User login
Management of Relapsed and Refractory Multiple Myeloma
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.

Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
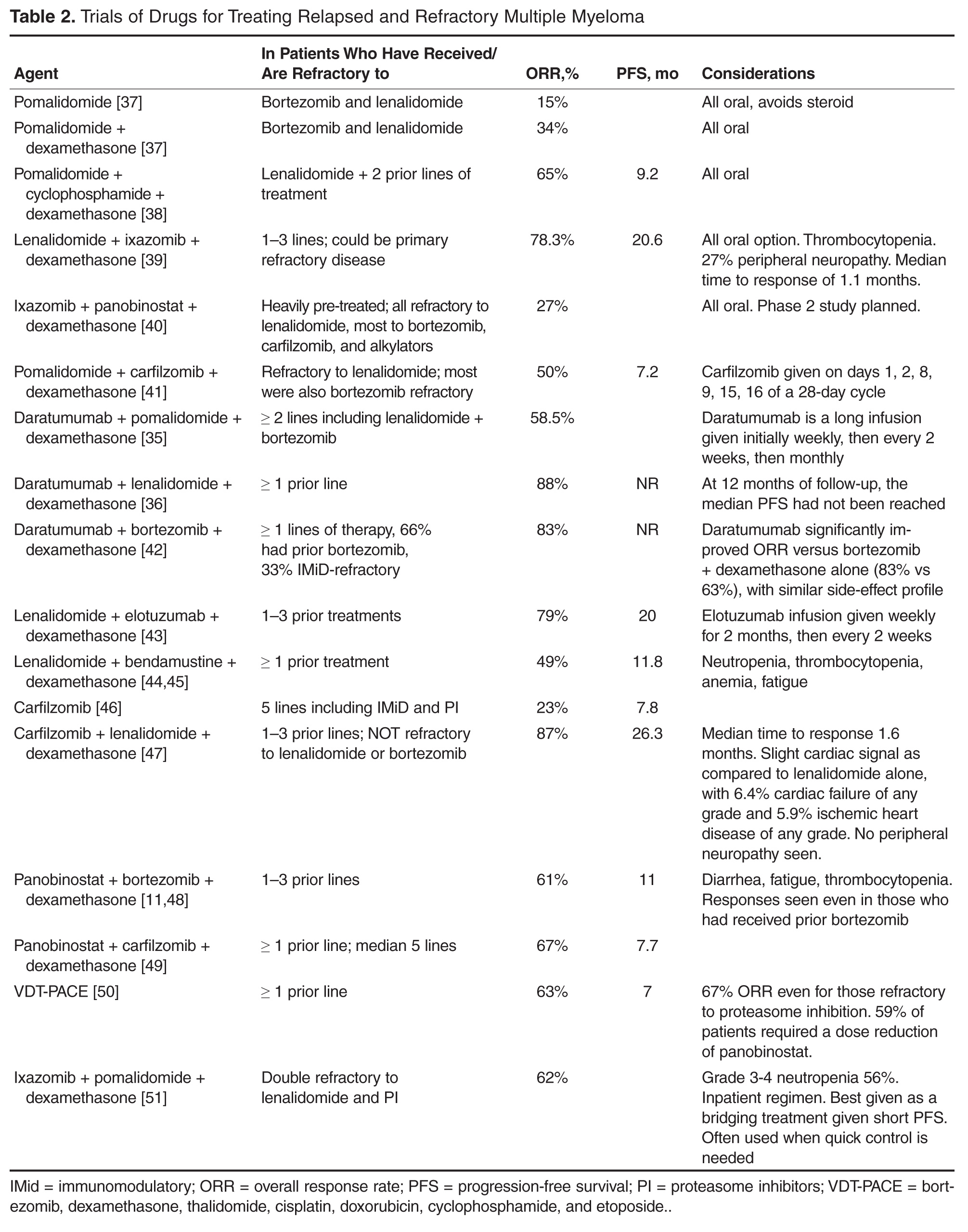
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, [email protected].
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.
Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, [email protected].
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.
Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, [email protected].
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.
Stabilized schizoaffective disorder; later confusion and depression appears
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.

How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.
How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.
How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
ADHD symptoms are stable, then a sudden relapse
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations


Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations
Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations
Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
Guide to Recognizing and Treating Sleep Disturbances in the Nursing Home
From the School of Aging Studies, University of South Florida, Tampa, FL (Dr. AA Gamaldo) and the Department of Neurology, Johns Hopkins Medicine, Baltimore, MD (Drs. Sloane, CE Gamaldo and Salas).
Abstract
- Objective: To provide guidance on identifying and treating sleep disturbances commonly encountered in older nursing home residents.
- Methods: Review of the literature in the context of 5 clinical cases.
- Results: Sleep disturbances continue to be a growing global epidemic, and public health initiatives have been aimed at improving sleep health across all ages. In older adults, sleep disturbances are often associated with the development and/or worsening of health conditions. Common sleep disturbances observed in older nursing home residents include obstructive sleep apnea, restless legs syndrome/Willis-Ekbom disease, circadian rhythm sleep-wake disorders, insomnia, and parasomnias. The symptoms and recommended treatment plans vary across the sleep disturbances. For many sleep disturbances, modification of residents’ daily activities and/or nursing home environment can be helpful.
- Conclusion: As the number of people residing in nursing homes increases, it is important for health care providers to be knowledgable about sleep disturbances in this population.
By 2030, almost 20% of the US population (approximately 72.1 million people) will be age 65 and older [1]. As many as 63% of older adults in the general population report sleep disturbances [2]. Specifically, older adults demonstrate difficulty with decreased total sleep duration, an increase in sleep fragmentation (ie, interruptions in nighttime sleep), and reduced total sleep time spent in rapid eye movement (REM) and slow wave sleep [3–5]. Poor sleep, either because of not getting enough sleep or having an undiagnosed and thus untreated sleep disorder, is associated with physical illness, impaired cognition, poor physical function, and mortality risk [6,7]. In fact, over 50% of individuals older than 65 years meet the diagnostic criteria for a sleep disorder, many of which are undiagnosed [6,7].
It is forecasted that we will see substantial increases in the rate of nursing home residence among the elderly [8]. The prevalence and severity of disturbed sleep is reportedly higher in NH residents [6,7]. Generally, NH residents tend to be on several medications for various medical disorders that may negatively impact sleep [7]. Reciprocally, sleep disruption may put NH residents at an increased risk for behavioral issues (eg, agitation) [9,10] as well as developing and/or exacerbating health conditions (eg, mood disorders, dementia, cardiovascular disease) [8]. Furthermore, NH residents exhibiting disturbed sleep, behavioral issues, and/or mood disorders are at an increased risk for being prescribed antipsychotic drugs [11], which are associated with adverse side effects and poorer quality of life [12]. Thus, the identification and management of sleep disturbances in the NH setting has become progressively more vital in efforts to optimize medical management of this population. This review identifies common sleep disturbances frequently underdiagnosed and undertreated among residents of NH facilities.
Case 1
A 73-year-old woman with a history of type 2 diabetes mellitus reports poor sleep quality with frequent awakenings during the night and excessive daytime sleepiness. She states that she can fall asleep within 5 minutes, but often is awoken throughout the night with a sensation of breathlessness. She has snored for many years, but the nursing staff at her NH facility has recently commented that her snoring has gone from intermittent to constant. She cannot remember the last time she has had restful sleep. She consumes 3 to 4 cups of caffeinated beverages daily to counter her sleepiness. She denies smoking or illicit drug or alcohol use. Her review of systems was notable for a 30-lb weight gain over the last year, and she reports increasing fatigue, irritability, and memory and concentration issues. Her current medication list includes metformin and amlodipine. Her examination is remarkable for a BMI of 31, large neck circumference (> 16), tonsillar enlargement, a crowded oropharynx, micrognathia, lungs clear to auscultation bilaterally, heart sounds of normal S1 and S2, and legs with trace pitting edema.
Case 1 Reflection: Sleep-Disordered Breathing


Although previous studies have observed high rates (60%–90%) of SDB in NH settings [16,17], one study observed that only 0.5% of nursing home residents carried a diagnosis with SDB, suggesting that SDB is being grossly underappreciated amongst NH residents over the age of 65 [18]. In order to evaluate for SBD, routine annual physical exams or medical chart reviews can elicit the risk factors for sleep apnea (eg, obesity [per BMI], male sex, postmenopausal women, family history of sleep apnea) as well as common comorbidities (eg, hypertension, coronary artery disease, and diabetes). Formal evaluation consists of a sleep evaluation with a sleep specialist and polysomnography (PSG; sleep study) that can be performed in the sleep center or at home depending on the patient’s history and other medical issues.
Case 1 Outcome

Case 2
An 85-year-old man with history of Alzheimer’s disease, major depression and arthritis, reports insomnia and “tingling in my legs” at bedtime. The patient cannot identify when the symptoms started but reports that his legs often jerk during sleep. He consumes a cup of coffee daily and has a previous 20 pack-year smoking history (he quit 40 years ago). On review of systems, he endorses fatigue. His current medication list includes fluoxetine, donepezil hydrochloride, ibuprofen as needed for arthritic pain, and a multivitamin. His examination was unremarkable, with a BMI of 26, neck circumference < 16, no tonsillar enlargement, normal (noncrowded) oropharynx, lungs clear to auscultation bilaterally, heart sounds demonstrating a normal S1 and S2, and legs without edema.
Case 2 Reflection: Restless Legs Syndrome/Willis-Ekbom Disease
Restless legs syndrome (RLS) also known as Willis-Ekbom disease, affects approximately 10 million adults in the United States alone [22]. RLS is a sensorimotor disorder that must satisfy the following 5 primary diagnostic criteria: (1) urge to move the legs with or without dysesthesias; (2) onset or exacerbation with rest or inactivity; (3) relief with movement; (4) symptoms are worse in the evening or at night (circadian component); (5) symptoms cannot be solely accounted for as consequence of another medical or behavioral condition. Other supporting clinical features can alert a clinician to the likelihood of a RLS diagnosis; these include positive family history, response to dopaminergic therapy, lack of profound daytime sleepiness, and presence of periodic limb movements during sleep (PLMS) [23–26]. In younger individuals, the symptoms present insidiously whereas older adults (> 50 years of age) will usually present with sudden onset [27].
Not only do patients lack the restorative sleep needed to ward off fatigue and restfulness, but patients also demonstrate higher rates of comorbidities (eg, anxiety, hypertension, depression) as well as large economic burden secondary to absenteeism and decreased on-the-job effectiveness [28,29]. As a results, patients with RLS experience significant reductions in quality of life related to this sensorimotor disorder [28].
No confirmatory laboratory test exists to diagnose RLS; however, patients suspected of having RLS should be evaluated with a basic metabolic panel, iron studies, and a thorough neurologic examination, as iron deficiency, kidney failure, uremia and peripheral neuropathy can lead to secondary RLS [30,31]. Evidence shows that RLS is common in NH residents [32] and may account for problematic behaviors, such as late night pacing [7]. Forty-five percent of community dwelling individuals over 65 years old exhibit a PLMS index (leg kicks per hour) of greater than 5 [33]. PLMS, while not a disorder in and of itself, can serve as a marker for potential disease. PLMS are characterized by intermittent episodes of stereotyped leg movements. PLMS typically do not awaken the patient from sleep and therefore do not contribute to insomnia or daytime sleepiness, representing a key clinical difference from RLS. It is important to note that PLMS are nonspecific and may be common in older adults that do not meet the diagnostic criteria for RLS.
Treatment of RLS is based on the frequency of symptoms and the level of functional impairment caused by the syndrome. RLS treatment recommendations should always espouse nonpharmacological interventions that include improving sleep practices, engagement in daily physical activity, targeted placement of sedentary activity in the morning when symptoms are less prominent, and concerted efforts to avoid the use of RLS-exacerbating medications (eg, selective serotonin reuptake inhibitor (SSRIs), neuroleptic agents, antihistamines) [28]. If there is an underlying condition contributing to RLS, such as metabolic disturbance or iron deficiency, then these conditions should be corrected before initiating RLS medications. Several medications are FDA-approved for treatment of RLS, including dopamine agonists (eg, ropinirole, rotigotine, pramipexole), dopamine precursor (eg, levodopa), glutamate-related (eg, gabapentin), benzodiazepines (eg, temazepam, clonazepam). Augmentation, the worsening of RLS symptoms, can occur in patients taking dopamine agonists. If this occurs, dopamine agents should be discontinued or switched to other agents (such as a long-acting dopamine agonist, gabapentin encarbil, as well as non-FDA approved therapies such as opioids). However, it is important to note that weaning off dopamine agents may result in mild but in most cases moderate and/or severe withdrawal from the medication, so counseling and close monitoring should be done.
Case 2 Outcome
Given the patient’s history of dementia, opioids, benzodiazepines and other delirium-inducing medications should be avoided. His antidepressive regimen, fluoxetine, should be re-evaluated as these medications have been associated with RLS exacerbation. In addition to SSRIs, medications associated with RLS are MAO inhibitors (selegeline, phenelzine), antipsychotics (risperdone, olanzapine), tricyclic antidepressants (mirtazapine), antihistamines (diphenhydramine, cimetidine), calcium channel blockers (verapamil, nifedipine, diltiazem), and phenytoin [34,35]. His treatment began with behavioral, nonpharmacological management, and blood testing for iron studies. His low iron level prompted initiation of oral supplementation, and he was asked to follow up in 3 months for reevaluation and possible initiation of low-dose dopamine agonists.
Case 3
A 73-year-old man with dementia is found to have very irregular sleep wake patterns with a variable bedtime and awakening time, often missing breakfast. He is found dozing off often during the day, particularly during times of inactivity. He has frequent awakenings during the night often calling for the staff to guide him back to bed. He has had some falls secondary to walking around his room. He has been prescribed various hypnotics without much benefit and instead, has suffered from some confusion while on these medications. His room is very dark and has no windows.
Case 3 Reflection: Circadian Rhythm Sleep-Wake Disorders
Circadian rhythm sleep-wake disorders (CRSWDs) are characterized by an individual’s natural propensity to want to go to sleep and be awake during a period that is undesirable personally and/or socially [36]. CRSWDs can be a result of the desynchronization of the 2 sleep processes: (1) homeostatic drive (regulates sleep intensity) and (2) circadian rhythm (maintains daytime alertness); [36]. CRSWDs can also be due to an individual’s naturally occurring sleep drives becoming misaligned with their social/personal sleep-wake demands (eg, employment schedule and socializing opportunities with family/friends). With increasing age, the circadian rhythm becomes less adept at functioning in a desynchronized pattern [7], which can result in daytime sleepiness and night time sleep fragmentation [7,37]. CRSWDs are highly prevalent in individuals with dementia [7,36]. As dementia progresses, the ability to maintain a balance of the 2 sleep process becomes more impaired [7]. As a result, individuals with dementia, particularly Alzheimer’s disease, are likely to experience agitation, irritability, and/or confusion during the evening and night, a behavioral problem referred to as “sundowning” [38].
There are several types of CRSWDs, including delayed sleep-phase syndrome, advanced sleep-phase syndrome, irregular sleep-wake disorder, non–24-hour sleep-wake disorder, shift work sleep disorder, and jet lag sleep disorder. However, the most common type of CRSWDs observed in older adults is advanced sleep-phase syndrome [39]. Due to excessive sleepiness in the early evening, affected individuals may report a need to shift to earlier and earlier bedtimes (~6 to 7 pm) and wake times (~3 to 4 am) [36]. For older affected adults, this can cause distress and frustration, particularly if their sleep phase prevents them from participating in evening activities (eg, socializing with family/friends) [36].
In the assessment of patients with suspected CRSWDs, sleep diaries (self-reported or caregiver) daily account of sleep and wake times over at least 1 week) and actigraphy (wrist-worn accelerometer designed to measure activity and inactivity at night) can be used, particularly in older adults with dementia [40,41].
CRSWD treatment may include behavioral modifications and/or pharmacological intervention. Behavioral modifications can consist of chronotherapy, relaxation training, and/or bright light therapy. Chronotherapy involves making gradual shifts in an individual’s sleep time to meet his/her desired sleep schedule. Relaxation training involves implementing behaviors/activities that reduce tension and enhance the smooth transition into sleep. Bright light therapy involves exposure to an appropriate intensity and duration of light, which is an important environmental cue to help the synchrony of the sleep-wake cycle [7]. Previous studies have observed that NH residents are exposed to a restricted amount of bright light during the daytime [42,43], but higher levels of artificial light at night (eg, hallway lighting) [7]. NH residents’ exposure to artificial bright light during the daytime has not only improved the residents’ sleep [44–46], but also has improved their cognitive functioning and reduced their depressive symptoms [47]. Thus, steps towards targeted light exposure in sync with the typical sleep-wake cycle (eg, mandated time in well-lit rooms during the day and during meals) for NH residents, particularly those with CRSWDs, could prove to be beneficial across several social, behavioral and neurocognitive domains. Lastly, NH residents exposed to at least 30 minutes of outdoor daylight and at least 3 occasions of low intensity physical activities for 10 to 15 minutes daily can potentially improve sleep-wake patterns [48]. Thus, it may be beneficial to have an intervention that couples bright light exposure and physical activity in the NH setting.
Pharmacological interventions can also be implemented to improve older residents’ symptoms. However, the medications prescribed should be used with caution and should not be used as part of a long-term treatment plan. Melatonin is a commonly used herbal supplement that can assist advancing the timing of the circadian rhythms in the evening but can delay the circadian rhythms in the morning [49]. Several brands of this herbal supplement can be purchased over-the-counter and are not regulated by the FDA. Since the amount of melatonin used in the herbal supplement varies by brand, caution should be used when selecting a brand [50]. Two FDA-approved drugs (modafinil and armodafinil) are currently being used to reduce daytime sleepiness and improve vigilance amongst adults, but limited research has explored the effectiveness of these medication for older adults specifically suffering with CRSWDS [36,51,52]. Other stimulants (eg, caffeine, amphetamines, and nonamphetamine-derived medications) are also currently being used to reduce daytime sleepiness in patients with CRSWDS. Stimulant use, particularly caffeine consumption, has also been associated with better cognitive functioning in older adults [53]. However, stimulants should be taken with caution, particularly in older adults, because stimulant use has been associated with potentially serious and fatal health sequalae (eg, tachycardia, heart failure, irreversible heart damage and hypertension) [36,54].
Case 3 Outcome
The patient was moved to a room with a window. An alarm clock was set for 7:30 in the morning and he was taken to breakfast, where he sat at a table near a window. Any time he appeared to be sleepy, he was encouraged to go for a walk or engage in other activities so daytime napping opportunities were limited. His environment was assessed for safety and bedrails were utilized to prevent falls.
