User login
Intimate partner violence: How you can help female survivors
Also known as “domestic violence” and “spouse abuse,” intimate partner violence (IPV) is now the term defined by the US Centers for Disease Control and Prevention to include physical violence, sexual violence, threats of physical or sexual violence, and psychological or emotional abuse by a current or former spouse, common-law spouse, nonmarital dating partner, or boyfriend or girlfriend of the same or opposite sex.1 Although IPV is often hidden or kept secret by those affected, it is a highly prevalent issue, especially in women. Knowing how to broach the subject and provide appropriate support in a caring and nonjudgmental manner are the keys to helping a woman move forward in her readiness and ability to improve her situation.
See related patient information
ONE IN THREE WOMEN EXPERIENCES IPV IN HER LIFE
As clinicians, we have all seen patients who have been affected by IPV—even if we did not realize it at the time. Indeed, 36% of women in the United States (approximately 42.4 million) have experienced rape, physical violence, or stalking by an intimate partner in their lifetime, and 6% (approximately 7 million) have experienced these forms of IPV within the past 12 months.2
ASSOCIATION WITH MURDER
From 30% to 70% of women who are murdered are killed by a current or former intimate partner.3,4 Of those killed by their partner, two-thirds had previously reported physical assault, and 83% had been threatened by the man who eventually killed them.4 In another study, 44% of IPV murder victims had presented to an emergency department within 2 years of their murder.5
PHYSICAL EFFECTS NOT ALWAYS APPARENT
Although 41% of women who experience IPV suffer physical injury from their attacks, only 28% of those who are injured seek medical care.6 Because injuries are often absent or no longer apparent when an IPV victim decides to get help, it is important to be aware of the clinical signs associated with IPV:
- Gastrointestinal disorders7
- Depression8
- Anxiety
- Chronic pain syndromes9
- Substance abuse
- Suicidal ideation.10
In women of childbearing age, IPV is associated with unintended pregnancy, sexually transmitted infections, condom non-use,11,12 inconsistent condom use,13 and fear of talking about condom use.11,12 Coerced sexual experiences (eg, sexual intercourse that was not wanted or consented to) are common, with 28% to 42% of college women reporting at least one such experience. In more than three quarters of women who have been sexually assaulted, the first experience occurred before age 25.14,15
One-quarter of women ages 16 to 29 have experienced reproductive coercion, which includes birth control sabotage or pregnancy coercion by the active male partner.16 Among women reporting birth control sabotage, 79% had also been victims of physical or sexual IPV.16
The cost of providing health care to women experiencing IPV is 1.4 to 2.5 times higher than that of the nonabused population. Studies have shown that female victims of both physical and nonphysical (eg, emotional or verbal) IPV are more likely to use emergency, mental health, and outpatient health care services. The economic toll of IPV, including health care and costs from lost productivity and premature death, ranges from $2.3 to $8.3 billion per year.17,18
ASK FEMALE PATIENTS ABOUT IPV
In the early 1990s, various medical organizations began issuing policy statements that endorsed screening for IPV.19–22 Since 1992, the Joint Commission on Accreditation of Healthcare Organizations has required hospitals and clinics to provide assistance to those experiencing IPV.23 Although the United States Preventive Services Task Force initially found insufficient evidence to support regular IPV screening in health care settings,24–27 the group reversed its position in 2012 after a review of more recent studies. The group now recommends that clinicians address IPV with all women of childbearing age.28
A Cochrane review found that IPV screening increased identification of IPV survivors.29 Female participants in many studies wanted clinicians to ask routinely about violence and to provide information on community and legal resources.30,31
How should we ask about IPV?
Although various sets of screening questions and tools are available, no one instrument is considered better than the others. However, women experiencing IPV have specific preferences regarding how they want clinicians to ask and talk about the topic. In one survey, women who had experienced IPV preferred that clinicians ask about it as part of the complete medical history, as long as it did not create “an atmosphere of interrogation.”32
The style in which a clinician asks about IPV may make a difference as well. In focus groups, immigrant Latina and Asian women who had experienced IPV stated that clinicians could facilitate open communication by initiating the discussion and exhibiting compassionate and supportive behavior during the visit.33 Being able to see the same clinician at each visit also enhanced clinician-patient communication.33
In a study of IPV screening in emergency room settings, most clinicians asked about IPV in a perfunctory, direct manner—generally some variant of, “Are you a victim of domestic violence?” In this study, patient IPV disclosure occurred more often when clinicians used an open-ended approach such as, “Tell me what happened,” or when clinicians probed for possible IPV (eg, “What do you think may be causing some of this stress?”).34
In a focus group, female IPV survivors described feeling stigmatized or invalidated when clinicians were condescending, judgmental, or dismissive.35 Nonjudgmental and supportive communication decreased the women’s sense of isolation and led to positive outcomes such as increased awareness of IPV as a problem, decreased isolation, and feeling that the clinician cared.35
When addressing IPV, clinicians should explain why they are asking about it because it allows the woman to understand the context of the inquiry and to feel more comfortable about disclosing IPV. If the query is a regular part of a general screening or history-taking, for example, they should frame the question to make that point apparent. For example, “Because we know that many women in the United States experience physical, sexual or emotional violence from their romantic partners, I like to ask all of my patients whether they have been hurt or have felt threatened or afraid in a current or past relationship.”
In situations in which clinicians are concerned about IPV with a particular patient, they should explicitly share their concerns and desire to help the patient. One IPV survivor offered this advice: “Just look at the patient like she is your friend. Call her by her name. For instance, say ‘Sally, is he hurting you? Are you having problems? If you need help, I have some [phone] numbers.’ Personalize the encounter.”
It is also important to address IPV in a manner that ensures the patient’s safety, confidentiality, and dignity. When having this type of sensitive conversation, the patient should ideally be clothed and alone—without others present, particularly her partner. Professional interpreters should be available to women who do not speak English. The clinician should maintain eye contact, smile to communicate friendliness, and use a supportive tone.36
Just asking may be an intervention
Qualitative studies have suggested that just the act of asking about IPV in a nonjudgmental and compassionate manner is helpful to women experiencing IPV.35,37 Doing so not only helps women recognize the abuse, but also begins to decrease their sense of isolation and increase their awareness of helpful resources. It also gives the patient a sense that the clinician cares about her situation.35 As a result, experts have begun to recommend that health clinicians view asking about IPV not merely as a screening tool, but as a potentially therapeutic intervention in and of itself.35,37,38
HOW TO HELP
What to do when a woman discloses IPV
Female survivors, advocates, and health care clinicians who care for abused women suggest responding to a positive disclosure of IPV by providing the following:
Validation. The IPV perpetrator will often attempt to justify the violence and abuse by shifting some of the blame or responsibility for the violence onto the victim. This “brainwashing” leads to self-blame and a diminished sense of self-worth. However, clinicians can help reverse this mindset by acknowledging the woman’s disclosure and emphasizing that she did not deserve the abuse or violence. An example of such a statement is, “I am so very sorry that you went through that with your partner. You definitely did not deserve that. No one should ever be hurt by or afraid of the people who are supposed to love them.” Providing validation helps women recognize that the violence was a problem they did not deserve.38,39
Support. Women who have experienced IPV appreciate feeling supported and cared for by their health care clinician. Even if the patient is not ready to take any definitive action regarding her relationship or situation, knowing that her clinician and the health care setting are resources and sources of support is both comforting and empowering.35,40 Clinicians can communicate this support by stating, “I want you to know that whatever happens and whatever you decide, we are here for you.”
Respect for autonomy. Women experiencing IPV best understand their own situation and its various complexities and so they know best what they can do, cannot do, or need to do. As such, clinicians must respect a woman’s autonomy and preserve her ability to express her own needs and desires and make her own decisions. Prescribing a plan of action or giving commands to IPV victims could further perpetuate their sense of disempowerment and lack of control over their life.

Information. Providing referral information or hotline numbers to community domestic violence programs is helpful. However, not all women feel safe or comfortable taking printed brochures or written information with them because their abusive partner may find it. Thus, clinicians should ask if the patient can take the information safely or offer to write the numbers down without labeling them if she is afraid. The National Domestic Violence hotline is a 24-hour toll-free resource that will help women locate and contact shelters and other support services in their own community. The number, 1-800-799-SAFE (7233), is easy to memorize and thus is an easy resource to pass along quickly and safely. Other national organizations such as Futures Without Violence (formerly known as the Family Violence Prevention Fund) and the National Coalition Against Violence also provide links to local resources (Table 1).
Safety planning. Discussing the need for a safety plan will help the patient prepare for future abusive episodes. Even women who report that they are no longer in an abusive relationship should be asked about their current safety needs and concerns because they often remain in contact with abusive partners even after a relationship has ended.
When discussing safety planning, clinicians should ask the woman if she is currently safe or if she needs shelter. If she intends to return to or is still in contact with her batterer, ask if she has a plan for what to do or how to escape if the violence occurs again. Advise the patient to:
- Hide money so that she can leave quickly
- Make copies of birth certificates, immunization records, Social Security Number, and other important documents and keep them hidden and accessible
- Make a spare car key
- Have a list of hotline numbers
- Develop a code with friends, family, and neighbors that will let them know she needs immediate help.
Studies show that discussing safety-promoting behaviors increases the number of them that are used by IPV victims.41 A detailed list that can be shared with patients is provided in the patient information page that accompanies this article.42 Examples of personalized safety plans are available from the National Center on Domestic and Sexual Violence at its website, www.ncdsv.org/images/NCDSV_DVSafetyPlan_updated2013.pdf.

Danger assessment. Several researchers have examined potential risk factors associated with increased risk of homicide.43 Table 2 lists some of the characteristics associated with an increased risk of homicide in IPV situations.42 From this work, a danger assessment tool and scoring system has been developed. This tool and training on how to use it are available for free at www.dangerassessment.org. Although there are currently no outcome data on the benefits or risks of using this instrument, its objective is to increase women’s awareness of their danger level and individualize their safety counseling.
Proper documentation. Victim advocates and lawyers working on behalf of IPV victims emphasize that documentation by a medical provider can help a woman with her legal case. This documentation should be clear, legible, and as detailed as possible. These details should include a patient’s own words set off by quotation marks, a description or body map illustrating associated injuries or physical signs corroborating the violence, and a description of the patient’s demeanor or signs of emotion. Clinicians should avoid legal terms such as “alleges” or “alleged perpetrator” and should either define or avoid abbreviations that may be considered ambiguous in a legal proceeding (eg, clinicians should write out the words “domestic violence” or “intimate partner violence” rather than using “DV” or “IPV”).44 Most states have passed laws that prevent insurance companies from discriminating against IPV victims; insurance companies can no longer deny women coverage for seeking care related to IPV.45
A nonthreatening physical examination. Women who have experienced physical or sexual violence may feel anxious or experience added trauma during certain portions of the physical examination. Women who have been raped or otherwise sexually abused may have difficulty tolerating a pelvic examination, for example. Others who have been choked or grabbed by the neck may experience distress during palpation of the thyroid or cervical lymph nodes. The oropharyngeal examination may be challenging for women who have experienced forced oral sex or who may have had objects such as the barrel of a gun forced into their mouth. Asking women who have experienced IPV what aspects of the physical examination they may find difficult and how this could be made easier allows the patient and clinician to work together to develop strategies for or alternatives to necessary examinations or testing.
Before starting an examination, clinicians should explain what will happen and then ask the patient for her permission before moving on to the next step. Doing so creates a sense of control in an otherwise challenging and potentially traumatizing examination.
Disclosure of mandatory IPV reporting. Many states require health care providers to report any injuries caused by a weapon or criminal act to police or other official institutions.45,46 Several of these states specify that this reporting should occur during an IPV evaluation.
Mandatory reporting of IPV to police is highly controversial.47–49 Opponents of these laws argue that they do not benefit victims because they do not respect autonomy, they violate patient-clinician confidentiality, and they violate informed consent.47 Female IPV victims indicate that they would be less likely to disclose IPV and seek help if they knew it had to be reported.50–53 In a survey of California physicians, 59% stated that they might not comply with reporting laws if the patient objected.54
In states with mandatory reporting laws, clinicians can advocate for their patients by disclosing whether IPV must be reported before beginning their examination. The Michigan Coalition Against Domestic Violence devised this sample statement:
“I do have to let you know that under state law, I am obligated to make a police report to (name police agency with jurisdiction over the facility), if I/we are treating someone for any injury sustained by means of violence (tailor this information to the specifics of your state law). So, if you would have any concerns about that, I would encourage you to speak with one of the domestic violence advocates/counselors at (DV agency), who are able to provide confidential services and help without being required to make a police report.”55
The Compendium of State Statutes and Polices on Domestic Violence and Health Care by Futures Without Violence is an excellent reference that describes current state laws and policies regarding domestic violence and includes descriptions of the specific characteristics of each state’s mandatory reporting laws and their implications for health care providers. A copy can be downloaded from www.futureswithout-violence.org/content/features/detail/1584/.
What not to do
Clinicians should not ignore or minimize the extent of IPV, make excuses for the batterer, or blame the woman.32,56,57 They also should not inadvertently blame or criticize the woman by asking her, “What did you do to deserve this?” or “Why don’t you just leave?”57
While leaving a violent relationship is an obvious solution, many women experiencing IPV are either unwilling or unable to do so. In fact, leaving the batterer can be just as dangerous as staying. In a North Carolina study, half of all women killed by their partner had divorced or broken up with the partner immediately before the murder.4
What to do when a woman does not disclose IPV
Women who have experienced IPV often deny the violence to others out of fear. So a denial of violence, even in highly suspicious cases, can be expected. Abused patients often want help but are confused and afraid to ask for it.58,59 Providing easy and anonymous access to IPV information and resources through posters, flyers, brochures, and booklets will allow any woman—regardless of disclosure—to obtain help.36
In one IPV trial, all women in a family planning clinic who were in the intervention group were given information about IPV and how it can affect sexual and reproductive health, whether or not they disclosed. Compared with women in the control group, women who received the intervention—both those who did and did not experience IPV—were 63% more likely than those in the control group to end a relationship because they perceived it to be unhealthy or unsafe. In this case, all women—not just those who were victims—benefited from receiving information about IPV.60
PROVIDERS’ SUPPORT MAKES A DIFFERENCE, IMPROVES OUTCOMES
Patients may seek help for and find safety from IPV in stages or steps, and it may take them several months or even years to do so.61–63 In this sense, then, IPV is an issue that is not likely to be “cured” or changed in a single medical visit. In addition, a single visit may not be associated with direct health and safety outcomes. However, a patient is more likely to make changes if she has supportive and informative interaction with a caring, nonjudgmental clinician who can increase her awareness of IPV and IPV resources and promote an improved sense of self-efficacy or perceived power.40
For example, in a study in which health clinicians expressed concern about potential IPV health effects and offered support services such as counseling, 67% of women used one of those resources at least once.40 In another study, women who talked to a health care provider about their abuse were almost four times more likely to use an IPV intervention (eg, advocacy, shelter, restraining order) than women who did not talk to their provider. Those who used an IPV intervention were 2.6 times more likely to leave their abusive relationship, and those who left reported improved physical health.64
Thus, health care providers can have a positive effect by addressing this complex and difficult issue. The power and influence of a provider’s kindness, empathy, and support cannot be overestimated in their ability to improve the safety and well-being of women dealing with IPV.
- Saltzman LE, Fanslow JL, McMahon PM, Shelley GA; National Center for Injury Prevention and Control; Centers for Disease Control and Prevention. Intimate partner violence surveillance: uniform definitions and recommended data elements, Version 1.0. Atlanta, GA; 1999. www.cdc.gov/ncipc/pub-res/ipv_surveillance/intimate%20partner%20violence.pdf. Accessed June 3, 2014.
- Black MC, Basile KC, Breiding MJ, et al; Center for Injury Prevention and Control; Centers for Disease Control and Prevention. The National Intimate Partner and Sexual Violence Survey (NISVS): 2010 Summary Report. Atlanta, GA. www.cdc.gov/violenceprevention/pdf/nisvs_executive_summary-a.pdf. Accessed June 3, 2014.
- Kellermann AL, Mercy JA. Men, women, and murder: gender-specific differences in rates of fatal violence and victimization. J Trauma 1992; 33:1–5.
- Moracco KE, Runywan CW, Butts JD. Femicide in North Carolina, 1991–1993: a statewide study of patterns and precursors. Homicide Studies 1998; 4:422–446.
- Wadman MC, Muelleman RL. Domestic violence homicides: ED use before victimization. Am J Emerg Med 1999; 17:689–691.
- Catalano SM; United States Bureau of Justice Statistics. Intimate partner violence in the United States: 2007. http://bjs.ojp.usdoj.gov/index.cfm?ty=pbdetail&iid=1000. Accessed February 19, 2014.
- Drossman DA, Talley NJ, Leserman J, Olden KW, Barreiro MA. Sexual and physical abuse and gastrointestinal illness. Review and recommendations. Ann Intern Med 1995; 123:782–794.
- Scholle SH, Rost KM, Golding JM. Physical abuse among depressed women. J Gen Intern Med 1998; 13:607–613.
- Walling MK, O’Hara MW, Reiter RC, Milburn AK, Lilly G, Vincent SD. Abuse history and chronic pain in women: II. A multivariate analysis of abuse and psychological morbidity. Obstet Gynecol 1994; 84:200–206.
- McCauley J, Kern DE, Kolodner K, Derogatis LR, Bass EB. Relation of low-severity violence to women’s health. J Gen Intern Med 1998; 13:687–691.
- Wingood GM, DiClemente RJ. The effects of an abusive primary partner on the condom use and sexual negotiation practices of African-American women. Am J Public Health 1997; 87:1016–1018.
- Sales JM, Salazar LF, Wingood GM, DiClemente RJ, Rose E, Crosby RA. The mediating role of partner communication skills on HIV/STD-associated risk behaviors in young African American females with a history of sexual violence. Arch Pediatr Adolesc Med 2008; 162:432–438.
- Davila YR, Brackley MH. Mexican and Mexican American women in a battered women’s shelter: barriers to condom negotiation for HIV/AIDS prevention. Issues Ment Health Nurs 1999; 20:333–355.
- Tjaden P, Thoennes N; National Institute of Justice; Centers for Disease Control and Prevention. Prevalence, incidence and consequences of violence against women: findings from the national violence against women survey. Washington, DC; 1998. https://www.ncjrs.gov/pdffiles/172837.pdf. Accessed June 3, 2014.
- Masho SW, Odor RK, Adera T. Sexual assault in Virginia: a population-based study. Womens Health Issues 2005; 15:157–166.
- Miller E, Decker MR, McCauley HL, et al. Pregnancy coercion, intimate partner violence and unintended pregnancy. Contraception 2010; 81:316–322.
- Bonomi AE, Anderson ML, Rivara FP, Thompson RS. Health care utilization and costs associated with physical and nonphysical-only intimate partner violence. Health Serv Res 2009; 44:1052–1067.
- Rivara FP, Anderson ML, Fishman P, et al. Healthcare utilization and costs for women with a history of intimate partner violence. Am J Prev Med 2007; 32:89–96.
- American Nurses Association. Position statement on physical violence against women. Washington, DC; 1991.
- Physicians and domestic violence. Ethical considerations. Council on Ethical and Judicial Affairs, American Medical Association. JAMA 1992; 267:3190–3193.
- The American College of Obstetricians and Gynecologists. Screening tools: domestic violence. http://www.acog.org/About_ACOG/ACOG_Departments/Violence_Against_Women/Screening_Tools__Domestic_Violence. Accessed June 3, 2014.
- Lee D, James L, Sawires P; The Family Violence Prevention Fund. Preventing domestic violence: clinical guidelines on routine screening. San Francisco, CA; 1999. http://new.vawnet.org/Assoc_Files_VAWnet/screpol.pdf. Accessed June 3, 2014.
- Joint Commission on Accreditation of Healthcare Organizations. Accreditation manual for hospitals. Chicago, IL; 1992.
- US Department of Health and Human Services. US Preventive Services Task Force. Guide to Clinical Preventive Services. 2nd ed. Washington, DC; 1996.
- Ramsay J, Richardson J, Carter YH, Davidson LL, Feder G. Should health professionals screen women for domestic violence? Systematic review. BMJ 2002; 325:314.
- Nelson HD, Nygren P, McInerney Y, Klein J; US Preventive Services Task Force. Screening women and elderly adults for family and intimate partner violence: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2004; 140:387–396.
- Chamberlain L. The USPSTF recommendation on intimate partner violence: what we can learn from it and what we can do about it. Fam Viol Prev Health Pract 2005; 1:1–24.
- Moyer VA; US Preventive Services Task Force. Screening for intimate partner violence and abuse of elderly and vulnerable adults: US Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158:478–486.
- Taft A, O’Doherty L, Hegarty K, Ramsay J, Davidson L, Feder G. Screening women for intimate partner violence in healthcare settings. Cochrane Database Syst Rev 2013; 4:CD007007.
- McNutt LA, Carlson BE, Gagen D, Winterbauer N. Reproductive violence screening in primary care: perspectives and experiences of patients and battered women. J Am Med Womens Assoc 1999; 54:85–90.
- Friedman LS, Samet JH, Roberts MS, Hudlin M, Hans P. Inquiry about victimization experiences. A survey of patient p and physician practices. Arch Intern Med 1992; 152:1186–1190.
- Hamberger LK, Ambuel B, Marbella A, Donze J. Physician interaction with battered women: the women’s perspective. Arch Fam Med 1998; 7:575–582.
- Rodriguez MA, Bauer HM, Flores-Ortiz Y, Szkupinski-Quiroga S. Factors affecting patient-physician communication for abused Latina and Asian immigrant women. J Fam Pract 1998; 47:309–311.
- Rhodes KV, Frankel RM, Levinthal N, Prenoveau E, Bailey J, Levinson W. “You’re not a victim of domestic violence, are you?” Provider patient communication about domestic violence”. Ann Intern Med 2007; 147:620–627.
- Chang JC, Decker M, Moracco KE, Martin SL, Petersen R, Frasier PY. What happens when health care providers ask about intimate partner violence? A description of consequences from the perspectives of female survivors. J Am Med Womens Assoc 2003; 58:76–81.
- Chang JC, Decker MR, Moracco KE, Martin SL, Petersen R, Frasier PY. Asking about intimate partner violence: advice from female survivors to health care providers. Patient Educ Couns 2005; 59:141–147.
- Hathaway JE, Willis G, Zimmer B. Listening to survivors’ voices: addressing partner abuse in the health care setting. Violence Against Women 2002; 8:687–716.
- Gerbert B, Caspers N, Bronstone A, Moe J, Abercrombie P. A qualitative analysis of how physicians with expertise in domestic violence approach the identification of victims. Ann Intern Med 1999; 131:578–584.
- Gerbert B, Caspers N, Milliken N, Berlin M, Bronstone A, Moe J. Interventions that help victims of domestic violence. A qualitative analysis of physicians’ experiences. J Fam Pract 2000; 49:889–895.
- McCaw B, Bauer HM, Berman WH, Mooney L, Holmberg M, Hunkeler E. Women referred for on-site domestic violence services in a managed care organization. Women Health 2002; 35:23–40.
- McFarlane J, Malecha A, Gist J, et al. An intervention to increase safety behaviors of abused women: results of a randomized clinical trial. Nurs Res 2002; 51:347–354.
- Chang JC. Domestic violence. In:Bieber EJ, Sanfilippo JS, Horowitz IR, editors. Clinical Gynecology. Philadelphia, PA; Elsevier, Inc; 2006:79–89.
- Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide in abusive relationships: results from a multisite case control study. Am J Public Health 2003; 93:1089–1097.
- Isaac NE, Enos V; National Institute of Justice. Documenting domestic violence: how health care providers can help victims. Research in Brief, 2001. https://www.ncjrs.gov/pdffiles1/nij/188564.pdf. Accessed June 3, 2014.
- Durborow N, Lizdas KC, O’Flaherty A, Marjavi A; Futures Without Violence. Compendium of state and US terrritory statutes and policies on domestic violence and health care, 2010. www.futureswithoutviolence.org/content/features/detail/1584/. Accessed June 3, 2014.
- Hyman A, Schillinger D, Lo B. Laws mandating reporting of domestic violence. Do they promote patient well-being? JAMA 1995; 273:1781–1787.
- Hyman A, Chez RA. Mandatory reporting of domestic violence by health care providers: a misguided approach. Womens Health Issues 1995; 5:208–213.
- Knight MA. Ethical debate: should doctors be more proactive as advocates for victims of violence? The police surgeon’s view: medical paternalism is unacceptable. BMJ 1995; 311:1620–1621.
- Hyman A; Futures Without Violence. Mandatory reporting of domestic violence by healthcare providers: a policy paper. San Francisco, CA; 1997. www.futureswithoutviolence.org/userfiles/file/HealthCare/mandatory_policypaper.pdf. Accessed June 3, 2014.
- Rodriguez MA, Craig AM, Mooney DR, Bauer HM. Patient attitudes about mandatory reporting of domestic violence. Implications for health care professionals. West J Med 1998; 169:337–341.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Sachs CJ, Koziol-McLain J, Glass N, Webster D, Campbell J. A population-based survey assessing support for mandatory domestic violence reporting by health care personnel. Women Health 2002; 35:121–133.
- Sullivan CM, Hagen LA. Survivors’ opinions about mandatory reporting of domestic violence and sexual assault by medical professionals. Affilia 2005; 20:1–16.
- Rodriguez MA, McLoughlin E, Bauer HM, Paredes V, Grumbach K. Mandatory reporting of intimate partner violence to police: views of physicians in California. Am J Public Health 1999; 89:575–578.
- Futures Without Violence. Mandatory reporting of domestic violence to law enforcement by health care providers: a guide for advocates working to respond to or amend reporting laws related to domestic violence. www.healthcaresaboutipv.org/wp-content/blogs.dir/3/files/2012/09/Mandatory_Reporting_of_DV_to_Law-Enforcement_by_HCP.pdf. Accessed June 3, 2014.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Gerbert B, Johnston K, Caspers N, Bleecker T, Woods A, Rosenbaum A. Experiences of battered women in health care settings: a qualitative study. Women Health 1996; 24:1–17.
- Chang JC, Cluss PA, Ranieri L, et al. Health care interventions for intimate partner violence: what women want. Womens Health Issues 2005; 15:21–30.
- Rodriguez MA, Quiroga SS, Bauer HM. Breaking the silence. Battered women’s perspectives on medical care. Arch Fam Med 1996; 5:153–158.
- Miller E, Decker MR, McCauley HL, et al. A family planning clinic partner violence intervention to reduce risk associated with reproductive coercion. Contraception 2011; 83:274–280.
- Landenburger K. A process of entrapment in and recovery from an abusive relationship. Issues Ment Health Nurs 1989; 10:209–227.
- Cluss PA, Chang JC, Hawker L, et al. The process of change for victims of intimate partner violence: support for a psychosocial readiness model. Womens Health Issues 2006; 16:262–274.
- Gerbert B, Abercrombie P, Caspers N, Love C, Bronstone A. How health care providers help battered women: the survivor’s perspective. Women Health 1999; 29:115–135.
- McCloskey LA, Lichter E, Williams C, Gerber M, Wittenberg E, Ganz M. Assessing intimate partner violence in health care settings leads to women’s receipt of interventions and improved health. Public Health Rep 2006; 121:435–444.
Also known as “domestic violence” and “spouse abuse,” intimate partner violence (IPV) is now the term defined by the US Centers for Disease Control and Prevention to include physical violence, sexual violence, threats of physical or sexual violence, and psychological or emotional abuse by a current or former spouse, common-law spouse, nonmarital dating partner, or boyfriend or girlfriend of the same or opposite sex.1 Although IPV is often hidden or kept secret by those affected, it is a highly prevalent issue, especially in women. Knowing how to broach the subject and provide appropriate support in a caring and nonjudgmental manner are the keys to helping a woman move forward in her readiness and ability to improve her situation.
See related patient information
ONE IN THREE WOMEN EXPERIENCES IPV IN HER LIFE
As clinicians, we have all seen patients who have been affected by IPV—even if we did not realize it at the time. Indeed, 36% of women in the United States (approximately 42.4 million) have experienced rape, physical violence, or stalking by an intimate partner in their lifetime, and 6% (approximately 7 million) have experienced these forms of IPV within the past 12 months.2
ASSOCIATION WITH MURDER
From 30% to 70% of women who are murdered are killed by a current or former intimate partner.3,4 Of those killed by their partner, two-thirds had previously reported physical assault, and 83% had been threatened by the man who eventually killed them.4 In another study, 44% of IPV murder victims had presented to an emergency department within 2 years of their murder.5
PHYSICAL EFFECTS NOT ALWAYS APPARENT
Although 41% of women who experience IPV suffer physical injury from their attacks, only 28% of those who are injured seek medical care.6 Because injuries are often absent or no longer apparent when an IPV victim decides to get help, it is important to be aware of the clinical signs associated with IPV:
- Gastrointestinal disorders7
- Depression8
- Anxiety
- Chronic pain syndromes9
- Substance abuse
- Suicidal ideation.10
In women of childbearing age, IPV is associated with unintended pregnancy, sexually transmitted infections, condom non-use,11,12 inconsistent condom use,13 and fear of talking about condom use.11,12 Coerced sexual experiences (eg, sexual intercourse that was not wanted or consented to) are common, with 28% to 42% of college women reporting at least one such experience. In more than three quarters of women who have been sexually assaulted, the first experience occurred before age 25.14,15
One-quarter of women ages 16 to 29 have experienced reproductive coercion, which includes birth control sabotage or pregnancy coercion by the active male partner.16 Among women reporting birth control sabotage, 79% had also been victims of physical or sexual IPV.16
The cost of providing health care to women experiencing IPV is 1.4 to 2.5 times higher than that of the nonabused population. Studies have shown that female victims of both physical and nonphysical (eg, emotional or verbal) IPV are more likely to use emergency, mental health, and outpatient health care services. The economic toll of IPV, including health care and costs from lost productivity and premature death, ranges from $2.3 to $8.3 billion per year.17,18
ASK FEMALE PATIENTS ABOUT IPV
In the early 1990s, various medical organizations began issuing policy statements that endorsed screening for IPV.19–22 Since 1992, the Joint Commission on Accreditation of Healthcare Organizations has required hospitals and clinics to provide assistance to those experiencing IPV.23 Although the United States Preventive Services Task Force initially found insufficient evidence to support regular IPV screening in health care settings,24–27 the group reversed its position in 2012 after a review of more recent studies. The group now recommends that clinicians address IPV with all women of childbearing age.28
A Cochrane review found that IPV screening increased identification of IPV survivors.29 Female participants in many studies wanted clinicians to ask routinely about violence and to provide information on community and legal resources.30,31
How should we ask about IPV?
Although various sets of screening questions and tools are available, no one instrument is considered better than the others. However, women experiencing IPV have specific preferences regarding how they want clinicians to ask and talk about the topic. In one survey, women who had experienced IPV preferred that clinicians ask about it as part of the complete medical history, as long as it did not create “an atmosphere of interrogation.”32
The style in which a clinician asks about IPV may make a difference as well. In focus groups, immigrant Latina and Asian women who had experienced IPV stated that clinicians could facilitate open communication by initiating the discussion and exhibiting compassionate and supportive behavior during the visit.33 Being able to see the same clinician at each visit also enhanced clinician-patient communication.33
In a study of IPV screening in emergency room settings, most clinicians asked about IPV in a perfunctory, direct manner—generally some variant of, “Are you a victim of domestic violence?” In this study, patient IPV disclosure occurred more often when clinicians used an open-ended approach such as, “Tell me what happened,” or when clinicians probed for possible IPV (eg, “What do you think may be causing some of this stress?”).34
In a focus group, female IPV survivors described feeling stigmatized or invalidated when clinicians were condescending, judgmental, or dismissive.35 Nonjudgmental and supportive communication decreased the women’s sense of isolation and led to positive outcomes such as increased awareness of IPV as a problem, decreased isolation, and feeling that the clinician cared.35
When addressing IPV, clinicians should explain why they are asking about it because it allows the woman to understand the context of the inquiry and to feel more comfortable about disclosing IPV. If the query is a regular part of a general screening or history-taking, for example, they should frame the question to make that point apparent. For example, “Because we know that many women in the United States experience physical, sexual or emotional violence from their romantic partners, I like to ask all of my patients whether they have been hurt or have felt threatened or afraid in a current or past relationship.”
In situations in which clinicians are concerned about IPV with a particular patient, they should explicitly share their concerns and desire to help the patient. One IPV survivor offered this advice: “Just look at the patient like she is your friend. Call her by her name. For instance, say ‘Sally, is he hurting you? Are you having problems? If you need help, I have some [phone] numbers.’ Personalize the encounter.”
It is also important to address IPV in a manner that ensures the patient’s safety, confidentiality, and dignity. When having this type of sensitive conversation, the patient should ideally be clothed and alone—without others present, particularly her partner. Professional interpreters should be available to women who do not speak English. The clinician should maintain eye contact, smile to communicate friendliness, and use a supportive tone.36
Just asking may be an intervention
Qualitative studies have suggested that just the act of asking about IPV in a nonjudgmental and compassionate manner is helpful to women experiencing IPV.35,37 Doing so not only helps women recognize the abuse, but also begins to decrease their sense of isolation and increase their awareness of helpful resources. It also gives the patient a sense that the clinician cares about her situation.35 As a result, experts have begun to recommend that health clinicians view asking about IPV not merely as a screening tool, but as a potentially therapeutic intervention in and of itself.35,37,38
HOW TO HELP
What to do when a woman discloses IPV
Female survivors, advocates, and health care clinicians who care for abused women suggest responding to a positive disclosure of IPV by providing the following:
Validation. The IPV perpetrator will often attempt to justify the violence and abuse by shifting some of the blame or responsibility for the violence onto the victim. This “brainwashing” leads to self-blame and a diminished sense of self-worth. However, clinicians can help reverse this mindset by acknowledging the woman’s disclosure and emphasizing that she did not deserve the abuse or violence. An example of such a statement is, “I am so very sorry that you went through that with your partner. You definitely did not deserve that. No one should ever be hurt by or afraid of the people who are supposed to love them.” Providing validation helps women recognize that the violence was a problem they did not deserve.38,39
Support. Women who have experienced IPV appreciate feeling supported and cared for by their health care clinician. Even if the patient is not ready to take any definitive action regarding her relationship or situation, knowing that her clinician and the health care setting are resources and sources of support is both comforting and empowering.35,40 Clinicians can communicate this support by stating, “I want you to know that whatever happens and whatever you decide, we are here for you.”
Respect for autonomy. Women experiencing IPV best understand their own situation and its various complexities and so they know best what they can do, cannot do, or need to do. As such, clinicians must respect a woman’s autonomy and preserve her ability to express her own needs and desires and make her own decisions. Prescribing a plan of action or giving commands to IPV victims could further perpetuate their sense of disempowerment and lack of control over their life.
Information. Providing referral information or hotline numbers to community domestic violence programs is helpful. However, not all women feel safe or comfortable taking printed brochures or written information with them because their abusive partner may find it. Thus, clinicians should ask if the patient can take the information safely or offer to write the numbers down without labeling them if she is afraid. The National Domestic Violence hotline is a 24-hour toll-free resource that will help women locate and contact shelters and other support services in their own community. The number, 1-800-799-SAFE (7233), is easy to memorize and thus is an easy resource to pass along quickly and safely. Other national organizations such as Futures Without Violence (formerly known as the Family Violence Prevention Fund) and the National Coalition Against Violence also provide links to local resources (Table 1).
Safety planning. Discussing the need for a safety plan will help the patient prepare for future abusive episodes. Even women who report that they are no longer in an abusive relationship should be asked about their current safety needs and concerns because they often remain in contact with abusive partners even after a relationship has ended.
When discussing safety planning, clinicians should ask the woman if she is currently safe or if she needs shelter. If she intends to return to or is still in contact with her batterer, ask if she has a plan for what to do or how to escape if the violence occurs again. Advise the patient to:
- Hide money so that she can leave quickly
- Make copies of birth certificates, immunization records, Social Security Number, and other important documents and keep them hidden and accessible
- Make a spare car key
- Have a list of hotline numbers
- Develop a code with friends, family, and neighbors that will let them know she needs immediate help.
Studies show that discussing safety-promoting behaviors increases the number of them that are used by IPV victims.41 A detailed list that can be shared with patients is provided in the patient information page that accompanies this article.42 Examples of personalized safety plans are available from the National Center on Domestic and Sexual Violence at its website, www.ncdsv.org/images/NCDSV_DVSafetyPlan_updated2013.pdf.
Danger assessment. Several researchers have examined potential risk factors associated with increased risk of homicide.43 Table 2 lists some of the characteristics associated with an increased risk of homicide in IPV situations.42 From this work, a danger assessment tool and scoring system has been developed. This tool and training on how to use it are available for free at www.dangerassessment.org. Although there are currently no outcome data on the benefits or risks of using this instrument, its objective is to increase women’s awareness of their danger level and individualize their safety counseling.
Proper documentation. Victim advocates and lawyers working on behalf of IPV victims emphasize that documentation by a medical provider can help a woman with her legal case. This documentation should be clear, legible, and as detailed as possible. These details should include a patient’s own words set off by quotation marks, a description or body map illustrating associated injuries or physical signs corroborating the violence, and a description of the patient’s demeanor or signs of emotion. Clinicians should avoid legal terms such as “alleges” or “alleged perpetrator” and should either define or avoid abbreviations that may be considered ambiguous in a legal proceeding (eg, clinicians should write out the words “domestic violence” or “intimate partner violence” rather than using “DV” or “IPV”).44 Most states have passed laws that prevent insurance companies from discriminating against IPV victims; insurance companies can no longer deny women coverage for seeking care related to IPV.45
A nonthreatening physical examination. Women who have experienced physical or sexual violence may feel anxious or experience added trauma during certain portions of the physical examination. Women who have been raped or otherwise sexually abused may have difficulty tolerating a pelvic examination, for example. Others who have been choked or grabbed by the neck may experience distress during palpation of the thyroid or cervical lymph nodes. The oropharyngeal examination may be challenging for women who have experienced forced oral sex or who may have had objects such as the barrel of a gun forced into their mouth. Asking women who have experienced IPV what aspects of the physical examination they may find difficult and how this could be made easier allows the patient and clinician to work together to develop strategies for or alternatives to necessary examinations or testing.
