User login
Measuring the MEWS and the Rothman Index
Bedside calculation of early warning system (EWS) scores is standard practice in many hospitals to predict clinical deterioration. These systems were designed for periodic hand‐scoring, typically using a half‐dozen variables dominated by vital signs. Most derive from the Modified Early Warning Score (MEWS).[1, 2] Despite years of modification, EWSs have had only modest impact on outcomes.[3, 4] Major improvement is possible only by adding more information than is contained in vital signs. Thus, the next generation of EWSs must analyze electronic medical records (EMRs). Analysis would be performed by computer, displayed automatically, and updated whenever new data are entered into the EMR. Such systems could deliver timely, accurate, longitudinally trended acuity information that could aid in earlier detection of declining patient condition as well as improving sensitivity and specificity of EWS alarms.
Advancing this endeavor along with others,[5, 6] we previously published a patient acuity metric, the Rothman Index (RI), which automatically updates when asynchronous vital signs, laboratory test results, Braden Scale,[7] cardiac rhythm, and nursing assessments are entered into the EMR.[8] Our goal was to enable clinicians to visualize changes in acuity by simple line graphs personalized to each patient at any point in time across the trajectory of care. In our model validation studies,[8] we made no attempt to identify generalizable thresholds, though others[9] have defined decision cut points for RI in a nonemergent context. To examine decision support feasibility in an emergent context, and to compare RI with a general EWS standard, we compare the accuracy of the RI with the MEWS in predicting hospital death within 24 hours.
METHODS
Site Description and Ethics
The institutional review board of Abington Memorial Hospital (Abington, PA) approved collection of retrospective data obtained from their 665‐bed, regional referral center and teaching hospital. Handling of patient information complied with the Health Insurance Portability and Accountability Act of 1996 regulations.
Patient Inclusion
The analysis included all patients, aged 18 years or older, admitted from July 2009 through June 2010, when there were sufficient data in the EMR to compute the RI. Obstetric and psychiatric patients were excluded because nursing documentation is insufficient in this dataset.
Data Collection/Data Sources
Clinical variables were extracted from the EMR (AllScripts Sunrise Clinical Manager, Chicago, IL) by SQL query and placed into a database. RI[8] and MEWS[1] were computed according to published methods. Table 1 shows definitions of standards for each nursing assessment,[8] and Table 2 identifies all clinical variables employed for each system. Briefly, RI utilizes 26 variables related to clinical care and routinely available in the EMR. These include vital signs, laboratory results, cardiac rhythms, and nursing assessments. Excess risk associated with any value of a variable is defined as percent absolute increase in 1‐year mortality relative to minimum 1‐year mortality identified for that variable. Excess risk is summed on a linear scale to reflect cumulative risk for individual patients at any given time. RI was computed at every new observation during a patient visit, when input values were available. Laboratory results are included when measured, but after 24 hours their weighting is reduced by 50%, and after 48 hours they are excluded. Data input intervals were a function of institutional patient care protocols and physician orders. All observations during a patient's stay were included in the analysis, per the method of Prytherch et al.[4] Because data did not contain the simplified alert/voice/pain/unresponsive (A/V/P/U) score, computation of MEWS used appropriate mapping of the Glasgow Coma Scale.[10] A corresponding MEWS was calculated for each RI. The relationship between RI and MEWS is inverse. RI ranges from 91 to 100, with lower scores indicating increasing acuity. MEWS ranges from 0 to 14, with higher scores indicating increasing acuity.
| |
| Cardiac | Pulse regular, rate 60100 bpm, skin warm and dry. Blood pressure 140/90 and no symptoms of hypotension. |
| Food/nutrition | No difficulty with chewing, swallowing, or manual dexterity. Patient consuming >50% of daily diet ordered as observed or stated. |
| Gastrointestinal | Abdomen soft and nontender. Bowel sounds present. No nausea or vomiting. Continent. Bowel pattern normal as observed or stated. |
| Genitourinary | Voids without difficulty. Continent. Urine clear, yellow to amber as observed or stated. Urinary catheter patent if present. |
| Musculoskeletal | Independently able to move all extremities and perform functional activities as observed or stated (includes assistive devices). |
| Neurological | Alert and oriented to person, place, time, situation. Speech is coherent. |
| Peripheral‐vascular | Extremities are normal or pink and warm. Peripheral pulses palpable. Capillary refill 3 seconds. No edema, numbness or tingling. |
| Psychosocial | Behavior appropriate to situation. Expressed concerns and fears being addressed. Adequate support system. |
| Respiratory | Respiration 1224/minute at rest, quiet and regular. Bilateral breath sounds clear. Nail beds and mucous membranes pink. Sputum clear, if present. |
| Safety/fall risk | Safety/fall risk factors not present. Not a risk to self or others. |
| Skin/tissue | Skin clean, dry, and intact with no reddened areas. Patient is alert, cooperative and able to reposition self independently. Braden Scale >15. |
| Input Variable | A: Alive in 24 Hours, Mean (SD) | B: Dead Within 24 Hours, Mean (SD) | P Value |
|---|---|---|---|
| |||
| Diastolic blood pressure, mm Hg | 66.8 (13.5) | 56.6 (16.8) | 0.0001 |
| Systolic blood pressure, mm Hga | 127.3 (23.8) | 105.2 (29.4) | 0.0001 |
| Temperature, Fa | 98.2 (1.1) | 98.2 (2.0) | 0.1165 |
| Respiration, breaths per minutea | 20.1 (4.7) | 23.6 (9.1) | 0.0001 |
| Heart rate, bpma | 81.1 (16.5) | 96.9 (22.2) | 0.0001 |
| Pulse oximetry, % O2 saturation | 96.3 (3.3) | 93.8 (10.1) | 0.0001 |
| Creatinine, mg/dL | 1.2 (1.2) | 1.8 (1.5) | 0.0001 |
| Blood urea nitrogen, mg/dL | 23.9 (17.9) | 42.1 (26.4) | 0.0001 |
| Serum chloride, mmol/L | 104.3 (5.4) | 106.9 (9.7) | 0.0001 |
| Serum potassium, mmol/L | 4.2 (0.5) | 4.4 (0.8) | 0.0001 |
| Serum sodium, mmol/L | 139.0 (4.1) | 140.7 (8.5) | 0.0001 |
| Hemoglobin, gm/dL | 11.2 (2.1) | 10.6 (2.1) | 0.0001 |
| White blood cell count, 103 cell/L | 9.9 (6.3) | 15.0 (10.9) | 0.0001 |
| Braden Scale, total points | 17.7 (3.4) | 12.2 (3.1) | 0.0001 |
| NURSING ASSESSMENTS | A: Alive in 24 Hours and Failed Standard | B: Dead Within 24 Hours and Failed Standard | P Value |
| Neurological | 38.7% | 91.4% | 0.0001 |
| Genitourinary | 46.6% | 90.0% | 0.0001 |
| Respiratory | 55.6% | 89.0% | 0.0001 |
| Peripheral vascular | 54.1% | 86.9% | 0.0001 |
| Food | 28.3% | 80.6% | 0.0001 |
| Skin | 56.3% | 75.0% | 0.0001 |
| Gastrointestinal | 49.3% | 75.0% | 0.0001 |
| Musculoskeletal | 50.3% | 72.4% | 0.0001 |
| Cardiac | 30.4% | 59.8% | 0.0001 |
| Psychosocial | 24.6% | 40.9% | 0.0001 |
| Safety | 25.5% | 29.0% | 0.0001 |
| A/V/P/U scorea | 96.3/2.1/1.4/0.2% | 88.6/21.6/4.6/5.3% | 0.0001 |
| Sinus rhythm (absent)b | 34.9% | 53.3% | 0.0001 |
Outcome Ascertainment
In‐hospital death was determined by merging the date and time of discharge with clinical inputs from the hospital's EMR. Data points were judged to be within 24 hours of death if the timestamp of the data point collection was within 24 hours of the discharge time with expired as the discharge disposition.
Statistical Methods
Demographics and input variables from the 2 groups of observations, those who were within 24 hours of death and those who were not, were compared using a t test with a Cochran and Cox[11] approximation of the probability level of the approximate t statistic for unequal variances. Mean, standard deviation, and P values are reported. Discrimination of RI and MEWS to predict 24‐hour mortality was estimated using area under the receiver operating characteristic (ROC) curve (AUC), and null hypothesis was tested using 2. Sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), positive and negative likelihood ratios (LR+, LR) were computed. Analyses were performed with SAS 9.3 (procedures ttest, freq, logistic, nlmixed; SAS Institute, Cary, NC). Typically MEWS=4 triggers a protocol to increase level of assessment and/or care, often a transfer to the intensive care unit (ICU). We denoted the point on ROC curve where MEWS=4 and identified an RI point of similar LR and sensitivity to compare false alarm rate. Then we identified an RI point of similar LR+ for comparison of LR and sensitivity.
RESULTS
A total of 1,794,910 observations during 32,472 patient visits were included; 617 patients died (1.9%). Physiological characteristics for all input variables used by RI or MEWS are shown in Table 2, comparing observations taken within 24 hours of death to all other observations.
RI versus MEWS demonstrated superior discrimination of 24‐hour mortality (AUC was 0.93 [95% confidence interval {CI}: 0.92‐0.93] vs 0.82 [95% CI: 0.82‐0.83]; difference, 0.11 [95% CI: 0.10‐0.11]; P0.0001). ROC curves for RI and MEWS are shown in Figure 1; the MEWS is subsumed by RI across the entire range. Further, paired comparisons at points of clinical importance are presented in Table 3 for LR+, LR, sensitivity, specificity, PPV, and NPV. In the first pair of columns, MEWS=4 (typical trigger point for alarms) is matched to RI using sensitivity or LR; the corresponding point is RI=16, which generates twice the LR+ and reduces false alarms by 53%. In the second pair of columns, MEWS=4 is matched to RI using PPV or LR+; the corresponding point is RI=30, which captures 54% more of those patients who will die within 24 hours.
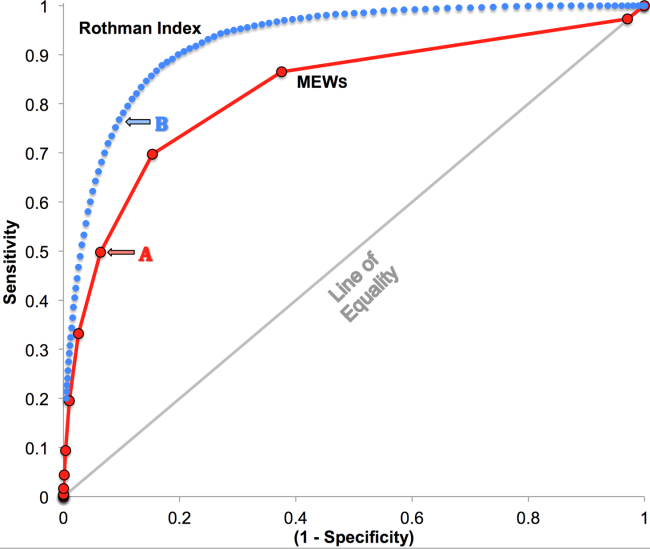
| Cut Points | MEWS=4 | RI=16a | MEWS=4 | RI=30b |
|---|---|---|---|---|
| ||||
| Likelihood ratio, positive | 7.8 | 16.9 | 7.8c | 7.9c |
| Likelihood ratio, negative | 0.54c | 0.53c | 0.54 | 0.26 |
| Sensitivity | 49.8% | 48.9% | 49.8% | 76.8% |
| Specificity | 93.6% | 97.1% | 93.6% | 90.4% |
| Positive predictive value | 5.2% | 10.6% | 5.2% | 5.3% |
| Negative predictive value | 99.6% | 99.6% | 99.6% | 99.8% |
DISCUSSION
We have shown that a general acuity metric (RI) computed using data routinely entered into an EMR outperforms MEWS in identifying hospitalized patients likely to die within 24 hours. At similar sensitivity, RI yields an LR+ more than 2‐fold greater, at a value often considered conclusive. MEWS is derived using 4 vital signs and a neurologic assessment. Such a focus on vital signs may limit responsiveness to changes in acuity, especially during early clinical deterioration. Indeed, threshold breach tools may inadvertently induce a false sense of an individual patient's condition and safety.[12] The present findings suggest the performance of RI over MEWS may be due to inclusion of nursing assessments, laboratory test results, and heart rhythm. Relative contributions of each category are: vital signs (35%), nursing assessments (34%), and laboratory test results (31%). We found in previous work that failed nursing assessments strongly correlate with mortality,[13] as illustrated in Table 2 by sharp differences between patients dying within 24 hours and those who did not.