Case 4
A 75-year-old woman with a history of anxiety and depression moved into the NH 4 months ago after suffering a stroke. She now reports difficulty falling asleep for many years, which has significantly worsened since moving to the NH. Currently, she has been getting only 3 to 4 hour of sleep per night. She reports mild but increasing daytime sleepiness and does not fall asleep until 1:00 am despite getting into bed at 10:30 pm. She occasionally reports arthritic pain that interferes with her sleep. The NH staff has mentioned that she will occasionally cry for her family when she appears to be asleep.
Case 4 Reflection: Insomnia
According to the International Classification of Sleep Disorders (ICSD-3) [39], insomnia is characterized as “a repeated difficulty with sleep initiation, duration, consolidation, or quality that occurs despite adequate opportunity and circumstances for sleep, and results in some form of daytime impairment.” Among the sleep disorders, insomnia is one of the most common sleep issues observed in sleep clinics [34]. Older adults with insomnia often have comorbid physical (eg, pulmonary disease, arthritis, chronic pain, cancer diabetes, Parkinson’s disease) and mental illness (eg, depression, panic disorder) [55]. Medications (eg, stimulants, respiratory medications, chemotherapy, decongestants, hormones, or psychotropics) may cause and exacerbate insomnia symptoms [55].
Since insomnia is a clinical diagnosis, there is no specific diagnostic tool or gold standard test to identify individuals suffering with insomnia. Insomnia screening usually involves a clinical interview, in which a health provider, preferably trained in sleep, conducts a physical examination and collects an in-depth history of a patients’ sleep problems [56]. Insomnia screening tools may also include having a patient complete a sleep diary or questionnaire, such as the insomnia severity index (ISI) [57] or Pittsburgh Sleep Quality Index (PSQI) [58].
Cognitive behavioral therapy for insomnia (CBT-I) and/or pharmacological intervention are typically used to treat insomnia in older adults. CBT-I is a combination of cognitive (eg, changing dysfunctional sleep attitudes/beliefs) and behavioral treatment (eg, adhering to a regular sleep schedule) [59]. CBT-I or a combination of CBT-I and pharmacological intervention is recommended as more effective long-term approach to insomnia treatment compared to pharmacological intervention alone [55]. CBT-I involves altering older adults’ misconceptions of their sleep and implementing behavioral techniques to their everyday life (eg, routine sleep-wake schedule, relaxation therapy). Several FDA-approved medications are available to treat insomnia; however, many commonly used medications to treat insomnia in older adults (ie, antihistamines, antidepressants, anticonvulsants, and anti-psychotics) pose more risks than benefits to their health and well-being [35,60–62]. Some of the more recent hypnotics (egm zolpidem, exzopiclone, and ramelteon) on the market have been shown to be safer and more effective pharmacological options [55]. In 2014, the FDA approved the first in class orexin receptor antagonist medication (suvorexant) to treat insomnia [63]. Unlike other medications to treat insomnia, suvorexant, via the blockade of the orexin neurotransmitter, effectively inhibits orexin (one of neurotransmitters involved in the activation pathways of the arousal system), so sleep can easily be induced and maintained [64, 65]. Furthermore, preliminary studies suggest that this medication may be associated with less severe side effects (ie, habituation) than the other approved medications on the market [64, 65]. In fact, in a recent clinical series that included both young and older insomnia patients, the most common adverse reaction to suvorexant was drowsiness [66].
Case 4 Outcome
The patient was initiated on basic CBT-I therapy strategies which included stimulus control therapy [67]; implementation of a consistent bedtime and awakening routine; reducing the use of TV, smart phone, or other electronic leisure devices 1 hour before bedtime; refraining from caffeine after lunchtime; improving the sleep environment; and relaxation techniques.
Case 5
The patient is a 65-year-old man diagnosed with Parkinson’s disease several years ago. Recently, he has often has been experiencing what appears to be very violent and terrifying dreams. While asleep, he often screams and shouts for help. In addition, he occasionally will punch, kick, and/or thrash around in bed at night, which the NH staff has noted as a concern for his safety.
Case 5 Reflection: Parasomnias
Parasomnias represent frequent arousals during sleep or in the wake-to-sleep transition due to abnormal motor movements, behaviors (eg, shouting, flailing, and leaping from bed) and/or sensory experiences (eg, “dreamlike” hallucinations) [68]. Motor movements that occur for parasomnia can be disruptive for the individual and potentially dangerous for the individual and/or bed partner. There are 3 primary types of parasomnias based on the stage of sleep that the event occurs: non-REM (NREM), REM, and other parasomnias during transitions of sleep [68]. The most commonly observed parasomnia seen in older adults is the REM-associated parasomnia or REM sleep behavior disorder (RBD), which is characterized by experiencing vivid, sometimes violent, dreams typically involving fighting an intruder or an animal to protect a loved one [69]. For RBD, disruptive behaviors typically occur during REM sleep [69]. RBD has been associated with neurodegenerative disorders (Parkinson’s disease and Lewy body disease), neurologic disorders (eg, brain tumors and stroke), other primary disorders (narcolepsy and periodic limb movement disorder), and well as some medications (eg, antidepressants and β-blockers) [68]. There is limited knowledge on the prevalence of parasomnias in NH settings. One study, however, reported that 31% of older NH residents experience parasomnias [70]. Evaluation for parasomnias generally involve a clinical evaluation by a sleep specialist and overnight sleep study (ie, polysomnography at a sleep center if there is a concern for sleep apnea or RBD [71].
Medications are not typically first-line for parasomnia. Instead education about improving sleep practices, addressing other underlying sleep disorders, and securing a safe sleep environment are first recommended. Pharmacologic treatment, particularly the use of clonazepam, is commonly used to treat RBD [72]. However, this medication should be used with caution for older adults with a dementia diagnosis, gait disorders, and OSA because the common side effects include sedation, confusion, memory dysfunction, and early morning motor incoordination [68]. Several alternative medications have also been used to treat RBD. For example, medications commonly used to Parkinson disease symptoms, such Levodopa and dopamine agonists, have also been used to treat RBD [73]. Zopiclone, a nonbenzodiazepine hypnotic agent, has also been shown to be as effective as clonazepam, but with less potential side effects [74]. Melatonin, a nutritional supplement, has also been used as a treatment and appears to alleviate some of the RBD symptoms and has fewer side effects [68]. Since melatonin is not regulated by the FDA, it has been suggested that this treatment be used with caution in the older population [73].
Case 5 Outcome
The patient was evaluated with video synchronized in lab PSG. It confirmed REM sleep without evidence of the normal atonia that should be apparent during REM. These PSG findings in combination with repeated accounts of dream enactment established the diagnosis of RBD. Patient was treated with low-dose clonazepam and closely monitored for potential side effects of daytime sedation. Bedroom environment was also carefully reconfigured for safety to avoid potential risk of injury during a dream enactment episode.
Conclusion
Sleep disturbances remain an underappreciated and undertreated health issue in NH residents. Nursing homes can help facilitate optimal sleep health and day functioning by providing mandatory natural light outlets, physical exercise opportunities, and minimal allowable time residents can spend in their bed/bedroom outside of their routine sleep period. Educating NH providers and staff on sleep medicine may benefit residents, but workload and restricted resources may hinder this. Education via mobile and internet based educational platforms and resources (Mysleep101) may be helpful in addressing education barriers [75]. Convenient and cost-effective methods to deliver sleep medicine education to NH health care providers should be part of our ongoing efforts to improve the viability, vitality and quality of life of our aging citizens.
Corresponding author: Alyssa Gamaldo, PhD, Univ. of South Florida, 13301 Bruce B. Downs Blvd, MHC 1340, Tampa, FL 33612, [email protected].
Financial disclosures: None.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2012: Key indicators of well-being. Washington, DC: U.S. Gov. Printing Office; 2012.
2. Almeida OP, Pfaff JJ. Sleep complaints among older general practice patients: association with depression. Br J Gen Pract 2005;55:864–866.
3. Wolkove N, Elkholy O, Baltzan M, et al. Sleep and aging: 1. Sleep disorders commonly found in older people. CMAJ 2007;176:1299–304.
4. Pace-Schott EF, Spencer RM. Age-related changes in the cognitive function of sleep. Prog Brain Res 2011;191:75–89.
5. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 2004;27:1255–73.
6. Jacobs D, Ancoli-Israel S, Parker L, et al. Twenty-four-hour sleep-wake patterns in a nursing home population. Psychol Aging 1989;4:352–6.
7. Neikrug AB, Ancoli-Israel S. Sleep disturbances in nursing homes. J Nutr Health Aging 2010;14:207–11.
8. Lakdawalla D, Goldman DP, Bhattacharya J, et al. Forecasting the nursing home population. Medical Care 2003;41:8-20.
9. Alessi CA, Yoon EJ, Schnelle JF, et al. A randomized trial of a combined physical activity and environmental intervention in nursing home residents: do sleep and agitation improve? J Am Geriatr Soc 1999;47:784–91.
10. Cohen-Mansfield J, Marx MS. The relationship between sleep disturbances and agitation in a nursing home. J Aging Health 1990;2:153–65.
11. Rolland Y, Andrieu S, Crochard A, et al. Psychotropic drug consumption at admission and discharge of nursing home residents. J Am Med Dir Assoc 2012;13:407 e407–412.
12. Salzman C. Antipsychotics in nursing homes. J Clin Psychopharmacol 2013;33:1–2.
13. Fleetham J, Ayas N, Bradley D, et al. Canadian Thoracic Society guidelines: diagnosis and treatment of sleep disordered breathing in adults. Can Respir J 2006;13:387–92.
14. International classification of sleep disorders: Chicago: American Academy of Sleep Medicine; 2005.
15. Gay PC. Complex sleep apnea: it really is a disease. J Clin Sleep Med 2008;4:403–5.
16. Ancoli-Israel S. Epidemiology of sleep disorders. In: Roth T, Roehrs TA, editors. Clinics in geriatric medicine. Philadelphia: WB Saunders; 1989:347–62.
17. Gehrman PR, Martin JL, Shochat T, et al. Sleep-disordered breathing and agitation in institutionalized adults with Alzheimer disease. Am J Geriatr Psychiatry 2003;11:426–33.
18. Resnick HE, Phillips B. Documentation of sleep apnea in nursing homes: United States 2004. J Am Med Dir Assoc 2008;9:260–4.
19. Park JG, Ramar K, Olson EJ. Updates on definition, consequences, and management of obstructive sleep apnea. Mayo Clin Proc 2011;86:549–54.
20. Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034–41.
21. McMillan A, Bratton DJ, Faria R, et al. Continuous positive airway pressure in older people with obstructive sleep apnoea syndrome (PREDICT): a 12-month, multicentre, randomised trial. Lancet Resp med 2014;2:804–12.
22. Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 2005;165:1286–92.
23. Allen RP, La Buda MC, Becker P, et al. Family history study of the restless legs syndrome. Sleep Med 2002;3 Suppl:S3–7.
24. Allen RP. Restless legs syndrome/Willis Ekbom disease: evaluation and treatment. Int Rev Psychiatry 2014;26:248–62.
25. Hening WA, Allen RP, Earley CJ, et al. An update on the dopaminergic treatment of restless legs syndrome and periodic limb movement disorder. Sleep 2004;27:560–83.
26. Gamaldo C, Benbrook AR, Allen RP, et al. Evaluating daytime alertness in individuals with Restless Legs Syndrome (RLS) compared to sleep restricted controls. Sleep Med 2009;10:134–8.
27. Allen RP, Earley CJ. Defining the phenotype of the restless legs syndrome (RLS) using age-of-symptom-onset. Sleep Med 2000;1:11–9.
28. Salas RE, Kwan AB. The real burden of restless legs syndrome: clinical and economic outcomes. Am J Manag Care 2012;18(9 Suppl):S207–212.
29. Silber MH, Ehrenberg BL, Allen RP, et al. An algorithm for the management of restless legs syndrome. Mayo Clin Proc 2004;79:916–22.
30. Hattan E, Chalk C, Postuma RB. Is there a higher risk of restless legs syndrome in peripheral neuropathy? Neurology 2009;72:955–60.
31. Winkelman JW, Chertow GM, Lazarus JM. Restless legs syndrome in end-stage renal disease. Am J Kidney Dis 1996;28:372–8.
32. Hornyak M, Trenkwalder C. Restless legs syndrome and periodic limb movement disorder in the elderly. J Psychosom Res 2004;56:543–8.
33. Ancoli-Israel S, Kripke DF, Klauber MR, et al. Periodic limb movements in sleep in community-dwelling elderly. Sleep 1991;14:496–500.
34. Ahmed QA. Effects of common medications used for sleep disorders. Crit Care Clin 2008;24:493–515, vi.
35. Rye DB, Trotti LM. Restless legs syndrome and periodic leg movements of sleep. Neurol clin 2012;30:1137–66.
36. Gamaldo CE, Chung Y, Kang YM, et al. Tick-tock-tick-tock: the impact of circadian rhythm disorders on cardiovascular health and wellness. J Am Soc Hypertens 2014;8:921–9.
37. Myers BL, Badia P. Changes in circadian rhythms and sleep quality with aging: mechanisms and interventions. Neurosci Biobehav Rev 1995;19:553–71.
38. National Institute on Aging. Alzheimer’s caregiving tips: sundowning. June 2013.
39. International classification of sleep disorders. 3rd ed. Darien, IL: AASD; 2014.
40. Barion A, Zee PC. A clinical approach to circadian rhythm sleep disorders. Sleep Med 2007;8:566–77.
41. Morgenthaler TI, Lee-Chiong T, Alessi C, et al. Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep 2007;30:1445–59.
42. Shochat T, Martin J, Marler M, et al. Illumination levels in nursing home patients: effects on sleep and activity rhythms. J Sleep Res 2000;9:373–9.
43. Ancoli-Israel S, Klauber MR, Jones DW, et al. Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 1997;20:18–23.
44. Satlin A, Volicer L, Ross V, et al. Bright light treatment of behavioral and sleep disturbances in patients with Alzheimer’s disease. Am J Psychiatry 1992;149:1028–32.
45. Koyama E, Matsubara H, Nakano T. Bright light treatment for sleep-wake disturbances in aged individuals with dementia. Psychiatry Clin Neurosci 1999;53:227–9.
46. Burns A, Allen H, Tomenson B, et al. Bright light therapy for agitation in dementia: a randomized controlled trial. Int Psychogeriatr 2009;21:711–21.
47. Riemersma-van der Lek RF, Swaab DF, Twisk J, et al. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA 2008;299:2642–55.
48. Martin JL, Marler MR, Harker JO, et al. A multicomponent nonpharmacological intervention improves activity rhythms among nursing home residents with disrupted sleep/wake patterns. J Gerontol Series A Biol Sci Med Sci 2007;62:67–72.
49. Lewy AJ, Ahmed S, Jackson JM, et al. Melatonin shifts human circadian rhythms according to a phase-response curve. Chronobiol Int 1992;9:380–92.
50. Williamson BL, Tomlinson AJ, Mishra PK, et al. Structural characterization of contaminants found in commercial preparations of melatonin: similarities to case-related compounds from L-tryptophan associated with eosinophilia-myalgia syndrome. Chem Res Toxicol 1998;11:234-40.
51. Czeisler CA, Walsh JK, Roth T, et al. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med 2005;353:476-86.
52. Howard R, Roth T, Drake CL. The effects of armodafinil on objective sleepiness and performance in a shift work disorder sample unselected for objective sleepiness. J Clin Psychopharmacol 2014;34:369–73.
53. Beydoun MA, Gamaldo AA, Beydoun HA, et al. Caffeine and alcohol intakes and overall nutrient adequacy are associated with longitudinal cognitive performance among U.S. adults. J Nutr 2014;144:890–901.
54. Pentel P. Toxicity of over-the-counter stimulants. JAMA 1984;252:1898–903.
55. Neikrug AB, Ancoli-Israel S. Sleep disorders in the older adult - a mini-review. Gerontology 2010;56:181–9.
56. Chesson A Jr, Hartse K, Anderson WM, et al. Practice parameters for the evaluation of chronic insomnia. An American Academy of Sleep Medicine report. Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep 2000;23:237–41.
57. Bastien CH, Vallieres A, Morin CM. Validation of the insomnia severity index as an outcome measure for insomnia research. Sleep Med 2001;2:297–307.
58. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
59. Trauer JM, Qian MY, Doyle JS, et al. Cognitive behavioral therapy for chronic insomnia: a systematic review and meta-analysis. Ann Intern Med 2015;163:191–204.
60. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults, June 13-15, 2005. Sleep 2005;28:1049–57.
61. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med 2008;4:487–504.
62. Bain KT. Management of chronic insomnia in elderly persons. Am J Geriatr Pharmacother 2006;4:168–92.
63. FDA approves new type of sleep drug, Belsomra. 13 Aug 2014. Available at fda.gov.
64. Dubey AK, Handu SS, Mediratta PK. Suvorexant: The first orexin receptor antagonist to treat insomnia. J Pharmacol Pharmacother 2015;6:118–21.
65. Patel KV, Aspesi AV, Evoy KE. Suvorexant: a dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann Pharmacother 2015;49:477–83.
66. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:461–71.
67. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. Case studies in insomnia. New York: Springer; 1991:19–28.
68. Gulyani S, Salas RE, Gamaldo CE. Sleep medicine pharmacotherapeutics overview: today, tomorrow, and the future (part 2: hypersomnia, parasomnia, and movement disorders). Chest 2013;143:242–51.
69. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11):2770–88.
70. Middelkoop HA, Kerkhof GA, Smilde-van den Doel DA, et al. Sleep and ageing: the effect of institutionalization on subjective and objective characteristics of sleep. Age Ageing 1994;23:411–7.
71. Tinuper P, Provini F, Bisulli F, et al. Movement disorders in sleep: guidelines for differentiating epileptic from non-epileptic motor phenomena arising from sleep. Sleep Med Rev 2007;11:255–67.
72. Aurora RN, Zak RS, Maganti RK, et al. Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med 2010;6:85–95.
73. Bloom HG, Ahmed I, Alessi CA, et al. Evidence-based recommendations for the assessment and management of sleep disorders in older persons. J Am Geriatr Soc 2009;57:761–89.
74. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
75. Doshi A, Gamaldo C, Kalloo A, et al. Implementing a clinical iPad application to detect sleep disorders is feasible across multiple non-sleep outpatient clinics (P7. 321). Neurology 2015;84(14 Suppl):P7.321.
From the School of Aging Studies, University of South Florida, Tampa, FL (Dr. AA Gamaldo) and the Department of Neurology, Johns Hopkins Medicine, Baltimore, MD (Drs. Sloane, CE Gamaldo and Salas).
Abstract
- Objective: To provide guidance on identifying and treating sleep disturbances commonly encountered in older nursing home residents.