Before starting an examination, clinicians should explain what will happen and then ask the patient for her permission before moving on to the next step. Doing so creates a sense of control in an otherwise challenging and potentially traumatizing examination.
Disclosure of mandatory IPV reporting. Many states require health care providers to report any injuries caused by a weapon or criminal act to police or other official institutions.45,46 Several of these states specify that this reporting should occur during an IPV evaluation.
Mandatory reporting of IPV to police is highly controversial.47–49 Opponents of these laws argue that they do not benefit victims because they do not respect autonomy, they violate patient-clinician confidentiality, and they violate informed consent.47 Female IPV victims indicate that they would be less likely to disclose IPV and seek help if they knew it had to be reported.50–53 In a survey of California physicians, 59% stated that they might not comply with reporting laws if the patient objected.54
In states with mandatory reporting laws, clinicians can advocate for their patients by disclosing whether IPV must be reported before beginning their examination. The Michigan Coalition Against Domestic Violence devised this sample statement:
“I do have to let you know that under state law, I am obligated to make a police report to (name police agency with jurisdiction over the facility), if I/we are treating someone for any injury sustained by means of violence (tailor this information to the specifics of your state law). So, if you would have any concerns about that, I would encourage you to speak with one of the domestic violence advocates/counselors at (DV agency), who are able to provide confidential services and help without being required to make a police report.”55
The Compendium of State Statutes and Polices on Domestic Violence and Health Care by Futures Without Violence is an excellent reference that describes current state laws and policies regarding domestic violence and includes descriptions of the specific characteristics of each state’s mandatory reporting laws and their implications for health care providers. A copy can be downloaded from www.futureswithout-violence.org/content/features/detail/1584/.
What not to do
Clinicians should not ignore or minimize the extent of IPV, make excuses for the batterer, or blame the woman.32,56,57 They also should not inadvertently blame or criticize the woman by asking her, “What did you do to deserve this?” or “Why don’t you just leave?”57
While leaving a violent relationship is an obvious solution, many women experiencing IPV are either unwilling or unable to do so. In fact, leaving the batterer can be just as dangerous as staying. In a North Carolina study, half of all women killed by their partner had divorced or broken up with the partner immediately before the murder.4
What to do when a woman does not disclose IPV
Women who have experienced IPV often deny the violence to others out of fear. So a denial of violence, even in highly suspicious cases, can be expected. Abused patients often want help but are confused and afraid to ask for it.58,59 Providing easy and anonymous access to IPV information and resources through posters, flyers, brochures, and booklets will allow any woman—regardless of disclosure—to obtain help.36
In one IPV trial, all women in a family planning clinic who were in the intervention group were given information about IPV and how it can affect sexual and reproductive health, whether or not they disclosed. Compared with women in the control group, women who received the intervention—both those who did and did not experience IPV—were 63% more likely than those in the control group to end a relationship because they perceived it to be unhealthy or unsafe. In this case, all women—not just those who were victims—benefited from receiving information about IPV.60
PROVIDERS’ SUPPORT MAKES A DIFFERENCE, IMPROVES OUTCOMES
Patients may seek help for and find safety from IPV in stages or steps, and it may take them several months or even years to do so.61–63 In this sense, then, IPV is an issue that is not likely to be “cured” or changed in a single medical visit. In addition, a single visit may not be associated with direct health and safety outcomes. However, a patient is more likely to make changes if she has supportive and informative interaction with a caring, nonjudgmental clinician who can increase her awareness of IPV and IPV resources and promote an improved sense of self-efficacy or perceived power.40
For example, in a study in which health clinicians expressed concern about potential IPV health effects and offered support services such as counseling, 67% of women used one of those resources at least once.40 In another study, women who talked to a health care provider about their abuse were almost four times more likely to use an IPV intervention (eg, advocacy, shelter, restraining order) than women who did not talk to their provider. Those who used an IPV intervention were 2.6 times more likely to leave their abusive relationship, and those who left reported improved physical health.64
Thus, health care providers can have a positive effect by addressing this complex and difficult issue. The power and influence of a provider’s kindness, empathy, and support cannot be overestimated in their ability to improve the safety and well-being of women dealing with IPV.
Also known as “domestic violence” and “spouse abuse,” intimate partner violence (IPV) is now the term defined by the US Centers for Disease Control and Prevention to include physical violence, sexual violence, threats of physical or sexual violence, and psychological or emotional abuse by a current or former spouse, common-law spouse, nonmarital dating partner, or boyfriend or girlfriend of the same or opposite sex.1 Although IPV is often hidden or kept secret by those affected, it is a highly prevalent issue, especially in women. Knowing how to broach the subject and provide appropriate support in a caring and nonjudgmental manner are the keys to helping a woman move forward in her readiness and ability to improve her situation.
See related patient information
ONE IN THREE WOMEN EXPERIENCES IPV IN HER LIFE
As clinicians, we have all seen patients who have been affected by IPV—even if we did not realize it at the time. Indeed, 36% of women in the United States (approximately 42.4 million) have experienced rape, physical violence, or stalking by an intimate partner in their lifetime, and 6% (approximately 7 million) have experienced these forms of IPV within the past 12 months.2
ASSOCIATION WITH MURDER
From 30% to 70% of women who are murdered are killed by a current or former intimate partner.3,4 Of those killed by their partner, two-thirds had previously reported physical assault, and 83% had been threatened by the man who eventually killed them.4 In another study, 44% of IPV murder victims had presented to an emergency department within 2 years of their murder.5
PHYSICAL EFFECTS NOT ALWAYS APPARENT
Although 41% of women who experience IPV suffer physical injury from their attacks, only 28% of those who are injured seek medical care.6 Because injuries are often absent or no longer apparent when an IPV victim decides to get help, it is important to be aware of the clinical signs associated with IPV:
- Gastrointestinal disorders7
- Depression8
- Anxiety
- Chronic pain syndromes9
- Substance abuse
- Suicidal ideation.10
In women of childbearing age, IPV is associated with unintended pregnancy, sexually transmitted infections, condom non-use,11,12 inconsistent condom use,13 and fear of talking about condom use.11,12 Coerced sexual experiences (eg, sexual intercourse that was not wanted or consented to) are common, with 28% to 42% of college women reporting at least one such experience. In more than three quarters of women who have been sexually assaulted, the first experience occurred before age 25.14,15
One-quarter of women ages 16 to 29 have experienced reproductive coercion, which includes birth control sabotage or pregnancy coercion by the active male partner.16 Among women reporting birth control sabotage, 79% had also been victims of physical or sexual IPV.16
The cost of providing health care to women experiencing IPV is 1.4 to 2.5 times higher than that of the nonabused population. Studies have shown that female victims of both physical and nonphysical (eg, emotional or verbal) IPV are more likely to use emergency, mental health, and outpatient health care services. The economic toll of IPV, including health care and costs from lost productivity and premature death, ranges from $2.3 to $8.3 billion per year.17,18
ASK FEMALE PATIENTS ABOUT IPV
In the early 1990s, various medical organizations began issuing policy statements that endorsed screening for IPV.19–22 Since 1992, the Joint Commission on Accreditation of Healthcare Organizations has required hospitals and clinics to provide assistance to those experiencing IPV.23 Although the United States Preventive Services Task Force initially found insufficient evidence to support regular IPV screening in health care settings,24–27 the group reversed its position in 2012 after a review of more recent studies. The group now recommends that clinicians address IPV with all women of childbearing age.28
A Cochrane review found that IPV screening increased identification of IPV survivors.29 Female participants in many studies wanted clinicians to ask routinely about violence and to provide information on community and legal resources.30,31
How should we ask about IPV?
Although various sets of screening questions and tools are available, no one instrument is considered better than the others. However, women experiencing IPV have specific preferences regarding how they want clinicians to ask and talk about the topic. In one survey, women who had experienced IPV preferred that clinicians ask about it as part of the complete medical history, as long as it did not create “an atmosphere of interrogation.”32
The style in which a clinician asks about IPV may make a difference as well. In focus groups, immigrant Latina and Asian women who had experienced IPV stated that clinicians could facilitate open communication by initiating the discussion and exhibiting compassionate and supportive behavior during the visit.33 Being able to see the same clinician at each visit also enhanced clinician-patient communication.33
In a study of IPV screening in emergency room settings, most clinicians asked about IPV in a perfunctory, direct manner—generally some variant of, “Are you a victim of domestic violence?” In this study, patient IPV disclosure occurred more often when clinicians used an open-ended approach such as, “Tell me what happened,” or when clinicians probed for possible IPV (eg, “What do you think may be causing some of this stress?”).34
In a focus group, female IPV survivors described feeling stigmatized or invalidated when clinicians were condescending, judgmental, or dismissive.35 Nonjudgmental and supportive communication decreased the women’s sense of isolation and led to positive outcomes such as increased awareness of IPV as a problem, decreased isolation, and feeling that the clinician cared.35
When addressing IPV, clinicians should explain why they are asking about it because it allows the woman to understand the context of the inquiry and to feel more comfortable about disclosing IPV. If the query is a regular part of a general screening or history-taking, for example, they should frame the question to make that point apparent. For example, “Because we know that many women in the United States experience physical, sexual or emotional violence from their romantic partners, I like to ask all of my patients whether they have been hurt or have felt threatened or afraid in a current or past relationship.”
In situations in which clinicians are concerned about IPV with a particular patient, they should explicitly share their concerns and desire to help the patient. One IPV survivor offered this advice: “Just look at the patient like she is your friend. Call her by her name. For instance, say ‘Sally, is he hurting you? Are you having problems? If you need help, I have some [phone] numbers.’ Personalize the encounter.”
It is also important to address IPV in a manner that ensures the patient’s safety, confidentiality, and dignity. When having this type of sensitive conversation, the patient should ideally be clothed and alone—without others present, particularly her partner. Professional interpreters should be available to women who do not speak English. The clinician should maintain eye contact, smile to communicate friendliness, and use a supportive tone.36
Just asking may be an intervention
Qualitative studies have suggested that just the act of asking about IPV in a nonjudgmental and compassionate manner is helpful to women experiencing IPV.35,37 Doing so not only helps women recognize the abuse, but also begins to decrease their sense of isolation and increase their awareness of helpful resources. It also gives the patient a sense that the clinician cares about her situation.35 As a result, experts have begun to recommend that health clinicians view asking about IPV not merely as a screening tool, but as a potentially therapeutic intervention in and of itself.35,37,38
HOW TO HELP
What to do when a woman discloses IPV
Female survivors, advocates, and health care clinicians who care for abused women suggest responding to a positive disclosure of IPV by providing the following:
Validation. The IPV perpetrator will often attempt to justify the violence and abuse by shifting some of the blame or responsibility for the violence onto the victim. This “brainwashing” leads to self-blame and a diminished sense of self-worth. However, clinicians can help reverse this mindset by acknowledging the woman’s disclosure and emphasizing that she did not deserve the abuse or violence. An example of such a statement is, “I am so very sorry that you went through that with your partner. You definitely did not deserve that. No one should ever be hurt by or afraid of the people who are supposed to love them.” Providing validation helps women recognize that the violence was a problem they did not deserve.38,39
Support. Women who have experienced IPV appreciate feeling supported and cared for by their health care clinician. Even if the patient is not ready to take any definitive action regarding her relationship or situation, knowing that her clinician and the health care setting are resources and sources of support is both comforting and empowering.35,40 Clinicians can communicate this support by stating, “I want you to know that whatever happens and whatever you decide, we are here for you.”
Respect for autonomy. Women experiencing IPV best understand their own situation and its various complexities and so they know best what they can do, cannot do, or need to do. As such, clinicians must respect a woman’s autonomy and preserve her ability to express her own needs and desires and make her own decisions. Prescribing a plan of action or giving commands to IPV victims could further perpetuate their sense of disempowerment and lack of control over their life.
Information. Providing referral information or hotline numbers to community domestic violence programs is helpful. However, not all women feel safe or comfortable taking printed brochures or written information with them because their abusive partner may find it. Thus, clinicians should ask if the patient can take the information safely or offer to write the numbers down without labeling them if she is afraid. The National Domestic Violence hotline is a 24-hour toll-free resource that will help women locate and contact shelters and other support services in their own community. The number, 1-800-799-SAFE (7233), is easy to memorize and thus is an easy resource to pass along quickly and safely. Other national organizations such as Futures Without Violence (formerly known as the Family Violence Prevention Fund) and the National Coalition Against Violence also provide links to local resources (Table 1).
Safety planning. Discussing the need for a safety plan will help the patient prepare for future abusive episodes. Even women who report that they are no longer in an abusive relationship should be asked about their current safety needs and concerns because they often remain in contact with abusive partners even after a relationship has ended.
When discussing safety planning, clinicians should ask the woman if she is currently safe or if she needs shelter. If she intends to return to or is still in contact with her batterer, ask if she has a plan for what to do or how to escape if the violence occurs again. Advise the patient to:
- Hide money so that she can leave quickly
- Make copies of birth certificates, immunization records, Social Security Number, and other important documents and keep them hidden and accessible
- Make a spare car key
- Have a list of hotline numbers
- Develop a code with friends, family, and neighbors that will let them know she needs immediate help.
Studies show that discussing safety-promoting behaviors increases the number of them that are used by IPV victims.41 A detailed list that can be shared with patients is provided in the patient information page that accompanies this article.42 Examples of personalized safety plans are available from the National Center on Domestic and Sexual Violence at its website, www.ncdsv.org/images/NCDSV_DVSafetyPlan_updated2013.pdf.
Danger assessment. Several researchers have examined potential risk factors associated with increased risk of homicide.43 Table 2 lists some of the characteristics associated with an increased risk of homicide in IPV situations.42 From this work, a danger assessment tool and scoring system has been developed. This tool and training on how to use it are available for free at www.dangerassessment.org. Although there are currently no outcome data on the benefits or risks of using this instrument, its objective is to increase women’s awareness of their danger level and individualize their safety counseling.
Proper documentation. Victim advocates and lawyers working on behalf of IPV victims emphasize that documentation by a medical provider can help a woman with her legal case. This documentation should be clear, legible, and as detailed as possible. These details should include a patient’s own words set off by quotation marks, a description or body map illustrating associated injuries or physical signs corroborating the violence, and a description of the patient’s demeanor or signs of emotion. Clinicians should avoid legal terms such as “alleges” or “alleged perpetrator” and should either define or avoid abbreviations that may be considered ambiguous in a legal proceeding (eg, clinicians should write out the words “domestic violence” or “intimate partner violence” rather than using “DV” or “IPV”).44 Most states have passed laws that prevent insurance companies from discriminating against IPV victims; insurance companies can no longer deny women coverage for seeking care related to IPV.45
A nonthreatening physical examination. Women who have experienced physical or sexual violence may feel anxious or experience added trauma during certain portions of the physical examination. Women who have been raped or otherwise sexually abused may have difficulty tolerating a pelvic examination, for example. Others who have been choked or grabbed by the neck may experience distress during palpation of the thyroid or cervical lymph nodes. The oropharyngeal examination may be challenging for women who have experienced forced oral sex or who may have had objects such as the barrel of a gun forced into their mouth. Asking women who have experienced IPV what aspects of the physical examination they may find difficult and how this could be made easier allows the patient and clinician to work together to develop strategies for or alternatives to necessary examinations or testing.
Before starting an examination, clinicians should explain what will happen and then ask the patient for her permission before moving on to the next step. Doing so creates a sense of control in an otherwise challenging and potentially traumatizing examination.
Disclosure of mandatory IPV reporting. Many states require health care providers to report any injuries caused by a weapon or criminal act to police or other official institutions.45,46 Several of these states specify that this reporting should occur during an IPV evaluation.
Mandatory reporting of IPV to police is highly controversial.47–49 Opponents of these laws argue that they do not benefit victims because they do not respect autonomy, they violate patient-clinician confidentiality, and they violate informed consent.47 Female IPV victims indicate that they would be less likely to disclose IPV and seek help if they knew it had to be reported.50–53 In a survey of California physicians, 59% stated that they might not comply with reporting laws if the patient objected.54
In states with mandatory reporting laws, clinicians can advocate for their patients by disclosing whether IPV must be reported before beginning their examination. The Michigan Coalition Against Domestic Violence devised this sample statement:
“I do have to let you know that under state law, I am obligated to make a police report to (name police agency with jurisdiction over the facility), if I/we are treating someone for any injury sustained by means of violence (tailor this information to the specifics of your state law). So, if you would have any concerns about that, I would encourage you to speak with one of the domestic violence advocates/counselors at (DV agency), who are able to provide confidential services and help without being required to make a police report.”55
The Compendium of State Statutes and Polices on Domestic Violence and Health Care by Futures Without Violence is an excellent reference that describes current state laws and policies regarding domestic violence and includes descriptions of the specific characteristics of each state’s mandatory reporting laws and their implications for health care providers. A copy can be downloaded from www.futureswithout-violence.org/content/features/detail/1584/.
What not to do
Clinicians should not ignore or minimize the extent of IPV, make excuses for the batterer, or blame the woman.32,56,57 They also should not inadvertently blame or criticize the woman by asking her, “What did you do to deserve this?” or “Why don’t you just leave?”57
While leaving a violent relationship is an obvious solution, many women experiencing IPV are either unwilling or unable to do so. In fact, leaving the batterer can be just as dangerous as staying. In a North Carolina study, half of all women killed by their partner had divorced or broken up with the partner immediately before the murder.4
What to do when a woman does not disclose IPV
Women who have experienced IPV often deny the violence to others out of fear. So a denial of violence, even in highly suspicious cases, can be expected. Abused patients often want help but are confused and afraid to ask for it.58,59 Providing easy and anonymous access to IPV information and resources through posters, flyers, brochures, and booklets will allow any woman—regardless of disclosure—to obtain help.36
In one IPV trial, all women in a family planning clinic who were in the intervention group were given information about IPV and how it can affect sexual and reproductive health, whether or not they disclosed. Compared with women in the control group, women who received the intervention—both those who did and did not experience IPV—were 63% more likely than those in the control group to end a relationship because they perceived it to be unhealthy or unsafe. In this case, all women—not just those who were victims—benefited from receiving information about IPV.60
PROVIDERS’ SUPPORT MAKES A DIFFERENCE, IMPROVES OUTCOMES
Patients may seek help for and find safety from IPV in stages or steps, and it may take them several months or even years to do so.61–63 In this sense, then, IPV is an issue that is not likely to be “cured” or changed in a single medical visit. In addition, a single visit may not be associated with direct health and safety outcomes. However, a patient is more likely to make changes if she has supportive and informative interaction with a caring, nonjudgmental clinician who can increase her awareness of IPV and IPV resources and promote an improved sense of self-efficacy or perceived power.40
For example, in a study in which health clinicians expressed concern about potential IPV health effects and offered support services such as counseling, 67% of women used one of those resources at least once.40 In another study, women who talked to a health care provider about their abuse were almost four times more likely to use an IPV intervention (eg, advocacy, shelter, restraining order) than women who did not talk to their provider. Those who used an IPV intervention were 2.6 times more likely to leave their abusive relationship, and those who left reported improved physical health.64
Thus, health care providers can have a positive effect by addressing this complex and difficult issue. The power and influence of a provider’s kindness, empathy, and support cannot be overestimated in their ability to improve the safety and well-being of women dealing with IPV.
- Saltzman LE, Fanslow JL, McMahon PM, Shelley GA; National Center for Injury Prevention and Control; Centers for Disease Control and Prevention. Intimate partner violence surveillance: uniform definitions and recommended data elements, Version 1.0. Atlanta, GA; 1999. www.cdc.gov/ncipc/pub-res/ipv_surveillance/intimate%20partner%20violence.pdf. Accessed June 3, 2014.
- Black MC, Basile KC, Breiding MJ, et al; Center for Injury Prevention and Control; Centers for Disease Control and Prevention. The National Intimate Partner and Sexual Violence Survey (NISVS): 2010 Summary Report. Atlanta, GA. www.cdc.gov/violenceprevention/pdf/nisvs_executive_summary-a.pdf. Accessed June 3, 2014.
- Kellermann AL, Mercy JA. Men, women, and murder: gender-specific differences in rates of fatal violence and victimization. J Trauma 1992; 33:1–5.
- Moracco KE, Runywan CW, Butts JD. Femicide in North Carolina, 1991–1993: a statewide study of patterns and precursors. Homicide Studies 1998; 4:422–446.
- Wadman MC, Muelleman RL. Domestic violence homicides: ED use before victimization. Am J Emerg Med 1999; 17:689–691.
- Catalano SM; United States Bureau of Justice Statistics. Intimate partner violence in the United States: 2007. http://bjs.ojp.usdoj.gov/index.cfm?ty=pbdetail&iid=1000. Accessed February 19, 2014.
- Drossman DA, Talley NJ, Leserman J, Olden KW, Barreiro MA. Sexual and physical abuse and gastrointestinal illness. Review and recommendations. Ann Intern Med 1995; 123:782–794.
- Scholle SH, Rost KM, Golding JM. Physical abuse among depressed women. J Gen Intern Med 1998; 13:607–613.
- Walling MK, O’Hara MW, Reiter RC, Milburn AK, Lilly G, Vincent SD. Abuse history and chronic pain in women: II. A multivariate analysis of abuse and psychological morbidity. Obstet Gynecol 1994; 84:200–206.
- McCauley J, Kern DE, Kolodner K, Derogatis LR, Bass EB. Relation of low-severity violence to women’s health. J Gen Intern Med 1998; 13:687–691.
- Wingood GM, DiClemente RJ. The effects of an abusive primary partner on the condom use and sexual negotiation practices of African-American women. Am J Public Health 1997; 87:1016–1018.
- Sales JM, Salazar LF, Wingood GM, DiClemente RJ, Rose E, Crosby RA. The mediating role of partner communication skills on HIV/STD-associated risk behaviors in young African American females with a history of sexual violence. Arch Pediatr Adolesc Med 2008; 162:432–438.
- Davila YR, Brackley MH. Mexican and Mexican American women in a battered women’s shelter: barriers to condom negotiation for HIV/AIDS prevention. Issues Ment Health Nurs 1999; 20:333–355.
- Tjaden P, Thoennes N; National Institute of Justice; Centers for Disease Control and Prevention. Prevalence, incidence and consequences of violence against women: findings from the national violence against women survey. Washington, DC; 1998. https://www.ncjrs.gov/pdffiles/172837.pdf. Accessed June 3, 2014.
- Masho SW, Odor RK, Adera T. Sexual assault in Virginia: a population-based study. Womens Health Issues 2005; 15:157–166.
- Miller E, Decker MR, McCauley HL, et al. Pregnancy coercion, intimate partner violence and unintended pregnancy. Contraception 2010; 81:316–322.
- Bonomi AE, Anderson ML, Rivara FP, Thompson RS. Health care utilization and costs associated with physical and nonphysical-only intimate partner violence. Health Serv Res 2009; 44:1052–1067.
- Rivara FP, Anderson ML, Fishman P, et al. Healthcare utilization and costs for women with a history of intimate partner violence. Am J Prev Med 2007; 32:89–96.
- American Nurses Association. Position statement on physical violence against women. Washington, DC; 1991.
- Physicians and domestic violence. Ethical considerations. Council on Ethical and Judicial Affairs, American Medical Association. JAMA 1992; 267:3190–3193.
- The American College of Obstetricians and Gynecologists. Screening tools: domestic violence. http://www.acog.org/About_ACOG/ACOG_Departments/Violence_Against_Women/Screening_Tools__Domestic_Violence. Accessed June 3, 2014.
- Lee D, James L, Sawires P; The Family Violence Prevention Fund. Preventing domestic violence: clinical guidelines on routine screening. San Francisco, CA; 1999. http://new.vawnet.org/Assoc_Files_VAWnet/screpol.pdf. Accessed June 3, 2014.
- Joint Commission on Accreditation of Healthcare Organizations. Accreditation manual for hospitals. Chicago, IL; 1992.
- US Department of Health and Human Services. US Preventive Services Task Force. Guide to Clinical Preventive Services. 2nd ed. Washington, DC; 1996.
- Ramsay J, Richardson J, Carter YH, Davidson LL, Feder G. Should health professionals screen women for domestic violence? Systematic review. BMJ 2002; 325:314.
- Nelson HD, Nygren P, McInerney Y, Klein J; US Preventive Services Task Force. Screening women and elderly adults for family and intimate partner violence: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2004; 140:387–396.
- Chamberlain L. The USPSTF recommendation on intimate partner violence: what we can learn from it and what we can do about it. Fam Viol Prev Health Pract 2005; 1:1–24.
- Moyer VA; US Preventive Services Task Force. Screening for intimate partner violence and abuse of elderly and vulnerable adults: US Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158:478–486.
- Taft A, O’Doherty L, Hegarty K, Ramsay J, Davidson L, Feder G. Screening women for intimate partner violence in healthcare settings. Cochrane Database Syst Rev 2013; 4:CD007007.
- McNutt LA, Carlson BE, Gagen D, Winterbauer N. Reproductive violence screening in primary care: perspectives and experiences of patients and battered women. J Am Med Womens Assoc 1999; 54:85–90.
- Friedman LS, Samet JH, Roberts MS, Hudlin M, Hans P. Inquiry about victimization experiences. A survey of patient p and physician practices. Arch Intern Med 1992; 152:1186–1190.
- Hamberger LK, Ambuel B, Marbella A, Donze J. Physician interaction with battered women: the women’s perspective. Arch Fam Med 1998; 7:575–582.
- Rodriguez MA, Bauer HM, Flores-Ortiz Y, Szkupinski-Quiroga S. Factors affecting patient-physician communication for abused Latina and Asian immigrant women. J Fam Pract 1998; 47:309–311.
- Rhodes KV, Frankel RM, Levinthal N, Prenoveau E, Bailey J, Levinson W. “You’re not a victim of domestic violence, are you?” Provider patient communication about domestic violence”. Ann Intern Med 2007; 147:620–627.
- Chang JC, Decker M, Moracco KE, Martin SL, Petersen R, Frasier PY. What happens when health care providers ask about intimate partner violence? A description of consequences from the perspectives of female survivors. J Am Med Womens Assoc 2003; 58:76–81.
- Chang JC, Decker MR, Moracco KE, Martin SL, Petersen R, Frasier PY. Asking about intimate partner violence: advice from female survivors to health care providers. Patient Educ Couns 2005; 59:141–147.
- Hathaway JE, Willis G, Zimmer B. Listening to survivors’ voices: addressing partner abuse in the health care setting. Violence Against Women 2002; 8:687–716.
- Gerbert B, Caspers N, Bronstone A, Moe J, Abercrombie P. A qualitative analysis of how physicians with expertise in domestic violence approach the identification of victims. Ann Intern Med 1999; 131:578–584.
- Gerbert B, Caspers N, Milliken N, Berlin M, Bronstone A, Moe J. Interventions that help victims of domestic violence. A qualitative analysis of physicians’ experiences. J Fam Pract 2000; 49:889–895.
- McCaw B, Bauer HM, Berman WH, Mooney L, Holmberg M, Hunkeler E. Women referred for on-site domestic violence services in a managed care organization. Women Health 2002; 35:23–40.
- McFarlane J, Malecha A, Gist J, et al. An intervention to increase safety behaviors of abused women: results of a randomized clinical trial. Nurs Res 2002; 51:347–354.
- Chang JC. Domestic violence. In:Bieber EJ, Sanfilippo JS, Horowitz IR, editors. Clinical Gynecology. Philadelphia, PA; Elsevier, Inc; 2006:79–89.
- Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide in abusive relationships: results from a multisite case control study. Am J Public Health 2003; 93:1089–1097.
- Isaac NE, Enos V; National Institute of Justice. Documenting domestic violence: how health care providers can help victims. Research in Brief, 2001. https://www.ncjrs.gov/pdffiles1/nij/188564.pdf. Accessed June 3, 2014.
- Durborow N, Lizdas KC, O’Flaherty A, Marjavi A; Futures Without Violence. Compendium of state and US terrritory statutes and policies on domestic violence and health care, 2010. www.futureswithoutviolence.org/content/features/detail/1584/. Accessed June 3, 2014.
- Hyman A, Schillinger D, Lo B. Laws mandating reporting of domestic violence. Do they promote patient well-being? JAMA 1995; 273:1781–1787.
- Hyman A, Chez RA. Mandatory reporting of domestic violence by health care providers: a misguided approach. Womens Health Issues 1995; 5:208–213.
- Knight MA. Ethical debate: should doctors be more proactive as advocates for victims of violence? The police surgeon’s view: medical paternalism is unacceptable. BMJ 1995; 311:1620–1621.
- Hyman A; Futures Without Violence. Mandatory reporting of domestic violence by healthcare providers: a policy paper. San Francisco, CA; 1997. www.futureswithoutviolence.org/userfiles/file/HealthCare/mandatory_policypaper.pdf. Accessed June 3, 2014.
- Rodriguez MA, Craig AM, Mooney DR, Bauer HM. Patient attitudes about mandatory reporting of domestic violence. Implications for health care professionals. West J Med 1998; 169:337–341.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Sachs CJ, Koziol-McLain J, Glass N, Webster D, Campbell J. A population-based survey assessing support for mandatory domestic violence reporting by health care personnel. Women Health 2002; 35:121–133.
- Sullivan CM, Hagen LA. Survivors’ opinions about mandatory reporting of domestic violence and sexual assault by medical professionals. Affilia 2005; 20:1–16.
- Rodriguez MA, McLoughlin E, Bauer HM, Paredes V, Grumbach K. Mandatory reporting of intimate partner violence to police: views of physicians in California. Am J Public Health 1999; 89:575–578.
- Futures Without Violence. Mandatory reporting of domestic violence to law enforcement by health care providers: a guide for advocates working to respond to or amend reporting laws related to domestic violence. www.healthcaresaboutipv.org/wp-content/blogs.dir/3/files/2012/09/Mandatory_Reporting_of_DV_to_Law-Enforcement_by_HCP.pdf. Accessed June 3, 2014.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Gerbert B, Johnston K, Caspers N, Bleecker T, Woods A, Rosenbaum A. Experiences of battered women in health care settings: a qualitative study. Women Health 1996; 24:1–17.
- Chang JC, Cluss PA, Ranieri L, et al. Health care interventions for intimate partner violence: what women want. Womens Health Issues 2005; 15:21–30.
- Rodriguez MA, Quiroga SS, Bauer HM. Breaking the silence. Battered women’s perspectives on medical care. Arch Fam Med 1996; 5:153–158.
- Miller E, Decker MR, McCauley HL, et al. A family planning clinic partner violence intervention to reduce risk associated with reproductive coercion. Contraception 2011; 83:274–280.
- Landenburger K. A process of entrapment in and recovery from an abusive relationship. Issues Ment Health Nurs 1989; 10:209–227.
- Cluss PA, Chang JC, Hawker L, et al. The process of change for victims of intimate partner violence: support for a psychosocial readiness model. Womens Health Issues 2006; 16:262–274.
- Gerbert B, Abercrombie P, Caspers N, Love C, Bronstone A. How health care providers help battered women: the survivor’s perspective. Women Health 1999; 29:115–135.
- McCloskey LA, Lichter E, Williams C, Gerber M, Wittenberg E, Ganz M. Assessing intimate partner violence in health care settings leads to women’s receipt of interventions and improved health. Public Health Rep 2006; 121:435–444.
- Saltzman LE, Fanslow JL, McMahon PM, Shelley GA; National Center for Injury Prevention and Control; Centers for Disease Control and Prevention. Intimate partner violence surveillance: uniform definitions and recommended data elements, Version 1.0. Atlanta, GA; 1999. www.cdc.gov/ncipc/pub-res/ipv_surveillance/intimate%20partner%20violence.pdf. Accessed June 3, 2014.
- Black MC, Basile KC, Breiding MJ, et al; Center for Injury Prevention and Control; Centers for Disease Control and Prevention. The National Intimate Partner and Sexual Violence Survey (NISVS): 2010 Summary Report. Atlanta, GA. www.cdc.gov/violenceprevention/pdf/nisvs_executive_summary-a.pdf. Accessed June 3, 2014.
- Kellermann AL, Mercy JA. Men, women, and murder: gender-specific differences in rates of fatal violence and victimization. J Trauma 1992; 33:1–5.
- Moracco KE, Runywan CW, Butts JD. Femicide in North Carolina, 1991–1993: a statewide study of patterns and precursors. Homicide Studies 1998; 4:422–446.
- Wadman MC, Muelleman RL. Domestic violence homicides: ED use before victimization. Am J Emerg Med 1999; 17:689–691.
- Catalano SM; United States Bureau of Justice Statistics. Intimate partner violence in the United States: 2007. http://bjs.ojp.usdoj.gov/index.cfm?ty=pbdetail&iid=1000. Accessed February 19, 2014.
- Drossman DA, Talley NJ, Leserman J, Olden KW, Barreiro MA. Sexual and physical abuse and gastrointestinal illness. Review and recommendations. Ann Intern Med 1995; 123:782–794.
- Scholle SH, Rost KM, Golding JM. Physical abuse among depressed women. J Gen Intern Med 1998; 13:607–613.
- Walling MK, O’Hara MW, Reiter RC, Milburn AK, Lilly G, Vincent SD. Abuse history and chronic pain in women: II. A multivariate analysis of abuse and psychological morbidity. Obstet Gynecol 1994; 84:200–206.
- McCauley J, Kern DE, Kolodner K, Derogatis LR, Bass EB. Relation of low-severity violence to women’s health. J Gen Intern Med 1998; 13:687–691.
- Wingood GM, DiClemente RJ. The effects of an abusive primary partner on the condom use and sexual negotiation practices of African-American women. Am J Public Health 1997; 87:1016–1018.
- Sales JM, Salazar LF, Wingood GM, DiClemente RJ, Rose E, Crosby RA. The mediating role of partner communication skills on HIV/STD-associated risk behaviors in young African American females with a history of sexual violence. Arch Pediatr Adolesc Med 2008; 162:432–438.
- Davila YR, Brackley MH. Mexican and Mexican American women in a battered women’s shelter: barriers to condom negotiation for HIV/AIDS prevention. Issues Ment Health Nurs 1999; 20:333–355.
- Tjaden P, Thoennes N; National Institute of Justice; Centers for Disease Control and Prevention. Prevalence, incidence and consequences of violence against women: findings from the national violence against women survey. Washington, DC; 1998. https://www.ncjrs.gov/pdffiles/172837.pdf. Accessed June 3, 2014.
- Masho SW, Odor RK, Adera T. Sexual assault in Virginia: a population-based study. Womens Health Issues 2005; 15:157–166.
- Miller E, Decker MR, McCauley HL, et al. Pregnancy coercion, intimate partner violence and unintended pregnancy. Contraception 2010; 81:316–322.
- Bonomi AE, Anderson ML, Rivara FP, Thompson RS. Health care utilization and costs associated with physical and nonphysical-only intimate partner violence. Health Serv Res 2009; 44:1052–1067.
- Rivara FP, Anderson ML, Fishman P, et al. Healthcare utilization and costs for women with a history of intimate partner violence. Am J Prev Med 2007; 32:89–96.
- American Nurses Association. Position statement on physical violence against women. Washington, DC; 1991.
- Physicians and domestic violence. Ethical considerations. Council on Ethical and Judicial Affairs, American Medical Association. JAMA 1992; 267:3190–3193.
- The American College of Obstetricians and Gynecologists. Screening tools: domestic violence. http://www.acog.org/About_ACOG/ACOG_Departments/Violence_Against_Women/Screening_Tools__Domestic_Violence. Accessed June 3, 2014.
- Lee D, James L, Sawires P; The Family Violence Prevention Fund. Preventing domestic violence: clinical guidelines on routine screening. San Francisco, CA; 1999. http://new.vawnet.org/Assoc_Files_VAWnet/screpol.pdf. Accessed June 3, 2014.
- Joint Commission on Accreditation of Healthcare Organizations. Accreditation manual for hospitals. Chicago, IL; 1992.
- US Department of Health and Human Services. US Preventive Services Task Force. Guide to Clinical Preventive Services. 2nd ed. Washington, DC; 1996.
- Ramsay J, Richardson J, Carter YH, Davidson LL, Feder G. Should health professionals screen women for domestic violence? Systematic review. BMJ 2002; 325:314.
- Nelson HD, Nygren P, McInerney Y, Klein J; US Preventive Services Task Force. Screening women and elderly adults for family and intimate partner violence: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2004; 140:387–396.
- Chamberlain L. The USPSTF recommendation on intimate partner violence: what we can learn from it and what we can do about it. Fam Viol Prev Health Pract 2005; 1:1–24.
- Moyer VA; US Preventive Services Task Force. Screening for intimate partner violence and abuse of elderly and vulnerable adults: US Preventive Services Task Force recommendation statement. Ann Intern Med 2013; 158:478–486.
- Taft A, O’Doherty L, Hegarty K, Ramsay J, Davidson L, Feder G. Screening women for intimate partner violence in healthcare settings. Cochrane Database Syst Rev 2013; 4:CD007007.
- McNutt LA, Carlson BE, Gagen D, Winterbauer N. Reproductive violence screening in primary care: perspectives and experiences of patients and battered women. J Am Med Womens Assoc 1999; 54:85–90.
- Friedman LS, Samet JH, Roberts MS, Hudlin M, Hans P. Inquiry about victimization experiences. A survey of patient p and physician practices. Arch Intern Med 1992; 152:1186–1190.
- Hamberger LK, Ambuel B, Marbella A, Donze J. Physician interaction with battered women: the women’s perspective. Arch Fam Med 1998; 7:575–582.
- Rodriguez MA, Bauer HM, Flores-Ortiz Y, Szkupinski-Quiroga S. Factors affecting patient-physician communication for abused Latina and Asian immigrant women. J Fam Pract 1998; 47:309–311.
- Rhodes KV, Frankel RM, Levinthal N, Prenoveau E, Bailey J, Levinson W. “You’re not a victim of domestic violence, are you?” Provider patient communication about domestic violence”. Ann Intern Med 2007; 147:620–627.
- Chang JC, Decker M, Moracco KE, Martin SL, Petersen R, Frasier PY. What happens when health care providers ask about intimate partner violence? A description of consequences from the perspectives of female survivors. J Am Med Womens Assoc 2003; 58:76–81.
