity of other viable treatment options—vitamin E is routinely prescribed for children with biopsy-proven NASH. However, the long- term risks of high-dose vitamin E therapy in children are largely unknown.
Polyunsaturated fats such as docosahexaenoic acid (DHA) [54] and probiotics such as VSL #3 [55] have showed efficacy reducing hepatic steatosis in small, randomized clinical trials. Both medications need to be further validated before they can be recommended for use in children. Conversely, ursodeoxycholic acid has not been found to be efficacious in children with NAFLD [56], whereas phase IIb data on cysteamine is expected soon. There are currently insufficient data to recommend bariatric surgery as treatment for NAFLD in adolescence.
Case Continued: Follow-up
After their biopsies, both patients with NASH (patients A and B) are started on vitamin E therapy. All 3 patients continue to report for follow-up visits without short-term complications, though they have still been unable to significantly reduce their body mass index and have a persistently elevated serum ALT.
Summary
NAFLD is a common condition in the United States with serious personal and public health ramifications. This case-based review highlights the diagnostic and management challenges in children with NAFLD and the unique role primary care providers play in caring for these patients.
Corresponding author: Bryan Rudolph, MD, Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, 3415 Bainbridge Ave., Bronx, NY 10467, [email protected].
Financial disclosures: None.
From the Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, Bronx, NY.
Abstract
- Objective: To review diagnostic challenges and management strategies in children with nonalcoholic fatty liver disease (NAFLD).
- Methods: Review of the literaure.
- Results: NAFLD is common in the United States and should be suspected in overweight or obese children with an elevated serum alanine aminotransferase level. The differential diagnosis for these patients is broad, however, and liver biopsy—the gold standard test—should be undertaken selectively after an appropriate workup. Patients should be counseled on lifestyle modifications, whereas vitamin E therapy can be initiated for those with biopsy-proven disease.
- Conclusion: Providers should have a high degree of suspicion for NAFLD, approaching the workup and diagnosis in an incremental, step-wise fashion. Further research is needed to standardize the diagnostic approach, identify reliable, noninvasive diagnostic measures, and develop novel treatment modalities.
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the Western world, affecting approximately 10% of children and a third of all adults in the United States [1–3]. It is a significant public health challenge and is estimated to soon be the number one indication for liver transplantation in adults.
NAFLD is a generic term encompassing 2 distinct conditions defined by their histopathology: simple steatosis and nonalcoholic steatohepatitis (NASH). Simple steatosis is characterized by predominantly macrovesicular—meaning large droplet—cytoplasmic lipid inclusions found in ≥ 5% of hepatocytes. NASH is defined as hepatic steatosis plus the additional features of inflammation, hepatocyte ballooning, and/or fibrosis. There are some adult data [4-6] and 1 retrospective pediatric study [7] demonstrating that over time, NAFLD may progress. That is, steatosis may progress to NASH and some patients with fibrosis will ultimately develop cirrhosis. If intervention is provided early in the histologic spectrum, NAFLD can be reversed [4,8] and late complications—such as cirrhosis, hepatocellular carcinoma, or liver transplantation—may be prevented.
It is important to highlight that the above definitions are based on histology and that a liver biopsy cannot be reasonably obtained in such a large percentage of the U.S. population. This case-based review will therefore focus primarily on the current diagnostic challenges facing health care providers as well as management strategies in children with presumed NAFLD.
Case Study
Initial Presentation
As you finish your charts at the end of a busy clinic day, you identify 3 patients who may have NAFLD:
History
All 3 patients presented to your office for a routine annual physical before the start of the school year and are asymptomatic. None of the patients has a family history of liver disease and their previously diagnosed comorbidities are listed in the table above. No patient is taking medications other than patient C, who is on metformin.
All 3 children have a smooth, velvety rash on their necks consistent with acanthosis nigricans with an otherwise normal physical exam. The liver and spleen are difficult to palpate but are seemingly normal.
What is the typical presentation for a child with NAFLD?
Most children with NAFLD are asymptomatic, though some may present with vague right upper quadrant abdominal pain. It is unclear, however, if the pain is caused by NAFLD or is rather an unrelated symptom that brings the child to the attention of a physician. In addition, hepatomegaly can be found in over 30% to 40% of patients [9]. For children without abdominal pain or hepatomegaly, most are recognized by an elevated serum alanine aminotransferase (ALT) or findings of increased liver echogenicity on ultrasonography.
Serum Alanine Aminotransferase
Serum aminotransferases are one of the more common screening tests for NAFLD. However, ALT is highly insensitive at commonly used thresholds and is also nonspecific. As documented in the SAFETY study, the upper limit of normal for ALT in healthy children should be set around 25 U/L in boys and 22 U/L in girls [10]. Yet even at these thresholds, the sensitivity of ALT to diagnose NAFLD is 80% in boys and 92% in girls, whereas specificity is 79% and 85%, respectively [10]. These findings are largely consistent with adult studies [11–14]. Furthermore, ALT does not correlate well with disease severity and children may still have NASH or significant fibrosis with normal values. In a well-characterized cohort of 91 children with biopsy-proven NAFLD, for example, early fibrosis was identified in 12% of children with a normal ALT (≤ 22 U/L for girls and ≤ 25 U/L in boys) [15]. Advanced fibrosis or cirrhosis was seen in 9% of children with an ALT up to 2 times this upper limit [15]. Thus, reliance on the serum ALT may significantly underestimate the prevalence and severity of liver injury.
Ultrasonography
Children with NAFLD typically have findings of increased hepatic echogenicity on abdominal ultrasonography. However, there are multiple limitations to sonography. First, ultrasound is insensitive for identifying mild steatosis if less than 30% of hepatocytes are affected [16,17]. Second, increased hepatic echogenicity is nonspecific and may be caused by inflammation, fibrosis, or intrahepatic accumulation of iron, copper, or glycogen. Third, there can be considerable inter- and intra-operator variability. And lastly, there is some evidence that ultrasounds do not add benefit to diagnosing children with NAFLD [18].
Which patients are at risk for developing hepatic steatosis and NASH?
Weight, Age, and Gender
There is a strong, direct correlation between body mass index (BMI) and NAFLD. The Study of Child and Adolescent Liver Epidemiology (SCALE)—a sentinel pediatric autopsy study of 742 children—found that 5% of normal weight children, 16% of overweight children, and 38% of obese children had NAFLD. The SCALE study also demonstrated an increasing prevalence with age, such that NAFLD was present in 17.3% of 15- to 19-year-olds but only in 0.2% of 2- to 4-year-olds [1]. With regards to gender, NAFLD is roughly twice as prevalent in males [18–20]. While the exact etiology of this difference is unclear, hormonal differences are a leading hypothesis.
Ethnicity
NAFLD is most common in Hispanics, followed by Asians, Caucasians, and African Americans. Research suggests that genetics may be largely responsible for these ethnic disparities. For example, the I148M allele of PNPLA3 (a single nucleotide polymorphism) is strongly associated with steatosis, NASH, and fibrosis [21] and is most common in Hispanics, with a 50% carrier frequency in some cohorts [22]. Conversely, African Americans are more likely to carry the S453I allele of PNPLA3, which is associated with decreased hepatic steatosis [22]. There is also considerable variability within ethnic groups. For example, Mexican-American children appear to be at the highest risk for steatosis or NASH among Hispanics, whereas Filipino-American children are believed to have higher disease prevalence than Cambodian or Vietnamese Americans [1].
Comorbidities
NAFLD is associated with obesity, insulin resistance and diabetes, cardiovascular disease, the metabolic syndrome [23], decreased quality of life [24,25], and obstructive sleep apnea (OSA). These associations generally hold even after controlling for the other confounders listed. It is important to note that these data come largely from cross-sectional studies and direct causation has yet to be determined.
Insulin resistance in particular is strongly associated with NAFLD—so much so, in fact, that some consider it to be the hepatic manifestation of the metabolic syndrome. Additionally, children with features of the metabolic syndrome are more likely to have advanced histologic features of NAFLD [23]. There are also intriguing data from small pediatric studies to suggest that OSA may contribute to the development of hepatic fibrosis. In one study of 25 children with biopsy-proven NAFLD, for example, the presence of OSA and hypoxemia correlated with the degree of hepatic fibrosis [26]. In a slightly larger study of 65 children, OSA was also strongly associated with significant hepatic fibrosis (odds ratio, 5.91; 95% confidence interval, 3.23–7.42; P < 0.001). The duration of hypoxemia also correlated with histologic findings of inflammation and circulating biomarkers of apoptosis and fibrogenesis [27].
Other Laboratory Tests
Several studies have documented an association between elevated gamma-glutamyl transferase (GGT) and hepatic fibrosis [28,29], though others have been conflicting [30,31]. Pediatric studies have also demonstrated an inverse correlation between NASH and total bilirubin [32], serum potassium [33], and serum ceruloplasmin [34]. In addition, there are a number of serum biomarkers or biomarker panels commercially available for use in adults. Because similar efficacy data are unavailable in children, however, serum biomarkers should be primarily used for research purposes only.
Who should be screened for NAFLD? And how?
Published professional society recommendations differ significantly with regards to screening. In 2007, the American Academy of Pediatrics suggested screening obese children over 10 years of age or overweight children with additional risk factors with biannual liver tests [35]. There were no management recommendations made for elevated aminotransferase levels other than for subspecialty referral. In 2012, the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) recommended obtaining an ultrasound and liver tests in every obese child [36]. One month later, however, the American Gastroenterological Association, American Association for the Study of Liver Disease, and the American College of Gastroenterology published joint guidelines without screening recommendations “due to a paucity of evidence” [37].
Because these statements conflict and are based heavily on expert opinion, one should consider the risks, benefits, and costs to screening large numbers of patients. Until additional research clarifies this controversy, we suggest that providers individualize their screening practices to their population and the risks of each individual patient. For example, we would consider screening children who are obese; Hispanic or Asian; have multiple features of the metabolic syndrome; and/or those who have a family history of NAFLD. Further, we recommend screening children for NAFLD with serum liver enzymes only and not with ultrasonography.
Case Continued: Laboratory Results
ALT and GGT tests are ordered and the results are as follows:
What is the differential for children with suspected NAFLD?
Autoimmune Hepatitis (AIH)
AIH is a progressive necro-inflammatory disorder of the liver characterized by elevated aminotransferases, positive autoantibodies, and distinctive histologic features. AIH is believed to occur in genetically predisposed patients in response to an environmental trigger. There is a female predominance and it can present in any age or ethnic group.
AIH is divided in 2 subtypes. Type 1 disease is characterized by a positive antinuclear (ANA) antibody and anti-smooth muscle antibody. More commonly, it presents in adolescence with an indolent course—many patients are asymptomatic until they develop features of cirrhosis and portal hypertension. Conversely, type 2 AIH is characterized by a positive liver kidney microsomal (LKM) antibody and tends to present acutely in young children. It is important to note that antibody titers can be falsely positive in a significant percentage of patients and, in such cases, are often mildly elevated [38]. We strongly suggest children with positive autoantibody titers be evaluated by a specialist.
Treatment should be started promptly to avoid progression to cirrhosis and should also done so in consultation with a pediatric gastroenterologist or hepatologist. The prognosis of AIH with immunosuppression is favorable, with long-term remission rates of approximately 80%. Transplantation is typically required in the remaining 10% to 20% [39].
Celiac Disease
Celiac disease is an autoimmune, inflammatory enteropathy caused by exposure to gluten in genetically susceptible individuals. Up to a third of all children presenting with celiac will have an elevated serum ALT [40]. Additional symptoms/features are both variable and nonspecific: abdominal pain, poor growth, diarrhea, or constipation, among others. Celiac is diagnosed by duodenal biopsy or a sufficiently elevated tissue transglutaminase antibody level [41]. Treatment with a strict gluten-free diet will resolve the enteropathy and normalize the serum aminotransferases.
Wilson’s Disease
Wilson’s disease is a metabolic disorder leading to copper deposition in the liver, brain, cornea, and kidneys. It is caused by an ATP7B gene mutation and inherited in an autosomal recessive fashion. Patients may present with asymptomatic liver disease, chronic hepatitis, acute liver failure, or with symptoms of portal hypertension. Neuropsychiatric symptoms may also be prominent. Screening tests include a serum ceruloplasmin and 24-hour urinary copper quantification. Because diagnosing Wilson’s disease can be challenging, however, further testing should occur in consultation with a pediatric gastroenterologist or hepatologist.
Viral Hepatitis
Chronic viral infections such as hepatitis B and C are still common etiologies of liver disease in the United States. However, universal vaccination and blood donor screening have reduced the risk of transmission; new antiviral agents will likely further decrease the prevalence and transmission risk over time. Acute viral hepatitis—cytomegalovirus, Epstein-Barr virus, hepatitis A, or hepatitis E—should also be considered in children who present with appropriate symptoms and an elevated ALT.
Drug-Induced
Drug-induced liver injury (DILI) can present with elevated serum aminotransferases (hepatocellular pattern), an elevated bilirubin (cholestatic pattern), or a mixed picture. Idiosyncratic DILI in children is commonly caused by antimicrobial or central nervous system agents and usually presents with a hepatocellular injury pattern. Substance abuse, including alcohol, is common and should also be investigated as the source of underlying liver disease.
Muscle Disease
Aspartate aminotransferase (AST) and ALT are present in hepatocytes, myocytes, and red blood cells, among other tissues. Thus, children with congenital myopathies or myositis can have elevated aminotransferases, typically with the AST higher than the ALT. In these patients, checking a creatine phosphokinase (CPK) level may lead to the correct diagnosis and limit unnecessary testing.
Other Metabolic Disorders
Myriad metabolic disorders present with liver disease and/or elevated serum aminotransferase levels. Individually, these conditions are rare but, collectively, are relatively common. Two of the more occult conditions—lysosomal acid lipase deficiency (LAL-D) and alpha-1 antitrypsin (A1A) deficiency—are discussed in further detail below.
LAL-D is an autosomal recessive disease resulting in the accumulation of cholesterol esters and triglycerides in lysosomes. Patients typically present with hepatomegaly and mildly elevated aminotransferases, an elevated LDL, low HDL cholesterol, and increased hepatic echogenicity on ultrasound. If a biopsy is obtained, microvesicular steatosis is predominant as opposed to macrovesicular steatosis found in NAFLD. The diagnosis of LAL-D can be made on a commercially available dry blood spot enzymatic assay or genetic testing and treatment has recently been FDA approved.
A1A deficiency is an autosomal recessive disease diagnosable by an alpha-1-antitrypsin phenotype. The clinical presentation is characterized by neonatal cholestasis in the infantile form and by hepatitis, cirrhosis and portal hypertension in older children. Classic symptoms of emphysema and chronic lung disease present in adulthood.
What further testing should be performed in children with suspected NAFLD?
