User login
News and Views that Matter to Pediatricians
The leading independent newspaper covering news and commentary in pediatrics.
Venetoclax shows activity against T-ALL in children
Data from a small retrospective study suggest that venetoclax-based regimens may have activity against relapsed or refractory T-lineage acute lymphoblastic leukemia (T-ALL) in children and young adults.
Among seven patients with T-ALL treated with venetoclax (Venclexta) in combination with chemotherapy, four had complete remissions and one had a CR with incomplete recovery of blood counts (CRi), and all four patients had undetectable minimal residual disease (MRD), reported pediatric hematology/oncology fellow Amber Gibson, MD, and colleagues from the University of Texas MD Anderson Cancer Center Children’s Cancer Hospital in Houston.
“This single-institution retrospective review found that venetoclax was safe and well tolerated in combination chemotherapy regimens, thrombocytopenia and neutropenia were the most common toxicities identified, [and] venetoclax should be considered for patients with refractory T-cell ALL and investigated as up-front therapy for this patient population,” they wrote in the abstract accompanying a poster presentation at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Children with relapsed T-ALL and T-lymphoblastic lymphoma (T-LL) have a dismal prognosis, with a 3-year event-free survival rate less than 10%, according to the researchers.
To see whether venetoclax, an inhibitor of the antiapoptotic protein B-cell lymphoma-2 (BCL-2), could improve outcomes for children with ALL, the investigators conducted a retrospective chart review of the safety and efficacy of venetoclax in young patients with relapsed/refractory ALL/LL who received the drug at their center.
They identified 10 patients aged 6-21 years (median, 18), 5 of whom had T-ALL (1 with early T-cell precursor ALL), 2 with T-LL, and 3 with B-lineage ALL (B-ALL).
The median number of prior lines of therapy was 3.5. Three of the 10 patients had received hematopoietic stem cell transplants, and the 3 patients with B-ALL had all received prior CD19-directed chimeric antigen receptor T-cell (CAR T) therapy. One of these patients received a dual CD19/CD22 CAR T product, one received CD19-directed blinotumumab.
There were no new safety signals with venetoclax, no treatment-related deaths, and no deaths within 30 days of starting venetoclax.
All 10 patients had grade 4 thrombocytopenias, 6 had grade 4 neutropenia, 3 had grade 4 febrile neutropenia, 2 had grade 4 anemia, and 1 each had grade 4 sepsis, pneumonia, or coagulopathy.
As noted, there were three CRs and one CRi, all in patients with T-ALL. All four of these patients were MRD negative by flow cytometry at a median of 22 days. The median duration of response was 17.4 months (range, 2-18 months).
At the most recent follow-up five patients were still alive, three without disease, one was still undergoing treatment, and one was alive following an allogeneic HSCT.
Early studies
Shilpa Shahani, MD, a pediatric oncologist and assistant clinical professor of pediatrics at City of Hope in Duarte, Calif., who was not involved in the study, said that there are early studies exploring the use of venetoclax in infants with ALL.
“Venetoclax is a BCL-2 inhibitor that is pretty well tolerated, but you can also have cytopenias with it,” she said.
She noted that it is not typically used in the frontline setting in pediatric populations, but may be considered for patients with difficult-to-treat disease or for whom the relatively good toxicity profile might be appropriate.
The MD Anderson investigators did not report a funding source. The authors and Dr. Shahani reported no relevant conflicts of interest.
Data from a small retrospective study suggest that venetoclax-based regimens may have activity against relapsed or refractory T-lineage acute lymphoblastic leukemia (T-ALL) in children and young adults.
Among seven patients with T-ALL treated with venetoclax (Venclexta) in combination with chemotherapy, four had complete remissions and one had a CR with incomplete recovery of blood counts (CRi), and all four patients had undetectable minimal residual disease (MRD), reported pediatric hematology/oncology fellow Amber Gibson, MD, and colleagues from the University of Texas MD Anderson Cancer Center Children’s Cancer Hospital in Houston.
“This single-institution retrospective review found that venetoclax was safe and well tolerated in combination chemotherapy regimens, thrombocytopenia and neutropenia were the most common toxicities identified, [and] venetoclax should be considered for patients with refractory T-cell ALL and investigated as up-front therapy for this patient population,” they wrote in the abstract accompanying a poster presentation at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Children with relapsed T-ALL and T-lymphoblastic lymphoma (T-LL) have a dismal prognosis, with a 3-year event-free survival rate less than 10%, according to the researchers.
To see whether venetoclax, an inhibitor of the antiapoptotic protein B-cell lymphoma-2 (BCL-2), could improve outcomes for children with ALL, the investigators conducted a retrospective chart review of the safety and efficacy of venetoclax in young patients with relapsed/refractory ALL/LL who received the drug at their center.
They identified 10 patients aged 6-21 years (median, 18), 5 of whom had T-ALL (1 with early T-cell precursor ALL), 2 with T-LL, and 3 with B-lineage ALL (B-ALL).
The median number of prior lines of therapy was 3.5. Three of the 10 patients had received hematopoietic stem cell transplants, and the 3 patients with B-ALL had all received prior CD19-directed chimeric antigen receptor T-cell (CAR T) therapy. One of these patients received a dual CD19/CD22 CAR T product, one received CD19-directed blinotumumab.
There were no new safety signals with venetoclax, no treatment-related deaths, and no deaths within 30 days of starting venetoclax.
All 10 patients had grade 4 thrombocytopenias, 6 had grade 4 neutropenia, 3 had grade 4 febrile neutropenia, 2 had grade 4 anemia, and 1 each had grade 4 sepsis, pneumonia, or coagulopathy.
As noted, there were three CRs and one CRi, all in patients with T-ALL. All four of these patients were MRD negative by flow cytometry at a median of 22 days. The median duration of response was 17.4 months (range, 2-18 months).
At the most recent follow-up five patients were still alive, three without disease, one was still undergoing treatment, and one was alive following an allogeneic HSCT.
Early studies
Shilpa Shahani, MD, a pediatric oncologist and assistant clinical professor of pediatrics at City of Hope in Duarte, Calif., who was not involved in the study, said that there are early studies exploring the use of venetoclax in infants with ALL.
“Venetoclax is a BCL-2 inhibitor that is pretty well tolerated, but you can also have cytopenias with it,” she said.
She noted that it is not typically used in the frontline setting in pediatric populations, but may be considered for patients with difficult-to-treat disease or for whom the relatively good toxicity profile might be appropriate.
The MD Anderson investigators did not report a funding source. The authors and Dr. Shahani reported no relevant conflicts of interest.
Data from a small retrospective study suggest that venetoclax-based regimens may have activity against relapsed or refractory T-lineage acute lymphoblastic leukemia (T-ALL) in children and young adults.
Among seven patients with T-ALL treated with venetoclax (Venclexta) in combination with chemotherapy, four had complete remissions and one had a CR with incomplete recovery of blood counts (CRi), and all four patients had undetectable minimal residual disease (MRD), reported pediatric hematology/oncology fellow Amber Gibson, MD, and colleagues from the University of Texas MD Anderson Cancer Center Children’s Cancer Hospital in Houston.
“This single-institution retrospective review found that venetoclax was safe and well tolerated in combination chemotherapy regimens, thrombocytopenia and neutropenia were the most common toxicities identified, [and] venetoclax should be considered for patients with refractory T-cell ALL and investigated as up-front therapy for this patient population,” they wrote in the abstract accompanying a poster presentation at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Children with relapsed T-ALL and T-lymphoblastic lymphoma (T-LL) have a dismal prognosis, with a 3-year event-free survival rate less than 10%, according to the researchers.
To see whether venetoclax, an inhibitor of the antiapoptotic protein B-cell lymphoma-2 (BCL-2), could improve outcomes for children with ALL, the investigators conducted a retrospective chart review of the safety and efficacy of venetoclax in young patients with relapsed/refractory ALL/LL who received the drug at their center.
They identified 10 patients aged 6-21 years (median, 18), 5 of whom had T-ALL (1 with early T-cell precursor ALL), 2 with T-LL, and 3 with B-lineage ALL (B-ALL).
The median number of prior lines of therapy was 3.5. Three of the 10 patients had received hematopoietic stem cell transplants, and the 3 patients with B-ALL had all received prior CD19-directed chimeric antigen receptor T-cell (CAR T) therapy. One of these patients received a dual CD19/CD22 CAR T product, one received CD19-directed blinotumumab.
There were no new safety signals with venetoclax, no treatment-related deaths, and no deaths within 30 days of starting venetoclax.
All 10 patients had grade 4 thrombocytopenias, 6 had grade 4 neutropenia, 3 had grade 4 febrile neutropenia, 2 had grade 4 anemia, and 1 each had grade 4 sepsis, pneumonia, or coagulopathy.
As noted, there were three CRs and one CRi, all in patients with T-ALL. All four of these patients were MRD negative by flow cytometry at a median of 22 days. The median duration of response was 17.4 months (range, 2-18 months).
At the most recent follow-up five patients were still alive, three without disease, one was still undergoing treatment, and one was alive following an allogeneic HSCT.
Early studies
Shilpa Shahani, MD, a pediatric oncologist and assistant clinical professor of pediatrics at City of Hope in Duarte, Calif., who was not involved in the study, said that there are early studies exploring the use of venetoclax in infants with ALL.
“Venetoclax is a BCL-2 inhibitor that is pretty well tolerated, but you can also have cytopenias with it,” she said.
She noted that it is not typically used in the frontline setting in pediatric populations, but may be considered for patients with difficult-to-treat disease or for whom the relatively good toxicity profile might be appropriate.
The MD Anderson investigators did not report a funding source. The authors and Dr. Shahani reported no relevant conflicts of interest.
FROM ASPHO 2021
How early can laser treatment for port wine stains in infants be initiated?
without any complications, results from a single-center study showed.
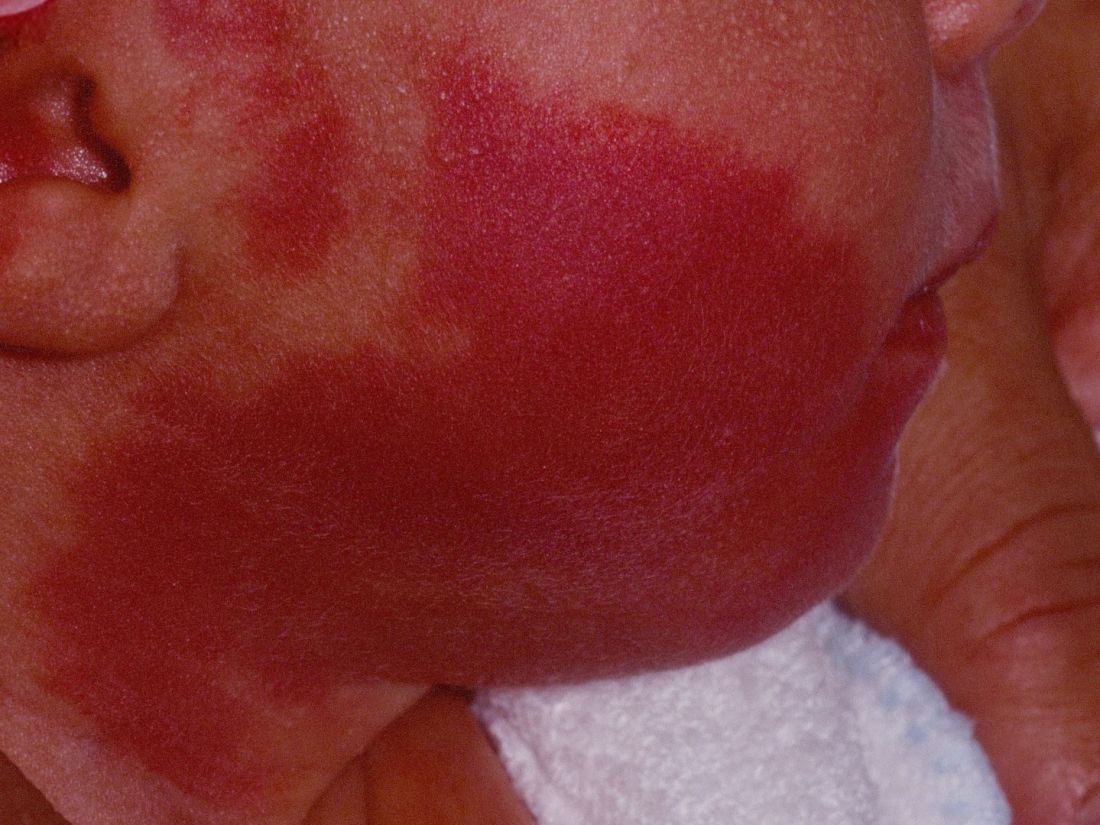
“The current modality of choice for the treatment of port wine birthmarks is pulsed dye laser,” Chelsea Grimes Fidai, MD, said during the annual conference of the American Society for Laser Medicine and Surgery. “When performed by a highly trained expert at efficient frequencies, PDL is a safe, effective treatment that is successful in the majority of patients. We know that earlier treatment yields maximal clearance. However, just how early can you initiate treatment?”
To find out, Dr. Fidai, Roy G. Geronemus, MD, and colleagues at the Laser and Skin Surgery Center of New York, conducted a retrospective chart review of 39 infants with port wine birthmarks who were treated with a 595-nm PDL between 2015 and 2020 at the center. Of the 39 infants, the average age at first treatment was 18 days, with a range from 5 to 29 days. The youngest patient was born prematurely at 35 weeks’ gestation and presented for his first treatment even before his expected due date. Most (74%) had facial lesions with the remaining distributed on the trunk or extremities. The average number of treatments was 15 over the course of 15 months.
The initial settings chosen for facial lesions were a 10-mm spot size, a fluence of 8.0 J/cm2, and a 1.5-millisecond pulse duration. For body lesions, the typical initial settings were a 12-mm spot size, a fluence of 6.7 J/cm2, and 1.5-millisecond pulse duration. Corneal eye shields were placed for all cases with port wine birthmarks approaching the eyelid. “We do recommend a treatment interval of every 2-3 weeks, with longer intervals for patients of darker skin type until the child is 2 years old, at which time the interval is increased to every 3-6 months,” said Dr. Fidai.
Patients in the study experienced the expected short-term side effects of erythema, edema, purpura, and mild transient postinflammatory hyperpigmentation, but there were no cases of atrophy, scarring, infection, or permanent pigmentary change.
“Families seeking early treatment of port wine birthmarks can be reassured that it can be safely initiated within the first few days after birth,” Dr. Fidai concluded. “This procedure can be quickly and confidently performed as an in-office procedure without any complications. The early intervention allows for treatment without general anesthesia and it maximizes the chance of significant clearance as early in life as possible.”
During a question-and-answer session, the abstract section chair, Albert Wolkerstorfer, MD, PhD, expressed concern about the effect of PDL on developing infants. “We do repeated treatments at this young age without any type of anesthesia,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam.
“Will that influence the development of the child, especially when I hear there might be 15 or 20 treatments done within the first year of life? I think this is a problem where we need to ask the experts in the field of pain management in children, like pediatric anesthesiologists, to find the right way, because I think that the results that you showed are fantastic. I don’t think we can achieve that at a later age, although there’s no direct comparison at this moment.”
Dr. Fidai said that she understood the concern, but pointed to a 2020 article by Dr. Geronemus and colleagues that assessed treatment tolerance and parental perspective of outpatient PDL treatment for port-wine birthmarks without general anesthesia in infants and toddlers. “The kids recover pretty quickly after the treatment,” she said. “There has never been any longstanding issue from the parents’ perspective.”
Dr. Fidai reported having no financial disclosures. Dr. Geronemus disclosed having financial conflicts with numerous device and pharmaceutical companies. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
without any complications, results from a single-center study showed.
“The current modality of choice for the treatment of port wine birthmarks is pulsed dye laser,” Chelsea Grimes Fidai, MD, said during the annual conference of the American Society for Laser Medicine and Surgery. “When performed by a highly trained expert at efficient frequencies, PDL is a safe, effective treatment that is successful in the majority of patients. We know that earlier treatment yields maximal clearance. However, just how early can you initiate treatment?”
To find out, Dr. Fidai, Roy G. Geronemus, MD, and colleagues at the Laser and Skin Surgery Center of New York, conducted a retrospective chart review of 39 infants with port wine birthmarks who were treated with a 595-nm PDL between 2015 and 2020 at the center. Of the 39 infants, the average age at first treatment was 18 days, with a range from 5 to 29 days. The youngest patient was born prematurely at 35 weeks’ gestation and presented for his first treatment even before his expected due date. Most (74%) had facial lesions with the remaining distributed on the trunk or extremities. The average number of treatments was 15 over the course of 15 months.