- Chang JC, Decker MR, Moracco KE, Martin SL, Petersen R, Frasier PY. Asking about intimate partner violence: advice from female survivors to health care providers. Patient Educ Couns 2005; 59:141–147.
- Hathaway JE, Willis G, Zimmer B. Listening to survivors’ voices: addressing partner abuse in the health care setting. Violence Against Women 2002; 8:687–716.
- Gerbert B, Caspers N, Bronstone A, Moe J, Abercrombie P. A qualitative analysis of how physicians with expertise in domestic violence approach the identification of victims. Ann Intern Med 1999; 131:578–584.
- Gerbert B, Caspers N, Milliken N, Berlin M, Bronstone A, Moe J. Interventions that help victims of domestic violence. A qualitative analysis of physicians’ experiences. J Fam Pract 2000; 49:889–895.
- McCaw B, Bauer HM, Berman WH, Mooney L, Holmberg M, Hunkeler E. Women referred for on-site domestic violence services in a managed care organization. Women Health 2002; 35:23–40.
- McFarlane J, Malecha A, Gist J, et al. An intervention to increase safety behaviors of abused women: results of a randomized clinical trial. Nurs Res 2002; 51:347–354.
- Chang JC. Domestic violence. In:Bieber EJ, Sanfilippo JS, Horowitz IR, editors. Clinical Gynecology. Philadelphia, PA; Elsevier, Inc; 2006:79–89.
- Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide in abusive relationships: results from a multisite case control study. Am J Public Health 2003; 93:1089–1097.
- Isaac NE, Enos V; National Institute of Justice. Documenting domestic violence: how health care providers can help victims. Research in Brief, 2001. https://www.ncjrs.gov/pdffiles1/nij/188564.pdf. Accessed June 3, 2014.
- Durborow N, Lizdas KC, O’Flaherty A, Marjavi A; Futures Without Violence. Compendium of state and US terrritory statutes and policies on domestic violence and health care, 2010. www.futureswithoutviolence.org/content/features/detail/1584/. Accessed June 3, 2014.
- Hyman A, Schillinger D, Lo B. Laws mandating reporting of domestic violence. Do they promote patient well-being? JAMA 1995; 273:1781–1787.
- Hyman A, Chez RA. Mandatory reporting of domestic violence by health care providers: a misguided approach. Womens Health Issues 1995; 5:208–213.
- Knight MA. Ethical debate: should doctors be more proactive as advocates for victims of violence? The police surgeon’s view: medical paternalism is unacceptable. BMJ 1995; 311:1620–1621.
- Hyman A; Futures Without Violence. Mandatory reporting of domestic violence by healthcare providers: a policy paper. San Francisco, CA; 1997. www.futureswithoutviolence.org/userfiles/file/HealthCare/mandatory_policypaper.pdf. Accessed June 3, 2014.
- Rodriguez MA, Craig AM, Mooney DR, Bauer HM. Patient attitudes about mandatory reporting of domestic violence. Implications for health care professionals. West J Med 1998; 169:337–341.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Sachs CJ, Koziol-McLain J, Glass N, Webster D, Campbell J. A population-based survey assessing support for mandatory domestic violence reporting by health care personnel. Women Health 2002; 35:121–133.
- Sullivan CM, Hagen LA. Survivors’ opinions about mandatory reporting of domestic violence and sexual assault by medical professionals. Affilia 2005; 20:1–16.
- Rodriguez MA, McLoughlin E, Bauer HM, Paredes V, Grumbach K. Mandatory reporting of intimate partner violence to police: views of physicians in California. Am J Public Health 1999; 89:575–578.
- Futures Without Violence. Mandatory reporting of domestic violence to law enforcement by health care providers: a guide for advocates working to respond to or amend reporting laws related to domestic violence. www.healthcaresaboutipv.org/wp-content/blogs.dir/3/files/2012/09/Mandatory_Reporting_of_DV_to_Law-Enforcement_by_HCP.pdf. Accessed June 3, 2014.
- Caralis PV, Musialowski R. Women’s experiences with domestic violence and their attitudes and expectations regarding medical care of abuse victims. South Med J 1997; 90:1075–1080.
- Gerbert B, Johnston K, Caspers N, Bleecker T, Woods A, Rosenbaum A. Experiences of battered women in health care settings: a qualitative study. Women Health 1996; 24:1–17.
- Chang JC, Cluss PA, Ranieri L, et al. Health care interventions for intimate partner violence: what women want. Womens Health Issues 2005; 15:21–30.
- Rodriguez MA, Quiroga SS, Bauer HM. Breaking the silence. Battered women’s perspectives on medical care. Arch Fam Med 1996; 5:153–158.
- Miller E, Decker MR, McCauley HL, et al. A family planning clinic partner violence intervention to reduce risk associated with reproductive coercion. Contraception 2011; 83:274–280.
- Landenburger K. A process of entrapment in and recovery from an abusive relationship. Issues Ment Health Nurs 1989; 10:209–227.
- Cluss PA, Chang JC, Hawker L, et al. The process of change for victims of intimate partner violence: support for a psychosocial readiness model. Womens Health Issues 2006; 16:262–274.
- Gerbert B, Abercrombie P, Caspers N, Love C, Bronstone A. How health care providers help battered women: the survivor’s perspective. Women Health 1999; 29:115–135.
- McCloskey LA, Lichter E, Williams C, Gerber M, Wittenberg E, Ganz M. Assessing intimate partner violence in health care settings leads to women’s receipt of interventions and improved health. Public Health Rep 2006; 121:435–444.
KEY POINTS
- Many victims of IPV will not present with injuries but may have medical or mental health issues related to their IPV experiences.
- Addressing IPV with female patients not only results in increased identification of survivors but is also associated with cognitive and emotional benefits.
- IPV information and resources should be provided to all women, regardless of IPV disclosure.
- Clinicians should respond to a patient’s IPV disclosure with validation, support, respect, and information.
- Clinicians must respect patients’ autonomy, as they are the ones who best understand their situation and know what they need. In some cases, leaving an abusive relationship can be more dangerous than staying.
Planning ahead: How to stay safe
If you are being abused, making a safety plan now may help you when you have to act quickly in the future. The following ideas are ways that other women have planned for their safety. Some of these ideas may work for you. You may come up with additional ideas for yourself. You know your own situation better than anyone else, so plan what will work best for you.
- Hide money or put it somewhere safe so you can leave quickly.
- Make copies of birth certificates, immunization records, Social Security numbers, and other important documents to keep in safe locations away from home such as at work, the homes of trusted family members or friends, or hidden in convenient locations.
- Hide a spare car key or bus or subway pass that you can grab quickly.
- Keep a list of hotline numbers, or memorize the 1-800-799-SAFE National Domestic Violence Hotline number.
- Develop a code with friends, family, or neighbors to let them know when you need help in an emergency. If you have children, teach them a signal (like a code word) that means they should call the police or go for help. You may want to have a special code for neighbors (like putting on a particular light or opening a certain window) that means you want them to call the police.
- Plan your exit. Know which doors, windows, stairwells, elevators, or fire escapes you can use if you have to leave quickly. Practice using them so that they feel familiar to you.
- Know how to reach the police and your local women’s shelter.
- Every day, think about where you can go immediately if you have to leave. Is a neighbor home today? A relative? A friend?
- Remove weapons from your home if you can.
- Something to think about: When you cannot get away and your partner becomes violent, which room is the safest for you to get to? Is there a room that has a phone and a lock on the door? Can you stay out of rooms with easy weapons, such as the kitchen?
- Try not to leave without your children. But if you have to leave your children with the abuser, call the police immediately after you escape.

This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Center for Consumer Health Information web site, www.clevelandclinic.org/health
If you are being abused, making a safety plan now may help you when you have to act quickly in the future. The following ideas are ways that other women have planned for their safety. Some of these ideas may work for you. You may come up with additional ideas for yourself. You know your own situation better than anyone else, so plan what will work best for you.
- Hide money or put it somewhere safe so you can leave quickly.
- Make copies of birth certificates, immunization records, Social Security numbers, and other important documents to keep in safe locations away from home such as at work, the homes of trusted family members or friends, or hidden in convenient locations.
- Hide a spare car key or bus or subway pass that you can grab quickly.
- Keep a list of hotline numbers, or memorize the 1-800-799-SAFE National Domestic Violence Hotline number.
- Develop a code with friends, family, or neighbors to let them know when you need help in an emergency. If you have children, teach them a signal (like a code word) that means they should call the police or go for help. You may want to have a special code for neighbors (like putting on a particular light or opening a certain window) that means you want them to call the police.
- Plan your exit. Know which doors, windows, stairwells, elevators, or fire escapes you can use if you have to leave quickly. Practice using them so that they feel familiar to you.
- Know how to reach the police and your local women’s shelter.
- Every day, think about where you can go immediately if you have to leave. Is a neighbor home today? A relative? A friend?
- Remove weapons from your home if you can.
- Something to think about: When you cannot get away and your partner becomes violent, which room is the safest for you to get to? Is there a room that has a phone and a lock on the door? Can you stay out of rooms with easy weapons, such as the kitchen?
- Try not to leave without your children. But if you have to leave your children with the abuser, call the police immediately after you escape.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Center for Consumer Health Information web site, www.clevelandclinic.org/health
If you are being abused, making a safety plan now may help you when you have to act quickly in the future. The following ideas are ways that other women have planned for their safety. Some of these ideas may work for you. You may come up with additional ideas for yourself. You know your own situation better than anyone else, so plan what will work best for you.
- Hide money or put it somewhere safe so you can leave quickly.
- Make copies of birth certificates, immunization records, Social Security numbers, and other important documents to keep in safe locations away from home such as at work, the homes of trusted family members or friends, or hidden in convenient locations.
- Hide a spare car key or bus or subway pass that you can grab quickly.
- Keep a list of hotline numbers, or memorize the 1-800-799-SAFE National Domestic Violence Hotline number.
- Develop a code with friends, family, or neighbors to let them know when you need help in an emergency. If you have children, teach them a signal (like a code word) that means they should call the police or go for help. You may want to have a special code for neighbors (like putting on a particular light or opening a certain window) that means you want them to call the police.
- Plan your exit. Know which doors, windows, stairwells, elevators, or fire escapes you can use if you have to leave quickly. Practice using them so that they feel familiar to you.
- Know how to reach the police and your local women’s shelter.
- Every day, think about where you can go immediately if you have to leave. Is a neighbor home today? A relative? A friend?
- Remove weapons from your home if you can.
- Something to think about: When you cannot get away and your partner becomes violent, which room is the safest for you to get to? Is there a room that has a phone and a lock on the door? Can you stay out of rooms with easy weapons, such as the kitchen?
- Try not to leave without your children. But if you have to leave your children with the abuser, call the police immediately after you escape.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Center for Consumer Health Information web site, www.clevelandclinic.org/health
Promoting higher blood pressure targets for frail older adults: A consensus guideline from Canada
Frail older adults deserve guidelines that take frailty into account while assessing the potential benefit and risks of treatment.
Specifically, our group—the Dalhousie Academic Detailing Service (ADS) and the Palliative and Therapeutic Harmonization (PATH) program—recommends that physicians strive to achieve more liberal treatment targets for elderly frail patients who have high blood pressure,1 as evidence does not support an aggressive approach in the frail elderly and the potential exists for harm.
This article reviews the evidence and reasoning that were used to develop and promote a guideline for drug treatment of hypertension in frail older adults. Our recommendations differ from other guidelines in that they focus as much on stopping or decreasing therapy as on starting or increasing it.
FRAILTY INCREASES THE RISK OF ADVERSE EFFECTS
The word frail, applied to older adults, describes those who have complex medical illnesses severe enough to compromise their ability to live independently.2 Many have multiple coexisting medical problems for which they take numerous drugs, in addition to dementia, impaired mobility, compromised functional ability, or a history of falling.

Frailty denotes vulnerability; it increases the risk of adverse effects from medical and surgical procedures,3 complicates drug therapy,4 prolongs hospital length of stay,5 leads to functional and cognitive decline,6 increases the risk of institutionalization,7 and reduces life expectancy8—all of which affect the benefit and harm of medical treatments.
Guidelines for treating hypertension9–11 now acknowledge that little evidence exists to support starting treatment for systolic blood pressure between 140 and 160 mm Hg or aiming for a target of less than 140 mm Hg for “very old” adults, commonly defined as over the age of 80. New guidelines loosen the treatment targets for the very old, but they do not specify targets for the frail and do not describe how to recognize or measure frailty.
RECOGNIZING AND MEASURING FRAILTY
A number of tools are available to recognize and measure frailty.12
The Fried frailty assessment13 has five items:
- Unintentional weight loss
- Self-reported exhaustion
- Weakness in grip
- Slow walking speed
- Low physical activity and energy expenditure.
People are deemed frail if they have three or more of these five. However, experts disagree about whether this system is too sensitive14 or not sensitive enough.15,16
The FRAIL questionnaire17 also has five items:
- Fatigue
- Resistance (inability to climb stairs)
- Ambulation (inability to walk 1 city block)
- Illness (more than 5 major illnesses)
- Weight loss.
People are deemed frail if they have at least three of these five items, and “prefrail” if they have two.
These and other tools are limited by being dichotomous: they classify people as being either frail or not frail18–20 but do not define the spectrum of frailty.
Other frailty assessments such as the Frailty Index21 identify frailty based on the number of accumulated health deficits but take a long time to complete, making them difficult to use in busy clinical settings.22–24
The Clinical Frailty Scale7 is a validated scale that categorizes frailty based on physical and functional indicators of health, such as cognition, function, and mobility, with scores that range from 1 (very fit) to 9 (terminally ill).7,12
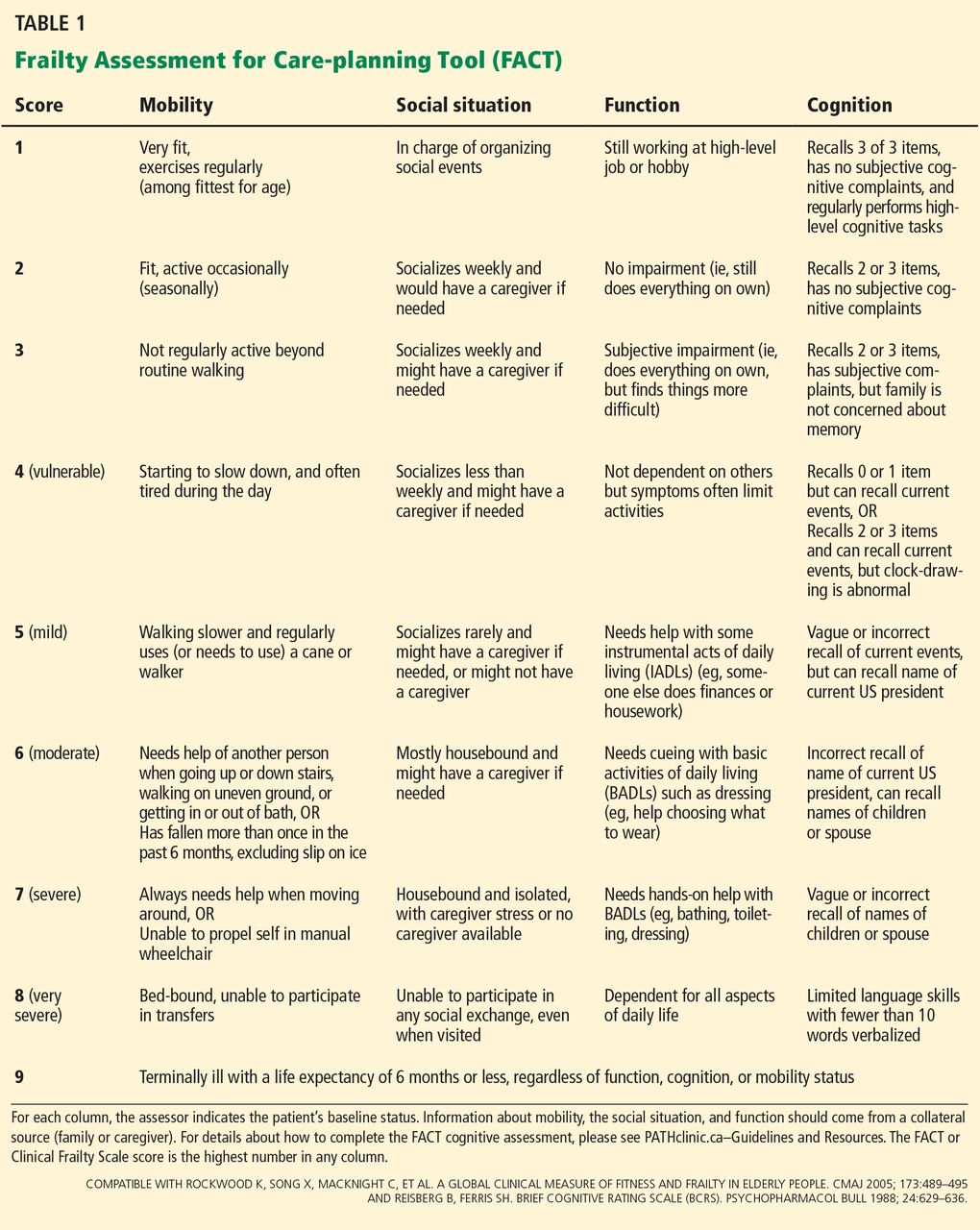
The Frailty Assessment for Care-planning Tool (FACT) uses scaling compatible with the Clinical Frailty Scale but has been developed for use as a practical and interpretable frailty screening tool for nonexperts (Table 1). The FACT assesses cognition, mobility, function, and the social situation, using a combination of caregiver report and objective measures. To assess cognition, a health care professional uses items from the Mini-Cog25 (ie, the ability to draw an analog clock face and then recall three unrelated items following the clock-drawing test) and the memory axis of the Brief Cognitive Rating Scale26 (ie, the ability to recall current events, the current US president, and the names of children or spouse). Mobility, function, and social circumstance scores are assigned according to the caregiver report of the patient’s baseline status.
The FACT can be completed in busy clinical settings. Once a caregiver is identified, it takes about 5 minutes to complete.
Our guideline27–31 is intended for those with a score of 7 or more on the Clinical Frailty Scale or FACT,7,12 a score we chose because it describes people who are severely frail with shortened life expectancy.8 At this level, people need help with all instrumental activities of daily living (eg, handling finances, medication management, household chores, and shopping) as well as with basic activities of daily living such as bathing or dressing.
REVIEWING THE LIMITED EVIDENCE
We found no studies that addressed the risks and benefits of treating hypertension in frail older adults; therefore, we concentrated on studies that enrolled individuals who were chronologically old but not frail. We reviewed prominent guidelines,9–11,32,33 the evidence base for these guidelines,34–44 and Cochrane reviews.45,46 A detailed description of the evidence used to build our recommendation can be found online.31
When we deliberated on treatment targets, we reviewed evidence from two types of randomized controlled trials47:
Drug treatment trials randomize patients to different treatments, such as placebo versus a drug or one drug compared with another drug. Patients in different treatment groups may achieve different blood pressures and clinical outcomes, and this information is then used to define optimal targets. However, it may be difficult to determine if the benefit came from lowering blood pressure or from some other effect of the drug, which can be independent of blood pressure lowering.
Treat-to-target trials randomize patients to different blood pressure goals, but the groups are treated with the same or similar drugs. Therefore, any identified benefit can be attributed to the differences in blood pressure rather than the medications used. Compared with a drug treatment trial, this type of trial provides stronger evidence about optimal targets.
We also considered the characteristics of frailty, the dilemma of polypharmacy, and the relevance of the available scientific evidence to those who are frail.
Drug treatment trials
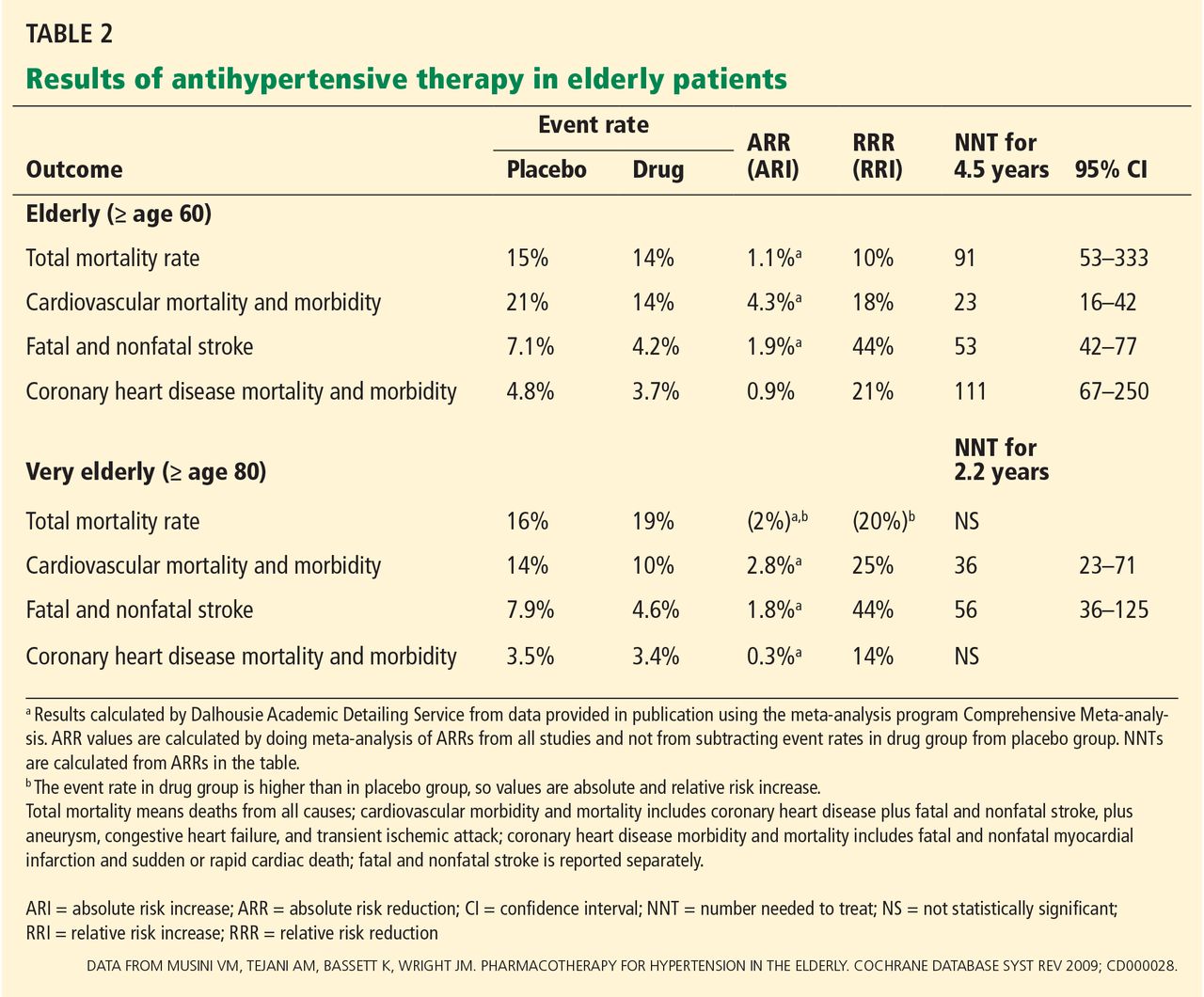
A Cochrane review45 of 15 studies with approximately 24,000 elderly participants found that treating hypertension decreased the rates of cardiovascular morbidity and mortality as well as fatal and nonfatal stroke in the “elderly” (defined as age ≥ 60) and “very elderly” (age ≥ 80). However, in the very elderly, all-cause mortality rates were not statistically significantly different with treatment compared with placebo. The mean duration of treatment was 4.5 years in the elderly and 2.2 years in the very elderly (Table 2). Of importance, all the trials enrolled only those individuals whose systolic blood pressure was at least 160 mm Hg at baseline.
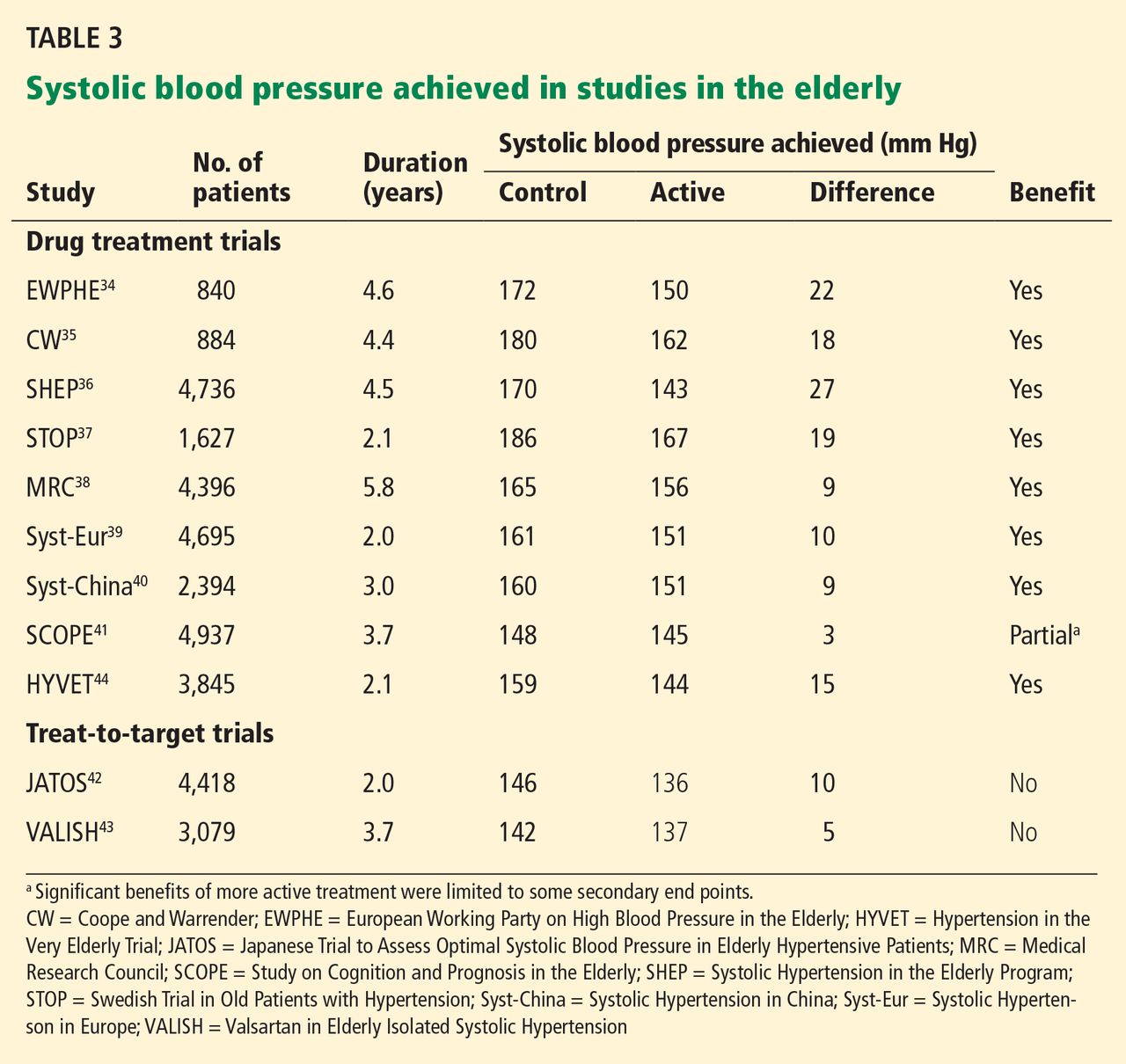
None of the studies were treat-to-target trials—patients were assigned either active medication or placebo. Thus, these trials provide evidence of benefit for treating hypertension in the elderly and very elderly but do not identify the optimal target. All of the drug treatment trials showed benefit, but none achieved a systolic pressure lower than 140 mm Hg with active treatment (Table 3). Therefore, these studies do not support a systolic target of less than 140 mm Hg in the elderly.
Treat-to-target trials: JATOS and VALISH
The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)42 and the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) study43 each enrolled more than 3,000 people age 65 or older (mean age approximately 75). Patients were randomized to either a strict systolic target of less than 140 mm Hg or a higher (more permissive) target of 140 to 160 mm Hg in JATOS and 140 to 149 mm Hg in VALISH.
In both trials, the group with strict targets achieved a systolic pressure of approximately 136 mm Hg, while the group with higher blood pressure targets achieved a systolic pressure of 146 mm Hg in JATOS and 142 mm Hg in VALISH. Despite these differences, there was no statistically significant difference in the primary outcome.
Thus, treat-to-target studies also fail to support a systolic target of less than 140 mm Hg in the elderly, although it is important to recognize the limitations of the studies. Approximately 15% of the participants had cardiovascular disease, so the applicability of the findings to patients with target-organ damage is uncertain. In addition, there were fewer efficacy outcome events than expected, which suggests that the studies were underpowered.
When to start drug treatment
In each of the drug treatment and treat-to-target trials, the inclusion criterion for study entry was a systolic blood pressure above 160 mm Hg, with a mean blood pressure at entry into the drug treatment trials of 182/95 mm Hg.46 Thus, data support starting treatment if the systolic blood pressure is above 160 mm Hg, but not lower.
Notably, in all but one study,46 at least two-thirds of the participants took no more than two antihypertensive medications. Since adverse events become more common as the number of medications increases, the benefit of adding a third drug to lower blood pressure is uncertain.
Evidence in the ‘very elderly’: HYVET
With the exception of the Hypertension in the Very Elderly Trial (HYVET),44 the mean age of elderly patients in the reported studies was between 67 and 76.
HYVET patients were age 80 and older (mean age 84) and were randomized to receive either indapamide (with or without perindopril) or placebo. The trial was stopped early at 2 years because the mortality rate was lower in the treatment group (10.1%) than in the placebo group (12.3%) (number needed to treat 46, 95% confidence interval 24–637, P = .02). There was no significant difference in the primary outcome of fatal and nonfatal stroke.
Notably, trials that are stopped early may overestimate treatment benefit.48
Evidence in frail older adults
While the above studies provide some information about managing hypertension in the elderly, the participants were generally healthy. HYVET44 specifically excluded those with a standing systolic blood pressure of less than 140 mm Hg and enrolled few patients with orthostasis (7.9% in the placebo group and 8.8% in the treatment group), a condition commonly associated with frailty. As such, these studies may be less relevant to the frail elderly, who are at higher risk of adverse drug events and have competing risks for morbidity and mortality.
Observational studies, in fact, raise questions about whether tight blood pressure control improves clinical outcomes for the very elderly. In the Leiden 85-plus study, lower systolic blood pressure was associated with lower cognitive scores, worse functional ability,49,50 and a higher mortality rate51 compared with higher systolic pressure, although it is uncertain whether these outcomes were indicative of underlying disease that could result in lower blood pressure or an effect of blood pressure-lowering.
The National Health and Nutrition Examination Survey52 found an association between blood pressure and mortality rate that varied by walking speed. For slower walkers (based on the 6-minute walk test), higher systolic pressures were not associated with a higher risk of death, suggesting that when older adults are frail (as indicated by their slow walking speed) they are less likely to benefit from aggressive treatment of hypertension.
People at high risk because of stroke
Because the evidence is limited, it is even more difficult to judge whether lowering blood pressure below 140 mm Hg is beneficial for frail patients who have a history of stroke, compared with the possibility that medications will cause adverse effects such as weakness, orthostasis, and falls. When reviewing the evidence to answer this question, we especially looked at outcomes that affect quality of life, such as nonfatal stroke leading to disability. In contrast, because the frail elderly have competing causes of mortality, we could not assume that a mortality benefit shown in nonfrail populations could be applied to frail populations.
The PROGRESS trial (Perindopril Protection Against Recurrent Stroke Study)53 was in patients with a history of stroke or transient ischemic attack and a mean age of 64, who were treated with either perindopril (with or without indapamide) or placebo.
At almost 4 years, the rate of disabling stroke was 2.7% in the treatment group and 4.3% in the placebo group, a relative risk reduction of 38% and an absolute risk reduction of 1.64% (number needed to treat 61, 95% confidence interval 39–139). The relative risk reduction for all strokes (fatal and nonfatal) was similar across a range of baseline systolic pressures, but the absolute risk reduction was greater in the prespecified subgroup that had hypertension at baseline (mean blood pressure 159/94 mm Hg) than in the normotensive subgroup (mean blood pressure 136/79 mm Hg), suggesting that treatment is most beneficial for those with higher systolic blood pressures. Also, the benefit was only demonstrated in the subgroup that received two antihypertensive medications; those who received perindopril alone showed no benefit.
This study involved relatively young patients in relatively good health except for their strokes. The extent to which the results can be extrapolated to older, frail adults is uncertain because of the time needed to achieve benefit and because of the added vulnerability of frailty, which could make treatment with two antihypertensive medications riskier.
PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes),54 another study in patients with previous stroke (mean age 66) showed no benefit over 2.5 years in the primary outcome of stroke using telmesartan 80 mg daily compared with placebo. This result is concordant with that of PROGRESS,53 in which patients who took only one medication did not show a significant decrease in the rate of stroke.
A possible reason for the lack of benefit from monotherapy was that the differences in blood pressure between the placebo group and the treatment group on monotherapy were small in both studies (3.8/2.0 mm Hg in PRoFESS, 5/3 mm Hg in PROGRESS). In contrast, patients on dual therapy in PROGRESS decreased their blood pressure by 12/5 mm Hg compared with placebo.
CURRENT HYPERTENSION GUIDELINES
Current guidelines make reference to the elderly, but we found none that made specific recommendations for the frail elderly.
JNC 8
In December 2013, members of the Eighth Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8) released new recommendations.32 One significant revision was to support higher blood pressure targets for older adults (age 60 and older). Whereas JNC 7 stated that lowering blood pressure below 140/90 mm Hg reduced cardiovascular complications,33 JNC 8 now acknowledges that there is no strong evidence to support blood pressure targets below 150/90 mm Hg for hypertensive persons without kidney disease or diabetes age 60 and older. Thus, in the general population age 60 and older, JNC 8 recommends starting antihypertensive treatment when blood pressure is 150/90 mm Hg or higher, and treating to a goal blood pressure of less than 150/90 mm Hg. JNC 8 makes no recommendation about how to adjust blood pressure targets for frailty or how to measure blood pressure.
American College of Cardiology and American Heart Association
In 2011, the American College of Cardiology and American Heart Association published a consensus document on the management of hypertension in the elderly.9
They acknowledged that the generally recommended blood pressure goal of lower than 140/90 mm Hg in uncomplicated elderly patients is based on expert opinion rather than on data from randomized controlled trials, but nevertheless recommended a target systolic pressure lower than 140 mm Hg for older adults, except for octogenarians.
For those over age 80, systolic levels of 140 to 145 mm Hg can be acceptable if tolerated and if the patient does not experience orthostasis when standing. Systolic pressure lower than 130 mm Hg and diastolic pressures lower than 65 mm Hg should be avoided in this age group.
The document acknowledges that systolic pressure may have to remain above 150 mm Hg if there is no response to four “well-selected drugs” or if there are unacceptable side effects. In these cases, the lowest “safely achieved” systolic blood pressure should be the goal.
Canadian Hypertension Education Program
The 2014 Canadian Hypertension Education Program (CHEP) report makes several recommendations for the “very elderly,” a group they define as over the age of 80. The CHEP website and resources include the following recommendations10:
- For the very elderly without diabetes or target-organ damage, drug therapy should be initiated when systolic blood pressure is higher than 160 mm Hg to reach a systolic blood pressure target lower than 150 mm Hg. This is a grade C level recommendation, indicating that it is based on low-quality trials, unvalidated surrogate outcomes, or results from nonrandomized observational studies.
- For the very elderly with macrovascular target-organ damage, antihypertensive therapy should be considered if systolic blood pressure readings average 140 mm Hg or higher (grade D for 140 to 160 mm Hg; grade A for higher than 160 mm Hg), although caution should be exercised in elderly patients who are frail. (Grade D recommendations are the weakest, as they are based on low-powered, imprecise studies or expert opinion, whereas grade A recommendations are based on the strongest evidence from high-quality randomized clinical trials.)
- Decisions regarding initiating and intensifying pharmacotherapy in the very elderly should be based on an individualized risk-benefit analysis.
The European Society of Hypertension and European Society of Cardiology
The 2013 guidelines from the European Society of Hypertension and the European Society of Cardiology11 recommend that for elderly patients under age 80, antihypertensive treatment may be considered at systolic values higher than 140 mm Hg and aimed at values lower than 140 mm Hg if the patient is fit and treatment is well tolerated.
For those over age 80 with an initial systolic pressure of 160 mm Hg or higher, the guidelines recommend lowering systolic pressure to between 150 and 140 mm Hg, provided the patient is in good physical and mental condition. In frail elderly patients, they recommend leaving decisions on antihypertensive therapy to the treating physician, based on monitoring of the clinical effects of treatment.11
The ADS/PATH guidelines
When finalizing our recommendations,1 we considered the characteristics of frailty and the following key points from the evidence:
- Although evidence from drug treatment trials indicates that there is benefit in treating healthy older adults who have hypertension, the benefit of treating frail older adults is unknown.
- Major trials enrolled elderly patients only if they had systolic blood pressures of at least 160 mm Hg. Therefore, evidence supports initiating pharmacotherapy at a systolic pressure of 160 mm Hg or higher.
- No evidence from randomized controlled trials supports a systolic target lower than 140 mm Hg in the elderly, and there is some evidence that such a target does not benefit.
- The benefit of adding a third medication to lower blood pressure has not been studied.
- Frailty makes the potential benefits of strict blood pressure targets even less certain and increases the possibility of harm from adverse drug events.
- The only study of very old adults, HYVET,44 enrolled relatively healthy older adults and few with orthostasis, while excluding those with a standing systolic blood pressure lower than 140 mm Hg.