For obese children with an elevated ALT or evidence of increased hepatic echogenicity, ESPGHAN recommends targeting the workup according to the child’s age [36]. According to their consensus statement, they recommend an upfront, thorough laboratory evaluation in children less than 10 years of age and consideration of a liver biopsy upon completion. For children over 10 years of age at low risk for NASH or fibrosis, additional laboratory evaluation is suggested 3 to 6 months after failed lifestyle interventions. In general, the recommended workup includes testing for conditions discussed in the section above such as viral hepatitis, AIH, Wilson’s disease, and others. If negative, ESPGHAN states that a liver biopsy should be “considered.”
The question of whether or not to obtain a liver biopsy is controversial, though there are several clear advantages to doing so. First, biopsy is the gold standard test for diagnosing NAFLD and there are no highly accurate, noninvasive tests currently approved for use in children. Second, biopsy is a more definitive means of ruling out competing diagnoses such as AIH. Third, biopsy may provide prognostic data. In a retrospective adult study of 136 patients, for example, those who presented with simple steatosis had a roughly 3% chance of progressing to cirrhosis within 10 years. If a patient within this cohort presented with NASH, however, the progression risk was approximately 30% within 5 years [42,43]. Fourth, due to potential side effects of medications, position papers recommend obtaining a liver biopsy prior to the initiation of pharmacotherapy [37]. Lastly, the risk for serious morbidity from a liver biopsy is low [44,45]. Alternatively, one must acknowledge the risks of liver biopsy: morbidity, sampling bias, invasiveness, cost, and sedation risks in children.
Case Continued: Biopsy Results
You refer your patients to a gastroenterologist. Tests for viral hepatitis, A1A deficiency, celiac disease, muscle disorders, Wilson’s disease, and AIH are negative. Ultimately, a liver biopsy is performed on all 3 children without complications. The results are presented below.
What is the treatment of NAFLD?
Lifestyle Modification
Lifestyle modifications are the mainstay of treatment for NAFLD. In adult studies, weight loss of more than 5% reduces hepatic steatosis whereas weight loss of more than 9% improves or eliminates NASH [47]. We recommend that children engage in age-appropriate, enjoyable, moderate- or vigorous-intensity aerobic activity for 60 minutes a day [48]. In addition, there should be a focus on reducing sedentary behavior by limiting screen time and a concerted effort to engage the family in lifestyle modifications.
Dietary interventions to treat NAFLD are less concrete but there is a growing body of literature to suggest that dietary fructose is particularly harmful. In adults, for example, fructose consumption is associated with the development of NAFLD [49] and hepatic fibrosis [50]. Recent data in adolescents has similarly documented an association between NAFLD incidence and energy-adjusted fructose intake [51]. It is worth highlighting that these clinical findings are also biologically plausible, as fructose is primarily metabolized within hepatocytes and has recently been shown to increase de novo lipogenesis [52,53]. In general, we suggest a well-balanced diet of unprocessed foods—that is, with limited added sugars—sufficient to induce gradual weight loss in older children or body weight maintenance in younger children.
Medications
Vitamin E is the only medication with proven efficacy in children, as demonstrated in the TONIC trial [20]. TONIC was a double-blind, multicenter, placebo-controlled study with 3 treatment arms: 800 IU of vitamin E daily, 1000 mg of metformin daily, or placebo. Metformin did not reduce the serum ALT or significantly improve liver histology and should therefore not be used for these indications. However, patients treated with vitamin E had a statistically significant improvement in the NAFLD activity score (a histologic grading system comprising steatosis, inflammation, and hepatocyte ballooning) and resolution of NASH when compared to placebo. For these reasons—as well as a paucity of other viable treatment options—vitamin E is routinely prescribed for children with biopsy-proven NASH. However, the long- term risks of high-dose vitamin E therapy in children are largely unknown.
Polyunsaturated fats such as docosahexaenoic acid (DHA) [54] and probiotics such as VSL #3 [55] have showed efficacy reducing hepatic steatosis in small, randomized clinical trials. Both medications need to be further validated before they can be recommended for use in children. Conversely, ursodeoxycholic acid has not been found to be efficacious in children with NAFLD [56], whereas phase IIb data on cysteamine is expected soon. There are currently insufficient data to recommend bariatric surgery as treatment for NAFLD in adolescence.
Case Continued: Follow-up
After their biopsies, both patients with NASH (patients A and B) are started on vitamin E therapy. All 3 patients continue to report for follow-up visits without short-term complications, though they have still been unable to significantly reduce their body mass index and have a persistently elevated serum ALT.
Summary
NAFLD is a common condition in the United States with serious personal and public health ramifications. This case-based review highlights the diagnostic and management challenges in children with NAFLD and the unique role primary care providers play in caring for these patients.
Corresponding author: Bryan Rudolph, MD, Albert Einstein College of Medicine, Division of Pediatric Gastroenterology and Nutrition, Children’s Hospital at Montefiore, 3415 Bainbridge Ave., Bronx, NY 10467, [email protected].
Financial disclosures: None.
1. Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388–93.
2. Welsh JA, Karpen S, Vos MB.Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr 2013;162:496–500.
3. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274–85.
4. McPherson S, Hardy T, Henderson E, et al. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 2015;62:1148–55.
5. Singh S, Allen AM, Wang Z, et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 2015;13:643–54.
6. Pais R, Charlotte F, Fedchuk L, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol 2013;59:550–6.
7. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, et al. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009;58:1538–44.
8. Mummadi RR, Kasturi KS, Chennareddygari S, et al. Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2008;6:1396–402.
9. Rashid M, Roberts EA. Nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2000;30:48–53.
10. Schwimmer JB, Dunn W, Norman GJ, et al. SAFETY study: alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010;138:1357–64.
11. Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med 2002;137:1–10.
12. Lee JK, Shim JH, Lee HC, et al. Estimation of the healthy upper limits for serum alanine aminotransferase in Asian populations with normal liver histology. Hepatology 2010;51:1577–83.
13. Kang HS, Um SH, Seo YS, et al. Healthy range for serum ALT and the clinical significance of "unhealthy" normal ALT levels in the Korean population. J Gastroenterol Hepatol 2011;26:292–9.
14. Zheng MH, Shi KQ, Fan YC, et al. Upper limits of normal for serum alanine aminotransferase levels in Chinese Han population. PLoS One 2012;7:e43736.
15. Molleston JP, Schwimmer JB, Yates KP, et al. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. J Pediatr 2014;164:707–13.
16. Dasarathy S, Dasarathy J, Khiyami A, et al. Validity of real time ultrasound in the diagnosis of hepatic steatosis: a prospective study. J Hepatol 2009;51:1061–7.
17. Nobili V, M. Pinzani M. Paediatric non-alcoholic fatty liver disease. Gut 2010;59:561–4.
18. Rudolph B, Rivas Y, Kulak S, et al. Yield of diagnostic tests in obese children with an elevated alanine aminotransferase. Acta Paediatr 2015;104:e557–63.
19. Nobili V, Manco M, Ciampalini P, et al. Metformin use in children with nonalcoholic fatty liver disease: an open-label, 24-month, observational pilot study. Clin Ther 2008;30:1168–76.
20. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659–68.
21. Krawczyk MP, Portincasa P, Lammert F. PNPLA3-associated steatohepatitis: toward a gene-based classification of fatty liver disease. Semin Liver Dis 2013;33:369–79.
22. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–5.
23. Patton HM, Yates K, Unalp-Arida A, et al. Association between metabolic syndrome and liver histology among children with nonalcoholic fatty liver disease. Am J Gastroenterol 2010;105:2093–102.
24. Kistler KD, Molleston J, Unalp A, et al., Symptoms and quality of life in obese children and adolescents with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2010;31:396–406.
25. Kerkar N, D'Urso C, Van Nostrand K, et al. Psychosocial outcomes for children with nonalcoholic fatty liver disease over time and compared with obese controls. J Pediatr Gastroenterol Nutr 2013;56:77–82.
26. Sundaram SS, Sokol RJ, Capocelli KE, et al. Obstructive sleep apnea and hypoxemia are associated with advanced liver histology in pediatric nonalcoholic fatty liver disease. J Pediatr 2014;164:699–706.
27. Nobili V, Cutrera R, Liccardo D, et al. Obstructive sleep apnea syndrome affects liver histology and inflammatory cell activation in pediatric nonalcoholic fatty liver disease, regardless of obesity/insulin resistance. Am J Respir Crit Care Med 2014;189:66–76.
28. Patton HM, Lavine JE, Van Natta ML, et al., Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008;135:1961–71.
29. Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641–9.
30. Nobili V, Parkes J, Bottazzo G, et al. Performance of ELF serum markers in predicting fibrosis stage in pediatric non-alcoholic fatty liver disease. Gastroenterology 2009;136:160–7.
31. Yang HR, Kim HR, Kim MJ, et al. Noninvasive parameters and hepatic fibrosis scores in children with nonalcoholic fatty liver disease. World J Gastroenterol 2012;18:1525–30.
32. Puri K, Nobili V, Melville K, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2013;57:114–8.
33. Tabbaa A, Shaker M, Lopez R, et al. Low serum potassium levels associated with disease severity in children with nonalcoholic fatty liver disease. Pediatr Gastroenterol Hepatol Nutr 2015;18:168–74.
34. Nobili V, Siotto M, Bedogni G, et al. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2013;56:370–5.
35. Barlow SE; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120 Suppl 4:S164–92.
36. Vajro P, Lenta S, Socha P, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2012;54:700–13.
37. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012;142:1592–609.
38. Vuppalanchi R, Gould RJ, Wilson LA, et al. Clinical significance of serum autoantibodies in patients with NAFLD: results from the nonalcoholic steatohepatitis clinical research network. Hepatol Int 2012;6:379–85.
39. Floreani A, Liberal R, Vergani D, et al. Autoimmune hepatitis: contrasts and comparisons in children and adults - a comprehensive review. J Autoimmun 2013;46:7–16.
40. Vajro P, Paolella G, Maggiore G, et al. Pediatric celiac disease, cryptogenic hypertransaminasemia, and autoimmune hepatitis. J Pediatr Gastroenterol Nutr 2013;56:663–70.
41. Husby S, Koletzko S, Korponay-Szabó IR, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54:136–60.
42. Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999;116:1413–9.
43. McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:521–33.
44. Ovchinsky N, Moreira RK, Lefkowitch JH, Lavine JE. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012;19:250–62.
45. Dezsőfi A, Baumann U, Dhawan A, et al. Liver biopsy in children: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2015;60:408–20.
46. Fusillo S, Rudolph B. Nonalcoholic fatty liver disease. Pediatr Rev 2015;36:198–205.
47. Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009;49:80–6.
48. School health guidelines to promote healthy eating and physical activity. MMWR Recomm Rep 2011;60(Rr-5):1–76.
49. Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–9.
50. Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1961–71.
51. O’Sullivan TA, Oddy WH, Bremner AP, et al. Lower fructose intake may help protect against development of nonalcoholic fatty liver in adolescents with obesity. J Pediatr Gastroenterol Nutr 2014;58:624–31.
52. Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 2008;138:1039–46.
53. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–34.
54. Nobili V, Alisi A, Della Corte C, et al., Docosahexaenoic acid for the treatment of fatty liver: randomised controlled trial in children. Nutr Metab Cardiovasc Dis 2013;23:1066–70.
55. Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2014;39:1276–85.
56. Vajro P, Franzese A, Valerio G, et al. Lack of efficacy of ursodeoxycholic acid for the treatment of liver abnormalities in obese children. J Pediatr 2000;136:739–43.
1. Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388–93.
2. Welsh JA, Karpen S, Vos MB.Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr 2013;162:496–500.
3. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274–85.
4. McPherson S, Hardy T, Henderson E, et al. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 2015;62:1148–55.
5. Singh S, Allen AM, Wang Z, et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 2015;13:643–54.
6. Pais R, Charlotte F, Fedchuk L, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol 2013;59:550–6.
7. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, et al. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009;58:1538–44.
8. Mummadi RR, Kasturi KS, Chennareddygari S, et al. Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2008;6:1396–402.
9. Rashid M, Roberts EA. Nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2000;30:48–53.
10. Schwimmer JB, Dunn W, Norman GJ, et al. SAFETY study: alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010;138:1357–64.
11. Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med 2002;137:1–10.
12. Lee JK, Shim JH, Lee HC, et al. Estimation of the healthy upper limits for serum alanine aminotransferase in Asian populations with normal liver histology. Hepatology 2010;51:1577–83.
13. Kang HS, Um SH, Seo YS, et al. Healthy range for serum ALT and the clinical significance of "unhealthy" normal ALT levels in the Korean population. J Gastroenterol Hepatol 2011;26:292–9.
14. Zheng MH, Shi KQ, Fan YC, et al. Upper limits of normal for serum alanine aminotransferase levels in Chinese Han population. PLoS One 2012;7:e43736.
15. Molleston JP, Schwimmer JB, Yates KP, et al. Histological abnormalities in children with nonalcoholic fatty liver disease and normal or mildly elevated alanine aminotransferase levels. J Pediatr 2014;164:707–13.
16. Dasarathy S, Dasarathy J, Khiyami A, et al. Validity of real time ultrasound in the diagnosis of hepatic steatosis: a prospective study. J Hepatol 2009;51:1061–7.
17. Nobili V, M. Pinzani M. Paediatric non-alcoholic fatty liver disease. Gut 2010;59:561–4.
18. Rudolph B, Rivas Y, Kulak S, et al. Yield of diagnostic tests in obese children with an elevated alanine aminotransferase. Acta Paediatr 2015;104:e557–63.
19. Nobili V, Manco M, Ciampalini P, et al. Metformin use in children with nonalcoholic fatty liver disease: an open-label, 24-month, observational pilot study. Clin Ther 2008;30:1168–76.
20. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659–68.
21. Krawczyk MP, Portincasa P, Lammert F. PNPLA3-associated steatohepatitis: toward a gene-based classification of fatty liver disease. Semin Liver Dis 2013;33:369–79.
22. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–5.
23. Patton HM, Yates K, Unalp-Arida A, et al. Association between metabolic syndrome and liver histology among children with nonalcoholic fatty liver disease. Am J Gastroenterol 2010;105:2093–102.
24. Kistler KD, Molleston J, Unalp A, et al., Symptoms and quality of life in obese children and adolescents with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2010;31:396–406.
25. Kerkar N, D'Urso C, Van Nostrand K, et al. Psychosocial outcomes for children with nonalcoholic fatty liver disease over time and compared with obese controls. J Pediatr Gastroenterol Nutr 2013;56:77–82.
26. Sundaram SS, Sokol RJ, Capocelli KE, et al. Obstructive sleep apnea and hypoxemia are associated with advanced liver histology in pediatric nonalcoholic fatty liver disease. J Pediatr 2014;164:699–706.
27. Nobili V, Cutrera R, Liccardo D, et al. Obstructive sleep apnea syndrome affects liver histology and inflammatory cell activation in pediatric nonalcoholic fatty liver disease, regardless of obesity/insulin resistance. Am J Respir Crit Care Med 2014;189:66–76.