The initial settings chosen for facial lesions were a 10-mm spot size, a fluence of 8.0 J/cm2, and a 1.5-millisecond pulse duration. For body lesions, the typical initial settings were a 12-mm spot size, a fluence of 6.7 J/cm2, and 1.5-millisecond pulse duration. Corneal eye shields were placed for all cases with port wine birthmarks approaching the eyelid. “We do recommend a treatment interval of every 2-3 weeks, with longer intervals for patients of darker skin type until the child is 2 years old, at which time the interval is increased to every 3-6 months,” said Dr. Fidai.
Patients in the study experienced the expected short-term side effects of erythema, edema, purpura, and mild transient postinflammatory hyperpigmentation, but there were no cases of atrophy, scarring, infection, or permanent pigmentary change.
“Families seeking early treatment of port wine birthmarks can be reassured that it can be safely initiated within the first few days after birth,” Dr. Fidai concluded. “This procedure can be quickly and confidently performed as an in-office procedure without any complications. The early intervention allows for treatment without general anesthesia and it maximizes the chance of significant clearance as early in life as possible.”
During a question-and-answer session, the abstract section chair, Albert Wolkerstorfer, MD, PhD, expressed concern about the effect of PDL on developing infants. “We do repeated treatments at this young age without any type of anesthesia,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam.
“Will that influence the development of the child, especially when I hear there might be 15 or 20 treatments done within the first year of life? I think this is a problem where we need to ask the experts in the field of pain management in children, like pediatric anesthesiologists, to find the right way, because I think that the results that you showed are fantastic. I don’t think we can achieve that at a later age, although there’s no direct comparison at this moment.”
Dr. Fidai said that she understood the concern, but pointed to a 2020 article by Dr. Geronemus and colleagues that assessed treatment tolerance and parental perspective of outpatient PDL treatment for port-wine birthmarks without general anesthesia in infants and toddlers. “The kids recover pretty quickly after the treatment,” she said. “There has never been any longstanding issue from the parents’ perspective.”
Dr. Fidai reported having no financial disclosures. Dr. Geronemus disclosed having financial conflicts with numerous device and pharmaceutical companies. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
without any complications, results from a single-center study showed.
“The current modality of choice for the treatment of port wine birthmarks is pulsed dye laser,” Chelsea Grimes Fidai, MD, said during the annual conference of the American Society for Laser Medicine and Surgery. “When performed by a highly trained expert at efficient frequencies, PDL is a safe, effective treatment that is successful in the majority of patients. We know that earlier treatment yields maximal clearance. However, just how early can you initiate treatment?”
To find out, Dr. Fidai, Roy G. Geronemus, MD, and colleagues at the Laser and Skin Surgery Center of New York, conducted a retrospective chart review of 39 infants with port wine birthmarks who were treated with a 595-nm PDL between 2015 and 2020 at the center. Of the 39 infants, the average age at first treatment was 18 days, with a range from 5 to 29 days. The youngest patient was born prematurely at 35 weeks’ gestation and presented for his first treatment even before his expected due date. Most (74%) had facial lesions with the remaining distributed on the trunk or extremities. The average number of treatments was 15 over the course of 15 months.
The initial settings chosen for facial lesions were a 10-mm spot size, a fluence of 8.0 J/cm2, and a 1.5-millisecond pulse duration. For body lesions, the typical initial settings were a 12-mm spot size, a fluence of 6.7 J/cm2, and 1.5-millisecond pulse duration. Corneal eye shields were placed for all cases with port wine birthmarks approaching the eyelid. “We do recommend a treatment interval of every 2-3 weeks, with longer intervals for patients of darker skin type until the child is 2 years old, at which time the interval is increased to every 3-6 months,” said Dr. Fidai.
Patients in the study experienced the expected short-term side effects of erythema, edema, purpura, and mild transient postinflammatory hyperpigmentation, but there were no cases of atrophy, scarring, infection, or permanent pigmentary change.
“Families seeking early treatment of port wine birthmarks can be reassured that it can be safely initiated within the first few days after birth,” Dr. Fidai concluded. “This procedure can be quickly and confidently performed as an in-office procedure without any complications. The early intervention allows for treatment without general anesthesia and it maximizes the chance of significant clearance as early in life as possible.”
During a question-and-answer session, the abstract section chair, Albert Wolkerstorfer, MD, PhD, expressed concern about the effect of PDL on developing infants. “We do repeated treatments at this young age without any type of anesthesia,” said Dr. Wolkerstorfer, a dermatologist at the Netherlands Institute for Pigment Disorders, department of dermatology, University of Amsterdam.
“Will that influence the development of the child, especially when I hear there might be 15 or 20 treatments done within the first year of life? I think this is a problem where we need to ask the experts in the field of pain management in children, like pediatric anesthesiologists, to find the right way, because I think that the results that you showed are fantastic. I don’t think we can achieve that at a later age, although there’s no direct comparison at this moment.”
Dr. Fidai said that she understood the concern, but pointed to a 2020 article by Dr. Geronemus and colleagues that assessed treatment tolerance and parental perspective of outpatient PDL treatment for port-wine birthmarks without general anesthesia in infants and toddlers. “The kids recover pretty quickly after the treatment,” she said. “There has never been any longstanding issue from the parents’ perspective.”
Dr. Fidai reported having no financial disclosures. Dr. Geronemus disclosed having financial conflicts with numerous device and pharmaceutical companies. Dr. Wolkerstorfer disclosed that he has received consulting fees from Lumenis and InCyte and equipment from Humeca and PerfAction Technologies. He has also received grant funding from Novartis and InCyte and he is a member of InCyte’s advisory board.
FROM ASLMS 2021
FDA panel endorses teplizumab for delaying type 1 diabetes
in at-risk individuals.

The 10-7 vote of the FDA’s endocrinologic and metabolic drugs advisory committee on May 27 reflected a difficult decision-making process on the part of many members to weigh the benefits of a potential 2-year delay in the onset of type 1 diabetes against both observed and theoretical risks, as well as what most considered to be insufficient data.
Regardless of their vote, nearly all panel members advised the FDA that the company should be required to conduct at least one additional larger long-term efficacy and safety trial to satisfy what they felt were major gaps in the data. Some advised that use of the drug be restricted to a very narrow group of recipients until efficacy and safety can be better established.
If approved, teplizumab, which interferes with T cell–mediated autoimmune destruction of pancreatic beta cells, would be the first disease-modifying therapy for impeding progression of type 1 diabetes. The proposed indication is for individuals who have two or more type 1 diabetes-associated autoantibodies and subclinical dysglycemia.
That “stage 2” or “at-risk” condition is associated with a nearly 100% lifetime risk of progression to clinical (“stage 3”) type 1 diabetes and a 75% risk of developing the disease within 5 years. As of now, most such individuals are first-degree relatives of people with type 1 diabetes identified through TrialNet.
What’s the evidence to support approval so far?
In 2019, a pivotal phase 2 randomized, placebo-controlled TN-10 trial involving 76 at-risk children and adults ages 8 years and older showed that a single 14-day treatment of daily intravenous infusions of teplizumab in 44 patients resulted in a significant median 2-year delay to onset of clinical type 1 diabetes, compared with 32 who received placebo. Further follow-up data continue to show that fewer patients who received teplizumab have progressed to clinical type 1 diabetes.
While most advisory panelists agreed that the TN-10 study demonstrated efficacy, several also said that the sample size was insufficient and at least one additional randomized trial should be conducted to replicate the findings.
Although the FDA typically requires companies to demonstrate a drug’s effectiveness with at least two separate clinical trials, the agency allows companies to substitute other forms of data for a second randomized clinical trial, such as study results for the drug in a closely related condition, mechanistic data, or knowledge of other drugs from the same class.
In this case, Provention’s submission included as “confirmatory” evidence a meta-analysis of data from five earlier randomized trials (three placebo controlled, two open label) of a total 942 individuals with newly diagnosed type 1 diabetes (“stage 3”) who received either one or two 14-day teplizumab courses (n = 729) or placebo. These showed consistent preservation of C-peptide, a surrogate marker of beta-cell function, along with lower mean insulin use.
Several panel members expressed dissatisfaction with those confirmatory data, noting the patient population was different from those for which the company is currently seeking the indication, and that C-peptide is an inadequate endpoint for demonstrating efficacy.
Safety: Adverse events mostly transient, but unanswered questions
Adverse events reported in at least 10% of teplizumab recipients included lymphopenia (76.8% vs. 9.4% placebo; relative risk, 8.2), leukopenia (82.1% vs. 24.1%; RR, 3.4), and rash (44.5% vs. 9.0%; RR, 4.9).
“Most adverse events related to teplizumab were mechanism-based, predictable, transient, and manageable,” Chief Medical Officer of Provention Bio, Eleanor Ramos, MD, said.
Among other safety issues that concerned the panel, diabetic ketoacidosis (DKA) was seen in 2.3% of 773 teplizumab recipients with new-onset type 1 diabetes versus just 1% among the 245 controls, a significant, nearly sixfold increase. No DKA occurred in the TN-10 trial. No clear explanation was offered for the imbalance in the meta-analysis.
Cytokine release syndrome occurred in 0.6% of patients who received teplizumab versus no controls, and infections in 3.4% versus 2.0%, respectively.
Approximately 10% of patients were not able to complete the treatment course because of protocol-directed withdrawal criteria, which included elevations in bilirubin or liver enzymes, or drops in platelet count, neutrophils, or hemoglobin, FDA reviewer Lauren Wood Heickman, MD, noted.
There was only one malignancy, a melanoma in a patient with a preexisting lesion, but malignancy is a theoretical concern with long-term immunosuppression, Dr. Heickman said.
Despite the concerns about the data, panel members expressed unanimous appreciation for the 18 people who spoke during public comments attesting to the lifelong burdens involved in living with type 1 diabetes who urged the FDA to approve teplizumab.
Many of them noted that even a 2-year reprieve from the burden of constant attention to managing blood glucose can make a major difference in the life of a young person. The speakers included physicians, parents of children with type 1 diabetes, adults who have the condition themselves and who worry about their children getting it, and researchers in the field.
Panel members describe ‘struggle’ with vote decision
Panel member Michael Blaha, MD, of Johns Hopkins University, Baltimore, voted in favor of teplizumab approval. However, he said, “I was very conflicted on this one and my ‘yes’ is very qualified. In my opinion the risk-benefit is very narrow, and I would only approve this drug for the exact indication of the trial. ... Patients who don’t fit the criteria could hopefully be enrolled in a second confirmatory trial.”
He also advised an extensive Risk Evaluation and Mitigation Strategies program to look for both short- and long-term adverse effects.
“My overall take on this is that I do think it’s a promising paradigm-shifting therapy that really needs to move forward, at least scientifically. I’m excited about it, but I have a lot of skepticism about the entire body of data to make any more than the most narrow of approval,” Dr. Blaha said.
Susan S. Ellenberg, PhD, professor of biostatistics, medical ethics, and health policy at the University of Pennsylvania, Philadelphia, voted yes but also with difficulty.
“I really struggled with it. ... I was pushed by the very encouraging results of what is admittedly a very small study and something I can’t feel is completely definitive. But I would not like to deny the kind of people that we heard from today the opportunity to weigh their own risks and benefits to try this. And I would certainly agree that a very, very rigorous postmarketing program, preferably including another controlled trial, should be carried out.”
But David M. Nathan, MD, director of the Diabetes Center and Clinical Research Center, Massachusetts General Hospital, Boston, voted no.
“I struggled with this vote, tremendously, having listened carefully to the patients with type 1 diabetes ... but that said, having done clinical research for 40 years in type 1 diabetes, I think we need more data, both in terms of efficacy and of safety. I would hate a number of years down the road to figure out that we actually caused more harm than good, especially keeping in mind that the treatment of type 1 diabetes is evolving rapidly.”
A different perspective came from Mara L. Becker, MD, vice chair of the department of pediatric rheumatology at Duke University, Durham, N.C. She voted yes, pointing out that she’s accustomed to prescribing biologics for chronic conditions in children.
“I was unconflicted in my vote, which was yes. I thought the data ... were convincing and the need is great. I would support a label for children [aged 8 years] and older with at least stage 2 disease ... and I would require postmarketing safety surveillance to understand what the long-term side effects could be, but I would still be in favor of it.”
FDA advisory panel committee members are vetted for conflicts of interest and waivers granted for participation if necessary; none were granted for this meeting.
A version of this article first appeared on Medscape.com.
in at-risk individuals.
The 10-7 vote of the FDA’s endocrinologic and metabolic drugs advisory committee on May 27 reflected a difficult decision-making process on the part of many members to weigh the benefits of a potential 2-year delay in the onset of type 1 diabetes against both observed and theoretical risks, as well as what most considered to be insufficient data.
Regardless of their vote, nearly all panel members advised the FDA that the company should be required to conduct at least one additional larger long-term efficacy and safety trial to satisfy what they felt were major gaps in the data. Some advised that use of the drug be restricted to a very narrow group of recipients until efficacy and safety can be better established.
If approved, teplizumab, which interferes with T cell–mediated autoimmune destruction of pancreatic beta cells, would be the first disease-modifying therapy for impeding progression of type 1 diabetes. The proposed indication is for individuals who have two or more type 1 diabetes-associated autoantibodies and subclinical dysglycemia.
That “stage 2” or “at-risk” condition is associated with a nearly 100% lifetime risk of progression to clinical (“stage 3”) type 1 diabetes and a 75% risk of developing the disease within 5 years. As of now, most such individuals are first-degree relatives of people with type 1 diabetes identified through TrialNet.
What’s the evidence to support approval so far?
In 2019, a pivotal phase 2 randomized, placebo-controlled TN-10 trial involving 76 at-risk children and adults ages 8 years and older showed that a single 14-day treatment of daily intravenous infusions of teplizumab in 44 patients resulted in a significant median 2-year delay to onset of clinical type 1 diabetes, compared with 32 who received placebo. Further follow-up data continue to show that fewer patients who received teplizumab have progressed to clinical type 1 diabetes.
While most advisory panelists agreed that the TN-10 study demonstrated efficacy, several also said that the sample size was insufficient and at least one additional randomized trial should be conducted to replicate the findings.
Although the FDA typically requires companies to demonstrate a drug’s effectiveness with at least two separate clinical trials, the agency allows companies to substitute other forms of data for a second randomized clinical trial, such as study results for the drug in a closely related condition, mechanistic data, or knowledge of other drugs from the same class.
In this case, Provention’s submission included as “confirmatory” evidence a meta-analysis of data from five earlier randomized trials (three placebo controlled, two open label) of a total 942 individuals with newly diagnosed type 1 diabetes (“stage 3”) who received either one or two 14-day teplizumab courses (n = 729) or placebo. These showed consistent preservation of C-peptide, a surrogate marker of beta-cell function, along with lower mean insulin use.
Several panel members expressed dissatisfaction with those confirmatory data, noting the patient population was different from those for which the company is currently seeking the indication, and that C-peptide is an inadequate endpoint for demonstrating efficacy.
Safety: Adverse events mostly transient, but unanswered questions
Adverse events reported in at least 10% of teplizumab recipients included lymphopenia (76.8% vs. 9.4% placebo; relative risk, 8.2), leukopenia (82.1% vs. 24.1%; RR, 3.4), and rash (44.5% vs. 9.0%; RR, 4.9).
“Most adverse events related to teplizumab were mechanism-based, predictable, transient, and manageable,” Chief Medical Officer of Provention Bio, Eleanor Ramos, MD, said.
Among other safety issues that concerned the panel, diabetic ketoacidosis (DKA) was seen in 2.3% of 773 teplizumab recipients with new-onset type 1 diabetes versus just 1% among the 245 controls, a significant, nearly sixfold increase. No DKA occurred in the TN-10 trial. No clear explanation was offered for the imbalance in the meta-analysis.
Cytokine release syndrome occurred in 0.6% of patients who received teplizumab versus no controls, and infections in 3.4% versus 2.0%, respectively.
Approximately 10% of patients were not able to complete the treatment course because of protocol-directed withdrawal criteria, which included elevations in bilirubin or liver enzymes, or drops in platelet count, neutrophils, or hemoglobin, FDA reviewer Lauren Wood Heickman, MD, noted.
There was only one malignancy, a melanoma in a patient with a preexisting lesion, but malignancy is a theoretical concern with long-term immunosuppression, Dr. Heickman said.