Sensitivity to detect early deterioration, especially when not evidenced by compromised vital signs, is crucial for acuity vigilance and preemptive interventions. Others[14] have demonstrated that our approach to longitudinal modeling of the acuity continuum is well positioned to investigate clinical pathophysiology preceding adverse events and to identify actionable trends in patients at high risk of complications and sepsis after colorectal operations. Future research may reveal both clinical and administrative advantages to having this real‐time acuity measure available for all patients during the entire hospital visit, with efficacy in applications beyond use as a trigger for EWS alarms.
Study limitations include retrospective design, single‐center cohort, no exclusion of expected hospital deaths, and EMR requirement. For MEWS, the Glasgow Coma Scale was mapped to A/V/P/U, which does not appear to affect results, as our c‐statistic is identical to the literature.[4] Any hospital with an EMR collects the data necessary for computation of RI values. The RI algorithms are available in software compatible with systems from numerous EMR manufacturers (eg, Epic, Cerner, McKesson, Siemens, AllScripts, Phillips).
The advent of the EMR in hospitals marries well with an EWS that leverages from additional data more information than is contained in vital signs, permitting complex numeric computations of acuity scores, a process simply not possible with paper systems. Further, the automatic recalculation of the score reduces the burden on clinicians, and broadens potential use over a wide range, from minute‐by‐minute recalculations when attached to sensors in the ICU, to comparative metrics of hospital performance, to nonclinical financial resource applications. This new information technology is guiding methods to achieve a significant performance increment over current EWS and may assist earlier detection of deterioration, providing a chance to avoid medical crises.[15]
Acknowledgements
The authors express their appreciation to Abington Memorial Hospital. Particular thanks are extended to Steven I. Rothman, MSEM, for extensive discussions and technical support. The authors thank Alan Solinger, PhD, for his assistance in reviewing the manuscript.
Disclosures: One author (RAS) declares no conflict of interest. Two authors (GDF, MJR) are employees and shareholders in PeraHealth, Inc. of Charlotte, North Carolina, a health information technology company that offers products utilizing the Rothman Index. All of the original research defining the Rothman Index was performed prior to the formation of the company and is now published in peer‐reviewed journals. The index is freely available to all qualified researchers and is currently installed at several major medical research centers and hospital systems. This present work is under the auspices and partly funded by an independent foundation, F.A.R. Institute of Sarasota, Florida. Early research defining the Rothman Index was funded by grants from Sarasota Memorial Healthcare Foundation and the Goldsmith Fund of Greenfield Foundation. Continuing research has been funded by the F.A.R. Institute.
- , , , . Validation of a modified Early Warning Score in medical admissions. QJM Mon J Assoc Physicians. 2001;94:521–526.
- , , . Monitoring vital signs using early warning scoring systems: a review of the literature. J Nurs Manag. 2011;19:311–330.
- , , , et al. A clinical deterioration prediction tool for internal medicine patients. Am J Med Qual. 2013;28:135–142.
- , , , . ViEWS—towards a national early warning score for detecting adult inpatient deterioration. Resuscitation. 2010;81:932–937.
- , , , , , . Early detection of impending physiologic deterioration among patients who are not in intensive care: development of predictive models using data from an automated electronic medical record. J Hosp Med. 2012;7:388–395.
- , , , et al. Predicting out of intensive care unit cardiopulmonary arrest or death using electronic medical record data. BMC Med Inform Decis Mak. 2013;13:28.
- , , , The Braden Scale for predicting pressure sore risk. Nurs Res. 1987;36:205–210.
- , , . Development and validation of a continuous measure of patient condition using the electronic medical record. J Biomed Inform. 2013;46:837–848.
- , , , , . Identifying patients at increased risk for unplanned readmission. Med Care. 2013;51:761–766.
- , , . Comparison of consciousness level assessment in the poisoned patient using the alert/verbal/painful/unresponsive scale and the Glasgow Coma Scale. Ann Emerg Med. 2004;44:108–113.
- , . Experimental Design. New York, NY: John Wiley 1950.
- , . Patterns of unexpected in‐hospital deaths: a root cause analysis. Patient Saf Surg. 2011;5:3.
- , , , . Clinical implications and validity of nursing assessments: a longitudinal measure of patient condition from analysis of the Electronic Medical Record. BMJ Open. 2012;2(4):pii: e000646.
- , , , . Automated analysis of electronic medical record data reflects the pathophysiology of operative complications. Surgery. 2013;154:918–926.
- , , Not getting better means getting worse—trends in Early Warning Scores suggest that there might only be a short time span to rescue those threatening to fall off a “physiological” cliff? Resuscitation. 2013;84:409–410.
Bedside calculation of early warning system (EWS) scores is standard practice in many hospitals to predict clinical deterioration. These systems were designed for periodic hand‐scoring, typically using a half‐dozen variables dominated by vital signs. Most derive from the Modified Early Warning Score (MEWS).[1, 2] Despite years of modification, EWSs have had only modest impact on outcomes.[3, 4] Major improvement is possible only by adding more information than is contained in vital signs. Thus, the next generation of EWSs must analyze electronic medical records (EMRs). Analysis would be performed by computer, displayed automatically, and updated whenever new data are entered into the EMR. Such systems could deliver timely, accurate, longitudinally trended acuity information that could aid in earlier detection of declining patient condition as well as improving sensitivity and specificity of EWS alarms.
Advancing this endeavor along with others,[5, 6] we previously published a patient acuity metric, the Rothman Index (RI), which automatically updates when asynchronous vital signs, laboratory test results, Braden Scale,[7] cardiac rhythm, and nursing assessments are entered into the EMR.[8] Our goal was to enable clinicians to visualize changes in acuity by simple line graphs personalized to each patient at any point in time across the trajectory of care. In our model validation studies,[8] we made no attempt to identify generalizable thresholds, though others[9] have defined decision cut points for RI in a nonemergent context. To examine decision support feasibility in an emergent context, and to compare RI with a general EWS standard, we compare the accuracy of the RI with the MEWS in predicting hospital death within 24 hours.
METHODS
Site Description and Ethics
The institutional review board of Abington Memorial Hospital (Abington, PA) approved collection of retrospective data obtained from their 665‐bed, regional referral center and teaching hospital. Handling of patient information complied with the Health Insurance Portability and Accountability Act of 1996 regulations.
Patient Inclusion
The analysis included all patients, aged 18 years or older, admitted from July 2009 through June 2010, when there were sufficient data in the EMR to compute the RI. Obstetric and psychiatric patients were excluded because nursing documentation is insufficient in this dataset.
Data Collection/Data Sources
Clinical variables were extracted from the EMR (AllScripts Sunrise Clinical Manager, Chicago, IL) by SQL query and placed into a database. RI[8] and MEWS[1] were computed according to published methods. Table 1 shows definitions of standards for each nursing assessment,[8] and Table 2 identifies all clinical variables employed for each system. Briefly, RI utilizes 26 variables related to clinical care and routinely available in the EMR. These include vital signs, laboratory results, cardiac rhythms, and nursing assessments. Excess risk associated with any value of a variable is defined as percent absolute increase in 1‐year mortality relative to minimum 1‐year mortality identified for that variable. Excess risk is summed on a linear scale to reflect cumulative risk for individual patients at any given time. RI was computed at every new observation during a patient visit, when input values were available. Laboratory results are included when measured, but after 24 hours their weighting is reduced by 50%, and after 48 hours they are excluded. Data input intervals were a function of institutional patient care protocols and physician orders. All observations during a patient's stay were included in the analysis, per the method of Prytherch et al.[4] Because data did not contain the simplified alert/voice/pain/unresponsive (A/V/P/U) score, computation of MEWS used appropriate mapping of the Glasgow Coma Scale.[10] A corresponding MEWS was calculated for each RI. The relationship between RI and MEWS is inverse. RI ranges from 91 to 100, with lower scores indicating increasing acuity. MEWS ranges from 0 to 14, with higher scores indicating increasing acuity.
| |
| Cardiac | Pulse regular, rate 60100 bpm, skin warm and dry. Blood pressure 140/90 and no symptoms of hypotension. |
| Food/nutrition | No difficulty with chewing, swallowing, or manual dexterity. Patient consuming >50% of daily diet ordered as observed or stated. |
| Gastrointestinal | Abdomen soft and nontender. Bowel sounds present. No nausea or vomiting. Continent. Bowel pattern normal as observed or stated. |
| Genitourinary | Voids without difficulty. Continent. Urine clear, yellow to amber as observed or stated. Urinary catheter patent if present. |
| Musculoskeletal | Independently able to move all extremities and perform functional activities as observed or stated (includes assistive devices). |
| Neurological | Alert and oriented to person, place, time, situation. Speech is coherent. |
| Peripheral‐vascular | Extremities are normal or pink and warm. Peripheral pulses palpable. Capillary refill 3 seconds. No edema, numbness or tingling. |
| Psychosocial | Behavior appropriate to situation. Expressed concerns and fears being addressed. Adequate support system. |
| Respiratory | Respiration 1224/minute at rest, quiet and regular. Bilateral breath sounds clear. Nail beds and mucous membranes pink. Sputum clear, if present. |
| Safety/fall risk | Safety/fall risk factors not present. Not a risk to self or others. |
| Skin/tissue | Skin clean, dry, and intact with no reddened areas. Patient is alert, cooperative and able to reposition self independently. Braden Scale >15. |
| Input Variable | A: Alive in 24 Hours, Mean (SD) | B: Dead Within 24 Hours, Mean (SD) | P Value |
|---|---|---|---|
| |||
| Diastolic blood pressure, mm Hg | 66.8 (13.5) | 56.6 (16.8) | 0.0001 |
| Systolic blood pressure, mm Hga | 127.3 (23.8) | 105.2 (29.4) | 0.0001 |
| Temperature, Fa | 98.2 (1.1) | 98.2 (2.0) | 0.1165 |
| Respiration, breaths per minutea | 20.1 (4.7) | 23.6 (9.1) | 0.0001 |
| Heart rate, bpma | 81.1 (16.5) | 96.9 (22.2) | 0.0001 |
| Pulse oximetry, % O2 saturation | 96.3 (3.3) | 93.8 (10.1) | 0.0001 |
| Creatinine, mg/dL | 1.2 (1.2) | 1.8 (1.5) | 0.0001 |
| Blood urea nitrogen, mg/dL | 23.9 (17.9) | 42.1 (26.4) | 0.0001 |
| Serum chloride, mmol/L | 104.3 (5.4) | 106.9 (9.7) | 0.0001 |
| Serum potassium, mmol/L | 4.2 (0.5) | 4.4 (0.8) | 0.0001 |
| Serum sodium, mmol/L | 139.0 (4.1) | 140.7 (8.5) | 0.0001 |
| Hemoglobin, gm/dL | 11.2 (2.1) | 10.6 (2.1) | 0.0001 |
| White blood cell count, 103 cell/L | 9.9 (6.3) | 15.0 (10.9) | 0.0001 |
| Braden Scale, total points | 17.7 (3.4) | 12.2 (3.1) | 0.0001 |
| NURSING ASSESSMENTS | A: Alive in 24 Hours and Failed Standard | B: Dead Within 24 Hours and Failed Standard | P Value |
| Neurological | 38.7% | 91.4% | 0.0001 |
| Genitourinary | 46.6% | 90.0% | 0.0001 |
| Respiratory | 55.6% | 89.0% | 0.0001 |
| Peripheral vascular | 54.1% | 86.9% | 0.0001 |
| Food | 28.3% | 80.6% | 0.0001 |
| Skin | 56.3% | 75.0% | 0.0001 |
| Gastrointestinal | 49.3% | 75.0% | 0.0001 |
| Musculoskeletal | 50.3% | 72.4% | 0.0001 |
| Cardiac | 30.4% | 59.8% | 0.0001 |
| Psychosocial | 24.6% | 40.9% | 0.0001 |
| Safety | 25.5% | 29.0% | 0.0001 |
| A/V/P/U scorea | 96.3/2.1/1.4/0.2% | 88.6/21.6/4.6/5.3% | 0.0001 |
| Sinus rhythm (absent)b | 34.9% | 53.3% | 0.0001 |
Outcome Ascertainment
In‐hospital death was determined by merging the date and time of discharge with clinical inputs from the hospital's EMR. Data points were judged to be within 24 hours of death if the timestamp of the data point collection was within 24 hours of the discharge time with expired as the discharge disposition.