OUR RECOMMENDATIONS
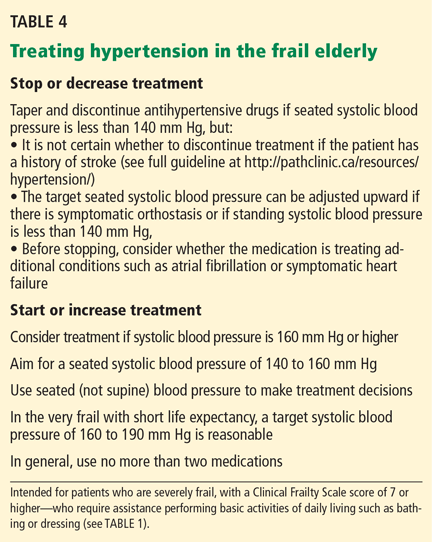
Based on the above, we advise against unnecessarily strict targets and recommend stopping antihypertensive medications that are used for the sole purpose of keeping the systolic blood pressure below 140 mm Hg. Our guidelines are unique in that they focus equally on when to stop and when to start medications. We concluded that without evidence of definitive benefit, “less is more” with frailty.55 We believe that if physicians and health professionals understand the limitations of the evidence, they can be more confident in stopping medications that lower blood pressure to an unnecessarily low level.
We recommend the following (Table 4):
Before treating
- Carefully review the risks and the potential but unproven benefits of treatment.
- To avoid overtreatment, treatment decisions should be based on blood pressure measurements in the seated (not supine) position, while also considering the presence of orthostasis.
- To evaluate orthostasis, measure blood pressure in the supine position, then immediately on standing, and again after 2 minutes. Ask the patient if he or she feels light-headed or dizzy when standing.
Stop treatment
- If the seated systolic blood pressure is less than 140 mm Hg, medications can be tapered and discontinued to achieve the targets described below.
- Before discontinuation, consider whether the medications are treating additional conditions such as rate control for atrial fibrillation or symptomatic management of heart failure.
- It is uncertain whether to discontinue treatment when there is a history of stroke. Consider that treatment with two medications resulted in an absolute risk reduction for disabling stroke of 1.64% over approximately 4 years for adults with previous stroke and a mean age of 64,57 an effect that may be more prominent at higher systolic pressures.
Start treatment
- Consider starting treatment when systolic pressure is 160 mm Hg or higher.
- Aim for a seated systolic pressure between 140 and 160 mm Hg if there are no adverse effects from treatment that affect quality of life.
- If there is symptomatic orthostasis or if standing systolic pressure is lower than 140 mm Hg, the target seated systolic pressure can be adjusted upwards.
- In the severely frail nearing the end of life, a target systolic pressure of 160 to 190 mm Hg is reasonable.
- The blood pressure target is the same in people with diabetes.
- In general, use no more than two medications.
Dissemination and implementation
The ADS/PATH guideline is intended for use by physicians and other health professionals (eg, pharmacists and nurses) who care for frail older adults or who work in long-term care facilities. Since creating our guideline, we have disseminated it to physicians, pharmacists, and other health professionals through academic detailing, large conferences, and interactive webinars.
While we do not have objective evidence of practice change, our evaluation data found that 34% of 403 family physicians who received academic detailing indicated that the guideline would change their practice, while 36% stated that the guideline confirmed their practice, an indication that family physicians are sensitive to the needs of the frail elderly.
Because health professionals may be wary of stopping medications and not meeting recommended targets, there may be barriers to adopting this guideline. However, our experience with the PATH program indicates that these barriers can be overcome using effective communication strategies between health professionals and consumers.
AN APPROACH APPROPRIATE TO FRAILTY
There is no direct evidence for systolic blood pressure targets in the frail elderly, so we applied evidence from the nonfrail elderly. Our recommendations differ somewhat from those of other groups, which recommend targets below 140 to 150 mm Hg for older adults, although some do advise caution in the elderly for whom a substantial fall in blood pressure might be poorly tolerated. Despite these messages, we believe that clearer guidance is needed to direct health practitioners toward models that acknowledge that frail patients are in a precarious balance of health and may be harmed by treatments that strive to lower blood pressure to unproven targets. For this reason, our guideline clearly indicates when to decrease or stop drug treatment.
After physicians and health professionals examine the evidence and more fully understand the benefits and harms of treating frail older adults, we are confident that they will be more comfortable stopping medications that lower blood pressure to an unnecessarily low level and instead use an approach that is more appropriate to frailty. We hope clinicians can use this guideline with the same enthusiasm applied to other guidelines, and we welcome discussion.
Acknowledgments: We would like to thank and acknowledge Tanya MacLeod and Kathryn Yuill for their review of and advice about the manuscript.
- Palliative and Therapeutic Harmonization program. Hypertension guidelines. Treating hypertension in frailty. http://pathclinic.ca/resources/hypertension/. Accessed May 2, 2014.
- Theou O, Rockwood MR, Mitnitski A, Rockwood K. Disability and co-morbidity in relation to frailty: how much do they overlap? Arch Gerontol Geriatr 2012; 55:e1–e8.
- Makary MA, Segev DL, Pronovost PJ, et al. Frailty as a predictor of surgical outcomes in older patients. J Am Coll Surg 2010; 210:901–908.
- Tinetti ME, Bogardus ST, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004; 351:2870–2874.
- Ekerstad N, Swahn E, Janzon M, et al. Frailty is independently associated with short-term outcomes for elderly patients with non-ST-segment elevation myocardial infarction. Circulation 2011; 124:2397–2404.
- Theou O, Rockwood K. Should frailty status always be considered when treating the elderly patient? Aging Health 2012; 8:261–271.
- Rockwood K, Song X, MacKnight C, et al. A global clinical measure of fitness and frailty in elderly people. CMAJ 2005; 173:489–495.
- Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr 2008; 8:24.
- Aronow WS, Fleg JL, Pepine CJ, et al; ACCF Task Force. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2011; 123:2434–2506.
- The Canadian Hypertension Education Program (CHEP). 2014 CHEP recommendations. www.hypertension.ca/en/. Accessed May 2, 2014.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013; 14:392–397.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Ensrud KE, Ewing SK, Cawthon PM, et al; Osteoporotic Fractures in Men Research Group. A comparison of frailty indexes for the prediction of falls, disability, fractures, and mortality in older men. J Am Geriatr Soc 2009; 57:492–498.
- Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 2009; 57:453–461.
- Bergman H, Ferrucci L, Guralnik J, et al. Frailty: an emerging research and clinical paradigm—issues and controversies. J Gerontol A Biol Sci Med Sci 2007; 62:731–737.
- Morley JE, Malmstrom TK, Miller DK. A simple frailty questionnaire (FRAIL) predicts outcomes in middle aged African Americans. J Nutr Health Aging 2012; 16:601–608.
- Strawbridge WJ, Shema SJ, Balfour JL, Higby HR, Kaplan GA. Antecedents of frailty over three decades in an older cohort. J Gerontol B Psychol Sci Soc Sci 1998; 53:S9–S16.
- Matthews M, Lucas A, Boland R, et al. Use of a questionnaire to screen for frailty in the elderly: an exploratory study. Aging Clin Exp Res 2004; 16:34–40.
- Salvi F, Morichi V, Grilli A, et al. Screening for frailty in elderly emergency department patients by using the Identification of Seniors At Risk (ISAR). J Nutr Health Aging 2012; 16:313–318.
- Mitnitski AB, Mogilner AJ, Rockwood K. Accumulation of deficits as a proxy measure of aging. ScientificWorldJournal 2001; 1:323–336.
- Kellen E, Bulens P, Deckx L, et al. Identifying an accurate pre-screening tool in geriatric oncology. Crit Rev Oncol Hematol 2010; 75:243–248.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Martin FC, Brighton P. Frailty: different tools for different purposes? Age Ageing 2008; 37:129–131.
- Borson S, Scanlan J, Brush M, Vitaliano P, Dokmak A. The mini-cog: a cognitive ‘vital signs’ measure for dementia screening in multi-lingual elderly. Int J Geriatr Psychiatry 2000; 15:1021–1027.
- Reisberg B, Ferris SH. Brief Cognitive Rating Scale (BCRS). Psychopharmacol Bull 1988; 24:629–636.
- Moorhouse P, Mallery LH. Palliative and therapeutic harmonization: a model for appropriate decision-making in frail older adults. J Am Geriatr Soc 2012; 60:2326–2332.
- Palliative and Therapeutic Harmonization Clinic (PATH). www.pathclinic.ca. Accessed May 2, 2014.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. http://cme.medicine.dal.ca/ADS.htm. Accessed January 8, 2014.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. Issues in hypertension 2011. http://cme.medicine.dal.ca/files/Hypertension%20book.pdf. Accessed May 2, 2014.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Amery A, Birkenhäger W, Brixko P, et al. Mortality and morbidity results from the European Working Party on High Blood Pressure in the Elderly trial. Lancet 1985; 1:1349–1354.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. Br Med J (Clin Res Ed) 1986; 293:1145–1151.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Dahlöf B, Lindholm LH, Hansson L, Scherstén B, Ekbom T, Wester PO. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- Medical Research Council trial of treatment of hypertension in older adults: principal results. MRC Working Party. BMJ 1992; 304:405–412.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Liu L, Wang JG, Gong L, Liu G, Staessen JA. Comparison of active treatment and placebo in older Chinese patients with isolated systolic hypertension. Systolic Hypertension in China (Syst-China) Collaborative Group. J Hypertens 1998; 16:1823–1829.
- Lithell H, Hansson L, Skoog I, et al; SCOPE Study Group. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double-blind intervention trial. J Hypertens 2003; 21:875–886.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Oparil S, Yarows SA, Patel S, Fang H, Zhang J, Satlin A. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet 2007; 370:221–229.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Musini VM, Tejani AM, Bassett K, Wright JM. Pharmacotherapy for hypertension in the elderly. Cochrane Database Syst Rev 2009;CD000028.
- He FJ, MacGregor GA. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst Rev 2004;CD004937.
- Allen M, Kelly K, Fleming I. Hypertension in elderly patients: recommended systolic targets are not evidence based [in French]. Can Fam Physician 2013; 59:19–24.
- Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ 2012; 344:e3863.
- Sabayan B, Oleksik AM, Maier AB, et al. High blood pressure and resilience to physical and cognitive decline in the oldest old: the Leiden 85-plus Study. J Am Geriatr Soc 2012; 60:2014–2019.
- Sabayan B, van Vliet P, de Ruijter W, Gussekloo J, de Craen AJ, Westendorp RG. High blood pressure, physical and cognitive function, and risk of stroke in the oldest old: the Leiden 85-plus Study. Stroke 2013; 44:15–20.
- Poortvliet RK, Blom JW, de Craen AJ, et al. Low blood pressure predicts increased mortality in very old age even without heart failure: the Leiden 85-plus Study. Eur J Heart Fail 2013; 15:528–533.
- Odden MC, Peralta CA, Haan MN, Covinsky KE. Rethinking the association of high blood pressure with mortality in elderly adults: the impact of frailty. Arch Intern Med 2012; 172:1162–1168.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Yusuf S, Diener HC, Sacco RL, et al; PRoFESS Study Group. Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med 2008; 359:1225–1237.
- Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010; 170:1648–1654.
Frail older adults deserve guidelines that take frailty into account while assessing the potential benefit and risks of treatment.
Specifically, our group—the Dalhousie Academic Detailing Service (ADS) and the Palliative and Therapeutic Harmonization (PATH) program—recommends that physicians strive to achieve more liberal treatment targets for elderly frail patients who have high blood pressure,1 as evidence does not support an aggressive approach in the frail elderly and the potential exists for harm.
This article reviews the evidence and reasoning that were used to develop and promote a guideline for drug treatment of hypertension in frail older adults. Our recommendations differ from other guidelines in that they focus as much on stopping or decreasing therapy as on starting or increasing it.
FRAILTY INCREASES THE RISK OF ADVERSE EFFECTS
The word frail, applied to older adults, describes those who have complex medical illnesses severe enough to compromise their ability to live independently.2 Many have multiple coexisting medical problems for which they take numerous drugs, in addition to dementia, impaired mobility, compromised functional ability, or a history of falling.
Frailty denotes vulnerability; it increases the risk of adverse effects from medical and surgical procedures,3 complicates drug therapy,4 prolongs hospital length of stay,5 leads to functional and cognitive decline,6 increases the risk of institutionalization,7 and reduces life expectancy8—all of which affect the benefit and harm of medical treatments.
Guidelines for treating hypertension9–11 now acknowledge that little evidence exists to support starting treatment for systolic blood pressure between 140 and 160 mm Hg or aiming for a target of less than 140 mm Hg for “very old” adults, commonly defined as over the age of 80. New guidelines loosen the treatment targets for the very old, but they do not specify targets for the frail and do not describe how to recognize or measure frailty.
RECOGNIZING AND MEASURING FRAILTY
A number of tools are available to recognize and measure frailty.12
The Fried frailty assessment13 has five items:
- Unintentional weight loss
- Self-reported exhaustion
- Weakness in grip
- Slow walking speed
- Low physical activity and energy expenditure.
People are deemed frail if they have three or more of these five. However, experts disagree about whether this system is too sensitive14 or not sensitive enough.15,16
The FRAIL questionnaire17 also has five items:
- Fatigue
- Resistance (inability to climb stairs)
- Ambulation (inability to walk 1 city block)
- Illness (more than 5 major illnesses)
- Weight loss.
People are deemed frail if they have at least three of these five items, and “prefrail” if they have two.
These and other tools are limited by being dichotomous: they classify people as being either frail or not frail18–20 but do not define the spectrum of frailty.
Other frailty assessments such as the Frailty Index21 identify frailty based on the number of accumulated health deficits but take a long time to complete, making them difficult to use in busy clinical settings.22–24
The Clinical Frailty Scale7 is a validated scale that categorizes frailty based on physical and functional indicators of health, such as cognition, function, and mobility, with scores that range from 1 (very fit) to 9 (terminally ill).7,12
The Frailty Assessment for Care-planning Tool (FACT) uses scaling compatible with the Clinical Frailty Scale but has been developed for use as a practical and interpretable frailty screening tool for nonexperts (Table 1). The FACT assesses cognition, mobility, function, and the social situation, using a combination of caregiver report and objective measures. To assess cognition, a health care professional uses items from the Mini-Cog25 (ie, the ability to draw an analog clock face and then recall three unrelated items following the clock-drawing test) and the memory axis of the Brief Cognitive Rating Scale26 (ie, the ability to recall current events, the current US president, and the names of children or spouse). Mobility, function, and social circumstance scores are assigned according to the caregiver report of the patient’s baseline status.
The FACT can be completed in busy clinical settings. Once a caregiver is identified, it takes about 5 minutes to complete.
Our guideline27–31 is intended for those with a score of 7 or more on the Clinical Frailty Scale or FACT,7,12 a score we chose because it describes people who are severely frail with shortened life expectancy.8 At this level, people need help with all instrumental activities of daily living (eg, handling finances, medication management, household chores, and shopping) as well as with basic activities of daily living such as bathing or dressing.
REVIEWING THE LIMITED EVIDENCE
We found no studies that addressed the risks and benefits of treating hypertension in frail older adults; therefore, we concentrated on studies that enrolled individuals who were chronologically old but not frail. We reviewed prominent guidelines,9–11,32,33 the evidence base for these guidelines,34–44 and Cochrane reviews.45,46 A detailed description of the evidence used to build our recommendation can be found online.31
When we deliberated on treatment targets, we reviewed evidence from two types of randomized controlled trials47:
Drug treatment trials randomize patients to different treatments, such as placebo versus a drug or one drug compared with another drug. Patients in different treatment groups may achieve different blood pressures and clinical outcomes, and this information is then used to define optimal targets. However, it may be difficult to determine if the benefit came from lowering blood pressure or from some other effect of the drug, which can be independent of blood pressure lowering.
Treat-to-target trials randomize patients to different blood pressure goals, but the groups are treated with the same or similar drugs. Therefore, any identified benefit can be attributed to the differences in blood pressure rather than the medications used. Compared with a drug treatment trial, this type of trial provides stronger evidence about optimal targets.
We also considered the characteristics of frailty, the dilemma of polypharmacy, and the relevance of the available scientific evidence to those who are frail.
Drug treatment trials
A Cochrane review45 of 15 studies with approximately 24,000 elderly participants found that treating hypertension decreased the rates of cardiovascular morbidity and mortality as well as fatal and nonfatal stroke in the “elderly” (defined as age ≥ 60) and “very elderly” (age ≥ 80). However, in the very elderly, all-cause mortality rates were not statistically significantly different with treatment compared with placebo. The mean duration of treatment was 4.5 years in the elderly and 2.2 years in the very elderly (Table 2). Of importance, all the trials enrolled only those individuals whose systolic blood pressure was at least 160 mm Hg at baseline.
None of the studies were treat-to-target trials—patients were assigned either active medication or placebo. Thus, these trials provide evidence of benefit for treating hypertension in the elderly and very elderly but do not identify the optimal target. All of the drug treatment trials showed benefit, but none achieved a systolic pressure lower than 140 mm Hg with active treatment (Table 3). Therefore, these studies do not support a systolic target of less than 140 mm Hg in the elderly.
Treat-to-target trials: JATOS and VALISH
The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)42 and the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) study43 each enrolled more than 3,000 people age 65 or older (mean age approximately 75). Patients were randomized to either a strict systolic target of less than 140 mm Hg or a higher (more permissive) target of 140 to 160 mm Hg in JATOS and 140 to 149 mm Hg in VALISH.
In both trials, the group with strict targets achieved a systolic pressure of approximately 136 mm Hg, while the group with higher blood pressure targets achieved a systolic pressure of 146 mm Hg in JATOS and 142 mm Hg in VALISH. Despite these differences, there was no statistically significant difference in the primary outcome.
Thus, treat-to-target studies also fail to support a systolic target of less than 140 mm Hg in the elderly, although it is important to recognize the limitations of the studies. Approximately 15% of the participants had cardiovascular disease, so the applicability of the findings to patients with target-organ damage is uncertain. In addition, there were fewer efficacy outcome events than expected, which suggests that the studies were underpowered.
When to start drug treatment
In each of the drug treatment and treat-to-target trials, the inclusion criterion for study entry was a systolic blood pressure above 160 mm Hg, with a mean blood pressure at entry into the drug treatment trials of 182/95 mm Hg.46 Thus, data support starting treatment if the systolic blood pressure is above 160 mm Hg, but not lower.
Notably, in all but one study,46 at least two-thirds of the participants took no more than two antihypertensive medications. Since adverse events become more common as the number of medications increases, the benefit of adding a third drug to lower blood pressure is uncertain.
Evidence in the ‘very elderly’: HYVET
With the exception of the Hypertension in the Very Elderly Trial (HYVET),44 the mean age of elderly patients in the reported studies was between 67 and 76.
HYVET patients were age 80 and older (mean age 84) and were randomized to receive either indapamide (with or without perindopril) or placebo. The trial was stopped early at 2 years because the mortality rate was lower in the treatment group (10.1%) than in the placebo group (12.3%) (number needed to treat 46, 95% confidence interval 24–637, P = .02). There was no significant difference in the primary outcome of fatal and nonfatal stroke.
Notably, trials that are stopped early may overestimate treatment benefit.48
Evidence in frail older adults
While the above studies provide some information about managing hypertension in the elderly, the participants were generally healthy. HYVET44 specifically excluded those with a standing systolic blood pressure of less than 140 mm Hg and enrolled few patients with orthostasis (7.9% in the placebo group and 8.8% in the treatment group), a condition commonly associated with frailty. As such, these studies may be less relevant to the frail elderly, who are at higher risk of adverse drug events and have competing risks for morbidity and mortality.
Observational studies, in fact, raise questions about whether tight blood pressure control improves clinical outcomes for the very elderly. In the Leiden 85-plus study, lower systolic blood pressure was associated with lower cognitive scores, worse functional ability,49,50 and a higher mortality rate51 compared with higher systolic pressure, although it is uncertain whether these outcomes were indicative of underlying disease that could result in lower blood pressure or an effect of blood pressure-lowering.
The National Health and Nutrition Examination Survey52 found an association between blood pressure and mortality rate that varied by walking speed. For slower walkers (based on the 6-minute walk test), higher systolic pressures were not associated with a higher risk of death, suggesting that when older adults are frail (as indicated by their slow walking speed) they are less likely to benefit from aggressive treatment of hypertension.
People at high risk because of stroke
Because the evidence is limited, it is even more difficult to judge whether lowering blood pressure below 140 mm Hg is beneficial for frail patients who have a history of stroke, compared with the possibility that medications will cause adverse effects such as weakness, orthostasis, and falls. When reviewing the evidence to answer this question, we especially looked at outcomes that affect quality of life, such as nonfatal stroke leading to disability. In contrast, because the frail elderly have competing causes of mortality, we could not assume that a mortality benefit shown in nonfrail populations could be applied to frail populations.
The PROGRESS trial (Perindopril Protection Against Recurrent Stroke Study)53 was in patients with a history of stroke or transient ischemic attack and a mean age of 64, who were treated with either perindopril (with or without indapamide) or placebo.
At almost 4 years, the rate of disabling stroke was 2.7% in the treatment group and 4.3% in the placebo group, a relative risk reduction of 38% and an absolute risk reduction of 1.64% (number needed to treat 61, 95% confidence interval 39–139). The relative risk reduction for all strokes (fatal and nonfatal) was similar across a range of baseline systolic pressures, but the absolute risk reduction was greater in the prespecified subgroup that had hypertension at baseline (mean blood pressure 159/94 mm Hg) than in the normotensive subgroup (mean blood pressure 136/79 mm Hg), suggesting that treatment is most beneficial for those with higher systolic blood pressures. Also, the benefit was only demonstrated in the subgroup that received two antihypertensive medications; those who received perindopril alone showed no benefit.
This study involved relatively young patients in relatively good health except for their strokes. The extent to which the results can be extrapolated to older, frail adults is uncertain because of the time needed to achieve benefit and because of the added vulnerability of frailty, which could make treatment with two antihypertensive medications riskier.
PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes),54 another study in patients with previous stroke (mean age 66) showed no benefit over 2.5 years in the primary outcome of stroke using telmesartan 80 mg daily compared with placebo. This result is concordant with that of PROGRESS,53 in which patients who took only one medication did not show a significant decrease in the rate of stroke.
A possible reason for the lack of benefit from monotherapy was that the differences in blood pressure between the placebo group and the treatment group on monotherapy were small in both studies (3.8/2.0 mm Hg in PRoFESS, 5/3 mm Hg in PROGRESS). In contrast, patients on dual therapy in PROGRESS decreased their blood pressure by 12/5 mm Hg compared with placebo.
CURRENT HYPERTENSION GUIDELINES
Current guidelines make reference to the elderly, but we found none that made specific recommendations for the frail elderly.
JNC 8
In December 2013, members of the Eighth Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8) released new recommendations.32 One significant revision was to support higher blood pressure targets for older adults (age 60 and older). Whereas JNC 7 stated that lowering blood pressure below 140/90 mm Hg reduced cardiovascular complications,33 JNC 8 now acknowledges that there is no strong evidence to support blood pressure targets below 150/90 mm Hg for hypertensive persons without kidney disease or diabetes age 60 and older. Thus, in the general population age 60 and older, JNC 8 recommends starting antihypertensive treatment when blood pressure is 150/90 mm Hg or higher, and treating to a goal blood pressure of less than 150/90 mm Hg. JNC 8 makes no recommendation about how to adjust blood pressure targets for frailty or how to measure blood pressure.
American College of Cardiology and American Heart Association
In 2011, the American College of Cardiology and American Heart Association published a consensus document on the management of hypertension in the elderly.9
They acknowledged that the generally recommended blood pressure goal of lower than 140/90 mm Hg in uncomplicated elderly patients is based on expert opinion rather than on data from randomized controlled trials, but nevertheless recommended a target systolic pressure lower than 140 mm Hg for older adults, except for octogenarians.
For those over age 80, systolic levels of 140 to 145 mm Hg can be acceptable if tolerated and if the patient does not experience orthostasis when standing. Systolic pressure lower than 130 mm Hg and diastolic pressures lower than 65 mm Hg should be avoided in this age group.
The document acknowledges that systolic pressure may have to remain above 150 mm Hg if there is no response to four “well-selected drugs” or if there are unacceptable side effects. In these cases, the lowest “safely achieved” systolic blood pressure should be the goal.
Canadian Hypertension Education Program
The 2014 Canadian Hypertension Education Program (CHEP) report makes several recommendations for the “very elderly,” a group they define as over the age of 80. The CHEP website and resources include the following recommendations10:
- For the very elderly without diabetes or target-organ damage, drug therapy should be initiated when systolic blood pressure is higher than 160 mm Hg to reach a systolic blood pressure target lower than 150 mm Hg. This is a grade C level recommendation, indicating that it is based on low-quality trials, unvalidated surrogate outcomes, or results from nonrandomized observational studies.
- For the very elderly with macrovascular target-organ damage, antihypertensive therapy should be considered if systolic blood pressure readings average 140 mm Hg or higher (grade D for 140 to 160 mm Hg; grade A for higher than 160 mm Hg), although caution should be exercised in elderly patients who are frail. (Grade D recommendations are the weakest, as they are based on low-powered, imprecise studies or expert opinion, whereas grade A recommendations are based on the strongest evidence from high-quality randomized clinical trials.)
- Decisions regarding initiating and intensifying pharmacotherapy in the very elderly should be based on an individualized risk-benefit analysis.
The European Society of Hypertension and European Society of Cardiology
The 2013 guidelines from the European Society of Hypertension and the European Society of Cardiology11 recommend that for elderly patients under age 80, antihypertensive treatment may be considered at systolic values higher than 140 mm Hg and aimed at values lower than 140 mm Hg if the patient is fit and treatment is well tolerated.
For those over age 80 with an initial systolic pressure of 160 mm Hg or higher, the guidelines recommend lowering systolic pressure to between 150 and 140 mm Hg, provided the patient is in good physical and mental condition. In frail elderly patients, they recommend leaving decisions on antihypertensive therapy to the treating physician, based on monitoring of the clinical effects of treatment.11
The ADS/PATH guidelines
When finalizing our recommendations,1 we considered the characteristics of frailty and the following key points from the evidence:
- Although evidence from drug treatment trials indicates that there is benefit in treating healthy older adults who have hypertension, the benefit of treating frail older adults is unknown.
- Major trials enrolled elderly patients only if they had systolic blood pressures of at least 160 mm Hg. Therefore, evidence supports initiating pharmacotherapy at a systolic pressure of 160 mm Hg or higher.
- No evidence from randomized controlled trials supports a systolic target lower than 140 mm Hg in the elderly, and there is some evidence that such a target does not benefit.
- The benefit of adding a third medication to lower blood pressure has not been studied.
- Frailty makes the potential benefits of strict blood pressure targets even less certain and increases the possibility of harm from adverse drug events.
- The only study of very old adults, HYVET,44 enrolled relatively healthy older adults and few with orthostasis, while excluding those with a standing systolic blood pressure lower than 140 mm Hg.
OUR RECOMMENDATIONS
Based on the above, we advise against unnecessarily strict targets and recommend stopping antihypertensive medications that are used for the sole purpose of keeping the systolic blood pressure below 140 mm Hg. Our guidelines are unique in that they focus equally on when to stop and when to start medications. We concluded that without evidence of definitive benefit, “less is more” with frailty.55 We believe that if physicians and health professionals understand the limitations of the evidence, they can be more confident in stopping medications that lower blood pressure to an unnecessarily low level.
We recommend the following (Table 4):
Before treating
- Carefully review the risks and the potential but unproven benefits of treatment.
- To avoid overtreatment, treatment decisions should be based on blood pressure measurements in the seated (not supine) position, while also considering the presence of orthostasis.
- To evaluate orthostasis, measure blood pressure in the supine position, then immediately on standing, and again after 2 minutes. Ask the patient if he or she feels light-headed or dizzy when standing.
Stop treatment
- If the seated systolic blood pressure is less than 140 mm Hg, medications can be tapered and discontinued to achieve the targets described below.
- Before discontinuation, consider whether the medications are treating additional conditions such as rate control for atrial fibrillation or symptomatic management of heart failure.
- It is uncertain whether to discontinue treatment when there is a history of stroke. Consider that treatment with two medications resulted in an absolute risk reduction for disabling stroke of 1.64% over approximately 4 years for adults with previous stroke and a mean age of 64,57 an effect that may be more prominent at higher systolic pressures.
Start treatment
- Consider starting treatment when systolic pressure is 160 mm Hg or higher.
- Aim for a seated systolic pressure between 140 and 160 mm Hg if there are no adverse effects from treatment that affect quality of life.
- If there is symptomatic orthostasis or if standing systolic pressure is lower than 140 mm Hg, the target seated systolic pressure can be adjusted upwards.
- In the severely frail nearing the end of life, a target systolic pressure of 160 to 190 mm Hg is reasonable.
- The blood pressure target is the same in people with diabetes.
- In general, use no more than two medications.
Dissemination and implementation
The ADS/PATH guideline is intended for use by physicians and other health professionals (eg, pharmacists and nurses) who care for frail older adults or who work in long-term care facilities. Since creating our guideline, we have disseminated it to physicians, pharmacists, and other health professionals through academic detailing, large conferences, and interactive webinars.
While we do not have objective evidence of practice change, our evaluation data found that 34% of 403 family physicians who received academic detailing indicated that the guideline would change their practice, while 36% stated that the guideline confirmed their practice, an indication that family physicians are sensitive to the needs of the frail elderly.
Because health professionals may be wary of stopping medications and not meeting recommended targets, there may be barriers to adopting this guideline. However, our experience with the PATH program indicates that these barriers can be overcome using effective communication strategies between health professionals and consumers.
AN APPROACH APPROPRIATE TO FRAILTY
There is no direct evidence for systolic blood pressure targets in the frail elderly, so we applied evidence from the nonfrail elderly. Our recommendations differ somewhat from those of other groups, which recommend targets below 140 to 150 mm Hg for older adults, although some do advise caution in the elderly for whom a substantial fall in blood pressure might be poorly tolerated. Despite these messages, we believe that clearer guidance is needed to direct health practitioners toward models that acknowledge that frail patients are in a precarious balance of health and may be harmed by treatments that strive to lower blood pressure to unproven targets. For this reason, our guideline clearly indicates when to decrease or stop drug treatment.
After physicians and health professionals examine the evidence and more fully understand the benefits and harms of treating frail older adults, we are confident that they will be more comfortable stopping medications that lower blood pressure to an unnecessarily low level and instead use an approach that is more appropriate to frailty. We hope clinicians can use this guideline with the same enthusiasm applied to other guidelines, and we welcome discussion.
Acknowledgments: We would like to thank and acknowledge Tanya MacLeod and Kathryn Yuill for their review of and advice about the manuscript.
Frail older adults deserve guidelines that take frailty into account while assessing the potential benefit and risks of treatment.
Specifically, our group—the Dalhousie Academic Detailing Service (ADS) and the Palliative and Therapeutic Harmonization (PATH) program—recommends that physicians strive to achieve more liberal treatment targets for elderly frail patients who have high blood pressure,1 as evidence does not support an aggressive approach in the frail elderly and the potential exists for harm.
This article reviews the evidence and reasoning that were used to develop and promote a guideline for drug treatment of hypertension in frail older adults. Our recommendations differ from other guidelines in that they focus as much on stopping or decreasing therapy as on starting or increasing it.
FRAILTY INCREASES THE RISK OF ADVERSE EFFECTS
The word frail, applied to older adults, describes those who have complex medical illnesses severe enough to compromise their ability to live independently.2 Many have multiple coexisting medical problems for which they take numerous drugs, in addition to dementia, impaired mobility, compromised functional ability, or a history of falling.
Frailty denotes vulnerability; it increases the risk of adverse effects from medical and surgical procedures,3 complicates drug therapy,4 prolongs hospital length of stay,5 leads to functional and cognitive decline,6 increases the risk of institutionalization,7 and reduces life expectancy8—all of which affect the benefit and harm of medical treatments.
Guidelines for treating hypertension9–11 now acknowledge that little evidence exists to support starting treatment for systolic blood pressure between 140 and 160 mm Hg or aiming for a target of less than 140 mm Hg for “very old” adults, commonly defined as over the age of 80. New guidelines loosen the treatment targets for the very old, but they do not specify targets for the frail and do not describe how to recognize or measure frailty.
RECOGNIZING AND MEASURING FRAILTY
A number of tools are available to recognize and measure frailty.12
The Fried frailty assessment13 has five items:
- Unintentional weight loss
- Self-reported exhaustion
- Weakness in grip
- Slow walking speed
- Low physical activity and energy expenditure.
People are deemed frail if they have three or more of these five. However, experts disagree about whether this system is too sensitive14 or not sensitive enough.15,16
The FRAIL questionnaire17 also has five items:
- Fatigue
- Resistance (inability to climb stairs)
- Ambulation (inability to walk 1 city block)
- Illness (more than 5 major illnesses)
- Weight loss.
People are deemed frail if they have at least three of these five items, and “prefrail” if they have two.
These and other tools are limited by being dichotomous: they classify people as being either frail or not frail18–20 but do not define the spectrum of frailty.
Other frailty assessments such as the Frailty Index21 identify frailty based on the number of accumulated health deficits but take a long time to complete, making them difficult to use in busy clinical settings.22–24
The Clinical Frailty Scale7 is a validated scale that categorizes frailty based on physical and functional indicators of health, such as cognition, function, and mobility, with scores that range from 1 (very fit) to 9 (terminally ill).7,12
The Frailty Assessment for Care-planning Tool (FACT) uses scaling compatible with the Clinical Frailty Scale but has been developed for use as a practical and interpretable frailty screening tool for nonexperts (Table 1). The FACT assesses cognition, mobility, function, and the social situation, using a combination of caregiver report and objective measures. To assess cognition, a health care professional uses items from the Mini-Cog25 (ie, the ability to draw an analog clock face and then recall three unrelated items following the clock-drawing test) and the memory axis of the Brief Cognitive Rating Scale26 (ie, the ability to recall current events, the current US president, and the names of children or spouse). Mobility, function, and social circumstance scores are assigned according to the caregiver report of the patient’s baseline status.
The FACT can be completed in busy clinical settings. Once a caregiver is identified, it takes about 5 minutes to complete.
Our guideline27–31 is intended for those with a score of 7 or more on the Clinical Frailty Scale or FACT,7,12 a score we chose because it describes people who are severely frail with shortened life expectancy.8 At this level, people need help with all instrumental activities of daily living (eg, handling finances, medication management, household chores, and shopping) as well as with basic activities of daily living such as bathing or dressing.
REVIEWING THE LIMITED EVIDENCE
We found no studies that addressed the risks and benefits of treating hypertension in frail older adults; therefore, we concentrated on studies that enrolled individuals who were chronologically old but not frail. We reviewed prominent guidelines,9–11,32,33 the evidence base for these guidelines,34–44 and Cochrane reviews.45,46 A detailed description of the evidence used to build our recommendation can be found online.31
When we deliberated on treatment targets, we reviewed evidence from two types of randomized controlled trials47:
Drug treatment trials randomize patients to different treatments, such as placebo versus a drug or one drug compared with another drug. Patients in different treatment groups may achieve different blood pressures and clinical outcomes, and this information is then used to define optimal targets. However, it may be difficult to determine if the benefit came from lowering blood pressure or from some other effect of the drug, which can be independent of blood pressure lowering.
Treat-to-target trials randomize patients to different blood pressure goals, but the groups are treated with the same or similar drugs. Therefore, any identified benefit can be attributed to the differences in blood pressure rather than the medications used. Compared with a drug treatment trial, this type of trial provides stronger evidence about optimal targets.
We also considered the characteristics of frailty, the dilemma of polypharmacy, and the relevance of the available scientific evidence to those who are frail.
Drug treatment trials
A Cochrane review45 of 15 studies with approximately 24,000 elderly participants found that treating hypertension decreased the rates of cardiovascular morbidity and mortality as well as fatal and nonfatal stroke in the “elderly” (defined as age ≥ 60) and “very elderly” (age ≥ 80). However, in the very elderly, all-cause mortality rates were not statistically significantly different with treatment compared with placebo. The mean duration of treatment was 4.5 years in the elderly and 2.2 years in the very elderly (Table 2). Of importance, all the trials enrolled only those individuals whose systolic blood pressure was at least 160 mm Hg at baseline.
None of the studies were treat-to-target trials—patients were assigned either active medication or placebo. Thus, these trials provide evidence of benefit for treating hypertension in the elderly and very elderly but do not identify the optimal target. All of the drug treatment trials showed benefit, but none achieved a systolic pressure lower than 140 mm Hg with active treatment (Table 3). Therefore, these studies do not support a systolic target of less than 140 mm Hg in the elderly.
Treat-to-target trials: JATOS and VALISH
The Japanese Trial to Assess Optimal Systolic Blood Pressure in Elderly Hypertensive Patients (JATOS)42 and the Valsartan in Elderly Isolated Systolic Hypertension (VALISH) study43 each enrolled more than 3,000 people age 65 or older (mean age approximately 75). Patients were randomized to either a strict systolic target of less than 140 mm Hg or a higher (more permissive) target of 140 to 160 mm Hg in JATOS and 140 to 149 mm Hg in VALISH.
In both trials, the group with strict targets achieved a systolic pressure of approximately 136 mm Hg, while the group with higher blood pressure targets achieved a systolic pressure of 146 mm Hg in JATOS and 142 mm Hg in VALISH. Despite these differences, there was no statistically significant difference in the primary outcome.
Thus, treat-to-target studies also fail to support a systolic target of less than 140 mm Hg in the elderly, although it is important to recognize the limitations of the studies. Approximately 15% of the participants had cardiovascular disease, so the applicability of the findings to patients with target-organ damage is uncertain. In addition, there were fewer efficacy outcome events than expected, which suggests that the studies were underpowered.
When to start drug treatment
In each of the drug treatment and treat-to-target trials, the inclusion criterion for study entry was a systolic blood pressure above 160 mm Hg, with a mean blood pressure at entry into the drug treatment trials of 182/95 mm Hg.46 Thus, data support starting treatment if the systolic blood pressure is above 160 mm Hg, but not lower.
Notably, in all but one study,46 at least two-thirds of the participants took no more than two antihypertensive medications. Since adverse events become more common as the number of medications increases, the benefit of adding a third drug to lower blood pressure is uncertain.