- Methods: Review of the literature in the context of 5 clinical cases.
- Results: Sleep disturbances continue to be a growing global epidemic, and public health initiatives have been aimed at improving sleep health across all ages. In older adults, sleep disturbances are often associated with the development and/or worsening of health conditions. Common sleep disturbances observed in older nursing home residents include obstructive sleep apnea, restless legs syndrome/Willis-Ekbom disease, circadian rhythm sleep-wake disorders, insomnia, and parasomnias. The symptoms and recommended treatment plans vary across the sleep disturbances. For many sleep disturbances, modification of residents’ daily activities and/or nursing home environment can be helpful.
- Conclusion: As the number of people residing in nursing homes increases, it is important for health care providers to be knowledgable about sleep disturbances in this population.
By 2030, almost 20% of the US population (approximately 72.1 million people) will be age 65 and older [1]. As many as 63% of older adults in the general population report sleep disturbances [2]. Specifically, older adults demonstrate difficulty with decreased total sleep duration, an increase in sleep fragmentation (ie, interruptions in nighttime sleep), and reduced total sleep time spent in rapid eye movement (REM) and slow wave sleep [3–5]. Poor sleep, either because of not getting enough sleep or having an undiagnosed and thus untreated sleep disorder, is associated with physical illness, impaired cognition, poor physical function, and mortality risk [6,7]. In fact, over 50% of individuals older than 65 years meet the diagnostic criteria for a sleep disorder, many of which are undiagnosed [6,7].
It is forecasted that we will see substantial increases in the rate of nursing home residence among the elderly [8]. The prevalence and severity of disturbed sleep is reportedly higher in NH residents [6,7]. Generally, NH residents tend to be on several medications for various medical disorders that may negatively impact sleep [7]. Reciprocally, sleep disruption may put NH residents at an increased risk for behavioral issues (eg, agitation) [9,10] as well as developing and/or exacerbating health conditions (eg, mood disorders, dementia, cardiovascular disease) [8]. Furthermore, NH residents exhibiting disturbed sleep, behavioral issues, and/or mood disorders are at an increased risk for being prescribed antipsychotic drugs [11], which are associated with adverse side effects and poorer quality of life [12]. Thus, the identification and management of sleep disturbances in the NH setting has become progressively more vital in efforts to optimize medical management of this population. This review identifies common sleep disturbances frequently underdiagnosed and undertreated among residents of NH facilities.
Case 1
A 73-year-old woman with a history of type 2 diabetes mellitus reports poor sleep quality with frequent awakenings during the night and excessive daytime sleepiness. She states that she can fall asleep within 5 minutes, but often is awoken throughout the night with a sensation of breathlessness. She has snored for many years, but the nursing staff at her NH facility has recently commented that her snoring has gone from intermittent to constant. She cannot remember the last time she has had restful sleep. She consumes 3 to 4 cups of caffeinated beverages daily to counter her sleepiness. She denies smoking or illicit drug or alcohol use. Her review of systems was notable for a 30-lb weight gain over the last year, and she reports increasing fatigue, irritability, and memory and concentration issues. Her current medication list includes metformin and amlodipine. Her examination is remarkable for a BMI of 31, large neck circumference (> 16), tonsillar enlargement, a crowded oropharynx, micrognathia, lungs clear to auscultation bilaterally, heart sounds of normal S1 and S2, and legs with trace pitting edema.
Case 1 Reflection: Sleep-Disordered Breathing
Although previous studies have observed high rates (60%–90%) of SDB in NH settings [16,17], one study observed that only 0.5% of nursing home residents carried a diagnosis with SDB, suggesting that SDB is being grossly underappreciated amongst NH residents over the age of 65 [18]. In order to evaluate for SBD, routine annual physical exams or medical chart reviews can elicit the risk factors for sleep apnea (eg, obesity [per BMI], male sex, postmenopausal women, family history of sleep apnea) as well as common comorbidities (eg, hypertension, coronary artery disease, and diabetes). Formal evaluation consists of a sleep evaluation with a sleep specialist and polysomnography (PSG; sleep study) that can be performed in the sleep center or at home depending on the patient’s history and other medical issues.
Case 1 Outcome
Case 2
An 85-year-old man with history of Alzheimer’s disease, major depression and arthritis, reports insomnia and “tingling in my legs” at bedtime. The patient cannot identify when the symptoms started but reports that his legs often jerk during sleep. He consumes a cup of coffee daily and has a previous 20 pack-year smoking history (he quit 40 years ago). On review of systems, he endorses fatigue. His current medication list includes fluoxetine, donepezil hydrochloride, ibuprofen as needed for arthritic pain, and a multivitamin. His examination was unremarkable, with a BMI of 26, neck circumference < 16, no tonsillar enlargement, normal (noncrowded) oropharynx, lungs clear to auscultation bilaterally, heart sounds demonstrating a normal S1 and S2, and legs without edema.
Case 2 Reflection: Restless Legs Syndrome/Willis-Ekbom Disease
Restless legs syndrome (RLS) also known as Willis-Ekbom disease, affects approximately 10 million adults in the United States alone [22]. RLS is a sensorimotor disorder that must satisfy the following 5 primary diagnostic criteria: (1) urge to move the legs with or without dysesthesias; (2) onset or exacerbation with rest or inactivity; (3) relief with movement; (4) symptoms are worse in the evening or at night (circadian component); (5) symptoms cannot be solely accounted for as consequence of another medical or behavioral condition. Other supporting clinical features can alert a clinician to the likelihood of a RLS diagnosis; these include positive family history, response to dopaminergic therapy, lack of profound daytime sleepiness, and presence of periodic limb movements during sleep (PLMS) [23–26]. In younger individuals, the symptoms present insidiously whereas older adults (> 50 years of age) will usually present with sudden onset [27].
Not only do patients lack the restorative sleep needed to ward off fatigue and restfulness, but patients also demonstrate higher rates of comorbidities (eg, anxiety, hypertension, depression) as well as large economic burden secondary to absenteeism and decreased on-the-job effectiveness [28,29]. As a results, patients with RLS experience significant reductions in quality of life related to this sensorimotor disorder [28].
No confirmatory laboratory test exists to diagnose RLS; however, patients suspected of having RLS should be evaluated with a basic metabolic panel, iron studies, and a thorough neurologic examination, as iron deficiency, kidney failure, uremia and peripheral neuropathy can lead to secondary RLS [30,31]. Evidence shows that RLS is common in NH residents [32] and may account for problematic behaviors, such as late night pacing [7]. Forty-five percent of community dwelling individuals over 65 years old exhibit a PLMS index (leg kicks per hour) of greater than 5 [33]. PLMS, while not a disorder in and of itself, can serve as a marker for potential disease. PLMS are characterized by intermittent episodes of stereotyped leg movements. PLMS typically do not awaken the patient from sleep and therefore do not contribute to insomnia or daytime sleepiness, representing a key clinical difference from RLS. It is important to note that PLMS are nonspecific and may be common in older adults that do not meet the diagnostic criteria for RLS.
Treatment of RLS is based on the frequency of symptoms and the level of functional impairment caused by the syndrome. RLS treatment recommendations should always espouse nonpharmacological interventions that include improving sleep practices, engagement in daily physical activity, targeted placement of sedentary activity in the morning when symptoms are less prominent, and concerted efforts to avoid the use of RLS-exacerbating medications (eg, selective serotonin reuptake inhibitor (SSRIs), neuroleptic agents, antihistamines) [28]. If there is an underlying condition contributing to RLS, such as metabolic disturbance or iron deficiency, then these conditions should be corrected before initiating RLS medications. Several medications are FDA-approved for treatment of RLS, including dopamine agonists (eg, ropinirole, rotigotine, pramipexole), dopamine precursor (eg, levodopa), glutamate-related (eg, gabapentin), benzodiazepines (eg, temazepam, clonazepam). Augmentation, the worsening of RLS symptoms, can occur in patients taking dopamine agonists. If this occurs, dopamine agents should be discontinued or switched to other agents (such as a long-acting dopamine agonist, gabapentin encarbil, as well as non-FDA approved therapies such as opioids). However, it is important to note that weaning off dopamine agents may result in mild but in most cases moderate and/or severe withdrawal from the medication, so counseling and close monitoring should be done.
Case 2 Outcome
Given the patient’s history of dementia, opioids, benzodiazepines and other delirium-inducing medications should be avoided. His antidepressive regimen, fluoxetine, should be re-evaluated as these medications have been associated with RLS exacerbation. In addition to SSRIs, medications associated with RLS are MAO inhibitors (selegeline, phenelzine), antipsychotics (risperdone, olanzapine), tricyclic antidepressants (mirtazapine), antihistamines (diphenhydramine, cimetidine), calcium channel blockers (verapamil, nifedipine, diltiazem), and phenytoin [34,35]. His treatment began with behavioral, nonpharmacological management, and blood testing for iron studies. His low iron level prompted initiation of oral supplementation, and he was asked to follow up in 3 months for reevaluation and possible initiation of low-dose dopamine agonists.
Case 3
A 73-year-old man with dementia is found to have very irregular sleep wake patterns with a variable bedtime and awakening time, often missing breakfast. He is found dozing off often during the day, particularly during times of inactivity. He has frequent awakenings during the night often calling for the staff to guide him back to bed. He has had some falls secondary to walking around his room. He has been prescribed various hypnotics without much benefit and instead, has suffered from some confusion while on these medications. His room is very dark and has no windows.
Case 3 Reflection: Circadian Rhythm Sleep-Wake Disorders
Circadian rhythm sleep-wake disorders (CRSWDs) are characterized by an individual’s natural propensity to want to go to sleep and be awake during a period that is undesirable personally and/or socially [36]. CRSWDs can be a result of the desynchronization of the 2 sleep processes: (1) homeostatic drive (regulates sleep intensity) and (2) circadian rhythm (maintains daytime alertness); [36]. CRSWDs can also be due to an individual’s naturally occurring sleep drives becoming misaligned with their social/personal sleep-wake demands (eg, employment schedule and socializing opportunities with family/friends). With increasing age, the circadian rhythm becomes less adept at functioning in a desynchronized pattern [7], which can result in daytime sleepiness and night time sleep fragmentation [7,37]. CRSWDs are highly prevalent in individuals with dementia [7,36]. As dementia progresses, the ability to maintain a balance of the 2 sleep process becomes more impaired [7]. As a result, individuals with dementia, particularly Alzheimer’s disease, are likely to experience agitation, irritability, and/or confusion during the evening and night, a behavioral problem referred to as “sundowning” [38].
There are several types of CRSWDs, including delayed sleep-phase syndrome, advanced sleep-phase syndrome, irregular sleep-wake disorder, non–24-hour sleep-wake disorder, shift work sleep disorder, and jet lag sleep disorder. However, the most common type of CRSWDs observed in older adults is advanced sleep-phase syndrome [39]. Due to excessive sleepiness in the early evening, affected individuals may report a need to shift to earlier and earlier bedtimes (~6 to 7 pm) and wake times (~3 to 4 am) [36]. For older affected adults, this can cause distress and frustration, particularly if their sleep phase prevents them from participating in evening activities (eg, socializing with family/friends) [36].
In the assessment of patients with suspected CRSWDs, sleep diaries (self-reported or caregiver) daily account of sleep and wake times over at least 1 week) and actigraphy (wrist-worn accelerometer designed to measure activity and inactivity at night) can be used, particularly in older adults with dementia [40,41].
CRSWD treatment may include behavioral modifications and/or pharmacological intervention. Behavioral modifications can consist of chronotherapy, relaxation training, and/or bright light therapy. Chronotherapy involves making gradual shifts in an individual’s sleep time to meet his/her desired sleep schedule. Relaxation training involves implementing behaviors/activities that reduce tension and enhance the smooth transition into sleep. Bright light therapy involves exposure to an appropriate intensity and duration of light, which is an important environmental cue to help the synchrony of the sleep-wake cycle [7]. Previous studies have observed that NH residents are exposed to a restricted amount of bright light during the daytime [42,43], but higher levels of artificial light at night (eg, hallway lighting) [7]. NH residents’ exposure to artificial bright light during the daytime has not only improved the residents’ sleep [44–46], but also has improved their cognitive functioning and reduced their depressive symptoms [47]. Thus, steps towards targeted light exposure in sync with the typical sleep-wake cycle (eg, mandated time in well-lit rooms during the day and during meals) for NH residents, particularly those with CRSWDs, could prove to be beneficial across several social, behavioral and neurocognitive domains. Lastly, NH residents exposed to at least 30 minutes of outdoor daylight and at least 3 occasions of low intensity physical activities for 10 to 15 minutes daily can potentially improve sleep-wake patterns [48]. Thus, it may be beneficial to have an intervention that couples bright light exposure and physical activity in the NH setting.
Pharmacological interventions can also be implemented to improve older residents’ symptoms. However, the medications prescribed should be used with caution and should not be used as part of a long-term treatment plan. Melatonin is a commonly used herbal supplement that can assist advancing the timing of the circadian rhythms in the evening but can delay the circadian rhythms in the morning [49]. Several brands of this herbal supplement can be purchased over-the-counter and are not regulated by the FDA. Since the amount of melatonin used in the herbal supplement varies by brand, caution should be used when selecting a brand [50]. Two FDA-approved drugs (modafinil and armodafinil) are currently being used to reduce daytime sleepiness and improve vigilance amongst adults, but limited research has explored the effectiveness of these medication for older adults specifically suffering with CRSWDS [36,51,52]. Other stimulants (eg, caffeine, amphetamines, and nonamphetamine-derived medications) are also currently being used to reduce daytime sleepiness in patients with CRSWDS. Stimulant use, particularly caffeine consumption, has also been associated with better cognitive functioning in older adults [53]. However, stimulants should be taken with caution, particularly in older adults, because stimulant use has been associated with potentially serious and fatal health sequalae (eg, tachycardia, heart failure, irreversible heart damage and hypertension) [36,54].
Case 3 Outcome
The patient was moved to a room with a window. An alarm clock was set for 7:30 in the morning and he was taken to breakfast, where he sat at a table near a window. Any time he appeared to be sleepy, he was encouraged to go for a walk or engage in other activities so daytime napping opportunities were limited. His environment was assessed for safety and bedrails were utilized to prevent falls.
Case 4
A 75-year-old woman with a history of anxiety and depression moved into the NH 4 months ago after suffering a stroke. She now reports difficulty falling asleep for many years, which has significantly worsened since moving to the NH. Currently, she has been getting only 3 to 4 hour of sleep per night. She reports mild but increasing daytime sleepiness and does not fall asleep until 1:00 am despite getting into bed at 10:30 pm. She occasionally reports arthritic pain that interferes with her sleep. The NH staff has mentioned that she will occasionally cry for her family when she appears to be asleep.
Case 4 Reflection: Insomnia
According to the International Classification of Sleep Disorders (ICSD-3) [39], insomnia is characterized as “a repeated difficulty with sleep initiation, duration, consolidation, or quality that occurs despite adequate opportunity and circumstances for sleep, and results in some form of daytime impairment.” Among the sleep disorders, insomnia is one of the most common sleep issues observed in sleep clinics [34]. Older adults with insomnia often have comorbid physical (eg, pulmonary disease, arthritis, chronic pain, cancer diabetes, Parkinson’s disease) and mental illness (eg, depression, panic disorder) [55]. Medications (eg, stimulants, respiratory medications, chemotherapy, decongestants, hormones, or psychotropics) may cause and exacerbate insomnia symptoms [55].
Since insomnia is a clinical diagnosis, there is no specific diagnostic tool or gold standard test to identify individuals suffering with insomnia. Insomnia screening usually involves a clinical interview, in which a health provider, preferably trained in sleep, conducts a physical examination and collects an in-depth history of a patients’ sleep problems [56]. Insomnia screening tools may also include having a patient complete a sleep diary or questionnaire, such as the insomnia severity index (ISI) [57] or Pittsburgh Sleep Quality Index (PSQI) [58].
Cognitive behavioral therapy for insomnia (CBT-I) and/or pharmacological intervention are typically used to treat insomnia in older adults. CBT-I is a combination of cognitive (eg, changing dysfunctional sleep attitudes/beliefs) and behavioral treatment (eg, adhering to a regular sleep schedule) [59]. CBT-I or a combination of CBT-I and pharmacological intervention is recommended as more effective long-term approach to insomnia treatment compared to pharmacological intervention alone [55]. CBT-I involves altering older adults’ misconceptions of their sleep and implementing behavioral techniques to their everyday life (eg, routine sleep-wake schedule, relaxation therapy). Several FDA-approved medications are available to treat insomnia; however, many commonly used medications to treat insomnia in older adults (ie, antihistamines, antidepressants, anticonvulsants, and anti-psychotics) pose more risks than benefits to their health and well-being [35,60–62]. Some of the more recent hypnotics (egm zolpidem, exzopiclone, and ramelteon) on the market have been shown to be safer and more effective pharmacological options [55]. In 2014, the FDA approved the first in class orexin receptor antagonist medication (suvorexant) to treat insomnia [63]. Unlike other medications to treat insomnia, suvorexant, via the blockade of the orexin neurotransmitter, effectively inhibits orexin (one of neurotransmitters involved in the activation pathways of the arousal system), so sleep can easily be induced and maintained [64, 65]. Furthermore, preliminary studies suggest that this medication may be associated with less severe side effects (ie, habituation) than the other approved medications on the market [64, 65]. In fact, in a recent clinical series that included both young and older insomnia patients, the most common adverse reaction to suvorexant was drowsiness [66].
Case 4 Outcome
The patient was initiated on basic CBT-I therapy strategies which included stimulus control therapy [67]; implementation of a consistent bedtime and awakening routine; reducing the use of TV, smart phone, or other electronic leisure devices 1 hour before bedtime; refraining from caffeine after lunchtime; improving the sleep environment; and relaxation techniques.
Case 5
The patient is a 65-year-old man diagnosed with Parkinson’s disease several years ago. Recently, he has often has been experiencing what appears to be very violent and terrifying dreams. While asleep, he often screams and shouts for help. In addition, he occasionally will punch, kick, and/or thrash around in bed at night, which the NH staff has noted as a concern for his safety.