Despite the concerns about the data, panel members expressed unanimous appreciation for the 18 people who spoke during public comments attesting to the lifelong burdens involved in living with type 1 diabetes who urged the FDA to approve teplizumab.
Many of them noted that even a 2-year reprieve from the burden of constant attention to managing blood glucose can make a major difference in the life of a young person. The speakers included physicians, parents of children with type 1 diabetes, adults who have the condition themselves and who worry about their children getting it, and researchers in the field.
Panel members describe ‘struggle’ with vote decision
Panel member Michael Blaha, MD, of Johns Hopkins University, Baltimore, voted in favor of teplizumab approval. However, he said, “I was very conflicted on this one and my ‘yes’ is very qualified. In my opinion the risk-benefit is very narrow, and I would only approve this drug for the exact indication of the trial. ... Patients who don’t fit the criteria could hopefully be enrolled in a second confirmatory trial.”
He also advised an extensive Risk Evaluation and Mitigation Strategies program to look for both short- and long-term adverse effects.
“My overall take on this is that I do think it’s a promising paradigm-shifting therapy that really needs to move forward, at least scientifically. I’m excited about it, but I have a lot of skepticism about the entire body of data to make any more than the most narrow of approval,” Dr. Blaha said.
Susan S. Ellenberg, PhD, professor of biostatistics, medical ethics, and health policy at the University of Pennsylvania, Philadelphia, voted yes but also with difficulty.
“I really struggled with it. ... I was pushed by the very encouraging results of what is admittedly a very small study and something I can’t feel is completely definitive. But I would not like to deny the kind of people that we heard from today the opportunity to weigh their own risks and benefits to try this. And I would certainly agree that a very, very rigorous postmarketing program, preferably including another controlled trial, should be carried out.”
But David M. Nathan, MD, director of the Diabetes Center and Clinical Research Center, Massachusetts General Hospital, Boston, voted no.
“I struggled with this vote, tremendously, having listened carefully to the patients with type 1 diabetes ... but that said, having done clinical research for 40 years in type 1 diabetes, I think we need more data, both in terms of efficacy and of safety. I would hate a number of years down the road to figure out that we actually caused more harm than good, especially keeping in mind that the treatment of type 1 diabetes is evolving rapidly.”
A different perspective came from Mara L. Becker, MD, vice chair of the department of pediatric rheumatology at Duke University, Durham, N.C. She voted yes, pointing out that she’s accustomed to prescribing biologics for chronic conditions in children.
“I was unconflicted in my vote, which was yes. I thought the data ... were convincing and the need is great. I would support a label for children [aged 8 years] and older with at least stage 2 disease ... and I would require postmarketing safety surveillance to understand what the long-term side effects could be, but I would still be in favor of it.”
FDA advisory panel committee members are vetted for conflicts of interest and waivers granted for participation if necessary; none were granted for this meeting.
A version of this article first appeared on Medscape.com.
in at-risk individuals.
The 10-7 vote of the FDA’s endocrinologic and metabolic drugs advisory committee on May 27 reflected a difficult decision-making process on the part of many members to weigh the benefits of a potential 2-year delay in the onset of type 1 diabetes against both observed and theoretical risks, as well as what most considered to be insufficient data.
Regardless of their vote, nearly all panel members advised the FDA that the company should be required to conduct at least one additional larger long-term efficacy and safety trial to satisfy what they felt were major gaps in the data. Some advised that use of the drug be restricted to a very narrow group of recipients until efficacy and safety can be better established.
If approved, teplizumab, which interferes with T cell–mediated autoimmune destruction of pancreatic beta cells, would be the first disease-modifying therapy for impeding progression of type 1 diabetes. The proposed indication is for individuals who have two or more type 1 diabetes-associated autoantibodies and subclinical dysglycemia.
That “stage 2” or “at-risk” condition is associated with a nearly 100% lifetime risk of progression to clinical (“stage 3”) type 1 diabetes and a 75% risk of developing the disease within 5 years. As of now, most such individuals are first-degree relatives of people with type 1 diabetes identified through TrialNet.
What’s the evidence to support approval so far?
In 2019, a pivotal phase 2 randomized, placebo-controlled TN-10 trial involving 76 at-risk children and adults ages 8 years and older showed that a single 14-day treatment of daily intravenous infusions of teplizumab in 44 patients resulted in a significant median 2-year delay to onset of clinical type 1 diabetes, compared with 32 who received placebo. Further follow-up data continue to show that fewer patients who received teplizumab have progressed to clinical type 1 diabetes.
While most advisory panelists agreed that the TN-10 study demonstrated efficacy, several also said that the sample size was insufficient and at least one additional randomized trial should be conducted to replicate the findings.
Although the FDA typically requires companies to demonstrate a drug’s effectiveness with at least two separate clinical trials, the agency allows companies to substitute other forms of data for a second randomized clinical trial, such as study results for the drug in a closely related condition, mechanistic data, or knowledge of other drugs from the same class.
In this case, Provention’s submission included as “confirmatory” evidence a meta-analysis of data from five earlier randomized trials (three placebo controlled, two open label) of a total 942 individuals with newly diagnosed type 1 diabetes (“stage 3”) who received either one or two 14-day teplizumab courses (n = 729) or placebo. These showed consistent preservation of C-peptide, a surrogate marker of beta-cell function, along with lower mean insulin use.
Several panel members expressed dissatisfaction with those confirmatory data, noting the patient population was different from those for which the company is currently seeking the indication, and that C-peptide is an inadequate endpoint for demonstrating efficacy.
Safety: Adverse events mostly transient, but unanswered questions
Adverse events reported in at least 10% of teplizumab recipients included lymphopenia (76.8% vs. 9.4% placebo; relative risk, 8.2), leukopenia (82.1% vs. 24.1%; RR, 3.4), and rash (44.5% vs. 9.0%; RR, 4.9).
“Most adverse events related to teplizumab were mechanism-based, predictable, transient, and manageable,” Chief Medical Officer of Provention Bio, Eleanor Ramos, MD, said.
Among other safety issues that concerned the panel, diabetic ketoacidosis (DKA) was seen in 2.3% of 773 teplizumab recipients with new-onset type 1 diabetes versus just 1% among the 245 controls, a significant, nearly sixfold increase. No DKA occurred in the TN-10 trial. No clear explanation was offered for the imbalance in the meta-analysis.
Cytokine release syndrome occurred in 0.6% of patients who received teplizumab versus no controls, and infections in 3.4% versus 2.0%, respectively.
Approximately 10% of patients were not able to complete the treatment course because of protocol-directed withdrawal criteria, which included elevations in bilirubin or liver enzymes, or drops in platelet count, neutrophils, or hemoglobin, FDA reviewer Lauren Wood Heickman, MD, noted.
There was only one malignancy, a melanoma in a patient with a preexisting lesion, but malignancy is a theoretical concern with long-term immunosuppression, Dr. Heickman said.
Despite the concerns about the data, panel members expressed unanimous appreciation for the 18 people who spoke during public comments attesting to the lifelong burdens involved in living with type 1 diabetes who urged the FDA to approve teplizumab.
Many of them noted that even a 2-year reprieve from the burden of constant attention to managing blood glucose can make a major difference in the life of a young person. The speakers included physicians, parents of children with type 1 diabetes, adults who have the condition themselves and who worry about their children getting it, and researchers in the field.
Panel members describe ‘struggle’ with vote decision
Panel member Michael Blaha, MD, of Johns Hopkins University, Baltimore, voted in favor of teplizumab approval. However, he said, “I was very conflicted on this one and my ‘yes’ is very qualified. In my opinion the risk-benefit is very narrow, and I would only approve this drug for the exact indication of the trial. ... Patients who don’t fit the criteria could hopefully be enrolled in a second confirmatory trial.”
He also advised an extensive Risk Evaluation and Mitigation Strategies program to look for both short- and long-term adverse effects.
“My overall take on this is that I do think it’s a promising paradigm-shifting therapy that really needs to move forward, at least scientifically. I’m excited about it, but I have a lot of skepticism about the entire body of data to make any more than the most narrow of approval,” Dr. Blaha said.
Susan S. Ellenberg, PhD, professor of biostatistics, medical ethics, and health policy at the University of Pennsylvania, Philadelphia, voted yes but also with difficulty.
“I really struggled with it. ... I was pushed by the very encouraging results of what is admittedly a very small study and something I can’t feel is completely definitive. But I would not like to deny the kind of people that we heard from today the opportunity to weigh their own risks and benefits to try this. And I would certainly agree that a very, very rigorous postmarketing program, preferably including another controlled trial, should be carried out.”
But David M. Nathan, MD, director of the Diabetes Center and Clinical Research Center, Massachusetts General Hospital, Boston, voted no.
“I struggled with this vote, tremendously, having listened carefully to the patients with type 1 diabetes ... but that said, having done clinical research for 40 years in type 1 diabetes, I think we need more data, both in terms of efficacy and of safety. I would hate a number of years down the road to figure out that we actually caused more harm than good, especially keeping in mind that the treatment of type 1 diabetes is evolving rapidly.”
A different perspective came from Mara L. Becker, MD, vice chair of the department of pediatric rheumatology at Duke University, Durham, N.C. She voted yes, pointing out that she’s accustomed to prescribing biologics for chronic conditions in children.
“I was unconflicted in my vote, which was yes. I thought the data ... were convincing and the need is great. I would support a label for children [aged 8 years] and older with at least stage 2 disease ... and I would require postmarketing safety surveillance to understand what the long-term side effects could be, but I would still be in favor of it.”
FDA advisory panel committee members are vetted for conflicts of interest and waivers granted for participation if necessary; none were granted for this meeting.
A version of this article first appeared on Medscape.com.
More and more doctors abandoning private practice
according to a new report.
These patterns likely reflect broader trends toward consolidation in health care, with both insurance companies and hospitals also having grown in size in recent years.
The latest biennial analysis of doctors’ practices by the American Medical Association showed an acceleration of a trend away from private practice, defined as a practice wholly owned by physicians. The 2020 results found less than half – 49.1 % – of doctors involved in patient care worked in a private practice, the AMA said in a report released in May 2021.
This marked the first time private practice was not the dominant approach since the AMA analysis began in 2012. What’s more, the trend appears to be gaining steam, with a drop of almost 5 percentage points from 54.0% in private practice in 2018. The percent of doctors in private practice declined at a slower rate in previous AMA surveys, slipping to 55.8% in 2016 from 56.8% in 2018 and 60.1% in 2012.
Employment and ownership structures have become so varied that no single approach or size of organization “can or should be considered the typical physician practice,” the report noted.
The AMA, for example, added to its 2020 benchmark survey an option to identify private equity organizations as employers. The survey found 4% of doctors involved in patient care worked in practices owned by these kinds of firms. Other options include practices wholly or jointly owned by hospital and health systems and insurers, as well as direct employment and contracting.
There are signs that the shift away from smaller private practices will continue, with younger doctors appearing more likely to seek employment.
The survey found 42% of doctors ages 55 and older were employed by someone else, compared with 51.2% of doctors ages 40-54 and 70% of physicians under the age of 40.
The AMA surveyed 3,500 U.S. doctors through the 2020 Physician Practice Benchmark Survey. The survey was conducted from September to October 2020, roughly 6 months into the COVID-19 pandemic, and therefore may not reflect its full impact.
“Physician practices were hit hard by the economic impact of the early pandemic as patient volume and revenues shrank while medical supply expenses spiked. The impact of these economic forces on physician practice arrangements is ongoing and may not be fully realized for some time,” AMA President Susan R. Bailey, MD, said in a statement.
In a survey released in 2020 by McKinsey & Company, 53% of independent doctors reported that they were worried about their practices surviving the stresses of the pandemic, this news organization reported.
Challenging environment
It’s not just money leading to the shift away from private practice, according to a 2020 report from the American Hospital Association, titled “Evolving Physician-Practice Ownership Models.”
Many recent graduates of medical schools have significant debt and are more likely to opt for employment, which offers more financial stability and work-life balance, the report said.
Doctors also need to keep up with expectations of their patients that have been shaped by advances in other sectors, like banking, the AHA noted. People are used to working on their own schedules, and want to make appointments through apps, get test results rapidly and on their mobile devices, and communicate with their providers virtually.
“It is challenging to meet these expectations and make the necessary technology investments as a solo or small group practice,” the AHA report said.
Hospitals face competition for doctors from insurers, which have been looking in some cases to directly employ more physicians, the AHA also noted. The report cites insurance giant UnitedHealth Group’s Optum unit as the most visible example of this trend.
On a January call about corporate earnings, David Wichmann, then chief executive of UnitedHealth, spoke about the firm’s “aim to reinvent health care delivery,” including efforts to have its own primary and multispecialty care practices.
“OptumCare entered 2021 with over 50,000 physicians and 1,400 clinics,” Mr. Wichmann said. “Over the course of this year, we expect to grow our employed and affiliated physicians by at least 10,000. This work of building local physician-led systems of care continues to be central to our mission. “
UnitedHealth’s new CEO is Andrew Witty, who had led the Optum unit.
Attractions of larger groups
Older doctors – those 55 years and up – were significantly more likely to work in small practices than those younger than 40, the 2020 survey found. Results showed 40.9% of doctors under 40 worked in practices of 10 or fewer colleagues, compared with 61.4% of those age 55 and older.
The large difference between age groups suggests that attrition is one reason for the shift in practice size. Retiring doctors who leave small practices are not being replaced on a one-for-one basis by younger doctors, AMA said. The same reason also appears to be a factor in the shift in practice ownership to larger systems.
Doctors in larger group practices can count on a stable business model, with a better ability to survive disruptive market trends, including those of a more extreme nature, like COVID-19, said Fred Horton, president of AMGA Consulting.
AMGA Consulting is a wholly-owned subsidiary of AMGA, formerly called American Medical Group Association. Its more than 400 members include well-known multispecialty groups and health care systems such as the Mayo Clinic, Cleveland Clinic, Geisinger, the Permanente Medical Group, and Intermountain Healthcare as well as many smaller physician practices.
Mr. Horton, who holds a master’s degree in health administration, said some doctors may want to participate in alternative payment programs offered by insurers, who are seeking to shift away from the fee-for-service model
“Larger organizations can dedicate more resources to continuous quality improvement,” Mr. Horton said. “This is especially important for physicians who are taking on risk-based contracts, as quality can directly impact how much they earn.”
For one oncologist, it was turning to alternative payment methods that helped him keep his private practice afloat.
Kashyap Patel, MD, chief executive of the Carolina Blood and Cancer Care Associates in Rock Hill, S.C., said he maintained the independence of his practice amid pressure from a large health system, which had been buying medical groups in the area. That began to interfere with referrals of patients from other doctors, which are key for cancer specialists, said Dr. Patel, who also is president of the Community Oncology Alliance.
In response, Dr. Patel worked with Blue Cross Blue Shield of South Carolina on an arrangement where his practice sought certifications from the National Committee for Quality Assurance to get better rates.
The effort has allowed Dr. Patel’s clinic to focus more on preventing hospitalizations and visits to the emergency room he said.
In Dr. Patel’s view, his patients benefit from his efforts to remain in independent practice. A switch to ownership by a large health care organization would have put them at risk for higher medical bills, jeopardizing their access to treatment, he said. The reason? Hospitals can charge more for services provided by doctors they employ.
“Nothing would change. I would be the same. The building would be the same, but the cost would go up,” Dr. Patel said.
For its part, the AHA has repeatedly challenged arguments that acquisitions and mergers result in higher costs for patients.
Instead, the AHA has raised alarms about consolidation of health insurers, a concern it shares with AMA. In a 2020 report examining competition among insurers, AMA noted doctors working in small practices can be put at a disadvantage if mergers and acquisitions leave an insurer with too much market power.
“Under antitrust law, independent physicians cannot negotiate collectively with health
Insurers,” the AMA said in the report. “This imbalance in relative size leaves most physicians with a weak bargaining position relative to commercial payers.”
AMA’s research on the effects of insurers’ wielding significant market clout has been used in effort to thwart mergers in this industry.
‘Dramatic restructuring’
The Federal Trade Commission also has taken note of the trends discussed in the new AMA report, saying that “U.S. physician markets are undergoing a dramatic restructuring.”
The FTC in January announced a study of the impact of the consolidation of doctors groups and health care facilities. FTC is seeking data for inpatient, outpatient, and doctors services in 15 states from 2015 through 2020. To gather this data, the commission has issued orders to six major insurers – Aetna, Anthem, Florida Blue, Cigna, Health Care Service Corporation and United Healthcare.