28. Patton HM, Lavine JE, Van Natta ML, et al., Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008;135:1961–71.
29. Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641–9.
30. Nobili V, Parkes J, Bottazzo G, et al. Performance of ELF serum markers in predicting fibrosis stage in pediatric non-alcoholic fatty liver disease. Gastroenterology 2009;136:160–7.
31. Yang HR, Kim HR, Kim MJ, et al. Noninvasive parameters and hepatic fibrosis scores in children with nonalcoholic fatty liver disease. World J Gastroenterol 2012;18:1525–30.
32. Puri K, Nobili V, Melville K, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2013;57:114–8.
33. Tabbaa A, Shaker M, Lopez R, et al. Low serum potassium levels associated with disease severity in children with nonalcoholic fatty liver disease. Pediatr Gastroenterol Hepatol Nutr 2015;18:168–74.
34. Nobili V, Siotto M, Bedogni G, et al. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2013;56:370–5.
35. Barlow SE; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120 Suppl 4:S164–92.
36. Vajro P, Lenta S, Socha P, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2012;54:700–13.
37. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012;142:1592–609.
38. Vuppalanchi R, Gould RJ, Wilson LA, et al. Clinical significance of serum autoantibodies in patients with NAFLD: results from the nonalcoholic steatohepatitis clinical research network. Hepatol Int 2012;6:379–85.
39. Floreani A, Liberal R, Vergani D, et al. Autoimmune hepatitis: contrasts and comparisons in children and adults - a comprehensive review. J Autoimmun 2013;46:7–16.
40. Vajro P, Paolella G, Maggiore G, et al. Pediatric celiac disease, cryptogenic hypertransaminasemia, and autoimmune hepatitis. J Pediatr Gastroenterol Nutr 2013;56:663–70.
41. Husby S, Koletzko S, Korponay-Szabó IR, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54:136–60.
42. Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999;116:1413–9.
43. McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:521–33.
44. Ovchinsky N, Moreira RK, Lefkowitch JH, Lavine JE. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012;19:250–62.
45. Dezsőfi A, Baumann U, Dhawan A, et al. Liver biopsy in children: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2015;60:408–20.
46. Fusillo S, Rudolph B. Nonalcoholic fatty liver disease. Pediatr Rev 2015;36:198–205.
47. Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009;49:80–6.
48. School health guidelines to promote healthy eating and physical activity. MMWR Recomm Rep 2011;60(Rr-5):1–76.
49. Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–9.
50. Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1961–71.
51. O’Sullivan TA, Oddy WH, Bremner AP, et al. Lower fructose intake may help protect against development of nonalcoholic fatty liver in adolescents with obesity. J Pediatr Gastroenterol Nutr 2014;58:624–31.
52. Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 2008;138:1039–46.
53. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–34.
54. Nobili V, Alisi A, Della Corte C, et al., Docosahexaenoic acid for the treatment of fatty liver: randomised controlled trial in children. Nutr Metab Cardiovasc Dis 2013;23:1066–70.
55. Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2014;39:1276–85.
56. Vajro P, Franzese A, Valerio G, et al. Lack of efficacy of ursodeoxycholic acid for the treatment of liver abnormalities in obese children. J Pediatr 2000;136:739–43.
Unresponsive and mute after he smoked ‘Spice’
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).

Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10

In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.

1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).
Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10
In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.
CASE Mute and nonresponsive
Mr. R, a 19-year-old African-American man, is brought to the emergency room (ER) because he has reduced oral intake and mutism, and is not attending to activities of daily living (ADL). His family reports gradual onset of symptoms over the past month after he began using “Spice,” a synthetic cannabinoid (Box1-8).
Mr. R has been using marijuana regularly for a few years. He has no history of psychiatric illness. The family history is positive for schizophrenia (mother).
Mr. R slowly stopped speaking and eating, and no longer responds to verbal stimulation. On examination, he responds only with unintelligible mumbling. Mr. R exhibits blunted affect and fails to maintain eye contact, looking to the side of the interviewer. He exhibits severe psychomotor retardation but without posturing or waxy flexibility. It takes him approximately 3 minutes to transfer between chairs, and he is incontinent of bladder and bowel.
Mr. R has not experienced a similar episode in the past, although he had exhibited brief paranoia while intoxicated with marijuana.
Before this episode, Mr. R had been moving between his grandmother’s and father’s homes and was attending high school classes. Recent stressful events include his brother’s incarceration and his father having re-entered his life after a long absence.
Which treatment would you initiate for Mr. R’s symptoms of catatonia?
a) dantrolene
b) a benzodiazepine
c) an antipsychotic
d) electroconvulsive therapy (ECT)
The authors’ observations
Catatonia is a common complication in a variety of psychiatric and medical contexts. It can be a feature of mood disorders, schizophrenia, metabolic disturbances, drug intoxication, neuroleptic malignant syndrome (NMS), and encephalopathy. The most common psychiatric comorbidity is bipolar disorder; as many as 25% of cases are caused by a medical or neurological condition.9 When accompanied by fever and autonomic instability, so-called malignant catatonia can lead to respiratory failure, coma, and death.
Catatonia is characterized by ≥3 of the elements outlined in Table 1.10
In DSM-5, catatonia is no longer considered a subtype of schizophrenia, but is a specifier in the following disorders: brief psychotic disorder, schizophreniform disorder, schizoaffective disorder, and substance-induced psychotic disorder. In addition, catatonia not otherwise specified is reserved for cases when the cause is not apparent; this diagnosis is intended to lead to greater recognition of catatonia and prompt initiation of treatment. DSM-5 stops short of classifying catatonia as an independent syndrome, however. Changes in clinical status can be charted with instruments such as the Bush-Francis Catatonia Rating Scale.
Workup and treatment
The initial workup of patients with catatonia is extensive. A basic metabolic panel can detect electrolyte disturbances and acute renal failure. Monitoring creatine kinase (CK) allows clinicians to assess for rhabdomyolysis. Patients should also undergo an infectious workup, including complete blood count (CBC) and chest radiography, because patients can develop pneumonia due to atelectasis or aspiration. Additional workup could include EEG, erythrocyte sedimentation rate, D-dimer, urinalysis, urine drug screen, antinuclear antibodies, magnetic resonance imaging, cerebrospinal fluid analysis, anti-N-methyl-D-aspartate receptor antibodies, and serum iron, which could predict development of NMS in patients treated with an antipsychotic.11
Treatment. In addition to supportive measures, the initial treatment of choice for catatonia is a benzodiazepine, lorazepam being the most commonly used agent; dramatic improvement in symptoms can be seen within minutes of IV administration. A high dosage of lorazepam (14 to 16 mg/d) sometimes is required for symptomatic relief. Zolpidem also has been used successfully to treat catatonia, although the supporting literature is less extensive.12
Antipsychotics generally are held during the initial stages of catatonia treatment because they can exacerbate symptoms and increase the likelihood of NMS. Glutamate antagonists, such as amantadine and memantine, also are being investigated for treating catatonia.9
ECT is effective but is reserved for when pharmacotherapy has failed or when a rapid response is required. ECT is associated with cognitive and medical complications, although current techniques have greatly mitigated the risks. Mortality is estimated to be 1 in every 10,000 patients or 1 for every 80,000 treatments, most often due to a cardiac or pulmonary cause.13 Patients receiving ECT could experience temporary anterograde amnesia and confusion as well as retrograde amnesia, particularly memories formed around the time of treatment.
Response to benzodiazepine therapy varies: Some patients experience significant improvement after 1 dose; others require a high dosage for an extended period. More than 70% of cases remit with benzodiazepines; ECT should be considered after several days or earlier if indicated.9 Some patients with catatonia require a slow benzodiazepine taper to prevent symptoms from recurring.
Patients with catatonia are at risk of dehydration and malnutrition, and might require IV fluids or parenteral nutrition. These patients also are at risk of constipation, ileus, decubitus ulcers, deep vein thrombosis, and pulmonary embolism. Encourage early ambulation and consider prescribing an antithrombotic. Some patients might require physical therapy to prevent or treat muscle contractures.
TREATMENT Benzodiazepines, ECT
Mr. R is admitted for stabilization of catatonic symptoms. A basic metabolic panel, CBC with differential, urine drug screen, urinalysis, folate level, thyroid-stimulating hormone level, vitamin B12, EEG, and a stool culture are unremarkable. Ammonia level is slightly elevated at 40 µmol/L.
Mr. R is started on IM lorazepam, 1 mg every 8 hours. Antipsychotics are held in part because of an elevated CK level (614 U/L). CK is rechecked daily and increases to 5,681 U/L by the second week. Internal medicine is consulted because Mr. R could develop NMS. However, the treatment team thinks that CK elevation is caused by immobility, because Mr. R remains afebrile, normotensive, and without leukocytosis.
After 4 days of treatment, Mr. R can follow simple commands. He nods or shakes his head when questioned. IV fluids are started because of limited oral intake. As the month progresses, Mr. R’s CK levels slowly trend downward, toward 500 U/L.
Mr. R progresses slowly with benzodiazepine therapy. He begins to ambulate, make eye contact, and look at interviewers. Lorazepam is slowly titrated to 4 mg IM every 8 hours. On hospital Day 20, his functioning reaches a plateau; Mr. R’s cognition continues to fluctuate with periods of unresponsiveness, immobility, and incontinence.
The treatment team obtains consent from the family to begin ECT. On hospital Day 24, bilateral transtemporal ECT is initiated and continued 3 times a week. Mr. R tolerates the procedure without complications. After the first treatment, he demonstrates spontaneous speech for the first time since admission. He continues to improve overall but has a variable clinical course.
By Day 30, Mr. R can state the day, month, year, and that he is in the “psych” unit. He remembers being on the unit for a long time and says that he had been attempting to talk but “it wasn’t coming out.” When further questioned about substance use, he admits to using Spice for the month before admission and marijuana regularly over several years. He denies using other illicit drugs or alcohol.
Mr. R is started on olanzapine, 2.5 mg/d, titrated to 15 mg/d. He becomes increasingly interactive, although with occasional bouts of confusion, and regains bladder and bowel control. He receives a total of 12 ECT treatments. The family is adamant that Mr. R should not receive more ECT treatments, and is not interested in maintenance therapy. Mr. R’s father and grandmother visit and believe that he is back to baseline functioning. After 51 days of inpatient treatment, Mr. R is discharged on olanzapine, 15 mg/d, and oral lorazepam, 1 mg/d.
Nine days later, Mr. R is brought to the ER because of unresponsiveness, poor oral intake, refusal of medication, bowel and bladder incontinence, and inability to perform ADL. His father reports that he administered olanzapine but, because he only recognized the brand name of lorazepam, he did not get that prescription filled. Mr. R slowly decompensates and, by the time of readmission, refuses all medications.
Over the next few months, Mr. R is readmitted several times for similar symptoms. Again, the family states they do not want further ECT; the father believes that these treatments have caused his son’s condition. Complicating the matter is that the father had been out of his son’s life for an extended period and is unaccustomed to his son’s display of psychiatric symptoms.
The authors’ observations
The use of ECT for drug-induced psychosis is not well described in the literature because substance abuse is exclusionary in many trials. The safety and efficacy of ECT has been established for adolescents with first-episode psychosis14 and with catatonia.15,16
The use of ECT in Spice-induced catatonia has been reported in 2 case studies.17,18
Case 1. A 36-year-old man with schizophrenia and Cannabis dependence was admitted for auditory hallucinations, disorganization, paranoia, and manic symptoms, which progressed to catatonia.17 His symptoms were profound, including psychomotor retardation, rigidity, posturing, waxy flexibility, and inability to perform ADL.
The patient later reported that, 3 weeks prior, he had stopped taking his psychotropic medications and started smoking “K2,” a synthetic cannabinoid, because it was cheaper and easier to obtain than Cannabis. He had never experienced disturbances in motor function or speech in the past, even during episodes of Cannabis use and medication non-adherence.
After clozapine and benzodiazepine treatment (as high as 12 mg/d of lorazepam) did not resolve his symptoms, the patient received 6 bilateral ECT treatments over 16 days, with complete resolution of catatonic symptoms. He showed marked improvement, including resumption of speech after the first treatment, although he required an additional 20 days of inpatient care. As in our case, exposure to synthetic cannabinoids was self-reported; no confirmatory tests were performed.
Case 2. A 17-year-old male with no history of psychosis exhibited catatonic symptoms after smoking an estimated 2 to 3 g/d of K2 over 2 months.18 Similar to the case of Mr. R, he plateaued after lorazepam treatment, and then received 6 ECT treatments, which resulted in complete resolution of symptoms. He was discharged with olanzapine.
As our patient, and the 2 cases cited, show, ECT seems to be an effective option for Spice-induced catatonia. Unlike those published cases, however, our patient achieved only brief resolution of symptoms after an acute course of ECT. There appears to be a subset of patients who require maintenance ECT or prolonged benzodiazepine therapy after Spice-induced catatonia.
1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
1. Cohen J, Morrison S, Greenberg J, et al. Clinical presentation of intoxication due to synthetic cannabinoids. Pediatrics. 2012;129(4):e1064-e1067.
2. Spaderna M, Addy PH, D’Souza DC. Spicing things up: synthetic cannabinoids. Psychopharmacology (Berl). 2013;228(4):525-540.
3. Johnston LD, O’Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use. 2012 Overview: key findings on adolescent drug use. http://monitoringthefuture.org/pubs/monographs/mtf-overview2012.pdf. Published February 2013. Accessed February 8, 2016.
4. Hu X, Primack BA, Barnett TE, et al. College students and use of K2: an emerging drug abuse in young persons. Subst Abuse Treat Prev Policy. 2011;6:16.
5. Hurst D, Loeffler G, McLay R. Psychosis associated with synthetic cannabinoid agonists: a case series. Am J Psychiatry. 2011;168(10):1119.
6. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Medi Biol Res. 2006;39(4):421-429.
7. Fadda P, Robinson L, Fratta W, et al. Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropsychopharmacology. 2004;47(8):1170-1179.
8. Large M, Sharma S, Compton MT, et al. Cannabis use and earlier onset of psychosis: a systemic meta-analysis. Arch Gen Psychiatry. 2011;68(6):555-561.
9. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181.
10. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
11. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome. Biol Psychiatry. 1998;44(6):499-507.
12. Thomas P, Rascle C, Mastain B, et al. Test for catatonia with zolpidem. Lancet. 1997;349(9053):702.
13. American Psychiatric Association. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, DC: American Psychiatric Publishing; 2001.
14. Zhang ZJ, Chen YC, Wang HN, et al. Electroconvulsive therapy improves antipsychotic and somnographic responses in adolescents with first-episode psychosis—a case-control study. Schizophr Res. 2012;137(1-3):97-103.