Case 5 Reflection: Parasomnias
Parasomnias represent frequent arousals during sleep or in the wake-to-sleep transition due to abnormal motor movements, behaviors (eg, shouting, flailing, and leaping from bed) and/or sensory experiences (eg, “dreamlike” hallucinations) [68]. Motor movements that occur for parasomnia can be disruptive for the individual and potentially dangerous for the individual and/or bed partner. There are 3 primary types of parasomnias based on the stage of sleep that the event occurs: non-REM (NREM), REM, and other parasomnias during transitions of sleep [68]. The most commonly observed parasomnia seen in older adults is the REM-associated parasomnia or REM sleep behavior disorder (RBD), which is characterized by experiencing vivid, sometimes violent, dreams typically involving fighting an intruder or an animal to protect a loved one [69]. For RBD, disruptive behaviors typically occur during REM sleep [69]. RBD has been associated with neurodegenerative disorders (Parkinson’s disease and Lewy body disease), neurologic disorders (eg, brain tumors and stroke), other primary disorders (narcolepsy and periodic limb movement disorder), and well as some medications (eg, antidepressants and β-blockers) [68]. There is limited knowledge on the prevalence of parasomnias in NH settings. One study, however, reported that 31% of older NH residents experience parasomnias [70]. Evaluation for parasomnias generally involve a clinical evaluation by a sleep specialist and overnight sleep study (ie, polysomnography at a sleep center if there is a concern for sleep apnea or RBD [71].
Medications are not typically first-line for parasomnia. Instead education about improving sleep practices, addressing other underlying sleep disorders, and securing a safe sleep environment are first recommended. Pharmacologic treatment, particularly the use of clonazepam, is commonly used to treat RBD [72]. However, this medication should be used with caution for older adults with a dementia diagnosis, gait disorders, and OSA because the common side effects include sedation, confusion, memory dysfunction, and early morning motor incoordination [68]. Several alternative medications have also been used to treat RBD. For example, medications commonly used to Parkinson disease symptoms, such Levodopa and dopamine agonists, have also been used to treat RBD [73]. Zopiclone, a nonbenzodiazepine hypnotic agent, has also been shown to be as effective as clonazepam, but with less potential side effects [74]. Melatonin, a nutritional supplement, has also been used as a treatment and appears to alleviate some of the RBD symptoms and has fewer side effects [68]. Since melatonin is not regulated by the FDA, it has been suggested that this treatment be used with caution in the older population [73].
Case 5 Outcome
The patient was evaluated with video synchronized in lab PSG. It confirmed REM sleep without evidence of the normal atonia that should be apparent during REM. These PSG findings in combination with repeated accounts of dream enactment established the diagnosis of RBD. Patient was treated with low-dose clonazepam and closely monitored for potential side effects of daytime sedation. Bedroom environment was also carefully reconfigured for safety to avoid potential risk of injury during a dream enactment episode.
Conclusion
Sleep disturbances remain an underappreciated and undertreated health issue in NH residents. Nursing homes can help facilitate optimal sleep health and day functioning by providing mandatory natural light outlets, physical exercise opportunities, and minimal allowable time residents can spend in their bed/bedroom outside of their routine sleep period. Educating NH providers and staff on sleep medicine may benefit residents, but workload and restricted resources may hinder this. Education via mobile and internet based educational platforms and resources (Mysleep101) may be helpful in addressing education barriers [75]. Convenient and cost-effective methods to deliver sleep medicine education to NH health care providers should be part of our ongoing efforts to improve the viability, vitality and quality of life of our aging citizens.
Corresponding author: Alyssa Gamaldo, PhD, Univ. of South Florida, 13301 Bruce B. Downs Blvd, MHC 1340, Tampa, FL 33612, [email protected].
Financial disclosures: None.
From the School of Aging Studies, University of South Florida, Tampa, FL (Dr. AA Gamaldo) and the Department of Neurology, Johns Hopkins Medicine, Baltimore, MD (Drs. Sloane, CE Gamaldo and Salas).
Abstract
- Objective: To provide guidance on identifying and treating sleep disturbances commonly encountered in older nursing home residents.
- Methods: Review of the literature in the context of 5 clinical cases.
- Results: Sleep disturbances continue to be a growing global epidemic, and public health initiatives have been aimed at improving sleep health across all ages. In older adults, sleep disturbances are often associated with the development and/or worsening of health conditions. Common sleep disturbances observed in older nursing home residents include obstructive sleep apnea, restless legs syndrome/Willis-Ekbom disease, circadian rhythm sleep-wake disorders, insomnia, and parasomnias. The symptoms and recommended treatment plans vary across the sleep disturbances. For many sleep disturbances, modification of residents’ daily activities and/or nursing home environment can be helpful.
- Conclusion: As the number of people residing in nursing homes increases, it is important for health care providers to be knowledgable about sleep disturbances in this population.
By 2030, almost 20% of the US population (approximately 72.1 million people) will be age 65 and older [1]. As many as 63% of older adults in the general population report sleep disturbances [2]. Specifically, older adults demonstrate difficulty with decreased total sleep duration, an increase in sleep fragmentation (ie, interruptions in nighttime sleep), and reduced total sleep time spent in rapid eye movement (REM) and slow wave sleep [3–5]. Poor sleep, either because of not getting enough sleep or having an undiagnosed and thus untreated sleep disorder, is associated with physical illness, impaired cognition, poor physical function, and mortality risk [6,7]. In fact, over 50% of individuals older than 65 years meet the diagnostic criteria for a sleep disorder, many of which are undiagnosed [6,7].
It is forecasted that we will see substantial increases in the rate of nursing home residence among the elderly [8]. The prevalence and severity of disturbed sleep is reportedly higher in NH residents [6,7]. Generally, NH residents tend to be on several medications for various medical disorders that may negatively impact sleep [7]. Reciprocally, sleep disruption may put NH residents at an increased risk for behavioral issues (eg, agitation) [9,10] as well as developing and/or exacerbating health conditions (eg, mood disorders, dementia, cardiovascular disease) [8]. Furthermore, NH residents exhibiting disturbed sleep, behavioral issues, and/or mood disorders are at an increased risk for being prescribed antipsychotic drugs [11], which are associated with adverse side effects and poorer quality of life [12]. Thus, the identification and management of sleep disturbances in the NH setting has become progressively more vital in efforts to optimize medical management of this population. This review identifies common sleep disturbances frequently underdiagnosed and undertreated among residents of NH facilities.
Case 1
A 73-year-old woman with a history of type 2 diabetes mellitus reports poor sleep quality with frequent awakenings during the night and excessive daytime sleepiness. She states that she can fall asleep within 5 minutes, but often is awoken throughout the night with a sensation of breathlessness. She has snored for many years, but the nursing staff at her NH facility has recently commented that her snoring has gone from intermittent to constant. She cannot remember the last time she has had restful sleep. She consumes 3 to 4 cups of caffeinated beverages daily to counter her sleepiness. She denies smoking or illicit drug or alcohol use. Her review of systems was notable for a 30-lb weight gain over the last year, and she reports increasing fatigue, irritability, and memory and concentration issues. Her current medication list includes metformin and amlodipine. Her examination is remarkable for a BMI of 31, large neck circumference (> 16), tonsillar enlargement, a crowded oropharynx, micrognathia, lungs clear to auscultation bilaterally, heart sounds of normal S1 and S2, and legs with trace pitting edema.
Case 1 Reflection: Sleep-Disordered Breathing
Although previous studies have observed high rates (60%–90%) of SDB in NH settings [16,17], one study observed that only 0.5% of nursing home residents carried a diagnosis with SDB, suggesting that SDB is being grossly underappreciated amongst NH residents over the age of 65 [18]. In order to evaluate for SBD, routine annual physical exams or medical chart reviews can elicit the risk factors for sleep apnea (eg, obesity [per BMI], male sex, postmenopausal women, family history of sleep apnea) as well as common comorbidities (eg, hypertension, coronary artery disease, and diabetes). Formal evaluation consists of a sleep evaluation with a sleep specialist and polysomnography (PSG; sleep study) that can be performed in the sleep center or at home depending on the patient’s history and other medical issues.
Case 1 Outcome
Case 2
An 85-year-old man with history of Alzheimer’s disease, major depression and arthritis, reports insomnia and “tingling in my legs” at bedtime. The patient cannot identify when the symptoms started but reports that his legs often jerk during sleep. He consumes a cup of coffee daily and has a previous 20 pack-year smoking history (he quit 40 years ago). On review of systems, he endorses fatigue. His current medication list includes fluoxetine, donepezil hydrochloride, ibuprofen as needed for arthritic pain, and a multivitamin. His examination was unremarkable, with a BMI of 26, neck circumference < 16, no tonsillar enlargement, normal (noncrowded) oropharynx, lungs clear to auscultation bilaterally, heart sounds demonstrating a normal S1 and S2, and legs without edema.
Case 2 Reflection: Restless Legs Syndrome/Willis-Ekbom Disease
Restless legs syndrome (RLS) also known as Willis-Ekbom disease, affects approximately 10 million adults in the United States alone [22]. RLS is a sensorimotor disorder that must satisfy the following 5 primary diagnostic criteria: (1) urge to move the legs with or without dysesthesias; (2) onset or exacerbation with rest or inactivity; (3) relief with movement; (4) symptoms are worse in the evening or at night (circadian component); (5) symptoms cannot be solely accounted for as consequence of another medical or behavioral condition. Other supporting clinical features can alert a clinician to the likelihood of a RLS diagnosis; these include positive family history, response to dopaminergic therapy, lack of profound daytime sleepiness, and presence of periodic limb movements during sleep (PLMS) [23–26]. In younger individuals, the symptoms present insidiously whereas older adults (> 50 years of age) will usually present with sudden onset [27].
Not only do patients lack the restorative sleep needed to ward off fatigue and restfulness, but patients also demonstrate higher rates of comorbidities (eg, anxiety, hypertension, depression) as well as large economic burden secondary to absenteeism and decreased on-the-job effectiveness [28,29]. As a results, patients with RLS experience significant reductions in quality of life related to this sensorimotor disorder [28].
No confirmatory laboratory test exists to diagnose RLS; however, patients suspected of having RLS should be evaluated with a basic metabolic panel, iron studies, and a thorough neurologic examination, as iron deficiency, kidney failure, uremia and peripheral neuropathy can lead to secondary RLS [30,31]. Evidence shows that RLS is common in NH residents [32] and may account for problematic behaviors, such as late night pacing [7]. Forty-five percent of community dwelling individuals over 65 years old exhibit a PLMS index (leg kicks per hour) of greater than 5 [33]. PLMS, while not a disorder in and of itself, can serve as a marker for potential disease. PLMS are characterized by intermittent episodes of stereotyped leg movements. PLMS typically do not awaken the patient from sleep and therefore do not contribute to insomnia or daytime sleepiness, representing a key clinical difference from RLS. It is important to note that PLMS are nonspecific and may be common in older adults that do not meet the diagnostic criteria for RLS.
Treatment of RLS is based on the frequency of symptoms and the level of functional impairment caused by the syndrome. RLS treatment recommendations should always espouse nonpharmacological interventions that include improving sleep practices, engagement in daily physical activity, targeted placement of sedentary activity in the morning when symptoms are less prominent, and concerted efforts to avoid the use of RLS-exacerbating medications (eg, selective serotonin reuptake inhibitor (SSRIs), neuroleptic agents, antihistamines) [28]. If there is an underlying condition contributing to RLS, such as metabolic disturbance or iron deficiency, then these conditions should be corrected before initiating RLS medications. Several medications are FDA-approved for treatment of RLS, including dopamine agonists (eg, ropinirole, rotigotine, pramipexole), dopamine precursor (eg, levodopa), glutamate-related (eg, gabapentin), benzodiazepines (eg, temazepam, clonazepam). Augmentation, the worsening of RLS symptoms, can occur in patients taking dopamine agonists. If this occurs, dopamine agents should be discontinued or switched to other agents (such as a long-acting dopamine agonist, gabapentin encarbil, as well as non-FDA approved therapies such as opioids). However, it is important to note that weaning off dopamine agents may result in mild but in most cases moderate and/or severe withdrawal from the medication, so counseling and close monitoring should be done.
Case 2 Outcome
Given the patient’s history of dementia, opioids, benzodiazepines and other delirium-inducing medications should be avoided. His antidepressive regimen, fluoxetine, should be re-evaluated as these medications have been associated with RLS exacerbation. In addition to SSRIs, medications associated with RLS are MAO inhibitors (selegeline, phenelzine), antipsychotics (risperdone, olanzapine), tricyclic antidepressants (mirtazapine), antihistamines (diphenhydramine, cimetidine), calcium channel blockers (verapamil, nifedipine, diltiazem), and phenytoin [34,35]. His treatment began with behavioral, nonpharmacological management, and blood testing for iron studies. His low iron level prompted initiation of oral supplementation, and he was asked to follow up in 3 months for reevaluation and possible initiation of low-dose dopamine agonists.
Case 3
A 73-year-old man with dementia is found to have very irregular sleep wake patterns with a variable bedtime and awakening time, often missing breakfast. He is found dozing off often during the day, particularly during times of inactivity. He has frequent awakenings during the night often calling for the staff to guide him back to bed. He has had some falls secondary to walking around his room. He has been prescribed various hypnotics without much benefit and instead, has suffered from some confusion while on these medications. His room is very dark and has no windows.
Case 3 Reflection: Circadian Rhythm Sleep-Wake Disorders
Circadian rhythm sleep-wake disorders (CRSWDs) are characterized by an individual’s natural propensity to want to go to sleep and be awake during a period that is undesirable personally and/or socially [36]. CRSWDs can be a result of the desynchronization of the 2 sleep processes: (1) homeostatic drive (regulates sleep intensity) and (2) circadian rhythm (maintains daytime alertness); [36]. CRSWDs can also be due to an individual’s naturally occurring sleep drives becoming misaligned with their social/personal sleep-wake demands (eg, employment schedule and socializing opportunities with family/friends). With increasing age, the circadian rhythm becomes less adept at functioning in a desynchronized pattern [7], which can result in daytime sleepiness and night time sleep fragmentation [7,37]. CRSWDs are highly prevalent in individuals with dementia [7,36]. As dementia progresses, the ability to maintain a balance of the 2 sleep process becomes more impaired [7]. As a result, individuals with dementia, particularly Alzheimer’s disease, are likely to experience agitation, irritability, and/or confusion during the evening and night, a behavioral problem referred to as “sundowning” [38].
There are several types of CRSWDs, including delayed sleep-phase syndrome, advanced sleep-phase syndrome, irregular sleep-wake disorder, non–24-hour sleep-wake disorder, shift work sleep disorder, and jet lag sleep disorder. However, the most common type of CRSWDs observed in older adults is advanced sleep-phase syndrome [39]. Due to excessive sleepiness in the early evening, affected individuals may report a need to shift to earlier and earlier bedtimes (~6 to 7 pm) and wake times (~3 to 4 am) [36]. For older affected adults, this can cause distress and frustration, particularly if their sleep phase prevents them from participating in evening activities (eg, socializing with family/friends) [36].
In the assessment of patients with suspected CRSWDs, sleep diaries (self-reported or caregiver) daily account of sleep and wake times over at least 1 week) and actigraphy (wrist-worn accelerometer designed to measure activity and inactivity at night) can be used, particularly in older adults with dementia [40,41].
CRSWD treatment may include behavioral modifications and/or pharmacological intervention. Behavioral modifications can consist of chronotherapy, relaxation training, and/or bright light therapy. Chronotherapy involves making gradual shifts in an individual’s sleep time to meet his/her desired sleep schedule. Relaxation training involves implementing behaviors/activities that reduce tension and enhance the smooth transition into sleep. Bright light therapy involves exposure to an appropriate intensity and duration of light, which is an important environmental cue to help the synchrony of the sleep-wake cycle [7]. Previous studies have observed that NH residents are exposed to a restricted amount of bright light during the daytime [42,43], but higher levels of artificial light at night (eg, hallway lighting) [7]. NH residents’ exposure to artificial bright light during the daytime has not only improved the residents’ sleep [44–46], but also has improved their cognitive functioning and reduced their depressive symptoms [47]. Thus, steps towards targeted light exposure in sync with the typical sleep-wake cycle (eg, mandated time in well-lit rooms during the day and during meals) for NH residents, particularly those with CRSWDs, could prove to be beneficial across several social, behavioral and neurocognitive domains. Lastly, NH residents exposed to at least 30 minutes of outdoor daylight and at least 3 occasions of low intensity physical activities for 10 to 15 minutes daily can potentially improve sleep-wake patterns [48]. Thus, it may be beneficial to have an intervention that couples bright light exposure and physical activity in the NH setting.
Pharmacological interventions can also be implemented to improve older residents’ symptoms. However, the medications prescribed should be used with caution and should not be used as part of a long-term treatment plan. Melatonin is a commonly used herbal supplement that can assist advancing the timing of the circadian rhythms in the evening but can delay the circadian rhythms in the morning [49]. Several brands of this herbal supplement can be purchased over-the-counter and are not regulated by the FDA. Since the amount of melatonin used in the herbal supplement varies by brand, caution should be used when selecting a brand [50]. Two FDA-approved drugs (modafinil and armodafinil) are currently being used to reduce daytime sleepiness and improve vigilance amongst adults, but limited research has explored the effectiveness of these medication for older adults specifically suffering with CRSWDS [36,51,52]. Other stimulants (eg, caffeine, amphetamines, and nonamphetamine-derived medications) are also currently being used to reduce daytime sleepiness in patients with CRSWDS. Stimulant use, particularly caffeine consumption, has also been associated with better cognitive functioning in older adults [53]. However, stimulants should be taken with caution, particularly in older adults, because stimulant use has been associated with potentially serious and fatal health sequalae (eg, tachycardia, heart failure, irreversible heart damage and hypertension) [36,54].
Case 3 Outcome
The patient was moved to a room with a window. An alarm clock was set for 7:30 in the morning and he was taken to breakfast, where he sat at a table near a window. Any time he appeared to be sleepy, he was encouraged to go for a walk or engage in other activities so daytime napping opportunities were limited. His environment was assessed for safety and bedrails were utilized to prevent falls.
Case 4
A 75-year-old woman with a history of anxiety and depression moved into the NH 4 months ago after suffering a stroke. She now reports difficulty falling asleep for many years, which has significantly worsened since moving to the NH. Currently, she has been getting only 3 to 4 hour of sleep per night. She reports mild but increasing daytime sleepiness and does not fall asleep until 1:00 am despite getting into bed at 10:30 pm. She occasionally reports arthritic pain that interferes with her sleep. The NH staff has mentioned that she will occasionally cry for her family when she appears to be asleep.
Case 4 Reflection: Insomnia
According to the International Classification of Sleep Disorders (ICSD-3) [39], insomnia is characterized as “a repeated difficulty with sleep initiation, duration, consolidation, or quality that occurs despite adequate opportunity and circumstances for sleep, and results in some form of daytime impairment.” Among the sleep disorders, insomnia is one of the most common sleep issues observed in sleep clinics [34]. Older adults with insomnia often have comorbid physical (eg, pulmonary disease, arthritis, chronic pain, cancer diabetes, Parkinson’s disease) and mental illness (eg, depression, panic disorder) [55]. Medications (eg, stimulants, respiratory medications, chemotherapy, decongestants, hormones, or psychotropics) may cause and exacerbate insomnia symptoms [55].