The FTC is concerned that acquired practices may have to alter their referral patterns to favor their affiliated hospital system over competing hospital systems. But FTC staff also said it might be that these acquisitions result in efficiencies, such as enhanced coordination of care between doctors and hospitals “that outweigh potential competitive harms.”
The research project will likely take several years to complete because of its scope, the FTC said. For that reason, the FTC said its Bureau of Economics will release a series of research papers examining different aspects of this inquiry rather than a single paper containing all of the analyses.
Private equity ‘roll-ups’
On the day the FTC announced the study of the impact of doctors groups, one of the panel’s commissioners argued for a closer look at how private equity firms make their purchases.
In a Jan. 15 tweet, FTC Commissioner Rohit Chopra said his agency needs to challenge their “roll-ups of small physician practices” as well as clinics and labs. This is a practice of using a series of acquisitions too small to trigger the federal threshold for a serious look from the FTC and Department of Justice. (The threshold for 2021 stands around the $92 million mark. This benchmark is known as Hart-Scott-Rodino notification after a 1976 law that set a reporting standard.)
Mr. Chopra attached to his Jan. 15 tweet a 2020 statement in which he called for stepped-up scrutiny of private-equity firms’ acquisitions of doctors’ practices. Mr. Chopra noted that private-equity firms have been buying practices focused on anesthesiology and emergency medicine, fields which triggered consumer complaints about surprise billing for emergency care.
“Given trends in today’s markets, it is critical that the FTC find new ways to ensure the agency has a rigorous, data-driven approach to market monitoring and enforcement,” Mr. Chopra wrote.
A version of this article first appeared on WebMD.com.
according to a new report.
These patterns likely reflect broader trends toward consolidation in health care, with both insurance companies and hospitals also having grown in size in recent years.
The latest biennial analysis of doctors’ practices by the American Medical Association showed an acceleration of a trend away from private practice, defined as a practice wholly owned by physicians. The 2020 results found less than half – 49.1 % – of doctors involved in patient care worked in a private practice, the AMA said in a report released in May 2021.
This marked the first time private practice was not the dominant approach since the AMA analysis began in 2012. What’s more, the trend appears to be gaining steam, with a drop of almost 5 percentage points from 54.0% in private practice in 2018. The percent of doctors in private practice declined at a slower rate in previous AMA surveys, slipping to 55.8% in 2016 from 56.8% in 2018 and 60.1% in 2012.
Employment and ownership structures have become so varied that no single approach or size of organization “can or should be considered the typical physician practice,” the report noted.
The AMA, for example, added to its 2020 benchmark survey an option to identify private equity organizations as employers. The survey found 4% of doctors involved in patient care worked in practices owned by these kinds of firms. Other options include practices wholly or jointly owned by hospital and health systems and insurers, as well as direct employment and contracting.
There are signs that the shift away from smaller private practices will continue, with younger doctors appearing more likely to seek employment.
The survey found 42% of doctors ages 55 and older were employed by someone else, compared with 51.2% of doctors ages 40-54 and 70% of physicians under the age of 40.
The AMA surveyed 3,500 U.S. doctors through the 2020 Physician Practice Benchmark Survey. The survey was conducted from September to October 2020, roughly 6 months into the COVID-19 pandemic, and therefore may not reflect its full impact.
“Physician practices were hit hard by the economic impact of the early pandemic as patient volume and revenues shrank while medical supply expenses spiked. The impact of these economic forces on physician practice arrangements is ongoing and may not be fully realized for some time,” AMA President Susan R. Bailey, MD, said in a statement.
In a survey released in 2020 by McKinsey & Company, 53% of independent doctors reported that they were worried about their practices surviving the stresses of the pandemic, this news organization reported.
Challenging environment
It’s not just money leading to the shift away from private practice, according to a 2020 report from the American Hospital Association, titled “Evolving Physician-Practice Ownership Models.”
Many recent graduates of medical schools have significant debt and are more likely to opt for employment, which offers more financial stability and work-life balance, the report said.
Doctors also need to keep up with expectations of their patients that have been shaped by advances in other sectors, like banking, the AHA noted. People are used to working on their own schedules, and want to make appointments through apps, get test results rapidly and on their mobile devices, and communicate with their providers virtually.
“It is challenging to meet these expectations and make the necessary technology investments as a solo or small group practice,” the AHA report said.
Hospitals face competition for doctors from insurers, which have been looking in some cases to directly employ more physicians, the AHA also noted. The report cites insurance giant UnitedHealth Group’s Optum unit as the most visible example of this trend.
On a January call about corporate earnings, David Wichmann, then chief executive of UnitedHealth, spoke about the firm’s “aim to reinvent health care delivery,” including efforts to have its own primary and multispecialty care practices.
“OptumCare entered 2021 with over 50,000 physicians and 1,400 clinics,” Mr. Wichmann said. “Over the course of this year, we expect to grow our employed and affiliated physicians by at least 10,000. This work of building local physician-led systems of care continues to be central to our mission. “
UnitedHealth’s new CEO is Andrew Witty, who had led the Optum unit.
Attractions of larger groups
Older doctors – those 55 years and up – were significantly more likely to work in small practices than those younger than 40, the 2020 survey found. Results showed 40.9% of doctors under 40 worked in practices of 10 or fewer colleagues, compared with 61.4% of those age 55 and older.
The large difference between age groups suggests that attrition is one reason for the shift in practice size. Retiring doctors who leave small practices are not being replaced on a one-for-one basis by younger doctors, AMA said. The same reason also appears to be a factor in the shift in practice ownership to larger systems.
Doctors in larger group practices can count on a stable business model, with a better ability to survive disruptive market trends, including those of a more extreme nature, like COVID-19, said Fred Horton, president of AMGA Consulting.
AMGA Consulting is a wholly-owned subsidiary of AMGA, formerly called American Medical Group Association. Its more than 400 members include well-known multispecialty groups and health care systems such as the Mayo Clinic, Cleveland Clinic, Geisinger, the Permanente Medical Group, and Intermountain Healthcare as well as many smaller physician practices.
Mr. Horton, who holds a master’s degree in health administration, said some doctors may want to participate in alternative payment programs offered by insurers, who are seeking to shift away from the fee-for-service model
“Larger organizations can dedicate more resources to continuous quality improvement,” Mr. Horton said. “This is especially important for physicians who are taking on risk-based contracts, as quality can directly impact how much they earn.”
For one oncologist, it was turning to alternative payment methods that helped him keep his private practice afloat.
Kashyap Patel, MD, chief executive of the Carolina Blood and Cancer Care Associates in Rock Hill, S.C., said he maintained the independence of his practice amid pressure from a large health system, which had been buying medical groups in the area. That began to interfere with referrals of patients from other doctors, which are key for cancer specialists, said Dr. Patel, who also is president of the Community Oncology Alliance.
In response, Dr. Patel worked with Blue Cross Blue Shield of South Carolina on an arrangement where his practice sought certifications from the National Committee for Quality Assurance to get better rates.
The effort has allowed Dr. Patel’s clinic to focus more on preventing hospitalizations and visits to the emergency room he said.
In Dr. Patel’s view, his patients benefit from his efforts to remain in independent practice. A switch to ownership by a large health care organization would have put them at risk for higher medical bills, jeopardizing their access to treatment, he said. The reason? Hospitals can charge more for services provided by doctors they employ.
“Nothing would change. I would be the same. The building would be the same, but the cost would go up,” Dr. Patel said.
For its part, the AHA has repeatedly challenged arguments that acquisitions and mergers result in higher costs for patients.
Instead, the AHA has raised alarms about consolidation of health insurers, a concern it shares with AMA. In a 2020 report examining competition among insurers, AMA noted doctors working in small practices can be put at a disadvantage if mergers and acquisitions leave an insurer with too much market power.
“Under antitrust law, independent physicians cannot negotiate collectively with health
Insurers,” the AMA said in the report. “This imbalance in relative size leaves most physicians with a weak bargaining position relative to commercial payers.”
AMA’s research on the effects of insurers’ wielding significant market clout has been used in effort to thwart mergers in this industry.
‘Dramatic restructuring’
The Federal Trade Commission also has taken note of the trends discussed in the new AMA report, saying that “U.S. physician markets are undergoing a dramatic restructuring.”
The FTC in January announced a study of the impact of the consolidation of doctors groups and health care facilities. FTC is seeking data for inpatient, outpatient, and doctors services in 15 states from 2015 through 2020. To gather this data, the commission has issued orders to six major insurers – Aetna, Anthem, Florida Blue, Cigna, Health Care Service Corporation and United Healthcare.
The FTC is concerned that acquired practices may have to alter their referral patterns to favor their affiliated hospital system over competing hospital systems. But FTC staff also said it might be that these acquisitions result in efficiencies, such as enhanced coordination of care between doctors and hospitals “that outweigh potential competitive harms.”
The research project will likely take several years to complete because of its scope, the FTC said. For that reason, the FTC said its Bureau of Economics will release a series of research papers examining different aspects of this inquiry rather than a single paper containing all of the analyses.
Private equity ‘roll-ups’
On the day the FTC announced the study of the impact of doctors groups, one of the panel’s commissioners argued for a closer look at how private equity firms make their purchases.
In a Jan. 15 tweet, FTC Commissioner Rohit Chopra said his agency needs to challenge their “roll-ups of small physician practices” as well as clinics and labs. This is a practice of using a series of acquisitions too small to trigger the federal threshold for a serious look from the FTC and Department of Justice. (The threshold for 2021 stands around the $92 million mark. This benchmark is known as Hart-Scott-Rodino notification after a 1976 law that set a reporting standard.)
Mr. Chopra attached to his Jan. 15 tweet a 2020 statement in which he called for stepped-up scrutiny of private-equity firms’ acquisitions of doctors’ practices. Mr. Chopra noted that private-equity firms have been buying practices focused on anesthesiology and emergency medicine, fields which triggered consumer complaints about surprise billing for emergency care.
“Given trends in today’s markets, it is critical that the FTC find new ways to ensure the agency has a rigorous, data-driven approach to market monitoring and enforcement,” Mr. Chopra wrote.
A version of this article first appeared on WebMD.com.
according to a new report.
These patterns likely reflect broader trends toward consolidation in health care, with both insurance companies and hospitals also having grown in size in recent years.
The latest biennial analysis of doctors’ practices by the American Medical Association showed an acceleration of a trend away from private practice, defined as a practice wholly owned by physicians. The 2020 results found less than half – 49.1 % – of doctors involved in patient care worked in a private practice, the AMA said in a report released in May 2021.
This marked the first time private practice was not the dominant approach since the AMA analysis began in 2012. What’s more, the trend appears to be gaining steam, with a drop of almost 5 percentage points from 54.0% in private practice in 2018. The percent of doctors in private practice declined at a slower rate in previous AMA surveys, slipping to 55.8% in 2016 from 56.8% in 2018 and 60.1% in 2012.
Employment and ownership structures have become so varied that no single approach or size of organization “can or should be considered the typical physician practice,” the report noted.
The AMA, for example, added to its 2020 benchmark survey an option to identify private equity organizations as employers. The survey found 4% of doctors involved in patient care worked in practices owned by these kinds of firms. Other options include practices wholly or jointly owned by hospital and health systems and insurers, as well as direct employment and contracting.
There are signs that the shift away from smaller private practices will continue, with younger doctors appearing more likely to seek employment.
The survey found 42% of doctors ages 55 and older were employed by someone else, compared with 51.2% of doctors ages 40-54 and 70% of physicians under the age of 40.
The AMA surveyed 3,500 U.S. doctors through the 2020 Physician Practice Benchmark Survey. The survey was conducted from September to October 2020, roughly 6 months into the COVID-19 pandemic, and therefore may not reflect its full impact.
“Physician practices were hit hard by the economic impact of the early pandemic as patient volume and revenues shrank while medical supply expenses spiked. The impact of these economic forces on physician practice arrangements is ongoing and may not be fully realized for some time,” AMA President Susan R. Bailey, MD, said in a statement.
In a survey released in 2020 by McKinsey & Company, 53% of independent doctors reported that they were worried about their practices surviving the stresses of the pandemic, this news organization reported.
Challenging environment
It’s not just money leading to the shift away from private practice, according to a 2020 report from the American Hospital Association, titled “Evolving Physician-Practice Ownership Models.”
Many recent graduates of medical schools have significant debt and are more likely to opt for employment, which offers more financial stability and work-life balance, the report said.
Doctors also need to keep up with expectations of their patients that have been shaped by advances in other sectors, like banking, the AHA noted. People are used to working on their own schedules, and want to make appointments through apps, get test results rapidly and on their mobile devices, and communicate with their providers virtually.
“It is challenging to meet these expectations and make the necessary technology investments as a solo or small group practice,” the AHA report said.
Hospitals face competition for doctors from insurers, which have been looking in some cases to directly employ more physicians, the AHA also noted. The report cites insurance giant UnitedHealth Group’s Optum unit as the most visible example of this trend.
On a January call about corporate earnings, David Wichmann, then chief executive of UnitedHealth, spoke about the firm’s “aim to reinvent health care delivery,” including efforts to have its own primary and multispecialty care practices.
“OptumCare entered 2021 with over 50,000 physicians and 1,400 clinics,” Mr. Wichmann said. “Over the course of this year, we expect to grow our employed and affiliated physicians by at least 10,000. This work of building local physician-led systems of care continues to be central to our mission. “
UnitedHealth’s new CEO is Andrew Witty, who had led the Optum unit.
Attractions of larger groups
Older doctors – those 55 years and up – were significantly more likely to work in small practices than those younger than 40, the 2020 survey found. Results showed 40.9% of doctors under 40 worked in practices of 10 or fewer colleagues, compared with 61.4% of those age 55 and older.
The large difference between age groups suggests that attrition is one reason for the shift in practice size. Retiring doctors who leave small practices are not being replaced on a one-for-one basis by younger doctors, AMA said. The same reason also appears to be a factor in the shift in practice ownership to larger systems.
Doctors in larger group practices can count on a stable business model, with a better ability to survive disruptive market trends, including those of a more extreme nature, like COVID-19, said Fred Horton, president of AMGA Consulting.
AMGA Consulting is a wholly-owned subsidiary of AMGA, formerly called American Medical Group Association. Its more than 400 members include well-known multispecialty groups and health care systems such as the Mayo Clinic, Cleveland Clinic, Geisinger, the Permanente Medical Group, and Intermountain Healthcare as well as many smaller physician practices.
Mr. Horton, who holds a master’s degree in health administration, said some doctors may want to participate in alternative payment programs offered by insurers, who are seeking to shift away from the fee-for-service model
“Larger organizations can dedicate more resources to continuous quality improvement,” Mr. Horton said. “This is especially important for physicians who are taking on risk-based contracts, as quality can directly impact how much they earn.”
For one oncologist, it was turning to alternative payment methods that helped him keep his private practice afloat.
Kashyap Patel, MD, chief executive of the Carolina Blood and Cancer Care Associates in Rock Hill, S.C., said he maintained the independence of his practice amid pressure from a large health system, which had been buying medical groups in the area. That began to interfere with referrals of patients from other doctors, which are key for cancer specialists, said Dr. Patel, who also is president of the Community Oncology Alliance.
In response, Dr. Patel worked with Blue Cross Blue Shield of South Carolina on an arrangement where his practice sought certifications from the National Committee for Quality Assurance to get better rates.
The effort has allowed Dr. Patel’s clinic to focus more on preventing hospitalizations and visits to the emergency room he said.
In Dr. Patel’s view, his patients benefit from his efforts to remain in independent practice. A switch to ownership by a large health care organization would have put them at risk for higher medical bills, jeopardizing their access to treatment, he said. The reason? Hospitals can charge more for services provided by doctors they employ.
“Nothing would change. I would be the same. The building would be the same, but the cost would go up,” Dr. Patel said.
For its part, the AHA has repeatedly challenged arguments that acquisitions and mergers result in higher costs for patients.
Instead, the AHA has raised alarms about consolidation of health insurers, a concern it shares with AMA. In a 2020 report examining competition among insurers, AMA noted doctors working in small practices can be put at a disadvantage if mergers and acquisitions leave an insurer with too much market power.
“Under antitrust law, independent physicians cannot negotiate collectively with health
Insurers,” the AMA said in the report. “This imbalance in relative size leaves most physicians with a weak bargaining position relative to commercial payers.”
AMA’s research on the effects of insurers’ wielding significant market clout has been used in effort to thwart mergers in this industry.
‘Dramatic restructuring’
The Federal Trade Commission also has taken note of the trends discussed in the new AMA report, saying that “U.S. physician markets are undergoing a dramatic restructuring.”