Statistical Methods
Demographics and input variables from the 2 groups of observations, those who were within 24 hours of death and those who were not, were compared using a t test with a Cochran and Cox[11] approximation of the probability level of the approximate t statistic for unequal variances. Mean, standard deviation, and P values are reported. Discrimination of RI and MEWS to predict 24‐hour mortality was estimated using area under the receiver operating characteristic (ROC) curve (AUC), and null hypothesis was tested using 2. Sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), positive and negative likelihood ratios (LR+, LR) were computed. Analyses were performed with SAS 9.3 (procedures ttest, freq, logistic, nlmixed; SAS Institute, Cary, NC). Typically MEWS=4 triggers a protocol to increase level of assessment and/or care, often a transfer to the intensive care unit (ICU). We denoted the point on ROC curve where MEWS=4 and identified an RI point of similar LR and sensitivity to compare false alarm rate. Then we identified an RI point of similar LR+ for comparison of LR and sensitivity.
RESULTS
A total of 1,794,910 observations during 32,472 patient visits were included; 617 patients died (1.9%). Physiological characteristics for all input variables used by RI or MEWS are shown in Table 2, comparing observations taken within 24 hours of death to all other observations.
RI versus MEWS demonstrated superior discrimination of 24‐hour mortality (AUC was 0.93 [95% confidence interval {CI}: 0.92‐0.93] vs 0.82 [95% CI: 0.82‐0.83]; difference, 0.11 [95% CI: 0.10‐0.11]; P0.0001). ROC curves for RI and MEWS are shown in Figure 1; the MEWS is subsumed by RI across the entire range. Further, paired comparisons at points of clinical importance are presented in Table 3 for LR+, LR, sensitivity, specificity, PPV, and NPV. In the first pair of columns, MEWS=4 (typical trigger point for alarms) is matched to RI using sensitivity or LR; the corresponding point is RI=16, which generates twice the LR+ and reduces false alarms by 53%. In the second pair of columns, MEWS=4 is matched to RI using PPV or LR+; the corresponding point is RI=30, which captures 54% more of those patients who will die within 24 hours.
| Cut Points | MEWS=4 | RI=16a | MEWS=4 | RI=30b |
|---|---|---|---|---|
| ||||
| Likelihood ratio, positive | 7.8 | 16.9 | 7.8c | 7.9c |
| Likelihood ratio, negative | 0.54c | 0.53c | 0.54 | 0.26 |
| Sensitivity | 49.8% | 48.9% | 49.8% | 76.8% |
| Specificity | 93.6% | 97.1% | 93.6% | 90.4% |
| Positive predictive value | 5.2% | 10.6% | 5.2% | 5.3% |
| Negative predictive value | 99.6% | 99.6% | 99.6% | 99.8% |
DISCUSSION
We have shown that a general acuity metric (RI) computed using data routinely entered into an EMR outperforms MEWS in identifying hospitalized patients likely to die within 24 hours. At similar sensitivity, RI yields an LR+ more than 2‐fold greater, at a value often considered conclusive. MEWS is derived using 4 vital signs and a neurologic assessment. Such a focus on vital signs may limit responsiveness to changes in acuity, especially during early clinical deterioration. Indeed, threshold breach tools may inadvertently induce a false sense of an individual patient's condition and safety.[12] The present findings suggest the performance of RI over MEWS may be due to inclusion of nursing assessments, laboratory test results, and heart rhythm. Relative contributions of each category are: vital signs (35%), nursing assessments (34%), and laboratory test results (31%). We found in previous work that failed nursing assessments strongly correlate with mortality,[13] as illustrated in Table 2 by sharp differences between patients dying within 24 hours and those who did not.
Sensitivity to detect early deterioration, especially when not evidenced by compromised vital signs, is crucial for acuity vigilance and preemptive interventions. Others[14] have demonstrated that our approach to longitudinal modeling of the acuity continuum is well positioned to investigate clinical pathophysiology preceding adverse events and to identify actionable trends in patients at high risk of complications and sepsis after colorectal operations. Future research may reveal both clinical and administrative advantages to having this real‐time acuity measure available for all patients during the entire hospital visit, with efficacy in applications beyond use as a trigger for EWS alarms.
Study limitations include retrospective design, single‐center cohort, no exclusion of expected hospital deaths, and EMR requirement. For MEWS, the Glasgow Coma Scale was mapped to A/V/P/U, which does not appear to affect results, as our c‐statistic is identical to the literature.[4] Any hospital with an EMR collects the data necessary for computation of RI values. The RI algorithms are available in software compatible with systems from numerous EMR manufacturers (eg, Epic, Cerner, McKesson, Siemens, AllScripts, Phillips).
The advent of the EMR in hospitals marries well with an EWS that leverages from additional data more information than is contained in vital signs, permitting complex numeric computations of acuity scores, a process simply not possible with paper systems. Further, the automatic recalculation of the score reduces the burden on clinicians, and broadens potential use over a wide range, from minute‐by‐minute recalculations when attached to sensors in the ICU, to comparative metrics of hospital performance, to nonclinical financial resource applications. This new information technology is guiding methods to achieve a significant performance increment over current EWS and may assist earlier detection of deterioration, providing a chance to avoid medical crises.[15]
Acknowledgements
The authors express their appreciation to Abington Memorial Hospital. Particular thanks are extended to Steven I. Rothman, MSEM, for extensive discussions and technical support. The authors thank Alan Solinger, PhD, for his assistance in reviewing the manuscript.
Disclosures: One author (RAS) declares no conflict of interest. Two authors (GDF, MJR) are employees and shareholders in PeraHealth, Inc. of Charlotte, North Carolina, a health information technology company that offers products utilizing the Rothman Index. All of the original research defining the Rothman Index was performed prior to the formation of the company and is now published in peer‐reviewed journals. The index is freely available to all qualified researchers and is currently installed at several major medical research centers and hospital systems. This present work is under the auspices and partly funded by an independent foundation, F.A.R. Institute of Sarasota, Florida. Early research defining the Rothman Index was funded by grants from Sarasota Memorial Healthcare Foundation and the Goldsmith Fund of Greenfield Foundation. Continuing research has been funded by the F.A.R. Institute.
Bedside calculation of early warning system (EWS) scores is standard practice in many hospitals to predict clinical deterioration. These systems were designed for periodic hand‐scoring, typically using a half‐dozen variables dominated by vital signs. Most derive from the Modified Early Warning Score (MEWS).[1, 2] Despite years of modification, EWSs have had only modest impact on outcomes.[3, 4] Major improvement is possible only by adding more information than is contained in vital signs. Thus, the next generation of EWSs must analyze electronic medical records (EMRs). Analysis would be performed by computer, displayed automatically, and updated whenever new data are entered into the EMR. Such systems could deliver timely, accurate, longitudinally trended acuity information that could aid in earlier detection of declining patient condition as well as improving sensitivity and specificity of EWS alarms.
Advancing this endeavor along with others,[5, 6] we previously published a patient acuity metric, the Rothman Index (RI), which automatically updates when asynchronous vital signs, laboratory test results, Braden Scale,[7] cardiac rhythm, and nursing assessments are entered into the EMR.[8] Our goal was to enable clinicians to visualize changes in acuity by simple line graphs personalized to each patient at any point in time across the trajectory of care. In our model validation studies,[8] we made no attempt to identify generalizable thresholds, though others[9] have defined decision cut points for RI in a nonemergent context. To examine decision support feasibility in an emergent context, and to compare RI with a general EWS standard, we compare the accuracy of the RI with the MEWS in predicting hospital death within 24 hours.
METHODS
Site Description and Ethics
The institutional review board of Abington Memorial Hospital (Abington, PA) approved collection of retrospective data obtained from their 665‐bed, regional referral center and teaching hospital. Handling of patient information complied with the Health Insurance Portability and Accountability Act of 1996 regulations.
Patient Inclusion
The analysis included all patients, aged 18 years or older, admitted from July 2009 through June 2010, when there were sufficient data in the EMR to compute the RI. Obstetric and psychiatric patients were excluded because nursing documentation is insufficient in this dataset.
Data Collection/Data Sources
Clinical variables were extracted from the EMR (AllScripts Sunrise Clinical Manager, Chicago, IL) by SQL query and placed into a database. RI[8] and MEWS[1] were computed according to published methods. Table 1 shows definitions of standards for each nursing assessment,[8] and Table 2 identifies all clinical variables employed for each system. Briefly, RI utilizes 26 variables related to clinical care and routinely available in the EMR. These include vital signs, laboratory results, cardiac rhythms, and nursing assessments. Excess risk associated with any value of a variable is defined as percent absolute increase in 1‐year mortality relative to minimum 1‐year mortality identified for that variable. Excess risk is summed on a linear scale to reflect cumulative risk for individual patients at any given time. RI was computed at every new observation during a patient visit, when input values were available. Laboratory results are included when measured, but after 24 hours their weighting is reduced by 50%, and after 48 hours they are excluded. Data input intervals were a function of institutional patient care protocols and physician orders. All observations during a patient's stay were included in the analysis, per the method of Prytherch et al.[4] Because data did not contain the simplified alert/voice/pain/unresponsive (A/V/P/U) score, computation of MEWS used appropriate mapping of the Glasgow Coma Scale.[10] A corresponding MEWS was calculated for each RI. The relationship between RI and MEWS is inverse. RI ranges from 91 to 100, with lower scores indicating increasing acuity. MEWS ranges from 0 to 14, with higher scores indicating increasing acuity.
| |
| Cardiac | Pulse regular, rate 60100 bpm, skin warm and dry. Blood pressure 140/90 and no symptoms of hypotension. |
| Food/nutrition | No difficulty with chewing, swallowing, or manual dexterity. Patient consuming >50% of daily diet ordered as observed or stated. |
| Gastrointestinal | Abdomen soft and nontender. Bowel sounds present. No nausea or vomiting. Continent. Bowel pattern normal as observed or stated. |
| Genitourinary | Voids without difficulty. Continent. Urine clear, yellow to amber as observed or stated. Urinary catheter patent if present. |
| Musculoskeletal | Independently able to move all extremities and perform functional activities as observed or stated (includes assistive devices). |
| Neurological | Alert and oriented to person, place, time, situation. Speech is coherent. |
| Peripheral‐vascular | Extremities are normal or pink and warm. Peripheral pulses palpable. Capillary refill 3 seconds. No edema, numbness or tingling. |
| Psychosocial | Behavior appropriate to situation. Expressed concerns and fears being addressed. Adequate support system. |
| Respiratory | Respiration 1224/minute at rest, quiet and regular. Bilateral breath sounds clear. Nail beds and mucous membranes pink. Sputum clear, if present. |
| Safety/fall risk | Safety/fall risk factors not present. Not a risk to self or others. |
| Skin/tissue | Skin clean, dry, and intact with no reddened areas. Patient is alert, cooperative and able to reposition self independently. Braden Scale >15. |
| Input Variable | A: Alive in 24 Hours, Mean (SD) | B: Dead Within 24 Hours, Mean (SD) | P Value |
|---|---|---|---|
| |||
| Diastolic blood pressure, mm Hg | 66.8 (13.5) | 56.6 (16.8) | 0.0001 |
| Systolic blood pressure, mm Hga | 127.3 (23.8) | 105.2 (29.4) | 0.0001 |
| Temperature, Fa | 98.2 (1.1) | 98.2 (2.0) | 0.1165 |
| Respiration, breaths per minutea | 20.1 (4.7) | 23.6 (9.1) | 0.0001 |
| Heart rate, bpma | 81.1 (16.5) | 96.9 (22.2) | 0.0001 |
| Pulse oximetry, % O2 saturation | 96.3 (3.3) | 93.8 (10.1) | 0.0001 |
| Creatinine, mg/dL | 1.2 (1.2) | 1.8 (1.5) | 0.0001 |
| Blood urea nitrogen, mg/dL | 23.9 (17.9) | 42.1 (26.4) | 0.0001 |
| Serum chloride, mmol/L | 104.3 (5.4) | 106.9 (9.7) | 0.0001 |
| Serum potassium, mmol/L | 4.2 (0.5) | 4.4 (0.8) | 0.0001 |
| Serum sodium, mmol/L | 139.0 (4.1) | 140.7 (8.5) | 0.0001 |
| Hemoglobin, gm/dL | 11.2 (2.1) | 10.6 (2.1) | 0.0001 |
| White blood cell count, 103 cell/L | 9.9 (6.3) | 15.0 (10.9) | 0.0001 |
| Braden Scale, total points | 17.7 (3.4) | 12.2 (3.1) | 0.0001 |
| NURSING ASSESSMENTS | A: Alive in 24 Hours and Failed Standard | B: Dead Within 24 Hours and Failed Standard | P Value |
| Neurological | 38.7% | 91.4% | 0.0001 |
| Genitourinary | 46.6% | 90.0% | 0.0001 |
| Respiratory | 55.6% | 89.0% | 0.0001 |
| Peripheral vascular | 54.1% | 86.9% | 0.0001 |
| Food | 28.3% | 80.6% | 0.0001 |
| Skin | 56.3% | 75.0% | 0.0001 |
| Gastrointestinal | 49.3% | 75.0% | 0.0001 |
| Musculoskeletal | 50.3% | 72.4% | 0.0001 |
| Cardiac | 30.4% | 59.8% | 0.0001 |
| Psychosocial | 24.6% | 40.9% | 0.0001 |
| Safety | 25.5% | 29.0% | 0.0001 |
| A/V/P/U scorea | 96.3/2.1/1.4/0.2% | 88.6/21.6/4.6/5.3% | 0.0001 |
| Sinus rhythm (absent)b | 34.9% | 53.3% | 0.0001 |
Outcome Ascertainment
In‐hospital death was determined by merging the date and time of discharge with clinical inputs from the hospital's EMR. Data points were judged to be within 24 hours of death if the timestamp of the data point collection was within 24 hours of the discharge time with expired as the discharge disposition.
