User login
Confused with ataxia and urinary and fecal incontinence
CASE Paranoia, ataxia
Ms. S, age 46, is admitted to the hospital for cellulitis and gait disturbance. She has been living in her car for the past week and presents to the local fire department to get help for housing. She is referred to this hospital where she was found to have cellulitis in her buttock secondary to urinary and fecal incontinence. She also was noted to have difficulty ambulating and a wide-based gait. Two weeks earlier, a hotel clerk found her on the floor, unable to get up. Ms. S was seen in a local emergency room (ER) and discharged after her glucose level was found to be normal.
At admission, she has an intact sensorium and is described as disheveled, illogical, rambling, and paranoid. Her mental status exam shows she is alert and oriented to person and time, with guarded and childlike behavior. Her affect/mood is irritable and oddly related, and her thought processes are concrete and simple with some thought-blocking and paranoid content. She denies thoughts of harming herself or others, and her insight is limited and judgment is poor.
Neurology is consulted to evaluate her gait disturbance. Ms. S has decreased muscle bulk in both calves, with brisk knee reflexes bilaterally. CT imaging shows nonspecific scattered periventricular white matter hypodensities consistent with microvascular ischemic diagnosis, but a demyelinating process could not be ruled out. Ms. S reports that the gait disturbance began in childhood, and that her grandmother had the same gait disturbance. Neurology recommends an electromyogram and MRI.
During her stay in the hospital, she is unwilling to cooperate with exams, declines to answer questions regarding her past, and appears suspicious of her acute care treatment team. The psychiatric team is consulted for evaluation of her paranoia and “seeming disorganization,” and she is transferred to the psychiatric unit. She appears to be repulsed by the fact that she was in a psychiatric ward stating, “I don’t belong here” and “I’m scared of the other people here.” She denies any psychiatric history, previous hospitalizations, or substance use, and no documentation of inpatient or outpatient care was found in the county’s computerized record system. Although she is willing to take a small dose of tranquilizer (eg, lorazepam) she refuses to take antipsychotic medications saying, “My mother told me not to take [antipsychotics]. I’m not psychotic.”
What is your diagnosis at this point?
a) normal pressure hydrocephalus
b) Charcot-Marie-Tooth disease
c) schizophrenia spectrum disorder
d) multiple sclerosis (MS)
e) vascular dementia
f) cord lesion compression
The authors’ observations
The neurology team initially suspected Charcot-Marie-Tooth disease because her clinical presentation included pes cavus, distal lower extremity weakness, and lower extremity muscle atrophy with a self-reported family history of similar gait disturbance, all of which are consistent with Charcot-Marie-Tooth disease.
Subcortical syndrome—a feature of vascular dementia—is characterized by focal motor deficits, gait disturbance, history of unsteadiness with frequent falls, urinary symptoms, personality and mood changes, and cognitive dysfunction.1-3 Subcortical syndrome is caused by chronic ischemia and lacunar infarctions that affect cerebral nuclei and white matter pathways.1 On imaging, subcortical vascular dementia is characterized by leukoaraiosis, which are hypointense spherical-like lesions on CT and hyperintense lesions in periventricular areas on T2 MRI.4
Although normal pressure hydrocephalus could be suspected given her clinical presentation of the Hakim-Adams triad (ie,“wacky, wobbly, and wet”), her head CT did not show any changes consistent with this condition.
Her clinical presentation does not align with schizophrenia spectrum disorder because of her history of higher functioning, acute later onset, and the absence of hallucinations, fixed delusions, or markedly disorganized speech. Although she is paranoid of her surroundings, her delusions were ill-formed. A cord lesion compression cannot be ruled out, and MRI is required urgently.
HISTORY High functioning
When asked, Ms. S states that she was admitted to the hospital because “someone who looked like a fake police officer [a member of the fire department] told me it was nice here.” She indicates that she initially thought it would be a nice place to live temporarily but later regretted coming after realizing that she was in a psychiatry unit. Available documentation from her recent hospitalization indicated that she was living in a motel on her own. Ms. S says that she works as an actress and has had minor roles in famous movies. She says that she studied at a well-known performance arts school and that her parents are famous musicians; however, she refuses to identify her parents or give permission to contact them—or any other collateral informant—because she is embarrassed about her current situation stating, “They would never believe it.”
During this interview, Ms. S appears confused as well as disorganized—which was a challenge to clearly delineate—disheveled, and guarded with hypoverbal and hypophonic speech. Her thought process is circumstantial, and she seems to be confabulating. She denies visual or auditory hallucinations but appears paranoid and states that she thinks we are experimenting on her. Except for the neurological exam, the rest of her physical exam is within normal limits. Urine toxicology screen and labs are negative except for a positive antinuclear antibody homogenous pattern with a titer of 1:640; B12 vitamin levels are not tested.
MRI is ordered, however, she does not consent to the scan saying, “It’s creepy, I don’t want people looking at my brain.” The team makes several attempts to encourage her for consent but she refuses. Because of the clinical urgency (ie, possible cord compression) and her refusal to provide a surrogate decision maker, the team felt the situation is urgent, confirmed by 2 physicians, which led them to perform the MRI on an emergent basis. The MRI reveals multiple periventricular, juxtacortical, infratentorial, and likely cervical spinal cord T2 hyperintense lesions (Figure).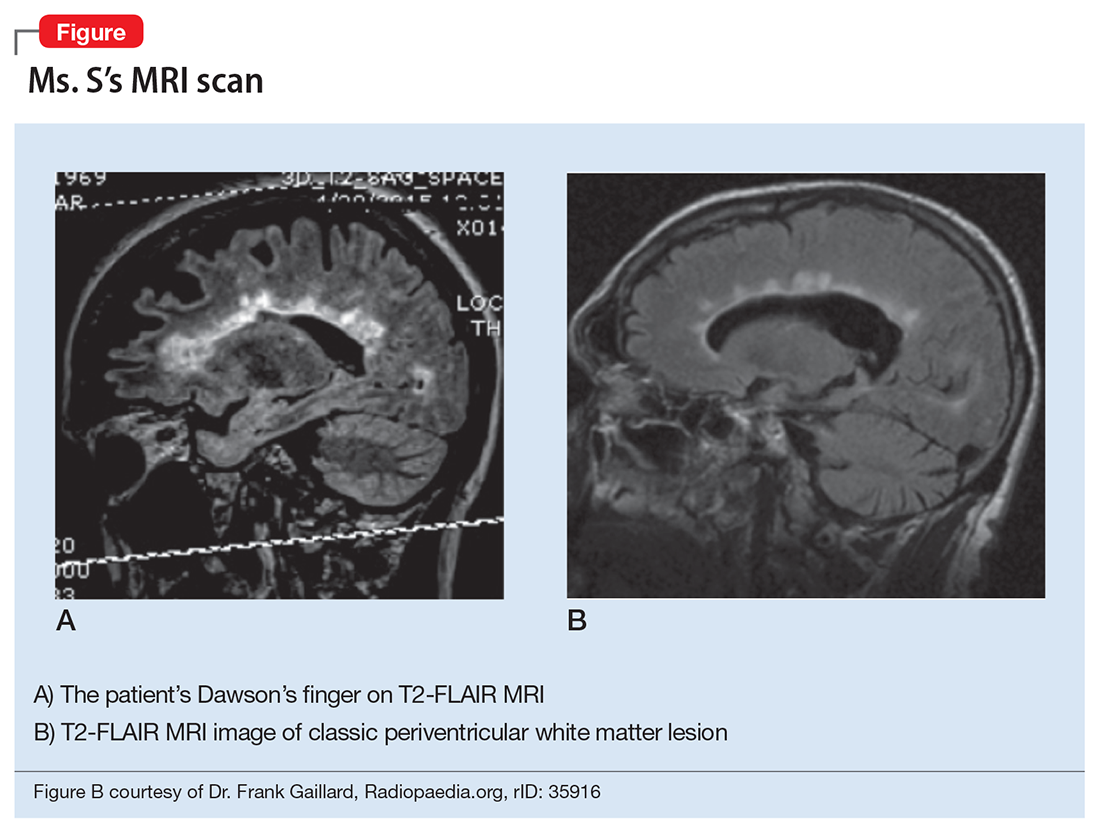
What would be your differential diagnosis at this time?
a) acute disseminated encephalomyelitis (ADEM)
b) systemic lupus erythematous
c) multiple sclerosis
d) vascular dementia
e) vitamin B deficiency
The authors’ observations
Psychosis in the presence of white matter demyelination could be associated with autoimmune, vascular, or nutritional disturbances. Deficiencies in vitamins B6, 9, and 12 (pyridoxine, folate, cobalamin) have been shown to cause neuropsychiatric symptoms and white matter lesions.5 Low levels of vitamins B6, 9, and 12 are associated with elevated homocysteine, which can cause small vessel ischemia leading to white matter lesions similar to changes seen in vascular dementia.5 The exact pathophysiology of ADEM is unclear, however, it is thought that after an infection, antiviral antibodies cross react with autoantigens on myelin causing an autoimmune demyelinating disease. Another hypothesized mechanism is that circulating immune complexes and humoral factors increase vascular permeability and inflammation thereby opening the blood–brain barrier. Once it is open, cells such as lymphocytes, phagocytes, and microglia cause gliosis and demyelination. Case reports have described ADEM associated with psychotic features.6
Likewise, systemic lupus erythematous has been associated with psychosis and neuropsychiatric symptoms in 14% to 75% of patients. Of these patients, 40% will experience neuropsychiatric symptoms before onset of lupus symptoms.7 One study found the most common MRI finding in neuropsychiatric systemic lupus erythematous was leukoaraiosis, which appeared in 57.1% of patients.8 Ms. S’s MRI results strongly suggest a diagnosis of MS.
EVALUATION Questionable story
Ms. S appears delusional and grandiose when she meets with the psychiatry team. She states that before her hospitalization, she was an actress and could ambulate, rent a motel room, and drive a car without assistance. However, during the examination, she cannot walk without 2 staff members for support, and overall her self-reported history sounds questionable. There were several pieces of evidence that corroborate portions of her story: (1) a screen actors guild card was found among personal belongings; (2) she was transported to the ER from a local motel; (3) she had recently visited another hospital and, at that time, was deemed stable enough to be discharged.
On the Montreal Cognitive Assessment (MoCA) Ms. S scored 19/30, with deficits mainly in executive/visuospatial and delayed recall memory. An alternate form of the MoCA is administered 1 day later, and she scores 20/30 with similar deficits. After obtaining medication consent, she is given risperidone, up to 2 mg/d, and becomes more cooperative with the treatment team.
The authors’ observations
Approximately 40% to 65% of MS patients experience cognitive impairment.9 Cognitive dysfunction in a depressed patient with MS might appear as pseudo-dementia, but other possible diagnoses include:
- true dementia
- encephalitis or infection
- medication- or substance-induced.
White matter demyelination is associated with subcortical dementia, which is characterized by slowness of information processing, forgetfulness, apathy, depression, and impaired cognition. According to meta-analyses, the most prominent neuropsychological deficits in MS are found in the areas of verbal fluency, information processing speed, working memory, and long-term memory.10 Relapsing-remitting type MS patients generally have less cognitive impairment than those with the chronic progressive type of the disease.
EVALUATION Cognitive deficits
Because of her acute condition and resistance to the evaluation, a modified screening neuropsychological battery is used. During the evaluation Ms. S is guarded and demonstrates paucity of speech; her responses are odd at times or contain word-substitution errors. Hand stiffness, tremor, and imprecision are noted during writing and drawing. Results of testing indicate average-range premorbid intellectual ability, with impairments in memory and information processing speed and a mild weakness in phonemic verbal fluency. Ms. S endorses statements reflecting paranoia and hostility on a self-report measure of emotional and personality functioning, consistent with her behavioral presentation. However, her responses on other subscales, including depression and psychotic symptoms, are within normal limits. Her cognitive deficits would be unusual if she had a psychiatric illness alone and are likely associated with her positive neuroimaging findings that suggest a demyelinating process. Overall, the results of the evaluation support a MS diagnosis.
The authors’ observations
Psychosis is found at a higher rate among MS patients (2% to 3%) than the general population (0.5% to 1%).9 Although rare, psychosis often can cloud the diagnosis of MS. Psychiatric symptoms that can occur in MS include:
- hallucinations and delusions (>50%)
- irritability and agitation (20%)
- grandiosity (15%)
- confusion, blunted affect, flight of ideas, depression, reduced self-care, and pressured speech (10%).11
A review of 10 studies found that depression was the most prevalent symptom in MS, and that schizophrenia occurred in up to 7% of MS patients.12 There are currently 3 theories about the relationship between psychosis and MS:
- MS and psychosis are thought to share the same pathophysiological process.
- Psychotic symptoms arise from regional demyelination simultaneously with MS.
- Psychosis is caused by medical treatment of MS.9
Other causes of psychiatric symptoms in MS include:
- depression associated with brain atrophy and lesions
- depression and anxiety as a result of chronic illness
- depression resulting from inflammatory changes
- corticosteroid treatment causing depression, mania, or psychosis.12
The link between psychosis and MS is still poorly understood and further investigation is needed.
How would you treat Ms. S?
a) haloperidol
b) risperidone
c) corticosteroids
d) selective serotonin reuptake inhibitors
Treating psychiatric symptoms in the context of MS
The literature, mainly case reports, suggests several treatment modalities for psychosis with MS. Clozapine has been shown to be beneficial in several case reports, and risperidone9 and ziprasidone13 also have been effective. Other studies recommended low-dose chlorpromazine.9
For MS patients with cognitive impairment, one study showed that interferon beta-1b (IFN-1b) treatment resulted in significant improvement in concentration, attention, visual learning, and recall after 1 year compared with control patients.9 However, there are also case reports of IFN-1b and glucocorticoid-induced psychosis in patients, which resolved after discontinuing treatment.9
Psychotic symptoms have been shown to resolve after corticosteroid treatment of MS.14 In another case report, mania and delusions subsided 3 days after IV methylprednisolone, whereas risperidone had no effect on psychotic features. However, it was unclear whether risperidone was discontinued when methylprednisolone was administered, therefore the specific effect of methylprednisolone is difficult to discern.15 Finally, in a case of a patient who has chronic MS for 16 years and presented with acute onset paranoid psychosis, symptoms resolved with aripiprazole, 10 to 20 mg/d.16 Because of the limited utility of case reports, there is a need for further research in medical management of psychiatric symptoms in MS.
1. de Groot JC, de Leeuw FE, Oudkerk M, et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000;47(2):145-151.
2. Tatemichi TK, Desmond DW, Prohovnik I, et al. Confusion and memory loss from capsular genu infarction: a thalamocortical disconnection syndrome? Neurology. 1992;42(10):1966-1979.
3. Staekenborg SS, van der Flier WM, van Straaten EC, et al. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke. 2008;39(2):317-322.
4. Mortimer A, Likeman M, Lewis T. Neuroimaging in dementia: a practical guide. Pract Neurol. 2013;13(2):92-103.
5. Xiong YY, Mok V. Age-related white matter changes. J Aging Res. 2011;2011:617927. doi:10.4061/2011/617927.
6. Habek M, Brinar M, Brinar VV, et al. Psychiatric manifestations of multiple sclerosis and acute disseminated encephalomyelitis. Clin Neurol Neurosug. 2006;108(3);290-294.
7. Benros ME, Eaton WW, Mortensen PB. The epidemiologic evidence linking autoimmune disease and psychosis. Biol Psychiatry. 2014;75(4);300-306.
8. Jeong HW, Her M, Bae JS, et al. Brain MRI in neuropsychiatric lupus: associations with the 1999 ACR case definitions. Rheumatol Int. 2014;35(5):861-869.
9. Haussleiter IS, Brüne M, Juckel G. Psychopathology in multiple sclerosis: diagnosis, prevalence and treatment. Ther Adv Neurol Disord. 2009;2(1):13-29.
10. Thornton AE, DeFreitas VG. The neuropsychology of multiple sclerosis. In: Grant I, Adams KM, eds. Neuropsychological assessment of neuropsychiatric and neuromedical disorders. New York, NY: Oxford University Press; 2009:280-305.
11. Kosmidis MH, Giannakou M, Messinis L, et al. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010;22(1):55-66.
12. Marrie RA, Reingold S, Cohen J, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. 2015;21(3):305-317.
13. Davids E, Hartwig U, Gastpar M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(4):734-744.
14. Thöne J, Kessler E. Improvement of neuropsychiatric symptoms in multiple sclerosis subsequent to high-dose corticosteroid treatment. Prim Care Companion J Clin Psychiatry. 2008;10(2):163-164.
15. Hoiter S, Maltete D, Bourre B, et al. A manic episode with psychotic features improved by methylprednisolone in a patient with multiple sclerosis. Gen Hosp Psychiatry. 2015;37(6):621.e1-621.e2.
16. Muzyk AJ, Christopher EJ, Gagliardi JP, et al. Use of aripiprazole in a patient with multiple sclerosis presenting with paranoid psychosis. J Psychiatr Pract. 2010;16(6):420-424.
CASE Paranoia, ataxia
Ms. S, age 46, is admitted to the hospital for cellulitis and gait disturbance. She has been living in her car for the past week and presents to the local fire department to get help for housing. She is referred to this hospital where she was found to have cellulitis in her buttock secondary to urinary and fecal incontinence. She also was noted to have difficulty ambulating and a wide-based gait. Two weeks earlier, a hotel clerk found her on the floor, unable to get up. Ms. S was seen in a local emergency room (ER) and discharged after her glucose level was found to be normal.
At admission, she has an intact sensorium and is described as disheveled, illogical, rambling, and paranoid. Her mental status exam shows she is alert and oriented to person and time, with guarded and childlike behavior. Her affect/mood is irritable and oddly related, and her thought processes are concrete and simple with some thought-blocking and paranoid content. She denies thoughts of harming herself or others, and her insight is limited and judgment is poor.
Neurology is consulted to evaluate her gait disturbance. Ms. S has decreased muscle bulk in both calves, with brisk knee reflexes bilaterally. CT imaging shows nonspecific scattered periventricular white matter hypodensities consistent with microvascular ischemic diagnosis, but a demyelinating process could not be ruled out. Ms. S reports that the gait disturbance began in childhood, and that her grandmother had the same gait disturbance. Neurology recommends an electromyogram and MRI.
During her stay in the hospital, she is unwilling to cooperate with exams, declines to answer questions regarding her past, and appears suspicious of her acute care treatment team. The psychiatric team is consulted for evaluation of her paranoia and “seeming disorganization,” and she is transferred to the psychiatric unit. She appears to be repulsed by the fact that she was in a psychiatric ward stating, “I don’t belong here” and “I’m scared of the other people here.” She denies any psychiatric history, previous hospitalizations, or substance use, and no documentation of inpatient or outpatient care was found in the county’s computerized record system. Although she is willing to take a small dose of tranquilizer (eg, lorazepam) she refuses to take antipsychotic medications saying, “My mother told me not to take [antipsychotics]. I’m not psychotic.”
What is your diagnosis at this point?
a) normal pressure hydrocephalus
b) Charcot-Marie-Tooth disease
c) schizophrenia spectrum disorder
d) multiple sclerosis (MS)
e) vascular dementia
f) cord lesion compression
The authors’ observations
The neurology team initially suspected Charcot-Marie-Tooth disease because her clinical presentation included pes cavus, distal lower extremity weakness, and lower extremity muscle atrophy with a self-reported family history of similar gait disturbance, all of which are consistent with Charcot-Marie-Tooth disease.
Subcortical syndrome—a feature of vascular dementia—is characterized by focal motor deficits, gait disturbance, history of unsteadiness with frequent falls, urinary symptoms, personality and mood changes, and cognitive dysfunction.1-3 Subcortical syndrome is caused by chronic ischemia and lacunar infarctions that affect cerebral nuclei and white matter pathways.1 On imaging, subcortical vascular dementia is characterized by leukoaraiosis, which are hypointense spherical-like lesions on CT and hyperintense lesions in periventricular areas on T2 MRI.4
Although normal pressure hydrocephalus could be suspected given her clinical presentation of the Hakim-Adams triad (ie,“wacky, wobbly, and wet”), her head CT did not show any changes consistent with this condition.
Her clinical presentation does not align with schizophrenia spectrum disorder because of her history of higher functioning, acute later onset, and the absence of hallucinations, fixed delusions, or markedly disorganized speech. Although she is paranoid of her surroundings, her delusions were ill-formed. A cord lesion compression cannot be ruled out, and MRI is required urgently.
HISTORY High functioning
When asked, Ms. S states that she was admitted to the hospital because “someone who looked like a fake police officer [a member of the fire department] told me it was nice here.” She indicates that she initially thought it would be a nice place to live temporarily but later regretted coming after realizing that she was in a psychiatry unit. Available documentation from her recent hospitalization indicated that she was living in a motel on her own. Ms. S says that she works as an actress and has had minor roles in famous movies. She says that she studied at a well-known performance arts school and that her parents are famous musicians; however, she refuses to identify her parents or give permission to contact them—or any other collateral informant—because she is embarrassed about her current situation stating, “They would never believe it.”
During this interview, Ms. S appears confused as well as disorganized—which was a challenge to clearly delineate—disheveled, and guarded with hypoverbal and hypophonic speech. Her thought process is circumstantial, and she seems to be confabulating. She denies visual or auditory hallucinations but appears paranoid and states that she thinks we are experimenting on her. Except for the neurological exam, the rest of her physical exam is within normal limits. Urine toxicology screen and labs are negative except for a positive antinuclear antibody homogenous pattern with a titer of 1:640; B12 vitamin levels are not tested.
MRI is ordered, however, she does not consent to the scan saying, “It’s creepy, I don’t want people looking at my brain.” The team makes several attempts to encourage her for consent but she refuses. Because of the clinical urgency (ie, possible cord compression) and her refusal to provide a surrogate decision maker, the team felt the situation is urgent, confirmed by 2 physicians, which led them to perform the MRI on an emergent basis. The MRI reveals multiple periventricular, juxtacortical, infratentorial, and likely cervical spinal cord T2 hyperintense lesions (Figure).