The FTC in January announced a study of the impact of the consolidation of doctors groups and health care facilities. FTC is seeking data for inpatient, outpatient, and doctors services in 15 states from 2015 through 2020. To gather this data, the commission has issued orders to six major insurers – Aetna, Anthem, Florida Blue, Cigna, Health Care Service Corporation and United Healthcare.
The FTC is concerned that acquired practices may have to alter their referral patterns to favor their affiliated hospital system over competing hospital systems. But FTC staff also said it might be that these acquisitions result in efficiencies, such as enhanced coordination of care between doctors and hospitals “that outweigh potential competitive harms.”
The research project will likely take several years to complete because of its scope, the FTC said. For that reason, the FTC said its Bureau of Economics will release a series of research papers examining different aspects of this inquiry rather than a single paper containing all of the analyses.
Private equity ‘roll-ups’
On the day the FTC announced the study of the impact of doctors groups, one of the panel’s commissioners argued for a closer look at how private equity firms make their purchases.
In a Jan. 15 tweet, FTC Commissioner Rohit Chopra said his agency needs to challenge their “roll-ups of small physician practices” as well as clinics and labs. This is a practice of using a series of acquisitions too small to trigger the federal threshold for a serious look from the FTC and Department of Justice. (The threshold for 2021 stands around the $92 million mark. This benchmark is known as Hart-Scott-Rodino notification after a 1976 law that set a reporting standard.)
Mr. Chopra attached to his Jan. 15 tweet a 2020 statement in which he called for stepped-up scrutiny of private-equity firms’ acquisitions of doctors’ practices. Mr. Chopra noted that private-equity firms have been buying practices focused on anesthesiology and emergency medicine, fields which triggered consumer complaints about surprise billing for emergency care.
“Given trends in today’s markets, it is critical that the FTC find new ways to ensure the agency has a rigorous, data-driven approach to market monitoring and enforcement,” Mr. Chopra wrote.
A version of this article first appeared on WebMD.com.
Sustained long-term benefit of gene therapy for SMA
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
, new long-term follow-up data show. At a median of 5.2 years since receiving the approved therapeutic dose, onasemnogene abeparvovec provided “sustained, durable efficacy, with all patients alive and without the need for permanent ventilation,” reported Jerry Mendell, MD, with the Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio, and colleagues.
The study was published online May 17 in JAMA Neurology.
Single infusion
SMA is a rare genetic disease that can lead to paralysis, breathing difficulty, and death. The disorder is caused by a mutation in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein critical for maintenance and function of motor neurons.
In 2019, Zolgensma was approved in the United States for children with SMA and younger than 2 years of age.
Zolgensma is an adeno-associated virus vector-based gene therapy that addresses the genetic root cause of SMA by replacing the defective or missing SMN1 gene to halt disease progression. A single, one-time intravenous infusion results in expression of the SMN protein motor neurons, which improves chances of survival, as well as muscle movement and function.
In the phase 1 START study, 15 infants with SMA type 1 were treated with either a low or therapeutic dose of Zolgensma at Nationwide Children’s Hospital between 2014 and 2017.
The START long-term follow-up study (START LTFU) is an ongoing, observational study assessing safety and durability of response over 15 years in 13 of the infants; three infants received the low dose and 10 received the approved high dose.
Prior to baseline, four patients (40%) in the therapeutic dose cohort required noninvasive ventilatory support, and six (60%) did not require regular ventilatory support, which did not change in long-term follow-up.
All 10 patients who received the therapeutic dose remained alive and without the need for permanent ventilation up to 6.2 years after dosing, Dr. Mendell and colleagues report.
These patients also maintained previously acquired motor milestones. Two patients attained the new milestone of “standing with assistance” without the use of nusinersen (Spinraza, Biogen).
Serious adverse events occurred in eight patients (62%), none of which resulted in study discontinuation or death. The most common serious adverse events were related to the underlying SMA disease process and included acute respiratory failure (31%), pneumonia (31%), dehydration (23%), respiratory distress (15%), and bronchiolitis (15%).
Importantly, the investigators noted, no new safety signals or “adverse events of special interest” emerged during follow-up, including liver function enzyme elevations, new incidences of malignancy or hematologic disorders, and new incidences or exacerbations of existing neurologic or autoimmune disorders.
The investigators acknowledged that this follow-up study is limited by the small sample size of the patient population and confounded by treatment with nusinersen in several patients. “However, given that the two patients who acquired the new motor milestone of standing with assistance did not receive nusinersen at any time, this benefit can be attributed solely to onasemnogene abeparvovec,” Dr. Mendell and colleagues said.
The study was supported by Novartis Gene Therapies. Dr. Mendell and several co-investigators have disclosed financial relationships with the company.
A version of this article first appeared on Medscape.com.
‘Overbasalization’ common in type 2 diabetes management
that impedes achievement of optimal glycemic control, new research suggests.
Such ‘overbasalization,’ defined as a hemoglobin A1c of greater than 8% despite use of more than 0.5 units/kg per day of basal insulin, was identified in about 40% of patients seen in a Florida primary care clinic during 2015-2018. The findings were published in the April 2021 issue of Clinical Diabetes by Kevin Cowart, PharmD, a diabetes care and education specialist at the University of South Florida, Tampa, and colleagues.
The literature suggests that once people with type 2 diabetes start basal insulin, the chance that they’ll achieve a given hemoglobin A1c target, i.e., less than 7%, diminishes significantly if that goal isn’t achieved within the first year of starting insulin, Dr. Cowart said in an interview.
“Our analysis suggests that overbasalization plays a role in patients with type 2 diabetes on basal insulin not achieving optimal glycemic control. Basal insulin is not designed to address postprandial hyperglycemia. I think there’s a clear need to address hesitancy in therapeutic progression beyond basal insulin. A lot of factors underlie the delays, with therapeutic inertia being one of them. It’s complex,” he said.
Overbasalization seen in large proportion of patients
The study comprised 655 adults diagnosed with type 2 diabetes for at least a year who received a prescription for a basal insulin (glargine U-100, glargine U-300, detemir, degludec U-100, degludec U-200, regular U-500, or NPH insulin).
The patients had a mean hemoglobin A1c of 8.4% and a mean basal insulin dose 0.4 units/kg per day. The prevalence of overbasalization was 38.1% for those with hemoglobin A1c above 8%, 42.7% for those with A1c of 9% or above, and 42% with A1c of 10% or greater.
Patient characteristics independently associated with overbasalization were age 35-54 years (odds ratio 1.89), age 65-80 years (0.44), A1c 9% or greater (13.97), and A1c 10% or greater (6.04). Having a prescription for insulin glargine U-100 was associated with a lower overbasalization risk (0.62). In multivariate analysis, only an A1c of 9% or greater remained significant.
Rozalina G. McCoy, MD, an endocrinologist and primary care clinician at the Mayo Clinic, Rochester, Minn., said in an interview that she sees [overbasalization] frequently in patients who are referred to her. “It’s kind of that wall that patients with type 2 diabetes hit because their A1c is high but their fasting blood sugars are normal. Sometimes it’s assumed that there’s a discrepancy, because people don’t always think about postprandial hyperglycemia.”
She also noted that there has been a push in recent years to simplify regimens, particularly in older patients.
“We really want to avoid rapid-acting insulin in older patients because we’re afraid of hypoglycemia, so we start them on basal and keep the noninsulins like metformin and sulfonylureas around. Initially those control the postprandial blood sugar but over time they’re no longer enough.”
Options exist for addressing postmeal blood sugar highs while minimizing lows
While in the past adding premeal insulin was the only option, today there are alternatives for addressing postmeal hyperglycemia, at least in the short term.
Dr. Cowart advised that the first step is to have patients self-monitor their blood glucose and titrate their basal insulin to address fasting hyperglycemia first. Once that appropriate dose is reached, if the patient’s hemoglobin A1c is still above target, the next step is to evaluate the need for postmeal control.
For patients who are at high cardiovascular risk, the next step might involve adding a sodium-glucose cotransporter 2 inhibitor (SGLT2i) or a glucagon-like peptide 1 receptor agonist (GLP-1RA) instead of premeal insulin. But for patients in whom overbasalization is the main concern, a GLP-1RA might be the better choice since it will have a greater impact on postprandial glucose levels, while an SGLT2i will have more effect on fasting blood sugar, he said.
Another option is to use a fixed-dose combination of basal insulin and a glucagon-like peptide 1 receptor agonist (GLP-1RA), provided there aren’t cost or formulary barriers. “We want to use the right combination of drugs and not use too much of one to lead to hypoglycemia,” Dr. Cowart said.
Dr. McCoy doesn’t use fixed-dose combinations because they don’t allow as much flexibility in dosing. To correct overbasalization, she also recommends adding either a GLP-1RA or SGLT2i instead of premeal insulin. However, she cautions, “you still have to monitor those patients because after a few years it still won’t be enough and you’ll have to add mealtime insulin.”
If cost or lack of coverage prevents a patient’s use of SLGT2i/GLP-1RAs, Dr. McCoy said that adding just one premeal injection of rapid-acting insulin before the largest meal of the day is one option. Another is to use twice-daily NPH insulin instead of analog basal insulin, since that does offer some postprandial coverage.
Dr. Cowart said his approach in cost barrier situations is to try to use patient assistance programs and to look into the patient’s formulary to see if there is step therapy or tier considerations, and maybe have a discussion with the insurance company. “We often have to navigate that, and it does take a significant amount of time and could potentially delay patients getting the right therapy when it’s warranted. That is an area where there is a particular role for pharmacists in helping to overcome that and get patients on the right drugs,” he explained.
Problem may be even more common; testing is key
Dr. McCoy said that the A1c cutoff of 8% used to define overbasalization in the study probably resulted in an underestimation of the problem, since many patients are experiencing nighttime hypoglycemia from the basal insulin. The lows bring down their A1c level, but they’re still experiencing postmeal highs.
“I think they’re missing a lot of people, to be honest. I see a lot of patients with A1cs that aren’t that bad, say 7.5%, and their fasting blood sugars are okay, but if you were to put a [continuous glucose monitor] on those patients, invariably there’s hypoglycemia at night that no one knew about.”
Of course, for insurance reasons, most people with type 2 diabetes don’t currently have access to continuous glucose monitors. And often those who are not taking multiple daily injections are limited to one fingerstick test strip a day.
Dr. McCoy says that if hypoglycemia is a concern she will write a prior authorization justifying more test strips.
“I state explicitly in my notes why I recommend frequent monitoring. If they’re on a sulfonylurea, they should be able to check more frequently because they can have hypoglycemia. Same thing with basal insulin.”
Dr. McCoy advises that patients test their blood sugar 2 hours after the largest meal on one day, and at other times on different days. “Blood glucose after a meal shouldn’t be more than 200 [mg/dL]. If it is, that’s not a failure of basal insulin. It’s doing its job. You just need a different agent.”
Dr. Cowart has no disclosures. Dr. McCoy receives funding from the National Institutes of Health.
that impedes achievement of optimal glycemic control, new research suggests.
Such ‘overbasalization,’ defined as a hemoglobin A1c of greater than 8% despite use of more than 0.5 units/kg per day of basal insulin, was identified in about 40% of patients seen in a Florida primary care clinic during 2015-2018. The findings were published in the April 2021 issue of Clinical Diabetes by Kevin Cowart, PharmD, a diabetes care and education specialist at the University of South Florida, Tampa, and colleagues.
The literature suggests that once people with type 2 diabetes start basal insulin, the chance that they’ll achieve a given hemoglobin A1c target, i.e., less than 7%, diminishes significantly if that goal isn’t achieved within the first year of starting insulin, Dr. Cowart said in an interview.
“Our analysis suggests that overbasalization plays a role in patients with type 2 diabetes on basal insulin not achieving optimal glycemic control. Basal insulin is not designed to address postprandial hyperglycemia. I think there’s a clear need to address hesitancy in therapeutic progression beyond basal insulin. A lot of factors underlie the delays, with therapeutic inertia being one of them. It’s complex,” he said.
Overbasalization seen in large proportion of patients
The study comprised 655 adults diagnosed with type 2 diabetes for at least a year who received a prescription for a basal insulin (glargine U-100, glargine U-300, detemir, degludec U-100, degludec U-200, regular U-500, or NPH insulin).
The patients had a mean hemoglobin A1c of 8.4% and a mean basal insulin dose 0.4 units/kg per day. The prevalence of overbasalization was 38.1% for those with hemoglobin A1c above 8%, 42.7% for those with A1c of 9% or above, and 42% with A1c of 10% or greater.
Patient characteristics independently associated with overbasalization were age 35-54 years (odds ratio 1.89), age 65-80 years (0.44), A1c 9% or greater (13.97), and A1c 10% or greater (6.04). Having a prescription for insulin glargine U-100 was associated with a lower overbasalization risk (0.62). In multivariate analysis, only an A1c of 9% or greater remained significant.
Rozalina G. McCoy, MD, an endocrinologist and primary care clinician at the Mayo Clinic, Rochester, Minn., said in an interview that she sees [overbasalization] frequently in patients who are referred to her. “It’s kind of that wall that patients with type 2 diabetes hit because their A1c is high but their fasting blood sugars are normal. Sometimes it’s assumed that there’s a discrepancy, because people don’t always think about postprandial hyperglycemia.”
She also noted that there has been a push in recent years to simplify regimens, particularly in older patients.
“We really want to avoid rapid-acting insulin in older patients because we’re afraid of hypoglycemia, so we start them on basal and keep the noninsulins like metformin and sulfonylureas around. Initially those control the postprandial blood sugar but over time they’re no longer enough.”
Options exist for addressing postmeal blood sugar highs while minimizing lows
While in the past adding premeal insulin was the only option, today there are alternatives for addressing postmeal hyperglycemia, at least in the short term.
Dr. Cowart advised that the first step is to have patients self-monitor their blood glucose and titrate their basal insulin to address fasting hyperglycemia first. Once that appropriate dose is reached, if the patient’s hemoglobin A1c is still above target, the next step is to evaluate the need for postmeal control.
For patients who are at high cardiovascular risk, the next step might involve adding a sodium-glucose cotransporter 2 inhibitor (SGLT2i) or a glucagon-like peptide 1 receptor agonist (GLP-1RA) instead of premeal insulin. But for patients in whom overbasalization is the main concern, a GLP-1RA might be the better choice since it will have a greater impact on postprandial glucose levels, while an SGLT2i will have more effect on fasting blood sugar, he said.
Another option is to use a fixed-dose combination of basal insulin and a glucagon-like peptide 1 receptor agonist (GLP-1RA), provided there aren’t cost or formulary barriers. “We want to use the right combination of drugs and not use too much of one to lead to hypoglycemia,” Dr. Cowart said.
Dr. McCoy doesn’t use fixed-dose combinations because they don’t allow as much flexibility in dosing. To correct overbasalization, she also recommends adding either a GLP-1RA or SGLT2i instead of premeal insulin. However, she cautions, “you still have to monitor those patients because after a few years it still won’t be enough and you’ll have to add mealtime insulin.”
If cost or lack of coverage prevents a patient’s use of SLGT2i/GLP-1RAs, Dr. McCoy said that adding just one premeal injection of rapid-acting insulin before the largest meal of the day is one option. Another is to use twice-daily NPH insulin instead of analog basal insulin, since that does offer some postprandial coverage.
Dr. Cowart said his approach in cost barrier situations is to try to use patient assistance programs and to look into the patient’s formulary to see if there is step therapy or tier considerations, and maybe have a discussion with the insurance company. “We often have to navigate that, and it does take a significant amount of time and could potentially delay patients getting the right therapy when it’s warranted. That is an area where there is a particular role for pharmacists in helping to overcome that and get patients on the right drugs,” he explained.
Problem may be even more common; testing is key
Dr. McCoy said that the A1c cutoff of 8% used to define overbasalization in the study probably resulted in an underestimation of the problem, since many patients are experiencing nighttime hypoglycemia from the basal insulin. The lows bring down their A1c level, but they’re still experiencing postmeal highs.
“I think they’re missing a lot of people, to be honest. I see a lot of patients with A1cs that aren’t that bad, say 7.5%, and their fasting blood sugars are okay, but if you were to put a [continuous glucose monitor] on those patients, invariably there’s hypoglycemia at night that no one knew about.”
Of course, for insurance reasons, most people with type 2 diabetes don’t currently have access to continuous glucose monitors. And often those who are not taking multiple daily injections are limited to one fingerstick test strip a day.