Evidence in the ‘very elderly’: HYVET
With the exception of the Hypertension in the Very Elderly Trial (HYVET),44 the mean age of elderly patients in the reported studies was between 67 and 76.
HYVET patients were age 80 and older (mean age 84) and were randomized to receive either indapamide (with or without perindopril) or placebo. The trial was stopped early at 2 years because the mortality rate was lower in the treatment group (10.1%) than in the placebo group (12.3%) (number needed to treat 46, 95% confidence interval 24–637, P = .02). There was no significant difference in the primary outcome of fatal and nonfatal stroke.
Notably, trials that are stopped early may overestimate treatment benefit.48
Evidence in frail older adults
While the above studies provide some information about managing hypertension in the elderly, the participants were generally healthy. HYVET44 specifically excluded those with a standing systolic blood pressure of less than 140 mm Hg and enrolled few patients with orthostasis (7.9% in the placebo group and 8.8% in the treatment group), a condition commonly associated with frailty. As such, these studies may be less relevant to the frail elderly, who are at higher risk of adverse drug events and have competing risks for morbidity and mortality.
Observational studies, in fact, raise questions about whether tight blood pressure control improves clinical outcomes for the very elderly. In the Leiden 85-plus study, lower systolic blood pressure was associated with lower cognitive scores, worse functional ability,49,50 and a higher mortality rate51 compared with higher systolic pressure, although it is uncertain whether these outcomes were indicative of underlying disease that could result in lower blood pressure or an effect of blood pressure-lowering.
The National Health and Nutrition Examination Survey52 found an association between blood pressure and mortality rate that varied by walking speed. For slower walkers (based on the 6-minute walk test), higher systolic pressures were not associated with a higher risk of death, suggesting that when older adults are frail (as indicated by their slow walking speed) they are less likely to benefit from aggressive treatment of hypertension.
People at high risk because of stroke
Because the evidence is limited, it is even more difficult to judge whether lowering blood pressure below 140 mm Hg is beneficial for frail patients who have a history of stroke, compared with the possibility that medications will cause adverse effects such as weakness, orthostasis, and falls. When reviewing the evidence to answer this question, we especially looked at outcomes that affect quality of life, such as nonfatal stroke leading to disability. In contrast, because the frail elderly have competing causes of mortality, we could not assume that a mortality benefit shown in nonfrail populations could be applied to frail populations.
The PROGRESS trial (Perindopril Protection Against Recurrent Stroke Study)53 was in patients with a history of stroke or transient ischemic attack and a mean age of 64, who were treated with either perindopril (with or without indapamide) or placebo.
At almost 4 years, the rate of disabling stroke was 2.7% in the treatment group and 4.3% in the placebo group, a relative risk reduction of 38% and an absolute risk reduction of 1.64% (number needed to treat 61, 95% confidence interval 39–139). The relative risk reduction for all strokes (fatal and nonfatal) was similar across a range of baseline systolic pressures, but the absolute risk reduction was greater in the prespecified subgroup that had hypertension at baseline (mean blood pressure 159/94 mm Hg) than in the normotensive subgroup (mean blood pressure 136/79 mm Hg), suggesting that treatment is most beneficial for those with higher systolic blood pressures. Also, the benefit was only demonstrated in the subgroup that received two antihypertensive medications; those who received perindopril alone showed no benefit.
This study involved relatively young patients in relatively good health except for their strokes. The extent to which the results can be extrapolated to older, frail adults is uncertain because of the time needed to achieve benefit and because of the added vulnerability of frailty, which could make treatment with two antihypertensive medications riskier.
PRoFESS (Prevention Regimen for Effectively Avoiding Second Strokes),54 another study in patients with previous stroke (mean age 66) showed no benefit over 2.5 years in the primary outcome of stroke using telmesartan 80 mg daily compared with placebo. This result is concordant with that of PROGRESS,53 in which patients who took only one medication did not show a significant decrease in the rate of stroke.
A possible reason for the lack of benefit from monotherapy was that the differences in blood pressure between the placebo group and the treatment group on monotherapy were small in both studies (3.8/2.0 mm Hg in PRoFESS, 5/3 mm Hg in PROGRESS). In contrast, patients on dual therapy in PROGRESS decreased their blood pressure by 12/5 mm Hg compared with placebo.
CURRENT HYPERTENSION GUIDELINES
Current guidelines make reference to the elderly, but we found none that made specific recommendations for the frail elderly.
JNC 8
In December 2013, members of the Eighth Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 8) released new recommendations.32 One significant revision was to support higher blood pressure targets for older adults (age 60 and older). Whereas JNC 7 stated that lowering blood pressure below 140/90 mm Hg reduced cardiovascular complications,33 JNC 8 now acknowledges that there is no strong evidence to support blood pressure targets below 150/90 mm Hg for hypertensive persons without kidney disease or diabetes age 60 and older. Thus, in the general population age 60 and older, JNC 8 recommends starting antihypertensive treatment when blood pressure is 150/90 mm Hg or higher, and treating to a goal blood pressure of less than 150/90 mm Hg. JNC 8 makes no recommendation about how to adjust blood pressure targets for frailty or how to measure blood pressure.
American College of Cardiology and American Heart Association
In 2011, the American College of Cardiology and American Heart Association published a consensus document on the management of hypertension in the elderly.9
They acknowledged that the generally recommended blood pressure goal of lower than 140/90 mm Hg in uncomplicated elderly patients is based on expert opinion rather than on data from randomized controlled trials, but nevertheless recommended a target systolic pressure lower than 140 mm Hg for older adults, except for octogenarians.
For those over age 80, systolic levels of 140 to 145 mm Hg can be acceptable if tolerated and if the patient does not experience orthostasis when standing. Systolic pressure lower than 130 mm Hg and diastolic pressures lower than 65 mm Hg should be avoided in this age group.
The document acknowledges that systolic pressure may have to remain above 150 mm Hg if there is no response to four “well-selected drugs” or if there are unacceptable side effects. In these cases, the lowest “safely achieved” systolic blood pressure should be the goal.
Canadian Hypertension Education Program
The 2014 Canadian Hypertension Education Program (CHEP) report makes several recommendations for the “very elderly,” a group they define as over the age of 80. The CHEP website and resources include the following recommendations10:
- For the very elderly without diabetes or target-organ damage, drug therapy should be initiated when systolic blood pressure is higher than 160 mm Hg to reach a systolic blood pressure target lower than 150 mm Hg. This is a grade C level recommendation, indicating that it is based on low-quality trials, unvalidated surrogate outcomes, or results from nonrandomized observational studies.
- For the very elderly with macrovascular target-organ damage, antihypertensive therapy should be considered if systolic blood pressure readings average 140 mm Hg or higher (grade D for 140 to 160 mm Hg; grade A for higher than 160 mm Hg), although caution should be exercised in elderly patients who are frail. (Grade D recommendations are the weakest, as they are based on low-powered, imprecise studies or expert opinion, whereas grade A recommendations are based on the strongest evidence from high-quality randomized clinical trials.)
- Decisions regarding initiating and intensifying pharmacotherapy in the very elderly should be based on an individualized risk-benefit analysis.
The European Society of Hypertension and European Society of Cardiology
The 2013 guidelines from the European Society of Hypertension and the European Society of Cardiology11 recommend that for elderly patients under age 80, antihypertensive treatment may be considered at systolic values higher than 140 mm Hg and aimed at values lower than 140 mm Hg if the patient is fit and treatment is well tolerated.
For those over age 80 with an initial systolic pressure of 160 mm Hg or higher, the guidelines recommend lowering systolic pressure to between 150 and 140 mm Hg, provided the patient is in good physical and mental condition. In frail elderly patients, they recommend leaving decisions on antihypertensive therapy to the treating physician, based on monitoring of the clinical effects of treatment.11
The ADS/PATH guidelines
When finalizing our recommendations,1 we considered the characteristics of frailty and the following key points from the evidence:
- Although evidence from drug treatment trials indicates that there is benefit in treating healthy older adults who have hypertension, the benefit of treating frail older adults is unknown.
- Major trials enrolled elderly patients only if they had systolic blood pressures of at least 160 mm Hg. Therefore, evidence supports initiating pharmacotherapy at a systolic pressure of 160 mm Hg or higher.
- No evidence from randomized controlled trials supports a systolic target lower than 140 mm Hg in the elderly, and there is some evidence that such a target does not benefit.
- The benefit of adding a third medication to lower blood pressure has not been studied.
- Frailty makes the potential benefits of strict blood pressure targets even less certain and increases the possibility of harm from adverse drug events.
- The only study of very old adults, HYVET,44 enrolled relatively healthy older adults and few with orthostasis, while excluding those with a standing systolic blood pressure lower than 140 mm Hg.
OUR RECOMMENDATIONS
Based on the above, we advise against unnecessarily strict targets and recommend stopping antihypertensive medications that are used for the sole purpose of keeping the systolic blood pressure below 140 mm Hg. Our guidelines are unique in that they focus equally on when to stop and when to start medications. We concluded that without evidence of definitive benefit, “less is more” with frailty.55 We believe that if physicians and health professionals understand the limitations of the evidence, they can be more confident in stopping medications that lower blood pressure to an unnecessarily low level.
We recommend the following (Table 4):
Before treating
- Carefully review the risks and the potential but unproven benefits of treatment.
- To avoid overtreatment, treatment decisions should be based on blood pressure measurements in the seated (not supine) position, while also considering the presence of orthostasis.
- To evaluate orthostasis, measure blood pressure in the supine position, then immediately on standing, and again after 2 minutes. Ask the patient if he or she feels light-headed or dizzy when standing.
Stop treatment
- If the seated systolic blood pressure is less than 140 mm Hg, medications can be tapered and discontinued to achieve the targets described below.
- Before discontinuation, consider whether the medications are treating additional conditions such as rate control for atrial fibrillation or symptomatic management of heart failure.
- It is uncertain whether to discontinue treatment when there is a history of stroke. Consider that treatment with two medications resulted in an absolute risk reduction for disabling stroke of 1.64% over approximately 4 years for adults with previous stroke and a mean age of 64,57 an effect that may be more prominent at higher systolic pressures.
Start treatment
- Consider starting treatment when systolic pressure is 160 mm Hg or higher.
- Aim for a seated systolic pressure between 140 and 160 mm Hg if there are no adverse effects from treatment that affect quality of life.
- If there is symptomatic orthostasis or if standing systolic pressure is lower than 140 mm Hg, the target seated systolic pressure can be adjusted upwards.
- In the severely frail nearing the end of life, a target systolic pressure of 160 to 190 mm Hg is reasonable.
- The blood pressure target is the same in people with diabetes.
- In general, use no more than two medications.
Dissemination and implementation
The ADS/PATH guideline is intended for use by physicians and other health professionals (eg, pharmacists and nurses) who care for frail older adults or who work in long-term care facilities. Since creating our guideline, we have disseminated it to physicians, pharmacists, and other health professionals through academic detailing, large conferences, and interactive webinars.
While we do not have objective evidence of practice change, our evaluation data found that 34% of 403 family physicians who received academic detailing indicated that the guideline would change their practice, while 36% stated that the guideline confirmed their practice, an indication that family physicians are sensitive to the needs of the frail elderly.
Because health professionals may be wary of stopping medications and not meeting recommended targets, there may be barriers to adopting this guideline. However, our experience with the PATH program indicates that these barriers can be overcome using effective communication strategies between health professionals and consumers.
AN APPROACH APPROPRIATE TO FRAILTY
There is no direct evidence for systolic blood pressure targets in the frail elderly, so we applied evidence from the nonfrail elderly. Our recommendations differ somewhat from those of other groups, which recommend targets below 140 to 150 mm Hg for older adults, although some do advise caution in the elderly for whom a substantial fall in blood pressure might be poorly tolerated. Despite these messages, we believe that clearer guidance is needed to direct health practitioners toward models that acknowledge that frail patients are in a precarious balance of health and may be harmed by treatments that strive to lower blood pressure to unproven targets. For this reason, our guideline clearly indicates when to decrease or stop drug treatment.
After physicians and health professionals examine the evidence and more fully understand the benefits and harms of treating frail older adults, we are confident that they will be more comfortable stopping medications that lower blood pressure to an unnecessarily low level and instead use an approach that is more appropriate to frailty. We hope clinicians can use this guideline with the same enthusiasm applied to other guidelines, and we welcome discussion.
Acknowledgments: We would like to thank and acknowledge Tanya MacLeod and Kathryn Yuill for their review of and advice about the manuscript.
- Palliative and Therapeutic Harmonization program. Hypertension guidelines. Treating hypertension in frailty. http://pathclinic.ca/resources/hypertension/. Accessed May 2, 2014.
- Theou O, Rockwood MR, Mitnitski A, Rockwood K. Disability and co-morbidity in relation to frailty: how much do they overlap? Arch Gerontol Geriatr 2012; 55:e1–e8.
- Makary MA, Segev DL, Pronovost PJ, et al. Frailty as a predictor of surgical outcomes in older patients. J Am Coll Surg 2010; 210:901–908.
- Tinetti ME, Bogardus ST, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004; 351:2870–2874.
- Ekerstad N, Swahn E, Janzon M, et al. Frailty is independently associated with short-term outcomes for elderly patients with non-ST-segment elevation myocardial infarction. Circulation 2011; 124:2397–2404.
- Theou O, Rockwood K. Should frailty status always be considered when treating the elderly patient? Aging Health 2012; 8:261–271.
- Rockwood K, Song X, MacKnight C, et al. A global clinical measure of fitness and frailty in elderly people. CMAJ 2005; 173:489–495.
- Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr 2008; 8:24.
- Aronow WS, Fleg JL, Pepine CJ, et al; ACCF Task Force. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2011; 123:2434–2506.
- The Canadian Hypertension Education Program (CHEP). 2014 CHEP recommendations. www.hypertension.ca/en/. Accessed May 2, 2014.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013; 14:392–397.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Ensrud KE, Ewing SK, Cawthon PM, et al; Osteoporotic Fractures in Men Research Group. A comparison of frailty indexes for the prediction of falls, disability, fractures, and mortality in older men. J Am Geriatr Soc 2009; 57:492–498.
- Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 2009; 57:453–461.
- Bergman H, Ferrucci L, Guralnik J, et al. Frailty: an emerging research and clinical paradigm—issues and controversies. J Gerontol A Biol Sci Med Sci 2007; 62:731–737.
- Morley JE, Malmstrom TK, Miller DK. A simple frailty questionnaire (FRAIL) predicts outcomes in middle aged African Americans. J Nutr Health Aging 2012; 16:601–608.
- Strawbridge WJ, Shema SJ, Balfour JL, Higby HR, Kaplan GA. Antecedents of frailty over three decades in an older cohort. J Gerontol B Psychol Sci Soc Sci 1998; 53:S9–S16.
- Matthews M, Lucas A, Boland R, et al. Use of a questionnaire to screen for frailty in the elderly: an exploratory study. Aging Clin Exp Res 2004; 16:34–40.
- Salvi F, Morichi V, Grilli A, et al. Screening for frailty in elderly emergency department patients by using the Identification of Seniors At Risk (ISAR). J Nutr Health Aging 2012; 16:313–318.
- Mitnitski AB, Mogilner AJ, Rockwood K. Accumulation of deficits as a proxy measure of aging. ScientificWorldJournal 2001; 1:323–336.
- Kellen E, Bulens P, Deckx L, et al. Identifying an accurate pre-screening tool in geriatric oncology. Crit Rev Oncol Hematol 2010; 75:243–248.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Martin FC, Brighton P. Frailty: different tools for different purposes? Age Ageing 2008; 37:129–131.
- Borson S, Scanlan J, Brush M, Vitaliano P, Dokmak A. The mini-cog: a cognitive ‘vital signs’ measure for dementia screening in multi-lingual elderly. Int J Geriatr Psychiatry 2000; 15:1021–1027.
- Reisberg B, Ferris SH. Brief Cognitive Rating Scale (BCRS). Psychopharmacol Bull 1988; 24:629–636.
- Moorhouse P, Mallery LH. Palliative and therapeutic harmonization: a model for appropriate decision-making in frail older adults. J Am Geriatr Soc 2012; 60:2326–2332.
- Palliative and Therapeutic Harmonization Clinic (PATH). www.pathclinic.ca. Accessed May 2, 2014.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. http://cme.medicine.dal.ca/ADS.htm. Accessed January 8, 2014.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. Issues in hypertension 2011. http://cme.medicine.dal.ca/files/Hypertension%20book.pdf. Accessed May 2, 2014.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Amery A, Birkenhäger W, Brixko P, et al. Mortality and morbidity results from the European Working Party on High Blood Pressure in the Elderly trial. Lancet 1985; 1:1349–1354.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. Br Med J (Clin Res Ed) 1986; 293:1145–1151.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Dahlöf B, Lindholm LH, Hansson L, Scherstén B, Ekbom T, Wester PO. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- Medical Research Council trial of treatment of hypertension in older adults: principal results. MRC Working Party. BMJ 1992; 304:405–412.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Liu L, Wang JG, Gong L, Liu G, Staessen JA. Comparison of active treatment and placebo in older Chinese patients with isolated systolic hypertension. Systolic Hypertension in China (Syst-China) Collaborative Group. J Hypertens 1998; 16:1823–1829.
- Lithell H, Hansson L, Skoog I, et al; SCOPE Study Group. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double-blind intervention trial. J Hypertens 2003; 21:875–886.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Oparil S, Yarows SA, Patel S, Fang H, Zhang J, Satlin A. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet 2007; 370:221–229.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Musini VM, Tejani AM, Bassett K, Wright JM. Pharmacotherapy for hypertension in the elderly. Cochrane Database Syst Rev 2009;CD000028.
- He FJ, MacGregor GA. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst Rev 2004;CD004937.
- Allen M, Kelly K, Fleming I. Hypertension in elderly patients: recommended systolic targets are not evidence based [in French]. Can Fam Physician 2013; 59:19–24.
- Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ 2012; 344:e3863.
- Sabayan B, Oleksik AM, Maier AB, et al. High blood pressure and resilience to physical and cognitive decline in the oldest old: the Leiden 85-plus Study. J Am Geriatr Soc 2012; 60:2014–2019.
- Sabayan B, van Vliet P, de Ruijter W, Gussekloo J, de Craen AJ, Westendorp RG. High blood pressure, physical and cognitive function, and risk of stroke in the oldest old: the Leiden 85-plus Study. Stroke 2013; 44:15–20.
- Poortvliet RK, Blom JW, de Craen AJ, et al. Low blood pressure predicts increased mortality in very old age even without heart failure: the Leiden 85-plus Study. Eur J Heart Fail 2013; 15:528–533.
- Odden MC, Peralta CA, Haan MN, Covinsky KE. Rethinking the association of high blood pressure with mortality in elderly adults: the impact of frailty. Arch Intern Med 2012; 172:1162–1168.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Yusuf S, Diener HC, Sacco RL, et al; PRoFESS Study Group. Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med 2008; 359:1225–1237.
- Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010; 170:1648–1654.
- Palliative and Therapeutic Harmonization program. Hypertension guidelines. Treating hypertension in frailty. http://pathclinic.ca/resources/hypertension/. Accessed May 2, 2014.
- Theou O, Rockwood MR, Mitnitski A, Rockwood K. Disability and co-morbidity in relation to frailty: how much do they overlap? Arch Gerontol Geriatr 2012; 55:e1–e8.
- Makary MA, Segev DL, Pronovost PJ, et al. Frailty as a predictor of surgical outcomes in older patients. J Am Coll Surg 2010; 210:901–908.
- Tinetti ME, Bogardus ST, Agostini JV. Potential pitfalls of disease-specific guidelines for patients with multiple conditions. N Engl J Med 2004; 351:2870–2874.
- Ekerstad N, Swahn E, Janzon M, et al. Frailty is independently associated with short-term outcomes for elderly patients with non-ST-segment elevation myocardial infarction. Circulation 2011; 124:2397–2404.
- Theou O, Rockwood K. Should frailty status always be considered when treating the elderly patient? Aging Health 2012; 8:261–271.
- Rockwood K, Song X, MacKnight C, et al. A global clinical measure of fitness and frailty in elderly people. CMAJ 2005; 173:489–495.
- Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr 2008; 8:24.
- Aronow WS, Fleg JL, Pepine CJ, et al; ACCF Task Force. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2011; 123:2434–2506.
- The Canadian Hypertension Education Program (CHEP). 2014 CHEP recommendations. www.hypertension.ca/en/. Accessed May 2, 2014.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013; 14:392–397.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Ensrud KE, Ewing SK, Cawthon PM, et al; Osteoporotic Fractures in Men Research Group. A comparison of frailty indexes for the prediction of falls, disability, fractures, and mortality in older men. J Am Geriatr Soc 2009; 57:492–498.
- Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 2009; 57:453–461.
- Bergman H, Ferrucci L, Guralnik J, et al. Frailty: an emerging research and clinical paradigm—issues and controversies. J Gerontol A Biol Sci Med Sci 2007; 62:731–737.
- Morley JE, Malmstrom TK, Miller DK. A simple frailty questionnaire (FRAIL) predicts outcomes in middle aged African Americans. J Nutr Health Aging 2012; 16:601–608.
- Strawbridge WJ, Shema SJ, Balfour JL, Higby HR, Kaplan GA. Antecedents of frailty over three decades in an older cohort. J Gerontol B Psychol Sci Soc Sci 1998; 53:S9–S16.
- Matthews M, Lucas A, Boland R, et al. Use of a questionnaire to screen for frailty in the elderly: an exploratory study. Aging Clin Exp Res 2004; 16:34–40.
- Salvi F, Morichi V, Grilli A, et al. Screening for frailty in elderly emergency department patients by using the Identification of Seniors At Risk (ISAR). J Nutr Health Aging 2012; 16:313–318.
- Mitnitski AB, Mogilner AJ, Rockwood K. Accumulation of deficits as a proxy measure of aging. ScientificWorldJournal 2001; 1:323–336.
- Kellen E, Bulens P, Deckx L, et al. Identifying an accurate pre-screening tool in geriatric oncology. Crit Rev Oncol Hematol 2010; 75:243–248.
- Rolfson DB, Majumdar SR, Tsuyuki RT, Tahir A, Rockwood K. Validity and reliability of the Edmonton Frail Scale. Age Ageing 2006; 35:526–529.
- Martin FC, Brighton P. Frailty: different tools for different purposes? Age Ageing 2008; 37:129–131.
- Borson S, Scanlan J, Brush M, Vitaliano P, Dokmak A. The mini-cog: a cognitive ‘vital signs’ measure for dementia screening in multi-lingual elderly. Int J Geriatr Psychiatry 2000; 15:1021–1027.
- Reisberg B, Ferris SH. Brief Cognitive Rating Scale (BCRS). Psychopharmacol Bull 1988; 24:629–636.
- Moorhouse P, Mallery LH. Palliative and therapeutic harmonization: a model for appropriate decision-making in frail older adults. J Am Geriatr Soc 2012; 60:2326–2332.
- Palliative and Therapeutic Harmonization Clinic (PATH). www.pathclinic.ca. Accessed May 2, 2014.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. http://cme.medicine.dal.ca/ADS.htm. Accessed January 8, 2014.
- Mallery LH, Moorhouse P. Respecting frailty. J Med Ethics 2011; 37:126–128.
- Dalhousie University Faculty of Medicine: Continuing Medical Education. Issues in hypertension 2011. http://cme.medicine.dal.ca/files/Hypertension%20book.pdf. Accessed May 2, 2014.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Amery A, Birkenhäger W, Brixko P, et al. Mortality and morbidity results from the European Working Party on High Blood Pressure in the Elderly trial. Lancet 1985; 1:1349–1354.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. Br Med J (Clin Res Ed) 1986; 293:1145–1151.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Dahlöf B, Lindholm LH, Hansson L, Scherstén B, Ekbom T, Wester PO. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- Medical Research Council trial of treatment of hypertension in older adults: principal results. MRC Working Party. BMJ 1992; 304:405–412.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Liu L, Wang JG, Gong L, Liu G, Staessen JA. Comparison of active treatment and placebo in older Chinese patients with isolated systolic hypertension. Systolic Hypertension in China (Syst-China) Collaborative Group. J Hypertens 1998; 16:1823–1829.
- Lithell H, Hansson L, Skoog I, et al; SCOPE Study Group. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double-blind intervention trial. J Hypertens 2003; 21:875–886.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Oparil S, Yarows SA, Patel S, Fang H, Zhang J, Satlin A. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet 2007; 370:221–229.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Musini VM, Tejani AM, Bassett K, Wright JM. Pharmacotherapy for hypertension in the elderly. Cochrane Database Syst Rev 2009;CD000028.
- He FJ, MacGregor GA. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst Rev 2004;CD004937.
- Allen M, Kelly K, Fleming I. Hypertension in elderly patients: recommended systolic targets are not evidence based [in French]. Can Fam Physician 2013; 59:19–24.
- Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ 2012; 344:e3863.
- Sabayan B, Oleksik AM, Maier AB, et al. High blood pressure and resilience to physical and cognitive decline in the oldest old: the Leiden 85-plus Study. J Am Geriatr Soc 2012; 60:2014–2019.
- Sabayan B, van Vliet P, de Ruijter W, Gussekloo J, de Craen AJ, Westendorp RG. High blood pressure, physical and cognitive function, and risk of stroke in the oldest old: the Leiden 85-plus Study. Stroke 2013; 44:15–20.
- Poortvliet RK, Blom JW, de Craen AJ, et al. Low blood pressure predicts increased mortality in very old age even without heart failure: the Leiden 85-plus Study. Eur J Heart Fail 2013; 15:528–533.
- Odden MC, Peralta CA, Haan MN, Covinsky KE. Rethinking the association of high blood pressure with mortality in elderly adults: the impact of frailty. Arch Intern Med 2012; 172:1162–1168.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Yusuf S, Diener HC, Sacco RL, et al; PRoFESS Study Group. Telmisartan to prevent recurrent stroke and cardiovascular events. N Engl J Med 2008; 359:1225–1237.
- Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medications in older adults: addressing polypharmacy. Arch Intern Med 2010; 170:1648–1654.
KEY POINTS
- For frail elderly patients, consider starting treatment if the systolic blood pressure is 160 mm Hg or higher.
- An appropriate target in this population is a seated systolic pressure between 140 and 160 mm Hg, as long as there is no orthostatic drop to less than 140 mm Hg upon standing from a lying position and treatment does not adversely affect quality of life.
- The blood pressure target does not need to be lower if the patient has diabetes. If the patient is severely frail and has a short life expectancy, a systolic target of 160 to 190 mm Hg may be reasonable.
- If the systolic pressure is below 140 mm Hg, antihypertensive medications can be reduced as long as they are not indicated for other conditions.
- In general, one should prescribe no more than two antihypertensive medications.
Patent foramen ovale and cryptogenic stroke: Many unanswered questions
Your patient has had an ischemic stroke, and so far you have found no obvious cause such as atrial fibrillation or carotid disease. Should you look for a patent foramen ovale (PFO)? And if you find it, what should you do?
This scenario continues to challenge primary care physicians and subspecialists and requires an understanding of the relationship between PFO and cryptogenic stroke, as well as familiarity with current data on the safety and effectiveness of the management options. PFO is known to be associated with cryptogenic stroke, but many questions remain, including:
- How can we tell if PFO is a culprit (“pathologic”) or an innocent bystander (“incidental”) in a patient who has had a cryptogenic stroke?
- Should stroke patients receive different medical therapy if they have a PFO? In particular, should they receive warfarin in addition to aspirin? And what about the novel oral anticoagulants?
- Which patients should undergo percutaneous closure of the PFO?
- Should we even be looking for PFO in stroke patients at this point, if we cannot say with certainty what we should do if we find it?
WHY IS THIS IMPORTANT?
Cerebrovascular disease is common and costly. The estimated yearly incidence of stroke in the United States is 795,000 events, at a cost of nearly $30 billion.1 The incidence of stroke in Europe is more than 1 million annually.2
During the diagnostic evaluation of stroke or transient ischemic attack (TIA), PFO is occasionally discovered incidentally by echocardiography. The management decisions that follow often fall to the primary care physician, who must decipher the conflicting data currently available and explain the options to the patient.
Although reviews have been published on this subject,3 several newer key trials and data on risk stratification warrant consideration.
DEFINITIONS
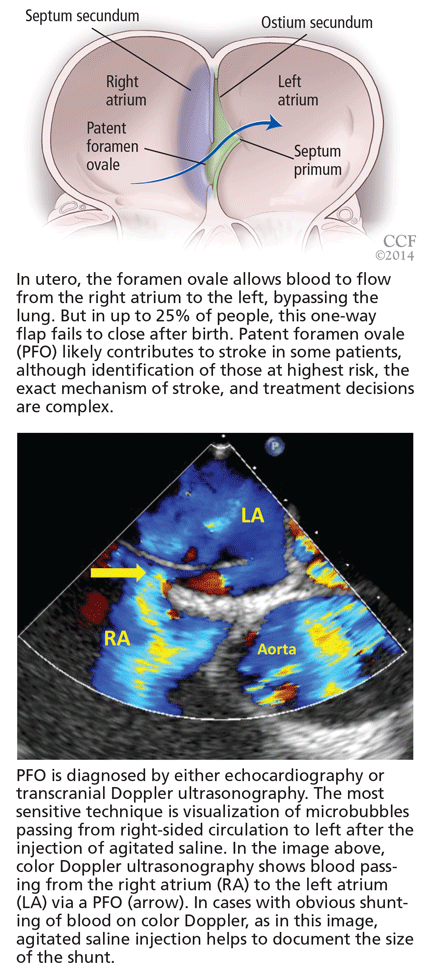
PFO is the failure of the septum primum to fuse with the septum secundum, so that a communication remains between the atria (Figure 1). The diagnosis is commonly made by echocardiography, when agitated saline is injected into the venous system and bubbles can be seen in the left atrium within three to five cardiac cycles (see video).
Atrial septal aneurysm is loosely defined as a septal excursion or bulging of at least 10 to 15 mm into the left and right atria during the cardiac cycle (Figure 2). The combination of PFO and atrial septal aneurysm may be more of a risk factor for stroke than PFO alone (see discussion below).
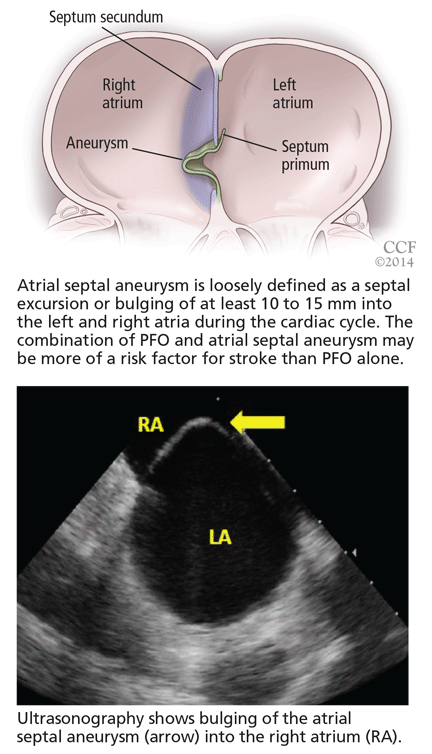
Cryptogenic stroke. The diagnostic workup of stroke fails to elucidate a clear cause in up to 40% of cases, which are thus called cryptogenic.4 The workup varies, but typically includes a search for a cardioembolic source and for atherosclerotic disease. Embolic sources are evaluated for by electrocardiography, transthoracic echocardiography, and possibly imaging of the aortic arch. Evaluation for atherosclerotic disease of the intracranial and extracranial arteries includes magnetic resonance angiography or, if that is unavailable, computed tomographic angiography or carotid Doppler ultrasonography. If no source is found, long-term cardiac monitoring may be used to detect paroxysmal atrial fibrillation, which may be more common than previously thought.
PFO AND CRYPTOGENIC STROKE ARE COMMON
As noted, there are approximately 800,000 strokes every year in the United States. If 25% to 40% of them are cryptogenic (the true prevalence warrants more evaluation),4,5 then 200,000 to 320,000 strokes are cryptogenic.
Autopsy studies indicate that 25% of the general population have a PFO, and if the prevalence is the same in people with cryptogenic stroke, that would equal 80,000 people with both cryptogenic stroke and PFO every year. However, the prevalence of PFO in patients with cryptogenic stroke appears to be significantly higher than in the general population.6 Although these numbers are crude estimates, they provide some insight into the prevalence of this clinical presentation.
HOW ARE CRYPTOGENIC STROKE AND PFO RELATED?
The exact relationship between PFO and cryptogenic stroke is unknown, although cases have been reported of thrombus in transit through a PFO, supporting paradoxical embolism as the plausible cause in stroke patients with PFO.7–9
There is clear evidence that the two conditions are associated by more than chance. Homma and Sacco6 reported that, in several studies, 93 (46%) of 202 patients under age 55 with cryptogenic stroke had PFOs, compared with 29 (11%) of 271 controls (P < .05 in all studies).6
In their evaluation of 23 case-control studies, Alsheikh-Ali et al10 found that the summary odds ratio (OR) for PFO in cryptogenic stroke vs PFO in control patients was 2.9 (95% confidence interval [CI] 2.1–4), largely driven by an OR of 5.1 (3.3–7.8) in those under age 55. Through Bayesian probability theory, this correlated with only a 33% probability that PFO in a patient with cryptogenic stroke was an innocent bystander rather than the culprit.10
IS PFO A RISK FACTOR FOR STROKE?
One of the more puzzling aspects of the relationship of PFO to cryptogenic stroke is that despite a clear association, there is little evidence that the relationship is causal.
Di Tullio et al11 followed 1,100 people who had no history of stroke and found that the risk of a first stroke in those with a PFO was not significantly higher than in those without a PFO, regardless of age, sex, or ethnic or racial group. At 80 months, the hazard ratio of stroke in people who had a PFO was 1.64 (95% CI 0.87–3.09).11 The findings were similar at 11 years, with a hazard ratio of 1.10 (95% CI 0.64–1.91).12
A prospective study of 585 patients found a similar risk of stroke in those with and without a PFO, with a hazard ratio of 1.46 (95% CI 0.74–2.88; P = .28).13
These prospective trials suggest that although previous studies have found a higher prevalence of PFOs in patients with cryptogenic stroke than in patients without stroke, there appears to be very little if any increased risk from baseline for a first stroke or TIA.
The lack of statistical significance in these trials should be interpreted with some caution, as a small increased risk is difficult to show if the event rate is low (approximately 10% of patients had events over 11 years in the study by Di Tullio et al12).
HOW DO WE KNOW IF A PFO IS A CULPRIT OR BYSTANDER?
Unfortunately, this is largely unanswered, though experts have suggested that echocardiographic features of the PFO, radiographic characteristics of the stroke, and clinical features of the patient may provide useful information.
‘High-risk’ features on echocardiography
Certain features of PFO may portend a high risk of cerebrovascular events. Both right-to-left shunting at rest and septal hypermobility were found in one study14 to be more common in patients with a PFO who had a stroke or TIA than in patients with a PFO but no cerebrovascular events. Also, patients who had these features and had a stroke had a higher risk of recurrence than stroke patients without these features (12.5% vs 4.3%, P = .05).14
Septal hypermobility and shunting at rest are easily diagnosed by echocardiography, and detecting these “high-risk” features would be useful if they could identify patients who would benefit from special therapy, such as percutaneous closure of the PFO.
Unfortunately, when investigators looked at these features in subgroup analysis of the major randomized controlled trials of percutaneous closure vs medical therapy, the results were mixed.
CLOSURE 1 (the Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale)15 found percutaneous closure to be no better than medical therapy, regardless of shunt size or the presence of atrial septal aneurysm.
Similarly, the PC trial (Clinical Trial Comparing Percutaneous Closure of Patent Foramen Ovale Using the Amplatzer PFO Occluder With Medical Treatment in Patients With Cryptogenic Embolism)16 found no statistically significant benefit of closure in those with atrial septal aneurysm.
In contrast, the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment)17 showed percutaneous closure to be beneficial in patients with atrial septal aneurysm or large shunt.
Radiographic characteristics of the stroke
Another area of interest in trying to identify culprit PFOs is the radiographic characteristics of the stroke.
In a study comparing patients with stroke related to atrial fibrillation vs patients with cryptogenic stroke and a known PFO, those in the latter group were more likely to have a single cortical infarction (34.2% vs 3.1%; P < .001) or multiple small scattered lesions (23.1% vs 5.9%; P < .01).18 Similarly, in a large database of patients with cryptogenic stroke and known PFO status, a superficially located stroke was associated with the presence of PFO (OR 1.54; P < .0001).19
Although these findings do not tell us with certainty that a patient’s PFO was the cause of his or her stroke, they provide guidance when dealing with the uncertainty of how to manage a patient with PFO. They may be useful in clinical practice, for example, when discussing treatment options with a young patient with cryptogenic stroke who has no risk factors and a superficial single infarct and who is found to have a PFO with a right-to-left shunting at rest.
Patient characteristics
Kent et al20 developed a 10-point index (the RoPE score) in an attempt to assign a probability to whether a stroke was PFO-related. Points were assigned for patients who were younger, who had a cortical stroke on neuroimaging, and who did not have diabetes, hypertension, smoking, or prior stroke or TIA. Patients with cryptogenic stroke with a higher RoPE score were more likely to have a PFO and thus had a higher likelihood that the index event was related to PFO. Of note, the patients with the highest likelihood of PFO-related stroke were the least likely to have a recurrence (RoPE score of 9 to 10; PFO-attributable fraction 88%; estimated 2-year recurrence rate 2%; 95% CI 0%–4%), whereas those with a low RoPE score have more traditional risk factors for stroke and thus are more likely to have a recurrence (RoPE 0 to 3; estimated 2-year recurrence rate 20%; 95% CI 12%–28%).20
Again, this sheds light on a difficulty faced by randomized controlled trials: the patients who may benefit from closure of a PFO may very well be those with the lowest recurrence rates without intervention.
The RoPE index was examined in an attempt to validate previously described morphologic criteria of “high-risk” PFO,21 though none of the previously described “high-risk” echocardiographic features (large physiologic size, hypermobile septum, shunt at rest) were more common in the group with presumed PFO-attributable stroke (RoPE score > 6). This underscores the difficulty of distinguishing pathologic PFO from incidental PFO.