What would be your differential diagnosis at this time?
a) acute disseminated encephalomyelitis (ADEM)
b) systemic lupus erythematous
c) multiple sclerosis
d) vascular dementia
e) vitamin B deficiency
The authors’ observations
Psychosis in the presence of white matter demyelination could be associated with autoimmune, vascular, or nutritional disturbances. Deficiencies in vitamins B6, 9, and 12 (pyridoxine, folate, cobalamin) have been shown to cause neuropsychiatric symptoms and white matter lesions.5 Low levels of vitamins B6, 9, and 12 are associated with elevated homocysteine, which can cause small vessel ischemia leading to white matter lesions similar to changes seen in vascular dementia.5 The exact pathophysiology of ADEM is unclear, however, it is thought that after an infection, antiviral antibodies cross react with autoantigens on myelin causing an autoimmune demyelinating disease. Another hypothesized mechanism is that circulating immune complexes and humoral factors increase vascular permeability and inflammation thereby opening the blood–brain barrier. Once it is open, cells such as lymphocytes, phagocytes, and microglia cause gliosis and demyelination. Case reports have described ADEM associated with psychotic features.6
Likewise, systemic lupus erythematous has been associated with psychosis and neuropsychiatric symptoms in 14% to 75% of patients. Of these patients, 40% will experience neuropsychiatric symptoms before onset of lupus symptoms.7 One study found the most common MRI finding in neuropsychiatric systemic lupus erythematous was leukoaraiosis, which appeared in 57.1% of patients.8 Ms. S’s MRI results strongly suggest a diagnosis of MS.
EVALUATION Questionable story
Ms. S appears delusional and grandiose when she meets with the psychiatry team. She states that before her hospitalization, she was an actress and could ambulate, rent a motel room, and drive a car without assistance. However, during the examination, she cannot walk without 2 staff members for support, and overall her self-reported history sounds questionable. There were several pieces of evidence that corroborate portions of her story: (1) a screen actors guild card was found among personal belongings; (2) she was transported to the ER from a local motel; (3) she had recently visited another hospital and, at that time, was deemed stable enough to be discharged.
On the Montreal Cognitive Assessment (MoCA) Ms. S scored 19/30, with deficits mainly in executive/visuospatial and delayed recall memory. An alternate form of the MoCA is administered 1 day later, and she scores 20/30 with similar deficits. After obtaining medication consent, she is given risperidone, up to 2 mg/d, and becomes more cooperative with the treatment team.
The authors’ observations
Approximately 40% to 65% of MS patients experience cognitive impairment.9 Cognitive dysfunction in a depressed patient with MS might appear as pseudo-dementia, but other possible diagnoses include:
- true dementia
- encephalitis or infection
- medication- or substance-induced.
White matter demyelination is associated with subcortical dementia, which is characterized by slowness of information processing, forgetfulness, apathy, depression, and impaired cognition. According to meta-analyses, the most prominent neuropsychological deficits in MS are found in the areas of verbal fluency, information processing speed, working memory, and long-term memory.10 Relapsing-remitting type MS patients generally have less cognitive impairment than those with the chronic progressive type of the disease.
EVALUATION Cognitive deficits
Because of her acute condition and resistance to the evaluation, a modified screening neuropsychological battery is used. During the evaluation Ms. S is guarded and demonstrates paucity of speech; her responses are odd at times or contain word-substitution errors. Hand stiffness, tremor, and imprecision are noted during writing and drawing. Results of testing indicate average-range premorbid intellectual ability, with impairments in memory and information processing speed and a mild weakness in phonemic verbal fluency. Ms. S endorses statements reflecting paranoia and hostility on a self-report measure of emotional and personality functioning, consistent with her behavioral presentation. However, her responses on other subscales, including depression and psychotic symptoms, are within normal limits. Her cognitive deficits would be unusual if she had a psychiatric illness alone and are likely associated with her positive neuroimaging findings that suggest a demyelinating process. Overall, the results of the evaluation support a MS diagnosis.
The authors’ observations
Psychosis is found at a higher rate among MS patients (2% to 3%) than the general population (0.5% to 1%).9 Although rare, psychosis often can cloud the diagnosis of MS. Psychiatric symptoms that can occur in MS include:
- hallucinations and delusions (>50%)
- irritability and agitation (20%)
- grandiosity (15%)
- confusion, blunted affect, flight of ideas, depression, reduced self-care, and pressured speech (10%).11
A review of 10 studies found that depression was the most prevalent symptom in MS, and that schizophrenia occurred in up to 7% of MS patients.12 There are currently 3 theories about the relationship between psychosis and MS:
- MS and psychosis are thought to share the same pathophysiological process.
- Psychotic symptoms arise from regional demyelination simultaneously with MS.
- Psychosis is caused by medical treatment of MS.9
Other causes of psychiatric symptoms in MS include:
- depression associated with brain atrophy and lesions
- depression and anxiety as a result of chronic illness
- depression resulting from inflammatory changes
- corticosteroid treatment causing depression, mania, or psychosis.12
The link between psychosis and MS is still poorly understood and further investigation is needed.
How would you treat Ms. S?
a) haloperidol
b) risperidone
c) corticosteroids
d) selective serotonin reuptake inhibitors
Treating psychiatric symptoms in the context of MS
The literature, mainly case reports, suggests several treatment modalities for psychosis with MS. Clozapine has been shown to be beneficial in several case reports, and risperidone9 and ziprasidone13 also have been effective. Other studies recommended low-dose chlorpromazine.9
For MS patients with cognitive impairment, one study showed that interferon beta-1b (IFN-1b) treatment resulted in significant improvement in concentration, attention, visual learning, and recall after 1 year compared with control patients.9 However, there are also case reports of IFN-1b and glucocorticoid-induced psychosis in patients, which resolved after discontinuing treatment.9
Psychotic symptoms have been shown to resolve after corticosteroid treatment of MS.14 In another case report, mania and delusions subsided 3 days after IV methylprednisolone, whereas risperidone had no effect on psychotic features. However, it was unclear whether risperidone was discontinued when methylprednisolone was administered, therefore the specific effect of methylprednisolone is difficult to discern.15 Finally, in a case of a patient who has chronic MS for 16 years and presented with acute onset paranoid psychosis, symptoms resolved with aripiprazole, 10 to 20 mg/d.16 Because of the limited utility of case reports, there is a need for further research in medical management of psychiatric symptoms in MS.
CASE Paranoia, ataxia
Ms. S, age 46, is admitted to the hospital for cellulitis and gait disturbance. She has been living in her car for the past week and presents to the local fire department to get help for housing. She is referred to this hospital where she was found to have cellulitis in her buttock secondary to urinary and fecal incontinence. She also was noted to have difficulty ambulating and a wide-based gait. Two weeks earlier, a hotel clerk found her on the floor, unable to get up. Ms. S was seen in a local emergency room (ER) and discharged after her glucose level was found to be normal.
At admission, she has an intact sensorium and is described as disheveled, illogical, rambling, and paranoid. Her mental status exam shows she is alert and oriented to person and time, with guarded and childlike behavior. Her affect/mood is irritable and oddly related, and her thought processes are concrete and simple with some thought-blocking and paranoid content. She denies thoughts of harming herself or others, and her insight is limited and judgment is poor.
Neurology is consulted to evaluate her gait disturbance. Ms. S has decreased muscle bulk in both calves, with brisk knee reflexes bilaterally. CT imaging shows nonspecific scattered periventricular white matter hypodensities consistent with microvascular ischemic diagnosis, but a demyelinating process could not be ruled out. Ms. S reports that the gait disturbance began in childhood, and that her grandmother had the same gait disturbance. Neurology recommends an electromyogram and MRI.
During her stay in the hospital, she is unwilling to cooperate with exams, declines to answer questions regarding her past, and appears suspicious of her acute care treatment team. The psychiatric team is consulted for evaluation of her paranoia and “seeming disorganization,” and she is transferred to the psychiatric unit. She appears to be repulsed by the fact that she was in a psychiatric ward stating, “I don’t belong here” and “I’m scared of the other people here.” She denies any psychiatric history, previous hospitalizations, or substance use, and no documentation of inpatient or outpatient care was found in the county’s computerized record system. Although she is willing to take a small dose of tranquilizer (eg, lorazepam) she refuses to take antipsychotic medications saying, “My mother told me not to take [antipsychotics]. I’m not psychotic.”
What is your diagnosis at this point?
a) normal pressure hydrocephalus
b) Charcot-Marie-Tooth disease
c) schizophrenia spectrum disorder
d) multiple sclerosis (MS)
e) vascular dementia
f) cord lesion compression
The authors’ observations
The neurology team initially suspected Charcot-Marie-Tooth disease because her clinical presentation included pes cavus, distal lower extremity weakness, and lower extremity muscle atrophy with a self-reported family history of similar gait disturbance, all of which are consistent with Charcot-Marie-Tooth disease.
Subcortical syndrome—a feature of vascular dementia—is characterized by focal motor deficits, gait disturbance, history of unsteadiness with frequent falls, urinary symptoms, personality and mood changes, and cognitive dysfunction.1-3 Subcortical syndrome is caused by chronic ischemia and lacunar infarctions that affect cerebral nuclei and white matter pathways.1 On imaging, subcortical vascular dementia is characterized by leukoaraiosis, which are hypointense spherical-like lesions on CT and hyperintense lesions in periventricular areas on T2 MRI.4
Although normal pressure hydrocephalus could be suspected given her clinical presentation of the Hakim-Adams triad (ie,“wacky, wobbly, and wet”), her head CT did not show any changes consistent with this condition.
Her clinical presentation does not align with schizophrenia spectrum disorder because of her history of higher functioning, acute later onset, and the absence of hallucinations, fixed delusions, or markedly disorganized speech. Although she is paranoid of her surroundings, her delusions were ill-formed. A cord lesion compression cannot be ruled out, and MRI is required urgently.
HISTORY High functioning
When asked, Ms. S states that she was admitted to the hospital because “someone who looked like a fake police officer [a member of the fire department] told me it was nice here.” She indicates that she initially thought it would be a nice place to live temporarily but later regretted coming after realizing that she was in a psychiatry unit. Available documentation from her recent hospitalization indicated that she was living in a motel on her own. Ms. S says that she works as an actress and has had minor roles in famous movies. She says that she studied at a well-known performance arts school and that her parents are famous musicians; however, she refuses to identify her parents or give permission to contact them—or any other collateral informant—because she is embarrassed about her current situation stating, “They would never believe it.”
During this interview, Ms. S appears confused as well as disorganized—which was a challenge to clearly delineate—disheveled, and guarded with hypoverbal and hypophonic speech. Her thought process is circumstantial, and she seems to be confabulating. She denies visual or auditory hallucinations but appears paranoid and states that she thinks we are experimenting on her. Except for the neurological exam, the rest of her physical exam is within normal limits. Urine toxicology screen and labs are negative except for a positive antinuclear antibody homogenous pattern with a titer of 1:640; B12 vitamin levels are not tested.
MRI is ordered, however, she does not consent to the scan saying, “It’s creepy, I don’t want people looking at my brain.” The team makes several attempts to encourage her for consent but she refuses. Because of the clinical urgency (ie, possible cord compression) and her refusal to provide a surrogate decision maker, the team felt the situation is urgent, confirmed by 2 physicians, which led them to perform the MRI on an emergent basis. The MRI reveals multiple periventricular, juxtacortical, infratentorial, and likely cervical spinal cord T2 hyperintense lesions (Figure).
What would be your differential diagnosis at this time?
a) acute disseminated encephalomyelitis (ADEM)
b) systemic lupus erythematous
c) multiple sclerosis
d) vascular dementia
e) vitamin B deficiency
The authors’ observations
Psychosis in the presence of white matter demyelination could be associated with autoimmune, vascular, or nutritional disturbances. Deficiencies in vitamins B6, 9, and 12 (pyridoxine, folate, cobalamin) have been shown to cause neuropsychiatric symptoms and white matter lesions.5 Low levels of vitamins B6, 9, and 12 are associated with elevated homocysteine, which can cause small vessel ischemia leading to white matter lesions similar to changes seen in vascular dementia.5 The exact pathophysiology of ADEM is unclear, however, it is thought that after an infection, antiviral antibodies cross react with autoantigens on myelin causing an autoimmune demyelinating disease. Another hypothesized mechanism is that circulating immune complexes and humoral factors increase vascular permeability and inflammation thereby opening the blood–brain barrier. Once it is open, cells such as lymphocytes, phagocytes, and microglia cause gliosis and demyelination. Case reports have described ADEM associated with psychotic features.6
Likewise, systemic lupus erythematous has been associated with psychosis and neuropsychiatric symptoms in 14% to 75% of patients. Of these patients, 40% will experience neuropsychiatric symptoms before onset of lupus symptoms.7 One study found the most common MRI finding in neuropsychiatric systemic lupus erythematous was leukoaraiosis, which appeared in 57.1% of patients.8 Ms. S’s MRI results strongly suggest a diagnosis of MS.
EVALUATION Questionable story
Ms. S appears delusional and grandiose when she meets with the psychiatry team. She states that before her hospitalization, she was an actress and could ambulate, rent a motel room, and drive a car without assistance. However, during the examination, she cannot walk without 2 staff members for support, and overall her self-reported history sounds questionable. There were several pieces of evidence that corroborate portions of her story: (1) a screen actors guild card was found among personal belongings; (2) she was transported to the ER from a local motel; (3) she had recently visited another hospital and, at that time, was deemed stable enough to be discharged.
On the Montreal Cognitive Assessment (MoCA) Ms. S scored 19/30, with deficits mainly in executive/visuospatial and delayed recall memory. An alternate form of the MoCA is administered 1 day later, and she scores 20/30 with similar deficits. After obtaining medication consent, she is given risperidone, up to 2 mg/d, and becomes more cooperative with the treatment team.
The authors’ observations
Approximately 40% to 65% of MS patients experience cognitive impairment.9 Cognitive dysfunction in a depressed patient with MS might appear as pseudo-dementia, but other possible diagnoses include:
- true dementia
- encephalitis or infection
- medication- or substance-induced.
White matter demyelination is associated with subcortical dementia, which is characterized by slowness of information processing, forgetfulness, apathy, depression, and impaired cognition. According to meta-analyses, the most prominent neuropsychological deficits in MS are found in the areas of verbal fluency, information processing speed, working memory, and long-term memory.10 Relapsing-remitting type MS patients generally have less cognitive impairment than those with the chronic progressive type of the disease.
EVALUATION Cognitive deficits
Because of her acute condition and resistance to the evaluation, a modified screening neuropsychological battery is used. During the evaluation Ms. S is guarded and demonstrates paucity of speech; her responses are odd at times or contain word-substitution errors. Hand stiffness, tremor, and imprecision are noted during writing and drawing. Results of testing indicate average-range premorbid intellectual ability, with impairments in memory and information processing speed and a mild weakness in phonemic verbal fluency. Ms. S endorses statements reflecting paranoia and hostility on a self-report measure of emotional and personality functioning, consistent with her behavioral presentation. However, her responses on other subscales, including depression and psychotic symptoms, are within normal limits. Her cognitive deficits would be unusual if she had a psychiatric illness alone and are likely associated with her positive neuroimaging findings that suggest a demyelinating process. Overall, the results of the evaluation support a MS diagnosis.
The authors’ observations
Psychosis is found at a higher rate among MS patients (2% to 3%) than the general population (0.5% to 1%).9 Although rare, psychosis often can cloud the diagnosis of MS. Psychiatric symptoms that can occur in MS include:
- hallucinations and delusions (>50%)
- irritability and agitation (20%)
- grandiosity (15%)
- confusion, blunted affect, flight of ideas, depression, reduced self-care, and pressured speech (10%).11
A review of 10 studies found that depression was the most prevalent symptom in MS, and that schizophrenia occurred in up to 7% of MS patients.12 There are currently 3 theories about the relationship between psychosis and MS:
- MS and psychosis are thought to share the same pathophysiological process.
- Psychotic symptoms arise from regional demyelination simultaneously with MS.
- Psychosis is caused by medical treatment of MS.9
Other causes of psychiatric symptoms in MS include:
- depression associated with brain atrophy and lesions
- depression and anxiety as a result of chronic illness
- depression resulting from inflammatory changes
- corticosteroid treatment causing depression, mania, or psychosis.12
The link between psychosis and MS is still poorly understood and further investigation is needed.
How would you treat Ms. S?
a) haloperidol
b) risperidone
c) corticosteroids
d) selective serotonin reuptake inhibitors
Treating psychiatric symptoms in the context of MS
The literature, mainly case reports, suggests several treatment modalities for psychosis with MS. Clozapine has been shown to be beneficial in several case reports, and risperidone9 and ziprasidone13 also have been effective. Other studies recommended low-dose chlorpromazine.9
For MS patients with cognitive impairment, one study showed that interferon beta-1b (IFN-1b) treatment resulted in significant improvement in concentration, attention, visual learning, and recall after 1 year compared with control patients.9 However, there are also case reports of IFN-1b and glucocorticoid-induced psychosis in patients, which resolved after discontinuing treatment.9
Psychotic symptoms have been shown to resolve after corticosteroid treatment of MS.14 In another case report, mania and delusions subsided 3 days after IV methylprednisolone, whereas risperidone had no effect on psychotic features. However, it was unclear whether risperidone was discontinued when methylprednisolone was administered, therefore the specific effect of methylprednisolone is difficult to discern.15 Finally, in a case of a patient who has chronic MS for 16 years and presented with acute onset paranoid psychosis, symptoms resolved with aripiprazole, 10 to 20 mg/d.16 Because of the limited utility of case reports, there is a need for further research in medical management of psychiatric symptoms in MS.
1. de Groot JC, de Leeuw FE, Oudkerk M, et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000;47(2):145-151.
2. Tatemichi TK, Desmond DW, Prohovnik I, et al. Confusion and memory loss from capsular genu infarction: a thalamocortical disconnection syndrome? Neurology. 1992;42(10):1966-1979.
3. Staekenborg SS, van der Flier WM, van Straaten EC, et al. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke. 2008;39(2):317-322.
4. Mortimer A, Likeman M, Lewis T. Neuroimaging in dementia: a practical guide. Pract Neurol. 2013;13(2):92-103.
5. Xiong YY, Mok V. Age-related white matter changes. J Aging Res. 2011;2011:617927. doi:10.4061/2011/617927.
6. Habek M, Brinar M, Brinar VV, et al. Psychiatric manifestations of multiple sclerosis and acute disseminated encephalomyelitis. Clin Neurol Neurosug. 2006;108(3);290-294.
7. Benros ME, Eaton WW, Mortensen PB. The epidemiologic evidence linking autoimmune disease and psychosis. Biol Psychiatry. 2014;75(4);300-306.
8. Jeong HW, Her M, Bae JS, et al. Brain MRI in neuropsychiatric lupus: associations with the 1999 ACR case definitions. Rheumatol Int. 2014;35(5):861-869.
9. Haussleiter IS, Brüne M, Juckel G. Psychopathology in multiple sclerosis: diagnosis, prevalence and treatment. Ther Adv Neurol Disord. 2009;2(1):13-29.
10. Thornton AE, DeFreitas VG. The neuropsychology of multiple sclerosis. In: Grant I, Adams KM, eds. Neuropsychological assessment of neuropsychiatric and neuromedical disorders. New York, NY: Oxford University Press; 2009:280-305.
11. Kosmidis MH, Giannakou M, Messinis L, et al. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010;22(1):55-66.
12. Marrie RA, Reingold S, Cohen J, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. 2015;21(3):305-317.
13. Davids E, Hartwig U, Gastpar M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(4):734-744.
14. Thöne J, Kessler E. Improvement of neuropsychiatric symptoms in multiple sclerosis subsequent to high-dose corticosteroid treatment. Prim Care Companion J Clin Psychiatry. 2008;10(2):163-164.
15. Hoiter S, Maltete D, Bourre B, et al. A manic episode with psychotic features improved by methylprednisolone in a patient with multiple sclerosis. Gen Hosp Psychiatry. 2015;37(6):621.e1-621.e2.
16. Muzyk AJ, Christopher EJ, Gagliardi JP, et al. Use of aripiprazole in a patient with multiple sclerosis presenting with paranoid psychosis. J Psychiatr Pract. 2010;16(6):420-424.
1. de Groot JC, de Leeuw FE, Oudkerk M, et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000;47(2):145-151.
2. Tatemichi TK, Desmond DW, Prohovnik I, et al. Confusion and memory loss from capsular genu infarction: a thalamocortical disconnection syndrome? Neurology. 1992;42(10):1966-1979.
3. Staekenborg SS, van der Flier WM, van Straaten EC, et al. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke. 2008;39(2):317-322.
4. Mortimer A, Likeman M, Lewis T. Neuroimaging in dementia: a practical guide. Pract Neurol. 2013;13(2):92-103.
5. Xiong YY, Mok V. Age-related white matter changes. J Aging Res. 2011;2011:617927. doi:10.4061/2011/617927.
6. Habek M, Brinar M, Brinar VV, et al. Psychiatric manifestations of multiple sclerosis and acute disseminated encephalomyelitis. Clin Neurol Neurosug. 2006;108(3);290-294.
7. Benros ME, Eaton WW, Mortensen PB. The epidemiologic evidence linking autoimmune disease and psychosis. Biol Psychiatry. 2014;75(4);300-306.
8. Jeong HW, Her M, Bae JS, et al. Brain MRI in neuropsychiatric lupus: associations with the 1999 ACR case definitions. Rheumatol Int. 2014;35(5):861-869.
9. Haussleiter IS, Brüne M, Juckel G. Psychopathology in multiple sclerosis: diagnosis, prevalence and treatment. Ther Adv Neurol Disord. 2009;2(1):13-29.
10. Thornton AE, DeFreitas VG. The neuropsychology of multiple sclerosis. In: Grant I, Adams KM, eds. Neuropsychological assessment of neuropsychiatric and neuromedical disorders. New York, NY: Oxford University Press; 2009:280-305.
11. Kosmidis MH, Giannakou M, Messinis L, et al. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010;22(1):55-66.
12. Marrie RA, Reingold S, Cohen J, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. 2015;21(3):305-317.
13. Davids E, Hartwig U, Gastpar M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(4):734-744.
14. Thöne J, Kessler E. Improvement of neuropsychiatric symptoms in multiple sclerosis subsequent to high-dose corticosteroid treatment. Prim Care Companion J Clin Psychiatry. 2008;10(2):163-164.
15. Hoiter S, Maltete D, Bourre B, et al. A manic episode with psychotic features improved by methylprednisolone in a patient with multiple sclerosis. Gen Hosp Psychiatry. 2015;37(6):621.e1-621.e2.