Statistical Methods
Demographics and input variables from the 2 groups of observations, those who were within 24 hours of death and those who were not, were compared using a t test with a Cochran and Cox[11] approximation of the probability level of the approximate t statistic for unequal variances. Mean, standard deviation, and P values are reported. Discrimination of RI and MEWS to predict 24‐hour mortality was estimated using area under the receiver operating characteristic (ROC) curve (AUC), and null hypothesis was tested using 2. Sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), positive and negative likelihood ratios (LR+, LR) were computed. Analyses were performed with SAS 9.3 (procedures ttest, freq, logistic, nlmixed; SAS Institute, Cary, NC). Typically MEWS=4 triggers a protocol to increase level of assessment and/or care, often a transfer to the intensive care unit (ICU). We denoted the point on ROC curve where MEWS=4 and identified an RI point of similar LR and sensitivity to compare false alarm rate. Then we identified an RI point of similar LR+ for comparison of LR and sensitivity.
RESULTS
A total of 1,794,910 observations during 32,472 patient visits were included; 617 patients died (1.9%). Physiological characteristics for all input variables used by RI or MEWS are shown in Table 2, comparing observations taken within 24 hours of death to all other observations.
RI versus MEWS demonstrated superior discrimination of 24‐hour mortality (AUC was 0.93 [95% confidence interval {CI}: 0.92‐0.93] vs 0.82 [95% CI: 0.82‐0.83]; difference, 0.11 [95% CI: 0.10‐0.11]; P0.0001). ROC curves for RI and MEWS are shown in Figure 1; the MEWS is subsumed by RI across the entire range. Further, paired comparisons at points of clinical importance are presented in Table 3 for LR+, LR, sensitivity, specificity, PPV, and NPV. In the first pair of columns, MEWS=4 (typical trigger point for alarms) is matched to RI using sensitivity or LR; the corresponding point is RI=16, which generates twice the LR+ and reduces false alarms by 53%. In the second pair of columns, MEWS=4 is matched to RI using PPV or LR+; the corresponding point is RI=30, which captures 54% more of those patients who will die within 24 hours.
| Cut Points | MEWS=4 | RI=16a | MEWS=4 | RI=30b |
|---|---|---|---|---|
| ||||
| Likelihood ratio, positive | 7.8 | 16.9 | 7.8c | 7.9c |
| Likelihood ratio, negative | 0.54c | 0.53c | 0.54 | 0.26 |
| Sensitivity | 49.8% | 48.9% | 49.8% | 76.8% |
| Specificity | 93.6% | 97.1% | 93.6% | 90.4% |
| Positive predictive value | 5.2% | 10.6% | 5.2% | 5.3% |
| Negative predictive value | 99.6% | 99.6% | 99.6% | 99.8% |
DISCUSSION
We have shown that a general acuity metric (RI) computed using data routinely entered into an EMR outperforms MEWS in identifying hospitalized patients likely to die within 24 hours. At similar sensitivity, RI yields an LR+ more than 2‐fold greater, at a value often considered conclusive. MEWS is derived using 4 vital signs and a neurologic assessment. Such a focus on vital signs may limit responsiveness to changes in acuity, especially during early clinical deterioration. Indeed, threshold breach tools may inadvertently induce a false sense of an individual patient's condition and safety.[12] The present findings suggest the performance of RI over MEWS may be due to inclusion of nursing assessments, laboratory test results, and heart rhythm. Relative contributions of each category are: vital signs (35%), nursing assessments (34%), and laboratory test results (31%). We found in previous work that failed nursing assessments strongly correlate with mortality,[13] as illustrated in Table 2 by sharp differences between patients dying within 24 hours and those who did not.
Sensitivity to detect early deterioration, especially when not evidenced by compromised vital signs, is crucial for acuity vigilance and preemptive interventions. Others[14] have demonstrated that our approach to longitudinal modeling of the acuity continuum is well positioned to investigate clinical pathophysiology preceding adverse events and to identify actionable trends in patients at high risk of complications and sepsis after colorectal operations. Future research may reveal both clinical and administrative advantages to having this real‐time acuity measure available for all patients during the entire hospital visit, with efficacy in applications beyond use as a trigger for EWS alarms.
Study limitations include retrospective design, single‐center cohort, no exclusion of expected hospital deaths, and EMR requirement. For MEWS, the Glasgow Coma Scale was mapped to A/V/P/U, which does not appear to affect results, as our c‐statistic is identical to the literature.[4] Any hospital with an EMR collects the data necessary for computation of RI values. The RI algorithms are available in software compatible with systems from numerous EMR manufacturers (eg, Epic, Cerner, McKesson, Siemens, AllScripts, Phillips).
The advent of the EMR in hospitals marries well with an EWS that leverages from additional data more information than is contained in vital signs, permitting complex numeric computations of acuity scores, a process simply not possible with paper systems. Further, the automatic recalculation of the score reduces the burden on clinicians, and broadens potential use over a wide range, from minute‐by‐minute recalculations when attached to sensors in the ICU, to comparative metrics of hospital performance, to nonclinical financial resource applications. This new information technology is guiding methods to achieve a significant performance increment over current EWS and may assist earlier detection of deterioration, providing a chance to avoid medical crises.[15]
Acknowledgements
The authors express their appreciation to Abington Memorial Hospital. Particular thanks are extended to Steven I. Rothman, MSEM, for extensive discussions and technical support. The authors thank Alan Solinger, PhD, for his assistance in reviewing the manuscript.
Disclosures: One author (RAS) declares no conflict of interest. Two authors (GDF, MJR) are employees and shareholders in PeraHealth, Inc. of Charlotte, North Carolina, a health information technology company that offers products utilizing the Rothman Index. All of the original research defining the Rothman Index was performed prior to the formation of the company and is now published in peer‐reviewed journals. The index is freely available to all qualified researchers and is currently installed at several major medical research centers and hospital systems. This present work is under the auspices and partly funded by an independent foundation, F.A.R. Institute of Sarasota, Florida. Early research defining the Rothman Index was funded by grants from Sarasota Memorial Healthcare Foundation and the Goldsmith Fund of Greenfield Foundation. Continuing research has been funded by the F.A.R. Institute.
- , , , . Validation of a modified Early Warning Score in medical admissions. QJM Mon J Assoc Physicians. 2001;94:521–526.
- , , . Monitoring vital signs using early warning scoring systems: a review of the literature. J Nurs Manag. 2011;19:311–330.
- , , , et al. A clinical deterioration prediction tool for internal medicine patients. Am J Med Qual. 2013;28:135–142.
- , , , . ViEWS—towards a national early warning score for detecting adult inpatient deterioration. Resuscitation. 2010;81:932–937.
- , , , , , . Early detection of impending physiologic deterioration among patients who are not in intensive care: development of predictive models using data from an automated electronic medical record. J Hosp Med. 2012;7:388–395.
- , , , et al. Predicting out of intensive care unit cardiopulmonary arrest or death using electronic medical record data. BMC Med Inform Decis Mak. 2013;13:28.
- , , , The Braden Scale for predicting pressure sore risk. Nurs Res. 1987;36:205–210.
- , , . Development and validation of a continuous measure of patient condition using the electronic medical record. J Biomed Inform. 2013;46:837–848.
- , , , , . Identifying patients at increased risk for unplanned readmission. Med Care. 2013;51:761–766.
- , , . Comparison of consciousness level assessment in the poisoned patient using the alert/verbal/painful/unresponsive scale and the Glasgow Coma Scale. Ann Emerg Med. 2004;44:108–113.
- , . Experimental Design. New York, NY: John Wiley 1950.
- , . Patterns of unexpected in‐hospital deaths: a root cause analysis. Patient Saf Surg. 2011;5:3.
- , , , . Clinical implications and validity of nursing assessments: a longitudinal measure of patient condition from analysis of the Electronic Medical Record. BMJ Open. 2012;2(4):pii: e000646.
- , , , . Automated analysis of electronic medical record data reflects the pathophysiology of operative complications. Surgery. 2013;154:918–926.
- , , Not getting better means getting worse—trends in Early Warning Scores suggest that there might only be a short time span to rescue those threatening to fall off a “physiological” cliff? Resuscitation. 2013;84:409–410.
- , , , . Validation of a modified Early Warning Score in medical admissions. QJM Mon J Assoc Physicians. 2001;94:521–526.
- , , . Monitoring vital signs using early warning scoring systems: a review of the literature. J Nurs Manag. 2011;19:311–330.
- , , , et al. A clinical deterioration prediction tool for internal medicine patients. Am J Med Qual. 2013;28:135–142.
- , , , . ViEWS—towards a national early warning score for detecting adult inpatient deterioration. Resuscitation. 2010;81:932–937.
- , , , , , . Early detection of impending physiologic deterioration among patients who are not in intensive care: development of predictive models using data from an automated electronic medical record. J Hosp Med. 2012;7:388–395.
- , , , et al. Predicting out of intensive care unit cardiopulmonary arrest or death using electronic medical record data. BMC Med Inform Decis Mak. 2013;13:28.
- , , , The Braden Scale for predicting pressure sore risk. Nurs Res. 1987;36:205–210.
- , , . Development and validation of a continuous measure of patient condition using the electronic medical record. J Biomed Inform. 2013;46:837–848.
- , , , , . Identifying patients at increased risk for unplanned readmission. Med Care. 2013;51:761–766.
- , , . Comparison of consciousness level assessment in the poisoned patient using the alert/verbal/painful/unresponsive scale and the Glasgow Coma Scale. Ann Emerg Med. 2004;44:108–113.
- , . Experimental Design. New York, NY: John Wiley 1950.
- , . Patterns of unexpected in‐hospital deaths: a root cause analysis. Patient Saf Surg. 2011;5:3.
- , , , . Clinical implications and validity of nursing assessments: a longitudinal measure of patient condition from analysis of the Electronic Medical Record. BMJ Open. 2012;2(4):pii: e000646.
- , , , . Automated analysis of electronic medical record data reflects the pathophysiology of operative complications. Surgery. 2013;154:918–926.
- , , Not getting better means getting worse—trends in Early Warning Scores suggest that there might only be a short time span to rescue those threatening to fall off a “physiological” cliff? Resuscitation. 2013;84:409–410.
Centers for Medicare & Medicaid Services (CMS) Allowing Specialty Society Registries To Submit Quality Data to PQRS
Hospitalists shouldn't get too excited over the recent decision by the Centers for Medicare & Medicaid Services (CMS) that allows specialty society-run clinical data registries to submit their own quality metrics under the Physician Quality Reporting System (PQRS).
CMS earlier this month agreed to let specialist medical societies draw up their own quality measures, but to qualify, societies must have a certified clinical data registry. SHM’s Public Policy Committee (PPC) and Performance Measurement and Reporting Committee (PMRC) consistently provide feedback to CMS on the current PQRS quality measures and is reviewing the potential value of a clinical data registry for SHM members in the future.
PPC and Team Hospitalist member Joshua Lenchus, DO, RPh, FACP, SFHM, says he and other hospitalist leaders will discuss CMS' decision, but he wonders whether the reporting system's average payment adjustment for foreseeable program years and hospitalist interest is high enough to make establishing a data registry worthwhile. “The question begs,” Dr. Lenchus says, “is the benefit worth the effort?”