Dr. McCoy says that if hypoglycemia is a concern she will write a prior authorization justifying more test strips.
“I state explicitly in my notes why I recommend frequent monitoring. If they’re on a sulfonylurea, they should be able to check more frequently because they can have hypoglycemia. Same thing with basal insulin.”
Dr. McCoy advises that patients test their blood sugar 2 hours after the largest meal on one day, and at other times on different days. “Blood glucose after a meal shouldn’t be more than 200 [mg/dL]. If it is, that’s not a failure of basal insulin. It’s doing its job. You just need a different agent.”
Dr. Cowart has no disclosures. Dr. McCoy receives funding from the National Institutes of Health.
that impedes achievement of optimal glycemic control, new research suggests.
Such ‘overbasalization,’ defined as a hemoglobin A1c of greater than 8% despite use of more than 0.5 units/kg per day of basal insulin, was identified in about 40% of patients seen in a Florida primary care clinic during 2015-2018. The findings were published in the April 2021 issue of Clinical Diabetes by Kevin Cowart, PharmD, a diabetes care and education specialist at the University of South Florida, Tampa, and colleagues.
The literature suggests that once people with type 2 diabetes start basal insulin, the chance that they’ll achieve a given hemoglobin A1c target, i.e., less than 7%, diminishes significantly if that goal isn’t achieved within the first year of starting insulin, Dr. Cowart said in an interview.
“Our analysis suggests that overbasalization plays a role in patients with type 2 diabetes on basal insulin not achieving optimal glycemic control. Basal insulin is not designed to address postprandial hyperglycemia. I think there’s a clear need to address hesitancy in therapeutic progression beyond basal insulin. A lot of factors underlie the delays, with therapeutic inertia being one of them. It’s complex,” he said.
Overbasalization seen in large proportion of patients
The study comprised 655 adults diagnosed with type 2 diabetes for at least a year who received a prescription for a basal insulin (glargine U-100, glargine U-300, detemir, degludec U-100, degludec U-200, regular U-500, or NPH insulin).
The patients had a mean hemoglobin A1c of 8.4% and a mean basal insulin dose 0.4 units/kg per day. The prevalence of overbasalization was 38.1% for those with hemoglobin A1c above 8%, 42.7% for those with A1c of 9% or above, and 42% with A1c of 10% or greater.
Patient characteristics independently associated with overbasalization were age 35-54 years (odds ratio 1.89), age 65-80 years (0.44), A1c 9% or greater (13.97), and A1c 10% or greater (6.04). Having a prescription for insulin glargine U-100 was associated with a lower overbasalization risk (0.62). In multivariate analysis, only an A1c of 9% or greater remained significant.
Rozalina G. McCoy, MD, an endocrinologist and primary care clinician at the Mayo Clinic, Rochester, Minn., said in an interview that she sees [overbasalization] frequently in patients who are referred to her. “It’s kind of that wall that patients with type 2 diabetes hit because their A1c is high but their fasting blood sugars are normal. Sometimes it’s assumed that there’s a discrepancy, because people don’t always think about postprandial hyperglycemia.”
She also noted that there has been a push in recent years to simplify regimens, particularly in older patients.
“We really want to avoid rapid-acting insulin in older patients because we’re afraid of hypoglycemia, so we start them on basal and keep the noninsulins like metformin and sulfonylureas around. Initially those control the postprandial blood sugar but over time they’re no longer enough.”
Options exist for addressing postmeal blood sugar highs while minimizing lows
While in the past adding premeal insulin was the only option, today there are alternatives for addressing postmeal hyperglycemia, at least in the short term.
Dr. Cowart advised that the first step is to have patients self-monitor their blood glucose and titrate their basal insulin to address fasting hyperglycemia first. Once that appropriate dose is reached, if the patient’s hemoglobin A1c is still above target, the next step is to evaluate the need for postmeal control.
For patients who are at high cardiovascular risk, the next step might involve adding a sodium-glucose cotransporter 2 inhibitor (SGLT2i) or a glucagon-like peptide 1 receptor agonist (GLP-1RA) instead of premeal insulin. But for patients in whom overbasalization is the main concern, a GLP-1RA might be the better choice since it will have a greater impact on postprandial glucose levels, while an SGLT2i will have more effect on fasting blood sugar, he said.
Another option is to use a fixed-dose combination of basal insulin and a glucagon-like peptide 1 receptor agonist (GLP-1RA), provided there aren’t cost or formulary barriers. “We want to use the right combination of drugs and not use too much of one to lead to hypoglycemia,” Dr. Cowart said.
Dr. McCoy doesn’t use fixed-dose combinations because they don’t allow as much flexibility in dosing. To correct overbasalization, she also recommends adding either a GLP-1RA or SGLT2i instead of premeal insulin. However, she cautions, “you still have to monitor those patients because after a few years it still won’t be enough and you’ll have to add mealtime insulin.”
If cost or lack of coverage prevents a patient’s use of SLGT2i/GLP-1RAs, Dr. McCoy said that adding just one premeal injection of rapid-acting insulin before the largest meal of the day is one option. Another is to use twice-daily NPH insulin instead of analog basal insulin, since that does offer some postprandial coverage.
Dr. Cowart said his approach in cost barrier situations is to try to use patient assistance programs and to look into the patient’s formulary to see if there is step therapy or tier considerations, and maybe have a discussion with the insurance company. “We often have to navigate that, and it does take a significant amount of time and could potentially delay patients getting the right therapy when it’s warranted. That is an area where there is a particular role for pharmacists in helping to overcome that and get patients on the right drugs,” he explained.
Problem may be even more common; testing is key
Dr. McCoy said that the A1c cutoff of 8% used to define overbasalization in the study probably resulted in an underestimation of the problem, since many patients are experiencing nighttime hypoglycemia from the basal insulin. The lows bring down their A1c level, but they’re still experiencing postmeal highs.
“I think they’re missing a lot of people, to be honest. I see a lot of patients with A1cs that aren’t that bad, say 7.5%, and their fasting blood sugars are okay, but if you were to put a [continuous glucose monitor] on those patients, invariably there’s hypoglycemia at night that no one knew about.”
Of course, for insurance reasons, most people with type 2 diabetes don’t currently have access to continuous glucose monitors. And often those who are not taking multiple daily injections are limited to one fingerstick test strip a day.
Dr. McCoy says that if hypoglycemia is a concern she will write a prior authorization justifying more test strips.
“I state explicitly in my notes why I recommend frequent monitoring. If they’re on a sulfonylurea, they should be able to check more frequently because they can have hypoglycemia. Same thing with basal insulin.”
Dr. McCoy advises that patients test their blood sugar 2 hours after the largest meal on one day, and at other times on different days. “Blood glucose after a meal shouldn’t be more than 200 [mg/dL]. If it is, that’s not a failure of basal insulin. It’s doing its job. You just need a different agent.”
Dr. Cowart has no disclosures. Dr. McCoy receives funding from the National Institutes of Health.
FROM CLINICAL DIABETES
New allergy guidelines call for end to food bans in schools
Children with food allergies often require diligent monitoring and a restricted diet to reduce allergic attacks, but there is little evidence available to support so-called “food bans” at schools and childcare centers.
Instead, a new practice guideline published earlier this month in the Journal of Allergy and Clinical Immunology calls for better allergy management training for staff, as well as increased epinephrine availability in educational environments. The guidelines were developed by an international panel of clinicians, school personnel, and parents.
The guidance at a glance
Rather than creating site-wide food prohibitions on nuts, dairy, and other allergenic foods, the practice guidance recommends centers and schools use “common-sense approaches” to reduce allergic reaction risk among school-aged children. According to the guideline authors, these strategies could include promoting handwashing, providing adult supervision during snacks and meals, and cleaning surfaces where food is either eaten or prepared.
Additionally, the new evidence-based guidance calls for schools and childcare centers to teach school personnel to recognize, prevent, and respond appropriately to food-related allergic reactions when they do occur.
The guidance also recommends that educational institutions require an up-to-date allergy ‘action plan’ from parents, designed for their children with allergies. These action plans can be integrated into the training of teachers and nurses to help manage potential allergic reactions.
Moreover, the guidance suggests schools should keep unassigned epinephrine autoinjectors in stock, both on site and even when traveling, where laws permit, rather than requiring students with allergies to bring in their own autoinjectors. Ultimately, this represents a more proactive approach to treating anaphylaxis, particularly in settings where treatment is urgently needed, such as when students are away from campus and participating in a school-designated trip or event.
Expert perspectives
Jennifer A. Dantzer, MD, MHS, allergist-immunologist and assistant professor of pediatrics at Johns Hopkins University, Baltimore, told this news organization via email that the practice guidelines offer an important starting point for ensuring quality of life of students, parents, and other school personnel.
While the Centers for Disease Control and Prevention published voluntary guidance for managing food allergies in schools back in 2013, there has since “been a lack of universal policies and procedures to manage the risk of allergic reactions in schools,” explained Dr. Dantzer. “The new guidelines are a good first step of using available evidence and all the key stakeholders, clinicians, school personnel, and families to figure out the best way to keep children with food allergies safe at school.”
Dr. Dantzer wasn’t involved in the creation of the new practice guidelines, but she shared how her clinical experience reinforces the need for the evidence-based recommendations. “Every single week we talk with families, both in clinic and in our research studies, about living with food allergies, and we recognize that every child is different,” she said. “We constantly work to advocate for each individual child with food allergies.”
Pediatric allergist Malika Gupta, MBBS, MD, said in an interview via email that the guidelines could assist in the creation of new nationwide policies for food allergy management at schools. “Also, the guidelines are labeled ‘conditional,’ which gives policymakers the ability to adapt to their specific circumstances and individuals, as well as make modifications according to regional trends,” she added.
Dr. Gupta, a clinical assistant professor in the Division of Allergy and Clinical Immunology at the University of Michigan, Ann Arbor, echoed the guideline panel’s sentiments regarding food bans, explaining that prohibiting certain foods could lend a “false sense of security” and could also “promote bullying and a sense of isolation for the food-allergic child.” In spite of the lack of evidence supporting food bans, Dr. Gupta noted that these bans can give families a sense of control and security. Ideally, more research should be performed to determine whether food bans actually work, she added.
In addition to promoting the new guidelines, allergists and pediatricians can also implement proactive allergy reaction mitigation strategies that work with school systems, according to Dr. Gupta. “In-clinic, we ensure all families have food allergy action plans for school and current epinephrine auto-injectors,” she said. “We also often have our food allergy nurses educate schools when food allergy awareness is a concern.”
Many of the 25 authors of the food allergy guidelines disclosed relevant financial relationships. The full list is with the original article. According to a footnote within the guidelines, “Panel members who were deemed to have a real, perceived, or potential conflict of interest were asked to abstain from voting on recommendations related to that interest.”
A version of this article first appeared on Medscape.com.
Children with food allergies often require diligent monitoring and a restricted diet to reduce allergic attacks, but there is little evidence available to support so-called “food bans” at schools and childcare centers.
Instead, a new practice guideline published earlier this month in the Journal of Allergy and Clinical Immunology calls for better allergy management training for staff, as well as increased epinephrine availability in educational environments. The guidelines were developed by an international panel of clinicians, school personnel, and parents.
The guidance at a glance
Rather than creating site-wide food prohibitions on nuts, dairy, and other allergenic foods, the practice guidance recommends centers and schools use “common-sense approaches” to reduce allergic reaction risk among school-aged children. According to the guideline authors, these strategies could include promoting handwashing, providing adult supervision during snacks and meals, and cleaning surfaces where food is either eaten or prepared.
Additionally, the new evidence-based guidance calls for schools and childcare centers to teach school personnel to recognize, prevent, and respond appropriately to food-related allergic reactions when they do occur.
The guidance also recommends that educational institutions require an up-to-date allergy ‘action plan’ from parents, designed for their children with allergies. These action plans can be integrated into the training of teachers and nurses to help manage potential allergic reactions.
Moreover, the guidance suggests schools should keep unassigned epinephrine autoinjectors in stock, both on site and even when traveling, where laws permit, rather than requiring students with allergies to bring in their own autoinjectors. Ultimately, this represents a more proactive approach to treating anaphylaxis, particularly in settings where treatment is urgently needed, such as when students are away from campus and participating in a school-designated trip or event.
Expert perspectives
Jennifer A. Dantzer, MD, MHS, allergist-immunologist and assistant professor of pediatrics at Johns Hopkins University, Baltimore, told this news organization via email that the practice guidelines offer an important starting point for ensuring quality of life of students, parents, and other school personnel.
While the Centers for Disease Control and Prevention published voluntary guidance for managing food allergies in schools back in 2013, there has since “been a lack of universal policies and procedures to manage the risk of allergic reactions in schools,” explained Dr. Dantzer. “The new guidelines are a good first step of using available evidence and all the key stakeholders, clinicians, school personnel, and families to figure out the best way to keep children with food allergies safe at school.”
Dr. Dantzer wasn’t involved in the creation of the new practice guidelines, but she shared how her clinical experience reinforces the need for the evidence-based recommendations. “Every single week we talk with families, both in clinic and in our research studies, about living with food allergies, and we recognize that every child is different,” she said. “We constantly work to advocate for each individual child with food allergies.”
Pediatric allergist Malika Gupta, MBBS, MD, said in an interview via email that the guidelines could assist in the creation of new nationwide policies for food allergy management at schools. “Also, the guidelines are labeled ‘conditional,’ which gives policymakers the ability to adapt to their specific circumstances and individuals, as well as make modifications according to regional trends,” she added.
Dr. Gupta, a clinical assistant professor in the Division of Allergy and Clinical Immunology at the University of Michigan, Ann Arbor, echoed the guideline panel’s sentiments regarding food bans, explaining that prohibiting certain foods could lend a “false sense of security” and could also “promote bullying and a sense of isolation for the food-allergic child.” In spite of the lack of evidence supporting food bans, Dr. Gupta noted that these bans can give families a sense of control and security. Ideally, more research should be performed to determine whether food bans actually work, she added.
In addition to promoting the new guidelines, allergists and pediatricians can also implement proactive allergy reaction mitigation strategies that work with school systems, according to Dr. Gupta. “In-clinic, we ensure all families have food allergy action plans for school and current epinephrine auto-injectors,” she said. “We also often have our food allergy nurses educate schools when food allergy awareness is a concern.”
Many of the 25 authors of the food allergy guidelines disclosed relevant financial relationships. The full list is with the original article. According to a footnote within the guidelines, “Panel members who were deemed to have a real, perceived, or potential conflict of interest were asked to abstain from voting on recommendations related to that interest.”
A version of this article first appeared on Medscape.com.
Children with food allergies often require diligent monitoring and a restricted diet to reduce allergic attacks, but there is little evidence available to support so-called “food bans” at schools and childcare centers.
Instead, a new practice guideline published earlier this month in the Journal of Allergy and Clinical Immunology calls for better allergy management training for staff, as well as increased epinephrine availability in educational environments. The guidelines were developed by an international panel of clinicians, school personnel, and parents.
The guidance at a glance
Rather than creating site-wide food prohibitions on nuts, dairy, and other allergenic foods, the practice guidance recommends centers and schools use “common-sense approaches” to reduce allergic reaction risk among school-aged children. According to the guideline authors, these strategies could include promoting handwashing, providing adult supervision during snacks and meals, and cleaning surfaces where food is either eaten or prepared.
Additionally, the new evidence-based guidance calls for schools and childcare centers to teach school personnel to recognize, prevent, and respond appropriately to food-related allergic reactions when they do occur.
The guidance also recommends that educational institutions require an up-to-date allergy ‘action plan’ from parents, designed for their children with allergies. These action plans can be integrated into the training of teachers and nurses to help manage potential allergic reactions.
Moreover, the guidance suggests schools should keep unassigned epinephrine autoinjectors in stock, both on site and even when traveling, where laws permit, rather than requiring students with allergies to bring in their own autoinjectors. Ultimately, this represents a more proactive approach to treating anaphylaxis, particularly in settings where treatment is urgently needed, such as when students are away from campus and participating in a school-designated trip or event.
Expert perspectives
Jennifer A. Dantzer, MD, MHS, allergist-immunologist and assistant professor of pediatrics at Johns Hopkins University, Baltimore, told this news organization via email that the practice guidelines offer an important starting point for ensuring quality of life of students, parents, and other school personnel.
While the Centers for Disease Control and Prevention published voluntary guidance for managing food allergies in schools back in 2013, there has since “been a lack of universal policies and procedures to manage the risk of allergic reactions in schools,” explained Dr. Dantzer. “The new guidelines are a good first step of using available evidence and all the key stakeholders, clinicians, school personnel, and families to figure out the best way to keep children with food allergies safe at school.”