Since insomnia is a clinical diagnosis, there is no specific diagnostic tool or gold standard test to identify individuals suffering with insomnia. Insomnia screening usually involves a clinical interview, in which a health provider, preferably trained in sleep, conducts a physical examination and collects an in-depth history of a patients’ sleep problems [56]. Insomnia screening tools may also include having a patient complete a sleep diary or questionnaire, such as the insomnia severity index (ISI) [57] or Pittsburgh Sleep Quality Index (PSQI) [58].
Cognitive behavioral therapy for insomnia (CBT-I) and/or pharmacological intervention are typically used to treat insomnia in older adults. CBT-I is a combination of cognitive (eg, changing dysfunctional sleep attitudes/beliefs) and behavioral treatment (eg, adhering to a regular sleep schedule) [59]. CBT-I or a combination of CBT-I and pharmacological intervention is recommended as more effective long-term approach to insomnia treatment compared to pharmacological intervention alone [55]. CBT-I involves altering older adults’ misconceptions of their sleep and implementing behavioral techniques to their everyday life (eg, routine sleep-wake schedule, relaxation therapy). Several FDA-approved medications are available to treat insomnia; however, many commonly used medications to treat insomnia in older adults (ie, antihistamines, antidepressants, anticonvulsants, and anti-psychotics) pose more risks than benefits to their health and well-being [35,60–62]. Some of the more recent hypnotics (egm zolpidem, exzopiclone, and ramelteon) on the market have been shown to be safer and more effective pharmacological options [55]. In 2014, the FDA approved the first in class orexin receptor antagonist medication (suvorexant) to treat insomnia [63]. Unlike other medications to treat insomnia, suvorexant, via the blockade of the orexin neurotransmitter, effectively inhibits orexin (one of neurotransmitters involved in the activation pathways of the arousal system), so sleep can easily be induced and maintained [64, 65]. Furthermore, preliminary studies suggest that this medication may be associated with less severe side effects (ie, habituation) than the other approved medications on the market [64, 65]. In fact, in a recent clinical series that included both young and older insomnia patients, the most common adverse reaction to suvorexant was drowsiness [66].
Case 4 Outcome
The patient was initiated on basic CBT-I therapy strategies which included stimulus control therapy [67]; implementation of a consistent bedtime and awakening routine; reducing the use of TV, smart phone, or other electronic leisure devices 1 hour before bedtime; refraining from caffeine after lunchtime; improving the sleep environment; and relaxation techniques.
Case 5
The patient is a 65-year-old man diagnosed with Parkinson’s disease several years ago. Recently, he has often has been experiencing what appears to be very violent and terrifying dreams. While asleep, he often screams and shouts for help. In addition, he occasionally will punch, kick, and/or thrash around in bed at night, which the NH staff has noted as a concern for his safety.
Case 5 Reflection: Parasomnias
Parasomnias represent frequent arousals during sleep or in the wake-to-sleep transition due to abnormal motor movements, behaviors (eg, shouting, flailing, and leaping from bed) and/or sensory experiences (eg, “dreamlike” hallucinations) [68]. Motor movements that occur for parasomnia can be disruptive for the individual and potentially dangerous for the individual and/or bed partner. There are 3 primary types of parasomnias based on the stage of sleep that the event occurs: non-REM (NREM), REM, and other parasomnias during transitions of sleep [68]. The most commonly observed parasomnia seen in older adults is the REM-associated parasomnia or REM sleep behavior disorder (RBD), which is characterized by experiencing vivid, sometimes violent, dreams typically involving fighting an intruder or an animal to protect a loved one [69]. For RBD, disruptive behaviors typically occur during REM sleep [69]. RBD has been associated with neurodegenerative disorders (Parkinson’s disease and Lewy body disease), neurologic disorders (eg, brain tumors and stroke), other primary disorders (narcolepsy and periodic limb movement disorder), and well as some medications (eg, antidepressants and β-blockers) [68]. There is limited knowledge on the prevalence of parasomnias in NH settings. One study, however, reported that 31% of older NH residents experience parasomnias [70]. Evaluation for parasomnias generally involve a clinical evaluation by a sleep specialist and overnight sleep study (ie, polysomnography at a sleep center if there is a concern for sleep apnea or RBD [71].
Medications are not typically first-line for parasomnia. Instead education about improving sleep practices, addressing other underlying sleep disorders, and securing a safe sleep environment are first recommended. Pharmacologic treatment, particularly the use of clonazepam, is commonly used to treat RBD [72]. However, this medication should be used with caution for older adults with a dementia diagnosis, gait disorders, and OSA because the common side effects include sedation, confusion, memory dysfunction, and early morning motor incoordination [68]. Several alternative medications have also been used to treat RBD. For example, medications commonly used to Parkinson disease symptoms, such Levodopa and dopamine agonists, have also been used to treat RBD [73]. Zopiclone, a nonbenzodiazepine hypnotic agent, has also been shown to be as effective as clonazepam, but with less potential side effects [74]. Melatonin, a nutritional supplement, has also been used as a treatment and appears to alleviate some of the RBD symptoms and has fewer side effects [68]. Since melatonin is not regulated by the FDA, it has been suggested that this treatment be used with caution in the older population [73].
Case 5 Outcome
The patient was evaluated with video synchronized in lab PSG. It confirmed REM sleep without evidence of the normal atonia that should be apparent during REM. These PSG findings in combination with repeated accounts of dream enactment established the diagnosis of RBD. Patient was treated with low-dose clonazepam and closely monitored for potential side effects of daytime sedation. Bedroom environment was also carefully reconfigured for safety to avoid potential risk of injury during a dream enactment episode.
Conclusion
Sleep disturbances remain an underappreciated and undertreated health issue in NH residents. Nursing homes can help facilitate optimal sleep health and day functioning by providing mandatory natural light outlets, physical exercise opportunities, and minimal allowable time residents can spend in their bed/bedroom outside of their routine sleep period. Educating NH providers and staff on sleep medicine may benefit residents, but workload and restricted resources may hinder this. Education via mobile and internet based educational platforms and resources (Mysleep101) may be helpful in addressing education barriers [75]. Convenient and cost-effective methods to deliver sleep medicine education to NH health care providers should be part of our ongoing efforts to improve the viability, vitality and quality of life of our aging citizens.
Corresponding author: Alyssa Gamaldo, PhD, Univ. of South Florida, 13301 Bruce B. Downs Blvd, MHC 1340, Tampa, FL 33612, [email protected].
Financial disclosures: None.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2012: Key indicators of well-being. Washington, DC: U.S. Gov. Printing Office; 2012.
2. Almeida OP, Pfaff JJ. Sleep complaints among older general practice patients: association with depression. Br J Gen Pract 2005;55:864–866.
3. Wolkove N, Elkholy O, Baltzan M, et al. Sleep and aging: 1. Sleep disorders commonly found in older people. CMAJ 2007;176:1299–304.
4. Pace-Schott EF, Spencer RM. Age-related changes in the cognitive function of sleep. Prog Brain Res 2011;191:75–89.
5. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 2004;27:1255–73.
6. Jacobs D, Ancoli-Israel S, Parker L, et al. Twenty-four-hour sleep-wake patterns in a nursing home population. Psychol Aging 1989;4:352–6.
7. Neikrug AB, Ancoli-Israel S. Sleep disturbances in nursing homes. J Nutr Health Aging 2010;14:207–11.
8. Lakdawalla D, Goldman DP, Bhattacharya J, et al. Forecasting the nursing home population. Medical Care 2003;41:8-20.
9. Alessi CA, Yoon EJ, Schnelle JF, et al. A randomized trial of a combined physical activity and environmental intervention in nursing home residents: do sleep and agitation improve? J Am Geriatr Soc 1999;47:784–91.
10. Cohen-Mansfield J, Marx MS. The relationship between sleep disturbances and agitation in a nursing home. J Aging Health 1990;2:153–65.
11. Rolland Y, Andrieu S, Crochard A, et al. Psychotropic drug consumption at admission and discharge of nursing home residents. J Am Med Dir Assoc 2012;13:407 e407–412.
12. Salzman C. Antipsychotics in nursing homes. J Clin Psychopharmacol 2013;33:1–2.
13. Fleetham J, Ayas N, Bradley D, et al. Canadian Thoracic Society guidelines: diagnosis and treatment of sleep disordered breathing in adults. Can Respir J 2006;13:387–92.
14. International classification of sleep disorders: Chicago: American Academy of Sleep Medicine; 2005.
15. Gay PC. Complex sleep apnea: it really is a disease. J Clin Sleep Med 2008;4:403–5.
16. Ancoli-Israel S. Epidemiology of sleep disorders. In: Roth T, Roehrs TA, editors. Clinics in geriatric medicine. Philadelphia: WB Saunders; 1989:347–62.
17. Gehrman PR, Martin JL, Shochat T, et al. Sleep-disordered breathing and agitation in institutionalized adults with Alzheimer disease. Am J Geriatr Psychiatry 2003;11:426–33.
18. Resnick HE, Phillips B. Documentation of sleep apnea in nursing homes: United States 2004. J Am Med Dir Assoc 2008;9:260–4.
19. Park JG, Ramar K, Olson EJ. Updates on definition, consequences, and management of obstructive sleep apnea. Mayo Clin Proc 2011;86:549–54.
20. Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034–41.
21. McMillan A, Bratton DJ, Faria R, et al. Continuous positive airway pressure in older people with obstructive sleep apnoea syndrome (PREDICT): a 12-month, multicentre, randomised trial. Lancet Resp med 2014;2:804–12.
22. Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 2005;165:1286–92.
23. Allen RP, La Buda MC, Becker P, et al. Family history study of the restless legs syndrome. Sleep Med 2002;3 Suppl:S3–7.
24. Allen RP. Restless legs syndrome/Willis Ekbom disease: evaluation and treatment. Int Rev Psychiatry 2014;26:248–62.
25. Hening WA, Allen RP, Earley CJ, et al. An update on the dopaminergic treatment of restless legs syndrome and periodic limb movement disorder. Sleep 2004;27:560–83.
26. Gamaldo C, Benbrook AR, Allen RP, et al. Evaluating daytime alertness in individuals with Restless Legs Syndrome (RLS) compared to sleep restricted controls. Sleep Med 2009;10:134–8.
27. Allen RP, Earley CJ. Defining the phenotype of the restless legs syndrome (RLS) using age-of-symptom-onset. Sleep Med 2000;1:11–9.
28. Salas RE, Kwan AB. The real burden of restless legs syndrome: clinical and economic outcomes. Am J Manag Care 2012;18(9 Suppl):S207–212.
29. Silber MH, Ehrenberg BL, Allen RP, et al. An algorithm for the management of restless legs syndrome. Mayo Clin Proc 2004;79:916–22.
30. Hattan E, Chalk C, Postuma RB. Is there a higher risk of restless legs syndrome in peripheral neuropathy? Neurology 2009;72:955–60.
31. Winkelman JW, Chertow GM, Lazarus JM. Restless legs syndrome in end-stage renal disease. Am J Kidney Dis 1996;28:372–8.
32. Hornyak M, Trenkwalder C. Restless legs syndrome and periodic limb movement disorder in the elderly. J Psychosom Res 2004;56:543–8.
33. Ancoli-Israel S, Kripke DF, Klauber MR, et al. Periodic limb movements in sleep in community-dwelling elderly. Sleep 1991;14:496–500.
34. Ahmed QA. Effects of common medications used for sleep disorders. Crit Care Clin 2008;24:493–515, vi.
35. Rye DB, Trotti LM. Restless legs syndrome and periodic leg movements of sleep. Neurol clin 2012;30:1137–66.
36. Gamaldo CE, Chung Y, Kang YM, et al. Tick-tock-tick-tock: the impact of circadian rhythm disorders on cardiovascular health and wellness. J Am Soc Hypertens 2014;8:921–9.
37. Myers BL, Badia P. Changes in circadian rhythms and sleep quality with aging: mechanisms and interventions. Neurosci Biobehav Rev 1995;19:553–71.
38. National Institute on Aging. Alzheimer’s caregiving tips: sundowning. June 2013.
39. International classification of sleep disorders. 3rd ed. Darien, IL: AASD; 2014.
40. Barion A, Zee PC. A clinical approach to circadian rhythm sleep disorders. Sleep Med 2007;8:566–77.
41. Morgenthaler TI, Lee-Chiong T, Alessi C, et al. Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep 2007;30:1445–59.
42. Shochat T, Martin J, Marler M, et al. Illumination levels in nursing home patients: effects on sleep and activity rhythms. J Sleep Res 2000;9:373–9.
43. Ancoli-Israel S, Klauber MR, Jones DW, et al. Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 1997;20:18–23.
44. Satlin A, Volicer L, Ross V, et al. Bright light treatment of behavioral and sleep disturbances in patients with Alzheimer’s disease. Am J Psychiatry 1992;149:1028–32.
45. Koyama E, Matsubara H, Nakano T. Bright light treatment for sleep-wake disturbances in aged individuals with dementia. Psychiatry Clin Neurosci 1999;53:227–9.
46. Burns A, Allen H, Tomenson B, et al. Bright light therapy for agitation in dementia: a randomized controlled trial. Int Psychogeriatr 2009;21:711–21.
47. Riemersma-van der Lek RF, Swaab DF, Twisk J, et al. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA 2008;299:2642–55.
48. Martin JL, Marler MR, Harker JO, et al. A multicomponent nonpharmacological intervention improves activity rhythms among nursing home residents with disrupted sleep/wake patterns. J Gerontol Series A Biol Sci Med Sci 2007;62:67–72.
49. Lewy AJ, Ahmed S, Jackson JM, et al. Melatonin shifts human circadian rhythms according to a phase-response curve. Chronobiol Int 1992;9:380–92.
50. Williamson BL, Tomlinson AJ, Mishra PK, et al. Structural characterization of contaminants found in commercial preparations of melatonin: similarities to case-related compounds from L-tryptophan associated with eosinophilia-myalgia syndrome. Chem Res Toxicol 1998;11:234-40.
51. Czeisler CA, Walsh JK, Roth T, et al. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med 2005;353:476-86.
52. Howard R, Roth T, Drake CL. The effects of armodafinil on objective sleepiness and performance in a shift work disorder sample unselected for objective sleepiness. J Clin Psychopharmacol 2014;34:369–73.
53. Beydoun MA, Gamaldo AA, Beydoun HA, et al. Caffeine and alcohol intakes and overall nutrient adequacy are associated with longitudinal cognitive performance among U.S. adults. J Nutr 2014;144:890–901.
54. Pentel P. Toxicity of over-the-counter stimulants. JAMA 1984;252:1898–903.
55. Neikrug AB, Ancoli-Israel S. Sleep disorders in the older adult - a mini-review. Gerontology 2010;56:181–9.
56. Chesson A Jr, Hartse K, Anderson WM, et al. Practice parameters for the evaluation of chronic insomnia. An American Academy of Sleep Medicine report. Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep 2000;23:237–41.
57. Bastien CH, Vallieres A, Morin CM. Validation of the insomnia severity index as an outcome measure for insomnia research. Sleep Med 2001;2:297–307.
58. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
59. Trauer JM, Qian MY, Doyle JS, et al. Cognitive behavioral therapy for chronic insomnia: a systematic review and meta-analysis. Ann Intern Med 2015;163:191–204.
60. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults, June 13-15, 2005. Sleep 2005;28:1049–57.
61. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med 2008;4:487–504.
62. Bain KT. Management of chronic insomnia in elderly persons. Am J Geriatr Pharmacother 2006;4:168–92.
63. FDA approves new type of sleep drug, Belsomra. 13 Aug 2014. Available at fda.gov.
64. Dubey AK, Handu SS, Mediratta PK. Suvorexant: The first orexin receptor antagonist to treat insomnia. J Pharmacol Pharmacother 2015;6:118–21.
65. Patel KV, Aspesi AV, Evoy KE. Suvorexant: a dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann Pharmacother 2015;49:477–83.
66. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:461–71.
67. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. Case studies in insomnia. New York: Springer; 1991:19–28.
68. Gulyani S, Salas RE, Gamaldo CE. Sleep medicine pharmacotherapeutics overview: today, tomorrow, and the future (part 2: hypersomnia, parasomnia, and movement disorders). Chest 2013;143:242–51.
69. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11):2770–88.
70. Middelkoop HA, Kerkhof GA, Smilde-van den Doel DA, et al. Sleep and ageing: the effect of institutionalization on subjective and objective characteristics of sleep. Age Ageing 1994;23:411–7.
71. Tinuper P, Provini F, Bisulli F, et al. Movement disorders in sleep: guidelines for differentiating epileptic from non-epileptic motor phenomena arising from sleep. Sleep Med Rev 2007;11:255–67.
72. Aurora RN, Zak RS, Maganti RK, et al. Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med 2010;6:85–95.
73. Bloom HG, Ahmed I, Alessi CA, et al. Evidence-based recommendations for the assessment and management of sleep disorders in older persons. J Am Geriatr Soc 2009;57:761–89.
74. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
75. Doshi A, Gamaldo C, Kalloo A, et al. Implementing a clinical iPad application to detect sleep disorders is feasible across multiple non-sleep outpatient clinics (P7. 321). Neurology 2015;84(14 Suppl):P7.321.
1. Federal Interagency Forum on Aging-Related Statistics. Older Americans 2012: Key indicators of well-being. Washington, DC: U.S. Gov. Printing Office; 2012.
2. Almeida OP, Pfaff JJ. Sleep complaints among older general practice patients: association with depression. Br J Gen Pract 2005;55:864–866.
3. Wolkove N, Elkholy O, Baltzan M, et al. Sleep and aging: 1. Sleep disorders commonly found in older people. CMAJ 2007;176:1299–304.
4. Pace-Schott EF, Spencer RM. Age-related changes in the cognitive function of sleep. Prog Brain Res 2011;191:75–89.
5. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 2004;27:1255–73.
6. Jacobs D, Ancoli-Israel S, Parker L, et al. Twenty-four-hour sleep-wake patterns in a nursing home population. Psychol Aging 1989;4:352–6.
7. Neikrug AB, Ancoli-Israel S. Sleep disturbances in nursing homes. J Nutr Health Aging 2010;14:207–11.
8. Lakdawalla D, Goldman DP, Bhattacharya J, et al. Forecasting the nursing home population. Medical Care 2003;41:8-20.
9. Alessi CA, Yoon EJ, Schnelle JF, et al. A randomized trial of a combined physical activity and environmental intervention in nursing home residents: do sleep and agitation improve? J Am Geriatr Soc 1999;47:784–91.
10. Cohen-Mansfield J, Marx MS. The relationship between sleep disturbances and agitation in a nursing home. J Aging Health 1990;2:153–65.