The 2014 Medicare physician fee schedule [PDF] reported that 26,515 medical practices with 266,521 eligible professionals participated in PQRS in 2011—or about 27% of eligible providers. SHM has encouraged its members to participate since the system's inception in 2007 to both take advantage of incentive payments that were available and to prepare for upcoming penalties for failure to report. Starting in 2015 and based on 2013 performance, there will be a penalty for not reporting PQRS quality measures.
Dr. Lenchus says PPC members will continue to monitor and advocate for quality metrics that are more in line with daily hospitalist duties. Similarly, SHM's Performance Measurement and Reporting Committee (PMRC) has been working to identify and ensure measures applicable to HM are included in PQRS.
"The committee is deeply concerned about the limited number of PQRS measures broadly applicable to hospitalists, and we are working to change this disparity," wrote Greg Seymann, MD, SFHM, chief of the division of hospital medicine at the University of California at San Diego and chair of SHM’s PMRC, and Josh Boswell, SHM’s senior manager of government relations in The Hospitalist last month.
Dr. Lenchus adds that while SHM and other societies can weigh in on the measures, CMS remains the final arbiter.
"Groups will submit whatever metrics they would like to be assessed against and those metrics will not be taken carte blanche, but rather will require CMS approval," he says.
Visit our website for more information about PQRS.
Hospitalists shouldn't get too excited over the recent decision by the Centers for Medicare & Medicaid Services (CMS) that allows specialty society-run clinical data registries to submit their own quality metrics under the Physician Quality Reporting System (PQRS).
CMS earlier this month agreed to let specialist medical societies draw up their own quality measures, but to qualify, societies must have a certified clinical data registry. SHM’s Public Policy Committee (PPC) and Performance Measurement and Reporting Committee (PMRC) consistently provide feedback to CMS on the current PQRS quality measures and is reviewing the potential value of a clinical data registry for SHM members in the future.
PPC and Team Hospitalist member Joshua Lenchus, DO, RPh, FACP, SFHM, says he and other hospitalist leaders will discuss CMS' decision, but he wonders whether the reporting system's average payment adjustment for foreseeable program years and hospitalist interest is high enough to make establishing a data registry worthwhile. “The question begs,” Dr. Lenchus says, “is the benefit worth the effort?”
The 2014 Medicare physician fee schedule [PDF] reported that 26,515 medical practices with 266,521 eligible professionals participated in PQRS in 2011—or about 27% of eligible providers. SHM has encouraged its members to participate since the system's inception in 2007 to both take advantage of incentive payments that were available and to prepare for upcoming penalties for failure to report. Starting in 2015 and based on 2013 performance, there will be a penalty for not reporting PQRS quality measures.
Dr. Lenchus says PPC members will continue to monitor and advocate for quality metrics that are more in line with daily hospitalist duties. Similarly, SHM's Performance Measurement and Reporting Committee (PMRC) has been working to identify and ensure measures applicable to HM are included in PQRS.
"The committee is deeply concerned about the limited number of PQRS measures broadly applicable to hospitalists, and we are working to change this disparity," wrote Greg Seymann, MD, SFHM, chief of the division of hospital medicine at the University of California at San Diego and chair of SHM’s PMRC, and Josh Boswell, SHM’s senior manager of government relations in The Hospitalist last month.
Dr. Lenchus adds that while SHM and other societies can weigh in on the measures, CMS remains the final arbiter.
"Groups will submit whatever metrics they would like to be assessed against and those metrics will not be taken carte blanche, but rather will require CMS approval," he says.
Visit our website for more information about PQRS.
Hospitalists shouldn't get too excited over the recent decision by the Centers for Medicare & Medicaid Services (CMS) that allows specialty society-run clinical data registries to submit their own quality metrics under the Physician Quality Reporting System (PQRS).
CMS earlier this month agreed to let specialist medical societies draw up their own quality measures, but to qualify, societies must have a certified clinical data registry. SHM’s Public Policy Committee (PPC) and Performance Measurement and Reporting Committee (PMRC) consistently provide feedback to CMS on the current PQRS quality measures and is reviewing the potential value of a clinical data registry for SHM members in the future.
PPC and Team Hospitalist member Joshua Lenchus, DO, RPh, FACP, SFHM, says he and other hospitalist leaders will discuss CMS' decision, but he wonders whether the reporting system's average payment adjustment for foreseeable program years and hospitalist interest is high enough to make establishing a data registry worthwhile. “The question begs,” Dr. Lenchus says, “is the benefit worth the effort?”
The 2014 Medicare physician fee schedule [PDF] reported that 26,515 medical practices with 266,521 eligible professionals participated in PQRS in 2011—or about 27% of eligible providers. SHM has encouraged its members to participate since the system's inception in 2007 to both take advantage of incentive payments that were available and to prepare for upcoming penalties for failure to report. Starting in 2015 and based on 2013 performance, there will be a penalty for not reporting PQRS quality measures.
Dr. Lenchus says PPC members will continue to monitor and advocate for quality metrics that are more in line with daily hospitalist duties. Similarly, SHM's Performance Measurement and Reporting Committee (PMRC) has been working to identify and ensure measures applicable to HM are included in PQRS.
"The committee is deeply concerned about the limited number of PQRS measures broadly applicable to hospitalists, and we are working to change this disparity," wrote Greg Seymann, MD, SFHM, chief of the division of hospital medicine at the University of California at San Diego and chair of SHM’s PMRC, and Josh Boswell, SHM’s senior manager of government relations in The Hospitalist last month.
Dr. Lenchus adds that while SHM and other societies can weigh in on the measures, CMS remains the final arbiter.
"Groups will submit whatever metrics they would like to be assessed against and those metrics will not be taken carte blanche, but rather will require CMS approval," he says.
Visit our website for more information about PQRS.
Infection Prevention Campaign Solicits Patient Participation
How would hospitalists feel if patients or families asked them to wash their hands when they entered the hospital room? A new campaign called "Infection Prevention and You," engages patients to help hospitals overcome one of the most persistent barriers to preventing hospital-acquired infections (HAIs)—healthcare professionals failing to practice proper hand hygiene.
Launched by the Association for Professionals in Infection Control and Epidemiology (APIC), the organization"s executives contend that everyone plays a role in infection prevention.
"We know that washing hands is important, and so many things have been tried," says Carol McLay, DrPH, MPH, RN, CIC, infection prevention consultant and chair of APIC's Communications Committee. "Patient empowerment is one of the newer approaches. Studies have shown that patients really like the idea, but often are afraid to speak up."
Dr. McLay says hand-washing advocacy is one piece of a larger campaign for preventing HAIs across settings of care.
"I would hope that physicians, including hospitalists, would view it as an opportunity to do the right thing, to serve as effective role models, and to say to their patients, 'Your health is important to me,'" she says.
"The aspiration of having anyone and everyone speak up and ask providers to apply hand hygiene is laudable," says hospitalist Ethan Cumbler, MD, FACP, who has spearheaded a multidisciplinary hand hygiene initiative at University of Colorado Hospital in Aurora. But he says it is naive to expect all providers to respond positively to being corrected in this way. "At first, we may bristle at being challenged on hand hygiene, but when we consider what kind of physicians we want to be, and what kind of culture we want to work in, I believe it is a challenge we will come to appreciate," Dr. Cumbler says.
Visit our website for more information about hospitalists and infection prevention.
How would hospitalists feel if patients or families asked them to wash their hands when they entered the hospital room? A new campaign called "Infection Prevention and You," engages patients to help hospitals overcome one of the most persistent barriers to preventing hospital-acquired infections (HAIs)—healthcare professionals failing to practice proper hand hygiene.
Launched by the Association for Professionals in Infection Control and Epidemiology (APIC), the organization"s executives contend that everyone plays a role in infection prevention.
"We know that washing hands is important, and so many things have been tried," says Carol McLay, DrPH, MPH, RN, CIC, infection prevention consultant and chair of APIC's Communications Committee. "Patient empowerment is one of the newer approaches. Studies have shown that patients really like the idea, but often are afraid to speak up."
Dr. McLay says hand-washing advocacy is one piece of a larger campaign for preventing HAIs across settings of care.
"I would hope that physicians, including hospitalists, would view it as an opportunity to do the right thing, to serve as effective role models, and to say to their patients, 'Your health is important to me,'" she says.
"The aspiration of having anyone and everyone speak up and ask providers to apply hand hygiene is laudable," says hospitalist Ethan Cumbler, MD, FACP, who has spearheaded a multidisciplinary hand hygiene initiative at University of Colorado Hospital in Aurora. But he says it is naive to expect all providers to respond positively to being corrected in this way. "At first, we may bristle at being challenged on hand hygiene, but when we consider what kind of physicians we want to be, and what kind of culture we want to work in, I believe it is a challenge we will come to appreciate," Dr. Cumbler says.
Visit our website for more information about hospitalists and infection prevention.
How would hospitalists feel if patients or families asked them to wash their hands when they entered the hospital room? A new campaign called "Infection Prevention and You," engages patients to help hospitals overcome one of the most persistent barriers to preventing hospital-acquired infections (HAIs)—healthcare professionals failing to practice proper hand hygiene.
Launched by the Association for Professionals in Infection Control and Epidemiology (APIC), the organization"s executives contend that everyone plays a role in infection prevention.
"We know that washing hands is important, and so many things have been tried," says Carol McLay, DrPH, MPH, RN, CIC, infection prevention consultant and chair of APIC's Communications Committee. "Patient empowerment is one of the newer approaches. Studies have shown that patients really like the idea, but often are afraid to speak up."
Dr. McLay says hand-washing advocacy is one piece of a larger campaign for preventing HAIs across settings of care.
"I would hope that physicians, including hospitalists, would view it as an opportunity to do the right thing, to serve as effective role models, and to say to their patients, 'Your health is important to me,'" she says.
"The aspiration of having anyone and everyone speak up and ask providers to apply hand hygiene is laudable," says hospitalist Ethan Cumbler, MD, FACP, who has spearheaded a multidisciplinary hand hygiene initiative at University of Colorado Hospital in Aurora. But he says it is naive to expect all providers to respond positively to being corrected in this way. "At first, we may bristle at being challenged on hand hygiene, but when we consider what kind of physicians we want to be, and what kind of culture we want to work in, I believe it is a challenge we will come to appreciate," Dr. Cumbler says.
Visit our website for more information about hospitalists and infection prevention.
‘JNC 8’ relaxes elderly systolic target below 150 mm Hg
The group of experts who had constituted the JNC 8 panel, a team assembled in 2008 by the National Heart, Lung, and Blood Institute to update official U.S. hypertension management guidelines, set the target blood pressure for the general population aged 60 years or older to less than 150/90 mm Hg, a major break from long-standing practice to treat such patients to a target systolic pressure of less than 140 mm Hg.
This decision, which the panel contends was driven by lack of clear evidence for extra benefit from the below–140 mm Hg target, will surely prove controversial, along with the panel’s relaxing of target blood pressures for patients with diabetes or chronic kidney disease to less than 140/90 mm Hg (increased from 130/80 mm Hg in the prior, JNC 7 guidelines). That controversy would be a fitting final curtain for the Eighth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8), a project that courted controversy by running years longer than anticipated and then generating several plot twists during the final months leading up to Dec. 18, when the former JNC 8 panel published its hypertension-management guideline (JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284427]).
The new target of a systolic pressure of less than 150 mm Hg for hypertensive patients aged 60 or older without diabetes or chronic kidney disease "is definitely controversial," said Dr. Paul A. James, cochairman of the panel and professor of family medicine at the University of Iowa in Iowa City. "There is A-level evidence that getting blood pressure below 150 mm Hg results in improved outcomes that really matter, but we have no evidence at this time to support going lower," to less than 140 mm Hg. "The good news is that the panel is comfortable that we don’t do harm," by treating patients to less than 140 mm Hg. "But why put patients at increased risk for medication adverse events when we don’t have strong evidence of benefit?" he said in an interview.
He stressed that his group released their conclusions and guideline on their own, identifying themselves as "the panel members appointed to the Eighth Joint National Committee (JNC 8)." Leaders from the National Heart, Lung, and Blood Institute announced last June that the agency was pulling out of the business of issuing cardiovascular-disease management guidelines, and would instead fund evidence reviews and partner with other organizations to issue guidelines. The NHLBI arranged for its cholesterol, obesity, and lifestyle guidelines to be released through the American Heart Association and American College of Cardiology, but no similar arrangement worked out for the JNC 8 panel, which became the former panel when the NHLBI officially dissolved it by late summer.