KEY TREATMENT CONSIDERATIONS FOR SECONDARY PREVENTION
Given the complicated relationship between PFO and cryptogenic stroke, there has been much debate over management strategies. The three options are surgical closure, percutaneous closure with a device, and medical therapy. The goal of all three is to prevent the recurrence of stroke or TIA.
Surgical closure has largely been supplanted by percutaneous closure, but is still done in specific situations such as when a PFO is found incidentally on transesophageal echocardiography during surgery for another cardiac condition. The data on such cases22 tend to support the argument that asymptomatic PFOs in the general population have a relatively benign natural history.
Thus, the two key questions about management that warrant discussion are: is anticoagulation superior to antiplatelet therapy? And is percutaneous closure superior to medical management?
Anticoagulant vs antiplatelet therapy
Whether to treat with aspirin or with a vitamin K antagonist has been a subject of debate, although there is no strong evidence to suggest that anticoagulation is superior to antiplatelet therapy.
The concern that aspirin alone is insufficient in some patients stems from a study by Mas et al,23 who followed 581 patients with cryptogenic stroke who had a PFO alone, a PFO with an atrial septal aneurysm, or neither. The rate of stroke recurrence at 4 years on aspirin therapy was 2.3% in those with a PFO alone, 15.2% in those with a PFO with an atrial septal aneurysm, and 4.2% in those with neither.
Many have concluded that aspirin therapy does not sufficiently protect those with both PFO and atrial septal aneurysm, given the high recurrence rate in this group. This might lead to the suggestion that anticoagulation could be of benefit in these patients.
However, the Patent Foramen Ovale in Cryptogenic Stroke Study (PiCSS)24 and the Spanish Multicenter Study Into Right-to-Left Shunt in Cryptogenic Stroke (CODICIA)25 found similar recurrence rates in patients with PFO and atrial septal aneurysm compared with those with only PFO. In these two studies, recurrence rates were similar regardless of whether patients were taking aspirin or warfarin.
In a study that followed 140 consecutive patients with both stroke and PFO, those treated in a nonrandomized fashion with antiplatelet agents had no difference in the recurrence rate compared with those treated with anticoagulation.26
Although uncertainty remains because no head-to-head randomized controlled trial has been done, some patients with PFO have other indications for anticoagulation, most commonly atrial fibrillation and venous thromboembolic disease.
There are currently no data on the use of novel oral anticoagulants in this setting.
Is percutaneous closure better than medical therapy?
When cryptogenic stroke is treated with antiplatelet therapy or anticoagulation therapy, the recurrence rate is the same whether or not the patient has a PFO.23–25 The belief that medical therapy offers adequate secondary protection is supported by a meta-analysis of 15 studies that found no increased risk of recurrent ischemic events in those with a PFO on medical therapy (antiplatelet or anticoagulant) vs those without a PFO (relative risk 1.1, 95% CI 0.8–1.5).27
Despite the conflicting evidence, percutaneous closure of PFO is still performed, mostly on a case-by-case basis. This has been supported by an apparent benefit in observational studies.
A systematic review of 52 single-arm studies and 7 comparative nonrandomized studies of patients with PFO and cryptogenic stroke found the rate of recurrent stroke to be 0.36 per 100 person-years with percutaneous closure vs 2.53 per 100 person-years with medical therapy.28 However, three long-awaited randomized controlled trials (CLOSURE 1, the PC trial, and RESPECT) failed to show a significant reduction in primary end points with percutaneous closure vs standard medical therapy.15–17
These trials had several limitations: event rates were low, medical therapy varied by provider, and enrollment was slowed by out-of-study percutaneous closure in patients perceived to be at high risk (though, as discussed above, high risk is difficult to determine).
Intention-to-treat analysis in RESPECT showed no benefit from percutaneous closure, but a favorable outcome was noted with closure in as-treated analysis (HR 0.27; 95% CI 0.1–0.75; P = .007) and per-protocol analysis (HR 0.37; 95% CI 0.14–0.96; P = .03) of the 980 randomized patients.17 This suggests some benefit, as does the CLOSURE 1 trial, in which 3 of the 12 recurrent strokes in the percutaneous closure group occurred before the device was implanted.15
The low event rates in these studies prompted several meta-analyses.29–35 However, only two suggested a benefit of percutaneous closure over medical therapy. In one recent meta-analysis,29 observational study data suggested benefit from percutaneous closure, whereas three randomized controlled trials failed to show a statistically significant benefit.
The conclusions of the meta-analyses must be interpreted with caution because of inherent differences in the randomized controlled trials, including the closure device used, inclusion criteria, study end points, and variations in medical therapy.
Devices differ
A meta-analysis by Khan et al35 showed a benefit of percutaneous closure when evaluating only studies using the Amplatzer PFO occluder (AGA Medical), as in RESPECT and the PC trial.35 As data accumulate, it is important to remember that there are differences between devices. Ongoing trials continue to investigate the Amplatzer device (NCT01550588) and the GORE HELEX Septal Occluder/GORE Septal Occluder (Gore Medical) (NCT00738894).
In another meta-analysis, Pineda et al31 found a benefit with closure in the as-treated analysis using data from all three randomized controlled trials (OR 0.62; 95% CI 0.41–0.94; P = .02).31 Although paradoxical embolism through the PFO as the mechanism of stroke has been questioned, this finding suggests that actual closure of a PFO may protect against further events, presumably by preventing paradoxical embolism.
Different closure devices have different side effects. The incidence of atrial fibrillation with the CardioSEAL STARFlex device (NMT Medical) is higher than with medical therapy (used in the CLOSURE trial15), whereas this risk was not statistically significantly increased in the PC trial16 and RESPECT,17 which used the Amplatzer device.
Benefit in those with atrial septal aneurysm?
Percutaneous closure has been shown to be safe and effective in patients with PFO and atrial septal aneurysm.36 There was some benefit of closure over medical therapy in a subgroup analysis from RESPECT in these patients, with a HR of 0.19 (95% CI 0.04–0.87, P = .02),17 although this was not seen in either CLOSURE 1 or the PC trial.
WHAT ARE THE RISKS OF PERCUTANEOUS CLOSURE?
Minor complications of percutaneous closure include bleeding, atrial arrhythmias, device embolization and fracture, and complications related to vascular access. Major complications include hemorrhage requiring transfusion, need for surgery, cardiac tamponade, pulmonary embolism, and death.
The cumulative rate of major complications in 10 observational studies was 1.5%, and the rate of minor complications was 7.9%.37 The RESPECT investigators reported a serious adverse event in 4.2% of patients (ranging in severity from chest tightness to cardiac tamponade).17
Another possible consequence of percutaneous closure is the need for chronic anticoagulation because of the increased risk of postprocedural atrial fibrillation seen in meta-analyses,29,31,32 though this may be device-specific.32
Percutaneous closure was considered successful—ie, to have nearly or completely eliminated shunting of blood through the defect—at 6 months of follow-up in 95.9% of patients in the PC trial, 93.5% in RESPECT, and 86.1% in CLOSURE 1.15–17
WHAT SHOULD WE BE DOING IN DAILY PRACTICE?
Give aspirin. Aspirin is effective in secondary stroke prevention, and data suggest that patients with PFO and cryptogenic stroke who receive aspirin therapy alone have a similar risk of recurrent events as patients without PFO.
Give warfarin if indicated. Evidence is insufficient to recommend vitamin K antagonist therapy in all patients with PFO and cryptogenic stroke. However, coexisting conditions that warrant anticoagulation must be taken into account.
Individualize. Given the lack of evidence to definitively guide management of patients with cryptogenic stroke and PFO, we need to individualize our approach, taking into account patient preferences, bleeding risk, ability to tolerate procedures, and the likelihood that the PFO is at fault.
No definitive answer on PFO closure. The most recent data suggest that closure may be beneficial, but key questions remain: Who will benefit? And what is the ideal medical therapy? Optimal management will only be established by the continued enrollment of appropriate patients into ongoing clinical trials.
Another question is whether it is possible to perform a randomized controlled trial with enough patients to definitively prove whether percutaneous closure is superior to medical therapy. Recent experience would suggest not.
In the meantime, we have some guidance from the American Heart Association and the American Stroke Association Council on Stroke38 based on the limited evidence available.
Consider patient preference. The physician should present the options to the patient in a balanced manner to enable him or her to make an informed decision. Patients can also be encouraged to seek additional information at websites such as www.stroke.org and www.nlm.nih.gov.
Referral to an interventional cardiologist for evaluation for closure is reasonable in patients with recurrent stroke, medication failure, complicated atrial septal anatomy such as PFO with aneurysm or large shunt, concurrent thromboembolic disease, or contraindications to anticoagulation.
MORE WORK NEEDED
Areas for further study include further identifying the characteristics of patients with PFO and cryptogenic stroke that might indicate who would benefit from percutaneous closure, elucidating the mechanism of stroke in these patients, and determining whether routine stroke evaluation should include echocardiography with a bubble study if there is no change in management based on the finding of PFO.39
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- Truelsen T, Piechowski-JóŸwiak B, Bonita R, Mathers C, Bogousslavsky J, Boysen G. Stroke incidence and prevalence in Europe: a review of available data. Eur J Neurol 2006; 13:581–598.
- Furlan AJ. Patent foramen ovale and stroke: to close or not to close? Cleve Clin J Med 2007; 74(suppl 1):S118–S120.
- Sacco RL, Ellenberg JH, Mohr JP, et al. Infarcts of undetermined cause: the NINCDS Stroke Data Bank. Ann Neurol 1989; 25:382–390.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German stroke data bank. Stroke 2001; 32:2559–2566.
- Homma S, Sacco RL. Patent foramen ovale and stroke. Circulation 2005; 112:1063–1072.
- Sattiraju S, Masri SC, Liao K, Missov E. Three-dimensional transesophageal echocardiography of a thrombus entrapped by a patent foramen ovale. Ann Thorac Surg 2012; 94:e101–e102.
- Schreiter SW, Phillips JH. Thromboembolus traversing a patent foramen ovale: resolution with anticoagulation. J Am Soc Echocardiogr 1994; 7:659–662.
- Hust MH, Staiger M, Braun B. Migration of paradoxic embolus through a patent foramen ovale diagnosed by echocardiography: successful thrombolysis. Am Heart J 1995; 129:620–622.
- Alsheikh-Ali AA, Thaler DE, Kent DM. Patent foramen ovale in cryptogenic stroke: incidental or pathogenic? Stroke 2009; 40:2349–2355.
- Di Tullio MR, Sacco RL, Sciacca RR, Jin Z, Homma S. Patent foramen ovale and the risk of ischemic stroke in a multiethnic population. J Am Coll Cardiol 2007; 49:797–802.
- Di Tullio MR, Jin Z, Russo C, et al. Patent foramen ovale, subclinical cerebrovascular disease, and ischemic stroke in a population-based cohort. J Am Coll Cardiol 2013; 62:35–41.
- Meissner I, Khandheria BK, Heit JA, et al. Patent foramen ovale: innocent or guilty? Evidence from a prospective population-based study. J Am Coll Cardiol 2006; 47:440–445.
- De Castro S, Cartoni D, Fiorelli M, et al. Morphological and functional characteristics of patent foramen ovale and their embolic implications. Stroke 2000; 31:2407–2413.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE I Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Kim BJ, Sohn H, Sun BJ, et al. Imaging characteristics of ischemic strokes related to patent foramen ovale. Stroke 2013; 44:3350–3356.
- Thaler DE, Ruthazer R, Di Angelantonio E, et al. Neuroimaging findings in cryptogenic stroke patients with and without patent foramen ovale. Stroke 2013; 44:675–680.
- Kent DM, Ruthazer R, Weimar C, et al. An index to identify stroke-related vs incidental patent foramen ovale in cryptogenic stroke. Neurology 2013; 81:619–625.
- Wessler BS, Thaler DE, Ruthazer R, et al. Transesophageal echocardiography in cryptogenic stroke and patent foramen ovale: analysis of putative high-risk features from the risk of paradoxical embolism database. Circ Cardiovasc Imaging 2014; 7:125–131.
- Krasuski RA, Hart SA, Allen D, et al. Prevalence and repair of intraoperatively diagnosed patent foramen ovale and association with perioperative outcomes and long-term survival. JAMA 2009; 302:290–297.
- Mas JL, Arquizan C, Lamy C, et al; Patent Foramen Ovale and Atrial Septal Aneurysm Study Group. Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysm, or both. N Engl J Med 2001; 345:1740–1746.
- Homma S, Sacco RL, Di Tullio MR, Sciacca RR, Mohr JP; PFO in Cryptogenic Stroke Study (PICSS) Investigators. Effect of medical treatment in stroke patients with patent foramen ovale: patent foramen ovale in Cryptogenic Stroke Study. Circulation 2002; 105:2625–2631.
- Serena J, Marti-Fàbregas J, Santamarina E, et al; CODICIA, Right-to-Left Shunt in Cryptogenic Stroke Study; Stroke Project of the Cerebrovascular Diseases Study Group, Spanish Society of Neurology. Recurrent stroke and massive right-to-left shunt: results from the prospective Spanish multicenter (CODICIA) study. Stroke 2008; 39:3131–3136.
- Bogousslavsky J, Garazi S, Jeanrenaud X, Aebischer N, Van Melle G. Stroke recurrence in patients with patent foramen ovale: the Lausanne Study. Lausanne Stroke with Paradoxal Embolism Study Group. Neurology 1996; 46:1301–1305.
- Almekhlafi MA, Wilton SB, Rabi DM, Ghali WA, Lorenzetti DL, Hill MD. Recurrent cerebral ischemia in medically treated patent foramen ovale: a meta-analysis. Neurology 2009; 73:89–97.
- Kitsios GD, Dahabreh IJ, Abu Dabrh AM, Thaler DE, Kent DM. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke 2012; 43:422–431.
- Wolfrum M, Froehlich GM, Knapp G, et al. Stroke prevention by percutaneous closure of patent foramen ovale: a systematic review and meta-analysis. Heart 2014; 100:389–395.
- Rengifo-Moreno P, Palacios IF, Junpaparp P, Witzke CF, Morris DL, Romero-Corral A. Patent foramen ovale transcatheter closure vs medical therapy on recurrent vascular events: a systematic review and meta-analysis of randomized controlled trials. Eur Heart J 2013; 34:3342–3352.
- Pineda AM, Nascimento FO, Yang SC, Kirtane AJ, Sommer RJ, Beohar N. A meta-analysis of transcatheter closure of patent foramen ovale versus medical therapy for prevention of recurrent thromboembolic events in patients with cryptogenic cerebrovascular events. Catheter Cardiovasc Interv 2013; 82:968–975.
- Kwong JS, Lam YY, Yu CM. Percutaneous closure of patent foramen ovale for cryptogenic stroke: a meta-analysis of randomized controlled trials. Int J Cardiol 2013; 168:4132–4148.
- Ntaios G, Papavasileiou V, Makaritsis K, Michel P. PFO closure vs medical therapy in cryptogenic stroke or transient ischemic attack: a systematic review and meta-analysis. Int J Cardiol 2013; 169:101–105.
- Nagaraja V, Raval J, Eslick GD, Burgess D, Denniss AR. Is transcatheter closure better than medical therapy for cryptogenic stroke with patent foramen ovale? A meta-analysis of randomised trials. Heart Lung Circ 2013; 22:903–909.
- Khan AR, Bin Abdulhak AA, Sheikh MA, et al. Device closure of patent foramen ovale versus medical therapy in cryptogenic stroke: a systematic review and meta-analysis. JACC Cardiovasc Interv 2013; 6:1316–1323.
- Wahl A, Krumsdorf U, Meier B, et al. Transcatheter treatment of atrial septal aneurysm associated with patent foramen ovale for prevention of recurrent paradoxical embolism in high-risk patients. J Am Coll Cardiol 2005; 45:377–380.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Sacco RL, Adams R, Albers G, et al; American Heart Association; American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke 2006; 37:577–617.
- Rana BS, Thomas MR, Calvert PA, Monaghan MJ, Hildick-Smith D. Echocardiographic evaluation of patent foramen ovale prior to device closure. JACC Cardiovasc Imaging 2010; 3:749–760.
Your patient has had an ischemic stroke, and so far you have found no obvious cause such as atrial fibrillation or carotid disease. Should you look for a patent foramen ovale (PFO)? And if you find it, what should you do?
This scenario continues to challenge primary care physicians and subspecialists and requires an understanding of the relationship between PFO and cryptogenic stroke, as well as familiarity with current data on the safety and effectiveness of the management options. PFO is known to be associated with cryptogenic stroke, but many questions remain, including:
- How can we tell if PFO is a culprit (“pathologic”) or an innocent bystander (“incidental”) in a patient who has had a cryptogenic stroke?
- Should stroke patients receive different medical therapy if they have a PFO? In particular, should they receive warfarin in addition to aspirin? And what about the novel oral anticoagulants?
- Which patients should undergo percutaneous closure of the PFO?
- Should we even be looking for PFO in stroke patients at this point, if we cannot say with certainty what we should do if we find it?
WHY IS THIS IMPORTANT?
Cerebrovascular disease is common and costly. The estimated yearly incidence of stroke in the United States is 795,000 events, at a cost of nearly $30 billion.1 The incidence of stroke in Europe is more than 1 million annually.2
During the diagnostic evaluation of stroke or transient ischemic attack (TIA), PFO is occasionally discovered incidentally by echocardiography. The management decisions that follow often fall to the primary care physician, who must decipher the conflicting data currently available and explain the options to the patient.
Although reviews have been published on this subject,3 several newer key trials and data on risk stratification warrant consideration.
DEFINITIONS
PFO is the failure of the septum primum to fuse with the septum secundum, so that a communication remains between the atria (Figure 1). The diagnosis is commonly made by echocardiography, when agitated saline is injected into the venous system and bubbles can be seen in the left atrium within three to five cardiac cycles (see video).
Atrial septal aneurysm is loosely defined as a septal excursion or bulging of at least 10 to 15 mm into the left and right atria during the cardiac cycle (Figure 2). The combination of PFO and atrial septal aneurysm may be more of a risk factor for stroke than PFO alone (see discussion below).
Cryptogenic stroke. The diagnostic workup of stroke fails to elucidate a clear cause in up to 40% of cases, which are thus called cryptogenic.4 The workup varies, but typically includes a search for a cardioembolic source and for atherosclerotic disease. Embolic sources are evaluated for by electrocardiography, transthoracic echocardiography, and possibly imaging of the aortic arch. Evaluation for atherosclerotic disease of the intracranial and extracranial arteries includes magnetic resonance angiography or, if that is unavailable, computed tomographic angiography or carotid Doppler ultrasonography. If no source is found, long-term cardiac monitoring may be used to detect paroxysmal atrial fibrillation, which may be more common than previously thought.
PFO AND CRYPTOGENIC STROKE ARE COMMON
As noted, there are approximately 800,000 strokes every year in the United States. If 25% to 40% of them are cryptogenic (the true prevalence warrants more evaluation),4,5 then 200,000 to 320,000 strokes are cryptogenic.
Autopsy studies indicate that 25% of the general population have a PFO, and if the prevalence is the same in people with cryptogenic stroke, that would equal 80,000 people with both cryptogenic stroke and PFO every year. However, the prevalence of PFO in patients with cryptogenic stroke appears to be significantly higher than in the general population.6 Although these numbers are crude estimates, they provide some insight into the prevalence of this clinical presentation.
HOW ARE CRYPTOGENIC STROKE AND PFO RELATED?
The exact relationship between PFO and cryptogenic stroke is unknown, although cases have been reported of thrombus in transit through a PFO, supporting paradoxical embolism as the plausible cause in stroke patients with PFO.7–9
There is clear evidence that the two conditions are associated by more than chance. Homma and Sacco6 reported that, in several studies, 93 (46%) of 202 patients under age 55 with cryptogenic stroke had PFOs, compared with 29 (11%) of 271 controls (P < .05 in all studies).6
In their evaluation of 23 case-control studies, Alsheikh-Ali et al10 found that the summary odds ratio (OR) for PFO in cryptogenic stroke vs PFO in control patients was 2.9 (95% confidence interval [CI] 2.1–4), largely driven by an OR of 5.1 (3.3–7.8) in those under age 55. Through Bayesian probability theory, this correlated with only a 33% probability that PFO in a patient with cryptogenic stroke was an innocent bystander rather than the culprit.10
IS PFO A RISK FACTOR FOR STROKE?
One of the more puzzling aspects of the relationship of PFO to cryptogenic stroke is that despite a clear association, there is little evidence that the relationship is causal.
Di Tullio et al11 followed 1,100 people who had no history of stroke and found that the risk of a first stroke in those with a PFO was not significantly higher than in those without a PFO, regardless of age, sex, or ethnic or racial group. At 80 months, the hazard ratio of stroke in people who had a PFO was 1.64 (95% CI 0.87–3.09).11 The findings were similar at 11 years, with a hazard ratio of 1.10 (95% CI 0.64–1.91).12
A prospective study of 585 patients found a similar risk of stroke in those with and without a PFO, with a hazard ratio of 1.46 (95% CI 0.74–2.88; P = .28).13
These prospective trials suggest that although previous studies have found a higher prevalence of PFOs in patients with cryptogenic stroke than in patients without stroke, there appears to be very little if any increased risk from baseline for a first stroke or TIA.
The lack of statistical significance in these trials should be interpreted with some caution, as a small increased risk is difficult to show if the event rate is low (approximately 10% of patients had events over 11 years in the study by Di Tullio et al12).
HOW DO WE KNOW IF A PFO IS A CULPRIT OR BYSTANDER?
Unfortunately, this is largely unanswered, though experts have suggested that echocardiographic features of the PFO, radiographic characteristics of the stroke, and clinical features of the patient may provide useful information.
‘High-risk’ features on echocardiography
Certain features of PFO may portend a high risk of cerebrovascular events. Both right-to-left shunting at rest and septal hypermobility were found in one study14 to be more common in patients with a PFO who had a stroke or TIA than in patients with a PFO but no cerebrovascular events. Also, patients who had these features and had a stroke had a higher risk of recurrence than stroke patients without these features (12.5% vs 4.3%, P = .05).14
Septal hypermobility and shunting at rest are easily diagnosed by echocardiography, and detecting these “high-risk” features would be useful if they could identify patients who would benefit from special therapy, such as percutaneous closure of the PFO.
Unfortunately, when investigators looked at these features in subgroup analysis of the major randomized controlled trials of percutaneous closure vs medical therapy, the results were mixed.
CLOSURE 1 (the Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale)15 found percutaneous closure to be no better than medical therapy, regardless of shunt size or the presence of atrial septal aneurysm.
Similarly, the PC trial (Clinical Trial Comparing Percutaneous Closure of Patent Foramen Ovale Using the Amplatzer PFO Occluder With Medical Treatment in Patients With Cryptogenic Embolism)16 found no statistically significant benefit of closure in those with atrial septal aneurysm.
In contrast, the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment)17 showed percutaneous closure to be beneficial in patients with atrial septal aneurysm or large shunt.
Radiographic characteristics of the stroke
Another area of interest in trying to identify culprit PFOs is the radiographic characteristics of the stroke.
In a study comparing patients with stroke related to atrial fibrillation vs patients with cryptogenic stroke and a known PFO, those in the latter group were more likely to have a single cortical infarction (34.2% vs 3.1%; P < .001) or multiple small scattered lesions (23.1% vs 5.9%; P < .01).18 Similarly, in a large database of patients with cryptogenic stroke and known PFO status, a superficially located stroke was associated with the presence of PFO (OR 1.54; P < .0001).19
Although these findings do not tell us with certainty that a patient’s PFO was the cause of his or her stroke, they provide guidance when dealing with the uncertainty of how to manage a patient with PFO. They may be useful in clinical practice, for example, when discussing treatment options with a young patient with cryptogenic stroke who has no risk factors and a superficial single infarct and who is found to have a PFO with a right-to-left shunting at rest.
Patient characteristics
Kent et al20 developed a 10-point index (the RoPE score) in an attempt to assign a probability to whether a stroke was PFO-related. Points were assigned for patients who were younger, who had a cortical stroke on neuroimaging, and who did not have diabetes, hypertension, smoking, or prior stroke or TIA. Patients with cryptogenic stroke with a higher RoPE score were more likely to have a PFO and thus had a higher likelihood that the index event was related to PFO. Of note, the patients with the highest likelihood of PFO-related stroke were the least likely to have a recurrence (RoPE score of 9 to 10; PFO-attributable fraction 88%; estimated 2-year recurrence rate 2%; 95% CI 0%–4%), whereas those with a low RoPE score have more traditional risk factors for stroke and thus are more likely to have a recurrence (RoPE 0 to 3; estimated 2-year recurrence rate 20%; 95% CI 12%–28%).20
Again, this sheds light on a difficulty faced by randomized controlled trials: the patients who may benefit from closure of a PFO may very well be those with the lowest recurrence rates without intervention.
The RoPE index was examined in an attempt to validate previously described morphologic criteria of “high-risk” PFO,21 though none of the previously described “high-risk” echocardiographic features (large physiologic size, hypermobile septum, shunt at rest) were more common in the group with presumed PFO-attributable stroke (RoPE score > 6). This underscores the difficulty of distinguishing pathologic PFO from incidental PFO.
KEY TREATMENT CONSIDERATIONS FOR SECONDARY PREVENTION
Given the complicated relationship between PFO and cryptogenic stroke, there has been much debate over management strategies. The three options are surgical closure, percutaneous closure with a device, and medical therapy. The goal of all three is to prevent the recurrence of stroke or TIA.
Surgical closure has largely been supplanted by percutaneous closure, but is still done in specific situations such as when a PFO is found incidentally on transesophageal echocardiography during surgery for another cardiac condition. The data on such cases22 tend to support the argument that asymptomatic PFOs in the general population have a relatively benign natural history.
Thus, the two key questions about management that warrant discussion are: is anticoagulation superior to antiplatelet therapy? And is percutaneous closure superior to medical management?
Anticoagulant vs antiplatelet therapy
Whether to treat with aspirin or with a vitamin K antagonist has been a subject of debate, although there is no strong evidence to suggest that anticoagulation is superior to antiplatelet therapy.
The concern that aspirin alone is insufficient in some patients stems from a study by Mas et al,23 who followed 581 patients with cryptogenic stroke who had a PFO alone, a PFO with an atrial septal aneurysm, or neither. The rate of stroke recurrence at 4 years on aspirin therapy was 2.3% in those with a PFO alone, 15.2% in those with a PFO with an atrial septal aneurysm, and 4.2% in those with neither.
Many have concluded that aspirin therapy does not sufficiently protect those with both PFO and atrial septal aneurysm, given the high recurrence rate in this group. This might lead to the suggestion that anticoagulation could be of benefit in these patients.
However, the Patent Foramen Ovale in Cryptogenic Stroke Study (PiCSS)24 and the Spanish Multicenter Study Into Right-to-Left Shunt in Cryptogenic Stroke (CODICIA)25 found similar recurrence rates in patients with PFO and atrial septal aneurysm compared with those with only PFO. In these two studies, recurrence rates were similar regardless of whether patients were taking aspirin or warfarin.
In a study that followed 140 consecutive patients with both stroke and PFO, those treated in a nonrandomized fashion with antiplatelet agents had no difference in the recurrence rate compared with those treated with anticoagulation.26
Although uncertainty remains because no head-to-head randomized controlled trial has been done, some patients with PFO have other indications for anticoagulation, most commonly atrial fibrillation and venous thromboembolic disease.
There are currently no data on the use of novel oral anticoagulants in this setting.
Is percutaneous closure better than medical therapy?
When cryptogenic stroke is treated with antiplatelet therapy or anticoagulation therapy, the recurrence rate is the same whether or not the patient has a PFO.23–25 The belief that medical therapy offers adequate secondary protection is supported by a meta-analysis of 15 studies that found no increased risk of recurrent ischemic events in those with a PFO on medical therapy (antiplatelet or anticoagulant) vs those without a PFO (relative risk 1.1, 95% CI 0.8–1.5).27
Despite the conflicting evidence, percutaneous closure of PFO is still performed, mostly on a case-by-case basis. This has been supported by an apparent benefit in observational studies.
A systematic review of 52 single-arm studies and 7 comparative nonrandomized studies of patients with PFO and cryptogenic stroke found the rate of recurrent stroke to be 0.36 per 100 person-years with percutaneous closure vs 2.53 per 100 person-years with medical therapy.28 However, three long-awaited randomized controlled trials (CLOSURE 1, the PC trial, and RESPECT) failed to show a significant reduction in primary end points with percutaneous closure vs standard medical therapy.15–17
These trials had several limitations: event rates were low, medical therapy varied by provider, and enrollment was slowed by out-of-study percutaneous closure in patients perceived to be at high risk (though, as discussed above, high risk is difficult to determine).
Intention-to-treat analysis in RESPECT showed no benefit from percutaneous closure, but a favorable outcome was noted with closure in as-treated analysis (HR 0.27; 95% CI 0.1–0.75; P = .007) and per-protocol analysis (HR 0.37; 95% CI 0.14–0.96; P = .03) of the 980 randomized patients.17 This suggests some benefit, as does the CLOSURE 1 trial, in which 3 of the 12 recurrent strokes in the percutaneous closure group occurred before the device was implanted.15
The low event rates in these studies prompted several meta-analyses.29–35 However, only two suggested a benefit of percutaneous closure over medical therapy. In one recent meta-analysis,29 observational study data suggested benefit from percutaneous closure, whereas three randomized controlled trials failed to show a statistically significant benefit.
The conclusions of the meta-analyses must be interpreted with caution because of inherent differences in the randomized controlled trials, including the closure device used, inclusion criteria, study end points, and variations in medical therapy.
Devices differ
A meta-analysis by Khan et al35 showed a benefit of percutaneous closure when evaluating only studies using the Amplatzer PFO occluder (AGA Medical), as in RESPECT and the PC trial.35 As data accumulate, it is important to remember that there are differences between devices. Ongoing trials continue to investigate the Amplatzer device (NCT01550588) and the GORE HELEX Septal Occluder/GORE Septal Occluder (Gore Medical) (NCT00738894).
In another meta-analysis, Pineda et al31 found a benefit with closure in the as-treated analysis using data from all three randomized controlled trials (OR 0.62; 95% CI 0.41–0.94; P = .02).31 Although paradoxical embolism through the PFO as the mechanism of stroke has been questioned, this finding suggests that actual closure of a PFO may protect against further events, presumably by preventing paradoxical embolism.
Different closure devices have different side effects. The incidence of atrial fibrillation with the CardioSEAL STARFlex device (NMT Medical) is higher than with medical therapy (used in the CLOSURE trial15), whereas this risk was not statistically significantly increased in the PC trial16 and RESPECT,17 which used the Amplatzer device.
Benefit in those with atrial septal aneurysm?
Percutaneous closure has been shown to be safe and effective in patients with PFO and atrial septal aneurysm.36 There was some benefit of closure over medical therapy in a subgroup analysis from RESPECT in these patients, with a HR of 0.19 (95% CI 0.04–0.87, P = .02),17 although this was not seen in either CLOSURE 1 or the PC trial.
WHAT ARE THE RISKS OF PERCUTANEOUS CLOSURE?
Minor complications of percutaneous closure include bleeding, atrial arrhythmias, device embolization and fracture, and complications related to vascular access. Major complications include hemorrhage requiring transfusion, need for surgery, cardiac tamponade, pulmonary embolism, and death.
The cumulative rate of major complications in 10 observational studies was 1.5%, and the rate of minor complications was 7.9%.37 The RESPECT investigators reported a serious adverse event in 4.2% of patients (ranging in severity from chest tightness to cardiac tamponade).17
Another possible consequence of percutaneous closure is the need for chronic anticoagulation because of the increased risk of postprocedural atrial fibrillation seen in meta-analyses,29,31,32 though this may be device-specific.32
Percutaneous closure was considered successful—ie, to have nearly or completely eliminated shunting of blood through the defect—at 6 months of follow-up in 95.9% of patients in the PC trial, 93.5% in RESPECT, and 86.1% in CLOSURE 1.15–17
WHAT SHOULD WE BE DOING IN DAILY PRACTICE?
Give aspirin. Aspirin is effective in secondary stroke prevention, and data suggest that patients with PFO and cryptogenic stroke who receive aspirin therapy alone have a similar risk of recurrent events as patients without PFO.
Give warfarin if indicated. Evidence is insufficient to recommend vitamin K antagonist therapy in all patients with PFO and cryptogenic stroke. However, coexisting conditions that warrant anticoagulation must be taken into account.
Individualize. Given the lack of evidence to definitively guide management of patients with cryptogenic stroke and PFO, we need to individualize our approach, taking into account patient preferences, bleeding risk, ability to tolerate procedures, and the likelihood that the PFO is at fault.
No definitive answer on PFO closure. The most recent data suggest that closure may be beneficial, but key questions remain: Who will benefit? And what is the ideal medical therapy? Optimal management will only be established by the continued enrollment of appropriate patients into ongoing clinical trials.
Another question is whether it is possible to perform a randomized controlled trial with enough patients to definitively prove whether percutaneous closure is superior to medical therapy. Recent experience would suggest not.
In the meantime, we have some guidance from the American Heart Association and the American Stroke Association Council on Stroke38 based on the limited evidence available.
Consider patient preference. The physician should present the options to the patient in a balanced manner to enable him or her to make an informed decision. Patients can also be encouraged to seek additional information at websites such as www.stroke.org and www.nlm.nih.gov.
Referral to an interventional cardiologist for evaluation for closure is reasonable in patients with recurrent stroke, medication failure, complicated atrial septal anatomy such as PFO with aneurysm or large shunt, concurrent thromboembolic disease, or contraindications to anticoagulation.
MORE WORK NEEDED
Areas for further study include further identifying the characteristics of patients with PFO and cryptogenic stroke that might indicate who would benefit from percutaneous closure, elucidating the mechanism of stroke in these patients, and determining whether routine stroke evaluation should include echocardiography with a bubble study if there is no change in management based on the finding of PFO.39
Your patient has had an ischemic stroke, and so far you have found no obvious cause such as atrial fibrillation or carotid disease. Should you look for a patent foramen ovale (PFO)? And if you find it, what should you do?
This scenario continues to challenge primary care physicians and subspecialists and requires an understanding of the relationship between PFO and cryptogenic stroke, as well as familiarity with current data on the safety and effectiveness of the management options. PFO is known to be associated with cryptogenic stroke, but many questions remain, including:
- How can we tell if PFO is a culprit (“pathologic”) or an innocent bystander (“incidental”) in a patient who has had a cryptogenic stroke?
- Should stroke patients receive different medical therapy if they have a PFO? In particular, should they receive warfarin in addition to aspirin? And what about the novel oral anticoagulants?
- Which patients should undergo percutaneous closure of the PFO?
- Should we even be looking for PFO in stroke patients at this point, if we cannot say with certainty what we should do if we find it?
WHY IS THIS IMPORTANT?
Cerebrovascular disease is common and costly. The estimated yearly incidence of stroke in the United States is 795,000 events, at a cost of nearly $30 billion.1 The incidence of stroke in Europe is more than 1 million annually.2
During the diagnostic evaluation of stroke or transient ischemic attack (TIA), PFO is occasionally discovered incidentally by echocardiography. The management decisions that follow often fall to the primary care physician, who must decipher the conflicting data currently available and explain the options to the patient.
Although reviews have been published on this subject,3 several newer key trials and data on risk stratification warrant consideration.
DEFINITIONS
PFO is the failure of the septum primum to fuse with the septum secundum, so that a communication remains between the atria (Figure 1). The diagnosis is commonly made by echocardiography, when agitated saline is injected into the venous system and bubbles can be seen in the left atrium within three to five cardiac cycles (see video).
Atrial septal aneurysm is loosely defined as a septal excursion or bulging of at least 10 to 15 mm into the left and right atria during the cardiac cycle (Figure 2). The combination of PFO and atrial septal aneurysm may be more of a risk factor for stroke than PFO alone (see discussion below).
Cryptogenic stroke. The diagnostic workup of stroke fails to elucidate a clear cause in up to 40% of cases, which are thus called cryptogenic.4 The workup varies, but typically includes a search for a cardioembolic source and for atherosclerotic disease. Embolic sources are evaluated for by electrocardiography, transthoracic echocardiography, and possibly imaging of the aortic arch. Evaluation for atherosclerotic disease of the intracranial and extracranial arteries includes magnetic resonance angiography or, if that is unavailable, computed tomographic angiography or carotid Doppler ultrasonography. If no source is found, long-term cardiac monitoring may be used to detect paroxysmal atrial fibrillation, which may be more common than previously thought.
PFO AND CRYPTOGENIC STROKE ARE COMMON
As noted, there are approximately 800,000 strokes every year in the United States. If 25% to 40% of them are cryptogenic (the true prevalence warrants more evaluation),4,5 then 200,000 to 320,000 strokes are cryptogenic.
Autopsy studies indicate that 25% of the general population have a PFO, and if the prevalence is the same in people with cryptogenic stroke, that would equal 80,000 people with both cryptogenic stroke and PFO every year. However, the prevalence of PFO in patients with cryptogenic stroke appears to be significantly higher than in the general population.6 Although these numbers are crude estimates, they provide some insight into the prevalence of this clinical presentation.
HOW ARE CRYPTOGENIC STROKE AND PFO RELATED?
The exact relationship between PFO and cryptogenic stroke is unknown, although cases have been reported of thrombus in transit through a PFO, supporting paradoxical embolism as the plausible cause in stroke patients with PFO.7–9
There is clear evidence that the two conditions are associated by more than chance. Homma and Sacco6 reported that, in several studies, 93 (46%) of 202 patients under age 55 with cryptogenic stroke had PFOs, compared with 29 (11%) of 271 controls (P < .05 in all studies).6
In their evaluation of 23 case-control studies, Alsheikh-Ali et al10 found that the summary odds ratio (OR) for PFO in cryptogenic stroke vs PFO in control patients was 2.9 (95% confidence interval [CI] 2.1–4), largely driven by an OR of 5.1 (3.3–7.8) in those under age 55. Through Bayesian probability theory, this correlated with only a 33% probability that PFO in a patient with cryptogenic stroke was an innocent bystander rather than the culprit.10
IS PFO A RISK FACTOR FOR STROKE?