15. Consoli A, Benmiloud M, Wachtel L, et al. Electroconvulsive therapy in adolescents with the catatonia syndrome: efficacy and ethics. J ECT. 2010;26(4):259-265.
16. Shoirah H, Hamoda HM. Electroconvulsive therapy in children and adolescents. Expert Rev Neurother. 2011;11(1):127-137.
17. Leibu E, Garakani A, McGonigle DP, et al. Electroconvulsive therapy (ECT) for catatonia in a patient with schizophrenia and synthetic cannabinoid abuse: a case report. J ECT. 2013;29(4):e61-e62. doi: 10.1097/YCT.0b013e318290fa36.
18. Smith DL, Roberts C. Synthetic marijuana use and development of catatonia in a 17-year-old male. Minn Med. 2014;97(5):38.
Polycystic Ovary Syndrome in Adolescents
From the Department of Pediatrics, Section of Endocrinology & Diabetes, Medical College of Wisconsin, Milwaukee, WI.
Abstract
- Objective: To review the diagnosis and management of polycystic ovary syndrome (PCOS) in adolescent patients.
- Methods: Review of the literature.
- Results: PCOS is a complex, heterogeneous disorder that frequently manifests during puberty. The symptoms of PCOS (ie, menstrual irregularities, hirsutism, and acne) tend to overlap with normal pubertal changes. Diagnostic criteria for PCOS in the adolescent age-group is still lacking. Current practice is to utilize adult diagnostic criteria, which raises the concern for misdiagnosis. The underlying etiology for the disorder is still unclear, but insulin resistance is present in both obese and non-obese PCOS patients. Although recognizing adolescents with PCOS is challenging, evaluating and managing patients for hyperandrogenemia and metabolic syndrome is imperative to prevent long-term reproductive and metabolic complications.
- Conclusion: PCOS is increasingly encountered during adolescence. Recognizing adolescent girls with PCOS is a challenge but important for preventing long-term adverse health outcomes.
Polycystic ovary syndrome (PCOS) is a complex disorder most commonly characterized by chronic anovulation and clinical and biochemical features of hyperandrogenemia. It affects 4% to 12% of reproductive-aged women [1,2]. In adolescents, the exact prevalence is unknown, but in a recent study the prevalence of a confirmed diagnosis of PCOS in adolescents aged 15 to 19 years was 0.56%, which increased to 1.14% when undiagnosed cases with documented symptoms qualifying for PCOS according to NIH criteria were included [3]. The primary underlying defect in PCOS remains unknown, but key features include insulin resistance, impaired gonadotropin dynamics, and androgen excess.
 Profound functional variations in the hypothalamic-pituitary-ovarian axis commonly seen during normal puberty may result in clinical and biochemical changes that mimic some of the features of PCOS. During the early stages of puberty, adolescent girls tend to have anovulatory menstrual cycles, higher androgen levels, and polycystic ovaries [4,5]. Thus, the clinical signs of hyperandrogenemia commonly seen in adults are less reliable in the adolescent age-group. Diagnostic criteria have been developed for adults and are based upon the various combinations of oligomenorrhea, unexplained hyperandrogenemia, and polycystic ovaries on imaging (Table 1) [6–8]. Applying these adult criteria in adolescent patients with suspected PCOS has always raised the concern of misdiagnosis as some of the changes seen in this age-group may likely be due to normal pubertal development. However, due to the paucity of data, the current practice is to utilize the adult diagnostic criteria. Because of the heterogeneous nature of the disorder, recognizing adolescents with PCOS may be challenging. However, early recognition and management is important to prevent some of the long-term reproductive and metabolic complications associated with this syndrome.
Profound functional variations in the hypothalamic-pituitary-ovarian axis commonly seen during normal puberty may result in clinical and biochemical changes that mimic some of the features of PCOS. During the early stages of puberty, adolescent girls tend to have anovulatory menstrual cycles, higher androgen levels, and polycystic ovaries [4,5]. Thus, the clinical signs of hyperandrogenemia commonly seen in adults are less reliable in the adolescent age-group. Diagnostic criteria have been developed for adults and are based upon the various combinations of oligomenorrhea, unexplained hyperandrogenemia, and polycystic ovaries on imaging (Table 1) [6–8]. Applying these adult criteria in adolescent patients with suspected PCOS has always raised the concern of misdiagnosis as some of the changes seen in this age-group may likely be due to normal pubertal development. However, due to the paucity of data, the current practice is to utilize the adult diagnostic criteria. Because of the heterogeneous nature of the disorder, recognizing adolescents with PCOS may be challenging. However, early recognition and management is important to prevent some of the long-term reproductive and metabolic complications associated with this syndrome.
Case Study
Initial Presentation
A 16-year-old female patient presents to the PCOS clinic for evaluation of obesity and amenorrhea.
History
The patient, who is otherwise healthy, began gaining weight at age 7. During this period, her weight increased from the 15th to (currently) the 90th percentile; her height remained constant (75th percentile). Menarche was at 12 years of age. Menstrual periods have been irregular since the onset of menarche and she has had no periods for the past 5 months. She noticed excessive hair growth on her face, chin, and neck soon after the onset of menarche. She has been shaving her facial hair once every 2–3 days.
The patient’s detailed diet history included eating 3 meals daily and snacks in-between meals. The patient was consuming sweet beverages regularly. There was minimal intake fruits and vegetables. The portion sizes for each meal were large. The patient had minimal physical activity and screen time was more than 2 hours daily.
Family history is significant for obesity and type 2 diabetes in her mother and maternal grandmother and is negative for PCOS.
Physical Examination
Vital signs were within normal limits. She was 5 ft 6 in tall and weighed 242 lb, with a body mass index (BMI) of 40 (99th percentile; Z-score 2.41). Physical examination showed coarse hair extending from the sideburns to the chin as well as from pubis symphysis to navel with evidence of hair removal. She had acanthosis nigricans on her neck, mild acne, and evidence of central obesity with pink striae marks on the abdomen. She was Tanner stage 5 for breast and pubic hair and there was no evidence of virilization (clitoral hypertrophy, deepening of the voice, severe hirsutism, male pattern baldness, and masculine habitus). Other physical examination findings were within normal limits.
What physical findings in this patient are suggestive of clinical hyperandrogenemia?
Physiologic irregular menstruation is a well known phenomenon in adolescent girls and is generally due to anovulatory cycles [9–12]. Menstrual cycles shorter than 19 days or longer than 90 days at any stage after menarche are considered abnormal. The menstrual irregularity that is commonly seen within the first 2–3 years after the first menarche can last up to 5 years [5]. However, the majority of girls establish 20- to 45-day cycles within the first 2 years [13].
Androgen excess, defined by the presence of clinical and/or biochemical hyperandrogenemia, should be considered in any adolescent girl who is 2 to 3 years’ post-menarche and presenting with irregular menstrual periods, coarse terminal hair in a male distribution pattern (hirsutism), or moderate to severe inflammatory acne. Hirsutism is androgen dependent [14–16] and must be distinguished from hypertrichosis, which is generalized excessive vellus hair growth present all over the body. Clinical hyperandrogenemia, which includes hirsutism, acne vulgaris, as well as androgenetic alopecia, is well correlated with elevated androgen levels; however, the severity of hirsutism does not correlate well with circulating androgen levels [17,18]. Mild hirsutism is often not associated with hyperandrogenemia in otherwise asymptomatic individuals,but it may be a sign of hyperandrogenemia in adolescents when associated with other features of PCOS, ie, menstrual irregularity [14–16, 19–22]. Defining hirsutism in early adolescence may be difficult since the sexual hair may still be developing, and laboratory evaluation should be considered (see below), especially in an overweight/obese adolescent girl presenting with oligomenorrhea. Ethnic variation due to decreased skin sensitivity to androgens can result in minimal hirsutism despite elevated plasma androgen levels and must be considered among certain Asian women. Women with PCOS from China, Japan, Thailand, and East and Southeast Asian countries tend to have low scores on hirsutism rating scales even with elevated plasma androgens levels [16,23].
Although having acne during puberty is not considered as a marker for hyperandrogenemia, patients with moderate to severe inflammatory acne that is poorly responsive to topical treatment should be evaluated for underlying hyperandrogenemia [19,24,25].
What laboratory tests should be obtained to when there is clinical suspicion of hyperandrogenemia?

Case Continued
The patient underwent laboratory assessment that included total and free testosterone levels, lipid panel, thyroid studies, prolactin level, comprehensive metabolic panel (CMP) and hemoglobin A1c (HbA1c). Due to lack of virilization, she was not tested for PCOS-like syndromes. Her total and free testosterone were 90 ng/dL (normal, < 41) and 24.7 pg/mL (normal, 0.5–3.9) respectively. Thyroid-stimulating hormone and prolactin levels were normal. She had normal lipid levels and CMP but HbA1c was 5.9% (pre-diabetic range). The results of a 2-hour oral glucose tolerance test revealed a level of 160 mg/dL, indicative of impaired glucose tolerance.
What is the pathophysiology and diagnostic criteria for PCOS in adolescents?
PCOS has diverse etiology and has been linked to both genetic and environmental factors affecting ovarian steroidogenesis [13,30]. While the familial clustering strongly supports the role of genetic factors, variability in phenotypic features within the same or different families indicates the importance of environmental contribution [31–34].
The exact underlying mechanism leading to disruption of ovulation is still unclear; however, hyperinsulinemia augmenting ovarian androgen production has been well recognized [35–37]. Insulin resistance is a characteristic finding in PCOS and occurs both in obese and lean patients [38,39]. Obesity further exacerbates the insulin resistance state in PCOS patients. Therefore, obese patients with PCOS have more severe hyperandrogenemia and consequences from it (hirsutism, menstrual abnormalities, and metabolic derangements) than normal-weight PCOS patients [40,41]. Similar to LH, insulin can stimulate ovarian theca cells directly and cause increased production of androgens [42]. Elevated androgen levels cause the irregular menstrual periods as well as clinical signs of hyperandrogenemia, such as hirsutism and acne.
Altered gonadotropin dynamics is another possible etiological factor that is linked with PCOS. Hyperinsulinemia affects the regulation of gonadotropin-releasing hormone (GnRH) pulse generator, causing hypersecretion of LH [43]. Obese peripubertal girls have been identified having altered LH secretion [44,45]. This results in increased LH levels relative to FSH. Normal FSH is required to stimulate ovarian folliculognensis; insufficient FSH levels cause anovulation and menstrual irregularities. Abnormal LH secretion and fasting insulin levels have been identified the independent predictors for hyperandrogenemia in some peripubertal obese girls [46].
In 2010 Carmina et al published new criteria to diagnose PCOS in adolescents [27].They recommended that in diagnosing PCOS in adolescents, all 3 previously mentioned criteria should be present: hyperandrogenemia, chronic anovulation, and polycystic ovaries. With the exception of worsening hirsutism, the new recommendations greatly emphasized biochemical hyperandrogenemia (elevated free testosterone levels using sensitive assays). Chronic anovulation was defined as persistence of menstrual irregularities 2 years post-menarche and pelvic ultrasound (USG) showing increased ovarian size (> 10 cm3). Normal physiological variations unrelated to hyperandrogenemia are common in adolescent ovaries and limits the usefulness of pelvic USG as a diagnostic criterion for PCOS [13,47,48]. Also, the prevalence of increased ovarian size in hyperandrogenemic adolescent patients was reported to be low, and its utility as a criterion for diagnosis needs to be further explored [49]. In our current practice we do not rely on pelvic USG findings to make a PCOS diagnosis.
Due to longstanding controversies and lack of consensus surrounding the accurate diagnostic criteria, a recent guideline was developed by experts in pediatric endocrinology and adolescent medicine invited by the Pediatric Endocrine Society to address these issues [13].The guideline committee assessed the literature in order to define which criteria have sufficient evidence to be used for diagnosis of PCOS in adolescents. They recommend that PCOS should be considered in an adolescent girl presenting with unexplained menstrual irregularities, moderate to severe hirsutism or acne, and elevated levels of serum androgens (total and free testosterone) using reliable assay with well-defined ranges. Although intrinsic insulin resistance unique to PCOS is well known, none of the current guidelines either for adolescent and adult women include it as part of the diagnostic criteria. Since longitudinal studies focusing on the natural history of PCOS in this age-group are lacking, the current recommendations focus on timely screening and treatment in symptomatic adolescent girls suspected of having PCOS.
When there are PCOS features but menstrual irregularity has not been present for at least 2 years, one can defer the diagnostic label and instead use the term at-risk for PCOS. Such patients should have frequent longitudinal re-evaluations and should be offered treatment for their symptoms [13].
How should adolescents with PCOS be managed?
The treatment of PCOS is symptom-directed and should be tailored according to the complaints of the individual patient. However, it also must focus on the core dysfunctions: anovulation, hyperandrogenemia, obesity, and insulin resistance. It also requires bridging patient expectations of regulating menses, lessening the troublesome clinical signs of hyperandrogenemia (hirsutism, acne), and obesity management with the health care provider’s goals of preventing endometrial hyperplasia and cancer, diabetes mellitus, and cardiovascular disease.
Regulating menstruation and reducing cutaneous manifestations of hyperandrogenemia is the priority for any adolescent with PCOS. Combined oral contraceptive pills (COCs) are the first line of medical treatment for most adolescents. COCs restore endometrial cycling and suppress androgen levels, and are therefore optimal in treating abnormal uterine bleeding, protecting against endometrial carcinoma, and alleviating cutaneous manifestations of hyperandrogenemia (hirsutism and acne). Progestin monotherapy is considered an alternative therapy in individuals with contraindications to COCs (ie, thromboembolic risk). Although it is not effective in lowering androgen levels thus does not help reduce hair growth and acne, progestin monotherapy protects the endometrium and reduces the risk of endometrial cancer [50].
The majority of patients with PCOS are overweight or obese. Regardless of BMI, patients with PCOS have profound intrinsic insulin resistance that gets worse with overweight or obesity. Weight reduction by restricting caloric intake and increasing physical exercise is vital and has shown to be effective in regulating menstrual cycles, but is difficult to achieve [51–53]. Metformin can regulate menstrual cycles and decrease androgen levels by improving insulin sensitivity [54,55]. The use of metformin in PCOS patients is still controversial and abnormal glucose tolerance is the only approved indication [61]. However, combing metformin with COCs and lifestyle modification in obese PCOS patients has been shown to be used more frequently in pediatric endocrine clinics [56]. COCs are the only agents that can lower testosterone levels and improve ovulation and hirsutism; these effects are seen less frequently with lifestyle modification or metformin, either used alone or in combination.
COC monotherapy is first-line therapy to treat hirsutism. Consider anti-androgen treatment for hirsutism if there is no improvement after 6–9 months of hormonal treatment [57]. Antiandrogens reduce hirsutism by decreasing androgen production and binding the androgen receptors in target tissue. Spironolactone is the most commonly used antiandrogen therapy in adolescent girls with PCOS. Given the risk of teratogenicity with antiandrogens if pregnancy occurs, it is recommended to use it in combination with COCs [57]. Cosmetic measures including direct hair removal and electrolysis should be discussed with patients as other options for treatment of hirsutism.