16. Muzyk AJ, Christopher EJ, Gagliardi JP, et al. Use of aripiprazole in a patient with multiple sclerosis presenting with paranoid psychosis. J Psychiatr Pract. 2010;16(6):420-424.
Worsening agitation and hallucinations: Could it be PTSD?
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.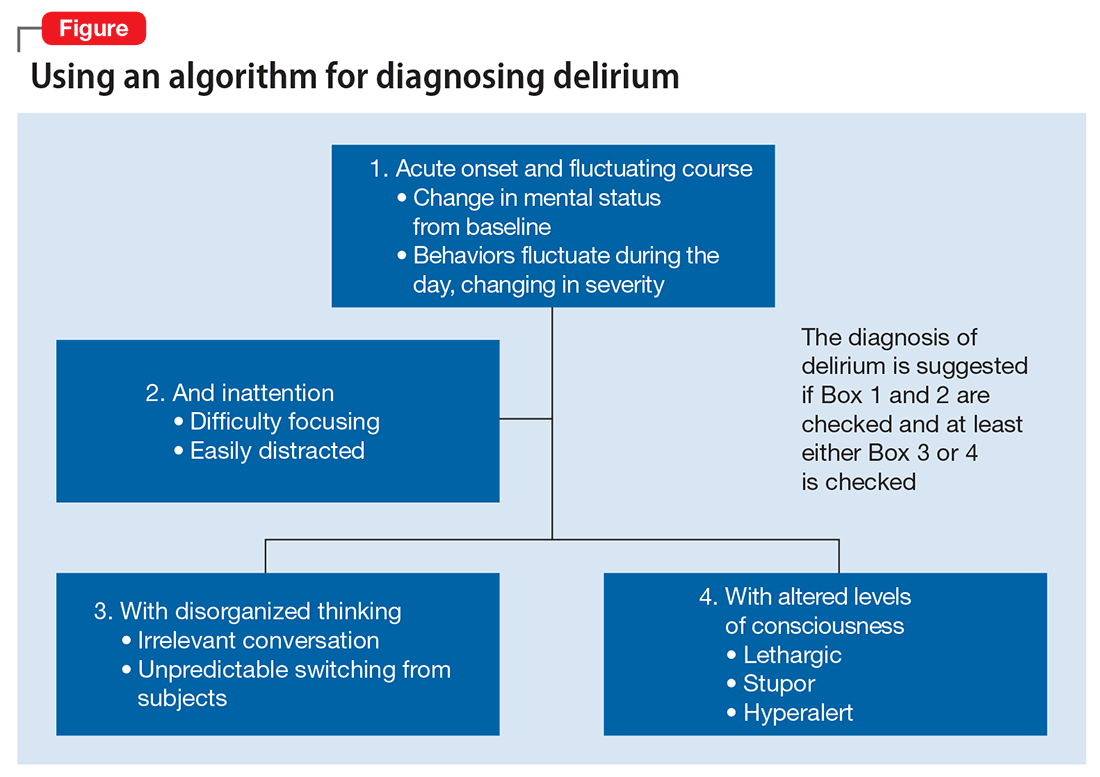
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
When to prescribe antidepressants to treat comorbid depression and pain disorders
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.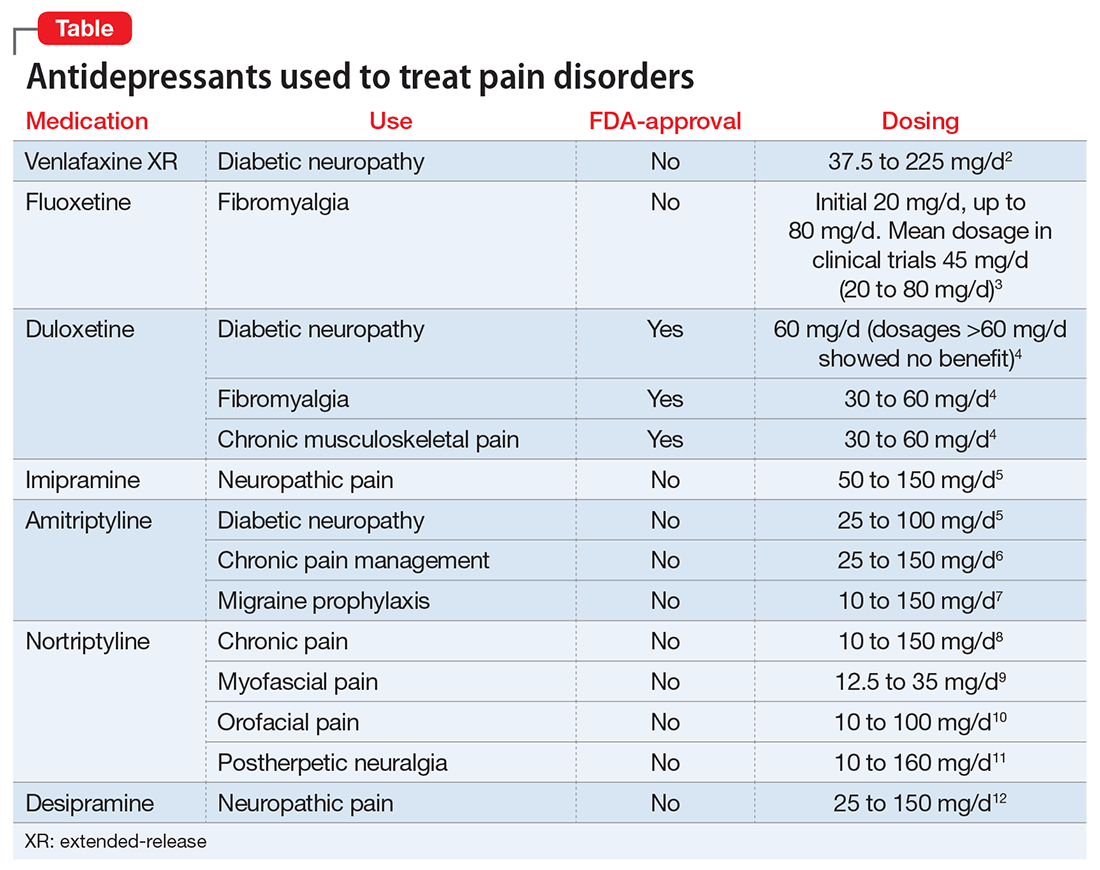
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
Current Therapeutic Approaches to Renal Cell Carcinoma
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
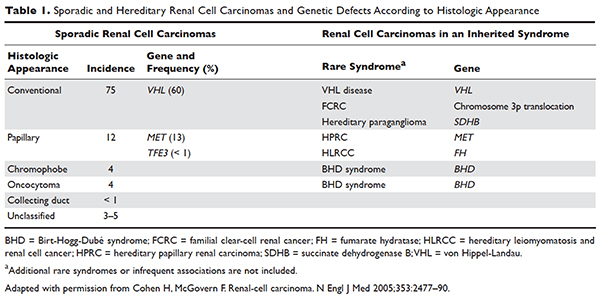
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
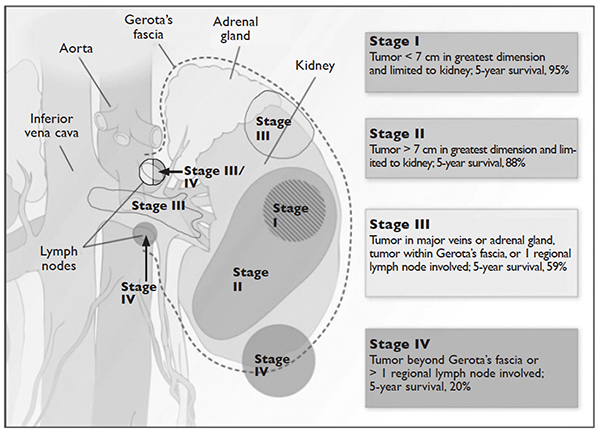
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
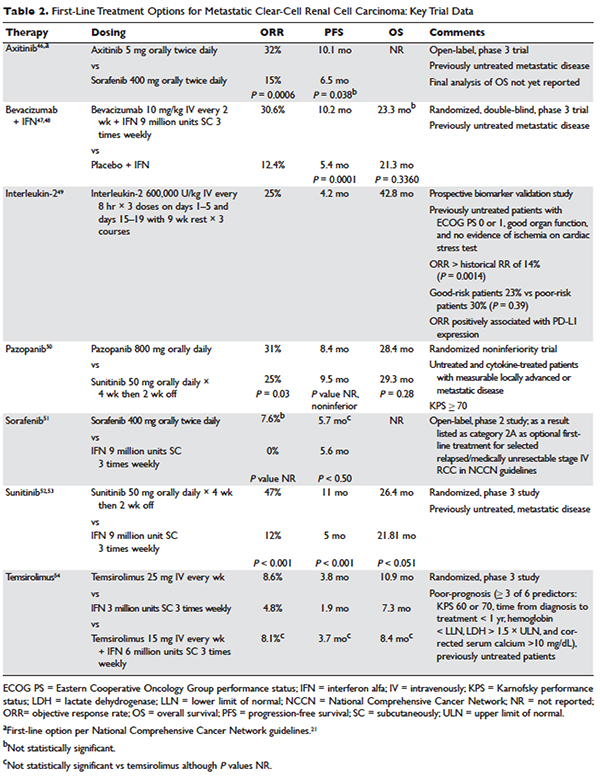
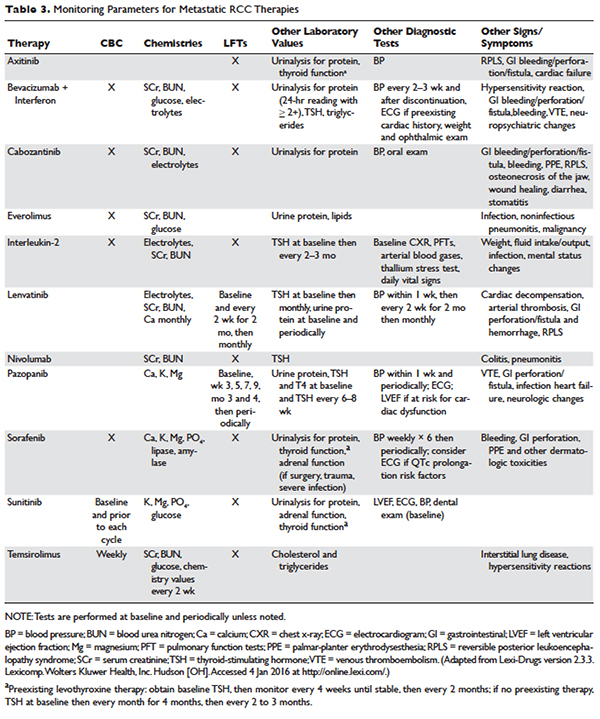
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
Soft Tissue Sarcoma: Diagnosis and Treatment
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).

A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”

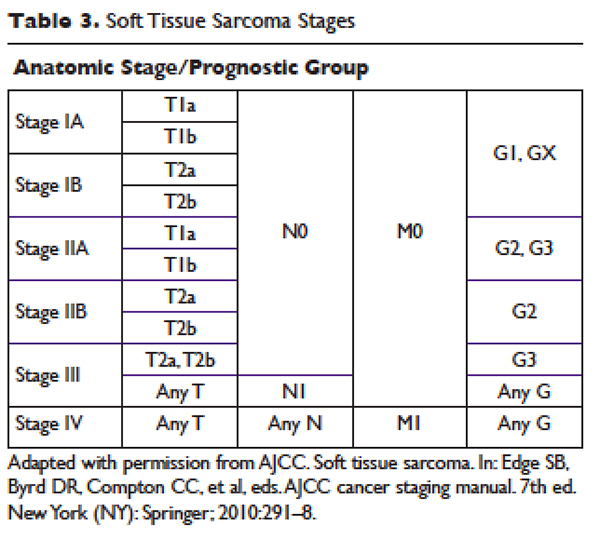
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Sickle Cell Disease
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).

The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).
The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).
The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
Using a Medical Interpreter with Persons of Limited English Proficiency
From the Department of Medicine, Mayo Clinic, Rochester, MN.
Abstract
- Objective: To provide an overview of important aspects of interpreting for medical visits for persons with limited English proficiency (LEP).
- Methods: Literature review.
- Results: When working with persons of LEP, providing a professional medical interpreter will facilitate optimal communication. Interpreters may work in different roles including as a conduit, cultural broker, clarifier, and advocate. In-person and remote (videoconferencing or telephonic) interpreting are available and one may be preferred depending on the medical visit. Clinicians should recognize that patients may have a preference for the interpreter’s gender and dialect and accommodations should be made if possible. Prior to the visit, the provider may want to clarify the goals of the medical encounter with the interpreter as some topics may be viewed differently in certain cultures. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient rather than to the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately to allow interpretation. Additionally, providers should assess patient understanding of what has been discussed. After the medical visit, providers should consider discussing with the interpreter any issues with communication or cultural factors noted to have affected the visit.
- Conclusion: Providers should utilize a professional medical interpreter for visits with persons with LEP. Appropriate communication techniques, including talking in first and second tenses and maintaining eye contact with the patient rather than the interpreter, are important for a successful visit. Realizing patients may have interpreter preferences is also important to facilitate patient-centered-care.
Key words: language barriers; quality of care; physician-patient communication; interpreter services.
The United States is a diverse country that includes many persons whose first language is not English. According to the U.S. Census Bureau, more than 63 million persons age 5 and above (about 51 million adults) reported speaking a language other than English at home. Also, about 25.7 million of the population age 5 and up (around 10.6 million adults) noted speaking English less than “very well” [1]. Protecting people from discrimination based on the language they speak is highlighted in Title VI of the Civil Rights Act of 1964 (which focuses on those receiving federal funding). President Clinton, furthermore, in 2000 signed Executive Order 13166, which encouraged federal agencies to provide appropriate access of their services to those with limited English proficiency (LEP) [2,3].
The benefits of using professional interpreters is well-documented. In addition to increased satisfaction with communication when professional medical interpreters are used [4], they also make fewer clinically significant interpretation errors compared to ad hoc interpreters (ie, untrained individuals such as bilingual staff member, family member, or friend who are asked to interpret) [5–7]. LEP patients who do not have a professional interpreter have less understanding of their medical issues, have less satisfaction of their medical care, and may have more tests ordered and be hospitalized more often compared to those who do utilize professional medical interpreters [8]. In addition to improved satisfaction and understanding of medical diagnoses, hospitalized persons requiring interpreters who utilized a professional medical interpreter on admission and discharge were noted to have a shorter length of stay than persons who required an interpreter and did not receive one [9].
Despite the documented benefits of using professional interpreters, they are underutilized. Reasons include underfunded medical interpreting services [10,11], lack of awareness of the risks involved with using an ad hoc interpreter [2,12], providers using their own or another worker’s limited second language skills to communicate rather than using a professional medical interpreter [13,14], perceived delay in obtaining a professional medical interpreter, and judging a medical situation as minor rather than complex [13]. In this article, the roles, importance, and considerations of using a professional medical interpreter are explored.
Case Study
Initial Presentation
A 23-year-old married Somali-speaking female who moved to the United States recently called a local primary care provider’s office to schedule an appointment. When asked the reason for the visit, she said the reason was private. The clinical assistant scheduled her with the next available provider.
When the patient arrived for her clinic appointment, a clinical assistant roomed her and her mother and asked the patient the reason for the visit. The patient remained quiet and her mother replied that she needed to speak with the doctor. When the female medical provider entered, she observed that the patient appeared anxious. When the doctor initiated conversation with the patient, she noted that her English was limited. Her mother tried to explain the reason for the visit saying her daughter was having severe pain. She then pointed towards her daughter’s lower abdomen. The clinician noted the limited English abilities of the patient and mother and used the interpreter line to request a Somali interpreter and placed the first available interpreter available on speaker.
How can patients who need an interpreter be identified?
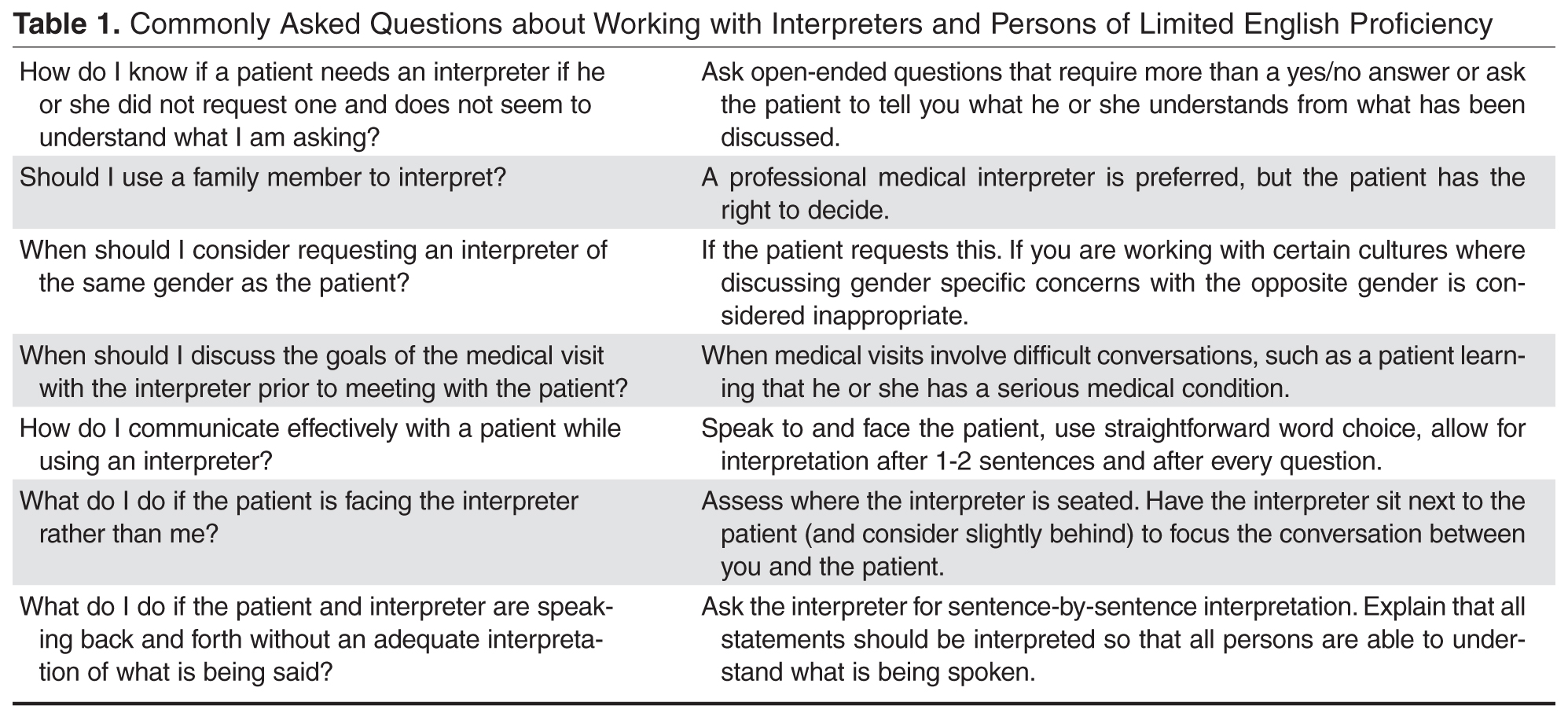
Health care systems can facilitate identifying patients in need of an interpreter by routinely collecting information on LEP status. The patient should be asked during registration if her or she speaks English and has a preferred language, and the answers should be recorded in the medical record [3]. If an interpreter is used during the hospital stay, it should be recorded to alert future providers to the need of an interpreter [15]. Patients may speak a dialect of a common language and this also should be noted, as an interpreter with a similar dialect to the patient should ideally be requested when necessary [16]. Furthermore, health care systems can measure rate of screening for language need at registration and rate of interpreter use during hospital stay to assess language need and adequacy of provision of interpreters to LEP patients [15].
It should be noted that patients may be wary about the presence of an interpreter. In one study involving pediatric oncologists and Spanish-speaking parents, the former reported concern regarding the accuracy of interpretation and the latter were concerned about missing out on important information even with the use of professional medical interpreters [17]. The concern for accuracy of interpreting was shared in a study by Chinese and Vietnamese Americans with LEP [18] as well as in a Swedish study involving Arabic-speaking persons. In the latter study, Arabic-speaking patients also felt uncomfortable speaking about bodily issues in the presence of interpreters [19]. In a study of Latina patients, there were concerns about confidentiality with interpreters [20].
What is the role of the interpreter? Should they offer emotional support to the patient?
Interpreter-as-conduit reflects a neutral, more literal information exchange and is preferred by certain medical providers who prioritize a more exact interpretation of the medical conversation. In this role, the interpreter assumes a more passive role and the emphasis is on the interpreter’s linguistic ability [21]. Providers need to be aware that word-for-word interpretation may not align with what is regarded as culturally sensitive care—such as when the term “cancer” is to be used. Also, word-for-word interpretation does not necessarily mean the patient will understand what is being interpreted if the terminology does not reflect the literacy level or dialect of the person with LEP [22].
Interpreters may also assume an active role, sometimes referred to as clarifier and cultural broker. Clarifying may be utilized, for example, when a medical provider is discussing complicated treatment options. This requires an interpreter to step out of a conduit role (if that is the preferred role) and confirm or clarify information to ensure accurate information exchange [21,23]. As a cultural broker, communication between provider and patient is exchanged in a manner that reflects consideration of the patient’s cultural background. Interpreters may explain, to the provider, the cultural reason for the patient’s perspective of what is causing or contributing to the illness. Cultural brokering may as well include communicating the medical terminology and disease explanation, given by the medical provider, in a way that the patient would understand. This role, additionally, can involve educating the provider about aspects of the culture that may influence the patient’s communication with him or her [22].
Furthermore, interpreters may fulfill an advocate role for patients by helping them understand the health care system and increasing patient empowerment by seeking information and services that the patient may not know to ask about [23].