11. Rolland Y, Andrieu S, Crochard A, et al. Psychotropic drug consumption at admission and discharge of nursing home residents. J Am Med Dir Assoc 2012;13:407 e407–412.
12. Salzman C. Antipsychotics in nursing homes. J Clin Psychopharmacol 2013;33:1–2.
13. Fleetham J, Ayas N, Bradley D, et al. Canadian Thoracic Society guidelines: diagnosis and treatment of sleep disordered breathing in adults. Can Respir J 2006;13:387–92.
14. International classification of sleep disorders: Chicago: American Academy of Sleep Medicine; 2005.
15. Gay PC. Complex sleep apnea: it really is a disease. J Clin Sleep Med 2008;4:403–5.
16. Ancoli-Israel S. Epidemiology of sleep disorders. In: Roth T, Roehrs TA, editors. Clinics in geriatric medicine. Philadelphia: WB Saunders; 1989:347–62.
17. Gehrman PR, Martin JL, Shochat T, et al. Sleep-disordered breathing and agitation in institutionalized adults with Alzheimer disease. Am J Geriatr Psychiatry 2003;11:426–33.
18. Resnick HE, Phillips B. Documentation of sleep apnea in nursing homes: United States 2004. J Am Med Dir Assoc 2008;9:260–4.
19. Park JG, Ramar K, Olson EJ. Updates on definition, consequences, and management of obstructive sleep apnea. Mayo Clin Proc 2011;86:549–54.
20. Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034–41.
21. McMillan A, Bratton DJ, Faria R, et al. Continuous positive airway pressure in older people with obstructive sleep apnoea syndrome (PREDICT): a 12-month, multicentre, randomised trial. Lancet Resp med 2014;2:804–12.
22. Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 2005;165:1286–92.
23. Allen RP, La Buda MC, Becker P, et al. Family history study of the restless legs syndrome. Sleep Med 2002;3 Suppl:S3–7.
24. Allen RP. Restless legs syndrome/Willis Ekbom disease: evaluation and treatment. Int Rev Psychiatry 2014;26:248–62.
25. Hening WA, Allen RP, Earley CJ, et al. An update on the dopaminergic treatment of restless legs syndrome and periodic limb movement disorder. Sleep 2004;27:560–83.
26. Gamaldo C, Benbrook AR, Allen RP, et al. Evaluating daytime alertness in individuals with Restless Legs Syndrome (RLS) compared to sleep restricted controls. Sleep Med 2009;10:134–8.
27. Allen RP, Earley CJ. Defining the phenotype of the restless legs syndrome (RLS) using age-of-symptom-onset. Sleep Med 2000;1:11–9.
28. Salas RE, Kwan AB. The real burden of restless legs syndrome: clinical and economic outcomes. Am J Manag Care 2012;18(9 Suppl):S207–212.
29. Silber MH, Ehrenberg BL, Allen RP, et al. An algorithm for the management of restless legs syndrome. Mayo Clin Proc 2004;79:916–22.
30. Hattan E, Chalk C, Postuma RB. Is there a higher risk of restless legs syndrome in peripheral neuropathy? Neurology 2009;72:955–60.
31. Winkelman JW, Chertow GM, Lazarus JM. Restless legs syndrome in end-stage renal disease. Am J Kidney Dis 1996;28:372–8.
32. Hornyak M, Trenkwalder C. Restless legs syndrome and periodic limb movement disorder in the elderly. J Psychosom Res 2004;56:543–8.
33. Ancoli-Israel S, Kripke DF, Klauber MR, et al. Periodic limb movements in sleep in community-dwelling elderly. Sleep 1991;14:496–500.
34. Ahmed QA. Effects of common medications used for sleep disorders. Crit Care Clin 2008;24:493–515, vi.
35. Rye DB, Trotti LM. Restless legs syndrome and periodic leg movements of sleep. Neurol clin 2012;30:1137–66.
36. Gamaldo CE, Chung Y, Kang YM, et al. Tick-tock-tick-tock: the impact of circadian rhythm disorders on cardiovascular health and wellness. J Am Soc Hypertens 2014;8:921–9.
37. Myers BL, Badia P. Changes in circadian rhythms and sleep quality with aging: mechanisms and interventions. Neurosci Biobehav Rev 1995;19:553–71.
38. National Institute on Aging. Alzheimer’s caregiving tips: sundowning. June 2013.
39. International classification of sleep disorders. 3rd ed. Darien, IL: AASD; 2014.
40. Barion A, Zee PC. A clinical approach to circadian rhythm sleep disorders. Sleep Med 2007;8:566–77.
41. Morgenthaler TI, Lee-Chiong T, Alessi C, et al. Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep 2007;30:1445–59.
42. Shochat T, Martin J, Marler M, et al. Illumination levels in nursing home patients: effects on sleep and activity rhythms. J Sleep Res 2000;9:373–9.
43. Ancoli-Israel S, Klauber MR, Jones DW, et al. Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 1997;20:18–23.
44. Satlin A, Volicer L, Ross V, et al. Bright light treatment of behavioral and sleep disturbances in patients with Alzheimer’s disease. Am J Psychiatry 1992;149:1028–32.
45. Koyama E, Matsubara H, Nakano T. Bright light treatment for sleep-wake disturbances in aged individuals with dementia. Psychiatry Clin Neurosci 1999;53:227–9.
46. Burns A, Allen H, Tomenson B, et al. Bright light therapy for agitation in dementia: a randomized controlled trial. Int Psychogeriatr 2009;21:711–21.
47. Riemersma-van der Lek RF, Swaab DF, Twisk J, et al. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA 2008;299:2642–55.
48. Martin JL, Marler MR, Harker JO, et al. A multicomponent nonpharmacological intervention improves activity rhythms among nursing home residents with disrupted sleep/wake patterns. J Gerontol Series A Biol Sci Med Sci 2007;62:67–72.
49. Lewy AJ, Ahmed S, Jackson JM, et al. Melatonin shifts human circadian rhythms according to a phase-response curve. Chronobiol Int 1992;9:380–92.
50. Williamson BL, Tomlinson AJ, Mishra PK, et al. Structural characterization of contaminants found in commercial preparations of melatonin: similarities to case-related compounds from L-tryptophan associated with eosinophilia-myalgia syndrome. Chem Res Toxicol 1998;11:234-40.
51. Czeisler CA, Walsh JK, Roth T, et al. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med 2005;353:476-86.
52. Howard R, Roth T, Drake CL. The effects of armodafinil on objective sleepiness and performance in a shift work disorder sample unselected for objective sleepiness. J Clin Psychopharmacol 2014;34:369–73.
53. Beydoun MA, Gamaldo AA, Beydoun HA, et al. Caffeine and alcohol intakes and overall nutrient adequacy are associated with longitudinal cognitive performance among U.S. adults. J Nutr 2014;144:890–901.
54. Pentel P. Toxicity of over-the-counter stimulants. JAMA 1984;252:1898–903.
55. Neikrug AB, Ancoli-Israel S. Sleep disorders in the older adult - a mini-review. Gerontology 2010;56:181–9.
56. Chesson A Jr, Hartse K, Anderson WM, et al. Practice parameters for the evaluation of chronic insomnia. An American Academy of Sleep Medicine report. Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep 2000;23:237–41.
57. Bastien CH, Vallieres A, Morin CM. Validation of the insomnia severity index as an outcome measure for insomnia research. Sleep Med 2001;2:297–307.
58. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
59. Trauer JM, Qian MY, Doyle JS, et al. Cognitive behavioral therapy for chronic insomnia: a systematic review and meta-analysis. Ann Intern Med 2015;163:191–204.
60. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults, June 13-15, 2005. Sleep 2005;28:1049–57.
61. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med 2008;4:487–504.
62. Bain KT. Management of chronic insomnia in elderly persons. Am J Geriatr Pharmacother 2006;4:168–92.
63. FDA approves new type of sleep drug, Belsomra. 13 Aug 2014. Available at fda.gov.
64. Dubey AK, Handu SS, Mediratta PK. Suvorexant: The first orexin receptor antagonist to treat insomnia. J Pharmacol Pharmacother 2015;6:118–21.
65. Patel KV, Aspesi AV, Evoy KE. Suvorexant: a dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann Pharmacother 2015;49:477–83.
66. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:461–71.
67. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. Case studies in insomnia. New York: Springer; 1991:19–28.
68. Gulyani S, Salas RE, Gamaldo CE. Sleep medicine pharmacotherapeutics overview: today, tomorrow, and the future (part 2: hypersomnia, parasomnia, and movement disorders). Chest 2013;143:242–51.
69. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11):2770–88.
70. Middelkoop HA, Kerkhof GA, Smilde-van den Doel DA, et al. Sleep and ageing: the effect of institutionalization on subjective and objective characteristics of sleep. Age Ageing 1994;23:411–7.
71. Tinuper P, Provini F, Bisulli F, et al. Movement disorders in sleep: guidelines for differentiating epileptic from non-epileptic motor phenomena arising from sleep. Sleep Med Rev 2007;11:255–67.
72. Aurora RN, Zak RS, Maganti RK, et al. Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med 2010;6:85–95.
73. Bloom HG, Ahmed I, Alessi CA, et al. Evidence-based recommendations for the assessment and management of sleep disorders in older persons. J Am Geriatr Soc 2009;57:761–89.
74. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
75. Doshi A, Gamaldo C, Kalloo A, et al. Implementing a clinical iPad application to detect sleep disorders is feasible across multiple non-sleep outpatient clinics (P7. 321). Neurology 2015;84(14 Suppl):P7.321.
Pregnant nearly a year? The patient has symptoms but evidence is lacking
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).

Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).
Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
Mrs. X, age 43, gravida 4 para 1, is a married woman of sub-Saharan African heritage with a history of idiopathic hypertension, uterine leiomyomas, and multiple spontaneous miscarriages. She has no psychiatric history and had never been evaluated by a mental health professional. Mrs. X is well known to the hospital’s emergency room and obstetrics and gynecology services for several presentations claiming to be pregnant, continuously, over the last 11 months, despite evidence—several negative serum beta human chorionic gonadotropin (ß-hCG) tests and transvaginal sonograms—to the contrary.
Mrs. X reports that after feeling ill for “a few days,” she began to believe that she was “losing [her] mucous plug” and needed urgent evaluation in preparation for the delivery of her “child.” She again is given a ß-hCG test, which is negative, as well as a negative transvaginal sonogram.
Mrs. X’s blood pressure is 220/113 mm Hg, and she emergently receives captopril, 25 mg sublingually, which lowers her systolic blood pressure to 194 mm Hg. The internal medicine team learns that Mrs. X stopped taking her blood pressure medications, lisinopril and hydrochlorothiazide, approximately 2 weeks earlier because she “didn’t want it [the antihypertensive agents] to hurt [her] baby.”
What explains Mrs. X’s belief that she is pregnant?
a) polycystic ovary syndrome (PCOS)
b) delusional disorder
c) bipolar I disorder
d) somatic symptom disorder
The authors’ observations
Pseudocyesis is a psychosomatic condition with an estimated incidence of 1 in 160 maternity admissions in many African countries and 1 in 22,000 in the United States.1 According to DSM-5, pseudocyesis is a false belief of being pregnant along with signs and symptoms of pregnancy.2
Pseudocyesis is more common in:
- developing countries
- areas of low socioeconomic status with minimal education
- societies that place great importance on childbirth
- areas with low access to care.3
The primary presenting symptoms are changes in menses, enlarging abdomen, awareness of fetal movement, enlarged and tender breasts, galactorrhea, and weight gain.4
The exact pathophysiology of the disorder has not been determined, but we believe the psychosomatic hypothesis offers the most compelling explanation. According to this hypothesis, intense social pressures, such as an overwhelming desire to become pregnant because of cultural considerations, personal reasons, or both, could alter the normal function of the hypothalamic-pituitary-ovarian axis,5 which could result in physical manifestations of pregnancy. Tarín et al1 found that rodents with chronic psychosocial stress had decreased brain norepinephrine and dopamine activity and elevated plasma levels of norepinephrine. This can translate to human models, in which a deficit or dysfunction of catecholaminergic activity in the brain could lead to increased pulsatile gonadotropin-releasing hormone, luteinizing hormone (LH), prolactin, and an elevated LH:follicle-stimulating hormone ratio.1 These endocrine changes could induce traits found in most women with pseudocyesis, such as hypomenorrhea or amenorrhea, diurnal or nocturnal hyperprolactinemia (or both), and galactorrhea.1
How would you approach Mrs. X’s care?
a) confront her with the negative pregnancy tests
b) admit her to the inpatient psychiatric unit
c) begin antipsychotic therapy
d) discharge her with outpatient follow-up
EVALUATION A curse on her
Although Mrs. X initially refused to see the psychiatry team, she is more receptive on hospital Day 3. Mrs. X reports that she and her husband had been trying to have a child since they were married 17 years earlier. She had a child with another man before she met her husband, causing her in-laws in Africa to become suspicious that she is intentionally not producing a child for her husband. She had 3 spontaneous abortions since her marriage; these added stress to the relationship because the couple would feel elated when learning of a pregnancy and increasingly devastated with each miscarriage.
Mrs. X reports that she and her husband have been seeing a number of reproductive endocrinologists for 7 years to try to become pregnant. She reports feeling that these physicians are not listening to her or giving her adequate treatment, which is why she has not been able to become pregnant. At the time of the evaluation, she reports that she is pregnant, and the tests have been negative because her mother-in-law placed a “curse” on her. This “curse” caused the baby to be invisible to the laboratory tests and sonograms.
During the psychiatric evaluation, Mrs. X displays her protuberant abdomen and says that she feels the fetus kicking. In addition, she also reports amenorrhea and breast tenderness and engorgement.
During her hospital stay, Mrs. X’s mental status exam does not demonstrate signs or symptoms of a mood disorder, bipolar disorder, or psychosis. Nonetheless, she remains delusional and holds to her fixed false belief of being pregnant. She refuses to be swayed by evidence that she is not pregnant. Despite this, clinicians build enough rapport that Mrs. X agrees to follow up with psychiatry in the outpatient clinic after discharge.
The internal medicine team is apprehensive that Mrs. X will continue to refuse antihypertensive medications out of concern that the medications would harm her pregnancy, as she had in the hospital. She remains hypertensive, with average systolic blood pressure in the 180 to 200 mm Hg range; however, after much discussion with her and her family members, she agrees to try amlodipine, 5 mg/d, a category C drug. She says that she will adhere to the medication if she does not experience any side effects.
Mrs. X is discharged on hospital Day 4 to outpatient follow-up.
The authors’ observations
When considering a diagnosis of pseudocyesis, the condition should be distinguished from others with similar presentations. Before beginning a psychiatric evaluation, a normal pregnancy must be ruled out. This is easily done with a positive urine or serum ß-hCG and an abdominal or transvaginal ultrasound. Pseudocyesis can be differentiated from:
- delusion of pregnancy (sometimes referred to as psychotic pregnancy)—a delusional disorder often seen in psychotic illness without any physical manifestations of pregnancy
- pseudopregnancy (sometimes referred to as erroneous pseudocyesis), another rare condition in which signs and symptoms of pregnancy are manifested1,6,7 but the patient does not have a delusion of pregnancy.
Pseudocyesis, in contrast, comprises the delusion of pregnancy and physical manifestations.2 These distinctions could be difficult to make clinically; for example, an increase in abdominal girth could be a result of pseudocyesis or obesity. In the setting of physical manifestations of pregnancy, a diagnosis of pseudocyesis is more likely (Table1).
Patients with pseudocyesis exhibit subjective and objective findings of pregnancy, such as abdominal distension, enlarged breasts, enhanced pigmentation, lordotic posture, cessation of menses, morning sickness, and weight gain.8,9 Furthermore, approximately 1% of pseudocyesis patients have false labor, as Mrs. X did.10 Typically, the duration of these symptoms range from a few weeks to 9 months. In some cases, symptoms can last longer11; at admission, Mrs. X reported that she was 11 months pregnant. She saw nothing wrong with this assertion, despite knowing that human gestation lasts 9 months.
In delusion of pregnancy, a patient might exhibit abdominal distension and cessation of menses but have no other objective findings of pregnancy.7 Rather than being a somatoform disorder such as pseudocyesis, a delusion of pregnancy is a symptom of psychosis or, rarely, dementia.12
Pseudopregnancy is a somatic state resembling pregnancy that can arise from a variety of medical conditions. A full medical workup and intensive mental status and cognitive evaluation are necessary for diagnostic clarity. Although the pathology and workup of delusional pregnancy is beyond the scope of this article, we suggest Seeman13 for a review and Chatterjee et al14 and Tarín et al1 for guidance on making the diagnosis.
Theories about pathophysiology
As with many psychosomatic conditions, the pathological process of pseudocyesis originally was thought of in a psychodynamic context. Several psychodynamic theories have been proposed, including instances in which the internal desire to be pregnant is strong enough to induce a series of physiological changes akin to the state of pregnancy.6
Other examiners of pseudocyesis have noted its development from fears and societal pressure, including the loss of companionship or “womanhood.”6,9 Last, the tenuous interplay of desire for a child and substantial fear of pregnancy appears to play a role in many cases.9-11 Rosenberg et al15 reported on a teenager with pseudocyesis who desired to be pregnant to appease her husband and family, but feared pregnancy and the implications of having a child at such a young age. As this team wrote, “this pregnancy sans child fulfilled the needs of the entire family, at least temporarily.”15
Prevailing modern theories behind the somatic presentations of these patients hinge on an imbalance of the hypothalamic-pituitary-adrenal axis.9 Although this remains the area of ongoing research, most literature has not shown a consistent change or trend in laboratory levels of hormones associated with pseudocyesis.16 Tarín et al,1 however, did show a similar hormonal profile between patients with pseudocyesis and those with PCOS. Although urine or serum pregnancy testing and ultrasonography are indicated to rule out pseudopregnancy, we see no benefit in obtaining other lab work in most cases beyond that of a general medical workup, because such evaluations are not helpful in diagnosis or treatment.
Mrs. X’s abdomen was protuberant and she displayed the typical linea nigra of pregnancy. Many authors have theorized the physiological mechanism behind the abdominal enlargement to include contraction of the diaphragm, which reduces the abdominal cavity and forces the bowel outwards. As abdominal fat increases, the patient becomes constipated, and the bowel becomes distended.10,16 Although the cause of our patient’s abdominal enlargement was not pursued, we note that the literature reported that the abdominal enlargement disappears when the patient is under general anesthesia.10,16,17
Characteristics of pseudocyesis
Bivin and Klinger’s 1937 compilation of >400 cases of pseudocyesis over nearly 200 years remains a landmark in the study of this condition.18 In their analysis, patients range in age from 20 to 44; >75% were married. The authors noted that many of the women they studied had borne children previously. Further social and psychological studies came from this breakthrough article, which shed light on the dynamics of pseudocyesis in many patients with the condition.