Obese patients with PCOS are at higher risk for metabolic syndrome, a constellation of features including glucose intolerance, central obesity, hypertension, and dyslipidemia. Hyperandrogenemia and insulin resistance are linked with metabolic syndrome in PCOS. Reducing hyperandrogenemia and insulin resistance could reverse metabolic derangements and further reduce the risk of cardiovascular disease [58].
Worsening insulin resistance with COCs in PCOS has raised the concern of long-term metabolic derangements and cardiovascular adverse effects. COCs tend to increase total cholesterol, triglyceride, and high-sensitivity C-reactive protein levels [59]. However, the long-term implications of these findings are not well understood, attributable to the lack of longitudinal studies, especially in women with PCOS receiving COCs. Newer COCs containing less androgenic progestin may have less deleterious effect on insulin resistance and lipid profile. Due to insufficient use in adolescent patients, a definitive conclusion about their long-term safety cannot be drawn. Thus, there remains a theoretical risk of COCs exacerbating the underlying metabolic derangements in PCOS that can lead to subsequent adverse cardiovascular events.
Adolescent girls with PCOS are also at an increased risk for depression and anxiety disorders. The 2013 Endocrine Society clinical practice guideline suggests that adolescent girls with PCOS should be screened for depression and anxiety by history [51].If symptoms are present, patients should receive appropriate psychological referral and treatment.
Case Continued
As she had no contraindications to COCs, the patient was started on COC therapy to regulate her menstrual periods and alleviate the symptoms of hirsutism. Due to impaired glucose tolerance test results and increased risk for type 2 diabetes, treatment with metformin was also initiated. The patient met with a dietician, who offered recommendations for adopting a healthy lifestyle and introduced her to the “3,2,1,0, blast off” model: 3 consistent meals, 2 hours or less of screen time, 1 hour or more of physical activity, and 0 sweetened beverages a day. The patient was also advised to increase daily consumption of fruits and vegetables. Results of the 2-item Patient Health Questionnaire (PHQ-2) for depression were negative.
At a follow-up visit 6 months later, the patient reported that her menstrual periods were regular. There was some improvement in hirsutism, requiring less shaving, and there was no increase in weight. Repeat laboratory evaluations showed normal free testosterone level, decreased HbA1c (5.2%), and improved random blood glucose (130 mg/dL). The patient was seen regularly and treatment results monitored. No side effects were seen over a 4.5-year period. As PCOS is a lifelong condition, at the age of 21 the patient was referred to an adult endocrine clinic for further management.
Corresponding author: Alvina R. Kansra, MD, Medical College of Wisconsin, 8701 Watertown Plank Rd., Wauwatosa, WI 53226, [email protected].
Financial disclosures: None.
1. Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome inthe Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999v;84:4006–11.
2. Azziz R, Woods KS, Reyna R, et al. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 2004;89:2745–9.
3. Christensen SB, Black MH, Smith N, et al. Prevalence of polycystic ovary syndrome in adolescents. Fertil Steril 2013;100:470–7.
4. Rosenfield RL. Clinical review: Adolescent anovulation: maturational mechanisms and implications. J Clin Endocrinol Metab 2013;98:3572–83.
5. Venturoli S, Porcu E, Fabbri R, et al. Menstrual irregularities in adolescents: hormonal pattern and ovarian morphology. Horm Res 1986;24:269–79.
6. Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. Boston: Blackwell Scientific; 1992.
7. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19–25.
8. Azziz R, Carmina E, Dewailly D, et al; Androgen Excess Society. Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab 2006;91:4237–45.
9. Treloar A, Boynton R, Benn B, Brown B. Variation of human menstrual cycle through reproductive life. Int J Fertil 1967;12:77–126.
10. Vollman RF. The menstrual cycle. Major Probl Obstet Gynecol 1977;7:1–193.
11. Diaz A, Laufer MR, Breech LL; American Academy of Pediatrics Committee on Adolescence; American College of Obstetricians and Gynecologists Committee on Adolescent Health Care. Menstruation in girls and adolescents:using the menstrual cycle as a vital sign. Pediatrics 2006;118:2245–50.
12. Metcalf MG, Skidmore DS, Lowry GF, Mackenzie JA. Incidence of ovulation in the years after the menarche. J Endocrinol 1983;97:213–9.
13. Witchel SF, Oberfield S, Rosenfield RL, et al. The diagnosis of polycystic ovary syndrome during adolescence. Horm Res Paediatr 2015 Apr 1.
14. Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000;21:363–92.
15. Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
16. Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012;18:146–70.
17. Rosenfield RL. The polycystic ovary morphology-polycystic ovary syndrome spectrum. J Pediatr Adolesc Gynecol 2015;28:412–9.
18. Yildiz BO, Bolour S, Woods K, et al. Visually scoring hirsutism. Hum Reprod Update 2010;16:51–64.
19. Chen WC, Zouboulis CC. Hormones and the pilosebaceous unit. Dermatoendocrinol 2009;1:81–6.
20. Hawryluk EB, English JC 3rd. Female adolescent hair disorders. J Pediatr Adolesc Gynecol 2009;22:271–81.
21. Souter I, Sanchez A, Perez M, et al. The prevalence of androgen excess among patients with minimal unwanted hair growth. Am J Obstet Gynecol 2004;191:1914–20.
22. Di Fede G, Mansueto P, Pepe G, et al. High prevalence of polycystic ovary syndrome in women with mild hirsutism and no other significant clinical symptoms. Fertil Steril 2010; 94:194–7.
23. Chan CNJ, Haines CJ, Chow CCF, et al. Polycystic ovarian syndrome in Hong Kong Chinese women: patient characteristics and diagnostic criteria. Hong Kong Med J 2005;11:336–41.
24. Lucky AW, Biro FM, Simbartl LA, et al. Predictors of severity of acne vulgaris in young adolescent girls: results of a five-year longitudinal study. J Pediatr 1997;130:30–9.
25. Eichenfield LF, Krakowski AC, Piggott C, et al. Evidence-based recommendations for the diagnosis and treatment of pediatric acne. Pediatrics 2013;131:S163–S186.
26. Silfen ME, Denburg MR, Manibo AM, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab 2003;88:4682–8.
27. Carmina E, Oberfield SE, Lobo RA. The diagnosis of polycystic ovary syndrome in adolescents. Am J Obstet Gynecol 2010;203:201.e1–5.
28. Rosenfield RL. The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics 2015;136:1154–65.
29. Cho LW, Jayagopal V, Kilpatrick ES, Holding S. The LH/FSH ratio has little use in diagnosing polycystic ovarian syndrome. Ann Clin Biochem 2006;43(Pt 3):217–9.
30. Rosenfield RL, Cooke DW, Radovick S. Puberty and its disorders in the female. In: Sperling M, editor. Pediatric Endocrinology. 4th ed. Philadelphia: Elsevier; 2014:569–663.
31. Givens JR. Familial polycystic ovarian disease. Endocrinol Metab Clin North Am 1988;17:771–83.
32. Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A 1998;95:14956–60.
33. Amato P, Simpson JL. The genetics of polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol 2005;18:707–18.
34. Prapas N, Karkanaki A, Prapas I, et al. Genetics of polycystic ovary syndrome. Hippokratia 2009;13:216–23.
35. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab 1980;50:113–6.
36. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 2012;33:981–1030.
37. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 1997;18:774–800.
38. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab 1983;57:356–9.
39. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989;38:1165–74.
40. Gambineri A, Pelusi C, Vicennati V, et al. Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord 2002;26:883–96.
41. Lewy VD, Danadian K, Witchel SF, Arslanian S. Early metabolic abnormalities in adolescent girls with polycystic ovarian syndrome. J Pediatr 2001;138:38–44.
42. Nestler JE, Jakubowicz DJ, de Vargas AF, et al. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 1998;83:2001–5.
43. Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update 2006;12:351–61.
44. McCartney CR, Prendergast KA, Chhabra S, et al. The association of obesity and hyperandrogenemia during the pubertal transition in girls: obesity as a potential factor in the genesis of postpubertal hyperandrogenism. J Clin Endocrinol Metab 2006;91:1714–22.
45. McCartney CR, Blank SK, Prendergast KA, et al. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab 2007;92:430–6.
46. Knudsen KL, Blank SK, Burt Solorzano C, et al. Hyperandrogenemia in obese peripubertal girls: correlates and potential etiological determinants. Obesity (Silver Spring) 2010;18:2118–24.
47. Venturoli S, Porcu E, Fabbri R, et al. Longitudinal change of sonographic ovarian aspects and endocrine parameters in irregular cycles of adolescence. Pediatr Res 1995;38:974–80.
48. Mortensen M, Rosenfield RL, Littlejohn E. Functional significance of polycystic-size ovaries in healthy adolescents. J Clin Endocrinol Metab 2006;91:3786–90.
49. Fruzzetti F, Campagna AM, Perini D, Carmina E. Ovarian volume in normal and hyperandrogenic adolescent women. Fertil Steril 2015;104:196–9.
50. Fearnley EJ, Marquart L, Spurdle AB, et al, the Australian Ovarian Cancer Study Group and the Australian National Endometrial Study Group. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an Austrialian case-control study. Cancer Causes Control 2010;21:2303–8.
51. Legro RS, Arslanian SA, Ehrmann DA, et al; Endocrine Society. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2013;98:4565-92.
52. Domecq JP, Prutsky G, Mullan RJ, et al. Lifestyle modification programs in polycystic ovary syndrome: systematic review and meta-analysis. J Clin Endocrinol Metab 2013;98:4655.
53. Lass N, Kleber M, Winkel K, et al. Effect of lifestyle intervention on features of polycystic ovarian syndrome, metabolic syndrome, and intima-media thickness in obese adolescent girls. J Clin Endocrinol Metab 2011;96:3533.
54. Costello M, Eden J. A systematic review of the reproductive system effects of metformin in patients with polycystic ovary syndrome. Fertil Steril 2003;79:1–13.
55. Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 2003;327:951–3.
56. Auble B, Elder D, Gross A, Hillman JB. Differences in the management of adolescents with polycystic ovary syndrome across pediatric specialties. J Pediatr Adolesc Gynecol 2013;26:234–8.
57. Martin KA, Chang J, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clincal practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
58. Geller DH, Pacaud D, Gordon CM, Misra M, for the Drug and Therapeutics Committee of the Pediatric Endocrine Society. State of the art review: emerging therapies: the use of insulin sensitizers in the treatment of adolescents with polycystic ovary syndrome (PCOS). Int J Ped Endocrinol 2011;2011:9.
59. Tfayli H, Ulnach JW, Lee S, et al. Drospirenon/ethinyl estradiol versus rosiglitazone treatment in overweight adolescents with polycystic ovary syndrome: comparison of metabolic, hormonal and cardiovascular risk factors. J Clin Endocrinol Metab 2011;96:1311–9.
From the Department of Pediatrics, Section of Endocrinology & Diabetes, Medical College of Wisconsin, Milwaukee, WI.
Abstract
- Objective: To review the diagnosis and management of polycystic ovary syndrome (PCOS) in adolescent patients.
- Methods: Review of the literature.
- Results: PCOS is a complex, heterogeneous disorder that frequently manifests during puberty. The symptoms of PCOS (ie, menstrual irregularities, hirsutism, and acne) tend to overlap with normal pubertal changes. Diagnostic criteria for PCOS in the adolescent age-group is still lacking. Current practice is to utilize adult diagnostic criteria, which raises the concern for misdiagnosis. The underlying etiology for the disorder is still unclear, but insulin resistance is present in both obese and non-obese PCOS patients. Although recognizing adolescents with PCOS is challenging, evaluating and managing patients for hyperandrogenemia and metabolic syndrome is imperative to prevent long-term reproductive and metabolic complications.
- Conclusion: PCOS is increasingly encountered during adolescence. Recognizing adolescent girls with PCOS is a challenge but important for preventing long-term adverse health outcomes.
Polycystic ovary syndrome (PCOS) is a complex disorder most commonly characterized by chronic anovulation and clinical and biochemical features of hyperandrogenemia. It affects 4% to 12% of reproductive-aged women [1,2]. In adolescents, the exact prevalence is unknown, but in a recent study the prevalence of a confirmed diagnosis of PCOS in adolescents aged 15 to 19 years was 0.56%, which increased to 1.14% when undiagnosed cases with documented symptoms qualifying for PCOS according to NIH criteria were included [3]. The primary underlying defect in PCOS remains unknown, but key features include insulin resistance, impaired gonadotropin dynamics, and androgen excess.
Profound functional variations in the hypothalamic-pituitary-ovarian axis commonly seen during normal puberty may result in clinical and biochemical changes that mimic some of the features of PCOS. During the early stages of puberty, adolescent girls tend to have anovulatory menstrual cycles, higher androgen levels, and polycystic ovaries [4,5]. Thus, the clinical signs of hyperandrogenemia commonly seen in adults are less reliable in the adolescent age-group. Diagnostic criteria have been developed for adults and are based upon the various combinations of oligomenorrhea, unexplained hyperandrogenemia, and polycystic ovaries on imaging (Table 1) [6–8]. Applying these adult criteria in adolescent patients with suspected PCOS has always raised the concern of misdiagnosis as some of the changes seen in this age-group may likely be due to normal pubertal development. However, due to the paucity of data, the current practice is to utilize the adult diagnostic criteria. Because of the heterogeneous nature of the disorder, recognizing adolescents with PCOS may be challenging. However, early recognition and management is important to prevent some of the long-term reproductive and metabolic complications associated with this syndrome.
Case Study
Initial Presentation
A 16-year-old female patient presents to the PCOS clinic for evaluation of obesity and amenorrhea.
History
The patient, who is otherwise healthy, began gaining weight at age 7. During this period, her weight increased from the 15th to (currently) the 90th percentile; her height remained constant (75th percentile). Menarche was at 12 years of age. Menstrual periods have been irregular since the onset of menarche and she has had no periods for the past 5 months. She noticed excessive hair growth on her face, chin, and neck soon after the onset of menarche. She has been shaving her facial hair once every 2–3 days.
The patient’s detailed diet history included eating 3 meals daily and snacks in-between meals. The patient was consuming sweet beverages regularly. There was minimal intake fruits and vegetables. The portion sizes for each meal were large. The patient had minimal physical activity and screen time was more than 2 hours daily.
Family history is significant for obesity and type 2 diabetes in her mother and maternal grandmother and is negative for PCOS.