The interpreter may offer emotional support during a medical visit, for example, where the diagnosis of cancer is conveyed. In such a case, an interpreter’s emotional support may be considered by providers to be appropriate. In contrast, with visits related to mental health evaluations, having the interpreter remain neutral, rather than being a more active participant by offering emotional support, may be preferred [24]. In addition to interpreters remaining more neutral during mental health visits, providers may prefer that interpreters not speak with the patient prior to the visit, depending on the mental health condition, as negative therapeutic consequences may occur [21]. Trust is an important element of the provider-patient relationship and, as such, there is concern on the part of some providers that if the trust of patients falls to the interpreter rather than to the provider, then therapeutic progress may be compromised [24]. In general, clarifying with interpreters the goals of the visit and expectations regarding speaking to the patient outside of the visit may ensure the provider-patient relationship is not diminished [21].
What are the disadvantages and caveats of using family members or bilingual staff as interpreters?
Although family members and other ad hoc interpreters may be present and willing to interpret, the risk of miscommunication is greater than with professional medical interpreters [5,6]. This risk of miscommunication extends to partially bilingual medical providers who do not utilize appropriate interpreter services [10,25]. Ad hoc interpreters may try to answer on behalf the patient [6,26] and may not have the appropriate medical terminology to correctly interpret what the provider is trying to communicate to the patient [6].
Professional medical interpreters are trained to facilitate communication of a spoken language in a medical setting [2,10]. Certification is offered by the National Board of Certification for Medical Interpreters and the Certification Commission for Healthcare Interpreters. In order to be certified certain requirements must be met which include a minimum of 40 hours of health care interpreter training (which includes medical terminology as well as roles and ethics involved in medical interpreting) as well as demonstrated oral proficiency in English as well as another chosen language (such as Spanish) [10].
In certain circumstances, patients may feel more comfortable disclosing personal details with a professional medical interpreter rather than in the presence of an ad hoc interpreter. For example, more details of traumatic events and psychological symptoms were spoken of in the presence of a professional, rather than an ad hoc, interpreter in medical interviews of asylum seekers requiring an interpreter in Switzerland. In the presence of ad hoc interpreters, more physical symptoms were disclosed rather than psychological [6].
Furthermore, in visits concerning sexuality or abuse issues, using family members as interpreters may violate privacy concerns of the patient [2,27]. Additionally, in certain cultures where respect for elders is very important, parents who use children as interpreters may feel that the structure of the family changes when he or she interprets on behalf of the parent [18]. Also, what children consider as embarrassing may not be interpreted to either the parent or to the care provider [8]. Furthermore, one should note there are ethical issues of using non-adult children as interpreters in situations involving confidentiality and privacy—by doing so, there may be resulting harmful effects on non-adult children [27,28].
Patients may at times decline the use of a professional medical interpreter and prefer to have a family member interpret; this preference should be documented in the patient’s medical chart [10]. Caution should be had using an ad hoc interpreter when obtaining informed consent [12].
What professional interpreting services are available to the clinician?
For the most part, access to interpreters via a telephone service is widely available [10]. The cost of providing interpreters in-person and/or remotely varies depending on the health care site [29–31] In general, considerations of using professional medical interpreters, whether remotely or in-person, involves accessibility and cost. There are certain sites that have explored having a shared network of interpreters available via the telephone and videoconference to reduce the cost of providing interpreters for individual hospitals [32]. While the costs of providing a person with LEP with interpretation varies depending on the health care site, the costs of not providing a professional medical interpreter should be considered as well, which include greater malpractice risk and potential medical errors [32]. In addition, the use of employees as interpreters takes time away from their respective jobs, which results in staff time lost [31].
In-person interpreting may be preferred for certain medical visits, as an in-person interpreter can interpret both verbal and nonverbal communication [16]. When emotional support is anticipated, in-person interpreting is usually preferred by providers [24]. There may be improved cultural competence when using an in-person interpreter, which may be important for certain visits such as those involving end-of life care discussions [4]. One concern may involve the comfort level of the patient if he or she personally knows the interpreter; this can occur in smaller ethnic communities [12]. Telephonic interpreting may be preferred in certain medical situations where confidentiality is desired [16].
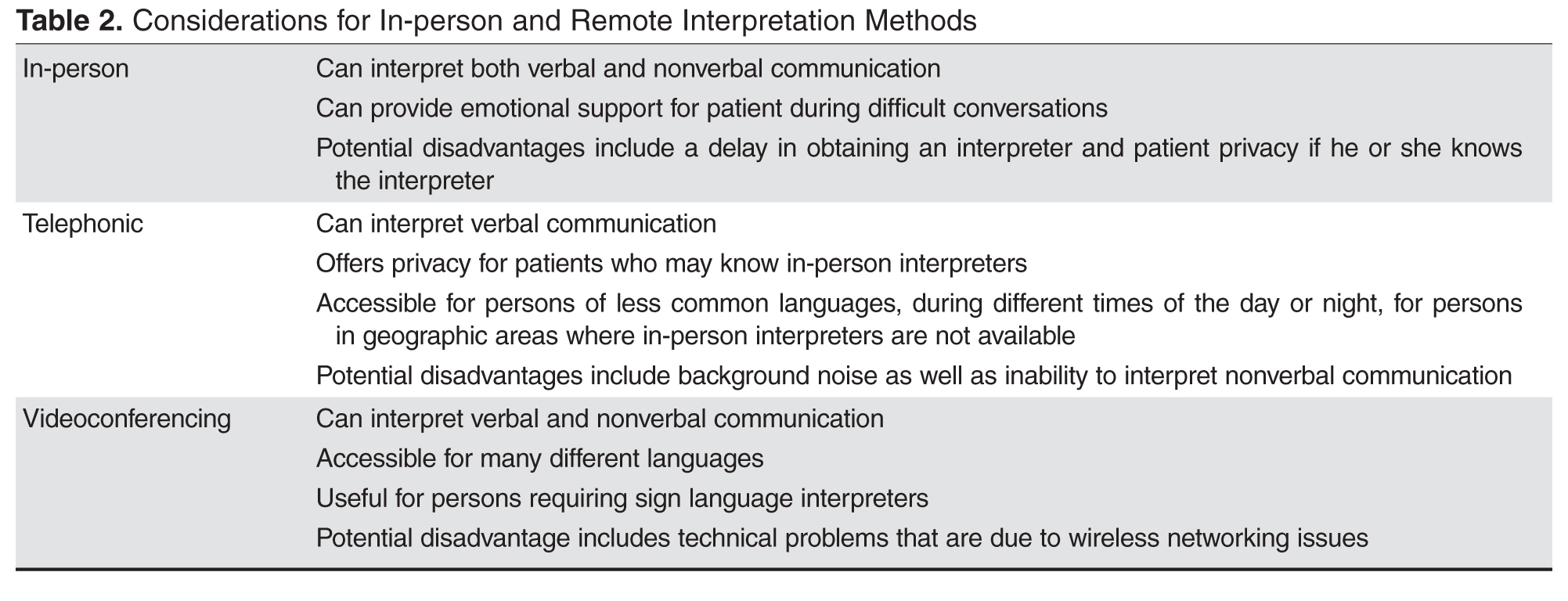
For providers working with persons who are deaf, options for interpreting include in-person sign language interpreters as well as remote videoconference interpretation [10].
- Are in-person interpreting and remote interpreting comparable?
In general, using in-person or remote interpreters does not significantly change patient satisfaction. In a study involving Spanish-speaking patients in a clinic setting, persons requiring an interpreter rated satisfaction of interpreting between in-person, videoconferencing, and telephonic methods highly with no significant differences. Of note, though, medical providers and interpreters preferred in-person rather than the 2 remote interpreting options [33]. In a different study involving Spanish, Chinese, Russian, or Vietnamese interpreters, satisfaction of information exchange was considered equal among the 3 interpreting modalities, although in-person interpreting was felt to establish rapport between clinician and patient with LEP better than telephonic and videoconferencing interpreting [35]. Additionally, in a study of providers in a clinic setting who worked with persons with LEP, no significant differences were noted in provider satisfaction of the medical visit, or in the quality of interpretation or communication, when using in-person versus remote videoconferencing interpretation. Providers, though, noted improved knowledge of the patient’s cultural beliefs when using in-person interpreting [4].
Regarding the question of a difference in understanding when using in-person versus remote interpreters, a study was done in a pediatric emergency department (ED) that compared in-person and telephonic interpretation. Family understanding of the discharge diagnosis was high (about 95%) regardless of whether an in-person or telephonic interpreter was utilized [36]. In a different study comparing telephone and video interpretation in a pediatric ED, while quality of communication and interpretation were rated similarly, the parents who used video interpretation were more likely to name their child’s diagnosis correctly [29].
Case Continued
The physician proceeded with introductions and explained that all conversations would be interpreted. She further stated that if there were any questions, the patient and mother should feel free to ask them. Via the interpreter, who was male, the provider began by asking about the nature of the abdominal pain. The patient looked to her mother and then down without answering. The mother nodded, but did not say more. The provider wondered if their reticence might be due to discomfort with discussing the issue through a male interpreter. The physician asked the patient if she would prefer a female interpreter and, once that was confirmed, she asked if she wanted an in-person or telephonic interpreter. The mother requested a female in-person interpreter.
How might gender-specific issues impact working with an interpreter?
As gender concordance of patient and physician [37] at times is desired, gender concordance of patient and interpreter [16,18] may also be important to optimize communication of gender-specific issues [16,18,37]. For example, an Arabic-speaking man from the Middle East may prefer to discuss sexuality-related concerns in the presence of a male rather than female interpreter [16]. An Arabic-speaking female who has a preference for female providers may prefer a female interpreter when discussing sexuality and undergoing a physical examination [19]. In one study, the majority of Somali females preferred female interpreters as well as female providers for breast, pelvic, and abdominal examinations [38]. If a same-gender interpreter cannot be present, an option is to have the interpreter either leave the room or step behind a curtain or turn away from the patient during a sensitive part of the physical examination [39].
What are recommended strategies for using a medical interpreter?
It is often helpful to have a brief discussion with the interpreter prior to the medical visit with the patient to speak about the general topics that will be discussed (especially if the topics involve sensitive issues or news that could be upsetting to the patient) and the goal of the visit [2, 11]. Certain topics may be viewed dissimilarly in different cultures, thus approaching the interpreter from the view point of cultural broker or liaison [10] may bring to light cultural factors that may influence the medical visit [40,41]. the name of the interpreter should be noted for documentation purposes [10].
To start the visit, introductions of everyone involved should take place with a brief disclosure about the role of the interpreter and assurance of confidentiality on the part of the interpreter [2,11]. Also, the provider should set the expectation that all statements said in the room will be interpreted so that all persons can understand what it being spoken [10].
There are several options for where each person should be positioned. In some medical visits, a triangle is pursued where the interpreter sits lateral to the provider, but this may lead to challenges in maintaining eye contact between the patient and provider. Another option is have the interpreter sit next to [10] and slightly behind the patient to improve eye contact between provider-patient and to maintain the patient-provider relationship [2,41]. When seated, the medical provider should try to sit at the same level as the patient [16]. Seating is different with persons requiring the use of sign language interpreters as the interpreter needs to be visible to the patient for communications purposes. One possibility is having the interpreter sit beside and slightly behind the provider; this positioning allows the patient to understand what is being communicated and also allows the patient to understand what is being communicated and allows him or her to see the provider during the conversation [40].
There are 2 main communication styles used by interpreters: consecutive interpreting, where the interpreter exchanges what has been said by the clinician or patient after each one has finished speaking, and simultaneous interpreting, where the interpreter translates as the person is speaking. Interpreters and medical providers may have a preference and it is important to clarify, if needed, which method is preferred [21].
The provider should face the patient and direct conversation to him or her rather than to the interpreter. Third-person statements should not be used, such as “tell her,” as this directs the conversation to the interpreter rather than to the patient [10]. By using the first and second person (when addressing the patient) and making eye contact, the relationship between the provider and patient is emphasized [14,40].
Choosing the right word is important to have meaningful communication. Interpreters advise that providers should understand that medical concepts may be unfamiliar to patients with LEP. Providers should use simpler words rather than medical terminology to discuss medical issues [42]. In general, straightforward word choice is recommended [16]. Providers are advised to not use acronyms or idioms. It is important to note that humor may be difficult to convey as well [10].
Clinicians are advised to speak clearly and not quickly and to use shorter sentences with appropriate pauses to allow time for the interpreter to interpret (if consecutive rather than simultaneous interpreting style is being used) [2,41,43,44]. In addition to limiting speech to one to two sentences at a time, asking one question at a time is important for optimal communication [43]. To improve information gathering, patients may respond better to open-ended questions [42], which is an aspect of patient-centered communication, as directive questioning often leads to shorter answers [43].
Furthermore, the provider should be aware that persons with LEP may know some English, so statements that one would not say to an English-speaking patient should not be said in the room with a person with LEP [10].
Encouraging the interpreter to clarify certain concepts, if necessary, may provide for improved information exchange [21] as well as encouraging the patient to ask questions during the medical visit may help elucidate potential areas of confusion [22,40,44]. Summarizing important concepts [40] and limiting the number of concepts discussed may increase patient understanding [10]. Additionally, asking the patient to repeat what was discussed in his or her words [10], rather than directly asking if he or she understands, will allow for more meaningful assessment of patient understanding [43].
Finally, recognition that interpreters may experience distress after certain visits, such as an oncologic medical encounters, is important and debriefing may be desired by the interpreter [22,45]. Also, discussing any communication concerns may be helpful [40,42] in addition to discussing certain cultural beliefs that impacted the visit may be educational for the provider [45].
Case Continued
After the female translator arrived, the physician asked the patient if she felt comfortable with her mother in the room for this medical visit. After the patient confirmed that she wanted her mother present, the physician tried to further clarify the reason for the medical visit. Her mother, appearing very concerned, began speaking quickly to the interpreter without stopping for interpretation. When the mother did stop speaking, the interpreter, rather than informing the provider what was spoken of by the mother, dialoged with the mother and the back and forth conversation continued.
What are strategies to optimize the medical visit when the provider is not satisfied with the flow of conversation?
If there are conversations occurring between the patient and interpreter with the exclusion of the provider, the provider should request sentence-by-sentence interpretation by the interpreter. If the interpreter is answering on behalf of the patient, providers should redirect communication to the patient [10]. At times, patients may speak for longer periods without stopping for the interpreter to provide accurate information exchange. The provider in this case may need to interrupt conversation to allow the interpreter time to convey what is being said [14].
If there are family members who know English, but the patient and/or others do not know English, there may be a risk of miscommunication if the exchange of medical information is done by a combination of family members and the interpreter, as the medical information may not accurately reflect what the clinician is trying to convey. The provider may need to redirect the conversation flow through the interpreter to make sure there is consistent information being communicated [22].
Case Continued
Finally, the provider interrupted. She emphasized with the patient, mother, and the interpreter that all that was being said should be interpreted. She asked the interpreter to sit next to the patient and mother (rather than lateral to the physician) so that eye contact between the patient and mother and the provider could be maintained thus supporting the patient-provider relationship. She then asked one question at a time to the patient. She needed to interrupt the conversation again when the mother started to speak to the interpreter without waiting for interpretation. The doctor reemphasized the need to allow time for the interpreter to adequately convey the information. After this the medical visit progressed successfully. Soon the provider found out that the mother was concerned that something serious could be happening to her daughter, as her daughter previously had a miscarriage. After hearing the mother’s concern, the provider was able to clarify with the daughter that the pain was suprapubic and she was having burning when she urinated. After further evaluation, the provider diagnosed a urinary tract infection. She told the patient about the diagnosis and provided her with appropriate medication and instructions on how to take it and for how long. The provider then asked the patient to tell her what she understood about the diagnosis and how to take the medication. The doctor then asked if either had any further questions. After the medical visit, the provider made sure that the patient’s chart reflected the need for a Somali interpreter with the notation that a female interpreter was preferred.
Conclusion
When working with persons with LEP, providing a professional medical interpreter will facilitate optimal communication. In-person and remote (videoconferencing or telephonic) interpreting are options. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient not the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately. Clinicians should remember that patients may have a preference in the gender and dialect of the interpreter and accommodations should be made if available. Finally, asking the patient to repeat back in his or her own words what has been discussed is important to make sure the patient understood what was communicated during the medical visit [10,16].
Corresponding author: Kimberly Schoonover, MD, 200 First Street SW, Rochester, MN 55905, [email protected].
Financial disclosures: None.
1. U.S. Census Bureau. American community survey. Accessed 11 Jul 2016 at www.census.gov/acs/www.
2. Wiener ESR, Ivonne M. Bridging language barriers: how to work with an interpreter. Clin Pediatr Emerg Med 2004;5:93–101.
3. Perez-Stable EJ, Karliner LS. What do we know about patient-clinician interactions with interpreters? J Gen Intern Med 2013;28:339–41.
4. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Clinician ratings of interpreter mediated visits in underserved primary care settings with ad hoc, in-person professional, and video conferencing modes. J Health Care Poor Underserved 2010;21:301–17.
5. Flores G, Abreu M, Barone CP, et al. Errors of medical interpretation and their potential clinical consequences: a comparison of professional versus ad hoc versus no interpreters. Ann Emerg Med 2012;60:545–53.
6. Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010;61:765–73.
7. Karliner LS, Jacobs EA, Chen AH, Mutha S. Do professional interpreters improve clinical care for patients with limited English proficiency? A systematic review of the literature. Health Serv Res 2007;42:727–54.
8. Flores G. The impact of medical interpreter services on the quality of health care: a systematic review. Med Care Res Rev 2005;62:255–99.
9. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
10. Juckett G, Unger K. Appropriate use of medical interpreters. Am Fam Physician 2014;90:476–80.
11. Schapira L, Vargas E, Hidalgo R, et al. Lost in translation: integrating medical interpreters into the multidisciplinary team. Oncologist 2008;13:586–92.
12. Gray B, Hilder J, Stubbe M. How to use interpreters in general practice: the development of a New Zealand toolkit. J Prim Health Care 2012;4:52–61, A1-8.
13. Hsieh E. Not just “getting by”: factors influencing providers’ choice of interpreters. J Gen Intern Med 2015;30:75–82.
14. Philips C. Using interpreters--a guide for GPs. Austr Fam Physician 2010;39:188–95.
15. Boscolo-Hightower A, Rafton SA, Tolman M, et al. Identifying families with limited English proficiency using a capture-recapture approach. Hosp Pediatr 2014;4:16–22.
16. Hadziabdic E, Hjelm K. Working with interpreters: practical advice for use of an interpreter in healthcare. Int J Evid Based Healthc 2013;11:69–76.
17. Abbe M, Simon C, Angiolillo A, et al. A survey of language barriers from the perspective of pediatric oncologists, interpreters, and parents. Pediatr Blood Cancer 2006;47:819–24.
18. Ngo-Metzger Q, Massagli MP, Clarridge BR, et al. Linguistic and cultural barriers to care. J Gen Intern Med 2003;18:44–52.
19. Hadziabdic E, Hjelm K. Arabic-speaking migrants’ experiences of the use of interpreters in healthcare: a qualitative explorative study. Int J Equity Health 2014;13:49.
20. Julliard K, Vivar J, Delgado C, et al. What Latina patients don’t tell their doctors: a qualitative study. Ann Family Med 2008;6:543–9.
21. Hsieh E, Ju H, Kong H. Dimensions of trust: the tensions and challenges in provider--interpreter trust. Qual Health Res 2010;20:170–81.
22. Butow PN, Lobb E, Jefford M, et al. A bridge between cultures: interpreters’ perspectives of consultations with migrant oncology patients. Support Care Cancer 2012;20:235-44.
23. Hsieh E. “I am not a robot!” Interpreters’ views of their roles in health care settings. Qual Health Res 2008;18:1367–83.
24. Hsieh E, Hong SJ. Not all are desired: providers’ views on interpreters’ emotional support for patients. Patient Educ Couns 2010;81:192–7.
25. Elderkin-Thompson V, Silver RC, Waitzkin H. When nurses double as interpreters: a study of Spanish-speaking patients in a US primary care setting. Soc Sci Med 2001;52:1343–58.
26. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Inaccurate language interpretation and its clinical significance in the medical encounters of Spanish-speaking Latinos. Med Care 2015;53:940–7.
27. Gray B, Hilder J, Donaldson H. Why do we not use trained interpreters for all patients with limited English proficiency? Is there a place for using family members? Austr J Prim Health 2011;17:240–9.
28. Jacobs B, Kroll L, Green J, David TJ. The hazards of using a child as an interpreter. J Royal Soc Med 1995;88:474P–5P.
29. Lion KC, Brown JC, Ebel BE, et al. Effect of telephone vs video interpretation on parent comprehension, communication, and utilization in the pediatric emergency department: a randomized clinical trial. JAMA Pediatr 2015;169:1117–25.
30. Wofford JL CC, Johnson DA, Brown MT. Providing a Spanish interpreter using low-cost videoconferencing in a community health centre: a pilot study using tablet computers. Inform Prim Care 2012;20:141–6.
31. Jacobs EA SL, Rathouz PJ. The impact of an enhanced interpreter service intervention on hospital costs and patient satisfaction. J Gen Intern Med 2007;22(Suppl 2):306–11.
32. Jacobs EA LG, Rathouz PJ, Fu P Jr. Shared networks of interpreter services, at relatively low cost, can help providers serve patients with limited english skills. Health Aff (Millwood) 2011;30:1930–8.
33. Locatis C, Williamson D, Gould-Kabler C, et al. Comparing in-person, video, and telephonic medical interpretation. J Gen Intern Med 2010;25:345–50.
34. Jones D, Gill P, Harrison R, et al. An exploratory study of language interpretation services provided by videoconferencing. J Telemed Telecare 2003;9:51–6.
35. Price EL, Perez-Stable EJ, Nickleach D, et al. Interpreter perspectives of in-person, telephonic, and videoconferencing medical interpretation in clinical encounters. Patient Educ Couns 2012;87:226–32.
36. Crossman KL, Wiener E, Roosevelt G, et al. Interpreters: telephonic, in-person interpretation and bilingual providers. Pediatrics 2010;125:e631–8.
37. Degni F, Suominen S, Essen B, et al. Communication and cultural issues in providing reproductive health care to immigrant women: health care providers’ experiences in meeting the needs of [corrected] Somali women living in Finland. J Immigr Minor Health 2012;14:330–43.
38. Odunukan OW, Abdulai RM, Hagi Salaad MF, et al. Provider and interpreter preferences among Somali women in a primary care setting. J Prim Care Comm Health 2015;6:105–10.
39. Poss JE, Beeman T. Effective use of interpreters in health care: guidelines for nurse managers and clinicians. Semin Nurse Manag 1999;7:166–71.