One of the more puzzling aspects of the relationship of PFO to cryptogenic stroke is that despite a clear association, there is little evidence that the relationship is causal.
Di Tullio et al11 followed 1,100 people who had no history of stroke and found that the risk of a first stroke in those with a PFO was not significantly higher than in those without a PFO, regardless of age, sex, or ethnic or racial group. At 80 months, the hazard ratio of stroke in people who had a PFO was 1.64 (95% CI 0.87–3.09).11 The findings were similar at 11 years, with a hazard ratio of 1.10 (95% CI 0.64–1.91).12
A prospective study of 585 patients found a similar risk of stroke in those with and without a PFO, with a hazard ratio of 1.46 (95% CI 0.74–2.88; P = .28).13
These prospective trials suggest that although previous studies have found a higher prevalence of PFOs in patients with cryptogenic stroke than in patients without stroke, there appears to be very little if any increased risk from baseline for a first stroke or TIA.
The lack of statistical significance in these trials should be interpreted with some caution, as a small increased risk is difficult to show if the event rate is low (approximately 10% of patients had events over 11 years in the study by Di Tullio et al12).
HOW DO WE KNOW IF A PFO IS A CULPRIT OR BYSTANDER?
Unfortunately, this is largely unanswered, though experts have suggested that echocardiographic features of the PFO, radiographic characteristics of the stroke, and clinical features of the patient may provide useful information.
‘High-risk’ features on echocardiography
Certain features of PFO may portend a high risk of cerebrovascular events. Both right-to-left shunting at rest and septal hypermobility were found in one study14 to be more common in patients with a PFO who had a stroke or TIA than in patients with a PFO but no cerebrovascular events. Also, patients who had these features and had a stroke had a higher risk of recurrence than stroke patients without these features (12.5% vs 4.3%, P = .05).14
Septal hypermobility and shunting at rest are easily diagnosed by echocardiography, and detecting these “high-risk” features would be useful if they could identify patients who would benefit from special therapy, such as percutaneous closure of the PFO.
Unfortunately, when investigators looked at these features in subgroup analysis of the major randomized controlled trials of percutaneous closure vs medical therapy, the results were mixed.
CLOSURE 1 (the Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale)15 found percutaneous closure to be no better than medical therapy, regardless of shunt size or the presence of atrial septal aneurysm.
Similarly, the PC trial (Clinical Trial Comparing Percutaneous Closure of Patent Foramen Ovale Using the Amplatzer PFO Occluder With Medical Treatment in Patients With Cryptogenic Embolism)16 found no statistically significant benefit of closure in those with atrial septal aneurysm.
In contrast, the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment)17 showed percutaneous closure to be beneficial in patients with atrial septal aneurysm or large shunt.
Radiographic characteristics of the stroke
Another area of interest in trying to identify culprit PFOs is the radiographic characteristics of the stroke.
In a study comparing patients with stroke related to atrial fibrillation vs patients with cryptogenic stroke and a known PFO, those in the latter group were more likely to have a single cortical infarction (34.2% vs 3.1%; P < .001) or multiple small scattered lesions (23.1% vs 5.9%; P < .01).18 Similarly, in a large database of patients with cryptogenic stroke and known PFO status, a superficially located stroke was associated with the presence of PFO (OR 1.54; P < .0001).19
Although these findings do not tell us with certainty that a patient’s PFO was the cause of his or her stroke, they provide guidance when dealing with the uncertainty of how to manage a patient with PFO. They may be useful in clinical practice, for example, when discussing treatment options with a young patient with cryptogenic stroke who has no risk factors and a superficial single infarct and who is found to have a PFO with a right-to-left shunting at rest.
Patient characteristics
Kent et al20 developed a 10-point index (the RoPE score) in an attempt to assign a probability to whether a stroke was PFO-related. Points were assigned for patients who were younger, who had a cortical stroke on neuroimaging, and who did not have diabetes, hypertension, smoking, or prior stroke or TIA. Patients with cryptogenic stroke with a higher RoPE score were more likely to have a PFO and thus had a higher likelihood that the index event was related to PFO. Of note, the patients with the highest likelihood of PFO-related stroke were the least likely to have a recurrence (RoPE score of 9 to 10; PFO-attributable fraction 88%; estimated 2-year recurrence rate 2%; 95% CI 0%–4%), whereas those with a low RoPE score have more traditional risk factors for stroke and thus are more likely to have a recurrence (RoPE 0 to 3; estimated 2-year recurrence rate 20%; 95% CI 12%–28%).20
Again, this sheds light on a difficulty faced by randomized controlled trials: the patients who may benefit from closure of a PFO may very well be those with the lowest recurrence rates without intervention.
The RoPE index was examined in an attempt to validate previously described morphologic criteria of “high-risk” PFO,21 though none of the previously described “high-risk” echocardiographic features (large physiologic size, hypermobile septum, shunt at rest) were more common in the group with presumed PFO-attributable stroke (RoPE score > 6). This underscores the difficulty of distinguishing pathologic PFO from incidental PFO.
KEY TREATMENT CONSIDERATIONS FOR SECONDARY PREVENTION
Given the complicated relationship between PFO and cryptogenic stroke, there has been much debate over management strategies. The three options are surgical closure, percutaneous closure with a device, and medical therapy. The goal of all three is to prevent the recurrence of stroke or TIA.
Surgical closure has largely been supplanted by percutaneous closure, but is still done in specific situations such as when a PFO is found incidentally on transesophageal echocardiography during surgery for another cardiac condition. The data on such cases22 tend to support the argument that asymptomatic PFOs in the general population have a relatively benign natural history.
Thus, the two key questions about management that warrant discussion are: is anticoagulation superior to antiplatelet therapy? And is percutaneous closure superior to medical management?
Anticoagulant vs antiplatelet therapy
Whether to treat with aspirin or with a vitamin K antagonist has been a subject of debate, although there is no strong evidence to suggest that anticoagulation is superior to antiplatelet therapy.
The concern that aspirin alone is insufficient in some patients stems from a study by Mas et al,23 who followed 581 patients with cryptogenic stroke who had a PFO alone, a PFO with an atrial septal aneurysm, or neither. The rate of stroke recurrence at 4 years on aspirin therapy was 2.3% in those with a PFO alone, 15.2% in those with a PFO with an atrial septal aneurysm, and 4.2% in those with neither.
Many have concluded that aspirin therapy does not sufficiently protect those with both PFO and atrial septal aneurysm, given the high recurrence rate in this group. This might lead to the suggestion that anticoagulation could be of benefit in these patients.
However, the Patent Foramen Ovale in Cryptogenic Stroke Study (PiCSS)24 and the Spanish Multicenter Study Into Right-to-Left Shunt in Cryptogenic Stroke (CODICIA)25 found similar recurrence rates in patients with PFO and atrial septal aneurysm compared with those with only PFO. In these two studies, recurrence rates were similar regardless of whether patients were taking aspirin or warfarin.
In a study that followed 140 consecutive patients with both stroke and PFO, those treated in a nonrandomized fashion with antiplatelet agents had no difference in the recurrence rate compared with those treated with anticoagulation.26
Although uncertainty remains because no head-to-head randomized controlled trial has been done, some patients with PFO have other indications for anticoagulation, most commonly atrial fibrillation and venous thromboembolic disease.
There are currently no data on the use of novel oral anticoagulants in this setting.
Is percutaneous closure better than medical therapy?
When cryptogenic stroke is treated with antiplatelet therapy or anticoagulation therapy, the recurrence rate is the same whether or not the patient has a PFO.23–25 The belief that medical therapy offers adequate secondary protection is supported by a meta-analysis of 15 studies that found no increased risk of recurrent ischemic events in those with a PFO on medical therapy (antiplatelet or anticoagulant) vs those without a PFO (relative risk 1.1, 95% CI 0.8–1.5).27
Despite the conflicting evidence, percutaneous closure of PFO is still performed, mostly on a case-by-case basis. This has been supported by an apparent benefit in observational studies.
A systematic review of 52 single-arm studies and 7 comparative nonrandomized studies of patients with PFO and cryptogenic stroke found the rate of recurrent stroke to be 0.36 per 100 person-years with percutaneous closure vs 2.53 per 100 person-years with medical therapy.28 However, three long-awaited randomized controlled trials (CLOSURE 1, the PC trial, and RESPECT) failed to show a significant reduction in primary end points with percutaneous closure vs standard medical therapy.15–17
These trials had several limitations: event rates were low, medical therapy varied by provider, and enrollment was slowed by out-of-study percutaneous closure in patients perceived to be at high risk (though, as discussed above, high risk is difficult to determine).
Intention-to-treat analysis in RESPECT showed no benefit from percutaneous closure, but a favorable outcome was noted with closure in as-treated analysis (HR 0.27; 95% CI 0.1–0.75; P = .007) and per-protocol analysis (HR 0.37; 95% CI 0.14–0.96; P = .03) of the 980 randomized patients.17 This suggests some benefit, as does the CLOSURE 1 trial, in which 3 of the 12 recurrent strokes in the percutaneous closure group occurred before the device was implanted.15
The low event rates in these studies prompted several meta-analyses.29–35 However, only two suggested a benefit of percutaneous closure over medical therapy. In one recent meta-analysis,29 observational study data suggested benefit from percutaneous closure, whereas three randomized controlled trials failed to show a statistically significant benefit.
The conclusions of the meta-analyses must be interpreted with caution because of inherent differences in the randomized controlled trials, including the closure device used, inclusion criteria, study end points, and variations in medical therapy.
Devices differ
A meta-analysis by Khan et al35 showed a benefit of percutaneous closure when evaluating only studies using the Amplatzer PFO occluder (AGA Medical), as in RESPECT and the PC trial.35 As data accumulate, it is important to remember that there are differences between devices. Ongoing trials continue to investigate the Amplatzer device (NCT01550588) and the GORE HELEX Septal Occluder/GORE Septal Occluder (Gore Medical) (NCT00738894).
In another meta-analysis, Pineda et al31 found a benefit with closure in the as-treated analysis using data from all three randomized controlled trials (OR 0.62; 95% CI 0.41–0.94; P = .02).31 Although paradoxical embolism through the PFO as the mechanism of stroke has been questioned, this finding suggests that actual closure of a PFO may protect against further events, presumably by preventing paradoxical embolism.
Different closure devices have different side effects. The incidence of atrial fibrillation with the CardioSEAL STARFlex device (NMT Medical) is higher than with medical therapy (used in the CLOSURE trial15), whereas this risk was not statistically significantly increased in the PC trial16 and RESPECT,17 which used the Amplatzer device.
Benefit in those with atrial septal aneurysm?
Percutaneous closure has been shown to be safe and effective in patients with PFO and atrial septal aneurysm.36 There was some benefit of closure over medical therapy in a subgroup analysis from RESPECT in these patients, with a HR of 0.19 (95% CI 0.04–0.87, P = .02),17 although this was not seen in either CLOSURE 1 or the PC trial.
WHAT ARE THE RISKS OF PERCUTANEOUS CLOSURE?
Minor complications of percutaneous closure include bleeding, atrial arrhythmias, device embolization and fracture, and complications related to vascular access. Major complications include hemorrhage requiring transfusion, need for surgery, cardiac tamponade, pulmonary embolism, and death.
The cumulative rate of major complications in 10 observational studies was 1.5%, and the rate of minor complications was 7.9%.37 The RESPECT investigators reported a serious adverse event in 4.2% of patients (ranging in severity from chest tightness to cardiac tamponade).17
Another possible consequence of percutaneous closure is the need for chronic anticoagulation because of the increased risk of postprocedural atrial fibrillation seen in meta-analyses,29,31,32 though this may be device-specific.32
Percutaneous closure was considered successful—ie, to have nearly or completely eliminated shunting of blood through the defect—at 6 months of follow-up in 95.9% of patients in the PC trial, 93.5% in RESPECT, and 86.1% in CLOSURE 1.15–17
WHAT SHOULD WE BE DOING IN DAILY PRACTICE?
Give aspirin. Aspirin is effective in secondary stroke prevention, and data suggest that patients with PFO and cryptogenic stroke who receive aspirin therapy alone have a similar risk of recurrent events as patients without PFO.
Give warfarin if indicated. Evidence is insufficient to recommend vitamin K antagonist therapy in all patients with PFO and cryptogenic stroke. However, coexisting conditions that warrant anticoagulation must be taken into account.
Individualize. Given the lack of evidence to definitively guide management of patients with cryptogenic stroke and PFO, we need to individualize our approach, taking into account patient preferences, bleeding risk, ability to tolerate procedures, and the likelihood that the PFO is at fault.
No definitive answer on PFO closure. The most recent data suggest that closure may be beneficial, but key questions remain: Who will benefit? And what is the ideal medical therapy? Optimal management will only be established by the continued enrollment of appropriate patients into ongoing clinical trials.
Another question is whether it is possible to perform a randomized controlled trial with enough patients to definitively prove whether percutaneous closure is superior to medical therapy. Recent experience would suggest not.
In the meantime, we have some guidance from the American Heart Association and the American Stroke Association Council on Stroke38 based on the limited evidence available.
Consider patient preference. The physician should present the options to the patient in a balanced manner to enable him or her to make an informed decision. Patients can also be encouraged to seek additional information at websites such as www.stroke.org and www.nlm.nih.gov.
Referral to an interventional cardiologist for evaluation for closure is reasonable in patients with recurrent stroke, medication failure, complicated atrial septal anatomy such as PFO with aneurysm or large shunt, concurrent thromboembolic disease, or contraindications to anticoagulation.
MORE WORK NEEDED
Areas for further study include further identifying the characteristics of patients with PFO and cryptogenic stroke that might indicate who would benefit from percutaneous closure, elucidating the mechanism of stroke in these patients, and determining whether routine stroke evaluation should include echocardiography with a bubble study if there is no change in management based on the finding of PFO.39
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- Truelsen T, Piechowski-JóŸwiak B, Bonita R, Mathers C, Bogousslavsky J, Boysen G. Stroke incidence and prevalence in Europe: a review of available data. Eur J Neurol 2006; 13:581–598.
- Furlan AJ. Patent foramen ovale and stroke: to close or not to close? Cleve Clin J Med 2007; 74(suppl 1):S118–S120.
- Sacco RL, Ellenberg JH, Mohr JP, et al. Infarcts of undetermined cause: the NINCDS Stroke Data Bank. Ann Neurol 1989; 25:382–390.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German stroke data bank. Stroke 2001; 32:2559–2566.
- Homma S, Sacco RL. Patent foramen ovale and stroke. Circulation 2005; 112:1063–1072.
- Sattiraju S, Masri SC, Liao K, Missov E. Three-dimensional transesophageal echocardiography of a thrombus entrapped by a patent foramen ovale. Ann Thorac Surg 2012; 94:e101–e102.
- Schreiter SW, Phillips JH. Thromboembolus traversing a patent foramen ovale: resolution with anticoagulation. J Am Soc Echocardiogr 1994; 7:659–662.
- Hust MH, Staiger M, Braun B. Migration of paradoxic embolus through a patent foramen ovale diagnosed by echocardiography: successful thrombolysis. Am Heart J 1995; 129:620–622.
- Alsheikh-Ali AA, Thaler DE, Kent DM. Patent foramen ovale in cryptogenic stroke: incidental or pathogenic? Stroke 2009; 40:2349–2355.
- Di Tullio MR, Sacco RL, Sciacca RR, Jin Z, Homma S. Patent foramen ovale and the risk of ischemic stroke in a multiethnic population. J Am Coll Cardiol 2007; 49:797–802.
- Di Tullio MR, Jin Z, Russo C, et al. Patent foramen ovale, subclinical cerebrovascular disease, and ischemic stroke in a population-based cohort. J Am Coll Cardiol 2013; 62:35–41.
- Meissner I, Khandheria BK, Heit JA, et al. Patent foramen ovale: innocent or guilty? Evidence from a prospective population-based study. J Am Coll Cardiol 2006; 47:440–445.
- De Castro S, Cartoni D, Fiorelli M, et al. Morphological and functional characteristics of patent foramen ovale and their embolic implications. Stroke 2000; 31:2407–2413.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE I Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Kim BJ, Sohn H, Sun BJ, et al. Imaging characteristics of ischemic strokes related to patent foramen ovale. Stroke 2013; 44:3350–3356.
- Thaler DE, Ruthazer R, Di Angelantonio E, et al. Neuroimaging findings in cryptogenic stroke patients with and without patent foramen ovale. Stroke 2013; 44:675–680.
- Kent DM, Ruthazer R, Weimar C, et al. An index to identify stroke-related vs incidental patent foramen ovale in cryptogenic stroke. Neurology 2013; 81:619–625.
- Wessler BS, Thaler DE, Ruthazer R, et al. Transesophageal echocardiography in cryptogenic stroke and patent foramen ovale: analysis of putative high-risk features from the risk of paradoxical embolism database. Circ Cardiovasc Imaging 2014; 7:125–131.
- Krasuski RA, Hart SA, Allen D, et al. Prevalence and repair of intraoperatively diagnosed patent foramen ovale and association with perioperative outcomes and long-term survival. JAMA 2009; 302:290–297.
- Mas JL, Arquizan C, Lamy C, et al; Patent Foramen Ovale and Atrial Septal Aneurysm Study Group. Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysm, or both. N Engl J Med 2001; 345:1740–1746.
- Homma S, Sacco RL, Di Tullio MR, Sciacca RR, Mohr JP; PFO in Cryptogenic Stroke Study (PICSS) Investigators. Effect of medical treatment in stroke patients with patent foramen ovale: patent foramen ovale in Cryptogenic Stroke Study. Circulation 2002; 105:2625–2631.
- Serena J, Marti-Fàbregas J, Santamarina E, et al; CODICIA, Right-to-Left Shunt in Cryptogenic Stroke Study; Stroke Project of the Cerebrovascular Diseases Study Group, Spanish Society of Neurology. Recurrent stroke and massive right-to-left shunt: results from the prospective Spanish multicenter (CODICIA) study. Stroke 2008; 39:3131–3136.
- Bogousslavsky J, Garazi S, Jeanrenaud X, Aebischer N, Van Melle G. Stroke recurrence in patients with patent foramen ovale: the Lausanne Study. Lausanne Stroke with Paradoxal Embolism Study Group. Neurology 1996; 46:1301–1305.
- Almekhlafi MA, Wilton SB, Rabi DM, Ghali WA, Lorenzetti DL, Hill MD. Recurrent cerebral ischemia in medically treated patent foramen ovale: a meta-analysis. Neurology 2009; 73:89–97.
- Kitsios GD, Dahabreh IJ, Abu Dabrh AM, Thaler DE, Kent DM. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke 2012; 43:422–431.
- Wolfrum M, Froehlich GM, Knapp G, et al. Stroke prevention by percutaneous closure of patent foramen ovale: a systematic review and meta-analysis. Heart 2014; 100:389–395.
- Rengifo-Moreno P, Palacios IF, Junpaparp P, Witzke CF, Morris DL, Romero-Corral A. Patent foramen ovale transcatheter closure vs medical therapy on recurrent vascular events: a systematic review and meta-analysis of randomized controlled trials. Eur Heart J 2013; 34:3342–3352.
- Pineda AM, Nascimento FO, Yang SC, Kirtane AJ, Sommer RJ, Beohar N. A meta-analysis of transcatheter closure of patent foramen ovale versus medical therapy for prevention of recurrent thromboembolic events in patients with cryptogenic cerebrovascular events. Catheter Cardiovasc Interv 2013; 82:968–975.
- Kwong JS, Lam YY, Yu CM. Percutaneous closure of patent foramen ovale for cryptogenic stroke: a meta-analysis of randomized controlled trials. Int J Cardiol 2013; 168:4132–4148.
- Ntaios G, Papavasileiou V, Makaritsis K, Michel P. PFO closure vs medical therapy in cryptogenic stroke or transient ischemic attack: a systematic review and meta-analysis. Int J Cardiol 2013; 169:101–105.
- Nagaraja V, Raval J, Eslick GD, Burgess D, Denniss AR. Is transcatheter closure better than medical therapy for cryptogenic stroke with patent foramen ovale? A meta-analysis of randomised trials. Heart Lung Circ 2013; 22:903–909.
- Khan AR, Bin Abdulhak AA, Sheikh MA, et al. Device closure of patent foramen ovale versus medical therapy in cryptogenic stroke: a systematic review and meta-analysis. JACC Cardiovasc Interv 2013; 6:1316–1323.
- Wahl A, Krumsdorf U, Meier B, et al. Transcatheter treatment of atrial septal aneurysm associated with patent foramen ovale for prevention of recurrent paradoxical embolism in high-risk patients. J Am Coll Cardiol 2005; 45:377–380.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Sacco RL, Adams R, Albers G, et al; American Heart Association; American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke 2006; 37:577–617.
- Rana BS, Thomas MR, Calvert PA, Monaghan MJ, Hildick-Smith D. Echocardiographic evaluation of patent foramen ovale prior to device closure. JACC Cardiovasc Imaging 2010; 3:749–760.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- Truelsen T, Piechowski-JóŸwiak B, Bonita R, Mathers C, Bogousslavsky J, Boysen G. Stroke incidence and prevalence in Europe: a review of available data. Eur J Neurol 2006; 13:581–598.
- Furlan AJ. Patent foramen ovale and stroke: to close or not to close? Cleve Clin J Med 2007; 74(suppl 1):S118–S120.
- Sacco RL, Ellenberg JH, Mohr JP, et al. Infarcts of undetermined cause: the NINCDS Stroke Data Bank. Ann Neurol 1989; 25:382–390.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German stroke data bank. Stroke 2001; 32:2559–2566.
- Homma S, Sacco RL. Patent foramen ovale and stroke. Circulation 2005; 112:1063–1072.
- Sattiraju S, Masri SC, Liao K, Missov E. Three-dimensional transesophageal echocardiography of a thrombus entrapped by a patent foramen ovale. Ann Thorac Surg 2012; 94:e101–e102.
- Schreiter SW, Phillips JH. Thromboembolus traversing a patent foramen ovale: resolution with anticoagulation. J Am Soc Echocardiogr 1994; 7:659–662.
- Hust MH, Staiger M, Braun B. Migration of paradoxic embolus through a patent foramen ovale diagnosed by echocardiography: successful thrombolysis. Am Heart J 1995; 129:620–622.
- Alsheikh-Ali AA, Thaler DE, Kent DM. Patent foramen ovale in cryptogenic stroke: incidental or pathogenic? Stroke 2009; 40:2349–2355.
- Di Tullio MR, Sacco RL, Sciacca RR, Jin Z, Homma S. Patent foramen ovale and the risk of ischemic stroke in a multiethnic population. J Am Coll Cardiol 2007; 49:797–802.
- Di Tullio MR, Jin Z, Russo C, et al. Patent foramen ovale, subclinical cerebrovascular disease, and ischemic stroke in a population-based cohort. J Am Coll Cardiol 2013; 62:35–41.
- Meissner I, Khandheria BK, Heit JA, et al. Patent foramen ovale: innocent or guilty? Evidence from a prospective population-based study. J Am Coll Cardiol 2006; 47:440–445.
- De Castro S, Cartoni D, Fiorelli M, et al. Morphological and functional characteristics of patent foramen ovale and their embolic implications. Stroke 2000; 31:2407–2413.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE I Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Kim BJ, Sohn H, Sun BJ, et al. Imaging characteristics of ischemic strokes related to patent foramen ovale. Stroke 2013; 44:3350–3356.
- Thaler DE, Ruthazer R, Di Angelantonio E, et al. Neuroimaging findings in cryptogenic stroke patients with and without patent foramen ovale. Stroke 2013; 44:675–680.
- Kent DM, Ruthazer R, Weimar C, et al. An index to identify stroke-related vs incidental patent foramen ovale in cryptogenic stroke. Neurology 2013; 81:619–625.
- Wessler BS, Thaler DE, Ruthazer R, et al. Transesophageal echocardiography in cryptogenic stroke and patent foramen ovale: analysis of putative high-risk features from the risk of paradoxical embolism database. Circ Cardiovasc Imaging 2014; 7:125–131.
- Krasuski RA, Hart SA, Allen D, et al. Prevalence and repair of intraoperatively diagnosed patent foramen ovale and association with perioperative outcomes and long-term survival. JAMA 2009; 302:290–297.
- Mas JL, Arquizan C, Lamy C, et al; Patent Foramen Ovale and Atrial Septal Aneurysm Study Group. Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysm, or both. N Engl J Med 2001; 345:1740–1746.
- Homma S, Sacco RL, Di Tullio MR, Sciacca RR, Mohr JP; PFO in Cryptogenic Stroke Study (PICSS) Investigators. Effect of medical treatment in stroke patients with patent foramen ovale: patent foramen ovale in Cryptogenic Stroke Study. Circulation 2002; 105:2625–2631.
- Serena J, Marti-Fàbregas J, Santamarina E, et al; CODICIA, Right-to-Left Shunt in Cryptogenic Stroke Study; Stroke Project of the Cerebrovascular Diseases Study Group, Spanish Society of Neurology. Recurrent stroke and massive right-to-left shunt: results from the prospective Spanish multicenter (CODICIA) study. Stroke 2008; 39:3131–3136.
- Bogousslavsky J, Garazi S, Jeanrenaud X, Aebischer N, Van Melle G. Stroke recurrence in patients with patent foramen ovale: the Lausanne Study. Lausanne Stroke with Paradoxal Embolism Study Group. Neurology 1996; 46:1301–1305.
- Almekhlafi MA, Wilton SB, Rabi DM, Ghali WA, Lorenzetti DL, Hill MD. Recurrent cerebral ischemia in medically treated patent foramen ovale: a meta-analysis. Neurology 2009; 73:89–97.
- Kitsios GD, Dahabreh IJ, Abu Dabrh AM, Thaler DE, Kent DM. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke 2012; 43:422–431.
- Wolfrum M, Froehlich GM, Knapp G, et al. Stroke prevention by percutaneous closure of patent foramen ovale: a systematic review and meta-analysis. Heart 2014; 100:389–395.
- Rengifo-Moreno P, Palacios IF, Junpaparp P, Witzke CF, Morris DL, Romero-Corral A. Patent foramen ovale transcatheter closure vs medical therapy on recurrent vascular events: a systematic review and meta-analysis of randomized controlled trials. Eur Heart J 2013; 34:3342–3352.
- Pineda AM, Nascimento FO, Yang SC, Kirtane AJ, Sommer RJ, Beohar N. A meta-analysis of transcatheter closure of patent foramen ovale versus medical therapy for prevention of recurrent thromboembolic events in patients with cryptogenic cerebrovascular events. Catheter Cardiovasc Interv 2013; 82:968–975.
- Kwong JS, Lam YY, Yu CM. Percutaneous closure of patent foramen ovale for cryptogenic stroke: a meta-analysis of randomized controlled trials. Int J Cardiol 2013; 168:4132–4148.
- Ntaios G, Papavasileiou V, Makaritsis K, Michel P. PFO closure vs medical therapy in cryptogenic stroke or transient ischemic attack: a systematic review and meta-analysis. Int J Cardiol 2013; 169:101–105.
- Nagaraja V, Raval J, Eslick GD, Burgess D, Denniss AR. Is transcatheter closure better than medical therapy for cryptogenic stroke with patent foramen ovale? A meta-analysis of randomised trials. Heart Lung Circ 2013; 22:903–909.
- Khan AR, Bin Abdulhak AA, Sheikh MA, et al. Device closure of patent foramen ovale versus medical therapy in cryptogenic stroke: a systematic review and meta-analysis. JACC Cardiovasc Interv 2013; 6:1316–1323.
- Wahl A, Krumsdorf U, Meier B, et al. Transcatheter treatment of atrial septal aneurysm associated with patent foramen ovale for prevention of recurrent paradoxical embolism in high-risk patients. J Am Coll Cardiol 2005; 45:377–380.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Sacco RL, Adams R, Albers G, et al; American Heart Association; American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke 2006; 37:577–617.
- Rana BS, Thomas MR, Calvert PA, Monaghan MJ, Hildick-Smith D. Echocardiographic evaluation of patent foramen ovale prior to device closure. JACC Cardiovasc Imaging 2010; 3:749–760.
KEY POINTS
- PFO is present in up to 25% of the general population, and it is even more common in young patients with cryptogenic stroke.
- PFO has not been shown to cause stroke or to significantly increase the risk of recurrent cerebrovascular events in patients treated with antiplatelet drugs.
- In patients with PFO, atrial septal aneurysm and large shunt size may confer increased risk of stroke.
- There is still no definitive evidence that closure of PFO is better than medical therapy in all patients with PFO and cryptogenic stroke.
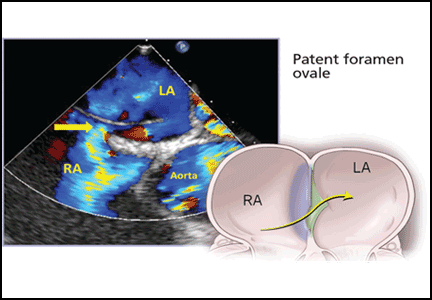
Patent foramen ovale and the risk of cryptogenic stroke
The article by Roth and Alli in this issue describes in depth more than 10 years of research that addresses the question, Should we close a patent foramen ovale (PFO) to prevent recurrent cryptogenic stroke?
There is no longer any doubt that PFO can be the pathway for thrombus from the venous circulation to go from the right atrium to the left atrium, bypassing the pulmonary capillary filtration bed, and entering the arterial side to produce a stroke, myocardial infarction, or peripheral embolus. Two questions remain: What should we do to prevent another episode? And is percutaneous closure of a PFO with the current devices preferable to medical therapy?
How much do we know about the risks and benefits of closure of PFO? I maintain that we know a great deal about interatrial shunt and paradoxical embolism as a cause of cryptogenic stroke. Prospective randomized clinical trials now give us data with which we can provide appropriate direction to our patients. Percutaneous closure is no longer an “experimental procedure,” as insurance companies claim. The experiment has been done, and the only issue is how one interprets the data from the randomized clinical trials.
The review by Roth and Alli comprehensively describes the observational studies, as well as the three randomized clinical trials done to determine whether PFO closure is preferable to medical therapy to prevent recurrent stroke in patients who have already had one cryptogenic stroke. If we understand some of the subtleties and differences between the trials, we can reach an appropriate conclusion as to what to recommend to our patients.
A review of 10 reports of transcatheter closure of PFO vs six reports of medical therapy for cryptogenic stroke showed a range of rates of recurrent stroke at 1 year—between 0% and 4.9% for transcatheter closure, and between 3.8% and 12% for medical therapy.1
These numbers are important because they were used to estimate the number of patients that would be necessary to study in a randomized clinical trial to demonstrate a benefit of PFO closure vs medical therapy. Unlike most studies of new devices, the PFO closure trials were done in an environment in which patients could get their PFO closed with other devices that were already approved by the US Food and Drug Administration (FDA) for closure of an atrial septal defect. This ability of patients to obtain PFO closure outside of the trial with an off-label device meant that the patients who agreed to be randomized tended to have lower risk for recurrence than patients studied in the observational populations. From a practical standpoint, this meant that the event rate in the patients who participated in the randomized clinical trials (1.7% per year) was lower than predicted from the observational studies.2,3
Another way of saying this is that the randomized clinical trials were underpowered to answer the question. A common way of dealing with this problem is to combine the results of different studies in a meta-analysis. This makes sense if the studies are assessing the same thing. This is not the case with the PFO closure trials. Although the topic of percutaneous PFO closure vs medical therapy was the same, the devices used were different.
In the CLOSURE trial (Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale),3 the device used was the STARFlex, which is no longer produced—and for good reasons. It is not as effective as the Amplatzer or Helex devices in completely closing the right-to-left shunt produced by a PFO. In addition, the CardioSEAL or STARFlex device increases the risk of atrial fibrillation, which was seen in 6% of the treated patients.3 This was the major cause of recurrent stroke in the CLOSURE trial. The CardioSEAL STARFlex device was also more thrombogenic.
In the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment),2 which used the Amplatzer PFO closure device, there was no increased incidence of atrial fibrillation in the device group compared with the control group. Therefore, it is not appropriate to combine the results of the CLOSURE trial with the results of the RESPECT trial and PC trial,4 both of which used the Amplatzer device.
Our patients want to know what the potential risks and benefits will be if they get their PFO closed with a specific device. They don’t want to know the average risk between two different devices.
However, if you do a meta-analysis of the RESPECT and PC trials, which used the same Amplatzer PFO occluder device, and combine the number of patients studied to increase the statistical power, then the benefit of PFO closure is significant even with an intention-to-treat analysis. By combining the two studies that assessed the same device, you reach a completely different interpretation than if you do a meta-analysis including the CLOSURE trial, which showed no benefit.
The medical community should not uncritically accept meta-analysis methodology. It is a marvelous case example of how scientific methods can be inappropriately used and two diametrically opposed conclusions reached if the meta-analysis combines two different types of devices vs a meta-analysis of just the Amplatzer device.
If we combine the numbers from the RESPECT and PC trials, there were 23 strokes in 691 patients (3.3%) in the medical groups and 10 strokes in 703 patients (1.4%) who underwent PFO closure. By chi square analysis of this intention-to-treat protocol, PFO closure provides a statistically significant reduction in preventing recurrent stroke (95% confidence interval 0.20–0.89, P = .02).
From the patient’s perspective, what is important is this: If I get my PFO closed with an Amplatzer PFO occluder device, what are the risks of the procedure, and what are the potential benefits compared with medical therapy? We can now answer that question definitively. I tell my patients, “The risks of the procedure are remarkably low (about 1%) in experienced hands, and the benefit is that your risk of recurrent stroke will be reduced 73%2 compared with medical therapy.” In the RESPECT Trial, the as-treated cohort consisted of 958 patients with 21 primary end-point events (5 in the closure group and 16 in the medical-therapy group). The rate of the primary end point was 0.39 events per 100 patient-years in the closure group vs 1.45 events per 100 patient-years in the medical-therapy group (hazard ratio 0.27; 95% confidence interval 0.10–0.75; P = .007).
Not all cryptogenic strokes in people who have a PFO are caused by paradoxical embolism. PFO may be an innocent bystander. In addition, not all people who have a paradoxical embolism will have a recurrent stroke. For example, if a young woman presents with a PFO and stroke, is it possible that she can prevent another stroke just by stopping her birth-control pills and not have her PFO closed? What is the risk of recurrent stroke if she were to become pregnant? We do not know the answers to these questions.
Your patients do not want to wait to find out if they are going to have another stroke. The meta-analysis of the randomized clinical trials for paradoxical embolism demonstrates that the closure devices are safe and effective. The FDA should approve the Amplatzer PFO occluder with an indication to prevent recurrent stroke in patients with PFO and an initial cryptogenic event.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
The article by Roth and Alli in this issue describes in depth more than 10 years of research that addresses the question, Should we close a patent foramen ovale (PFO) to prevent recurrent cryptogenic stroke?
There is no longer any doubt that PFO can be the pathway for thrombus from the venous circulation to go from the right atrium to the left atrium, bypassing the pulmonary capillary filtration bed, and entering the arterial side to produce a stroke, myocardial infarction, or peripheral embolus. Two questions remain: What should we do to prevent another episode? And is percutaneous closure of a PFO with the current devices preferable to medical therapy?
How much do we know about the risks and benefits of closure of PFO? I maintain that we know a great deal about interatrial shunt and paradoxical embolism as a cause of cryptogenic stroke. Prospective randomized clinical trials now give us data with which we can provide appropriate direction to our patients. Percutaneous closure is no longer an “experimental procedure,” as insurance companies claim. The experiment has been done, and the only issue is how one interprets the data from the randomized clinical trials.
The review by Roth and Alli comprehensively describes the observational studies, as well as the three randomized clinical trials done to determine whether PFO closure is preferable to medical therapy to prevent recurrent stroke in patients who have already had one cryptogenic stroke. If we understand some of the subtleties and differences between the trials, we can reach an appropriate conclusion as to what to recommend to our patients.
A review of 10 reports of transcatheter closure of PFO vs six reports of medical therapy for cryptogenic stroke showed a range of rates of recurrent stroke at 1 year—between 0% and 4.9% for transcatheter closure, and between 3.8% and 12% for medical therapy.1
These numbers are important because they were used to estimate the number of patients that would be necessary to study in a randomized clinical trial to demonstrate a benefit of PFO closure vs medical therapy. Unlike most studies of new devices, the PFO closure trials were done in an environment in which patients could get their PFO closed with other devices that were already approved by the US Food and Drug Administration (FDA) for closure of an atrial septal defect. This ability of patients to obtain PFO closure outside of the trial with an off-label device meant that the patients who agreed to be randomized tended to have lower risk for recurrence than patients studied in the observational populations. From a practical standpoint, this meant that the event rate in the patients who participated in the randomized clinical trials (1.7% per year) was lower than predicted from the observational studies.2,3
Another way of saying this is that the randomized clinical trials were underpowered to answer the question. A common way of dealing with this problem is to combine the results of different studies in a meta-analysis. This makes sense if the studies are assessing the same thing. This is not the case with the PFO closure trials. Although the topic of percutaneous PFO closure vs medical therapy was the same, the devices used were different.
In the CLOSURE trial (Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale),3 the device used was the STARFlex, which is no longer produced—and for good reasons. It is not as effective as the Amplatzer or Helex devices in completely closing the right-to-left shunt produced by a PFO. In addition, the CardioSEAL or STARFlex device increases the risk of atrial fibrillation, which was seen in 6% of the treated patients.3 This was the major cause of recurrent stroke in the CLOSURE trial. The CardioSEAL STARFlex device was also more thrombogenic.
In the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment),2 which used the Amplatzer PFO closure device, there was no increased incidence of atrial fibrillation in the device group compared with the control group. Therefore, it is not appropriate to combine the results of the CLOSURE trial with the results of the RESPECT trial and PC trial,4 both of which used the Amplatzer device.
Our patients want to know what the potential risks and benefits will be if they get their PFO closed with a specific device. They don’t want to know the average risk between two different devices.