Physical Examination
Vital signs were within normal limits. She was 5 ft 6 in tall and weighed 242 lb, with a body mass index (BMI) of 40 (99th percentile; Z-score 2.41). Physical examination showed coarse hair extending from the sideburns to the chin as well as from pubis symphysis to navel with evidence of hair removal. She had acanthosis nigricans on her neck, mild acne, and evidence of central obesity with pink striae marks on the abdomen. She was Tanner stage 5 for breast and pubic hair and there was no evidence of virilization (clitoral hypertrophy, deepening of the voice, severe hirsutism, male pattern baldness, and masculine habitus). Other physical examination findings were within normal limits.
What physical findings in this patient are suggestive of clinical hyperandrogenemia?
Physiologic irregular menstruation is a well known phenomenon in adolescent girls and is generally due to anovulatory cycles [9–12]. Menstrual cycles shorter than 19 days or longer than 90 days at any stage after menarche are considered abnormal. The menstrual irregularity that is commonly seen within the first 2–3 years after the first menarche can last up to 5 years [5]. However, the majority of girls establish 20- to 45-day cycles within the first 2 years [13].
Androgen excess, defined by the presence of clinical and/or biochemical hyperandrogenemia, should be considered in any adolescent girl who is 2 to 3 years’ post-menarche and presenting with irregular menstrual periods, coarse terminal hair in a male distribution pattern (hirsutism), or moderate to severe inflammatory acne. Hirsutism is androgen dependent [14–16] and must be distinguished from hypertrichosis, which is generalized excessive vellus hair growth present all over the body. Clinical hyperandrogenemia, which includes hirsutism, acne vulgaris, as well as androgenetic alopecia, is well correlated with elevated androgen levels; however, the severity of hirsutism does not correlate well with circulating androgen levels [17,18]. Mild hirsutism is often not associated with hyperandrogenemia in otherwise asymptomatic individuals,but it may be a sign of hyperandrogenemia in adolescents when associated with other features of PCOS, ie, menstrual irregularity [14–16, 19–22]. Defining hirsutism in early adolescence may be difficult since the sexual hair may still be developing, and laboratory evaluation should be considered (see below), especially in an overweight/obese adolescent girl presenting with oligomenorrhea. Ethnic variation due to decreased skin sensitivity to androgens can result in minimal hirsutism despite elevated plasma androgen levels and must be considered among certain Asian women. Women with PCOS from China, Japan, Thailand, and East and Southeast Asian countries tend to have low scores on hirsutism rating scales even with elevated plasma androgens levels [16,23].
Although having acne during puberty is not considered as a marker for hyperandrogenemia, patients with moderate to severe inflammatory acne that is poorly responsive to topical treatment should be evaluated for underlying hyperandrogenemia [19,24,25].
What laboratory tests should be obtained to when there is clinical suspicion of hyperandrogenemia?
Case Continued
The patient underwent laboratory assessment that included total and free testosterone levels, lipid panel, thyroid studies, prolactin level, comprehensive metabolic panel (CMP) and hemoglobin A1c (HbA1c). Due to lack of virilization, she was not tested for PCOS-like syndromes. Her total and free testosterone were 90 ng/dL (normal, < 41) and 24.7 pg/mL (normal, 0.5–3.9) respectively. Thyroid-stimulating hormone and prolactin levels were normal. She had normal lipid levels and CMP but HbA1c was 5.9% (pre-diabetic range). The results of a 2-hour oral glucose tolerance test revealed a level of 160 mg/dL, indicative of impaired glucose tolerance.
What is the pathophysiology and diagnostic criteria for PCOS in adolescents?
PCOS has diverse etiology and has been linked to both genetic and environmental factors affecting ovarian steroidogenesis [13,30]. While the familial clustering strongly supports the role of genetic factors, variability in phenotypic features within the same or different families indicates the importance of environmental contribution [31–34].
The exact underlying mechanism leading to disruption of ovulation is still unclear; however, hyperinsulinemia augmenting ovarian androgen production has been well recognized [35–37]. Insulin resistance is a characteristic finding in PCOS and occurs both in obese and lean patients [38,39]. Obesity further exacerbates the insulin resistance state in PCOS patients. Therefore, obese patients with PCOS have more severe hyperandrogenemia and consequences from it (hirsutism, menstrual abnormalities, and metabolic derangements) than normal-weight PCOS patients [40,41]. Similar to LH, insulin can stimulate ovarian theca cells directly and cause increased production of androgens [42]. Elevated androgen levels cause the irregular menstrual periods as well as clinical signs of hyperandrogenemia, such as hirsutism and acne.
Altered gonadotropin dynamics is another possible etiological factor that is linked with PCOS. Hyperinsulinemia affects the regulation of gonadotropin-releasing hormone (GnRH) pulse generator, causing hypersecretion of LH [43]. Obese peripubertal girls have been identified having altered LH secretion [44,45]. This results in increased LH levels relative to FSH. Normal FSH is required to stimulate ovarian folliculognensis; insufficient FSH levels cause anovulation and menstrual irregularities. Abnormal LH secretion and fasting insulin levels have been identified the independent predictors for hyperandrogenemia in some peripubertal obese girls [46].
In 2010 Carmina et al published new criteria to diagnose PCOS in adolescents [27].They recommended that in diagnosing PCOS in adolescents, all 3 previously mentioned criteria should be present: hyperandrogenemia, chronic anovulation, and polycystic ovaries. With the exception of worsening hirsutism, the new recommendations greatly emphasized biochemical hyperandrogenemia (elevated free testosterone levels using sensitive assays). Chronic anovulation was defined as persistence of menstrual irregularities 2 years post-menarche and pelvic ultrasound (USG) showing increased ovarian size (> 10 cm3). Normal physiological variations unrelated to hyperandrogenemia are common in adolescent ovaries and limits the usefulness of pelvic USG as a diagnostic criterion for PCOS [13,47,48]. Also, the prevalence of increased ovarian size in hyperandrogenemic adolescent patients was reported to be low, and its utility as a criterion for diagnosis needs to be further explored [49]. In our current practice we do not rely on pelvic USG findings to make a PCOS diagnosis.
Due to longstanding controversies and lack of consensus surrounding the accurate diagnostic criteria, a recent guideline was developed by experts in pediatric endocrinology and adolescent medicine invited by the Pediatric Endocrine Society to address these issues [13].The guideline committee assessed the literature in order to define which criteria have sufficient evidence to be used for diagnosis of PCOS in adolescents. They recommend that PCOS should be considered in an adolescent girl presenting with unexplained menstrual irregularities, moderate to severe hirsutism or acne, and elevated levels of serum androgens (total and free testosterone) using reliable assay with well-defined ranges. Although intrinsic insulin resistance unique to PCOS is well known, none of the current guidelines either for adolescent and adult women include it as part of the diagnostic criteria. Since longitudinal studies focusing on the natural history of PCOS in this age-group are lacking, the current recommendations focus on timely screening and treatment in symptomatic adolescent girls suspected of having PCOS.
When there are PCOS features but menstrual irregularity has not been present for at least 2 years, one can defer the diagnostic label and instead use the term at-risk for PCOS. Such patients should have frequent longitudinal re-evaluations and should be offered treatment for their symptoms [13].
How should adolescents with PCOS be managed?
The treatment of PCOS is symptom-directed and should be tailored according to the complaints of the individual patient. However, it also must focus on the core dysfunctions: anovulation, hyperandrogenemia, obesity, and insulin resistance. It also requires bridging patient expectations of regulating menses, lessening the troublesome clinical signs of hyperandrogenemia (hirsutism, acne), and obesity management with the health care provider’s goals of preventing endometrial hyperplasia and cancer, diabetes mellitus, and cardiovascular disease.
Regulating menstruation and reducing cutaneous manifestations of hyperandrogenemia is the priority for any adolescent with PCOS. Combined oral contraceptive pills (COCs) are the first line of medical treatment for most adolescents. COCs restore endometrial cycling and suppress androgen levels, and are therefore optimal in treating abnormal uterine bleeding, protecting against endometrial carcinoma, and alleviating cutaneous manifestations of hyperandrogenemia (hirsutism and acne). Progestin monotherapy is considered an alternative therapy in individuals with contraindications to COCs (ie, thromboembolic risk). Although it is not effective in lowering androgen levels thus does not help reduce hair growth and acne, progestin monotherapy protects the endometrium and reduces the risk of endometrial cancer [50].
The majority of patients with PCOS are overweight or obese. Regardless of BMI, patients with PCOS have profound intrinsic insulin resistance that gets worse with overweight or obesity. Weight reduction by restricting caloric intake and increasing physical exercise is vital and has shown to be effective in regulating menstrual cycles, but is difficult to achieve [51–53]. Metformin can regulate menstrual cycles and decrease androgen levels by improving insulin sensitivity [54,55]. The use of metformin in PCOS patients is still controversial and abnormal glucose tolerance is the only approved indication [61]. However, combing metformin with COCs and lifestyle modification in obese PCOS patients has been shown to be used more frequently in pediatric endocrine clinics [56]. COCs are the only agents that can lower testosterone levels and improve ovulation and hirsutism; these effects are seen less frequently with lifestyle modification or metformin, either used alone or in combination.
COC monotherapy is first-line therapy to treat hirsutism. Consider anti-androgen treatment for hirsutism if there is no improvement after 6–9 months of hormonal treatment [57]. Antiandrogens reduce hirsutism by decreasing androgen production and binding the androgen receptors in target tissue. Spironolactone is the most commonly used antiandrogen therapy in adolescent girls with PCOS. Given the risk of teratogenicity with antiandrogens if pregnancy occurs, it is recommended to use it in combination with COCs [57]. Cosmetic measures including direct hair removal and electrolysis should be discussed with patients as other options for treatment of hirsutism.
Obese patients with PCOS are at higher risk for metabolic syndrome, a constellation of features including glucose intolerance, central obesity, hypertension, and dyslipidemia. Hyperandrogenemia and insulin resistance are linked with metabolic syndrome in PCOS. Reducing hyperandrogenemia and insulin resistance could reverse metabolic derangements and further reduce the risk of cardiovascular disease [58].
Worsening insulin resistance with COCs in PCOS has raised the concern of long-term metabolic derangements and cardiovascular adverse effects. COCs tend to increase total cholesterol, triglyceride, and high-sensitivity C-reactive protein levels [59]. However, the long-term implications of these findings are not well understood, attributable to the lack of longitudinal studies, especially in women with PCOS receiving COCs. Newer COCs containing less androgenic progestin may have less deleterious effect on insulin resistance and lipid profile. Due to insufficient use in adolescent patients, a definitive conclusion about their long-term safety cannot be drawn. Thus, there remains a theoretical risk of COCs exacerbating the underlying metabolic derangements in PCOS that can lead to subsequent adverse cardiovascular events.
Adolescent girls with PCOS are also at an increased risk for depression and anxiety disorders. The 2013 Endocrine Society clinical practice guideline suggests that adolescent girls with PCOS should be screened for depression and anxiety by history [51].If symptoms are present, patients should receive appropriate psychological referral and treatment.
Case Continued
As she had no contraindications to COCs, the patient was started on COC therapy to regulate her menstrual periods and alleviate the symptoms of hirsutism. Due to impaired glucose tolerance test results and increased risk for type 2 diabetes, treatment with metformin was also initiated. The patient met with a dietician, who offered recommendations for adopting a healthy lifestyle and introduced her to the “3,2,1,0, blast off” model: 3 consistent meals, 2 hours or less of screen time, 1 hour or more of physical activity, and 0 sweetened beverages a day. The patient was also advised to increase daily consumption of fruits and vegetables. Results of the 2-item Patient Health Questionnaire (PHQ-2) for depression were negative.
At a follow-up visit 6 months later, the patient reported that her menstrual periods were regular. There was some improvement in hirsutism, requiring less shaving, and there was no increase in weight. Repeat laboratory evaluations showed normal free testosterone level, decreased HbA1c (5.2%), and improved random blood glucose (130 mg/dL). The patient was seen regularly and treatment results monitored. No side effects were seen over a 4.5-year period. As PCOS is a lifelong condition, at the age of 21 the patient was referred to an adult endocrine clinic for further management.
Corresponding author: Alvina R. Kansra, MD, Medical College of Wisconsin, 8701 Watertown Plank Rd., Wauwatosa, WI 53226, [email protected].
Financial disclosures: None.
From the Department of Pediatrics, Section of Endocrinology & Diabetes, Medical College of Wisconsin, Milwaukee, WI.
Abstract
- Objective: To review the diagnosis and management of polycystic ovary syndrome (PCOS) in adolescent patients.
- Methods: Review of the literature.
- Results: PCOS is a complex, heterogeneous disorder that frequently manifests during puberty. The symptoms of PCOS (ie, menstrual irregularities, hirsutism, and acne) tend to overlap with normal pubertal changes. Diagnostic criteria for PCOS in the adolescent age-group is still lacking. Current practice is to utilize adult diagnostic criteria, which raises the concern for misdiagnosis. The underlying etiology for the disorder is still unclear, but insulin resistance is present in both obese and non-obese PCOS patients. Although recognizing adolescents with PCOS is challenging, evaluating and managing patients for hyperandrogenemia and metabolic syndrome is imperative to prevent long-term reproductive and metabolic complications.
- Conclusion: PCOS is increasingly encountered during adolescence. Recognizing adolescent girls with PCOS is a challenge but important for preventing long-term adverse health outcomes.
Polycystic ovary syndrome (PCOS) is a complex disorder most commonly characterized by chronic anovulation and clinical and biochemical features of hyperandrogenemia. It affects 4% to 12% of reproductive-aged women [1,2]. In adolescents, the exact prevalence is unknown, but in a recent study the prevalence of a confirmed diagnosis of PCOS in adolescents aged 15 to 19 years was 0.56%, which increased to 1.14% when undiagnosed cases with documented symptoms qualifying for PCOS according to NIH criteria were included [3]. The primary underlying defect in PCOS remains unknown, but key features include insulin resistance, impaired gonadotropin dynamics, and androgen excess.
Profound functional variations in the hypothalamic-pituitary-ovarian axis commonly seen during normal puberty may result in clinical and biochemical changes that mimic some of the features of PCOS. During the early stages of puberty, adolescent girls tend to have anovulatory menstrual cycles, higher androgen levels, and polycystic ovaries [4,5]. Thus, the clinical signs of hyperandrogenemia commonly seen in adults are less reliable in the adolescent age-group. Diagnostic criteria have been developed for adults and are based upon the various combinations of oligomenorrhea, unexplained hyperandrogenemia, and polycystic ovaries on imaging (Table 1) [6–8]. Applying these adult criteria in adolescent patients with suspected PCOS has always raised the concern of misdiagnosis as some of the changes seen in this age-group may likely be due to normal pubertal development. However, due to the paucity of data, the current practice is to utilize the adult diagnostic criteria. Because of the heterogeneous nature of the disorder, recognizing adolescents with PCOS may be challenging. However, early recognition and management is important to prevent some of the long-term reproductive and metabolic complications associated with this syndrome.