40. Phelan M, Parkman S. How to work with an interpreter. BMJ 1995;311:555–7.
41. Lubrano di Ciccone B, Brown RF, Gueguen JA, et al. Interviewing patients using interpreters in an oncology setting: initial evaluation of a communication skills module. Ann Oncol 2010;21:27–32.
42. Hudelson P. Improving patient-provider communication: insights from interpreters. Fam Pract 2005;22:311–6.
43. Hudelson P, Dao MD, Perron NJ, Bischoff A. Interpreter-mediated diabetes consultations: a qualitative analysis of physician communication practices. BMC Fam Pract 2013;14:163.
44. Green AR, Ngo-Metzger Q, Legedza AT, et al. Interpreter services, language concordance, and health care quality. Experiences of Asian Americans with limited English proficiency. J Gen Intern Med 2005;20:1050–6.
45. Schenker Y, Smith AK, Arnold RM, Fernandez A. “Her husband doesn’t speak much English”: conducting a family meeting with an interpreter. J Palliat Med 2012;15:494–8.
From the Department of Medicine, Mayo Clinic, Rochester, MN.
Abstract
- Objective: To provide an overview of important aspects of interpreting for medical visits for persons with limited English proficiency (LEP).
- Methods: Literature review.
- Results: When working with persons of LEP, providing a professional medical interpreter will facilitate optimal communication. Interpreters may work in different roles including as a conduit, cultural broker, clarifier, and advocate. In-person and remote (videoconferencing or telephonic) interpreting are available and one may be preferred depending on the medical visit. Clinicians should recognize that patients may have a preference for the interpreter’s gender and dialect and accommodations should be made if possible. Prior to the visit, the provider may want to clarify the goals of the medical encounter with the interpreter as some topics may be viewed differently in certain cultures. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient rather than to the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately to allow interpretation. Additionally, providers should assess patient understanding of what has been discussed. After the medical visit, providers should consider discussing with the interpreter any issues with communication or cultural factors noted to have affected the visit.
- Conclusion: Providers should utilize a professional medical interpreter for visits with persons with LEP. Appropriate communication techniques, including talking in first and second tenses and maintaining eye contact with the patient rather than the interpreter, are important for a successful visit. Realizing patients may have interpreter preferences is also important to facilitate patient-centered-care.
Key words: language barriers; quality of care; physician-patient communication; interpreter services.
The United States is a diverse country that includes many persons whose first language is not English. According to the U.S. Census Bureau, more than 63 million persons age 5 and above (about 51 million adults) reported speaking a language other than English at home. Also, about 25.7 million of the population age 5 and up (around 10.6 million adults) noted speaking English less than “very well” [1]. Protecting people from discrimination based on the language they speak is highlighted in Title VI of the Civil Rights Act of 1964 (which focuses on those receiving federal funding). President Clinton, furthermore, in 2000 signed Executive Order 13166, which encouraged federal agencies to provide appropriate access of their services to those with limited English proficiency (LEP) [2,3].
The benefits of using professional interpreters is well-documented. In addition to increased satisfaction with communication when professional medical interpreters are used [4], they also make fewer clinically significant interpretation errors compared to ad hoc interpreters (ie, untrained individuals such as bilingual staff member, family member, or friend who are asked to interpret) [5–7]. LEP patients who do not have a professional interpreter have less understanding of their medical issues, have less satisfaction of their medical care, and may have more tests ordered and be hospitalized more often compared to those who do utilize professional medical interpreters [8]. In addition to improved satisfaction and understanding of medical diagnoses, hospitalized persons requiring interpreters who utilized a professional medical interpreter on admission and discharge were noted to have a shorter length of stay than persons who required an interpreter and did not receive one [9].
Despite the documented benefits of using professional interpreters, they are underutilized. Reasons include underfunded medical interpreting services [10,11], lack of awareness of the risks involved with using an ad hoc interpreter [2,12], providers using their own or another worker’s limited second language skills to communicate rather than using a professional medical interpreter [13,14], perceived delay in obtaining a professional medical interpreter, and judging a medical situation as minor rather than complex [13]. In this article, the roles, importance, and considerations of using a professional medical interpreter are explored.
Case Study
Initial Presentation
A 23-year-old married Somali-speaking female who moved to the United States recently called a local primary care provider’s office to schedule an appointment. When asked the reason for the visit, she said the reason was private. The clinical assistant scheduled her with the next available provider.
When the patient arrived for her clinic appointment, a clinical assistant roomed her and her mother and asked the patient the reason for the visit. The patient remained quiet and her mother replied that she needed to speak with the doctor. When the female medical provider entered, she observed that the patient appeared anxious. When the doctor initiated conversation with the patient, she noted that her English was limited. Her mother tried to explain the reason for the visit saying her daughter was having severe pain. She then pointed towards her daughter’s lower abdomen. The clinician noted the limited English abilities of the patient and mother and used the interpreter line to request a Somali interpreter and placed the first available interpreter available on speaker.
How can patients who need an interpreter be identified?
Health care systems can facilitate identifying patients in need of an interpreter by routinely collecting information on LEP status. The patient should be asked during registration if her or she speaks English and has a preferred language, and the answers should be recorded in the medical record [3]. If an interpreter is used during the hospital stay, it should be recorded to alert future providers to the need of an interpreter [15]. Patients may speak a dialect of a common language and this also should be noted, as an interpreter with a similar dialect to the patient should ideally be requested when necessary [16]. Furthermore, health care systems can measure rate of screening for language need at registration and rate of interpreter use during hospital stay to assess language need and adequacy of provision of interpreters to LEP patients [15].
It should be noted that patients may be wary about the presence of an interpreter. In one study involving pediatric oncologists and Spanish-speaking parents, the former reported concern regarding the accuracy of interpretation and the latter were concerned about missing out on important information even with the use of professional medical interpreters [17]. The concern for accuracy of interpreting was shared in a study by Chinese and Vietnamese Americans with LEP [18] as well as in a Swedish study involving Arabic-speaking persons. In the latter study, Arabic-speaking patients also felt uncomfortable speaking about bodily issues in the presence of interpreters [19]. In a study of Latina patients, there were concerns about confidentiality with interpreters [20].
What is the role of the interpreter? Should they offer emotional support to the patient?
Interpreter-as-conduit reflects a neutral, more literal information exchange and is preferred by certain medical providers who prioritize a more exact interpretation of the medical conversation. In this role, the interpreter assumes a more passive role and the emphasis is on the interpreter’s linguistic ability [21]. Providers need to be aware that word-for-word interpretation may not align with what is regarded as culturally sensitive care—such as when the term “cancer” is to be used. Also, word-for-word interpretation does not necessarily mean the patient will understand what is being interpreted if the terminology does not reflect the literacy level or dialect of the person with LEP [22].
Interpreters may also assume an active role, sometimes referred to as clarifier and cultural broker. Clarifying may be utilized, for example, when a medical provider is discussing complicated treatment options. This requires an interpreter to step out of a conduit role (if that is the preferred role) and confirm or clarify information to ensure accurate information exchange [21,23]. As a cultural broker, communication between provider and patient is exchanged in a manner that reflects consideration of the patient’s cultural background. Interpreters may explain, to the provider, the cultural reason for the patient’s perspective of what is causing or contributing to the illness. Cultural brokering may as well include communicating the medical terminology and disease explanation, given by the medical provider, in a way that the patient would understand. This role, additionally, can involve educating the provider about aspects of the culture that may influence the patient’s communication with him or her [22].
Furthermore, interpreters may fulfill an advocate role for patients by helping them understand the health care system and increasing patient empowerment by seeking information and services that the patient may not know to ask about [23].
The interpreter may offer emotional support during a medical visit, for example, where the diagnosis of cancer is conveyed. In such a case, an interpreter’s emotional support may be considered by providers to be appropriate. In contrast, with visits related to mental health evaluations, having the interpreter remain neutral, rather than being a more active participant by offering emotional support, may be preferred [24]. In addition to interpreters remaining more neutral during mental health visits, providers may prefer that interpreters not speak with the patient prior to the visit, depending on the mental health condition, as negative therapeutic consequences may occur [21]. Trust is an important element of the provider-patient relationship and, as such, there is concern on the part of some providers that if the trust of patients falls to the interpreter rather than to the provider, then therapeutic progress may be compromised [24]. In general, clarifying with interpreters the goals of the visit and expectations regarding speaking to the patient outside of the visit may ensure the provider-patient relationship is not diminished [21].
What are the disadvantages and caveats of using family members or bilingual staff as interpreters?
Although family members and other ad hoc interpreters may be present and willing to interpret, the risk of miscommunication is greater than with professional medical interpreters [5,6]. This risk of miscommunication extends to partially bilingual medical providers who do not utilize appropriate interpreter services [10,25]. Ad hoc interpreters may try to answer on behalf the patient [6,26] and may not have the appropriate medical terminology to correctly interpret what the provider is trying to communicate to the patient [6].
Professional medical interpreters are trained to facilitate communication of a spoken language in a medical setting [2,10]. Certification is offered by the National Board of Certification for Medical Interpreters and the Certification Commission for Healthcare Interpreters. In order to be certified certain requirements must be met which include a minimum of 40 hours of health care interpreter training (which includes medical terminology as well as roles and ethics involved in medical interpreting) as well as demonstrated oral proficiency in English as well as another chosen language (such as Spanish) [10].
In certain circumstances, patients may feel more comfortable disclosing personal details with a professional medical interpreter rather than in the presence of an ad hoc interpreter. For example, more details of traumatic events and psychological symptoms were spoken of in the presence of a professional, rather than an ad hoc, interpreter in medical interviews of asylum seekers requiring an interpreter in Switzerland. In the presence of ad hoc interpreters, more physical symptoms were disclosed rather than psychological [6].
Furthermore, in visits concerning sexuality or abuse issues, using family members as interpreters may violate privacy concerns of the patient [2,27]. Additionally, in certain cultures where respect for elders is very important, parents who use children as interpreters may feel that the structure of the family changes when he or she interprets on behalf of the parent [18]. Also, what children consider as embarrassing may not be interpreted to either the parent or to the care provider [8]. Furthermore, one should note there are ethical issues of using non-adult children as interpreters in situations involving confidentiality and privacy—by doing so, there may be resulting harmful effects on non-adult children [27,28].
Patients may at times decline the use of a professional medical interpreter and prefer to have a family member interpret; this preference should be documented in the patient’s medical chart [10]. Caution should be had using an ad hoc interpreter when obtaining informed consent [12].
What professional interpreting services are available to the clinician?
For the most part, access to interpreters via a telephone service is widely available [10]. The cost of providing interpreters in-person and/or remotely varies depending on the health care site [29–31] In general, considerations of using professional medical interpreters, whether remotely or in-person, involves accessibility and cost. There are certain sites that have explored having a shared network of interpreters available via the telephone and videoconference to reduce the cost of providing interpreters for individual hospitals [32]. While the costs of providing a person with LEP with interpretation varies depending on the health care site, the costs of not providing a professional medical interpreter should be considered as well, which include greater malpractice risk and potential medical errors [32]. In addition, the use of employees as interpreters takes time away from their respective jobs, which results in staff time lost [31].
In-person interpreting may be preferred for certain medical visits, as an in-person interpreter can interpret both verbal and nonverbal communication [16]. When emotional support is anticipated, in-person interpreting is usually preferred by providers [24]. There may be improved cultural competence when using an in-person interpreter, which may be important for certain visits such as those involving end-of life care discussions [4]. One concern may involve the comfort level of the patient if he or she personally knows the interpreter; this can occur in smaller ethnic communities [12]. Telephonic interpreting may be preferred in certain medical situations where confidentiality is desired [16].
For providers working with persons who are deaf, options for interpreting include in-person sign language interpreters as well as remote videoconference interpretation [10].
- Are in-person interpreting and remote interpreting comparable?
In general, using in-person or remote interpreters does not significantly change patient satisfaction. In a study involving Spanish-speaking patients in a clinic setting, persons requiring an interpreter rated satisfaction of interpreting between in-person, videoconferencing, and telephonic methods highly with no significant differences. Of note, though, medical providers and interpreters preferred in-person rather than the 2 remote interpreting options [33]. In a different study involving Spanish, Chinese, Russian, or Vietnamese interpreters, satisfaction of information exchange was considered equal among the 3 interpreting modalities, although in-person interpreting was felt to establish rapport between clinician and patient with LEP better than telephonic and videoconferencing interpreting [35]. Additionally, in a study of providers in a clinic setting who worked with persons with LEP, no significant differences were noted in provider satisfaction of the medical visit, or in the quality of interpretation or communication, when using in-person versus remote videoconferencing interpretation. Providers, though, noted improved knowledge of the patient’s cultural beliefs when using in-person interpreting [4].
Regarding the question of a difference in understanding when using in-person versus remote interpreters, a study was done in a pediatric emergency department (ED) that compared in-person and telephonic interpretation. Family understanding of the discharge diagnosis was high (about 95%) regardless of whether an in-person or telephonic interpreter was utilized [36]. In a different study comparing telephone and video interpretation in a pediatric ED, while quality of communication and interpretation were rated similarly, the parents who used video interpretation were more likely to name their child’s diagnosis correctly [29].
Case Continued
The physician proceeded with introductions and explained that all conversations would be interpreted. She further stated that if there were any questions, the patient and mother should feel free to ask them. Via the interpreter, who was male, the provider began by asking about the nature of the abdominal pain. The patient looked to her mother and then down without answering. The mother nodded, but did not say more. The provider wondered if their reticence might be due to discomfort with discussing the issue through a male interpreter. The physician asked the patient if she would prefer a female interpreter and, once that was confirmed, she asked if she wanted an in-person or telephonic interpreter. The mother requested a female in-person interpreter.
How might gender-specific issues impact working with an interpreter?
As gender concordance of patient and physician [37] at times is desired, gender concordance of patient and interpreter [16,18] may also be important to optimize communication of gender-specific issues [16,18,37]. For example, an Arabic-speaking man from the Middle East may prefer to discuss sexuality-related concerns in the presence of a male rather than female interpreter [16]. An Arabic-speaking female who has a preference for female providers may prefer a female interpreter when discussing sexuality and undergoing a physical examination [19]. In one study, the majority of Somali females preferred female interpreters as well as female providers for breast, pelvic, and abdominal examinations [38]. If a same-gender interpreter cannot be present, an option is to have the interpreter either leave the room or step behind a curtain or turn away from the patient during a sensitive part of the physical examination [39].
What are recommended strategies for using a medical interpreter?
It is often helpful to have a brief discussion with the interpreter prior to the medical visit with the patient to speak about the general topics that will be discussed (especially if the topics involve sensitive issues or news that could be upsetting to the patient) and the goal of the visit [2, 11]. Certain topics may be viewed dissimilarly in different cultures, thus approaching the interpreter from the view point of cultural broker or liaison [10] may bring to light cultural factors that may influence the medical visit [40,41]. the name of the interpreter should be noted for documentation purposes [10].
To start the visit, introductions of everyone involved should take place with a brief disclosure about the role of the interpreter and assurance of confidentiality on the part of the interpreter [2,11]. Also, the provider should set the expectation that all statements said in the room will be interpreted so that all persons can understand what it being spoken [10].
There are several options for where each person should be positioned. In some medical visits, a triangle is pursued where the interpreter sits lateral to the provider, but this may lead to challenges in maintaining eye contact between the patient and provider. Another option is have the interpreter sit next to [10] and slightly behind the patient to improve eye contact between provider-patient and to maintain the patient-provider relationship [2,41]. When seated, the medical provider should try to sit at the same level as the patient [16]. Seating is different with persons requiring the use of sign language interpreters as the interpreter needs to be visible to the patient for communications purposes. One possibility is having the interpreter sit beside and slightly behind the provider; this positioning allows the patient to understand what is being communicated and also allows the patient to understand what is being communicated and allows him or her to see the provider during the conversation [40].
There are 2 main communication styles used by interpreters: consecutive interpreting, where the interpreter exchanges what has been said by the clinician or patient after each one has finished speaking, and simultaneous interpreting, where the interpreter translates as the person is speaking. Interpreters and medical providers may have a preference and it is important to clarify, if needed, which method is preferred [21].
The provider should face the patient and direct conversation to him or her rather than to the interpreter. Third-person statements should not be used, such as “tell her,” as this directs the conversation to the interpreter rather than to the patient [10]. By using the first and second person (when addressing the patient) and making eye contact, the relationship between the provider and patient is emphasized [14,40].
Choosing the right word is important to have meaningful communication. Interpreters advise that providers should understand that medical concepts may be unfamiliar to patients with LEP. Providers should use simpler words rather than medical terminology to discuss medical issues [42]. In general, straightforward word choice is recommended [16]. Providers are advised to not use acronyms or idioms. It is important to note that humor may be difficult to convey as well [10].
Clinicians are advised to speak clearly and not quickly and to use shorter sentences with appropriate pauses to allow time for the interpreter to interpret (if consecutive rather than simultaneous interpreting style is being used) [2,41,43,44]. In addition to limiting speech to one to two sentences at a time, asking one question at a time is important for optimal communication [43]. To improve information gathering, patients may respond better to open-ended questions [42], which is an aspect of patient-centered communication, as directive questioning often leads to shorter answers [43].
Furthermore, the provider should be aware that persons with LEP may know some English, so statements that one would not say to an English-speaking patient should not be said in the room with a person with LEP [10].
Encouraging the interpreter to clarify certain concepts, if necessary, may provide for improved information exchange [21] as well as encouraging the patient to ask questions during the medical visit may help elucidate potential areas of confusion [22,40,44]. Summarizing important concepts [40] and limiting the number of concepts discussed may increase patient understanding [10]. Additionally, asking the patient to repeat what was discussed in his or her words [10], rather than directly asking if he or she understands, will allow for more meaningful assessment of patient understanding [43].
Finally, recognition that interpreters may experience distress after certain visits, such as an oncologic medical encounters, is important and debriefing may be desired by the interpreter [22,45]. Also, discussing any communication concerns may be helpful [40,42] in addition to discussing certain cultural beliefs that impacted the visit may be educational for the provider [45].
Case Continued
After the female translator arrived, the physician asked the patient if she felt comfortable with her mother in the room for this medical visit. After the patient confirmed that she wanted her mother present, the physician tried to further clarify the reason for the medical visit. Her mother, appearing very concerned, began speaking quickly to the interpreter without stopping for interpretation. When the mother did stop speaking, the interpreter, rather than informing the provider what was spoken of by the mother, dialoged with the mother and the back and forth conversation continued.
What are strategies to optimize the medical visit when the provider is not satisfied with the flow of conversation?
If there are conversations occurring between the patient and interpreter with the exclusion of the provider, the provider should request sentence-by-sentence interpretation by the interpreter. If the interpreter is answering on behalf of the patient, providers should redirect communication to the patient [10]. At times, patients may speak for longer periods without stopping for the interpreter to provide accurate information exchange. The provider in this case may need to interrupt conversation to allow the interpreter time to convey what is being said [14].
If there are family members who know English, but the patient and/or others do not know English, there may be a risk of miscommunication if the exchange of medical information is done by a combination of family members and the interpreter, as the medical information may not accurately reflect what the clinician is trying to convey. The provider may need to redirect the conversation flow through the interpreter to make sure there is consistent information being communicated [22].
Case Continued
Finally, the provider interrupted. She emphasized with the patient, mother, and the interpreter that all that was being said should be interpreted. She asked the interpreter to sit next to the patient and mother (rather than lateral to the physician) so that eye contact between the patient and mother and the provider could be maintained thus supporting the patient-provider relationship. She then asked one question at a time to the patient. She needed to interrupt the conversation again when the mother started to speak to the interpreter without waiting for interpretation. The doctor reemphasized the need to allow time for the interpreter to adequately convey the information. After this the medical visit progressed successfully. Soon the provider found out that the mother was concerned that something serious could be happening to her daughter, as her daughter previously had a miscarriage. After hearing the mother’s concern, the provider was able to clarify with the daughter that the pain was suprapubic and she was having burning when she urinated. After further evaluation, the provider diagnosed a urinary tract infection. She told the patient about the diagnosis and provided her with appropriate medication and instructions on how to take it and for how long. The provider then asked the patient to tell her what she understood about the diagnosis and how to take the medication. The doctor then asked if either had any further questions. After the medical visit, the provider made sure that the patient’s chart reflected the need for a Somali interpreter with the notation that a female interpreter was preferred.
Conclusion
When working with persons with LEP, providing a professional medical interpreter will facilitate optimal communication. In-person and remote (videoconferencing or telephonic) interpreting are options. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient not the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately. Clinicians should remember that patients may have a preference in the gender and dialect of the interpreter and accommodations should be made if available. Finally, asking the patient to repeat back in his or her own words what has been discussed is important to make sure the patient understood what was communicated during the medical visit [10,16].
Corresponding author: Kimberly Schoonover, MD, 200 First Street SW, Rochester, MN 55905, [email protected].
Financial disclosures: None.
From the Department of Medicine, Mayo Clinic, Rochester, MN.
Abstract
- Objective: To provide an overview of important aspects of interpreting for medical visits for persons with limited English proficiency (LEP).
- Methods: Literature review.
- Results: When working with persons of LEP, providing a professional medical interpreter will facilitate optimal communication. Interpreters may work in different roles including as a conduit, cultural broker, clarifier, and advocate. In-person and remote (videoconferencing or telephonic) interpreting are available and one may be preferred depending on the medical visit. Clinicians should recognize that patients may have a preference for the interpreter’s gender and dialect and accommodations should be made if possible. Prior to the visit, the provider may want to clarify the goals of the medical encounter with the interpreter as some topics may be viewed differently in certain cultures. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient rather than to the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately to allow interpretation. Additionally, providers should assess patient understanding of what has been discussed. After the medical visit, providers should consider discussing with the interpreter any issues with communication or cultural factors noted to have affected the visit.
- Conclusion: Providers should utilize a professional medical interpreter for visits with persons with LEP. Appropriate communication techniques, including talking in first and second tenses and maintaining eye contact with the patient rather than the interpreter, are important for a successful visit. Realizing patients may have interpreter preferences is also important to facilitate patient-centered-care.
Key words: language barriers; quality of care; physician-patient communication; interpreter services.