The former JNC 8 panel applied "a very narrow interpretation" of the clinical evidence where the evidence is very incomplete, commented Dr. Michael A. Weber, professor of medicine at State University of New York, Brooklyn. "The purpose of guidelines is for a group of experts to be guided as far as they can by the evidence, and then use their judgment and experience to make recommendations that in the best interests of patients." He cited findings from the ACCOMPLISH, INVEST, and VALUE trials that show benefits from treating patients older than 60 years to a systolic pressure of less than 140 mm Hg, though he admitted that in each of these studies the findings did not come from primary, prespecified analyses.

Dr. Weber led a panel organized by the American Society of Hypertension and International Society of Hypertension that released its own set of hypertension diagnosis and management guidelines a day earlier, on Dec. 17 (J. Clin. Hypertension 2013 [doi:10.1111/ch.1223]). Where they overlap, the guidelines from ASH/ISH and from the former JNC 8 panel are mostly the same, with the systolic target for the general population aged 60-79 years being the main area of contention, Dr. Weber said. The ASH/ISH guideline set a systolic target of less than 150 mm Hg for the general hypertensive population aged 80 years or older.
The former-JNC 8 panel also qualified their 150 mm Hg–target by adding that if general population patients aged 60 years or older are on stable, well-tolerated antihypertensive treatment and have a systolic pressure of less than 140 mm Hg, changing treatment and aiming for a higher systolic pressure is not recommended.
The target of less than 150 mm Hg for these patients also had defenders. "They made a reasonable recommendation for the elderly based on the evidence," said Dr. John M. Flack, professor and chief of medicine at Wayne State University in Detroit. But he took the JNC 8 panel to task for relaxing the systolic and diastolic pressure targets for patients with either diabetes or chronic kidney disease from the prior target of less than 130/80 mm Hg to new targets of less than 140/90 mm Hg. "Relaxing blood pressure targets in high-risk groups when so much progress has been made over the last decade is going to be very controversial," he said in an interview. The new ASH-ISH hypertension guideline also set a blood pressure target of less than 140/90 mm Hg for patients with diabetes or chronic kidney disease.
The guideline from the former JNC 8 panel "will produce a lot of discussion, and the main target will be whether the 150 mm Hg target is right or not," commented Dr. Eric D. Peterson, professor of medicine at Duke University in Durham, N.C. In an editorial that accompanied the published guideline, Dr. Peterson and his associates also noted that the hypertension goals specified in authoritative guidelines had a magnified importance these days because they often are incorporated into "performance measures" to which physicians can be often held rigidly accountable.(JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284430]).
"I chair the ACC/AHA Task Force on Performance Measures, and we will be in a bind because the current performance measures call for a blood pressure target of less than 140/90 mm Hg," he said in an interview. The ACC/AHA task force is one of the main contributors of performance measures for cardiovascular disease to the U.S. clearing house for performance measures, the National Quality Forum. "The Task Force will need to respond to this guideline in some way," he said, but the Task Force takes into account the range of current guidelines that exist and their backup evidence, so how it will decide on this issue remains uncertain.
"My concern is not so much with the number they came up with as with how it will be used by physicians in the community," Dr. Peterson said. On one hand, you don’t want physicians to get carried away and feel they need to treat all their patients to below some magical number." As he pointed out in his editorial, the counterbalancing problem is that there is always a gap between the hypertension treatment goals and what is often achieved in practice. If that relationship remains and the accepted goal for patients aged 60-79 years becomes less than 150 mm Hg, then many U.S. patients in this group may end up treated but with systolic pressures above 150 mm Hg.
Dr. James and Dr. Peterson said that they had no disclosures. Dr. Weber said that he has been a consultant to Novartis, Takeda, and Forest. Dr. Flack said that he has been a consultant to Novartis, Medtronic, and Back Beat Hypertension and received funding from Novartis and Medtronic.
On Twitter @mitchelzoler
The group of experts who had constituted the JNC 8 panel, a team assembled in 2008 by the National Heart, Lung, and Blood Institute to update official U.S. hypertension management guidelines, set the target blood pressure for the general population aged 60 years or older to less than 150/90 mm Hg, a major break from long-standing practice to treat such patients to a target systolic pressure of less than 140 mm Hg.
This decision, which the panel contends was driven by lack of clear evidence for extra benefit from the below–140 mm Hg target, will surely prove controversial, along with the panel’s relaxing of target blood pressures for patients with diabetes or chronic kidney disease to less than 140/90 mm Hg (increased from 130/80 mm Hg in the prior, JNC 7 guidelines). That controversy would be a fitting final curtain for the Eighth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8), a project that courted controversy by running years longer than anticipated and then generating several plot twists during the final months leading up to Dec. 18, when the former JNC 8 panel published its hypertension-management guideline (JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284427]).
The new target of a systolic pressure of less than 150 mm Hg for hypertensive patients aged 60 or older without diabetes or chronic kidney disease "is definitely controversial," said Dr. Paul A. James, cochairman of the panel and professor of family medicine at the University of Iowa in Iowa City. "There is A-level evidence that getting blood pressure below 150 mm Hg results in improved outcomes that really matter, but we have no evidence at this time to support going lower," to less than 140 mm Hg. "The good news is that the panel is comfortable that we don’t do harm," by treating patients to less than 140 mm Hg. "But why put patients at increased risk for medication adverse events when we don’t have strong evidence of benefit?" he said in an interview.
He stressed that his group released their conclusions and guideline on their own, identifying themselves as "the panel members appointed to the Eighth Joint National Committee (JNC 8)." Leaders from the National Heart, Lung, and Blood Institute announced last June that the agency was pulling out of the business of issuing cardiovascular-disease management guidelines, and would instead fund evidence reviews and partner with other organizations to issue guidelines. The NHLBI arranged for its cholesterol, obesity, and lifestyle guidelines to be released through the American Heart Association and American College of Cardiology, but no similar arrangement worked out for the JNC 8 panel, which became the former panel when the NHLBI officially dissolved it by late summer.
The former JNC 8 panel applied "a very narrow interpretation" of the clinical evidence where the evidence is very incomplete, commented Dr. Michael A. Weber, professor of medicine at State University of New York, Brooklyn. "The purpose of guidelines is for a group of experts to be guided as far as they can by the evidence, and then use their judgment and experience to make recommendations that in the best interests of patients." He cited findings from the ACCOMPLISH, INVEST, and VALUE trials that show benefits from treating patients older than 60 years to a systolic pressure of less than 140 mm Hg, though he admitted that in each of these studies the findings did not come from primary, prespecified analyses.
Dr. Weber led a panel organized by the American Society of Hypertension and International Society of Hypertension that released its own set of hypertension diagnosis and management guidelines a day earlier, on Dec. 17 (J. Clin. Hypertension 2013 [doi:10.1111/ch.1223]). Where they overlap, the guidelines from ASH/ISH and from the former JNC 8 panel are mostly the same, with the systolic target for the general population aged 60-79 years being the main area of contention, Dr. Weber said. The ASH/ISH guideline set a systolic target of less than 150 mm Hg for the general hypertensive population aged 80 years or older.
The former-JNC 8 panel also qualified their 150 mm Hg–target by adding that if general population patients aged 60 years or older are on stable, well-tolerated antihypertensive treatment and have a systolic pressure of less than 140 mm Hg, changing treatment and aiming for a higher systolic pressure is not recommended.
The target of less than 150 mm Hg for these patients also had defenders. "They made a reasonable recommendation for the elderly based on the evidence," said Dr. John M. Flack, professor and chief of medicine at Wayne State University in Detroit. But he took the JNC 8 panel to task for relaxing the systolic and diastolic pressure targets for patients with either diabetes or chronic kidney disease from the prior target of less than 130/80 mm Hg to new targets of less than 140/90 mm Hg. "Relaxing blood pressure targets in high-risk groups when so much progress has been made over the last decade is going to be very controversial," he said in an interview. The new ASH-ISH hypertension guideline also set a blood pressure target of less than 140/90 mm Hg for patients with diabetes or chronic kidney disease.
The guideline from the former JNC 8 panel "will produce a lot of discussion, and the main target will be whether the 150 mm Hg target is right or not," commented Dr. Eric D. Peterson, professor of medicine at Duke University in Durham, N.C. In an editorial that accompanied the published guideline, Dr. Peterson and his associates also noted that the hypertension goals specified in authoritative guidelines had a magnified importance these days because they often are incorporated into "performance measures" to which physicians can be often held rigidly accountable.(JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284430]).
"I chair the ACC/AHA Task Force on Performance Measures, and we will be in a bind because the current performance measures call for a blood pressure target of less than 140/90 mm Hg," he said in an interview. The ACC/AHA task force is one of the main contributors of performance measures for cardiovascular disease to the U.S. clearing house for performance measures, the National Quality Forum. "The Task Force will need to respond to this guideline in some way," he said, but the Task Force takes into account the range of current guidelines that exist and their backup evidence, so how it will decide on this issue remains uncertain.
"My concern is not so much with the number they came up with as with how it will be used by physicians in the community," Dr. Peterson said. On one hand, you don’t want physicians to get carried away and feel they need to treat all their patients to below some magical number." As he pointed out in his editorial, the counterbalancing problem is that there is always a gap between the hypertension treatment goals and what is often achieved in practice. If that relationship remains and the accepted goal for patients aged 60-79 years becomes less than 150 mm Hg, then many U.S. patients in this group may end up treated but with systolic pressures above 150 mm Hg.
Dr. James and Dr. Peterson said that they had no disclosures. Dr. Weber said that he has been a consultant to Novartis, Takeda, and Forest. Dr. Flack said that he has been a consultant to Novartis, Medtronic, and Back Beat Hypertension and received funding from Novartis and Medtronic.
On Twitter @mitchelzoler
The group of experts who had constituted the JNC 8 panel, a team assembled in 2008 by the National Heart, Lung, and Blood Institute to update official U.S. hypertension management guidelines, set the target blood pressure for the general population aged 60 years or older to less than 150/90 mm Hg, a major break from long-standing practice to treat such patients to a target systolic pressure of less than 140 mm Hg.
This decision, which the panel contends was driven by lack of clear evidence for extra benefit from the below–140 mm Hg target, will surely prove controversial, along with the panel’s relaxing of target blood pressures for patients with diabetes or chronic kidney disease to less than 140/90 mm Hg (increased from 130/80 mm Hg in the prior, JNC 7 guidelines). That controversy would be a fitting final curtain for the Eighth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8), a project that courted controversy by running years longer than anticipated and then generating several plot twists during the final months leading up to Dec. 18, when the former JNC 8 panel published its hypertension-management guideline (JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284427]).
The new target of a systolic pressure of less than 150 mm Hg for hypertensive patients aged 60 or older without diabetes or chronic kidney disease "is definitely controversial," said Dr. Paul A. James, cochairman of the panel and professor of family medicine at the University of Iowa in Iowa City. "There is A-level evidence that getting blood pressure below 150 mm Hg results in improved outcomes that really matter, but we have no evidence at this time to support going lower," to less than 140 mm Hg. "The good news is that the panel is comfortable that we don’t do harm," by treating patients to less than 140 mm Hg. "But why put patients at increased risk for medication adverse events when we don’t have strong evidence of benefit?" he said in an interview.
He stressed that his group released their conclusions and guideline on their own, identifying themselves as "the panel members appointed to the Eighth Joint National Committee (JNC 8)." Leaders from the National Heart, Lung, and Blood Institute announced last June that the agency was pulling out of the business of issuing cardiovascular-disease management guidelines, and would instead fund evidence reviews and partner with other organizations to issue guidelines. The NHLBI arranged for its cholesterol, obesity, and lifestyle guidelines to be released through the American Heart Association and American College of Cardiology, but no similar arrangement worked out for the JNC 8 panel, which became the former panel when the NHLBI officially dissolved it by late summer.
The former JNC 8 panel applied "a very narrow interpretation" of the clinical evidence where the evidence is very incomplete, commented Dr. Michael A. Weber, professor of medicine at State University of New York, Brooklyn. "The purpose of guidelines is for a group of experts to be guided as far as they can by the evidence, and then use their judgment and experience to make recommendations that in the best interests of patients." He cited findings from the ACCOMPLISH, INVEST, and VALUE trials that show benefits from treating patients older than 60 years to a systolic pressure of less than 140 mm Hg, though he admitted that in each of these studies the findings did not come from primary, prespecified analyses.