Case Study
Initial Presentation
A 16-year-old female patient presents to the PCOS clinic for evaluation of obesity and amenorrhea.
History
The patient, who is otherwise healthy, began gaining weight at age 7. During this period, her weight increased from the 15th to (currently) the 90th percentile; her height remained constant (75th percentile). Menarche was at 12 years of age. Menstrual periods have been irregular since the onset of menarche and she has had no periods for the past 5 months. She noticed excessive hair growth on her face, chin, and neck soon after the onset of menarche. She has been shaving her facial hair once every 2–3 days.
The patient’s detailed diet history included eating 3 meals daily and snacks in-between meals. The patient was consuming sweet beverages regularly. There was minimal intake fruits and vegetables. The portion sizes for each meal were large. The patient had minimal physical activity and screen time was more than 2 hours daily.
Family history is significant for obesity and type 2 diabetes in her mother and maternal grandmother and is negative for PCOS.
Physical Examination
Vital signs were within normal limits. She was 5 ft 6 in tall and weighed 242 lb, with a body mass index (BMI) of 40 (99th percentile; Z-score 2.41). Physical examination showed coarse hair extending from the sideburns to the chin as well as from pubis symphysis to navel with evidence of hair removal. She had acanthosis nigricans on her neck, mild acne, and evidence of central obesity with pink striae marks on the abdomen. She was Tanner stage 5 for breast and pubic hair and there was no evidence of virilization (clitoral hypertrophy, deepening of the voice, severe hirsutism, male pattern baldness, and masculine habitus). Other physical examination findings were within normal limits.
What physical findings in this patient are suggestive of clinical hyperandrogenemia?
Physiologic irregular menstruation is a well known phenomenon in adolescent girls and is generally due to anovulatory cycles [9–12]. Menstrual cycles shorter than 19 days or longer than 90 days at any stage after menarche are considered abnormal. The menstrual irregularity that is commonly seen within the first 2–3 years after the first menarche can last up to 5 years [5]. However, the majority of girls establish 20- to 45-day cycles within the first 2 years [13].
Androgen excess, defined by the presence of clinical and/or biochemical hyperandrogenemia, should be considered in any adolescent girl who is 2 to 3 years’ post-menarche and presenting with irregular menstrual periods, coarse terminal hair in a male distribution pattern (hirsutism), or moderate to severe inflammatory acne. Hirsutism is androgen dependent [14–16] and must be distinguished from hypertrichosis, which is generalized excessive vellus hair growth present all over the body. Clinical hyperandrogenemia, which includes hirsutism, acne vulgaris, as well as androgenetic alopecia, is well correlated with elevated androgen levels; however, the severity of hirsutism does not correlate well with circulating androgen levels [17,18]. Mild hirsutism is often not associated with hyperandrogenemia in otherwise asymptomatic individuals,but it may be a sign of hyperandrogenemia in adolescents when associated with other features of PCOS, ie, menstrual irregularity [14–16, 19–22]. Defining hirsutism in early adolescence may be difficult since the sexual hair may still be developing, and laboratory evaluation should be considered (see below), especially in an overweight/obese adolescent girl presenting with oligomenorrhea. Ethnic variation due to decreased skin sensitivity to androgens can result in minimal hirsutism despite elevated plasma androgen levels and must be considered among certain Asian women. Women with PCOS from China, Japan, Thailand, and East and Southeast Asian countries tend to have low scores on hirsutism rating scales even with elevated plasma androgens levels [16,23].
Although having acne during puberty is not considered as a marker for hyperandrogenemia, patients with moderate to severe inflammatory acne that is poorly responsive to topical treatment should be evaluated for underlying hyperandrogenemia [19,24,25].
What laboratory tests should be obtained to when there is clinical suspicion of hyperandrogenemia?
Case Continued
The patient underwent laboratory assessment that included total and free testosterone levels, lipid panel, thyroid studies, prolactin level, comprehensive metabolic panel (CMP) and hemoglobin A1c (HbA1c). Due to lack of virilization, she was not tested for PCOS-like syndromes. Her total and free testosterone were 90 ng/dL (normal, < 41) and 24.7 pg/mL (normal, 0.5–3.9) respectively. Thyroid-stimulating hormone and prolactin levels were normal. She had normal lipid levels and CMP but HbA1c was 5.9% (pre-diabetic range). The results of a 2-hour oral glucose tolerance test revealed a level of 160 mg/dL, indicative of impaired glucose tolerance.
What is the pathophysiology and diagnostic criteria for PCOS in adolescents?
PCOS has diverse etiology and has been linked to both genetic and environmental factors affecting ovarian steroidogenesis [13,30]. While the familial clustering strongly supports the role of genetic factors, variability in phenotypic features within the same or different families indicates the importance of environmental contribution [31–34].
The exact underlying mechanism leading to disruption of ovulation is still unclear; however, hyperinsulinemia augmenting ovarian androgen production has been well recognized [35–37]. Insulin resistance is a characteristic finding in PCOS and occurs both in obese and lean patients [38,39]. Obesity further exacerbates the insulin resistance state in PCOS patients. Therefore, obese patients with PCOS have more severe hyperandrogenemia and consequences from it (hirsutism, menstrual abnormalities, and metabolic derangements) than normal-weight PCOS patients [40,41]. Similar to LH, insulin can stimulate ovarian theca cells directly and cause increased production of androgens [42]. Elevated androgen levels cause the irregular menstrual periods as well as clinical signs of hyperandrogenemia, such as hirsutism and acne.
Altered gonadotropin dynamics is another possible etiological factor that is linked with PCOS. Hyperinsulinemia affects the regulation of gonadotropin-releasing hormone (GnRH) pulse generator, causing hypersecretion of LH [43]. Obese peripubertal girls have been identified having altered LH secretion [44,45]. This results in increased LH levels relative to FSH. Normal FSH is required to stimulate ovarian folliculognensis; insufficient FSH levels cause anovulation and menstrual irregularities. Abnormal LH secretion and fasting insulin levels have been identified the independent predictors for hyperandrogenemia in some peripubertal obese girls [46].
In 2010 Carmina et al published new criteria to diagnose PCOS in adolescents [27].They recommended that in diagnosing PCOS in adolescents, all 3 previously mentioned criteria should be present: hyperandrogenemia, chronic anovulation, and polycystic ovaries. With the exception of worsening hirsutism, the new recommendations greatly emphasized biochemical hyperandrogenemia (elevated free testosterone levels using sensitive assays). Chronic anovulation was defined as persistence of menstrual irregularities 2 years post-menarche and pelvic ultrasound (USG) showing increased ovarian size (> 10 cm3). Normal physiological variations unrelated to hyperandrogenemia are common in adolescent ovaries and limits the usefulness of pelvic USG as a diagnostic criterion for PCOS [13,47,48]. Also, the prevalence of increased ovarian size in hyperandrogenemic adolescent patients was reported to be low, and its utility as a criterion for diagnosis needs to be further explored [49]. In our current practice we do not rely on pelvic USG findings to make a PCOS diagnosis.
Due to longstanding controversies and lack of consensus surrounding the accurate diagnostic criteria, a recent guideline was developed by experts in pediatric endocrinology and adolescent medicine invited by the Pediatric Endocrine Society to address these issues [13].The guideline committee assessed the literature in order to define which criteria have sufficient evidence to be used for diagnosis of PCOS in adolescents. They recommend that PCOS should be considered in an adolescent girl presenting with unexplained menstrual irregularities, moderate to severe hirsutism or acne, and elevated levels of serum androgens (total and free testosterone) using reliable assay with well-defined ranges. Although intrinsic insulin resistance unique to PCOS is well known, none of the current guidelines either for adolescent and adult women include it as part of the diagnostic criteria. Since longitudinal studies focusing on the natural history of PCOS in this age-group are lacking, the current recommendations focus on timely screening and treatment in symptomatic adolescent girls suspected of having PCOS.
When there are PCOS features but menstrual irregularity has not been present for at least 2 years, one can defer the diagnostic label and instead use the term at-risk for PCOS. Such patients should have frequent longitudinal re-evaluations and should be offered treatment for their symptoms [13].
How should adolescents with PCOS be managed?
The treatment of PCOS is symptom-directed and should be tailored according to the complaints of the individual patient. However, it also must focus on the core dysfunctions: anovulation, hyperandrogenemia, obesity, and insulin resistance. It also requires bridging patient expectations of regulating menses, lessening the troublesome clinical signs of hyperandrogenemia (hirsutism, acne), and obesity management with the health care provider’s goals of preventing endometrial hyperplasia and cancer, diabetes mellitus, and cardiovascular disease.
Regulating menstruation and reducing cutaneous manifestations of hyperandrogenemia is the priority for any adolescent with PCOS. Combined oral contraceptive pills (COCs) are the first line of medical treatment for most adolescents. COCs restore endometrial cycling and suppress androgen levels, and are therefore optimal in treating abnormal uterine bleeding, protecting against endometrial carcinoma, and alleviating cutaneous manifestations of hyperandrogenemia (hirsutism and acne). Progestin monotherapy is considered an alternative therapy in individuals with contraindications to COCs (ie, thromboembolic risk). Although it is not effective in lowering androgen levels thus does not help reduce hair growth and acne, progestin monotherapy protects the endometrium and reduces the risk of endometrial cancer [50].
The majority of patients with PCOS are overweight or obese. Regardless of BMI, patients with PCOS have profound intrinsic insulin resistance that gets worse with overweight or obesity. Weight reduction by restricting caloric intake and increasing physical exercise is vital and has shown to be effective in regulating menstrual cycles, but is difficult to achieve [51–53]. Metformin can regulate menstrual cycles and decrease androgen levels by improving insulin sensitivity [54,55]. The use of metformin in PCOS patients is still controversial and abnormal glucose tolerance is the only approved indication [61]. However, combing metformin with COCs and lifestyle modification in obese PCOS patients has been shown to be used more frequently in pediatric endocrine clinics [56]. COCs are the only agents that can lower testosterone levels and improve ovulation and hirsutism; these effects are seen less frequently with lifestyle modification or metformin, either used alone or in combination.
COC monotherapy is first-line therapy to treat hirsutism. Consider anti-androgen treatment for hirsutism if there is no improvement after 6–9 months of hormonal treatment [57]. Antiandrogens reduce hirsutism by decreasing androgen production and binding the androgen receptors in target tissue. Spironolactone is the most commonly used antiandrogen therapy in adolescent girls with PCOS. Given the risk of teratogenicity with antiandrogens if pregnancy occurs, it is recommended to use it in combination with COCs [57]. Cosmetic measures including direct hair removal and electrolysis should be discussed with patients as other options for treatment of hirsutism.
Obese patients with PCOS are at higher risk for metabolic syndrome, a constellation of features including glucose intolerance, central obesity, hypertension, and dyslipidemia. Hyperandrogenemia and insulin resistance are linked with metabolic syndrome in PCOS. Reducing hyperandrogenemia and insulin resistance could reverse metabolic derangements and further reduce the risk of cardiovascular disease [58].
Worsening insulin resistance with COCs in PCOS has raised the concern of long-term metabolic derangements and cardiovascular adverse effects. COCs tend to increase total cholesterol, triglyceride, and high-sensitivity C-reactive protein levels [59]. However, the long-term implications of these findings are not well understood, attributable to the lack of longitudinal studies, especially in women with PCOS receiving COCs. Newer COCs containing less androgenic progestin may have less deleterious effect on insulin resistance and lipid profile. Due to insufficient use in adolescent patients, a definitive conclusion about their long-term safety cannot be drawn. Thus, there remains a theoretical risk of COCs exacerbating the underlying metabolic derangements in PCOS that can lead to subsequent adverse cardiovascular events.
Adolescent girls with PCOS are also at an increased risk for depression and anxiety disorders. The 2013 Endocrine Society clinical practice guideline suggests that adolescent girls with PCOS should be screened for depression and anxiety by history [51].If symptoms are present, patients should receive appropriate psychological referral and treatment.
Case Continued
As she had no contraindications to COCs, the patient was started on COC therapy to regulate her menstrual periods and alleviate the symptoms of hirsutism. Due to impaired glucose tolerance test results and increased risk for type 2 diabetes, treatment with metformin was also initiated. The patient met with a dietician, who offered recommendations for adopting a healthy lifestyle and introduced her to the “3,2,1,0, blast off” model: 3 consistent meals, 2 hours or less of screen time, 1 hour or more of physical activity, and 0 sweetened beverages a day. The patient was also advised to increase daily consumption of fruits and vegetables. Results of the 2-item Patient Health Questionnaire (PHQ-2) for depression were negative.
At a follow-up visit 6 months later, the patient reported that her menstrual periods were regular. There was some improvement in hirsutism, requiring less shaving, and there was no increase in weight. Repeat laboratory evaluations showed normal free testosterone level, decreased HbA1c (5.2%), and improved random blood glucose (130 mg/dL). The patient was seen regularly and treatment results monitored. No side effects were seen over a 4.5-year period. As PCOS is a lifelong condition, at the age of 21 the patient was referred to an adult endocrine clinic for further management.
Corresponding author: Alvina R. Kansra, MD, Medical College of Wisconsin, 8701 Watertown Plank Rd., Wauwatosa, WI 53226, [email protected].
Financial disclosures: None.
1. Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome inthe Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999v;84:4006–11.
2. Azziz R, Woods KS, Reyna R, et al. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 2004;89:2745–9.
3. Christensen SB, Black MH, Smith N, et al. Prevalence of polycystic ovary syndrome in adolescents. Fertil Steril 2013;100:470–7.
4. Rosenfield RL. Clinical review: Adolescent anovulation: maturational mechanisms and implications. J Clin Endocrinol Metab 2013;98:3572–83.
5. Venturoli S, Porcu E, Fabbri R, et al. Menstrual irregularities in adolescents: hormonal pattern and ovarian morphology. Horm Res 1986;24:269–79.
6. Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. Boston: Blackwell Scientific; 1992.
7. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19–25.
8. Azziz R, Carmina E, Dewailly D, et al; Androgen Excess Society. Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab 2006;91:4237–45.
9. Treloar A, Boynton R, Benn B, Brown B. Variation of human menstrual cycle through reproductive life. Int J Fertil 1967;12:77–126.
10. Vollman RF. The menstrual cycle. Major Probl Obstet Gynecol 1977;7:1–193.
11. Diaz A, Laufer MR, Breech LL; American Academy of Pediatrics Committee on Adolescence; American College of Obstetricians and Gynecologists Committee on Adolescent Health Care. Menstruation in girls and adolescents:using the menstrual cycle as a vital sign. Pediatrics 2006;118:2245–50.
12. Metcalf MG, Skidmore DS, Lowry GF, Mackenzie JA. Incidence of ovulation in the years after the menarche. J Endocrinol 1983;97:213–9.
13. Witchel SF, Oberfield S, Rosenfield RL, et al. The diagnosis of polycystic ovary syndrome during adolescence. Horm Res Paediatr 2015 Apr 1.
14. Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000;21:363–92.
15. Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
16. Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012;18:146–70.
17. Rosenfield RL. The polycystic ovary morphology-polycystic ovary syndrome spectrum. J Pediatr Adolesc Gynecol 2015;28:412–9.
18. Yildiz BO, Bolour S, Woods K, et al. Visually scoring hirsutism. Hum Reprod Update 2010;16:51–64.
19. Chen WC, Zouboulis CC. Hormones and the pilosebaceous unit. Dermatoendocrinol 2009;1:81–6.
20. Hawryluk EB, English JC 3rd. Female adolescent hair disorders. J Pediatr Adolesc Gynecol 2009;22:271–81.
21. Souter I, Sanchez A, Perez M, et al. The prevalence of androgen excess among patients with minimal unwanted hair growth. Am J Obstet Gynecol 2004;191:1914–20.
22. Di Fede G, Mansueto P, Pepe G, et al. High prevalence of polycystic ovary syndrome in women with mild hirsutism and no other significant clinical symptoms. Fertil Steril 2010; 94:194–7.
23. Chan CNJ, Haines CJ, Chow CCF, et al. Polycystic ovarian syndrome in Hong Kong Chinese women: patient characteristics and diagnostic criteria. Hong Kong Med J 2005;11:336–41.
24. Lucky AW, Biro FM, Simbartl LA, et al. Predictors of severity of acne vulgaris in young adolescent girls: results of a five-year longitudinal study. J Pediatr 1997;130:30–9.
25. Eichenfield LF, Krakowski AC, Piggott C, et al. Evidence-based recommendations for the diagnosis and treatment of pediatric acne. Pediatrics 2013;131:S163–S186.
26. Silfen ME, Denburg MR, Manibo AM, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab 2003;88:4682–8.
27. Carmina E, Oberfield SE, Lobo RA. The diagnosis of polycystic ovary syndrome in adolescents. Am J Obstet Gynecol 2010;203:201.e1–5.
28. Rosenfield RL. The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics 2015;136:1154–65.
29. Cho LW, Jayagopal V, Kilpatrick ES, Holding S. The LH/FSH ratio has little use in diagnosing polycystic ovarian syndrome. Ann Clin Biochem 2006;43(Pt 3):217–9.
30. Rosenfield RL, Cooke DW, Radovick S. Puberty and its disorders in the female. In: Sperling M, editor. Pediatric Endocrinology. 4th ed. Philadelphia: Elsevier; 2014:569–663.
31. Givens JR. Familial polycystic ovarian disease. Endocrinol Metab Clin North Am 1988;17:771–83.
32. Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A 1998;95:14956–60.
33. Amato P, Simpson JL. The genetics of polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol 2005;18:707–18.
34. Prapas N, Karkanaki A, Prapas I, et al. Genetics of polycystic ovary syndrome. Hippokratia 2009;13:216–23.
35. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab 1980;50:113–6.
36. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 2012;33:981–1030.
37. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 1997;18:774–800.
38. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab 1983;57:356–9.
39. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989;38:1165–74.
40. Gambineri A, Pelusi C, Vicennati V, et al. Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord 2002;26:883–96.
41. Lewy VD, Danadian K, Witchel SF, Arslanian S. Early metabolic abnormalities in adolescent girls with polycystic ovarian syndrome. J Pediatr 2001;138:38–44.
42. Nestler JE, Jakubowicz DJ, de Vargas AF, et al. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 1998;83:2001–5.
43. Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update 2006;12:351–61.
44. McCartney CR, Prendergast KA, Chhabra S, et al. The association of obesity and hyperandrogenemia during the pubertal transition in girls: obesity as a potential factor in the genesis of postpubertal hyperandrogenism. J Clin Endocrinol Metab 2006;91:1714–22.
45. McCartney CR, Blank SK, Prendergast KA, et al. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab 2007;92:430–6.
46. Knudsen KL, Blank SK, Burt Solorzano C, et al. Hyperandrogenemia in obese peripubertal girls: correlates and potential etiological determinants. Obesity (Silver Spring) 2010;18:2118–24.
47. Venturoli S, Porcu E, Fabbri R, et al. Longitudinal change of sonographic ovarian aspects and endocrine parameters in irregular cycles of adolescence. Pediatr Res 1995;38:974–80.
48. Mortensen M, Rosenfield RL, Littlejohn E. Functional significance of polycystic-size ovaries in healthy adolescents. J Clin Endocrinol Metab 2006;91:3786–90.
49. Fruzzetti F, Campagna AM, Perini D, Carmina E. Ovarian volume in normal and hyperandrogenic adolescent women. Fertil Steril 2015;104:196–9.
50. Fearnley EJ, Marquart L, Spurdle AB, et al, the Australian Ovarian Cancer Study Group and the Australian National Endometrial Study Group. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an Austrialian case-control study. Cancer Causes Control 2010;21:2303–8.
51. Legro RS, Arslanian SA, Ehrmann DA, et al; Endocrine Society. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2013;98:4565-92.
52. Domecq JP, Prutsky G, Mullan RJ, et al. Lifestyle modification programs in polycystic ovary syndrome: systematic review and meta-analysis. J Clin Endocrinol Metab 2013;98:4655.
53. Lass N, Kleber M, Winkel K, et al. Effect of lifestyle intervention on features of polycystic ovarian syndrome, metabolic syndrome, and intima-media thickness in obese adolescent girls. J Clin Endocrinol Metab 2011;96:3533.
54. Costello M, Eden J. A systematic review of the reproductive system effects of metformin in patients with polycystic ovary syndrome. Fertil Steril 2003;79:1–13.
55. Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 2003;327:951–3.
56. Auble B, Elder D, Gross A, Hillman JB. Differences in the management of adolescents with polycystic ovary syndrome across pediatric specialties. J Pediatr Adolesc Gynecol 2013;26:234–8.
57. Martin KA, Chang J, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clincal practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
58. Geller DH, Pacaud D, Gordon CM, Misra M, for the Drug and Therapeutics Committee of the Pediatric Endocrine Society. State of the art review: emerging therapies: the use of insulin sensitizers in the treatment of adolescents with polycystic ovary syndrome (PCOS). Int J Ped Endocrinol 2011;2011:9.
59. Tfayli H, Ulnach JW, Lee S, et al. Drospirenon/ethinyl estradiol versus rosiglitazone treatment in overweight adolescents with polycystic ovary syndrome: comparison of metabolic, hormonal and cardiovascular risk factors. J Clin Endocrinol Metab 2011;96:1311–9.
1. Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome inthe Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999v;84:4006–11.
2. Azziz R, Woods KS, Reyna R, et al. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 2004;89:2745–9.
3. Christensen SB, Black MH, Smith N, et al. Prevalence of polycystic ovary syndrome in adolescents. Fertil Steril 2013;100:470–7.
4. Rosenfield RL. Clinical review: Adolescent anovulation: maturational mechanisms and implications. J Clin Endocrinol Metab 2013;98:3572–83.
5. Venturoli S, Porcu E, Fabbri R, et al. Menstrual irregularities in adolescents: hormonal pattern and ovarian morphology. Horm Res 1986;24:269–79.
6. Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. Boston: Blackwell Scientific; 1992.
7. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19–25.
8. Azziz R, Carmina E, Dewailly D, et al; Androgen Excess Society. Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab 2006;91:4237–45.
9. Treloar A, Boynton R, Benn B, Brown B. Variation of human menstrual cycle through reproductive life. Int J Fertil 1967;12:77–126.
10. Vollman RF. The menstrual cycle. Major Probl Obstet Gynecol 1977;7:1–193.
11. Diaz A, Laufer MR, Breech LL; American Academy of Pediatrics Committee on Adolescence; American College of Obstetricians and Gynecologists Committee on Adolescent Health Care. Menstruation in girls and adolescents:using the menstrual cycle as a vital sign. Pediatrics 2006;118:2245–50.
12. Metcalf MG, Skidmore DS, Lowry GF, Mackenzie JA. Incidence of ovulation in the years after the menarche. J Endocrinol 1983;97:213–9.
13. Witchel SF, Oberfield S, Rosenfield RL, et al. The diagnosis of polycystic ovary syndrome during adolescence. Horm Res Paediatr 2015 Apr 1.
14. Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000;21:363–92.
15. Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
16. Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012;18:146–70.
17. Rosenfield RL. The polycystic ovary morphology-polycystic ovary syndrome spectrum. J Pediatr Adolesc Gynecol 2015;28:412–9.
18. Yildiz BO, Bolour S, Woods K, et al. Visually scoring hirsutism. Hum Reprod Update 2010;16:51–64.
19. Chen WC, Zouboulis CC. Hormones and the pilosebaceous unit. Dermatoendocrinol 2009;1:81–6.
20. Hawryluk EB, English JC 3rd. Female adolescent hair disorders. J Pediatr Adolesc Gynecol 2009;22:271–81.
21. Souter I, Sanchez A, Perez M, et al. The prevalence of androgen excess among patients with minimal unwanted hair growth. Am J Obstet Gynecol 2004;191:1914–20.
22. Di Fede G, Mansueto P, Pepe G, et al. High prevalence of polycystic ovary syndrome in women with mild hirsutism and no other significant clinical symptoms. Fertil Steril 2010; 94:194–7.
23. Chan CNJ, Haines CJ, Chow CCF, et al. Polycystic ovarian syndrome in Hong Kong Chinese women: patient characteristics and diagnostic criteria. Hong Kong Med J 2005;11:336–41.
24. Lucky AW, Biro FM, Simbartl LA, et al. Predictors of severity of acne vulgaris in young adolescent girls: results of a five-year longitudinal study. J Pediatr 1997;130:30–9.
25. Eichenfield LF, Krakowski AC, Piggott C, et al. Evidence-based recommendations for the diagnosis and treatment of pediatric acne. Pediatrics 2013;131:S163–S186.
26. Silfen ME, Denburg MR, Manibo AM, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab 2003;88:4682–8.
27. Carmina E, Oberfield SE, Lobo RA. The diagnosis of polycystic ovary syndrome in adolescents. Am J Obstet Gynecol 2010;203:201.e1–5.
28. Rosenfield RL. The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics 2015;136:1154–65.
29. Cho LW, Jayagopal V, Kilpatrick ES, Holding S. The LH/FSH ratio has little use in diagnosing polycystic ovarian syndrome. Ann Clin Biochem 2006;43(Pt 3):217–9.
30. Rosenfield RL, Cooke DW, Radovick S. Puberty and its disorders in the female. In: Sperling M, editor. Pediatric Endocrinology. 4th ed. Philadelphia: Elsevier; 2014:569–663.
31. Givens JR. Familial polycystic ovarian disease. Endocrinol Metab Clin North Am 1988;17:771–83.
32. Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A 1998;95:14956–60.
33. Amato P, Simpson JL. The genetics of polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol 2005;18:707–18.
34. Prapas N, Karkanaki A, Prapas I, et al. Genetics of polycystic ovary syndrome. Hippokratia 2009;13:216–23.
35. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab 1980;50:113–6.
36. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 2012;33:981–1030.
37. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 1997;18:774–800.
38. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab 1983;57:356–9.
39. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989;38:1165–74.
40. Gambineri A, Pelusi C, Vicennati V, et al. Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord 2002;26:883–96.
41. Lewy VD, Danadian K, Witchel SF, Arslanian S. Early metabolic abnormalities in adolescent girls with polycystic ovarian syndrome. J Pediatr 2001;138:38–44.
42. Nestler JE, Jakubowicz DJ, de Vargas AF, et al. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 1998;83:2001–5.
43. Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update 2006;12:351–61.
44. McCartney CR, Prendergast KA, Chhabra S, et al. The association of obesity and hyperandrogenemia during the pubertal transition in girls: obesity as a potential factor in the genesis of postpubertal hyperandrogenism. J Clin Endocrinol Metab 2006;91:1714–22.
45. McCartney CR, Blank SK, Prendergast KA, et al. Obesity and sex steroid changes across puberty: evidence for marked hyperandrogenemia in pre- and early pubertal obese girls. J Clin Endocrinol Metab 2007;92:430–6.
46. Knudsen KL, Blank SK, Burt Solorzano C, et al. Hyperandrogenemia in obese peripubertal girls: correlates and potential etiological determinants. Obesity (Silver Spring) 2010;18:2118–24.
47. Venturoli S, Porcu E, Fabbri R, et al. Longitudinal change of sonographic ovarian aspects and endocrine parameters in irregular cycles of adolescence. Pediatr Res 1995;38:974–80.
48. Mortensen M, Rosenfield RL, Littlejohn E. Functional significance of polycystic-size ovaries in healthy adolescents. J Clin Endocrinol Metab 2006;91:3786–90.
49. Fruzzetti F, Campagna AM, Perini D, Carmina E. Ovarian volume in normal and hyperandrogenic adolescent women. Fertil Steril 2015;104:196–9.
50. Fearnley EJ, Marquart L, Spurdle AB, et al, the Australian Ovarian Cancer Study Group and the Australian National Endometrial Study Group. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an Austrialian case-control study. Cancer Causes Control 2010;21:2303–8.
51. Legro RS, Arslanian SA, Ehrmann DA, et al; Endocrine Society. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2013;98:4565-92.
52. Domecq JP, Prutsky G, Mullan RJ, et al. Lifestyle modification programs in polycystic ovary syndrome: systematic review and meta-analysis. J Clin Endocrinol Metab 2013;98:4655.
53. Lass N, Kleber M, Winkel K, et al. Effect of lifestyle intervention on features of polycystic ovarian syndrome, metabolic syndrome, and intima-media thickness in obese adolescent girls. J Clin Endocrinol Metab 2011;96:3533.
54. Costello M, Eden J. A systematic review of the reproductive system effects of metformin in patients with polycystic ovary syndrome. Fertil Steril 2003;79:1–13.
55. Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 2003;327:951–3.
56. Auble B, Elder D, Gross A, Hillman JB. Differences in the management of adolescents with polycystic ovary syndrome across pediatric specialties. J Pediatr Adolesc Gynecol 2013;26:234–8.
57. Martin KA, Chang J, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clincal practice guideline. J Clin Endocrinol Metab 2008;93:1105–20.
58. Geller DH, Pacaud D, Gordon CM, Misra M, for the Drug and Therapeutics Committee of the Pediatric Endocrine Society. State of the art review: emerging therapies: the use of insulin sensitizers in the treatment of adolescents with polycystic ovary syndrome (PCOS). Int J Ped Endocrinol 2011;2011:9.
59. Tfayli H, Ulnach JW, Lee S, et al. Drospirenon/ethinyl estradiol versus rosiglitazone treatment in overweight adolescents with polycystic ovary syndrome: comparison of metabolic, hormonal and cardiovascular risk factors. J Clin Endocrinol Metab 2011;96:1311–9.