The United States is a diverse country that includes many persons whose first language is not English. According to the U.S. Census Bureau, more than 63 million persons age 5 and above (about 51 million adults) reported speaking a language other than English at home. Also, about 25.7 million of the population age 5 and up (around 10.6 million adults) noted speaking English less than “very well” [1]. Protecting people from discrimination based on the language they speak is highlighted in Title VI of the Civil Rights Act of 1964 (which focuses on those receiving federal funding). President Clinton, furthermore, in 2000 signed Executive Order 13166, which encouraged federal agencies to provide appropriate access of their services to those with limited English proficiency (LEP) [2,3].
The benefits of using professional interpreters is well-documented. In addition to increased satisfaction with communication when professional medical interpreters are used [4], they also make fewer clinically significant interpretation errors compared to ad hoc interpreters (ie, untrained individuals such as bilingual staff member, family member, or friend who are asked to interpret) [5–7]. LEP patients who do not have a professional interpreter have less understanding of their medical issues, have less satisfaction of their medical care, and may have more tests ordered and be hospitalized more often compared to those who do utilize professional medical interpreters [8]. In addition to improved satisfaction and understanding of medical diagnoses, hospitalized persons requiring interpreters who utilized a professional medical interpreter on admission and discharge were noted to have a shorter length of stay than persons who required an interpreter and did not receive one [9].
Despite the documented benefits of using professional interpreters, they are underutilized. Reasons include underfunded medical interpreting services [10,11], lack of awareness of the risks involved with using an ad hoc interpreter [2,12], providers using their own or another worker’s limited second language skills to communicate rather than using a professional medical interpreter [13,14], perceived delay in obtaining a professional medical interpreter, and judging a medical situation as minor rather than complex [13]. In this article, the roles, importance, and considerations of using a professional medical interpreter are explored.
Case Study
Initial Presentation
A 23-year-old married Somali-speaking female who moved to the United States recently called a local primary care provider’s office to schedule an appointment. When asked the reason for the visit, she said the reason was private. The clinical assistant scheduled her with the next available provider.
When the patient arrived for her clinic appointment, a clinical assistant roomed her and her mother and asked the patient the reason for the visit. The patient remained quiet and her mother replied that she needed to speak with the doctor. When the female medical provider entered, she observed that the patient appeared anxious. When the doctor initiated conversation with the patient, she noted that her English was limited. Her mother tried to explain the reason for the visit saying her daughter was having severe pain. She then pointed towards her daughter’s lower abdomen. The clinician noted the limited English abilities of the patient and mother and used the interpreter line to request a Somali interpreter and placed the first available interpreter available on speaker.
How can patients who need an interpreter be identified?
Health care systems can facilitate identifying patients in need of an interpreter by routinely collecting information on LEP status. The patient should be asked during registration if her or she speaks English and has a preferred language, and the answers should be recorded in the medical record [3]. If an interpreter is used during the hospital stay, it should be recorded to alert future providers to the need of an interpreter [15]. Patients may speak a dialect of a common language and this also should be noted, as an interpreter with a similar dialect to the patient should ideally be requested when necessary [16]. Furthermore, health care systems can measure rate of screening for language need at registration and rate of interpreter use during hospital stay to assess language need and adequacy of provision of interpreters to LEP patients [15].
It should be noted that patients may be wary about the presence of an interpreter. In one study involving pediatric oncologists and Spanish-speaking parents, the former reported concern regarding the accuracy of interpretation and the latter were concerned about missing out on important information even with the use of professional medical interpreters [17]. The concern for accuracy of interpreting was shared in a study by Chinese and Vietnamese Americans with LEP [18] as well as in a Swedish study involving Arabic-speaking persons. In the latter study, Arabic-speaking patients also felt uncomfortable speaking about bodily issues in the presence of interpreters [19]. In a study of Latina patients, there were concerns about confidentiality with interpreters [20].
What is the role of the interpreter? Should they offer emotional support to the patient?
Interpreter-as-conduit reflects a neutral, more literal information exchange and is preferred by certain medical providers who prioritize a more exact interpretation of the medical conversation. In this role, the interpreter assumes a more passive role and the emphasis is on the interpreter’s linguistic ability [21]. Providers need to be aware that word-for-word interpretation may not align with what is regarded as culturally sensitive care—such as when the term “cancer” is to be used. Also, word-for-word interpretation does not necessarily mean the patient will understand what is being interpreted if the terminology does not reflect the literacy level or dialect of the person with LEP [22].
Interpreters may also assume an active role, sometimes referred to as clarifier and cultural broker. Clarifying may be utilized, for example, when a medical provider is discussing complicated treatment options. This requires an interpreter to step out of a conduit role (if that is the preferred role) and confirm or clarify information to ensure accurate information exchange [21,23]. As a cultural broker, communication between provider and patient is exchanged in a manner that reflects consideration of the patient’s cultural background. Interpreters may explain, to the provider, the cultural reason for the patient’s perspective of what is causing or contributing to the illness. Cultural brokering may as well include communicating the medical terminology and disease explanation, given by the medical provider, in a way that the patient would understand. This role, additionally, can involve educating the provider about aspects of the culture that may influence the patient’s communication with him or her [22].
Furthermore, interpreters may fulfill an advocate role for patients by helping them understand the health care system and increasing patient empowerment by seeking information and services that the patient may not know to ask about [23].
The interpreter may offer emotional support during a medical visit, for example, where the diagnosis of cancer is conveyed. In such a case, an interpreter’s emotional support may be considered by providers to be appropriate. In contrast, with visits related to mental health evaluations, having the interpreter remain neutral, rather than being a more active participant by offering emotional support, may be preferred [24]. In addition to interpreters remaining more neutral during mental health visits, providers may prefer that interpreters not speak with the patient prior to the visit, depending on the mental health condition, as negative therapeutic consequences may occur [21]. Trust is an important element of the provider-patient relationship and, as such, there is concern on the part of some providers that if the trust of patients falls to the interpreter rather than to the provider, then therapeutic progress may be compromised [24]. In general, clarifying with interpreters the goals of the visit and expectations regarding speaking to the patient outside of the visit may ensure the provider-patient relationship is not diminished [21].
What are the disadvantages and caveats of using family members or bilingual staff as interpreters?
Although family members and other ad hoc interpreters may be present and willing to interpret, the risk of miscommunication is greater than with professional medical interpreters [5,6]. This risk of miscommunication extends to partially bilingual medical providers who do not utilize appropriate interpreter services [10,25]. Ad hoc interpreters may try to answer on behalf the patient [6,26] and may not have the appropriate medical terminology to correctly interpret what the provider is trying to communicate to the patient [6].
Professional medical interpreters are trained to facilitate communication of a spoken language in a medical setting [2,10]. Certification is offered by the National Board of Certification for Medical Interpreters and the Certification Commission for Healthcare Interpreters. In order to be certified certain requirements must be met which include a minimum of 40 hours of health care interpreter training (which includes medical terminology as well as roles and ethics involved in medical interpreting) as well as demonstrated oral proficiency in English as well as another chosen language (such as Spanish) [10].
In certain circumstances, patients may feel more comfortable disclosing personal details with a professional medical interpreter rather than in the presence of an ad hoc interpreter. For example, more details of traumatic events and psychological symptoms were spoken of in the presence of a professional, rather than an ad hoc, interpreter in medical interviews of asylum seekers requiring an interpreter in Switzerland. In the presence of ad hoc interpreters, more physical symptoms were disclosed rather than psychological [6].
Furthermore, in visits concerning sexuality or abuse issues, using family members as interpreters may violate privacy concerns of the patient [2,27]. Additionally, in certain cultures where respect for elders is very important, parents who use children as interpreters may feel that the structure of the family changes when he or she interprets on behalf of the parent [18]. Also, what children consider as embarrassing may not be interpreted to either the parent or to the care provider [8]. Furthermore, one should note there are ethical issues of using non-adult children as interpreters in situations involving confidentiality and privacy—by doing so, there may be resulting harmful effects on non-adult children [27,28].
Patients may at times decline the use of a professional medical interpreter and prefer to have a family member interpret; this preference should be documented in the patient’s medical chart [10]. Caution should be had using an ad hoc interpreter when obtaining informed consent [12].
What professional interpreting services are available to the clinician?
For the most part, access to interpreters via a telephone service is widely available [10]. The cost of providing interpreters in-person and/or remotely varies depending on the health care site [29–31] In general, considerations of using professional medical interpreters, whether remotely or in-person, involves accessibility and cost. There are certain sites that have explored having a shared network of interpreters available via the telephone and videoconference to reduce the cost of providing interpreters for individual hospitals [32]. While the costs of providing a person with LEP with interpretation varies depending on the health care site, the costs of not providing a professional medical interpreter should be considered as well, which include greater malpractice risk and potential medical errors [32]. In addition, the use of employees as interpreters takes time away from their respective jobs, which results in staff time lost [31].
In-person interpreting may be preferred for certain medical visits, as an in-person interpreter can interpret both verbal and nonverbal communication [16]. When emotional support is anticipated, in-person interpreting is usually preferred by providers [24]. There may be improved cultural competence when using an in-person interpreter, which may be important for certain visits such as those involving end-of life care discussions [4]. One concern may involve the comfort level of the patient if he or she personally knows the interpreter; this can occur in smaller ethnic communities [12]. Telephonic interpreting may be preferred in certain medical situations where confidentiality is desired [16].
For providers working with persons who are deaf, options for interpreting include in-person sign language interpreters as well as remote videoconference interpretation [10].
- Are in-person interpreting and remote interpreting comparable?
In general, using in-person or remote interpreters does not significantly change patient satisfaction. In a study involving Spanish-speaking patients in a clinic setting, persons requiring an interpreter rated satisfaction of interpreting between in-person, videoconferencing, and telephonic methods highly with no significant differences. Of note, though, medical providers and interpreters preferred in-person rather than the 2 remote interpreting options [33]. In a different study involving Spanish, Chinese, Russian, or Vietnamese interpreters, satisfaction of information exchange was considered equal among the 3 interpreting modalities, although in-person interpreting was felt to establish rapport between clinician and patient with LEP better than telephonic and videoconferencing interpreting [35]. Additionally, in a study of providers in a clinic setting who worked with persons with LEP, no significant differences were noted in provider satisfaction of the medical visit, or in the quality of interpretation or communication, when using in-person versus remote videoconferencing interpretation. Providers, though, noted improved knowledge of the patient’s cultural beliefs when using in-person interpreting [4].
Regarding the question of a difference in understanding when using in-person versus remote interpreters, a study was done in a pediatric emergency department (ED) that compared in-person and telephonic interpretation. Family understanding of the discharge diagnosis was high (about 95%) regardless of whether an in-person or telephonic interpreter was utilized [36]. In a different study comparing telephone and video interpretation in a pediatric ED, while quality of communication and interpretation were rated similarly, the parents who used video interpretation were more likely to name their child’s diagnosis correctly [29].
Case Continued
The physician proceeded with introductions and explained that all conversations would be interpreted. She further stated that if there were any questions, the patient and mother should feel free to ask them. Via the interpreter, who was male, the provider began by asking about the nature of the abdominal pain. The patient looked to her mother and then down without answering. The mother nodded, but did not say more. The provider wondered if their reticence might be due to discomfort with discussing the issue through a male interpreter. The physician asked the patient if she would prefer a female interpreter and, once that was confirmed, she asked if she wanted an in-person or telephonic interpreter. The mother requested a female in-person interpreter.
How might gender-specific issues impact working with an interpreter?
As gender concordance of patient and physician [37] at times is desired, gender concordance of patient and interpreter [16,18] may also be important to optimize communication of gender-specific issues [16,18,37]. For example, an Arabic-speaking man from the Middle East may prefer to discuss sexuality-related concerns in the presence of a male rather than female interpreter [16]. An Arabic-speaking female who has a preference for female providers may prefer a female interpreter when discussing sexuality and undergoing a physical examination [19]. In one study, the majority of Somali females preferred female interpreters as well as female providers for breast, pelvic, and abdominal examinations [38]. If a same-gender interpreter cannot be present, an option is to have the interpreter either leave the room or step behind a curtain or turn away from the patient during a sensitive part of the physical examination [39].
What are recommended strategies for using a medical interpreter?
It is often helpful to have a brief discussion with the interpreter prior to the medical visit with the patient to speak about the general topics that will be discussed (especially if the topics involve sensitive issues or news that could be upsetting to the patient) and the goal of the visit [2, 11]. Certain topics may be viewed dissimilarly in different cultures, thus approaching the interpreter from the view point of cultural broker or liaison [10] may bring to light cultural factors that may influence the medical visit [40,41]. the name of the interpreter should be noted for documentation purposes [10].
To start the visit, introductions of everyone involved should take place with a brief disclosure about the role of the interpreter and assurance of confidentiality on the part of the interpreter [2,11]. Also, the provider should set the expectation that all statements said in the room will be interpreted so that all persons can understand what it being spoken [10].
There are several options for where each person should be positioned. In some medical visits, a triangle is pursued where the interpreter sits lateral to the provider, but this may lead to challenges in maintaining eye contact between the patient and provider. Another option is have the interpreter sit next to [10] and slightly behind the patient to improve eye contact between provider-patient and to maintain the patient-provider relationship [2,41]. When seated, the medical provider should try to sit at the same level as the patient [16]. Seating is different with persons requiring the use of sign language interpreters as the interpreter needs to be visible to the patient for communications purposes. One possibility is having the interpreter sit beside and slightly behind the provider; this positioning allows the patient to understand what is being communicated and also allows the patient to understand what is being communicated and allows him or her to see the provider during the conversation [40].
There are 2 main communication styles used by interpreters: consecutive interpreting, where the interpreter exchanges what has been said by the clinician or patient after each one has finished speaking, and simultaneous interpreting, where the interpreter translates as the person is speaking. Interpreters and medical providers may have a preference and it is important to clarify, if needed, which method is preferred [21].
The provider should face the patient and direct conversation to him or her rather than to the interpreter. Third-person statements should not be used, such as “tell her,” as this directs the conversation to the interpreter rather than to the patient [10]. By using the first and second person (when addressing the patient) and making eye contact, the relationship between the provider and patient is emphasized [14,40].
Choosing the right word is important to have meaningful communication. Interpreters advise that providers should understand that medical concepts may be unfamiliar to patients with LEP. Providers should use simpler words rather than medical terminology to discuss medical issues [42]. In general, straightforward word choice is recommended [16]. Providers are advised to not use acronyms or idioms. It is important to note that humor may be difficult to convey as well [10].
Clinicians are advised to speak clearly and not quickly and to use shorter sentences with appropriate pauses to allow time for the interpreter to interpret (if consecutive rather than simultaneous interpreting style is being used) [2,41,43,44]. In addition to limiting speech to one to two sentences at a time, asking one question at a time is important for optimal communication [43]. To improve information gathering, patients may respond better to open-ended questions [42], which is an aspect of patient-centered communication, as directive questioning often leads to shorter answers [43].
Furthermore, the provider should be aware that persons with LEP may know some English, so statements that one would not say to an English-speaking patient should not be said in the room with a person with LEP [10].
Encouraging the interpreter to clarify certain concepts, if necessary, may provide for improved information exchange [21] as well as encouraging the patient to ask questions during the medical visit may help elucidate potential areas of confusion [22,40,44]. Summarizing important concepts [40] and limiting the number of concepts discussed may increase patient understanding [10]. Additionally, asking the patient to repeat what was discussed in his or her words [10], rather than directly asking if he or she understands, will allow for more meaningful assessment of patient understanding [43].
Finally, recognition that interpreters may experience distress after certain visits, such as an oncologic medical encounters, is important and debriefing may be desired by the interpreter [22,45]. Also, discussing any communication concerns may be helpful [40,42] in addition to discussing certain cultural beliefs that impacted the visit may be educational for the provider [45].
Case Continued
After the female translator arrived, the physician asked the patient if she felt comfortable with her mother in the room for this medical visit. After the patient confirmed that she wanted her mother present, the physician tried to further clarify the reason for the medical visit. Her mother, appearing very concerned, began speaking quickly to the interpreter without stopping for interpretation. When the mother did stop speaking, the interpreter, rather than informing the provider what was spoken of by the mother, dialoged with the mother and the back and forth conversation continued.
What are strategies to optimize the medical visit when the provider is not satisfied with the flow of conversation?
If there are conversations occurring between the patient and interpreter with the exclusion of the provider, the provider should request sentence-by-sentence interpretation by the interpreter. If the interpreter is answering on behalf of the patient, providers should redirect communication to the patient [10]. At times, patients may speak for longer periods without stopping for the interpreter to provide accurate information exchange. The provider in this case may need to interrupt conversation to allow the interpreter time to convey what is being said [14].
If there are family members who know English, but the patient and/or others do not know English, there may be a risk of miscommunication if the exchange of medical information is done by a combination of family members and the interpreter, as the medical information may not accurately reflect what the clinician is trying to convey. The provider may need to redirect the conversation flow through the interpreter to make sure there is consistent information being communicated [22].
Case Continued
Finally, the provider interrupted. She emphasized with the patient, mother, and the interpreter that all that was being said should be interpreted. She asked the interpreter to sit next to the patient and mother (rather than lateral to the physician) so that eye contact between the patient and mother and the provider could be maintained thus supporting the patient-provider relationship. She then asked one question at a time to the patient. She needed to interrupt the conversation again when the mother started to speak to the interpreter without waiting for interpretation. The doctor reemphasized the need to allow time for the interpreter to adequately convey the information. After this the medical visit progressed successfully. Soon the provider found out that the mother was concerned that something serious could be happening to her daughter, as her daughter previously had a miscarriage. After hearing the mother’s concern, the provider was able to clarify with the daughter that the pain was suprapubic and she was having burning when she urinated. After further evaluation, the provider diagnosed a urinary tract infection. She told the patient about the diagnosis and provided her with appropriate medication and instructions on how to take it and for how long. The provider then asked the patient to tell her what she understood about the diagnosis and how to take the medication. The doctor then asked if either had any further questions. After the medical visit, the provider made sure that the patient’s chart reflected the need for a Somali interpreter with the notation that a female interpreter was preferred.
Conclusion
When working with persons with LEP, providing a professional medical interpreter will facilitate optimal communication. In-person and remote (videoconferencing or telephonic) interpreting are options. When using an interpreter, the provider should maintain eye contact with and direct speech to the patient not the interpreter. The provider should speak clearly, avoid complex terminology, and pause appropriately. Clinicians should remember that patients may have a preference in the gender and dialect of the interpreter and accommodations should be made if available. Finally, asking the patient to repeat back in his or her own words what has been discussed is important to make sure the patient understood what was communicated during the medical visit [10,16].
Corresponding author: Kimberly Schoonover, MD, 200 First Street SW, Rochester, MN 55905, [email protected].
Financial disclosures: None.
1. U.S. Census Bureau. American community survey. Accessed 11 Jul 2016 at www.census.gov/acs/www.
2. Wiener ESR, Ivonne M. Bridging language barriers: how to work with an interpreter. Clin Pediatr Emerg Med 2004;5:93–101.
3. Perez-Stable EJ, Karliner LS. What do we know about patient-clinician interactions with interpreters? J Gen Intern Med 2013;28:339–41.
4. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Clinician ratings of interpreter mediated visits in underserved primary care settings with ad hoc, in-person professional, and video conferencing modes. J Health Care Poor Underserved 2010;21:301–17.
5. Flores G, Abreu M, Barone CP, et al. Errors of medical interpretation and their potential clinical consequences: a comparison of professional versus ad hoc versus no interpreters. Ann Emerg Med 2012;60:545–53.
6. Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010;61:765–73.
7. Karliner LS, Jacobs EA, Chen AH, Mutha S. Do professional interpreters improve clinical care for patients with limited English proficiency? A systematic review of the literature. Health Serv Res 2007;42:727–54.
8. Flores G. The impact of medical interpreter services on the quality of health care: a systematic review. Med Care Res Rev 2005;62:255–99.
9. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
10. Juckett G, Unger K. Appropriate use of medical interpreters. Am Fam Physician 2014;90:476–80.
11. Schapira L, Vargas E, Hidalgo R, et al. Lost in translation: integrating medical interpreters into the multidisciplinary team. Oncologist 2008;13:586–92.
12. Gray B, Hilder J, Stubbe M. How to use interpreters in general practice: the development of a New Zealand toolkit. J Prim Health Care 2012;4:52–61, A1-8.
13. Hsieh E. Not just “getting by”: factors influencing providers’ choice of interpreters. J Gen Intern Med 2015;30:75–82.
14. Philips C. Using interpreters--a guide for GPs. Austr Fam Physician 2010;39:188–95.
15. Boscolo-Hightower A, Rafton SA, Tolman M, et al. Identifying families with limited English proficiency using a capture-recapture approach. Hosp Pediatr 2014;4:16–22.
16. Hadziabdic E, Hjelm K. Working with interpreters: practical advice for use of an interpreter in healthcare. Int J Evid Based Healthc 2013;11:69–76.
17. Abbe M, Simon C, Angiolillo A, et al. A survey of language barriers from the perspective of pediatric oncologists, interpreters, and parents. Pediatr Blood Cancer 2006;47:819–24.
18. Ngo-Metzger Q, Massagli MP, Clarridge BR, et al. Linguistic and cultural barriers to care. J Gen Intern Med 2003;18:44–52.
19. Hadziabdic E, Hjelm K. Arabic-speaking migrants’ experiences of the use of interpreters in healthcare: a qualitative explorative study. Int J Equity Health 2014;13:49.
20. Julliard K, Vivar J, Delgado C, et al. What Latina patients don’t tell their doctors: a qualitative study. Ann Family Med 2008;6:543–9.
21. Hsieh E, Ju H, Kong H. Dimensions of trust: the tensions and challenges in provider--interpreter trust. Qual Health Res 2010;20:170–81.
22. Butow PN, Lobb E, Jefford M, et al. A bridge between cultures: interpreters’ perspectives of consultations with migrant oncology patients. Support Care Cancer 2012;20:235-44.
23. Hsieh E. “I am not a robot!” Interpreters’ views of their roles in health care settings. Qual Health Res 2008;18:1367–83.
24. Hsieh E, Hong SJ. Not all are desired: providers’ views on interpreters’ emotional support for patients. Patient Educ Couns 2010;81:192–7.
25. Elderkin-Thompson V, Silver RC, Waitzkin H. When nurses double as interpreters: a study of Spanish-speaking patients in a US primary care setting. Soc Sci Med 2001;52:1343–58.
26. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Inaccurate language interpretation and its clinical significance in the medical encounters of Spanish-speaking Latinos. Med Care 2015;53:940–7.