Dr. Dantzer wasn’t involved in the creation of the new practice guidelines, but she shared how her clinical experience reinforces the need for the evidence-based recommendations. “Every single week we talk with families, both in clinic and in our research studies, about living with food allergies, and we recognize that every child is different,” she said. “We constantly work to advocate for each individual child with food allergies.”
Pediatric allergist Malika Gupta, MBBS, MD, said in an interview via email that the guidelines could assist in the creation of new nationwide policies for food allergy management at schools. “Also, the guidelines are labeled ‘conditional,’ which gives policymakers the ability to adapt to their specific circumstances and individuals, as well as make modifications according to regional trends,” she added.
Dr. Gupta, a clinical assistant professor in the Division of Allergy and Clinical Immunology at the University of Michigan, Ann Arbor, echoed the guideline panel’s sentiments regarding food bans, explaining that prohibiting certain foods could lend a “false sense of security” and could also “promote bullying and a sense of isolation for the food-allergic child.” In spite of the lack of evidence supporting food bans, Dr. Gupta noted that these bans can give families a sense of control and security. Ideally, more research should be performed to determine whether food bans actually work, she added.
In addition to promoting the new guidelines, allergists and pediatricians can also implement proactive allergy reaction mitigation strategies that work with school systems, according to Dr. Gupta. “In-clinic, we ensure all families have food allergy action plans for school and current epinephrine auto-injectors,” she said. “We also often have our food allergy nurses educate schools when food allergy awareness is a concern.”
Many of the 25 authors of the food allergy guidelines disclosed relevant financial relationships. The full list is with the original article. According to a footnote within the guidelines, “Panel members who were deemed to have a real, perceived, or potential conflict of interest were asked to abstain from voting on recommendations related to that interest.”
A version of this article first appeared on Medscape.com.
COVID-19 vaccination rate rising quickly among adolescents
With nearly half of all Americans having received at least one dose of a COVID-19 vaccine, the youngest eligible group is beginning to overcome its late start, according to data from the Centers for Disease Control and Prevention.
As of May 24, 49.4% of the U.S. population – that’s almost 164 million people – has received at least one dose of vaccine. The corresponding figure for children aged 12-15 years is 14.4%, but that’s up from only 0.6% just 3 weeks before. Among children aged 16-17, who’ve been getting vaccinated since early April in some states, the proportion receiving at least one dose went from 24.9% to 33.9% over those same 3 weeks, the CDC said on its COVID Data Tracker site.
The comparatively rapid increase among the younger group of eligible children can be seen over the last 14 days. To put that into perspective, only those aged 25-39 years were higher at 21.9%, while 18-24 (12.1%), 40-49 (13.4%), 50-64 (18.2%), 65-74 (5.3%), and ≥75 (2.9%) were all lower.
The 12- to 15-year-olds are further behind when it comes to full vaccination status, however, with just 0.6% having received both doses of a two-dose vaccine or one dose of the single-shot variety, compared with 21.6% for those aged 16-17 years. Children aged 12-15 make up 5% of the total U.S. population but just 0.1% of all those who have been fully vaccinated versus 2.5% and 1.4%, respectively, for those aged 16-17, the CDC reported.
With nearly half of all Americans having received at least one dose of a COVID-19 vaccine, the youngest eligible group is beginning to overcome its late start, according to data from the Centers for Disease Control and Prevention.
As of May 24, 49.4% of the U.S. population – that’s almost 164 million people – has received at least one dose of vaccine. The corresponding figure for children aged 12-15 years is 14.4%, but that’s up from only 0.6% just 3 weeks before. Among children aged 16-17, who’ve been getting vaccinated since early April in some states, the proportion receiving at least one dose went from 24.9% to 33.9% over those same 3 weeks, the CDC said on its COVID Data Tracker site.
The comparatively rapid increase among the younger group of eligible children can be seen over the last 14 days. To put that into perspective, only those aged 25-39 years were higher at 21.9%, while 18-24 (12.1%), 40-49 (13.4%), 50-64 (18.2%), 65-74 (5.3%), and ≥75 (2.9%) were all lower.
The 12- to 15-year-olds are further behind when it comes to full vaccination status, however, with just 0.6% having received both doses of a two-dose vaccine or one dose of the single-shot variety, compared with 21.6% for those aged 16-17 years. Children aged 12-15 make up 5% of the total U.S. population but just 0.1% of all those who have been fully vaccinated versus 2.5% and 1.4%, respectively, for those aged 16-17, the CDC reported.
With nearly half of all Americans having received at least one dose of a COVID-19 vaccine, the youngest eligible group is beginning to overcome its late start, according to data from the Centers for Disease Control and Prevention.
As of May 24, 49.4% of the U.S. population – that’s almost 164 million people – has received at least one dose of vaccine. The corresponding figure for children aged 12-15 years is 14.4%, but that’s up from only 0.6% just 3 weeks before. Among children aged 16-17, who’ve been getting vaccinated since early April in some states, the proportion receiving at least one dose went from 24.9% to 33.9% over those same 3 weeks, the CDC said on its COVID Data Tracker site.
The comparatively rapid increase among the younger group of eligible children can be seen over the last 14 days. To put that into perspective, only those aged 25-39 years were higher at 21.9%, while 18-24 (12.1%), 40-49 (13.4%), 50-64 (18.2%), 65-74 (5.3%), and ≥75 (2.9%) were all lower.
The 12- to 15-year-olds are further behind when it comes to full vaccination status, however, with just 0.6% having received both doses of a two-dose vaccine or one dose of the single-shot variety, compared with 21.6% for those aged 16-17 years. Children aged 12-15 make up 5% of the total U.S. population but just 0.1% of all those who have been fully vaccinated versus 2.5% and 1.4%, respectively, for those aged 16-17, the CDC reported.
Tezepelumab reduces serious exacerbations in severe asthma
Results from the NAVIGATOR study of tezepelumab showed that treatment of adults and adolescents with severe, uncontrolled asthma with the new biologic led to a large reduction in exacerbations requiring hospital stays and ED visits.
Tezepelumab, codeveloped by Amgen and AstraZeneca, has a novel mechanism of action. It blocks thymic stromal lymphopoietin (TSLP), which is a cytokine produced by epithelial cells. TSLP levels correlate with airway obstruction, severity of disease, and glucocorticoid resistance. TSLP is involved in T2 inflammation within the airway, but also plays a role in the interactions between airway cells and immune cells, which doesn’t rely only solely on T2 inflammation. That broad mechanism of action distinguishes tezepelumab from most other biologics for the treatment of asthma, which are more targeted.
“By working at the top of the cascade, tezepelumab helps stop inflammation at a key source. Clinical trials with tezepelumab showed a clinical benefit in patients irrespective of their baseline biomarker level, including patients with low eosinophil levels at baseline,” said Jean-Pierre Llanos-Ackert, MD, who is executive medical director and global medical affairs lead for tezepelumab at Amgen.

The primary endpoint data look robust, according to Praveen Akuthota, MD, who is an associate professor of medicine at the University of California, San Diego, and comoderated the session at the American Thoracic Society’s virtual international conference, where the research was presented. The study was also published on May 13, 2021, in the New England Journal of Medicine. The conference session included updated results.
The drug holds promise, but more study is needed. “The question really will be, how is this drug different from the existing biologics? How much better is this drug in patients who have borderline T2 biomarkers, or even low T2. The study does show some efficacy in patients whose T2 signals may not be as robust. We’ll have to see with ongoing longitudinal data, how this drug positions, compared to the other agents. It’s obviously exciting, though, to have another option, given that we know what our current armamentarium of agents there are still nonresponders,” said Dr. Akuthota in an interview.
The other comoderator in the session, Laura Crotty Alexander, MD, commented: “It seems like it might work possibly even better than some that are directly covering one pathway only. Hopefully, this agent will be efficacious in a broader population than some of the more targeted biologics.” Dr. Alexander is an associate professor of medicine at the University of California, San Diego, and section chief of pulmonary critical care at the Veterans Affairs San Diego Healthcare System.
She pointed out that physicians often think of asthma patients in broad brush terms, as high or low T2, or T2 high and Th1 or neutrophilic or obese, but many patients present a more complicated picture. “There is some overlap across those phenotypes, such that an agent that works really well for one group doesn’t mean that it won’t have an impact, especially clinically, on some of these other phenotypes,” said Dr. Alexander.
Dr. Akuthota agreed. “Having options for patients whose biomarkers are not maybe as clear is, I think, important.”
Promising results
The study included 1,059 patients aged 12-80 who received 210 mg tezepelumab or placebo. Over 52 weeks, the treatment group had a 79% reduction in exacerbations requiring hospitalization or an ED visit, compared with placebo (rate ratio, 0.21; 95% confidence interval, 0.12-0.37), and an 85% reduction in exacerbations requiring hospitalization (RR, 0.15; 95% CI, 0.07-0.33). The drug increased the time to first exacerbation requiring hospitalization that required hospitalization or an ED visit, reducing risk by 65% (hazard ratio, 0.35; 95% CI, 0.22-0.56).
Fewer patients in the treatment group than placebo used asthma-related health care resources, including: ED visits (32 vs. 94), unscheduled visit to a specialist (285 vs. 406), telephone calls to a health care provider (234 vs. 599), ambulance transport (5 vs. 22), and home visits from a health care provider (18 vs. 22). Fewer patients in the tezepelumab group had hospital stays (3.2% vs. 7.0%), and they had a lower total number of hospital days (108 vs. 497) and days in the ICU (0 vs. 31).
The study was funded by Amgen and AstraZeneca. Dr. Llanos-Ackert is an employee of Amgen. Dr. Alexander has no relevant financial disclosures. Dr. Akuthota has consulted for AstraZeneca and participated in their clinical trials.
Results from the NAVIGATOR study of tezepelumab showed that treatment of adults and adolescents with severe, uncontrolled asthma with the new biologic led to a large reduction in exacerbations requiring hospital stays and ED visits.
Tezepelumab, codeveloped by Amgen and AstraZeneca, has a novel mechanism of action. It blocks thymic stromal lymphopoietin (TSLP), which is a cytokine produced by epithelial cells. TSLP levels correlate with airway obstruction, severity of disease, and glucocorticoid resistance. TSLP is involved in T2 inflammation within the airway, but also plays a role in the interactions between airway cells and immune cells, which doesn’t rely only solely on T2 inflammation. That broad mechanism of action distinguishes tezepelumab from most other biologics for the treatment of asthma, which are more targeted.
“By working at the top of the cascade, tezepelumab helps stop inflammation at a key source. Clinical trials with tezepelumab showed a clinical benefit in patients irrespective of their baseline biomarker level, including patients with low eosinophil levels at baseline,” said Jean-Pierre Llanos-Ackert, MD, who is executive medical director and global medical affairs lead for tezepelumab at Amgen.
The primary endpoint data look robust, according to Praveen Akuthota, MD, who is an associate professor of medicine at the University of California, San Diego, and comoderated the session at the American Thoracic Society’s virtual international conference, where the research was presented. The study was also published on May 13, 2021, in the New England Journal of Medicine. The conference session included updated results.
The drug holds promise, but more study is needed. “The question really will be, how is this drug different from the existing biologics? How much better is this drug in patients who have borderline T2 biomarkers, or even low T2. The study does show some efficacy in patients whose T2 signals may not be as robust. We’ll have to see with ongoing longitudinal data, how this drug positions, compared to the other agents. It’s obviously exciting, though, to have another option, given that we know what our current armamentarium of agents there are still nonresponders,” said Dr. Akuthota in an interview.
The other comoderator in the session, Laura Crotty Alexander, MD, commented: “It seems like it might work possibly even better than some that are directly covering one pathway only. Hopefully, this agent will be efficacious in a broader population than some of the more targeted biologics.” Dr. Alexander is an associate professor of medicine at the University of California, San Diego, and section chief of pulmonary critical care at the Veterans Affairs San Diego Healthcare System.
She pointed out that physicians often think of asthma patients in broad brush terms, as high or low T2, or T2 high and Th1 or neutrophilic or obese, but many patients present a more complicated picture. “There is some overlap across those phenotypes, such that an agent that works really well for one group doesn’t mean that it won’t have an impact, especially clinically, on some of these other phenotypes,” said Dr. Alexander.
Dr. Akuthota agreed. “Having options for patients whose biomarkers are not maybe as clear is, I think, important.”
Promising results
The study included 1,059 patients aged 12-80 who received 210 mg tezepelumab or placebo. Over 52 weeks, the treatment group had a 79% reduction in exacerbations requiring hospitalization or an ED visit, compared with placebo (rate ratio, 0.21; 95% confidence interval, 0.12-0.37), and an 85% reduction in exacerbations requiring hospitalization (RR, 0.15; 95% CI, 0.07-0.33). The drug increased the time to first exacerbation requiring hospitalization that required hospitalization or an ED visit, reducing risk by 65% (hazard ratio, 0.35; 95% CI, 0.22-0.56).
Fewer patients in the treatment group than placebo used asthma-related health care resources, including: ED visits (32 vs. 94), unscheduled visit to a specialist (285 vs. 406), telephone calls to a health care provider (234 vs. 599), ambulance transport (5 vs. 22), and home visits from a health care provider (18 vs. 22). Fewer patients in the tezepelumab group had hospital stays (3.2% vs. 7.0%), and they had a lower total number of hospital days (108 vs. 497) and days in the ICU (0 vs. 31).
The study was funded by Amgen and AstraZeneca. Dr. Llanos-Ackert is an employee of Amgen. Dr. Alexander has no relevant financial disclosures. Dr. Akuthota has consulted for AstraZeneca and participated in their clinical trials.
Results from the NAVIGATOR study of tezepelumab showed that treatment of adults and adolescents with severe, uncontrolled asthma with the new biologic led to a large reduction in exacerbations requiring hospital stays and ED visits.
Tezepelumab, codeveloped by Amgen and AstraZeneca, has a novel mechanism of action. It blocks thymic stromal lymphopoietin (TSLP), which is a cytokine produced by epithelial cells. TSLP levels correlate with airway obstruction, severity of disease, and glucocorticoid resistance. TSLP is involved in T2 inflammation within the airway, but also plays a role in the interactions between airway cells and immune cells, which doesn’t rely only solely on T2 inflammation. That broad mechanism of action distinguishes tezepelumab from most other biologics for the treatment of asthma, which are more targeted.
“By working at the top of the cascade, tezepelumab helps stop inflammation at a key source. Clinical trials with tezepelumab showed a clinical benefit in patients irrespective of their baseline biomarker level, including patients with low eosinophil levels at baseline,” said Jean-Pierre Llanos-Ackert, MD, who is executive medical director and global medical affairs lead for tezepelumab at Amgen.
The primary endpoint data look robust, according to Praveen Akuthota, MD, who is an associate professor of medicine at the University of California, San Diego, and comoderated the session at the American Thoracic Society’s virtual international conference, where the research was presented. The study was also published on May 13, 2021, in the New England Journal of Medicine. The conference session included updated results.
The drug holds promise, but more study is needed. “The question really will be, how is this drug different from the existing biologics? How much better is this drug in patients who have borderline T2 biomarkers, or even low T2. The study does show some efficacy in patients whose T2 signals may not be as robust. We’ll have to see with ongoing longitudinal data, how this drug positions, compared to the other agents. It’s obviously exciting, though, to have another option, given that we know what our current armamentarium of agents there are still nonresponders,” said Dr. Akuthota in an interview.
The other comoderator in the session, Laura Crotty Alexander, MD, commented: “It seems like it might work possibly even better than some that are directly covering one pathway only. Hopefully, this agent will be efficacious in a broader population than some of the more targeted biologics.” Dr. Alexander is an associate professor of medicine at the University of California, San Diego, and section chief of pulmonary critical care at the Veterans Affairs San Diego Healthcare System.
She pointed out that physicians often think of asthma patients in broad brush terms, as high or low T2, or T2 high and Th1 or neutrophilic or obese, but many patients present a more complicated picture. “There is some overlap across those phenotypes, such that an agent that works really well for one group doesn’t mean that it won’t have an impact, especially clinically, on some of these other phenotypes,” said Dr. Alexander.
Dr. Akuthota agreed. “Having options for patients whose biomarkers are not maybe as clear is, I think, important.”