According to Koic,11 pseudocyesis is a form of conversion disorder with underlying depression. This theory is based on literature reports of patients displaying similar personal, cultural, and social factors. These similarities, although not comprehensive, are paramount in both the diagnosis and treatment of this condition.
Often, pseudocyesis presents in patients with lower education and socioeconomic status.1,3,11 This is particularly true in developing nations in sub-Saharan Africa and the Indian subcontinent. Case reports, cross-sectional, and longitudinal studies from these developing nations in particular note the extremely high stress placed on women to produce children for their husbands and family in male-dominated society; it is common for a woman to be rejected by her husband and family if she is unable to reproduce.3
The effect of a lower level of education on development of pseudocyesis appears to be multifactorial:
- Lack of understanding of the human body and reproductive health can lead to misperception of signs of pregnancy and bodily changes
- Low education correlates with poor earnings and worse prenatal care; delayed or no prenatal care also has been associated with an increased incidence of pseudocyesis.3
In Ouj’s study of pseudocyesis in Nigeria, the author postulated that an educated woman does not endure the same stress of fertility as an uneducated woman; she is already respected in her society and will not be rejected if she does not have children.3
Mrs. X’s ethnic background and continued close ties with sub-Saharan Africa are notable: Her background is one that is typically associated with pseudocyesis. She is from an developing country, did not complete higher education, was ostracized by her mother-in-law because of her inability to conceive, and was told several times, during her visits to Ghana, that she was indeed pregnant.
Mrs. X noted a strong desire to conceive for her husband and family and carried with her perhaps an even stronger fear of loss of marriage and female identity—which has been bolstered by the importance placed on the woman’s raison d’être in the family by her cultural upbringing.3,6,9-11,15 What Mrs. X never made clear, however, was whether she wanted another child at her age and in the setting of having many friends and rewarding full-time employment.
Epidemiology of pseudocyesis worldwide has been evaluated in a handful of studies. As compiled by Cohen,8 the prevalence of pseudocyesis in Boston, Massachusetts, was 1/22,000 births, whereas it was dramatically higher in Sudan (1/160 women who had previously been managed for reproductive failure).1 This discrepancy in prevalance is consistent with current theories on patient characteristics that lead to increased incidence of pseudocyesis in underdeveloped nations. A 1951 study at an academic hospital in Philadelphia, Pennsylvania, noted 27 cases of pseudocyesis in maternity admissions during the study period—an incidence of 1 in 250.19 Of note, 85% of cases were of African American heritage; in 89% of cases, the woman had been trying to conceive for as long as 17 years.
Avoiding confrontation
Initially, Mrs. X was resistant to talking with a psychiatrist; this is consistent with studies showing that a patient can be suspicious and even hostile when a clinician attempts to engage her in mental health treatment.10,16 The patient interprets the physical sensations she experiences during pseudocyesis, for example, as a real pregnancy, a perception that is contradicted by medical testing.
It is important to understand this conflict and to avoid confronting the patient directly about false beliefs; confrontation has been shown to be detrimental to patient recovery. Instead, offer the patient alternatives to her symptoms (ie, sensations of abdominal movement also can be caused by indigestion), while not directly discounting her experiences.6,9 Indeed, from early on in the study of pseudocyesis, there have been many reports of resolution of symptoms when the physician helped the patient understand that she is not pregnant.20,21
OUTCOME Supportive therapy
Mrs. X is seen for outpatient psychiatry follow-up several weeks after hospitalization. She acknowledges that, although she still thought pregnancy is possible, she is willing to entertain the idea that there could be another medical explanation for her symptoms.
Mrs. X is provided with supportive therapy techniques, and her marital and societal stressors are discussed. Psychotropic medications are considered, but eventually deemed unnecessary; the treatment team is concerned that Mrs. X, who remains wary of mental health providers, would view the offer of medication as offensive.
Mrs. X is seen in the gynecology clinic approximately 2 weeks later; there, a diagnosis of secondary anovulation is made and a workup for PCOS initiated.
Subsequent review of the medical record states that, during further follow-up with gynecology, Mrs. X no longer believes that she is pregnant.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
1. Tarín JJ, Hermenegildo C, García-Pérez MA, et al. Endocrinology and physiology of pseudocyesis. Reprod Biol Endocrinol. 2013;11:39.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Ouj U. Pseudocyesis in a rural southeast Nigerian community. J Obstet Gynaecol Res. 2009;35(4):660-665.
4. Signer SF, Weinstein RP, Munoz RA, et al. Pseudocyesis in organic mood disorders. Six cases. Psychosomatics. 1992;33(3):316-323.
5. Omer H, Elizur Y, Barnea T, et al. Psychological variables and premature labour: a possible solution for some methodological problems. J Psychosom Res. 1986;30(5):559-565.
6. Starkman MN, Marshall JC, La Ferla J, et al. Pseudocyesis: psychologic and neuroendocrine interrelationships. Psychosom Med. 1985;47(1):46-57.
7. Yadav T, Balhara YP, Kataria DK. Pseudocyesis versus delusion of pregnancy: differential diagnoses to be kept in mind. Indian J Psychol Med. 2012;34(1):82-84.
8. Cohen LM. A current perspective of pseudocyesis. Am J Psychiatry. 1982;139(9):1140-1144.
9. Brown E, Barglow P. Pseudocyesis. A paradigm for psychophysiological interactions. Arch Gen Psychiatry. 1971;24(3):221-229.
10. Small GW. Pseudocyesis: an overview. Can J Psychiatry. 1986;31(5):452-457.
11. Koi´c E, Mu´zin´c L, Đordevic V, et al. Pseudocyesis and couvade syndrome. Drustvena Istrazivanja. 2002;11:1031-1047.
12. Bhattacharyya S, Chaturvedi SK. Metamorphosis of delusion of pregnancy. Can J Psychiatry. 2001;46(6):561-562.
13. Seeman MV. Pseudocyesis, delusional pregnancy, and psychosis: the birth of a delusion. World J Clin Cases. 2014;2(8):338-344.
14. Chatterjee SS, Nath N, Dasgupta G, et al. Delusion of pregnancy and other pregnancy-mimicking conditions: dissecting through differential diagnosis. Medical Journal of Dr. D.Y. Patil University. 2014;7(3):369-372.
15. Rosenberg HK, Coleman BG, Croop J, et al. Pseudocyesis in an adolescent patient. Clin Pediatr (Phila). 1983;22(10):708-712.
16. O’Grady JP, Rosenthal M. Pseudocyesis: a modern perspective on an old disorder. Obstet Gynecol Surv. 1989;44(7):500-511.
17. Whelan CI, Stewart DE. Pseudocyesis–a review and report of six cases. Int J Psychiatry Med. 1990;20(1):97-108.
18. Bivin GD, Klinger MP. Pseudocyesis. Bloomington, IN: Principia Press; 1937.
19. Fried PH, Rakoff AE, Schopbach RR, et al. Pseudocyesis; a psychosomatic study in gynecology. J Am Med Assoc. 1951;145(17):1329-1335.
20. Dunbar F. Emotions and bodily changes. 3rd ed. New York, NY: Columbia University Press; 1947.
21. Steinberg A, Pastor N, Winheld EB, et al. Psychoendocrine relationship in pseudocyesis. Psychosom Med. 1946;8(3):176-179.
Current Therapeutic Approaches to Renal Cell Carcinoma
From the Department of Medicine, Carole and Ray Neag Comprehensive Cancer Center, UConn Health, Farmington, CT (Dr. Namakydoust and Dr. Clement) and the UConn School of Pharmacy, Storrs, CT (Dr. Holle).
Abstract
- Objective: To review therapeutic options for the treatment of renal cell carcinoma (RCC).
- Methods: Review of the literature in the context of a clinical case.
- Results: RCC accounts for 90% of all renal tumors. For RCC patients with nondistant metastases, preferred treatment is curative-intent radical nephrectomy or partial nephrectomy; oncologic outcomes for the 2 procedures are similar. For patients who are deemed not to be surgical candidates, ablative techniques such as cryoablation and radiofrequency ablation may be considered. Systemic therapy for metastatic RCC is based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. First-line treatment options for patients with metastatic clear-cell RCC include biologic agents such as high-dose interleukin-2 immune therapy, as well as targeted therapies including tyrosine kinase inhibitors (TKIs) and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis. Second-line therapies in this setting include TKIs and nivolumab (PD-1 inhibitor). If TKIs were used as first-line therapy, mTOR inhibitors can be used in the second line. In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options. Best supportive care should always be provided along with initial and subsequent therapies.
- Conclusion: Multiple treatment options are now available for patients with metastatic or unresectable RCC. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors [1]. Approximately 55,000 new RCC cases are diagnosed each year [1]. Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured sur-gically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [2]. Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
Epidemiology and Classification
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs [2]. Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure [3]. Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics [2]. Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%) [2]. Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis.
Familial Syndromes
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs [4].
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS) [4]. Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common [5]. VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models [6–8]. HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease [4–8].
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome [9]. In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
Case Study
Initial Presentation
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further workup is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes [10]. Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease [11]. Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion [12]. Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
Surgical Intervention
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8% [14]. Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as either an open or laparoscopic procedure, the latter of which may be performed robotically [15]. Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach) [16]. The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact [16,17].
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma [15].
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events [18]. Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC [19].
Adjuvant Therapy
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy [20]. Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection [21]. Therefore, observation remains the standard of care after nephrectomy.
Renal Tumor Ablation
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates [22]. There have been no studies performed directly comparing the modalities.
Cryoablation. Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high-pressure gas (argon, nitrogen) is passed through the probe and it cools once it enters a lower pressure region. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor [23,24].
RFA. Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) from a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum [24].
Microwave ablation. Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA [24]. The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years [25]. However, a study by Castle and colleagues [26] demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible electroporation. Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe [27]. Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings [24].
Active Surveillance
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter [28,29]. The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance [30]. The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews [31–33].
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion [34]. The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016 [21,35]. These guidelines use TNM staging to risk-stratify patients and recommend follow up.
Case Continued
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
Prognostic Models
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [13]. Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse [36,37]. Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa [38]. The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors) [39]. Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively [40].
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy [41]. This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients were not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level [42].
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant medial survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively) [43]. The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available [44,45]. Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
Case Continued
Based on the MSKCC prognostic factor model, the patient is deemed to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology [57,58]. In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus [59]. Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option [21].
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features [60]. Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response [61]. A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient [62].
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients [63].
Biologics
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology [64]. Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients [65].
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study [66,67]. However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF Monoclonal Antibodies
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo [68]. In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SQ] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001) [47,48]. Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
Tyrosine Kinase Inhibitors
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib. Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues [52,53] compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SQ 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy [69–71].
Pazopanib. Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4% [72]. This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy [50]. In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib. Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy [73]. A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029) [51]. Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib. Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving one prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted [74]. In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2) [46]. The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
Cabozantinib
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues [75] compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% CI 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66; 95% CI 0.53 to 0.83; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC [76].
mTOR Inhibitors
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis [77]. Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model) [54]. The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SQ 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SQ 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hypercholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval [78]. The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + mTOR Inhibitor
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor, everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40; 95% CI 0.24 to 0.68; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66, 95% CI 0.30 to 1.10, P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61; 95% CI 0.38 to 0.98; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC [55].
Case Continued
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 Blockade
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized anti-bodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy [79]. Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC [80]. Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues [81] demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
Case Conclusion
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
Future Directions
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Corresponding author: Jessica Clement, MD, UConn Health, 263 Farmington Avenue, Farmington, CT 06030, [email protected].
Financial disclosures: None.
1. Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
2. Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
3. Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
4. Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90
5. Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
6. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
7. Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
8. Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
9. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
10. Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
11. Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
12. Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28:253–61.
13. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
14. O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
15. McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
16. Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
17. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49:1374–403.
18. Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
19. Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
20. Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
21. NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
22. El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
23. Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
24. Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
25. Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
26. Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
27. Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
28. Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
29. Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
30. Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
31. Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
32. Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
33. Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
34. Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
35. Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
36. Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
37. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
38. Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
39. Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
40. Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
41. Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
42. Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
43. Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
44. Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
45. Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
46. Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
47. Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
48. Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
49. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
50. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
51. Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
52. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
53. Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
54. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
55. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
56. Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
57. Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
58. Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
59. Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
60. Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
61. Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
62. Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
63. Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
64. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
65. Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
66. Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
67. Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
68. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
69. Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
70. Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
71. Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
72. Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
73. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
74. Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
75. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
76. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
77. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
78. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
79. Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
80. Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
81. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
From the Department of Medicine, Carole and Ray Neag Comprehensive Cancer Center, UConn Health, Farmington, CT (Dr. Namakydoust and Dr. Clement) and the UConn School of Pharmacy, Storrs, CT (Dr. Holle).
Abstract
- Objective: To review therapeutic options for the treatment of renal cell carcinoma (RCC).
- Methods: Review of the literature in the context of a clinical case.
- Results: RCC accounts for 90% of all renal tumors. For RCC patients with nondistant metastases, preferred treatment is curative-intent radical nephrectomy or partial nephrectomy; oncologic outcomes for the 2 procedures are similar. For patients who are deemed not to be surgical candidates, ablative techniques such as cryoablation and radiofrequency ablation may be considered. Systemic therapy for metastatic RCC is based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. First-line treatment options for patients with metastatic clear-cell RCC include biologic agents such as high-dose interleukin-2 immune therapy, as well as targeted therapies including tyrosine kinase inhibitors (TKIs) and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis. Second-line therapies in this setting include TKIs and nivolumab (PD-1 inhibitor). If TKIs were used as first-line therapy, mTOR inhibitors can be used in the second line. In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options. Best supportive care should always be provided along with initial and subsequent therapies.
- Conclusion: Multiple treatment options are now available for patients with metastatic or unresectable RCC. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors [1]. Approximately 55,000 new RCC cases are diagnosed each year [1]. Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured sur-gically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [2]. Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
Epidemiology and Classification
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs [2]. Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure [3]. Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics [2]. Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%) [2]. Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis.
Familial Syndromes
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs [4].
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS) [4]. Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common [5]. VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models [6–8]. HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease [4–8].
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome [9]. In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
Case Study
Initial Presentation
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further workup is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes [10]. Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease [11]. Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion [12]. Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
Surgical Intervention
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8% [14]. Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as either an open or laparoscopic procedure, the latter of which may be performed robotically [15]. Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach) [16]. The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact [16,17].
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma [15].
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events [18]. Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC [19].
Adjuvant Therapy
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy [20]. Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection [21]. Therefore, observation remains the standard of care after nephrectomy.
Renal Tumor Ablation
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates [22]. There have been no studies performed directly comparing the modalities.
Cryoablation. Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high-pressure gas (argon, nitrogen) is passed through the probe and it cools once it enters a lower pressure region. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor [23,24].
RFA. Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) from a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum [24].
Microwave ablation. Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA [24]. The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years [25]. However, a study by Castle and colleagues [26] demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible electroporation. Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe [27]. Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings [24].
Active Surveillance
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter [28,29]. The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance [30]. The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews [31–33].
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion [34]. The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016 [21,35]. These guidelines use TNM staging to risk-stratify patients and recommend follow up.
Case Continued
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
Prognostic Models
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [13]. Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse [36,37]. Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa [38]. The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors) [39]. Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively [40].
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy [41]. This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients were not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level [42].
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant medial survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively) [43]. The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available [44,45]. Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
Case Continued
Based on the MSKCC prognostic factor model, the patient is deemed to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology [57,58]. In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus [59]. Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option [21].
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features [60]. Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response [61]. A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient [62].
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients [63].
Biologics
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology [64]. Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients [65].
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study [66,67]. However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF Monoclonal Antibodies
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo [68]. In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SQ] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001) [47,48]. Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
Tyrosine Kinase Inhibitors
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib. Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues [52,53] compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SQ 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy [69–71].
Pazopanib. Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4% [72]. This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy [50]. In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib. Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy [73]. A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029) [51]. Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib. Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving one prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted [74]. In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2) [46]. The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
Cabozantinib
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues [75] compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% CI 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66; 95% CI 0.53 to 0.83; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC [76].
mTOR Inhibitors
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis [77]. Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model) [54]. The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SQ 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SQ 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hypercholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval [78]. The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + mTOR Inhibitor
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor, everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40; 95% CI 0.24 to 0.68; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66, 95% CI 0.30 to 1.10, P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61; 95% CI 0.38 to 0.98; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC [55].
Case Continued
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 Blockade
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized anti-bodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy [79]. Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC [80]. Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues [81] demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
Case Conclusion
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
Future Directions
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Corresponding author: Jessica Clement, MD, UConn Health, 263 Farmington Avenue, Farmington, CT 06030, [email protected].
Financial disclosures: None.
From the Department of Medicine, Carole and Ray Neag Comprehensive Cancer Center, UConn Health, Farmington, CT (Dr. Namakydoust and Dr. Clement) and the UConn School of Pharmacy, Storrs, CT (Dr. Holle).
Abstract
- Objective: To review therapeutic options for the treatment of renal cell carcinoma (RCC).
- Methods: Review of the literature in the context of a clinical case.
- Results: RCC accounts for 90% of all renal tumors. For RCC patients with nondistant metastases, preferred treatment is curative-intent radical nephrectomy or partial nephrectomy; oncologic outcomes for the 2 procedures are similar. For patients who are deemed not to be surgical candidates, ablative techniques such as cryoablation and radiofrequency ablation may be considered. Systemic therapy for metastatic RCC is based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. First-line treatment options for patients with metastatic clear-cell RCC include biologic agents such as high-dose interleukin-2 immune therapy, as well as targeted therapies including tyrosine kinase inhibitors (TKIs) and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis. Second-line therapies in this setting include TKIs and nivolumab (PD-1 inhibitor). If TKIs were used as first-line therapy, mTOR inhibitors can be used in the second line. In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options. Best supportive care should always be provided along with initial and subsequent therapies.
- Conclusion: Multiple treatment options are now available for patients with metastatic or unresectable RCC. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors [1]. Approximately 55,000 new RCC cases are diagnosed each year [1]. Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured sur-gically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [2]. Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
Epidemiology and Classification
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs [2]. Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure [3]. Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics [2]. Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%) [2]. Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis.
Familial Syndromes
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs [4].
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS) [4]. Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common [5]. VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models [6–8]. HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease [4–8].
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome [9]. In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
Case Study
Initial Presentation
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further workup is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes [10]. Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease [11]. Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion [12]. Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
Surgical Intervention
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8% [14]. Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as either an open or laparoscopic procedure, the latter of which may be performed robotically [15]. Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach) [16]. The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact [16,17].
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma [15].
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events [18]. Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC [19].
Adjuvant Therapy
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy [20]. Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection [21]. Therefore, observation remains the standard of care after nephrectomy.
Renal Tumor Ablation
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates [22]. There have been no studies performed directly comparing the modalities.
Cryoablation. Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high-pressure gas (argon, nitrogen) is passed through the probe and it cools once it enters a lower pressure region. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor [23,24].