27. Gray B, Hilder J, Donaldson H. Why do we not use trained interpreters for all patients with limited English proficiency? Is there a place for using family members? Austr J Prim Health 2011;17:240–9.
28. Jacobs B, Kroll L, Green J, David TJ. The hazards of using a child as an interpreter. J Royal Soc Med 1995;88:474P–5P.
29. Lion KC, Brown JC, Ebel BE, et al. Effect of telephone vs video interpretation on parent comprehension, communication, and utilization in the pediatric emergency department: a randomized clinical trial. JAMA Pediatr 2015;169:1117–25.
30. Wofford JL CC, Johnson DA, Brown MT. Providing a Spanish interpreter using low-cost videoconferencing in a community health centre: a pilot study using tablet computers. Inform Prim Care 2012;20:141–6.
31. Jacobs EA SL, Rathouz PJ. The impact of an enhanced interpreter service intervention on hospital costs and patient satisfaction. J Gen Intern Med 2007;22(Suppl 2):306–11.
32. Jacobs EA LG, Rathouz PJ, Fu P Jr. Shared networks of interpreter services, at relatively low cost, can help providers serve patients with limited english skills. Health Aff (Millwood) 2011;30:1930–8.
33. Locatis C, Williamson D, Gould-Kabler C, et al. Comparing in-person, video, and telephonic medical interpretation. J Gen Intern Med 2010;25:345–50.
34. Jones D, Gill P, Harrison R, et al. An exploratory study of language interpretation services provided by videoconferencing. J Telemed Telecare 2003;9:51–6.
35. Price EL, Perez-Stable EJ, Nickleach D, et al. Interpreter perspectives of in-person, telephonic, and videoconferencing medical interpretation in clinical encounters. Patient Educ Couns 2012;87:226–32.
36. Crossman KL, Wiener E, Roosevelt G, et al. Interpreters: telephonic, in-person interpretation and bilingual providers. Pediatrics 2010;125:e631–8.
37. Degni F, Suominen S, Essen B, et al. Communication and cultural issues in providing reproductive health care to immigrant women: health care providers’ experiences in meeting the needs of [corrected] Somali women living in Finland. J Immigr Minor Health 2012;14:330–43.
38. Odunukan OW, Abdulai RM, Hagi Salaad MF, et al. Provider and interpreter preferences among Somali women in a primary care setting. J Prim Care Comm Health 2015;6:105–10.
39. Poss JE, Beeman T. Effective use of interpreters in health care: guidelines for nurse managers and clinicians. Semin Nurse Manag 1999;7:166–71.
40. Phelan M, Parkman S. How to work with an interpreter. BMJ 1995;311:555–7.
41. Lubrano di Ciccone B, Brown RF, Gueguen JA, et al. Interviewing patients using interpreters in an oncology setting: initial evaluation of a communication skills module. Ann Oncol 2010;21:27–32.
42. Hudelson P. Improving patient-provider communication: insights from interpreters. Fam Pract 2005;22:311–6.
43. Hudelson P, Dao MD, Perron NJ, Bischoff A. Interpreter-mediated diabetes consultations: a qualitative analysis of physician communication practices. BMC Fam Pract 2013;14:163.
44. Green AR, Ngo-Metzger Q, Legedza AT, et al. Interpreter services, language concordance, and health care quality. Experiences of Asian Americans with limited English proficiency. J Gen Intern Med 2005;20:1050–6.
45. Schenker Y, Smith AK, Arnold RM, Fernandez A. “Her husband doesn’t speak much English”: conducting a family meeting with an interpreter. J Palliat Med 2012;15:494–8.
1. U.S. Census Bureau. American community survey. Accessed 11 Jul 2016 at www.census.gov/acs/www.
2. Wiener ESR, Ivonne M. Bridging language barriers: how to work with an interpreter. Clin Pediatr Emerg Med 2004;5:93–101.
3. Perez-Stable EJ, Karliner LS. What do we know about patient-clinician interactions with interpreters? J Gen Intern Med 2013;28:339–41.
4. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Clinician ratings of interpreter mediated visits in underserved primary care settings with ad hoc, in-person professional, and video conferencing modes. J Health Care Poor Underserved 2010;21:301–17.
5. Flores G, Abreu M, Barone CP, et al. Errors of medical interpretation and their potential clinical consequences: a comparison of professional versus ad hoc versus no interpreters. Ann Emerg Med 2012;60:545–53.
6. Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010;61:765–73.
7. Karliner LS, Jacobs EA, Chen AH, Mutha S. Do professional interpreters improve clinical care for patients with limited English proficiency? A systematic review of the literature. Health Serv Res 2007;42:727–54.
8. Flores G. The impact of medical interpreter services on the quality of health care: a systematic review. Med Care Res Rev 2005;62:255–99.
9. Lindholm M, Hargraves JL, Ferguson WJ, Reed G. Professional language interpretation and inpatient length of stay and readmission rates. J Gen Intern Med 2012;27:1294–9.
10. Juckett G, Unger K. Appropriate use of medical interpreters. Am Fam Physician 2014;90:476–80.
11. Schapira L, Vargas E, Hidalgo R, et al. Lost in translation: integrating medical interpreters into the multidisciplinary team. Oncologist 2008;13:586–92.
12. Gray B, Hilder J, Stubbe M. How to use interpreters in general practice: the development of a New Zealand toolkit. J Prim Health Care 2012;4:52–61, A1-8.
13. Hsieh E. Not just “getting by”: factors influencing providers’ choice of interpreters. J Gen Intern Med 2015;30:75–82.
14. Philips C. Using interpreters--a guide for GPs. Austr Fam Physician 2010;39:188–95.
15. Boscolo-Hightower A, Rafton SA, Tolman M, et al. Identifying families with limited English proficiency using a capture-recapture approach. Hosp Pediatr 2014;4:16–22.
16. Hadziabdic E, Hjelm K. Working with interpreters: practical advice for use of an interpreter in healthcare. Int J Evid Based Healthc 2013;11:69–76.
17. Abbe M, Simon C, Angiolillo A, et al. A survey of language barriers from the perspective of pediatric oncologists, interpreters, and parents. Pediatr Blood Cancer 2006;47:819–24.
18. Ngo-Metzger Q, Massagli MP, Clarridge BR, et al. Linguistic and cultural barriers to care. J Gen Intern Med 2003;18:44–52.
19. Hadziabdic E, Hjelm K. Arabic-speaking migrants’ experiences of the use of interpreters in healthcare: a qualitative explorative study. Int J Equity Health 2014;13:49.
20. Julliard K, Vivar J, Delgado C, et al. What Latina patients don’t tell their doctors: a qualitative study. Ann Family Med 2008;6:543–9.
21. Hsieh E, Ju H, Kong H. Dimensions of trust: the tensions and challenges in provider--interpreter trust. Qual Health Res 2010;20:170–81.
22. Butow PN, Lobb E, Jefford M, et al. A bridge between cultures: interpreters’ perspectives of consultations with migrant oncology patients. Support Care Cancer 2012;20:235-44.
23. Hsieh E. “I am not a robot!” Interpreters’ views of their roles in health care settings. Qual Health Res 2008;18:1367–83.
24. Hsieh E, Hong SJ. Not all are desired: providers’ views on interpreters’ emotional support for patients. Patient Educ Couns 2010;81:192–7.
25. Elderkin-Thompson V, Silver RC, Waitzkin H. When nurses double as interpreters: a study of Spanish-speaking patients in a US primary care setting. Soc Sci Med 2001;52:1343–58.
26. Napoles AM, Santoyo-Olsson J, Karliner LS, et al. Inaccurate language interpretation and its clinical significance in the medical encounters of Spanish-speaking Latinos. Med Care 2015;53:940–7.
27. Gray B, Hilder J, Donaldson H. Why do we not use trained interpreters for all patients with limited English proficiency? Is there a place for using family members? Austr J Prim Health 2011;17:240–9.
28. Jacobs B, Kroll L, Green J, David TJ. The hazards of using a child as an interpreter. J Royal Soc Med 1995;88:474P–5P.
29. Lion KC, Brown JC, Ebel BE, et al. Effect of telephone vs video interpretation on parent comprehension, communication, and utilization in the pediatric emergency department: a randomized clinical trial. JAMA Pediatr 2015;169:1117–25.
30. Wofford JL CC, Johnson DA, Brown MT. Providing a Spanish interpreter using low-cost videoconferencing in a community health centre: a pilot study using tablet computers. Inform Prim Care 2012;20:141–6.
31. Jacobs EA SL, Rathouz PJ. The impact of an enhanced interpreter service intervention on hospital costs and patient satisfaction. J Gen Intern Med 2007;22(Suppl 2):306–11.
32. Jacobs EA LG, Rathouz PJ, Fu P Jr. Shared networks of interpreter services, at relatively low cost, can help providers serve patients with limited english skills. Health Aff (Millwood) 2011;30:1930–8.
33. Locatis C, Williamson D, Gould-Kabler C, et al. Comparing in-person, video, and telephonic medical interpretation. J Gen Intern Med 2010;25:345–50.
34. Jones D, Gill P, Harrison R, et al. An exploratory study of language interpretation services provided by videoconferencing. J Telemed Telecare 2003;9:51–6.
35. Price EL, Perez-Stable EJ, Nickleach D, et al. Interpreter perspectives of in-person, telephonic, and videoconferencing medical interpretation in clinical encounters. Patient Educ Couns 2012;87:226–32.
36. Crossman KL, Wiener E, Roosevelt G, et al. Interpreters: telephonic, in-person interpretation and bilingual providers. Pediatrics 2010;125:e631–8.
37. Degni F, Suominen S, Essen B, et al. Communication and cultural issues in providing reproductive health care to immigrant women: health care providers’ experiences in meeting the needs of [corrected] Somali women living in Finland. J Immigr Minor Health 2012;14:330–43.
38. Odunukan OW, Abdulai RM, Hagi Salaad MF, et al. Provider and interpreter preferences among Somali women in a primary care setting. J Prim Care Comm Health 2015;6:105–10.
39. Poss JE, Beeman T. Effective use of interpreters in health care: guidelines for nurse managers and clinicians. Semin Nurse Manag 1999;7:166–71.
40. Phelan M, Parkman S. How to work with an interpreter. BMJ 1995;311:555–7.
41. Lubrano di Ciccone B, Brown RF, Gueguen JA, et al. Interviewing patients using interpreters in an oncology setting: initial evaluation of a communication skills module. Ann Oncol 2010;21:27–32.
42. Hudelson P. Improving patient-provider communication: insights from interpreters. Fam Pract 2005;22:311–6.
43. Hudelson P, Dao MD, Perron NJ, Bischoff A. Interpreter-mediated diabetes consultations: a qualitative analysis of physician communication practices. BMC Fam Pract 2013;14:163.
44. Green AR, Ngo-Metzger Q, Legedza AT, et al. Interpreter services, language concordance, and health care quality. Experiences of Asian Americans with limited English proficiency. J Gen Intern Med 2005;20:1050–6.
45. Schenker Y, Smith AK, Arnold RM, Fernandez A. “Her husband doesn’t speak much English”: conducting a family meeting with an interpreter. J Palliat Med 2012;15:494–8.
Managing clozapine-induced neutropenia and agranulocytosis
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to 
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
Suicidal and asking for money for food
CASE Suicidal and hungry
Mr. L, age 59, attempts suicide by taking approximately 20 acetaminophen tablets of unknown dosage. He immediately comes to the emergency department where blood work reveals a 4-hour acetaminophen level of 94.8 μg/mL (therapeutic range, 10 to 30 μg/mL; toxic range, >150 μg/mL); administration of N-acetylcysteine is unnecessary. Mr. L is admitted to general medical services for monitoring and is transferred to our unit for psychiatric evaluation and management.
During our initial interview, Mr. L, who has a developmental disability, is grossly oriented and generally cooperative, reporting depressed mood with an irritable affect. He is preoccupied with having limited funds and repeatedly states he is worried that he can’t buy food, but says that the hospital could help by providing for him. Mr. L states that his depressed mood is directly related to his financial situation and, that if he had more money, he would not be suicidal. He cites worsening visual impairment that requires surgery as an additional stressor.
On several occasions, Mr. L states that the only way to help him is to give him $600 so that he can buy food and pay for medical treatment. Mr. L says he does not feel supported by his family, despite having a sister who lives nearby.
What would you include in the differential diagnosis for Mr. L?
a) major depressive disorder (MDD)
b) depression secondary to a medical condition
c) neurocognitive disorder
d) adjustment disorder with depressive features
e) factitious disorder
The authors’ observations
Our differential diagnosis included MDD, adjustment disorder, neurocognitive disorder, and factitious disorder. He did not meet criteria for MDD because he did not have excessive guilt, loss of interest, change in sleep or appetite, psychomotor dysregulation, or change in energy level. Although suicidal behavior could indicate MDD, the fact that he immediately walked to the hospital after taking an excessive amount of acetaminophen suggests that he did not want to die. Further, he attributed his suicidal thoughts to environmental stressors. Similarly, we ruled out adjustment disorder because he had no reported or observed changes in mood or anxiety. Although financial difficulties might have overwhelmed his limited coping abilities, he took too much acetaminophen to ensure that he was hospitalized. His motivation for seeking hospitalization ruled out factitious disorder. Mr. L has a developmental disability, but information obtained from collateral sources ruled out an acute change to cognitive functioning.
HISTORY Repeated admissions
Mr. L has a history of a psychiatric hospitalization 3 weeks prior to this admission. He presented to an emergency department stating that his blood glucose was low. Mr. L was noted to be confused and anxious and said he was convinced he was going to die. At that time, his thought content was hyper-religious and he claimed he could hear the devil. Mr. L was hospitalized and started on low-dosage risperidone. At discharge, he declined referral for outpatient mental health treatment because he denied having a mental illness. However, he was amenable to follow up at a wellness clinic.
Mr. L has worked at a local supermarket for 19 years and has lived independently throughout his adult life. After he returned to the community, he was repeatedly absent from work, which further exacerbated his financial strain. He attended a follow-up outpatient appointment but reported, “They didn’t help me,” although it was unclear what he meant.
Between admissions to our hospital, Mr. L had 2 visits to an emergency department, the first time saying he felt depressed and the second reporting he attempted suicide by taking 5 acetaminophen tablets. On both occasions he requested placement in a residential facility but was discharged home after an initial assessment. Emergency room records indicated that Mr. L stated, “If you cannot give me money for food, then there is no use and I would rather die.”
What is the most likely DSM-5 diagnosis for Mr. L?
a) schizophrenia
b) malingering
c) brief psychotic disorder
d) dependent personality disorder
The authors’ observations
Malingering in DSM-5 is defined as the “intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.”1 These external incentives include financial compensation, avoiding military duties, evading criminal charges, and avoiding work, and are collectively considered as secondary gain. Although not considered a diagnosis in the strictest sense, clinicians must differentiate malingering from other psychiatric disorders. In the literature, case reports describe patients who feigned an array of symptoms including those of posttraumatic stress disorder, paraphilias, cognitive dysfunction, depression, anxiety, and psychosis.2-5
In Mr. L’s case, malingering presented as suicidal behavior with an inadvertently high fatality risk. Notably, Mr. L came to an emergency room a few days before this admission after swallowing 5 acetaminophen tablets in a suicide attempt, which did not lead to a medical or psychiatric hospitalization. In an attempt to ensure admission, Mr. L then took a potentially lethal dose of 20 acetaminophen tablets. In our assessment and according to his statements, the primary motivation for the suicide attempt was to obtain reliable food and housing. Mr. L’s developmental disability might have contributed to a relative lack of understanding of the consequences of his actions. In addition, poor overall communication and coping skills led to an exaggerated response to psychosocial stressors.
Malingering and suicide attempts
Few studies have investigated malingering in regards to suicide and other psychiatric emergencies. In a study of 227 consecutive psychiatric emergencies assessed for evidence of malingering, 13% were thought to be feigned or exaggerated.6 Interestingly, the most commonly reported secondary gain was food and shelter, similar to Mr. L. This study did not report the types of psychiatric emergencies, therefore suicidal actions associated with malingering could not be evaluated.
In another study, 40 patients hospitalized for suicidal ideation (n = 29, 72%) or suicidal gestures (n = 11, 28%) in a large, urban tertiary care center were evaluated for malingering by anonymous report of feigned or exaggerated symptoms.7 Most of these patients were diagnosed with a mood disorder (28%) and/or an adjustment disorder (53%). Four (10%) admitted to malingering. Among the malingerers, reasons for feigning illness included:
- wanting to be hospitalized
- wanting to make someone angry or feel sorry
- gaining access to detoxification programs
- getting treatment for emotional problems.
Interestingly, an analysis of demographic factors associated with malingering reveals an association with suicide attempts but not persistent suicidal ideations. This could be because of selection bias; patients who reported a suicide attempt might be more likely to be hospitalized.
A follow-up study8 evaluated 50 additional consecutive psychiatric inpatients admitted to the same tertiary care hospital for suicide risk. Unlike the previous study, a larger proportion of these patients had made a suicide attempt (n = 21, 42%) and a greater number had made a previous suicide attempt (n = 33, 66%). Primary mood disorders comprised most of the psychiatric diagnoses (n = 28, 56%). In this study, the exact nature of the suicide gestures was not documented, leaving open the question of lethality of the attempts. These studies do not suggest that those who malinger are not at risk for suicide, only that these patients tend to exaggerate the severity of their ideations or behaviors.
OUTCOME Reluctantly discharged
We contact Mr. L’s siblings, who offer to provide temporary housing and financial support and assist him with medical needs. This abated Mr. L’s suicidal ideation; however, he wishes to remain in the hospital with the goals of obtaining eyeglasses and dentures. We explain that psychiatric hospitalization is no longer indicated and he is discharged.
Which of the following is the most effective management strategy for malingering?
a) direct confrontation of the malingering patient
b) immediate discharge once malingering is identified
c) evaluation for possible comorbid psychiatric conditions
d) neuropsychiatric consultation
The authors’ observations


The challenges of treating patients who malinger include clinician uncertainty in making the diagnosis and high variability in occurrence across settings (Table 1). Current estimates indicate that 4% to 8% of medical and psychiatric cases not involved in litigation or compensation have an element of feigned symptoms.3,9 The rate could be higher in specific circumstances such as medicolegal disputes and criminal cases.10
The societal impact of malingering is significant. Therefore, identifying these patients is an important clinical intervention that can have a wide impact.11 However, it is also important to acknowledge that genuine psychiatric illness could be comorbid with malingering. Although differentiating a patient’s true from feigned symptoms can be difficult, it is critical to carefully evaluate the patient in order to provide the best treatment.
It seems that physicians can detect malingering, but documentation often is not provided. In the Rissmiller et al study,7 all 4 cases of malingering were identified retrospectively by study psychiatrists; however, none of their medical records included documentation of malingering, a finding also reported in the Yates et al study.6 Also concerning, the clinicians suspected malingering in some patients who were not feigning symptoms, suggesting that a relatively high threshold is necessary for making the diagnosis.
How to help patients who malinger
Identifying malingering in patients with obvious secondary gain is important to prevent exposure to potential adverse effects of medication and unnecessary use of medical resources. In addition, obtaining collateral information, records from previous admissions or outpatient treatment, and psychological testing adds to the body of evidence suggesting malingering. We also recommend a comprehensive psychosocial evaluation to identify the presence of secondary gain.
Management of malingering (Table 2) includes building a strong therapeutic alliance, exploring reasons for feigning symptoms, open discussion of inciting external factors such as interpersonal conflict or difficulties at work, and/or confrontation.10 In addition, supportive psychotherapy might help strengthen coping mechanisms and problem solving strategies, thereby removing the need for secondary gain.12 Additionally, face-saving mechanisms that allow the patient to discard their feigned symptoms, or enable the person to alter his (her) history, could be to his benefit. Lastly, and importantly, clinicians should focus efforts on ruling out or effectively treating comorbid psychiatric conditions.
From a risk management standpoint, include all available data to support the malingering diagnosis in your progress notes and discharge summaries. A clinician seeking to discharge a patient suspected of malingering who is still endorsing suicidal or homicidal intent will benefit from administrative review, including legal counsel to mitigate risk, and be more confident discharging somebody assessed to be malingering.
We recognize that certain patients could trigger countertransference reactions that impel clinicians to take on a significant caretaking role. Patients skillful at deception could manifest a desire to rescue or save them. In these instances, clinicians should examine why and how these feelings have come about, particularly if there is evidence that the individual could be attempting to use the interaction to achieve secondary gain. Awareness of these feelings could help with the diagnostic formulation. Moreover, a clinician who has such strong feelings might be tempted to abet a patient in achieving the secondary gain, or protect him (her) from the natural consequences of individual’s deception (eg, not discharging a hospitalized patient). This is counter-therapeutic and reinforces maladaptive behaviors and coping processes.13
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington DC: American Psychiatric Association; 2013.
2. Fedoroff JP, Hanson A, McGuire M, et al. Simulated paraphilias: a preliminary study of patients who imitate or exaggerate paraphilic symptoms and behaviors. J Forensic Sci. 1992;37(3):902-911.
3. Mittenberg W, Patton C, Canyock EM, et al. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol. 2002;24(8):1094-1102.
4. Waite S, Geddes A. Malingered psychosis leading to involuntary psychiatric hospitalization. Australas Psychiatry. 2006;14(4):419-421.
5. Hall RC, Hall RC. Malingering of PTSD: forensic and diagnostic consideration, characteristics of malingerers and clinical presentations. Gen Hosp Psychiatry. 2006;28(6):525-535.
6. Yates BD, Nordquist CR, Shultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv. 1996;47(9):998-1000.
7. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicidal ideators and attempters. Crisis. 1998;19(2):62-66.
8. Rissmiller D, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric inpatients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
9. Sullivan K, Lange RT, Dawes S. Methods of detecting malingering and estimated symptom exaggeration base rates in Australia. Journal of Forensic Neuropsychology. 2007;4(4):49-70.
10. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
11. Chafetz M, Underhill J. Estimated costs of malingered disability. Arch Clin Neuropsychol. 2013;28(7):633-639.
12. Peebles R, Sabel
13. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
CASE Suicidal and hungry
Mr. L, age 59, attempts suicide by taking approximately 20 acetaminophen tablets of unknown dosage. He immediately comes to the emergency department where blood work reveals a 4-hour acetaminophen level of 94.8 μg/mL (therapeutic range, 10 to 30 μg/mL; toxic range, >150 μg/mL); administration of N-acetylcysteine is unnecessary. Mr. L is admitted to general medical services for monitoring and is transferred to our unit for psychiatric evaluation and management.