Promising results
The study included 1,059 patients aged 12-80 who received 210 mg tezepelumab or placebo. Over 52 weeks, the treatment group had a 79% reduction in exacerbations requiring hospitalization or an ED visit, compared with placebo (rate ratio, 0.21; 95% confidence interval, 0.12-0.37), and an 85% reduction in exacerbations requiring hospitalization (RR, 0.15; 95% CI, 0.07-0.33). The drug increased the time to first exacerbation requiring hospitalization that required hospitalization or an ED visit, reducing risk by 65% (hazard ratio, 0.35; 95% CI, 0.22-0.56).
Fewer patients in the treatment group than placebo used asthma-related health care resources, including: ED visits (32 vs. 94), unscheduled visit to a specialist (285 vs. 406), telephone calls to a health care provider (234 vs. 599), ambulance transport (5 vs. 22), and home visits from a health care provider (18 vs. 22). Fewer patients in the tezepelumab group had hospital stays (3.2% vs. 7.0%), and they had a lower total number of hospital days (108 vs. 497) and days in the ICU (0 vs. 31).
The study was funded by Amgen and AstraZeneca. Dr. Llanos-Ackert is an employee of Amgen. Dr. Alexander has no relevant financial disclosures. Dr. Akuthota has consulted for AstraZeneca and participated in their clinical trials.
FROM ATS 2021
Skip routine probiotics for preemies, AAP says
The American Academy of Pediatrics now recommends against the routine administration of probiotics to preterm infants, particularly the most vulnerable (those whose birth weight is <1,000 g), for the treatment or prevention of necrotizing enterocolitis (NEC) and late-onset sepsis.
Although probiotics are increasingly given to preterm infants, the AAP notes that the data on their safety and efficacy are inconsistent. In addition, the supplements are not subject to approval by the Food and Drug Administration.
Therefore, the academy advises clinicians to use extreme caution in selecting preterm neonates to receive these microorganisms and recommends obtaining informed consent from parents after carefully discussing the risks. It also recommends that centers using probiotics conduct surveillance, inasmuch as probiotics can alter a center’s flora, potentially affecting all patients. Such centers should also carefully document outcomes, adverse events, and safety.
The AAP’s clinical report, published online May 24 in Pediatrics, highlights wide differences between commercially available formulations and a lack of regulatory standards in this country.
Absent FDA-approved drug labeling, these nutritional supplements cannot be marketed as treatment or prophylaxis, but that has scarcely stopped their use. “Despite lack of availability of a pharmaceutical-grade product, the number of preterm infants receiving probiotics in the United States and Canada is steadily increasing,” wrote Brenda Poindexter, MD, FAAP, chief of neonatology at Children’s Healthcare of Atlanta, and members of the AAP’s Committee on Fetus and Newborn.
Analyses of U.S. collaborative databases indicate that approximately 10% of neonates of extremely low gestational age receive a probiotic preparation in the neonatal intensive care unit (NICU). The use of these preparations varies widely across institutions.
“NEC is a devastating morbidity of prematurity, and it’s multifactorial. Some babies only given mother’s milk still get NEC, and the decision to use these products is a very nuanced one,” Dr. Poindexter said in an interview. “I suspect some people will disagree with the report, and we tried to give folks some wiggle room.”
Evidence from other countries suggests that probiotics can be protective against NEC, she added, “so not to have a reliable product in this country is very frustrating.”
Dr. Poindexter and colleagues pointed to a 2015 study that found that only 1 of 16 commercial products tested contained the exact organisms listed on their labels. One product contained none of the species listed on the label.
In light of increasing use, the AAP emphasizes the need for development of pharmaceutical-grade probiotics that would be rigorously evaluated for safety and efficacy.
The infant microbiome
Over the past decade, the gut microbiome has been increasingly recognized as a factor relevant to health and disease in preterm infants, the authors noted. Differences in the intestinal microbiota between full-term and preterm infants are substantial. The microbiome of preterm infants tends to include fewer bacterial species and greater proportions of potentially pathogenic strains.
Evidence of benefit from probiotics has been mixed. Some studies and pooled systematic analyses suggest a significant benefit. However, Dr. Poindexter and colleagues noted that some researchers express concerns about the study methods used, such as pooling results from trials that tested different probiotic strains or that had few infants in the highest risk category.
Whereas the potential for probiotic-related infection appears low, there does seem to be some risk for sepsis associated with colonization by a strain in a given product or from contamination with a pathogen during manufacturing, the report explains.
At least one trial found that a third of infants randomly assigned to receive placebo showed evidence of the probiotic strain.
“However, it may be difficult to distinguish the change in the infant from the change in the resident flora of the NICU,” the AAP panel wrote.
In addition, there have recently been several recalls of dietary supplement–grade probiotics for contamination, which have raised concerns. Pathogens include Salmonella, Rhizopus, and Penicillium species. Fatal gastrointestinal mucormycosis has also been reported in a preterm infant who received ABC Dophilus powder that was contaminated with the microfungus Rhizopus oryzae.
Other safety considerations, according to the authors, are the unknown longer-term effect of probiotics on preterm infants and the unknown impact of microorganisms on the microbiome over time.
Last year, the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition published a position paper with a conditional recommendation for selected probiotics to reduce NEC rates. “These guidelines would be applicable in the U.S. if we had products manufactured in a way that could guarantee that what’s on the label is in the bottle,” Dr. Poindexter said.
Asked for her perspective on the AAP clinical report, Erica Wymore, MD, assistant professor of neonatal and perinatal medicine at the University of Colorado at Denver, Aurora, called it “an excellent review of the current literature” that shows the inadequacy of data on the composition, dosage, timing, duration, and use of single-strain vs. multiple-strain probiotics to reduce NEC. Her clinical center, Children’s Hospital Colorado, does not administer probiotics to preterm babies.
Although guidelines can improve outcomes, said Dr. Wymore, who was not involved in the AAP report, improvement observed with probiotics results from more stringent care in centers that experience high NEC rates. “It’s hard to know if it’s due to the probiotics if they already have a high rate of NEC,” Dr. Wymore said.
She echoed the AAP’s position and stressed the need for extreme caution in giving these products to vulnerable infants with immature immune systems. Before that can be safely done, she said, “we need more FDA oversight of product composition [and] pharmaceutical-grade products, and more studies to determine efficacy.”
Added Dr. Poindexter: “Hopefully, this report will inform clinicians of the risks of using non–pharmaceutical-grade products and encourage industry to actually develop probiotics for neonates that we can feel comfortable using.”
The report received no external funding. Dr. Poindexter and Dr. Wymore have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The American Academy of Pediatrics now recommends against the routine administration of probiotics to preterm infants, particularly the most vulnerable (those whose birth weight is <1,000 g), for the treatment or prevention of necrotizing enterocolitis (NEC) and late-onset sepsis.
Although probiotics are increasingly given to preterm infants, the AAP notes that the data on their safety and efficacy are inconsistent. In addition, the supplements are not subject to approval by the Food and Drug Administration.
Therefore, the academy advises clinicians to use extreme caution in selecting preterm neonates to receive these microorganisms and recommends obtaining informed consent from parents after carefully discussing the risks. It also recommends that centers using probiotics conduct surveillance, inasmuch as probiotics can alter a center’s flora, potentially affecting all patients. Such centers should also carefully document outcomes, adverse events, and safety.
The AAP’s clinical report, published online May 24 in Pediatrics, highlights wide differences between commercially available formulations and a lack of regulatory standards in this country.
Absent FDA-approved drug labeling, these nutritional supplements cannot be marketed as treatment or prophylaxis, but that has scarcely stopped their use. “Despite lack of availability of a pharmaceutical-grade product, the number of preterm infants receiving probiotics in the United States and Canada is steadily increasing,” wrote Brenda Poindexter, MD, FAAP, chief of neonatology at Children’s Healthcare of Atlanta, and members of the AAP’s Committee on Fetus and Newborn.
Analyses of U.S. collaborative databases indicate that approximately 10% of neonates of extremely low gestational age receive a probiotic preparation in the neonatal intensive care unit (NICU). The use of these preparations varies widely across institutions.
“NEC is a devastating morbidity of prematurity, and it’s multifactorial. Some babies only given mother’s milk still get NEC, and the decision to use these products is a very nuanced one,” Dr. Poindexter said in an interview. “I suspect some people will disagree with the report, and we tried to give folks some wiggle room.”
Evidence from other countries suggests that probiotics can be protective against NEC, she added, “so not to have a reliable product in this country is very frustrating.”
Dr. Poindexter and colleagues pointed to a 2015 study that found that only 1 of 16 commercial products tested contained the exact organisms listed on their labels. One product contained none of the species listed on the label.
In light of increasing use, the AAP emphasizes the need for development of pharmaceutical-grade probiotics that would be rigorously evaluated for safety and efficacy.
The infant microbiome
Over the past decade, the gut microbiome has been increasingly recognized as a factor relevant to health and disease in preterm infants, the authors noted. Differences in the intestinal microbiota between full-term and preterm infants are substantial. The microbiome of preterm infants tends to include fewer bacterial species and greater proportions of potentially pathogenic strains.
Evidence of benefit from probiotics has been mixed. Some studies and pooled systematic analyses suggest a significant benefit. However, Dr. Poindexter and colleagues noted that some researchers express concerns about the study methods used, such as pooling results from trials that tested different probiotic strains or that had few infants in the highest risk category.
Whereas the potential for probiotic-related infection appears low, there does seem to be some risk for sepsis associated with colonization by a strain in a given product or from contamination with a pathogen during manufacturing, the report explains.
At least one trial found that a third of infants randomly assigned to receive placebo showed evidence of the probiotic strain.
“However, it may be difficult to distinguish the change in the infant from the change in the resident flora of the NICU,” the AAP panel wrote.
In addition, there have recently been several recalls of dietary supplement–grade probiotics for contamination, which have raised concerns. Pathogens include Salmonella, Rhizopus, and Penicillium species. Fatal gastrointestinal mucormycosis has also been reported in a preterm infant who received ABC Dophilus powder that was contaminated with the microfungus Rhizopus oryzae.
Other safety considerations, according to the authors, are the unknown longer-term effect of probiotics on preterm infants and the unknown impact of microorganisms on the microbiome over time.
Last year, the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition published a position paper with a conditional recommendation for selected probiotics to reduce NEC rates. “These guidelines would be applicable in the U.S. if we had products manufactured in a way that could guarantee that what’s on the label is in the bottle,” Dr. Poindexter said.
Asked for her perspective on the AAP clinical report, Erica Wymore, MD, assistant professor of neonatal and perinatal medicine at the University of Colorado at Denver, Aurora, called it “an excellent review of the current literature” that shows the inadequacy of data on the composition, dosage, timing, duration, and use of single-strain vs. multiple-strain probiotics to reduce NEC. Her clinical center, Children’s Hospital Colorado, does not administer probiotics to preterm babies.
Although guidelines can improve outcomes, said Dr. Wymore, who was not involved in the AAP report, improvement observed with probiotics results from more stringent care in centers that experience high NEC rates. “It’s hard to know if it’s due to the probiotics if they already have a high rate of NEC,” Dr. Wymore said.
She echoed the AAP’s position and stressed the need for extreme caution in giving these products to vulnerable infants with immature immune systems. Before that can be safely done, she said, “we need more FDA oversight of product composition [and] pharmaceutical-grade products, and more studies to determine efficacy.”
Added Dr. Poindexter: “Hopefully, this report will inform clinicians of the risks of using non–pharmaceutical-grade products and encourage industry to actually develop probiotics for neonates that we can feel comfortable using.”
The report received no external funding. Dr. Poindexter and Dr. Wymore have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The American Academy of Pediatrics now recommends against the routine administration of probiotics to preterm infants, particularly the most vulnerable (those whose birth weight is <1,000 g), for the treatment or prevention of necrotizing enterocolitis (NEC) and late-onset sepsis.
Although probiotics are increasingly given to preterm infants, the AAP notes that the data on their safety and efficacy are inconsistent. In addition, the supplements are not subject to approval by the Food and Drug Administration.
Therefore, the academy advises clinicians to use extreme caution in selecting preterm neonates to receive these microorganisms and recommends obtaining informed consent from parents after carefully discussing the risks. It also recommends that centers using probiotics conduct surveillance, inasmuch as probiotics can alter a center’s flora, potentially affecting all patients. Such centers should also carefully document outcomes, adverse events, and safety.
The AAP’s clinical report, published online May 24 in Pediatrics, highlights wide differences between commercially available formulations and a lack of regulatory standards in this country.
Absent FDA-approved drug labeling, these nutritional supplements cannot be marketed as treatment or prophylaxis, but that has scarcely stopped their use. “Despite lack of availability of a pharmaceutical-grade product, the number of preterm infants receiving probiotics in the United States and Canada is steadily increasing,” wrote Brenda Poindexter, MD, FAAP, chief of neonatology at Children’s Healthcare of Atlanta, and members of the AAP’s Committee on Fetus and Newborn.
Analyses of U.S. collaborative databases indicate that approximately 10% of neonates of extremely low gestational age receive a probiotic preparation in the neonatal intensive care unit (NICU). The use of these preparations varies widely across institutions.
“NEC is a devastating morbidity of prematurity, and it’s multifactorial. Some babies only given mother’s milk still get NEC, and the decision to use these products is a very nuanced one,” Dr. Poindexter said in an interview. “I suspect some people will disagree with the report, and we tried to give folks some wiggle room.”
Evidence from other countries suggests that probiotics can be protective against NEC, she added, “so not to have a reliable product in this country is very frustrating.”
Dr. Poindexter and colleagues pointed to a 2015 study that found that only 1 of 16 commercial products tested contained the exact organisms listed on their labels. One product contained none of the species listed on the label.
In light of increasing use, the AAP emphasizes the need for development of pharmaceutical-grade probiotics that would be rigorously evaluated for safety and efficacy.
The infant microbiome
Over the past decade, the gut microbiome has been increasingly recognized as a factor relevant to health and disease in preterm infants, the authors noted. Differences in the intestinal microbiota between full-term and preterm infants are substantial. The microbiome of preterm infants tends to include fewer bacterial species and greater proportions of potentially pathogenic strains.
Evidence of benefit from probiotics has been mixed. Some studies and pooled systematic analyses suggest a significant benefit. However, Dr. Poindexter and colleagues noted that some researchers express concerns about the study methods used, such as pooling results from trials that tested different probiotic strains or that had few infants in the highest risk category.
Whereas the potential for probiotic-related infection appears low, there does seem to be some risk for sepsis associated with colonization by a strain in a given product or from contamination with a pathogen during manufacturing, the report explains.
At least one trial found that a third of infants randomly assigned to receive placebo showed evidence of the probiotic strain.
“However, it may be difficult to distinguish the change in the infant from the change in the resident flora of the NICU,” the AAP panel wrote.
In addition, there have recently been several recalls of dietary supplement–grade probiotics for contamination, which have raised concerns. Pathogens include Salmonella, Rhizopus, and Penicillium species. Fatal gastrointestinal mucormycosis has also been reported in a preterm infant who received ABC Dophilus powder that was contaminated with the microfungus Rhizopus oryzae.
Other safety considerations, according to the authors, are the unknown longer-term effect of probiotics on preterm infants and the unknown impact of microorganisms on the microbiome over time.
Last year, the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition published a position paper with a conditional recommendation for selected probiotics to reduce NEC rates. “These guidelines would be applicable in the U.S. if we had products manufactured in a way that could guarantee that what’s on the label is in the bottle,” Dr. Poindexter said.
Asked for her perspective on the AAP clinical report, Erica Wymore, MD, assistant professor of neonatal and perinatal medicine at the University of Colorado at Denver, Aurora, called it “an excellent review of the current literature” that shows the inadequacy of data on the composition, dosage, timing, duration, and use of single-strain vs. multiple-strain probiotics to reduce NEC. Her clinical center, Children’s Hospital Colorado, does not administer probiotics to preterm babies.
Although guidelines can improve outcomes, said Dr. Wymore, who was not involved in the AAP report, improvement observed with probiotics results from more stringent care in centers that experience high NEC rates. “It’s hard to know if it’s due to the probiotics if they already have a high rate of NEC,” Dr. Wymore said.
She echoed the AAP’s position and stressed the need for extreme caution in giving these products to vulnerable infants with immature immune systems. Before that can be safely done, she said, “we need more FDA oversight of product composition [and] pharmaceutical-grade products, and more studies to determine efficacy.”
Added Dr. Poindexter: “Hopefully, this report will inform clinicians of the risks of using non–pharmaceutical-grade products and encourage industry to actually develop probiotics for neonates that we can feel comfortable using.”
The report received no external funding. Dr. Poindexter and Dr. Wymore have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.