RFA. Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) from a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum [24].
Microwave ablation. Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA [24]. The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years [25]. However, a study by Castle and colleagues [26] demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible electroporation. Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe [27]. Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings [24].
Active Surveillance
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter [28,29]. The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance [30]. The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews [31–33].
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion [34]. The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016 [21,35]. These guidelines use TNM staging to risk-stratify patients and recommend follow up.
Case Continued
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
Prognostic Models
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20% [13]. Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse [36,37]. Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa [38]. The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors) [39]. Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively [40].
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy [41]. This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients were not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level [42].
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant medial survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively) [43]. The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available [44,45]. Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
Case Continued
Based on the MSKCC prognostic factor model, the patient is deemed to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology [57,58]. In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus [59]. Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option [21].
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features [60]. Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response [61]. A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient [62].
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients [63].
Biologics
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology [64]. Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients [65].
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study [66,67]. However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF Monoclonal Antibodies
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo [68]. In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SQ] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001) [47,48]. Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
Tyrosine Kinase Inhibitors
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib. Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues [52,53] compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SQ 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy [69–71].
Pazopanib. Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4% [72]. This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy [50]. In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib. Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy [73]. A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029) [51]. Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib. Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving one prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted [74]. In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2) [46]. The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
Cabozantinib
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues [75] compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% CI 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66; 95% CI 0.53 to 0.83; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC [76].
mTOR Inhibitors
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis [77]. Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model) [54]. The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SQ 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SQ 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hypercholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval [78]. The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + mTOR Inhibitor
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor, everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40; 95% CI 0.24 to 0.68; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66, 95% CI 0.30 to 1.10, P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61; 95% CI 0.38 to 0.98; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC [55].
Case Continued
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 Blockade
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized anti-bodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy [79]. Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC [80]. Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues [81] demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
Case Conclusion
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
Future Directions
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
Corresponding author: Jessica Clement, MD, UConn Health, 263 Farmington Avenue, Farmington, CT 06030, [email protected].
Financial disclosures: None.
1. Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
2. Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
3. Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
4. Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90
5. Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
6. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
7. Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
8. Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
9. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
10. Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
11. Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
12. Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28:253–61.
13. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
14. O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
15. McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
16. Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
17. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49:1374–403.
18. Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
19. Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
20. Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
21. NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
22. El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
23. Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
24. Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
25. Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
26. Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
27. Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
28. Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
29. Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
30. Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
31. Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
32. Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
33. Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
34. Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
35. Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
36. Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
37. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
38. Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
39. Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
40. Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
41. Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
42. Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
43. Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
44. Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
45. Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
46. Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
47. Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
48. Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
49. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
50. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
51. Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
52. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
53. Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
54. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
55. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
56. Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
57. Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
58. Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
59. Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
60. Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
61. Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
62. Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
63. Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
64. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
65. Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
66. Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
67. Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
68. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
69. Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
70. Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
71. Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
72. Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
73. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
74. Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
75. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
76. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
77. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
78. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
79. Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
80. Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
81. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
1. Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
2. Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
3. Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
4. Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90
5. Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
6. Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
7. Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
8. Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
9. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
10. Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
11. Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
12. Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28:253–61.
13. Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
14. O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
15. McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
16. Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
17. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49:1374–403.
18. Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
19. Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
20. Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
21. NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
22. El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
23. Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
24. Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
25. Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
26. Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
27. Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
28. Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
29. Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
30. Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
31. Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
32. Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
33. Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
34. Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
35. Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
36. Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
37. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
38. Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
39. Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
40. Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
41. Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
42. Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
43. Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
44. Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
45. Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
46. Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
47. Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
48. Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
49. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
50. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
51. Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
52. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
53. Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
54. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
55. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
56. Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
57. Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
58. Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
59. Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
60. Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
61. Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
62. Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
63. Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
64. McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
65. Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
66. Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
67. Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
68. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
69. Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
70. Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
71. Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
72. Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
73. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
74. Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
75. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
76. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
77. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
78. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
79. Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
80. Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
81. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
When to adjust the dosing of psychotropics in patients with renal impairment
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
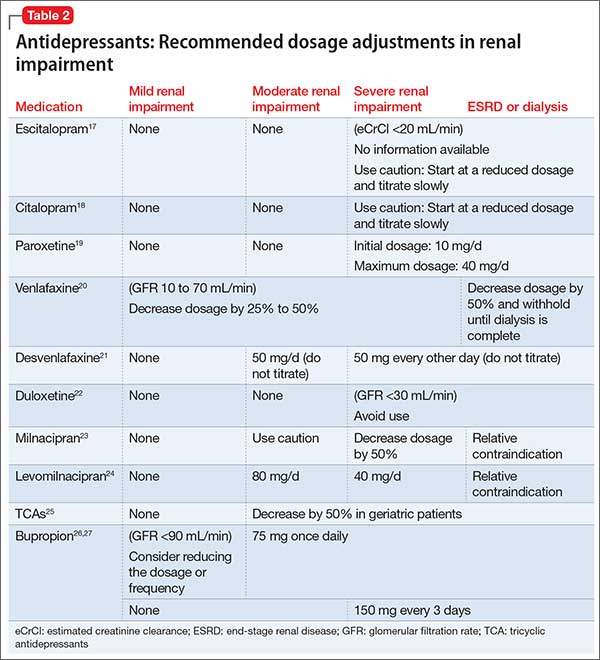
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
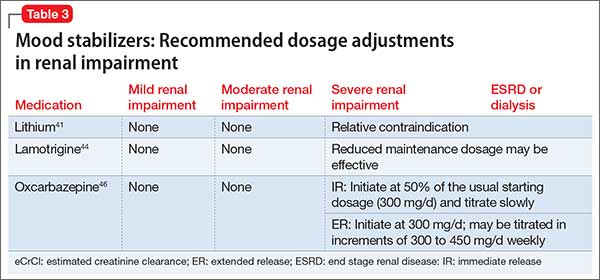
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Psychosis in treated neurosyphilis: Is now the time to stop his antipsychotic?
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
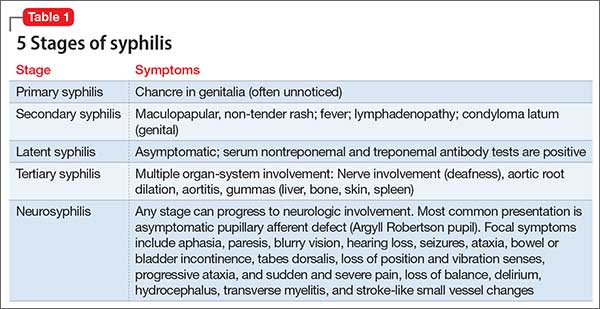
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
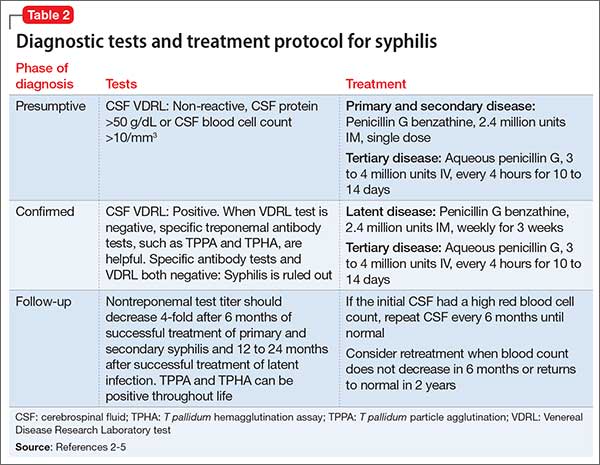
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
Using lipid guidelines to manage metabolic syndrome for patients taking an antipsychotic
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?

Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
No evidence of pregnancy, but she is suicidal and depressed after ‘my baby died’
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.

Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.
Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
CASE Depressed after she says her baby died
Ms. R, age 50, is an African-American woman who presents to a psychiatric hospital under an involuntary commitment executed by local law enforcement. Her sister called the authorities because Ms. R reportedly told her that she is “very depressed” and wants to “end [her] life” by taking an overdose of medications after the death of her newborn 1 week earlier.
Ms. R states that she delivered a child at “full term” in the emergency department of an outside community hospital, and that her current psychiatric symptoms began after the child died from “SIDS” [sudden infant death syndrome] shortly after birth.
Ms. R describes depressive symptoms including depressed mood, anhedonia, decreased energy, feelings of guilt, decreased concentration, poor sleep, and suicidal ideation. She denies substance use or a medical condition that could have induced these symptoms, and denies symptoms of mania, anxiety, or psychosis at admission or during the previous year.
Ms. R reports a history of manic episodes that includes periods of elevated mood or irritability, impulsivity, increased energy, excessive spending despite negative consequences, lack of need for sleep, rapid thoughts, and rapid speech that impaired her social and occupational functioning. Her most recent manic episode was approximately 3 years before this admission. She reports a previous suicide attempt and a history of physical abuse from a former intimate partner.
Neither the findings of a physical examination nor the results of a screening test for serum β-human chorionic gonadotropin (βHCG) are consistent with pregnancy. Ms. R’s medical record reveals that she was hospitalized for a “cardiac workup” a week earlier and requested investigation of possible pregnancy, which was negative. Records also reveal that she had a hysterectomy 10 years earlier.
Although Ms. R’s sister and boyfriend support her claim of pregnancy, the patient’s young adult son refutes it and states that she “does stuff like this for attention.” Her son also reports receiving a forged sonogram picture that his mother found online 1 month earlier. Ms. R presents an obituary from a local newspaper for the child but, on further investigation, the photograph of the infant was discovered to be of another child, also obtained online. Ms. R’s family denies knowledge of potential external reward Ms. R could gain by claiming to be pregnant.
Which of the following diagnoses can be considered after Ms. R’s initial presentation?
a) somatic symptom disorder
b) major depressive disorder
c) bipolar I disorder
d) delusional disorder
The authors’ observations
Ms. R reported the recent death of a newborn that was incompatible with her medical history. Her family members revealed that Ms. R made an active effort to deceive them about the reported pregnancy. She also exhibited symptoms of a major depressive episode in the context of previous manic episodes and expressed suicidal ideation.
The first step in the diagnostic pathway was to rule out possible medical explanations, including pregnancy, which could account for the patient’s symptoms. Although the serum βHCG level usually returns to non-pregnant levels 2 to 4 weeks after delivery, it can take even longer in some women.1 The absence of βHCG along with the recorded history of hysterectomy indicated that Ms. R was not pregnant at the time of testing or within the preceding few weeks. Once medical anomalies and substance use were ruled out, further classification of the psychiatric condition was undertaken.
One aspect of establishing a diagnosis for Ms. R is determining the presence of psychosis (eg, delusional thinking) (Table 1). Ms. R deliberately fabricated evidence of her pregnancy and manipulated family members, which indicated a low likelihood of delusions and supported a diagnostic alternative to psychosis.
Ms. R has a well-described history of manic episodes with current symptoms of a major depressive episode. The treatment team makes a diagnosis of bipolar I disorder, most recent episode depressed. The depressive symptoms Ms. R described were consistent with bipolar depression but did not explain her report of a pregnancy that is inconsistent with reality.
As is the case with Ms. R, diagnostic clarity often requires observation and evaluation over time. Building a strong therapeutic relationship with Ms. R in the context of an appropriate treatment plan allows the treatment team to explore the origin, motivations, and evolution of her thought content while managing her illness.
Confronting a patient about her false claims is likely to result in which of the following?
a) spontaneous resolution of symptoms
b) improved therapeutic alliance
c) degradation of the patient’s coping mechanism
d) violent outbursts by the patient
EVALUATION Confrontation
At admission, Ms. R remains resolute that she was pregnant and is suffering immense psychological distress secondary to the death of her child. Early in the treatment course, she is confronted with evidence indicating that her pregnancy was impossible. Shortly after this interaction, nursing staff alerts the treating physician that Ms. R experienced a “seizure-like spell” characterized by gross non-stereotyped jerking of the upper extremities, intact orientation, retention of bowel and bladder function, and coherent speech consistent with a diagnosis of pseudoseizure.2
Ms. R is transferred to a tertiary care facility for neurologic evaluation and observation. Ms. R repeatedly presents a photograph that she claims to be of her deceased child and implores the allied treatment team to advocate for discharge. Evaluation of Ms. R’s neurologic symptoms revealed no medical explanation for the “seizure-like spell” and she is transferred to the inpatient psychiatric hospital.
Upon return to the inpatient psychiatric unit, Ms. R receives intensive psychological exploration of her symptoms, thought content, and the foundation of her pregnancy claim. Within days, she acknowledges that the pregnancy was “not real” and that she was conscious of this fact in the months prior to hospitalization. She cites turmoil in her romantic relationship as the primary stimulus for her actions.
The authors’ observations
Ms. R’s reported pregnancy was not a delusion, but rather a deceitful exposition constructed with appropriate reality testing and a conscious awareness of the manipulation. This eliminated delusions as the explanation of her pregnancy claim. Although Ms. R initially rejected evidence refuting her belief of pregnancy, she recognized and accepted reality with appropriate intervention.
Factitious disorder vs malingering
Factitious disorder and malingering can present with intentional induction or report of symptoms or signs of a physical abnormality:
Factitious disorder imposed on the self is a willful misrepresentation or fabrication of signs or symptoms of an illness by a person in the absence of obvious personal gain that cannot be explained by a separate physical or mental illness (Table 2).3,4
Malingering is the intentional production or exaggeration of physical or psychological signs or symptoms with obvious secondary gain.
Malingering can be excluded in Ms. R’s case: She did not gain external reward by falsely reporting pregnancy. Although DSM-IV-TR (Table 2) assumes that the motivation for the patient with factitious disorder is to assume the sick role, DSM-5 merely states that the she (he) should present themselves as ill, impaired, or injured.3,4
Ms. R’s treatment team diagnosed factitious disorder imposed on self after careful exclusion of other causes for her symptoms. Bipolar I disorder, most recent episode depressed, also was diagnosed after considering Ms. R’s previous history of manic episodes and depressive symptoms at presentation.
Factitious disorder and other psychiatric conditions often are comorbid. Bipolar disorder, as in Ms. R’s case, as well as major depressive disorder commonly are comorbid with factitious disorder. It is also important to note that factitious disorder often occurs in the context of a personality disorder.5
Which of the following medications are FDA-approved for treating factitious disorder?
a) olanzapine-fluoxetine combination
b) lurasidone
c) valproic acid
d) all of the above
e) no medications are approved for treating factitious disorder
TREATMENT Support, drug therapy
Treatment of Ms. R’s factitious disorder consists of psychological interventions via psychotherapy and strengthening of social support. She participates in daily individual therapy sessions as well as several group therapy activities. Ms. R engages with her social worker to facilitate a successful transition to an appropriate support network and access community resources to aid her wellness.
The treatment team feels that her diagnosis of bipolar I disorder, most recent episode depressed, warrants pharmacologic intervention. Ms. R agrees to begin a mood stabilizer, valproic acid, instead of medications FDA-approved to treat bipolar depression, such as lurasidone or quetiapine, because she reports good efficacy and tolerability when she took it during a major depressive episode approximately 4 years earlier.
Valproic acid is started at 250 mg/d and increased to 1,000 mg/d. Ms. R tolerates the medication without observed or reported adverse effects.
The authors’ observations
Managing factitious disorder can be challenging; patients can evoke strong feelings of countertransference during treatment.3,6,7 Providers might feel that the patient does not need to be treated, or that the patient is “not really sick.” This may induce anger and animosity toward the patient (therapeutic nihilism).8 These negative emotions are likely to disrupt the patient–provider relationship and exacerbate the patient’s symptoms.
It is generally accepted that the patient should be made aware of the treatment plan, in an indirect and tactful way, so that the patient does not feel “outed.” Unmasking the patient—the process of instilling insight—is a delicate step and can be a stressful time for the patient.9 A confrontational approach often places the patient’s sick role in doubt and does not address the pathological aspect of the disorder.
It is rare for a patient to admit to fabricating symptoms; confronted, the patient is likely to double their efforts to maintain the rouse of a fictional disease.10,11 It is important for the treatment team to be aware that patients frequently leave the treatment facility against medical advice, seek a different provider, or even pursue legal action for defamation against the treating physician.
Treating comorbid medical and psychiatric conditions is important for successful management of a patient with factitious disorder. Initiating valproic acid to address Ms. R’s bipolar depression contributed to her overall psychiatric stability. Initial treatment with a medication that is FDA-approved for treating bipolar depression, such as lurasidone, quetiapine, or olanzapine-fluoxetine combination, should be considered as an alternative. We chose valproic acid for Ms. R because of its previous efficacy, good tolerability, and the patient’s high level of comfort with the medication.
Which of the following are risk factors for factitious disorder?
a) lengthy medical treatments or hospitalizations as a child
b) female sex
c) experience as a health care worker
d) all of the above
OUTCOME Stabilization
Successful treatment during Ms. R’s inpatient psychiatric admission results in improved insight, remission of suicidal ideation, and stabilization of mood lability. She is discharged to the care of her family with a plan to follow up with a psychotherapist and psychiatrist. Continued administration of valproic acid continues to be effective after discharge.
Ms. R engages in frequent follow-up with outpatient psychiatric services. She remains engaged in psychotherapy and psychiatric care 1 year after discharge. Ms. R has made no report of pregnancy or required hospitalization during this time. She expresses trust in the mental health care system and acknowledges the role treatment played in her improvement.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.
1. Reyes FI, Winter JS, Faiman C. Postpartum disappearance of chorionic gonadotropin from the maternal and neonatal circulations. Am J Obstet Gynecol. 1985;153(5):486-489.
2. Avbersek A, Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? J Neurol Neurosurg Psychiatry. 2010;81(7):719-725.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Kapfhammer HP, Rothenhausler HM, Dietrich E, et al. Artifactual disorders—between deception and self-mutilation. Experiences in consultation psychiatry at a university clinic [in German]. Nervenarzt. 1998;69(5):401-409.
6. Feldman MD, Feldman JM. Tangled in the web: countertransference in the therapy of factitious disorders. Int J Psychiatry Med. 1995;25(4):389-399.
7. Wedel KR. A therapeutic confrontation approach to treating patients with factitious illness. Soc Work. 1971;16(2):69-73.
8. Feldman MD, Hamilton JC, Deemer HN. Factitious disorder. In: Phillips KA, ed. Somatoform and factitious disorder. Washington, DC: American Psychiatric Press; 2001:129-159.
9. Scher LM, Knudsen P, Leamon M. Somatic symptom and related disorders. In: Hales RE, Yudofsky SC, Weiss Roberts L, eds. The American Publishing Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing; 2014:531-556.
10. Lipsitt DR. Introduction. In: Feldman MD, Eisendrath SJ, eds. The spectrum of factitious disorders. Washington, DC: American Psychiatric Press; 1996:xix-xxviii.
11. van der Feltz-Cornelis CM. Confronting patients about a factitious disorder [in Dutch]. Ned Tidjschr Geneeskd. 2000;144(12):545-548.