During our initial interview, Mr. L, who has a developmental disability, is grossly oriented and generally cooperative, reporting depressed mood with an irritable affect. He is preoccupied with having limited funds and repeatedly states he is worried that he can’t buy food, but says that the hospital could help by providing for him. Mr. L states that his depressed mood is directly related to his financial situation and, that if he had more money, he would not be suicidal. He cites worsening visual impairment that requires surgery as an additional stressor.
On several occasions, Mr. L states that the only way to help him is to give him $600 so that he can buy food and pay for medical treatment. Mr. L says he does not feel supported by his family, despite having a sister who lives nearby.
What would you include in the differential diagnosis for Mr. L?
a) major depressive disorder (MDD)
b) depression secondary to a medical condition
c) neurocognitive disorder
d) adjustment disorder with depressive features
e) factitious disorder
The authors’ observations
Our differential diagnosis included MDD, adjustment disorder, neurocognitive disorder, and factitious disorder. He did not meet criteria for MDD because he did not have excessive guilt, loss of interest, change in sleep or appetite, psychomotor dysregulation, or change in energy level. Although suicidal behavior could indicate MDD, the fact that he immediately walked to the hospital after taking an excessive amount of acetaminophen suggests that he did not want to die. Further, he attributed his suicidal thoughts to environmental stressors. Similarly, we ruled out adjustment disorder because he had no reported or observed changes in mood or anxiety. Although financial difficulties might have overwhelmed his limited coping abilities, he took too much acetaminophen to ensure that he was hospitalized. His motivation for seeking hospitalization ruled out factitious disorder. Mr. L has a developmental disability, but information obtained from collateral sources ruled out an acute change to cognitive functioning.
HISTORY Repeated admissions
Mr. L has a history of a psychiatric hospitalization 3 weeks prior to this admission. He presented to an emergency department stating that his blood glucose was low. Mr. L was noted to be confused and anxious and said he was convinced he was going to die. At that time, his thought content was hyper-religious and he claimed he could hear the devil. Mr. L was hospitalized and started on low-dosage risperidone. At discharge, he declined referral for outpatient mental health treatment because he denied having a mental illness. However, he was amenable to follow up at a wellness clinic.
Mr. L has worked at a local supermarket for 19 years and has lived independently throughout his adult life. After he returned to the community, he was repeatedly absent from work, which further exacerbated his financial strain. He attended a follow-up outpatient appointment but reported, “They didn’t help me,” although it was unclear what he meant.
Between admissions to our hospital, Mr. L had 2 visits to an emergency department, the first time saying he felt depressed and the second reporting he attempted suicide by taking 5 acetaminophen tablets. On both occasions he requested placement in a residential facility but was discharged home after an initial assessment. Emergency room records indicated that Mr. L stated, “If you cannot give me money for food, then there is no use and I would rather die.”
What is the most likely DSM-5 diagnosis for Mr. L?
a) schizophrenia
b) malingering
c) brief psychotic disorder
d) dependent personality disorder
The authors’ observations
Malingering in DSM-5 is defined as the “intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.”1 These external incentives include financial compensation, avoiding military duties, evading criminal charges, and avoiding work, and are collectively considered as secondary gain. Although not considered a diagnosis in the strictest sense, clinicians must differentiate malingering from other psychiatric disorders. In the literature, case reports describe patients who feigned an array of symptoms including those of posttraumatic stress disorder, paraphilias, cognitive dysfunction, depression, anxiety, and psychosis.2-5
In Mr. L’s case, malingering presented as suicidal behavior with an inadvertently high fatality risk. Notably, Mr. L came to an emergency room a few days before this admission after swallowing 5 acetaminophen tablets in a suicide attempt, which did not lead to a medical or psychiatric hospitalization. In an attempt to ensure admission, Mr. L then took a potentially lethal dose of 20 acetaminophen tablets. In our assessment and according to his statements, the primary motivation for the suicide attempt was to obtain reliable food and housing. Mr. L’s developmental disability might have contributed to a relative lack of understanding of the consequences of his actions. In addition, poor overall communication and coping skills led to an exaggerated response to psychosocial stressors.
Malingering and suicide attempts
Few studies have investigated malingering in regards to suicide and other psychiatric emergencies. In a study of 227 consecutive psychiatric emergencies assessed for evidence of malingering, 13% were thought to be feigned or exaggerated.6 Interestingly, the most commonly reported secondary gain was food and shelter, similar to Mr. L. This study did not report the types of psychiatric emergencies, therefore suicidal actions associated with malingering could not be evaluated.
In another study, 40 patients hospitalized for suicidal ideation (n = 29, 72%) or suicidal gestures (n = 11, 28%) in a large, urban tertiary care center were evaluated for malingering by anonymous report of feigned or exaggerated symptoms.7 Most of these patients were diagnosed with a mood disorder (28%) and/or an adjustment disorder (53%). Four (10%) admitted to malingering. Among the malingerers, reasons for feigning illness included:
- wanting to be hospitalized
- wanting to make someone angry or feel sorry
- gaining access to detoxification programs
- getting treatment for emotional problems.
Interestingly, an analysis of demographic factors associated with malingering reveals an association with suicide attempts but not persistent suicidal ideations. This could be because of selection bias; patients who reported a suicide attempt might be more likely to be hospitalized.
A follow-up study8 evaluated 50 additional consecutive psychiatric inpatients admitted to the same tertiary care hospital for suicide risk. Unlike the previous study, a larger proportion of these patients had made a suicide attempt (n = 21, 42%) and a greater number had made a previous suicide attempt (n = 33, 66%). Primary mood disorders comprised most of the psychiatric diagnoses (n = 28, 56%). In this study, the exact nature of the suicide gestures was not documented, leaving open the question of lethality of the attempts. These studies do not suggest that those who malinger are not at risk for suicide, only that these patients tend to exaggerate the severity of their ideations or behaviors.
OUTCOME Reluctantly discharged
We contact Mr. L’s siblings, who offer to provide temporary housing and financial support and assist him with medical needs. This abated Mr. L’s suicidal ideation; however, he wishes to remain in the hospital with the goals of obtaining eyeglasses and dentures. We explain that psychiatric hospitalization is no longer indicated and he is discharged.
Which of the following is the most effective management strategy for malingering?
a) direct confrontation of the malingering patient
b) immediate discharge once malingering is identified
c) evaluation for possible comorbid psychiatric conditions
d) neuropsychiatric consultation
The authors’ observations
The challenges of treating patients who malinger include clinician uncertainty in making the diagnosis and high variability in occurrence across settings (Table 1). Current estimates indicate that 4% to 8% of medical and psychiatric cases not involved in litigation or compensation have an element of feigned symptoms.3,9 The rate could be higher in specific circumstances such as medicolegal disputes and criminal cases.10
The societal impact of malingering is significant. Therefore, identifying these patients is an important clinical intervention that can have a wide impact.11 However, it is also important to acknowledge that genuine psychiatric illness could be comorbid with malingering. Although differentiating a patient’s true from feigned symptoms can be difficult, it is critical to carefully evaluate the patient in order to provide the best treatment.
It seems that physicians can detect malingering, but documentation often is not provided. In the Rissmiller et al study,7 all 4 cases of malingering were identified retrospectively by study psychiatrists; however, none of their medical records included documentation of malingering, a finding also reported in the Yates et al study.6 Also concerning, the clinicians suspected malingering in some patients who were not feigning symptoms, suggesting that a relatively high threshold is necessary for making the diagnosis.
How to help patients who malinger
Identifying malingering in patients with obvious secondary gain is important to prevent exposure to potential adverse effects of medication and unnecessary use of medical resources. In addition, obtaining collateral information, records from previous admissions or outpatient treatment, and psychological testing adds to the body of evidence suggesting malingering. We also recommend a comprehensive psychosocial evaluation to identify the presence of secondary gain.
Management of malingering (Table 2) includes building a strong therapeutic alliance, exploring reasons for feigning symptoms, open discussion of inciting external factors such as interpersonal conflict or difficulties at work, and/or confrontation.10 In addition, supportive psychotherapy might help strengthen coping mechanisms and problem solving strategies, thereby removing the need for secondary gain.12 Additionally, face-saving mechanisms that allow the patient to discard their feigned symptoms, or enable the person to alter his (her) history, could be to his benefit. Lastly, and importantly, clinicians should focus efforts on ruling out or effectively treating comorbid psychiatric conditions.
From a risk management standpoint, include all available data to support the malingering diagnosis in your progress notes and discharge summaries. A clinician seeking to discharge a patient suspected of malingering who is still endorsing suicidal or homicidal intent will benefit from administrative review, including legal counsel to mitigate risk, and be more confident discharging somebody assessed to be malingering.
We recognize that certain patients could trigger countertransference reactions that impel clinicians to take on a significant caretaking role. Patients skillful at deception could manifest a desire to rescue or save them. In these instances, clinicians should examine why and how these feelings have come about, particularly if there is evidence that the individual could be attempting to use the interaction to achieve secondary gain. Awareness of these feelings could help with the diagnostic formulation. Moreover, a clinician who has such strong feelings might be tempted to abet a patient in achieving the secondary gain, or protect him (her) from the natural consequences of individual’s deception (eg, not discharging a hospitalized patient). This is counter-therapeutic and reinforces maladaptive behaviors and coping processes.13
CASE Suicidal and hungry
Mr. L, age 59, attempts suicide by taking approximately 20 acetaminophen tablets of unknown dosage. He immediately comes to the emergency department where blood work reveals a 4-hour acetaminophen level of 94.8 μg/mL (therapeutic range, 10 to 30 μg/mL; toxic range, >150 μg/mL); administration of N-acetylcysteine is unnecessary. Mr. L is admitted to general medical services for monitoring and is transferred to our unit for psychiatric evaluation and management.
During our initial interview, Mr. L, who has a developmental disability, is grossly oriented and generally cooperative, reporting depressed mood with an irritable affect. He is preoccupied with having limited funds and repeatedly states he is worried that he can’t buy food, but says that the hospital could help by providing for him. Mr. L states that his depressed mood is directly related to his financial situation and, that if he had more money, he would not be suicidal. He cites worsening visual impairment that requires surgery as an additional stressor.
On several occasions, Mr. L states that the only way to help him is to give him $600 so that he can buy food and pay for medical treatment. Mr. L says he does not feel supported by his family, despite having a sister who lives nearby.
What would you include in the differential diagnosis for Mr. L?
a) major depressive disorder (MDD)
b) depression secondary to a medical condition
c) neurocognitive disorder
d) adjustment disorder with depressive features
e) factitious disorder
The authors’ observations
Our differential diagnosis included MDD, adjustment disorder, neurocognitive disorder, and factitious disorder. He did not meet criteria for MDD because he did not have excessive guilt, loss of interest, change in sleep or appetite, psychomotor dysregulation, or change in energy level. Although suicidal behavior could indicate MDD, the fact that he immediately walked to the hospital after taking an excessive amount of acetaminophen suggests that he did not want to die. Further, he attributed his suicidal thoughts to environmental stressors. Similarly, we ruled out adjustment disorder because he had no reported or observed changes in mood or anxiety. Although financial difficulties might have overwhelmed his limited coping abilities, he took too much acetaminophen to ensure that he was hospitalized. His motivation for seeking hospitalization ruled out factitious disorder. Mr. L has a developmental disability, but information obtained from collateral sources ruled out an acute change to cognitive functioning.
HISTORY Repeated admissions
Mr. L has a history of a psychiatric hospitalization 3 weeks prior to this admission. He presented to an emergency department stating that his blood glucose was low. Mr. L was noted to be confused and anxious and said he was convinced he was going to die. At that time, his thought content was hyper-religious and he claimed he could hear the devil. Mr. L was hospitalized and started on low-dosage risperidone. At discharge, he declined referral for outpatient mental health treatment because he denied having a mental illness. However, he was amenable to follow up at a wellness clinic.
Mr. L has worked at a local supermarket for 19 years and has lived independently throughout his adult life. After he returned to the community, he was repeatedly absent from work, which further exacerbated his financial strain. He attended a follow-up outpatient appointment but reported, “They didn’t help me,” although it was unclear what he meant.
Between admissions to our hospital, Mr. L had 2 visits to an emergency department, the first time saying he felt depressed and the second reporting he attempted suicide by taking 5 acetaminophen tablets. On both occasions he requested placement in a residential facility but was discharged home after an initial assessment. Emergency room records indicated that Mr. L stated, “If you cannot give me money for food, then there is no use and I would rather die.”
What is the most likely DSM-5 diagnosis for Mr. L?
a) schizophrenia
b) malingering
c) brief psychotic disorder
d) dependent personality disorder
The authors’ observations
Malingering in DSM-5 is defined as the “intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.”1 These external incentives include financial compensation, avoiding military duties, evading criminal charges, and avoiding work, and are collectively considered as secondary gain. Although not considered a diagnosis in the strictest sense, clinicians must differentiate malingering from other psychiatric disorders. In the literature, case reports describe patients who feigned an array of symptoms including those of posttraumatic stress disorder, paraphilias, cognitive dysfunction, depression, anxiety, and psychosis.2-5
In Mr. L’s case, malingering presented as suicidal behavior with an inadvertently high fatality risk. Notably, Mr. L came to an emergency room a few days before this admission after swallowing 5 acetaminophen tablets in a suicide attempt, which did not lead to a medical or psychiatric hospitalization. In an attempt to ensure admission, Mr. L then took a potentially lethal dose of 20 acetaminophen tablets. In our assessment and according to his statements, the primary motivation for the suicide attempt was to obtain reliable food and housing. Mr. L’s developmental disability might have contributed to a relative lack of understanding of the consequences of his actions. In addition, poor overall communication and coping skills led to an exaggerated response to psychosocial stressors.
Malingering and suicide attempts
Few studies have investigated malingering in regards to suicide and other psychiatric emergencies. In a study of 227 consecutive psychiatric emergencies assessed for evidence of malingering, 13% were thought to be feigned or exaggerated.6 Interestingly, the most commonly reported secondary gain was food and shelter, similar to Mr. L. This study did not report the types of psychiatric emergencies, therefore suicidal actions associated with malingering could not be evaluated.
In another study, 40 patients hospitalized for suicidal ideation (n = 29, 72%) or suicidal gestures (n = 11, 28%) in a large, urban tertiary care center were evaluated for malingering by anonymous report of feigned or exaggerated symptoms.7 Most of these patients were diagnosed with a mood disorder (28%) and/or an adjustment disorder (53%). Four (10%) admitted to malingering. Among the malingerers, reasons for feigning illness included:
- wanting to be hospitalized
- wanting to make someone angry or feel sorry
- gaining access to detoxification programs
- getting treatment for emotional problems.
Interestingly, an analysis of demographic factors associated with malingering reveals an association with suicide attempts but not persistent suicidal ideations. This could be because of selection bias; patients who reported a suicide attempt might be more likely to be hospitalized.
A follow-up study8 evaluated 50 additional consecutive psychiatric inpatients admitted to the same tertiary care hospital for suicide risk. Unlike the previous study, a larger proportion of these patients had made a suicide attempt (n = 21, 42%) and a greater number had made a previous suicide attempt (n = 33, 66%). Primary mood disorders comprised most of the psychiatric diagnoses (n = 28, 56%). In this study, the exact nature of the suicide gestures was not documented, leaving open the question of lethality of the attempts. These studies do not suggest that those who malinger are not at risk for suicide, only that these patients tend to exaggerate the severity of their ideations or behaviors.
OUTCOME Reluctantly discharged
We contact Mr. L’s siblings, who offer to provide temporary housing and financial support and assist him with medical needs. This abated Mr. L’s suicidal ideation; however, he wishes to remain in the hospital with the goals of obtaining eyeglasses and dentures. We explain that psychiatric hospitalization is no longer indicated and he is discharged.
Which of the following is the most effective management strategy for malingering?
a) direct confrontation of the malingering patient
b) immediate discharge once malingering is identified
c) evaluation for possible comorbid psychiatric conditions
d) neuropsychiatric consultation
The authors’ observations
The challenges of treating patients who malinger include clinician uncertainty in making the diagnosis and high variability in occurrence across settings (Table 1). Current estimates indicate that 4% to 8% of medical and psychiatric cases not involved in litigation or compensation have an element of feigned symptoms.3,9 The rate could be higher in specific circumstances such as medicolegal disputes and criminal cases.10
The societal impact of malingering is significant. Therefore, identifying these patients is an important clinical intervention that can have a wide impact.11 However, it is also important to acknowledge that genuine psychiatric illness could be comorbid with malingering. Although differentiating a patient’s true from feigned symptoms can be difficult, it is critical to carefully evaluate the patient in order to provide the best treatment.
It seems that physicians can detect malingering, but documentation often is not provided. In the Rissmiller et al study,7 all 4 cases of malingering were identified retrospectively by study psychiatrists; however, none of their medical records included documentation of malingering, a finding also reported in the Yates et al study.6 Also concerning, the clinicians suspected malingering in some patients who were not feigning symptoms, suggesting that a relatively high threshold is necessary for making the diagnosis.
How to help patients who malinger
Identifying malingering in patients with obvious secondary gain is important to prevent exposure to potential adverse effects of medication and unnecessary use of medical resources. In addition, obtaining collateral information, records from previous admissions or outpatient treatment, and psychological testing adds to the body of evidence suggesting malingering. We also recommend a comprehensive psychosocial evaluation to identify the presence of secondary gain.
Management of malingering (Table 2) includes building a strong therapeutic alliance, exploring reasons for feigning symptoms, open discussion of inciting external factors such as interpersonal conflict or difficulties at work, and/or confrontation.10 In addition, supportive psychotherapy might help strengthen coping mechanisms and problem solving strategies, thereby removing the need for secondary gain.12 Additionally, face-saving mechanisms that allow the patient to discard their feigned symptoms, or enable the person to alter his (her) history, could be to his benefit. Lastly, and importantly, clinicians should focus efforts on ruling out or effectively treating comorbid psychiatric conditions.
From a risk management standpoint, include all available data to support the malingering diagnosis in your progress notes and discharge summaries. A clinician seeking to discharge a patient suspected of malingering who is still endorsing suicidal or homicidal intent will benefit from administrative review, including legal counsel to mitigate risk, and be more confident discharging somebody assessed to be malingering.
We recognize that certain patients could trigger countertransference reactions that impel clinicians to take on a significant caretaking role. Patients skillful at deception could manifest a desire to rescue or save them. In these instances, clinicians should examine why and how these feelings have come about, particularly if there is evidence that the individual could be attempting to use the interaction to achieve secondary gain. Awareness of these feelings could help with the diagnostic formulation. Moreover, a clinician who has such strong feelings might be tempted to abet a patient in achieving the secondary gain, or protect him (her) from the natural consequences of individual’s deception (eg, not discharging a hospitalized patient). This is counter-therapeutic and reinforces maladaptive behaviors and coping processes.13
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington DC: American Psychiatric Association; 2013.
2. Fedoroff JP, Hanson A, McGuire M, et al. Simulated paraphilias: a preliminary study of patients who imitate or exaggerate paraphilic symptoms and behaviors. J Forensic Sci. 1992;37(3):902-911.
3. Mittenberg W, Patton C, Canyock EM, et al. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol. 2002;24(8):1094-1102.
4. Waite S, Geddes A. Malingered psychosis leading to involuntary psychiatric hospitalization. Australas Psychiatry. 2006;14(4):419-421.
5. Hall RC, Hall RC. Malingering of PTSD: forensic and diagnostic consideration, characteristics of malingerers and clinical presentations. Gen Hosp Psychiatry. 2006;28(6):525-535.
6. Yates BD, Nordquist CR, Shultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv. 1996;47(9):998-1000.
7. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicidal ideators and attempters. Crisis. 1998;19(2):62-66.
8. Rissmiller D, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric inpatients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
9. Sullivan K, Lange RT, Dawes S. Methods of detecting malingering and estimated symptom exaggeration base rates in Australia. Journal of Forensic Neuropsychology. 2007;4(4):49-70.
10. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
11. Chafetz M, Underhill J. Estimated costs of malingered disability. Arch Clin Neuropsychol. 2013;28(7):633-639.
12. Peebles R, Sabel
13. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington DC: American Psychiatric Association; 2013.
2. Fedoroff JP, Hanson A, McGuire M, et al. Simulated paraphilias: a preliminary study of patients who imitate or exaggerate paraphilic symptoms and behaviors. J Forensic Sci. 1992;37(3):902-911.
3. Mittenberg W, Patton C, Canyock EM, et al. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol. 2002;24(8):1094-1102.
4. Waite S, Geddes A. Malingered psychosis leading to involuntary psychiatric hospitalization. Australas Psychiatry. 2006;14(4):419-421.
5. Hall RC, Hall RC. Malingering of PTSD: forensic and diagnostic consideration, characteristics of malingerers and clinical presentations. Gen Hosp Psychiatry. 2006;28(6):525-535.
6. Yates BD, Nordquist CR, Shultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv. 1996;47(9):998-1000.
7. Rissmiller DJ, Wayslow A, Madison H, et al. Prevalence of malingering in inpatient suicidal ideators and attempters. Crisis. 1998;19(2):62-66.
8. Rissmiller D, Steer RA, Friedman M, et al. Prevalence of malingering in suicidal psychiatric inpatients: a replication. Psychol Rep. 1999;84(3 pt 1):726-730.
9. Sullivan K, Lange RT, Dawes S. Methods of detecting malingering and estimated symptom exaggeration base rates in Australia. Journal of Forensic Neuropsychology. 2007;4(4):49-70.
10. Bass C, Halligan P. Factitious disorders and malingering: challenges for clinical assessment and management. Lancet. 2014;383(9926):1422-1432.
11. Chafetz M, Underhill J. Estimated costs of malingered disability. Arch Clin Neuropsychol. 2013;28(7):633-639.
12. Peebles R, Sabel
13. Malone RD, Lange CL. A clinical approach to the malingering patient. J Am Acad Psychoanal Dyn Psychiatry. 2007;35(1):13-21.
A girl repeatedly jabs her finger up her nose: Compulsion or self-injury?
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).

In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.

Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).
In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.
Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).
In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.
Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.