Dr. Weber led a panel organized by the American Society of Hypertension and International Society of Hypertension that released its own set of hypertension diagnosis and management guidelines a day earlier, on Dec. 17 (J. Clin. Hypertension 2013 [doi:10.1111/ch.1223]). Where they overlap, the guidelines from ASH/ISH and from the former JNC 8 panel are mostly the same, with the systolic target for the general population aged 60-79 years being the main area of contention, Dr. Weber said. The ASH/ISH guideline set a systolic target of less than 150 mm Hg for the general hypertensive population aged 80 years or older.
The former-JNC 8 panel also qualified their 150 mm Hg–target by adding that if general population patients aged 60 years or older are on stable, well-tolerated antihypertensive treatment and have a systolic pressure of less than 140 mm Hg, changing treatment and aiming for a higher systolic pressure is not recommended.
The target of less than 150 mm Hg for these patients also had defenders. "They made a reasonable recommendation for the elderly based on the evidence," said Dr. John M. Flack, professor and chief of medicine at Wayne State University in Detroit. But he took the JNC 8 panel to task for relaxing the systolic and diastolic pressure targets for patients with either diabetes or chronic kidney disease from the prior target of less than 130/80 mm Hg to new targets of less than 140/90 mm Hg. "Relaxing blood pressure targets in high-risk groups when so much progress has been made over the last decade is going to be very controversial," he said in an interview. The new ASH-ISH hypertension guideline also set a blood pressure target of less than 140/90 mm Hg for patients with diabetes or chronic kidney disease.
The guideline from the former JNC 8 panel "will produce a lot of discussion, and the main target will be whether the 150 mm Hg target is right or not," commented Dr. Eric D. Peterson, professor of medicine at Duke University in Durham, N.C. In an editorial that accompanied the published guideline, Dr. Peterson and his associates also noted that the hypertension goals specified in authoritative guidelines had a magnified importance these days because they often are incorporated into "performance measures" to which physicians can be often held rigidly accountable.(JAMA 2013 Dec. 18 [doi:10.1001/jama.2013.284430]).
"I chair the ACC/AHA Task Force on Performance Measures, and we will be in a bind because the current performance measures call for a blood pressure target of less than 140/90 mm Hg," he said in an interview. The ACC/AHA task force is one of the main contributors of performance measures for cardiovascular disease to the U.S. clearing house for performance measures, the National Quality Forum. "The Task Force will need to respond to this guideline in some way," he said, but the Task Force takes into account the range of current guidelines that exist and their backup evidence, so how it will decide on this issue remains uncertain.
"My concern is not so much with the number they came up with as with how it will be used by physicians in the community," Dr. Peterson said. On one hand, you don’t want physicians to get carried away and feel they need to treat all their patients to below some magical number." As he pointed out in his editorial, the counterbalancing problem is that there is always a gap between the hypertension treatment goals and what is often achieved in practice. If that relationship remains and the accepted goal for patients aged 60-79 years becomes less than 150 mm Hg, then many U.S. patients in this group may end up treated but with systolic pressures above 150 mm Hg.
Dr. James and Dr. Peterson said that they had no disclosures. Dr. Weber said that he has been a consultant to Novartis, Takeda, and Forest. Dr. Flack said that he has been a consultant to Novartis, Medtronic, and Back Beat Hypertension and received funding from Novartis and Medtronic.
On Twitter @mitchelzoler
FROM JAMA

Locked Knee Caused by Lateral Meniscal Capsular Disruption: Verification by Magnetic Resonance Imaging and Arthroscopy
Endovascular coiling aids pelvic congestion syndrome
CHICAGO – Endovascular coiling should be offered to women with pelvic congestion syndrome as an effective treatment.
"The technical success rate is high, pain scores were significantly improved, and most importantly, the patient satisfaction with resolution of their symptoms is very high," Dr. Axel Thors said at the annual meeting of the Midwestern Vascular Surgical Society.

He reported on a 4-year review involving 15 women with pelvic congestion syndrome (PCS) who underwent endovenous coil embolization (n = 14) or stenting of the iliac vein (n = 1).
The diagnosis of PCS was made clinically by the presence of chronic pelvic pain for 6 months or more, sensations of pelvic fullness, dyspareunia, or perineal varicosities. There was no evidence of nutcracker syndrome or perirenal varicosities. Other pathologies had been previously ruled out.
"By the time these women got to us, we were probably the last provider they had seen and they had all undergone extensive evaluation for their pelvic pain, all the way from their primary providers to the ob.gyns.," said Dr. Thors of Ohio State University, Columbus.
Their average age was 36 years. Fourteen patients had a previous pregnancy, with an average parity of two.
Twelve patients presented with symptomatic vulvar varices and three with imaging or laproscopic findings of tubo-ovarian varices. All had complaints of chronic pelvic pain.
"Lower extremity venous insufficiency was closely associated with the incidence [of PCS], as was chronic dyspareunia," Dr. Thors said.
Gonadal vein venograms were performed during normal breath and the Valsalva maneuver. Embolization was performed if there was gonadal vein incompetence, congestion of the ovarian venous plexus, uterine venous congestion, cross-pelvic congestion, or marked enlargement of gonadal veins (minimum 6 mm). The average venality size was 7.3 mm.
In all, 13 gonadal veins were embolized with an average of three coils, ranging in size from 6 mm to 12 mm, Dr. Thors said.
Four gonadal veins were occluded using an Amplatzer plug (range 12-18 mm). One iliac vein was stented with a 16 mm by 60 mm stent.
Lower-extremity venous insufficiency was treated with ablation and subsequently followed clinically, he said.
Pain scores on a 10-point visual analog scale declined significantly from baseline for eight evaluable patients for pelvic pain (9.3 vs. 1.8), dyspareunia (8.875 vs. 1.5), painful vulvar varices (9.2 vs. 1.2), and lower extremity venous insufficiency (7 vs. 1), he said.
Two patients had recurrence, and their baseline pain score of 1.2 increased to 4.0 after a mean of 21 months.
All eight patients reported that they were "satisfied" or "very satisfied" with their procedure.
"Patients with chronic pelvic pain, vulvar varices, multiparity, and lower extremity venous insufficiency should be offered endovascular evaluation and treatment," Dr. Thors concluded.
Audience members said that the study represents an important concept in the management of these patients. It is a validation of a very old treatment that sometimes is not offered because of a lack of knowledge or perceived lack of data. A 2012 Agency for Healthcare Research and Quality review estimated that outpatient management of chronic pelvic pain cost $1.2 billion annually. The AHRQ review of 36 studies concluded that there is insufficient evidence to demonstrate the effectiveness of surgical approaches for chronic pelvic pain.
Dr. Thors and his coauthors reported having no financial disclosures.
Pelvic venous congestion is misunderstood and frequently overlooked. Unfortunately pelvic pain is multifactorial. Even with significant reflux findings and encouraging results these patients, much like patients with other areas of venous insufficiency, frequently recur if followed longitudinally. Good markers to predict who will benefit from intervention and which interventions should be undertaken do not exist. This is an area that needs further study and development of standard outcome measures that can be followed sequentially.
Dr. Joann M. Lohr is associate program director, Good Samaritan Hospital Vascular Surgery Program She is also an associate medical editor for Vascular Specialist.
Pelvic venous congestion is misunderstood and frequently overlooked. Unfortunately pelvic pain is multifactorial. Even with significant reflux findings and encouraging results these patients, much like patients with other areas of venous insufficiency, frequently recur if followed longitudinally. Good markers to predict who will benefit from intervention and which interventions should be undertaken do not exist. This is an area that needs further study and development of standard outcome measures that can be followed sequentially.
Dr. Joann M. Lohr is associate program director, Good Samaritan Hospital Vascular Surgery Program She is also an associate medical editor for Vascular Specialist.
Pelvic venous congestion is misunderstood and frequently overlooked. Unfortunately pelvic pain is multifactorial. Even with significant reflux findings and encouraging results these patients, much like patients with other areas of venous insufficiency, frequently recur if followed longitudinally. Good markers to predict who will benefit from intervention and which interventions should be undertaken do not exist. This is an area that needs further study and development of standard outcome measures that can be followed sequentially.
Dr. Joann M. Lohr is associate program director, Good Samaritan Hospital Vascular Surgery Program She is also an associate medical editor for Vascular Specialist.
CHICAGO – Endovascular coiling should be offered to women with pelvic congestion syndrome as an effective treatment.
"The technical success rate is high, pain scores were significantly improved, and most importantly, the patient satisfaction with resolution of their symptoms is very high," Dr. Axel Thors said at the annual meeting of the Midwestern Vascular Surgical Society.
He reported on a 4-year review involving 15 women with pelvic congestion syndrome (PCS) who underwent endovenous coil embolization (n = 14) or stenting of the iliac vein (n = 1).
The diagnosis of PCS was made clinically by the presence of chronic pelvic pain for 6 months or more, sensations of pelvic fullness, dyspareunia, or perineal varicosities. There was no evidence of nutcracker syndrome or perirenal varicosities. Other pathologies had been previously ruled out.
"By the time these women got to us, we were probably the last provider they had seen and they had all undergone extensive evaluation for their pelvic pain, all the way from their primary providers to the ob.gyns.," said Dr. Thors of Ohio State University, Columbus.
Their average age was 36 years. Fourteen patients had a previous pregnancy, with an average parity of two.
Twelve patients presented with symptomatic vulvar varices and three with imaging or laproscopic findings of tubo-ovarian varices. All had complaints of chronic pelvic pain.
"Lower extremity venous insufficiency was closely associated with the incidence [of PCS], as was chronic dyspareunia," Dr. Thors said.
Gonadal vein venograms were performed during normal breath and the Valsalva maneuver. Embolization was performed if there was gonadal vein incompetence, congestion of the ovarian venous plexus, uterine venous congestion, cross-pelvic congestion, or marked enlargement of gonadal veins (minimum 6 mm). The average venality size was 7.3 mm.
In all, 13 gonadal veins were embolized with an average of three coils, ranging in size from 6 mm to 12 mm, Dr. Thors said.
Four gonadal veins were occluded using an Amplatzer plug (range 12-18 mm). One iliac vein was stented with a 16 mm by 60 mm stent.
Lower-extremity venous insufficiency was treated with ablation and subsequently followed clinically, he said.
Pain scores on a 10-point visual analog scale declined significantly from baseline for eight evaluable patients for pelvic pain (9.3 vs. 1.8), dyspareunia (8.875 vs. 1.5), painful vulvar varices (9.2 vs. 1.2), and lower extremity venous insufficiency (7 vs. 1), he said.
Two patients had recurrence, and their baseline pain score of 1.2 increased to 4.0 after a mean of 21 months.
All eight patients reported that they were "satisfied" or "very satisfied" with their procedure.
"Patients with chronic pelvic pain, vulvar varices, multiparity, and lower extremity venous insufficiency should be offered endovascular evaluation and treatment," Dr. Thors concluded.
Audience members said that the study represents an important concept in the management of these patients. It is a validation of a very old treatment that sometimes is not offered because of a lack of knowledge or perceived lack of data. A 2012 Agency for Healthcare Research and Quality review estimated that outpatient management of chronic pelvic pain cost $1.2 billion annually. The AHRQ review of 36 studies concluded that there is insufficient evidence to demonstrate the effectiveness of surgical approaches for chronic pelvic pain.
Dr. Thors and his coauthors reported having no financial disclosures.
CHICAGO – Endovascular coiling should be offered to women with pelvic congestion syndrome as an effective treatment.
"The technical success rate is high, pain scores were significantly improved, and most importantly, the patient satisfaction with resolution of their symptoms is very high," Dr. Axel Thors said at the annual meeting of the Midwestern Vascular Surgical Society.
He reported on a 4-year review involving 15 women with pelvic congestion syndrome (PCS) who underwent endovenous coil embolization (n = 14) or stenting of the iliac vein (n = 1).
The diagnosis of PCS was made clinically by the presence of chronic pelvic pain for 6 months or more, sensations of pelvic fullness, dyspareunia, or perineal varicosities. There was no evidence of nutcracker syndrome or perirenal varicosities. Other pathologies had been previously ruled out.
"By the time these women got to us, we were probably the last provider they had seen and they had all undergone extensive evaluation for their pelvic pain, all the way from their primary providers to the ob.gyns.," said Dr. Thors of Ohio State University, Columbus.
Their average age was 36 years. Fourteen patients had a previous pregnancy, with an average parity of two.
Twelve patients presented with symptomatic vulvar varices and three with imaging or laproscopic findings of tubo-ovarian varices. All had complaints of chronic pelvic pain.
"Lower extremity venous insufficiency was closely associated with the incidence [of PCS], as was chronic dyspareunia," Dr. Thors said.
Gonadal vein venograms were performed during normal breath and the Valsalva maneuver. Embolization was performed if there was gonadal vein incompetence, congestion of the ovarian venous plexus, uterine venous congestion, cross-pelvic congestion, or marked enlargement of gonadal veins (minimum 6 mm). The average venality size was 7.3 mm.
In all, 13 gonadal veins were embolized with an average of three coils, ranging in size from 6 mm to 12 mm, Dr. Thors said.
Four gonadal veins were occluded using an Amplatzer plug (range 12-18 mm). One iliac vein was stented with a 16 mm by 60 mm stent.
Lower-extremity venous insufficiency was treated with ablation and subsequently followed clinically, he said.
Pain scores on a 10-point visual analog scale declined significantly from baseline for eight evaluable patients for pelvic pain (9.3 vs. 1.8), dyspareunia (8.875 vs. 1.5), painful vulvar varices (9.2 vs. 1.2), and lower extremity venous insufficiency (7 vs. 1), he said.
Two patients had recurrence, and their baseline pain score of 1.2 increased to 4.0 after a mean of 21 months.
All eight patients reported that they were "satisfied" or "very satisfied" with their procedure.
"Patients with chronic pelvic pain, vulvar varices, multiparity, and lower extremity venous insufficiency should be offered endovascular evaluation and treatment," Dr. Thors concluded.
Audience members said that the study represents an important concept in the management of these patients. It is a validation of a very old treatment that sometimes is not offered because of a lack of knowledge or perceived lack of data. A 2012 Agency for Healthcare Research and Quality review estimated that outpatient management of chronic pelvic pain cost $1.2 billion annually. The AHRQ review of 36 studies concluded that there is insufficient evidence to demonstrate the effectiveness of surgical approaches for chronic pelvic pain.
Dr. Thors and his coauthors reported having no financial disclosures.
AT MIDWESTERN VASCULAR 2013
Major finding: Key numerical finding (e.g., number needed to treat to prevent one death/event; number lived or died as result of intervention). Maximum 10 words/1 sentence.
Data source: Review of 15 women treated for pelvic congestion syndrome.
Disclosures: Dr. Thors and his coauthors reported having no financial disclosures.

Consider small-fiber neuropathies in systemic lupus erythematosus
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
FROM ARTHRITIS AND RHEUMATISM
Major finding: A total of 14 patients, or 17% of 82 with peripheral neuropathies, had biopsy-proven small-fiber neuropathies and often presented with unorthodox patterns of pain.
Data source: A retrospective analysis of data regarding 2,097 consecutive patients with SLE registered in the Johns Hopkins Lupus Cohort during a 25-year period, including 82 who developed peripheral neuropathies related to the disease.
Disclosures: This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Electronic Consult Experience: Making Health Care More Accessible and Convenient for Veterans
Focusing on statins
I have been thinking about the recent cholesterol management guidelines offered by the American Heart Association and American College of Cardiology experts (J. Am. Coll. Cardiol. 2013;doi:10.1016/j.jacc.2013.11.002) and how they affect my approach to my patients. I am quick to agree to the first three points and the end of LDL targeted therapy in the guidelines, which focus now on the intensity of statins therapy in patients who have already expressed the complications of atherosclerotic cardiovascular disease (ASCVD).
However, I do question a cardiovascular prevention program that, for low-risk individuals with an LDL cholesterol level above 190 mg/dL, is largely driven by statin therapy based on a risk prediction model using age, sex, hypertension, smoking, HDL, and LDL cholesterol elevation. Of all risk factors, smoking and LDL are the only ones that we can modify. Although we have made a major attack on smoking, it would seem that the key to survival is that all of us should take a statin.
There is an abundant source of data on the benefit of statin therapy in patients who have already expressed ASCVD. Although data are limited in regard to very-low-risk groups without evidence of ASCVD, a meta-analysis by the Cholesterol Treatment Trialist Collaborators indicates that the lowering of LDL cholesterol by 40 mg/dL results in an approximate 12% decrease in vascular mortality and 20% decrease in cardiac deaths, regardless of regardless of risk category (Lancet 2012;380;581-90). This benefit was observed even in low-risk individuals despite the slight excess risk of hemorrhagic strokes and diabetes.
The prediction model appears to be the major point of controversy. Along with thousands other Americans, I went to the AHA website to see what my risk score was. I found that by modifying a few factors I could move from less than a 7.5% risk of a stroke or a heart attack in the next 10 years to a risk of well over that. I was not reassured that I was in the company of more than 45 million fellow Americans. Critics of the risk model suggest that based on a number of epidemiologic surveys, the risk model may double the number of individuals to whom the prevention guidelines apply (Lancet 2013;382:1762-5). If we expand the population so broadly, are we going to be a society of statin pill poppers?
Our attempts in the last half-century to develop prevention therapy for hypertension and diabetes have only been marginally successful. The cardiorenal scourge of hypertension remains, despite a plethora of effective drugs that have had little effect on chronic renal disease. Although therapy for diabetes has been supremely effective in treating the acute and chronic metabolic aspects of diabetes, insulin therapy has not been successful in preventing the long-term expression of the cardiovascular, ophthalmic, and renal events. And now we are trying to assess the role of statins for the prevention of cardiovascular events.
In comparison to hypertension and diabetes, statin therapy has the potential to be a sea change in the prevention of ASCVD by lowering serum cholesterol and thereby limiting the growth of the atherosclerotic plaque. A number of clinical trials support the cardiovascular benefit of statin therapy and its effect on lowering serum cholesterol. Although it is clear that we need to reflect on the reliability of the current risk factor model, the current guidelines are an important step forward in the integration of statin therapy into the prevention of cardiovascular disease.
However, talking to patients and telling them that they have greater than a 7.5% risk of having a stroke or a heart attack in the next 10 years remains an abstract concept. The guideline committee now urges me to sit down with my patients and have a heart-to-heart talk about risk and how to decrease it by changing their dangerous lifestyles rather than taking statins for the rest of their lives. When it comes down to it, lifestyle change loses and statins win.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
I have been thinking about the recent cholesterol management guidelines offered by the American Heart Association and American College of Cardiology experts (J. Am. Coll. Cardiol. 2013;doi:10.1016/j.jacc.2013.11.002) and how they affect my approach to my patients. I am quick to agree to the first three points and the end of LDL targeted therapy in the guidelines, which focus now on the intensity of statins therapy in patients who have already expressed the complications of atherosclerotic cardiovascular disease (ASCVD).
However, I do question a cardiovascular prevention program that, for low-risk individuals with an LDL cholesterol level above 190 mg/dL, is largely driven by statin therapy based on a risk prediction model using age, sex, hypertension, smoking, HDL, and LDL cholesterol elevation. Of all risk factors, smoking and LDL are the only ones that we can modify. Although we have made a major attack on smoking, it would seem that the key to survival is that all of us should take a statin.
There is an abundant source of data on the benefit of statin therapy in patients who have already expressed ASCVD. Although data are limited in regard to very-low-risk groups without evidence of ASCVD, a meta-analysis by the Cholesterol Treatment Trialist Collaborators indicates that the lowering of LDL cholesterol by 40 mg/dL results in an approximate 12% decrease in vascular mortality and 20% decrease in cardiac deaths, regardless of regardless of risk category (Lancet 2012;380;581-90). This benefit was observed even in low-risk individuals despite the slight excess risk of hemorrhagic strokes and diabetes.
The prediction model appears to be the major point of controversy. Along with thousands other Americans, I went to the AHA website to see what my risk score was. I found that by modifying a few factors I could move from less than a 7.5% risk of a stroke or a heart attack in the next 10 years to a risk of well over that. I was not reassured that I was in the company of more than 45 million fellow Americans. Critics of the risk model suggest that based on a number of epidemiologic surveys, the risk model may double the number of individuals to whom the prevention guidelines apply (Lancet 2013;382:1762-5). If we expand the population so broadly, are we going to be a society of statin pill poppers?
Our attempts in the last half-century to develop prevention therapy for hypertension and diabetes have only been marginally successful. The cardiorenal scourge of hypertension remains, despite a plethora of effective drugs that have had little effect on chronic renal disease. Although therapy for diabetes has been supremely effective in treating the acute and chronic metabolic aspects of diabetes, insulin therapy has not been successful in preventing the long-term expression of the cardiovascular, ophthalmic, and renal events. And now we are trying to assess the role of statins for the prevention of cardiovascular events.
In comparison to hypertension and diabetes, statin therapy has the potential to be a sea change in the prevention of ASCVD by lowering serum cholesterol and thereby limiting the growth of the atherosclerotic plaque. A number of clinical trials support the cardiovascular benefit of statin therapy and its effect on lowering serum cholesterol. Although it is clear that we need to reflect on the reliability of the current risk factor model, the current guidelines are an important step forward in the integration of statin therapy into the prevention of cardiovascular disease.
However, talking to patients and telling them that they have greater than a 7.5% risk of having a stroke or a heart attack in the next 10 years remains an abstract concept. The guideline committee now urges me to sit down with my patients and have a heart-to-heart talk about risk and how to decrease it by changing their dangerous lifestyles rather than taking statins for the rest of their lives. When it comes down to it, lifestyle change loses and statins win.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
I have been thinking about the recent cholesterol management guidelines offered by the American Heart Association and American College of Cardiology experts (J. Am. Coll. Cardiol. 2013;doi:10.1016/j.jacc.2013.11.002) and how they affect my approach to my patients. I am quick to agree to the first three points and the end of LDL targeted therapy in the guidelines, which focus now on the intensity of statins therapy in patients who have already expressed the complications of atherosclerotic cardiovascular disease (ASCVD).
However, I do question a cardiovascular prevention program that, for low-risk individuals with an LDL cholesterol level above 190 mg/dL, is largely driven by statin therapy based on a risk prediction model using age, sex, hypertension, smoking, HDL, and LDL cholesterol elevation. Of all risk factors, smoking and LDL are the only ones that we can modify. Although we have made a major attack on smoking, it would seem that the key to survival is that all of us should take a statin.
There is an abundant source of data on the benefit of statin therapy in patients who have already expressed ASCVD. Although data are limited in regard to very-low-risk groups without evidence of ASCVD, a meta-analysis by the Cholesterol Treatment Trialist Collaborators indicates that the lowering of LDL cholesterol by 40 mg/dL results in an approximate 12% decrease in vascular mortality and 20% decrease in cardiac deaths, regardless of regardless of risk category (Lancet 2012;380;581-90). This benefit was observed even in low-risk individuals despite the slight excess risk of hemorrhagic strokes and diabetes.
The prediction model appears to be the major point of controversy. Along with thousands other Americans, I went to the AHA website to see what my risk score was. I found that by modifying a few factors I could move from less than a 7.5% risk of a stroke or a heart attack in the next 10 years to a risk of well over that. I was not reassured that I was in the company of more than 45 million fellow Americans. Critics of the risk model suggest that based on a number of epidemiologic surveys, the risk model may double the number of individuals to whom the prevention guidelines apply (Lancet 2013;382:1762-5). If we expand the population so broadly, are we going to be a society of statin pill poppers?
Our attempts in the last half-century to develop prevention therapy for hypertension and diabetes have only been marginally successful. The cardiorenal scourge of hypertension remains, despite a plethora of effective drugs that have had little effect on chronic renal disease. Although therapy for diabetes has been supremely effective in treating the acute and chronic metabolic aspects of diabetes, insulin therapy has not been successful in preventing the long-term expression of the cardiovascular, ophthalmic, and renal events. And now we are trying to assess the role of statins for the prevention of cardiovascular events.
In comparison to hypertension and diabetes, statin therapy has the potential to be a sea change in the prevention of ASCVD by lowering serum cholesterol and thereby limiting the growth of the atherosclerotic plaque. A number of clinical trials support the cardiovascular benefit of statin therapy and its effect on lowering serum cholesterol. Although it is clear that we need to reflect on the reliability of the current risk factor model, the current guidelines are an important step forward in the integration of statin therapy into the prevention of cardiovascular disease.
However, talking to patients and telling them that they have greater than a 7.5% risk of having a stroke or a heart attack in the next 10 years remains an abstract concept. The guideline committee now urges me to sit down with my patients and have a heart-to-heart talk about risk and how to decrease it by changing their dangerous lifestyles rather than taking statins for the rest of their lives. When it comes down to it, lifestyle change loses and statins win.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