However, if you do a meta-analysis of the RESPECT and PC trials, which used the same Amplatzer PFO occluder device, and combine the number of patients studied to increase the statistical power, then the benefit of PFO closure is significant even with an intention-to-treat analysis. By combining the two studies that assessed the same device, you reach a completely different interpretation than if you do a meta-analysis including the CLOSURE trial, which showed no benefit.
The medical community should not uncritically accept meta-analysis methodology. It is a marvelous case example of how scientific methods can be inappropriately used and two diametrically opposed conclusions reached if the meta-analysis combines two different types of devices vs a meta-analysis of just the Amplatzer device.
If we combine the numbers from the RESPECT and PC trials, there were 23 strokes in 691 patients (3.3%) in the medical groups and 10 strokes in 703 patients (1.4%) who underwent PFO closure. By chi square analysis of this intention-to-treat protocol, PFO closure provides a statistically significant reduction in preventing recurrent stroke (95% confidence interval 0.20–0.89, P = .02).
From the patient’s perspective, what is important is this: If I get my PFO closed with an Amplatzer PFO occluder device, what are the risks of the procedure, and what are the potential benefits compared with medical therapy? We can now answer that question definitively. I tell my patients, “The risks of the procedure are remarkably low (about 1%) in experienced hands, and the benefit is that your risk of recurrent stroke will be reduced 73%2 compared with medical therapy.” In the RESPECT Trial, the as-treated cohort consisted of 958 patients with 21 primary end-point events (5 in the closure group and 16 in the medical-therapy group). The rate of the primary end point was 0.39 events per 100 patient-years in the closure group vs 1.45 events per 100 patient-years in the medical-therapy group (hazard ratio 0.27; 95% confidence interval 0.10–0.75; P = .007).
Not all cryptogenic strokes in people who have a PFO are caused by paradoxical embolism. PFO may be an innocent bystander. In addition, not all people who have a paradoxical embolism will have a recurrent stroke. For example, if a young woman presents with a PFO and stroke, is it possible that she can prevent another stroke just by stopping her birth-control pills and not have her PFO closed? What is the risk of recurrent stroke if she were to become pregnant? We do not know the answers to these questions.
Your patients do not want to wait to find out if they are going to have another stroke. The meta-analysis of the randomized clinical trials for paradoxical embolism demonstrates that the closure devices are safe and effective. The FDA should approve the Amplatzer PFO occluder with an indication to prevent recurrent stroke in patients with PFO and an initial cryptogenic event.
The article by Roth and Alli in this issue describes in depth more than 10 years of research that addresses the question, Should we close a patent foramen ovale (PFO) to prevent recurrent cryptogenic stroke?
There is no longer any doubt that PFO can be the pathway for thrombus from the venous circulation to go from the right atrium to the left atrium, bypassing the pulmonary capillary filtration bed, and entering the arterial side to produce a stroke, myocardial infarction, or peripheral embolus. Two questions remain: What should we do to prevent another episode? And is percutaneous closure of a PFO with the current devices preferable to medical therapy?
How much do we know about the risks and benefits of closure of PFO? I maintain that we know a great deal about interatrial shunt and paradoxical embolism as a cause of cryptogenic stroke. Prospective randomized clinical trials now give us data with which we can provide appropriate direction to our patients. Percutaneous closure is no longer an “experimental procedure,” as insurance companies claim. The experiment has been done, and the only issue is how one interprets the data from the randomized clinical trials.
The review by Roth and Alli comprehensively describes the observational studies, as well as the three randomized clinical trials done to determine whether PFO closure is preferable to medical therapy to prevent recurrent stroke in patients who have already had one cryptogenic stroke. If we understand some of the subtleties and differences between the trials, we can reach an appropriate conclusion as to what to recommend to our patients.
A review of 10 reports of transcatheter closure of PFO vs six reports of medical therapy for cryptogenic stroke showed a range of rates of recurrent stroke at 1 year—between 0% and 4.9% for transcatheter closure, and between 3.8% and 12% for medical therapy.1
These numbers are important because they were used to estimate the number of patients that would be necessary to study in a randomized clinical trial to demonstrate a benefit of PFO closure vs medical therapy. Unlike most studies of new devices, the PFO closure trials were done in an environment in which patients could get their PFO closed with other devices that were already approved by the US Food and Drug Administration (FDA) for closure of an atrial septal defect. This ability of patients to obtain PFO closure outside of the trial with an off-label device meant that the patients who agreed to be randomized tended to have lower risk for recurrence than patients studied in the observational populations. From a practical standpoint, this meant that the event rate in the patients who participated in the randomized clinical trials (1.7% per year) was lower than predicted from the observational studies.2,3
Another way of saying this is that the randomized clinical trials were underpowered to answer the question. A common way of dealing with this problem is to combine the results of different studies in a meta-analysis. This makes sense if the studies are assessing the same thing. This is not the case with the PFO closure trials. Although the topic of percutaneous PFO closure vs medical therapy was the same, the devices used were different.
In the CLOSURE trial (Evaluation of the STARFlex Septal Closure System in Patients With a Stroke and/or Transient Ischemic Attack Due to Presumed Paradoxical Embolism Through a Patent Foramen Ovale),3 the device used was the STARFlex, which is no longer produced—and for good reasons. It is not as effective as the Amplatzer or Helex devices in completely closing the right-to-left shunt produced by a PFO. In addition, the CardioSEAL or STARFlex device increases the risk of atrial fibrillation, which was seen in 6% of the treated patients.3 This was the major cause of recurrent stroke in the CLOSURE trial. The CardioSEAL STARFlex device was also more thrombogenic.
In the RESPECT trial (Randomized Evaluation of Recurrent Stroke Comparing PFO Closure to Established Current Standard of Care Treatment),2 which used the Amplatzer PFO closure device, there was no increased incidence of atrial fibrillation in the device group compared with the control group. Therefore, it is not appropriate to combine the results of the CLOSURE trial with the results of the RESPECT trial and PC trial,4 both of which used the Amplatzer device.
Our patients want to know what the potential risks and benefits will be if they get their PFO closed with a specific device. They don’t want to know the average risk between two different devices.
However, if you do a meta-analysis of the RESPECT and PC trials, which used the same Amplatzer PFO occluder device, and combine the number of patients studied to increase the statistical power, then the benefit of PFO closure is significant even with an intention-to-treat analysis. By combining the two studies that assessed the same device, you reach a completely different interpretation than if you do a meta-analysis including the CLOSURE trial, which showed no benefit.
The medical community should not uncritically accept meta-analysis methodology. It is a marvelous case example of how scientific methods can be inappropriately used and two diametrically opposed conclusions reached if the meta-analysis combines two different types of devices vs a meta-analysis of just the Amplatzer device.
If we combine the numbers from the RESPECT and PC trials, there were 23 strokes in 691 patients (3.3%) in the medical groups and 10 strokes in 703 patients (1.4%) who underwent PFO closure. By chi square analysis of this intention-to-treat protocol, PFO closure provides a statistically significant reduction in preventing recurrent stroke (95% confidence interval 0.20–0.89, P = .02).
From the patient’s perspective, what is important is this: If I get my PFO closed with an Amplatzer PFO occluder device, what are the risks of the procedure, and what are the potential benefits compared with medical therapy? We can now answer that question definitively. I tell my patients, “The risks of the procedure are remarkably low (about 1%) in experienced hands, and the benefit is that your risk of recurrent stroke will be reduced 73%2 compared with medical therapy.” In the RESPECT Trial, the as-treated cohort consisted of 958 patients with 21 primary end-point events (5 in the closure group and 16 in the medical-therapy group). The rate of the primary end point was 0.39 events per 100 patient-years in the closure group vs 1.45 events per 100 patient-years in the medical-therapy group (hazard ratio 0.27; 95% confidence interval 0.10–0.75; P = .007).
Not all cryptogenic strokes in people who have a PFO are caused by paradoxical embolism. PFO may be an innocent bystander. In addition, not all people who have a paradoxical embolism will have a recurrent stroke. For example, if a young woman presents with a PFO and stroke, is it possible that she can prevent another stroke just by stopping her birth-control pills and not have her PFO closed? What is the risk of recurrent stroke if she were to become pregnant? We do not know the answers to these questions.
Your patients do not want to wait to find out if they are going to have another stroke. The meta-analysis of the randomized clinical trials for paradoxical embolism demonstrates that the closure devices are safe and effective. The FDA should approve the Amplatzer PFO occluder with an indication to prevent recurrent stroke in patients with PFO and an initial cryptogenic event.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
- Khairy P, O’Donnell CP, Landzberg MJ. Transcatheter closure versus medical therapy of patent foramen ovale and presumed paradoxical thromboemboli: a systematic review. Ann Intern Med 2003; 139:753–760.
- Carroll JD, Saver JL, Thaler DE, et al; RESPECT Investigators. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med 2013; 368:1092–1100.
- Furlan AJ, Reisman M, Massaro J, et al; CLOSURE Investigators. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med 2012; 366:991–999.
- Meier B, Kalesan B, Mattle HP, et al; PC Trial Investigators. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med 2013; 368:1083–1091.
Swelling and pain 2 weeks after a dog bite
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing

Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
A 48-year-old man with gout, multiple sclerosis, and previously treated methicillin-resistant Staphylococcus aureus (MRSA) infection presented to the emergency room with pain and significant swelling at the site of a dog bite on his left forearm. He had been bitten 2 weeks earlier by a friend’s dog, and the bite had punctured the skin. He also had red streaking on the skin of the left arm from the wrist to the elbow, and he reported feeling “feverish” and having night sweats.
At first, the bite had seemed to improve, then swelling and pain had developed and increased. He reported this to his primary care physician, along with the information that he had previously had an anaphylactic reaction to penicillin and a cephalosporin. His physician, considering a penicillin allergy, started him on ciprofloxacin (Cipro) plus clindamycin (Cleocin). The patient took this for 5 days, but without improvement. The appearance of the red streaking on his left forearm prompted his presentation to our emergency room.
ORGANISMS IN DOG BITES
1. Which is the most common cause of infected dog bite?
- Pasteurella canis
- Streptococci and S aureus
- Erysipelothrix rhusiopathiae
- Capnocytophaga canimorsus
- Eikenella corrodens
Streptococci (50%) and S aureus (20% to 40%) are the organisms most commonly responsible for infected dog bites, as they are for other skin and soft-tissue infections.1P canis is unique to dog bite infections but accounts for only 18%.2E rhusiopathiae is an unusual isolate from cat and dog bites and is more commonly isolated from the mouths of fish and aquatic mammals. C canimorsus is a normal inhabitant of the oral cavity of dogs and cats but an unusual cause of wound infection from a dog bite. It is notable for sepsis and central nervous system infections uniquely associated with veterinarians, dog owners, kennel workers, and mail carriers.3E corrodens infection is more common with human bites.4
THE EVALUATION BEGINS
On examination, the patient had marked edema of the left forearm and pain in the joints of the left hand. His temperature was 100.2°F (37.9°C). Because of the duration and severity of symptoms, the examining physician was concerned about septic arthritis of the wrist, and the patient was admitted to the hospital.
In the hospital, our patient was thermodynamically stable without documented fever or chills. There was no open wound to culture, and blood cultures were negative. Marked edema and joint involvement raised suspicion of erysipeloid. This “cousin” of erysipelas often involves the underlying joint, is associated with edema, and produces systemic manifestations of fever and arthralgia.
Radiography of the left forearm and hand demonstrated multiple foci of demineralization within the carpal bones and proximal radius, attributed to disuse. Magnetic resonance imaging (MRI) the next day showed multiple bone infarcts in the carpal bones and the distal radius, with synovitis and fluid in the carpal joints and without adjacent osteomyelitis. Fluid was also seen in the soft tissues in the ulnar aspect of the left wrist, and tenosynovitis involving the flexor carpi radialis tendon was noted.
Arthrocentesis of his left radiocarpal joint produced synovial fluid negative for crystals and negative on Gram stain; the fluid was also sent for culture. The patient’s tetanus immunization was current, and the dog was known to have been immunized against rabies.
ANTIBIOTICS FOR INFECTED DOG BITES
2. Which antibiotic regimen would you choose for this patient?
- Oral amoxicillin and clavulanate
- Meropenem
- Vancomycin, clindamycin, aztreonam
- Clindamycin and levofloxacin
- Clindamycin and trimethoprim-sulfamethoxazole
Oral amoxicillin and clavulanate (Augmentin) is a judicious choice for prophylactic treatment of deep bites in the early stages of infection. However, our patient’s wound was no longer in the early stages of infection, and he had a history of an adverse reaction to penicillin.
Meropenem (Merrem IV) cross-reacts minimally with penicillin allergy and is reported to be safe in patients with a history of anaphylactic reactions to penicillin,5 but overuse of carbapenems has led to the development of carbapenem-resistant strains of Klebsiella, Stenotrophomonas, and Acinetobacter organisms.
Given the rise of MRSA infections and the common involvement of staphylococci, streptococci, and anaerobic bacteria in complicated dog bites, the combination of vancomycin and clindamycin is a good choice, and aztreonam (Azactam) would add empiric coverage of gram-negative enteric organisms.
Levofloxacin (Levaquin) also covers gramnegative enteric organisms, but Fusobacterium canifelinum, a common anaerobe in the oral flora of dogs and cats, is intrinsically resistant to fluoroquinolones.
Clindamycin and levofloxacin would be a good step-down oral regimen. Pasteurella multocida has variable sensitivity to the commonly used agents dicloxacillin (Dynapen), cephalexin (Keflex), macrolides, and clindamycin, but it is a less likely pathogen at this late stage and could be covered with levofloxacin alone.
C canimorsus is resistant to trimethoprim-sulfamethoxazole (Bactrim) and cephalexin, but is well covered by clindamycin.6
CASE CONTINUED
Our patient was started on intravenous vancomycin, clindamycin, and aztreonam for coverage of dog-mouth flora. Blood cultures and cultures of synovial fluid of the left wrist were negative. Vancomycin was discontinued after 48 hours when blood cultures did not grow staphylococcal organisms, but clindamycin and aztreonam were continued for a total of 8 days to treat possible infection with anaerobic and gram-negative enteric pathogens.
To test for autonomic dysfunction, a plastic pen case drawn lightly across each forearm revealed a loss of tactile adherence (ie, areas where moist, sweaty skin impeded the movement of the pen case) on the affected forearm, a sign of underlying nerve injury. The affected forearm was sensitive to light touch, with pain out of proportion to the stimulus.
ARRIVING AT THE DIAGNOSIS
Based on the wide distribution of inflammation, autonomic dysfunction (shown by differences in temperature and sweating), radiographic evidence of demineralization, hyperesthesia, and lack of improvement in pain and swelling after two courses of antibiotics, the patient’s clinical course was determined to be consistent with complex regional pain syndrome type 1, previously referred to as reflex sympathetic dystrophy.
Symptoms of complex regional pain syndrome traditionally include pain, regional edema, joint stiffness, muscular atrophy, vasomotor disturbances (causing temperature variability and erythema), regional diaphoresis, and localized skeletal demineralization on radiography.
Complex regional pain syndrome type 1 occurs as regional pain and inflammation as an excessive sympathetic reaction to an often minor insult, without nerve injury. When the syndrome occurs in a patient with obvious partial nerve injury, it is categorized as type 2 (formerly known as causalgia). The two types are clinically indistinguishable and are not uncommon. About 10% of all patients with complex regional pain syndrome have obvious nerve injury (complex regional pain syndrome type 2). In a study of 109 patients with Colles fracture, 25% developed symptoms of complex regional pain syndrome.7
Complex regional pain syndrome is difficult to diagnose, as it resembles many other ailments, such as gout, infection, bone tumor, stress fracture, and arthritis. Its pathophysiology is poorly understood, but it is believed to result from a “short circuit” in the reflex arc between somatic afferent sensory fibers and autonomic sympathetic efferent fibers, and this is thought to explain the increased sympathetic stimulation.
Although the pathophysiology is likely the same in type 1 and type 2, electromyography with a nerve conduction study is a reliable way to detect nerve damage and thus distinguish between the two types of complex regional pain syndrome.8
Our understanding of this syndrome is evolving. A recent study using sensory testing showed that 33% of patients with type 1 had combinations of increased and decreased thresholds for the detection of thermal, vibratory, and mechanical stimuli in the distribution of discrete peripheral nerves, suggesting that the patients actually had type 2.9
CONFIRMING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
3. Which of the following is the best way to confirm complex regional pain syndrome type 1?
- Erythrocyte sedimentation rate, C-reactive protein, and complete blood cell count
- Plain radiography of the hand and forearm
- Three-phase technetium bone scan
- The Budapest diagnostic criteria
- MRI
- Autonomic testing
Complex regional pain syndrome type 1 is a clinical diagnosis. Diagnostic studies lack sensitivity and specificity but may confirm complex regional pain syndrome type 1 or rule out other diagnoses. The Budapest diagnostic criteria10 (Table 1) may be the best way to confirm this diagnosis. The criteria are as follows: continuing pain disproportionate to an inciting event, coupled with three of four symptoms plus at least one sign from sensory, vasomotor, sudomotor, and motor-trophic categories.
Laboratory tests are not helpful because acute-phase reactants and blood counts remain normal in these patients.
Plain radiography is not sensitive in early diagnosis, but at 2 weeks it may show patchy areas of osteopenia in adjacent bones throughout the region, as well as subsequent diffuse demineralization.
Three-phase bone scanning is more sensitive than plain radiography, with 75% of patients showing regional disparities in blood flow in early sequences and increased bone uptake in the later sequences.
MRI is a sensitive early test, as it better defines focal areas of bone loss and increased T2 bone signal in adjacent bone, as well as early soft-tissue changes. Computed tomography does not show early specific changes in muscle, tendon, or bone and so is not recommended.
THE EVALUATION CONTINUES
The admitting diagnosis was septic arthritis, and our patient underwent computed tomography, which showed focal demineralization that could have represented bone infarcts or infection, confounding the diagnosis of complex regional pain syndrome.
Autonomic nerve testing can help distinguish complex regional pain syndrome from other disorders. Complex regional pain syndrome is characterized by increased sympathetic activity and results in increased sweat output. Autonomic testing includes resting sweat output, resting skin temperature, and quantitative sudomotor axon reflex testing. In one study, an increase in resting sweat output used in conjunction with quantitative sudomotor axon reflex testing predicted the diagnosis of complex regional pain syndrome with a specificity of 98%.11 However, autonomic testing is limited to academic centers and is not readily available.
TREATING COMPLEX REGIONAL PAIN SYNDROME TYPE 1
4. Which is the best first-line therapy for complex regional pain syndrome type 1?
- Stellate ganglion nerve block
- Occupational therapy to splint the wrist and forearm
- Oral corticosteroids
- Physical therapy to prevent loss of joint motion
- Tricyclic antidepressant drugs (eg, amitriptyline), pregabalin, and bisphosphonates
Physical therapy should be started early in all patients, with range-of-motion exercises to prevent contracture and enhance mobility.
Stellate ganglion nerve block has been used to counter severe sympathetic hyperactivity, but it also may aggravate symptoms of complex regional pain syndrome and so remains a controversial treatment.
Immobilization and splinting should be avoided, as this will augment edema, pain, and contracture of joints.
Corticosteroids do not shorten the course or assuage symptoms and may increase edema.
Amitriptyline (Elavil) and pregabalin (Lyrica) have been used successfully to counter extended courses of allodynia and hyperalgesia. Bisphosphonates may decrease bone loss and pain and may be needed should the course be complicated by myositis ossificans, a form of dystrophic bone formation in juxtaposed tendon and muscle related to neuroactivation of fibroblasts and osteoblasts.
THE COURSE OF COMPLEX REGIONAL PAIN SYNDROME
Traditionally, type 1 was divided into three stages—an early inflammatory stage, a dystrophic stage, and a late atrophic stage.12 Although there is no evidence to support a consistent three-stage evolution, the severity of symptoms may help determine the best approach to management.13
Patients initially exhibit burning or throbbing pain, diffuse aching, sensitivity to touch or cold (allodynia), localized edema, and vasomotor disturbances of variable intensity that may produce altered color and temperature. Topical capsaicin cream; a tricyclic antidepressant; an anticonvulsant such as gabapentin (Neurontin), pregabalin, or lamotrigine (Lamictal); or a nonsteroidal anti-inflammatory drug should be tried first. Some of these treatments are poorly tolerated in elderly patients. If pain persists, nasal calcitonin may be added. Trigger-point injections with an anesthetic or glucocorticoid may be tried.
The management of early complex regional pain syndrome is sometimes supplemented with systemic corticosteroids, but reviews of randomized controlled trials have failed to show efficacy.14
Later in the course, patients may suffer persistent soft-tissue edema, accompanied by thickening of the skin and periarticular soft tissues, muscle wasting, and the skin changes of brawny edema. Regional blockade of sympathetic ganglions, epidural administration of clonidine, implantable peripheral nerve stimulators, and spinal cord stimulators have all been applied by experts in pain management and may provide benefit. Progression of the syndrome may include cyanosis, mottling, increased sweating, abnormal hair growth, and diffuse swelling in nonarticular tissue.
It is always acceptable to refer to an experienced pain management specialist, and a multidisciplinary approach is recommended at the outset.12
OUR PATIENT’S CARE CONTINUED
Our patient’s forearm and wrist were placed in a sling to keep his left arm elevated when active. This helped control sympathetic vascular edema and throbbing pain. Physical therapy with range-of-motion exercises prevented contracture.
He was discharged home on limited oxycodone as needed, with close follow-up by his primary care physician to monitor his pain symptoms. The pain and swelling slowly improved over the next 2 months, but he suffered a fall, twisting his left wrist. This minor injury was followed by more intense pain and swelling of the forearm, hand, and wrist.
COMORBIDITIES
5. Which of the following statements about conditions associated with complex regional pain syndrome most likely applies to our patient?
- Gout is likely following minor trauma
- Minor trauma or surgical bone biopsy may reactivate complex regional pain syndrome
- Septic hip arthritis due to MRSA may have reemerged and seeded the wrist
- Patients with multiple sclerosis have a propensity for complex regional pain syndrome
- Complex regional pain syndrome type 1 begets type 2
Gout does follow minor injury, but our patient’s uric acid was well controlled on allopurinol (Zyloprim), and gout is unlikely to be causing polyarticular swelling of the hand, wrist, and forearm.
Minor trauma, sometimes inconsequential enough to have been completely forgotten, may either initiate complex regional pain syndrome or, as seen here, reactivate it. Bone changes seen on MRI sometimes trigger surgical bone biopsy, only to reactivate the dysesthesia and sympathetic vascular reaction. Surgery should be avoided. Trauma and surgery are causative rather than associative comorbidities.
Sepsis due to MRSA after total hip arthroplasty may be reactivated, especially in the setting of immunosuppressive treatment. But the diffuse bone changes seen in multiple carpal, radial, and ulnar bones suggest generalized vascular and sympathetic disarray, most consistent with complex regional pain syndrome type 1.
AN ASSOCIATION WITH MULTIPLE SCLEROSIS?
Multiple sclerosis and other central neuropathic conditions such as stroke are associated with complex regional pain syndrome type 1.15,16
A hypothetical cause for the higher prevalence of complex regional pain syndrome in patients with multiple sclerosis may be demyelination resulting in aberrant signaling and overreaction to distal pain receptors. Demyelination of neurons within the autonomic or spinothalamic tracts potentially increases susceptibility to development of the pain syndrome.
Our patient had an apparent stimulus for the development of the syndrome, ie, the initial dog bite, and the wrist injury later may have caused peripheral nerve injury. Such injury may lead to release of vasodilatory neuropeptides including substance P from stimulated cutaneous nerves with cell bodies in the dorsal root ganglia. Excessive vasodilation and increased vascular permeability result in the affected limb becoming edematous and causing cutaneous nerves to be further activated. Stimulated cutaneous neurons normally have an inhibitory influence on sympathetic activity at the level of entry of the dorsal root ganglia in the cord. In complex regional pain syndrome, this inhibition is lost, resulting in a hyperactive somatosympathetic reflex.17 Underlying multiple sclerosis may have contributed to the loss of inhibition by the cutaneous nerves on the sympathetic system.
CASE CONCLUDED
We continued to closely follow this patient, who was on a self-directed program of physical therapy. One year after the original dog bite, the complex regional pain syndrome had completely resolved.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
- Talan DA, Citron DM, Abrahamian FM, Moran GJ, Goldstein EJ. Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group. N Engl J Med 1999; 340:85–92.
- Holst E, Rollof J, Larsson L, Nielsen JP. Characterization and distribution of Pasteurella species recovered from infected humans. J Clin Microbiol 1992; 30:2984–2987.
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents 2007; 29:367–373.
- Paul K, Patel SS. Eikenella corrodens infections in children and adolescents: case reports and review of the literature. Clin Infect Dis 2001; 33:54–61.
- Cunha BA, Hamid NS, Krol V, Eisenstein L. Safety of meropenem in patients reporting penicillin allergy: lack of allergic cross reactions. J Chemother 2008; 20:233–237.
- Verghese A, Hamati F, Berk S, Franzus B, Berk S, Smith JK. Susceptibility of dysgonic fermenter 2 to antimicrobial agents in vitro. Antimicrob Agents Chemother 1988; 32:78–80.
- Atkins RM, Duckworth T, Kanis JA. Algodystrophy following Colles’ fracture. J Hand Surg Br 1989; 14:161–164.
- Rommel O, Malin JP, Zenz M, Jänig W. Quantitative sensory testing, neurophysiological and psychological examination in patients with complex regional pain syndrome and hemisensory deficits. Pain 2001; 93:279–293.
- Sethna NF, Meier PM, Zurakowski D, Berde CB. Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007; 131:153–161.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8:326–331.
- Chelimsky TC, Low PA, Naessens JM, Wilson PR, Amadio PC, O’Brien PC. Value of autonomic testing in reflex sympathetic dystrophy. Mayo Clin Proc 1995; 70:1029–1040.
- Stanton-Hicks MD, Burton AW, Bruehl SP, et al. An updated interdisciplinary clinical pathway for CRPS: report of an expert panel. Pain Pract 2002; 2:1–16.
- Brummett CM, Cohen SP, eds. Managing pain: essentials of diagnosis and treatment. New York; Oxford University Press; 2013.
- Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: a systematic review. Clin J Pain 2012; 28:355–363.
- Schwartzman RJ, Gurusinghe C, Gracely E. Prevalence of complex regional pain syndrome in a cohort of multiple sclerosis patients. Pain Physician 2008; 11:133–136.
- Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103:199–207.
- Kurvers HA, Jacobs MJ, Beuk RJ, et al. Reflex sympathetic dystrophy: evolution of microcirculatory disturbances in time. Pain 1995; 60:333–340.
Double trouble: Simultaneous complications of therapeutic thoracentesis
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
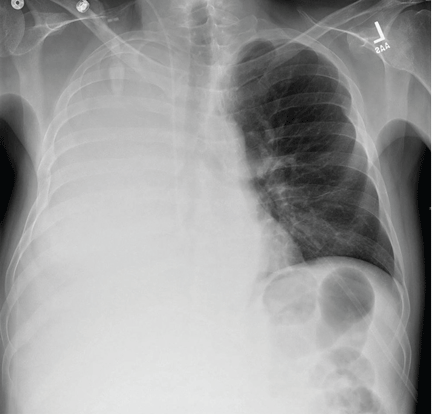
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
A 51-year-old man with end-stage liver disease from alcohol abuse presented with worsening dyspnea on exertion. He had a history of ascites requiring diuretic therapy and intermittent paracentesis, as well as symptomatic hepatic hydrothorax requiring thoracentesis. Chest radiography showed a large right hydrothorax (Figure 1).
The patient underwent high-volume thoracentesis, and 3.2 L of clear fluid was removed. Chest radiography after the procedure revealed a right-sided pneumothorax (Figure 2, arrow). The patient was mildly short of breath and was treated with high-flow oxygen. Later the same day, his shortness of breath worsened, and repeat chest radiography showed an unchanged pneumothorax that was now complicated by reexpansion pulmonary edema after thoracentesis (Figure 3, star). The reexpansion pulmonary edema resolved by the following day, and the pneumothorax resolved after placement of a pig-tail catheter into the pleural space (Figure 4).
Iatrogenic pneumothorax after thoracentesis occurs in 6% of cases.1 Iatrogenic reexpansion pulmonary edema after thoracentesis occurs in fewer than 1% of cases.2,3 Simultaneous pneumothorax and reexpansion pulmonary edema arising from the same procedure appears to be extremely rare.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Ragozzino MW, Greene R. Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest 1991; 99:506–508.
- Dias OM, Teixeira LR, Vargas FS. Reexpansion pulmonary edema after therapeutic thoracentesis. Clinics (Sao Paulo) 2010; 65:1387–1389.
Changes to practice may help avoid ‘double trouble’
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
Large-volume thoracentesis is defined as the drainage of more than 1 L of fluid. Inherent in this procedure is the removal of a large amount of fluid from a cavity with a rigid wall, which leads to changes in pleural pressure and to expansion of the lung. Two specific complications occur, pneumothorax and reexpansion pulmonary edema. The images submitted for the Clinical Picture article by Drs. Apter and Aronowitz in this issue of the Journal highlight these complications.
Retrospective studies have found an association between the amount of fluid drained and the incidence of pneumothorax.1,2 Although technical issues may account for it (eg, needle injury to the lung that leads to postprocedural pneumothorax), the available evidence suggests that it has more to do with the drainage of larger volumes than the lung can expand to fill.3,4 That is, the patient’s lung cannot expand,5 so drainage creates a vacuum, and air enters the pleural space3 through the lung parenchyma, or perhaps from around the drainage catheter.
In a series of patients who underwent therapeutic thoracentesis,3 23 (8.7%) of 265 patients had pneumothorax. Interestingly, some patients had only symptoms, some had only excessively negative pressures (< 25 cm H2O), some had both, and some had neither. Thus, there does not seem to be a reliable sign or symptom of an unexpanding lung, but pleural manometry may help increase its detection.6 This technique, however, is rarely used in clinical practice.
Another consequence of therapeutic thoracentesis is reexpansion pulmonary edema. This rare condition occurs only after large-volume thoracentesis or evacuation of a moderate to large pneumothorax.7 The pathophysiology behind this is controversial.8 As with pneumothorax, a large case series did not find a correlation between volume removed or pleural pressures and reexpansion pulmonary edema.7 Experimental data and analysis of case series8–10 suggest that the duration of lung collapse and the speed of drainage and negative pressure applied contribute to the development of edema. Vacuum bottles are often used to speed drainage and to contain the large amount of fluid drained. These bottles have an initial negative pressure of about −723 mm Hg (personal communication with Baxter Healthcare Product information line), which may lead to rapid changes in lung volume and perhaps to higher negative pleural pressures.
Given the risks discussed above, we believe it is appropriate to avoid vacuum bottles and instead to use the syringe and one-way valve supplied in most thoracentesis kits. Further, pleural manometry to detect changes in pressure that suggest an unexpandable lung may lead to the appropriate early termination of a planned large-volume thoracentesis.3 The complications reported by Drs. Apter and Aronowitz are relatively rare and, at this point, unpredictable; therefore, generating high-quality evidence for prediction or management will be difficult. In the meantime, understanding the physiologic changes in the lung and the pleural space when draining large effusions from the chest may help avoid double trouble.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.
- Josephson T, Nordenskjold CA, Larsson J, Rosenberg LU, Kaijser M. Amount drained at ultrasound-guided thoracentesis and risk of pneumothorax. Acta Radiol 2009; 50:42–47.
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170:332–339.
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130:1173–1184.
- Huggins JT, Sahn SA, Heidecker J, Ravenel JG, Doelken P. Characteristics of trapped lung: pleural fluid analysis, manometry, and air-contrast chest CT. Chest 2007; 131:206–213.
- Woodring JH, Baker MD, Stark P. Pneumothorax ex vacuo. Chest 1996; 110:1102–1105.
- Feller-Kopman D. Therapeutic thoracentesis: the role of ultrasound and pleural manometry. Curr Opin Pulmon Med 2007; 13:312–318.
- Feller-Kopman D, Berkowitz D, Boiselle P, Ernst A. Large-volume thoracentesis and the risk of reexpansion pulmonary edema. Ann Thorac Surg 2007; 84:1656–1661.
- Tarver RD, Broderick LS, Conces DJ, Jr. Reexpansion pulmonary edema. J Thorac Imag 1996; 11:198–209.
- Murphy K, Tomlanovich MC. Unilateral pulmonary edema after drainage of a spontaneous pneumothorax: case report and review of the world literature. J Emerg Med 1983; 1:29–36.
- Pavlin J, Cheney FW Unilateral pulmonary edema in rabbits after reexpansion of collapsed lung. J Appl Physiol Respir Environ Exerc Physiol 1979; 46:31–35.
Series Introduction: Doing the right thing to control health care costs
Health care costs in the United States are rising at an unsustainable rate, currently approaching 20% of the nation’s gross domestic product.1 The reasons for the rapidly increasing costs are many and complex and include new devices and drugs, greater intensity of care in the last years of life, and most perniciously, wasted care.
In its 2010 report The Healthcare Imperative: Lowering costs and Improving Outcomes, the Institute of Medicine estimated that we spend $765 billion annually on wasted care, defined as care that provides no value to the patient.2 Identified causes of wasted care include inefficiently delivered services, excessive pricing, and missed opportunities for prevention. Unnecessary services provided by physicians account for $210 billion annually, accounting for 30% of “wasted care.” Chief culprits are unnecessary imaging procedures and diagnostic tests. These two categories of physician-provided services have skyrocketed, with a cumulative increase of approximately 90% from 2000 to 2009.3
Despite our extensive use of diagnostic imaging and other testing, the US population does not benefit from better health or longer life than other industrialized nations. For example, US male life expectancy from birth is the lowest of 21 high-income countries despite greater use of health care resources, such as an 84% higher rate of magnetic resonance imaging testing per 1,000 population.4
These costs are generated directly by physicians. As aptly put by Walt Kelly’s cartoon character Pogo, “We have met the enemy, and he is us.”
COST AND VALUE
This economic crisis is not all about cost, but about value. The distinction between cost and value is important and provides a framework for physicians striving to be good shepherds of health care resources.
An expensive imaging procedure or diagnostic test may be a good value if its net benefit outweighs or at least justifies the cost. A computed tomographic angiogram provides good value for patients with an intermediate probability of pulmonary embolism in its ability to identify those who may benefit from potentially life-saving therapy.
Conversely, inexpensive tests may provide little value if they provide no patient benefit or even lead to downstream harm such as unnecessary additional testing or therapy. An example might be preoperative electrocardiography in a patient at low risk and without symptoms. Not uncommonly, unexpected electrocardiographic abnormalities are pursued with additional diagnostic tests, even though there is no evidence that patients without symptoms and at low risk benefit from this additional diagnostic scrutiny.
Because some high-cost interventions provide benefit and low-cost interventions may not, efforts to control cost should focus on value, not just cost.
REASONS FOR EXCESSIVE TESTING
Many reasons are offered for excessive testing, including assuaging concerns about diagnostic uncertainty, lack of confidence in diagnostic skills, meeting patient expectations, and lack of time to educate patients about the appropriate use of imaging and diagnostic testing.5 Both attending physicians and residents have knowledge gaps that contribute to overuse of testing.6 Physicians also report deliberate overtesting in a misguided attempt to prevent malpractice claims,5 an unproven defensive strategy that may be associated with more harm than benefit.
EDUCATIONAL INITIATIVES TO CONTROL COSTS
To meet this growing need for clinical guidance and education, regulatory agencies, professional societies, consumer groups, and foundations have prioritized high-value care as an important strategic objective. For example, cost-effective care has been incorporated into the training milestones reported to the Accreditation Council for Graduate Medical Education by internal medicine residency programs. The American College of Physicians (ACP) and the Alliance of Academic Internal Medicine have developed a curriculum to teach high-value care to internal medicine residents, and the ACP has released an interactive online curriculum for practicing physicians. The American Board of Internal Medicine Foundation launched its Choosing Wisely campaign, which asks professional societies to create lists of “things physicians and patients should question” to help make wise decisions about appropriate care. Consumer Reports has joined both the ACP and the American Board of Internal Medicine Foundation to promote high-value care to its consumer audience.
‘SMART TESTING’: THE JOURNAL’S CONTRIBUTION TO CONTROLLING COST
In this issue, Cleveland Clinic Journal of Medicine initiates its contribution to high-value care with a new series—“Smart Testing.”7 The series offers short, clinically engaging vignettes and discussions on the appropriate use of imaging procedures and other diagnostic tests. The vignettes depict common situations in clinical practice, and the discussions focus on identifying and incorporating evidence-based recommendations most likely to provide optimal patient outcome and value. This laudable goal of the Journal is reminiscent of the exhortation by Samuel Clemens (Mark Twain): “Always do right. This will gratify some people and astonish the rest.”
Physicians want to do the right thing, and with the help of the Journal, we can gratify ourselves and society with our efforts to deliver high-value care.
- Centers for Medicare & Medicaid Services, Office of the Actuary, National Health Statistics Group. www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthEx-pendData/downloads/tables.pdf. Accessed June 2, 2014.
- Institute of Medicine (US) Roundtable on Evidence-Based Medicine; Yong PL, Saunders RS, Olsen LA, editors. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Washington, DC: National Academies Press (US); 2010. www.ncbi.nlm.nih.gov/books/NBK53920/. Accessed June 2, 2014.
- Reinhardt UE. Fees, volume, and spending at Medicare. Economix. December 24, 2010. http://economix.blogs.nytimes.com/2010/12/24/fees-volume-and-spending-at-medicare/?_php=true&_type=blogs&_r=0. Accessed June 2, 2014.
- National Research Council (US); Institute of Medicine (US); Woolf SH, Aron L, eds. US Health in International Perspective: Shorter Lives, Poorer Health. Washington, DC: National Academies Press (US); 2013. www.ncbi.nlm.nih.gov/books/NBK115854/. Accessed June 2, 2014.
- Sirovich BE, Woloshin S, Schwartz LM. Too little? Too much? Primary care physicians’ views on US health care: a brief report. Arch Intern Med 2011; 171:1582–1585.
- Dine CJ, Miller J, Fuld A, Bellini LM, Iwashyna TJ. Educating physicians-in-training about resource utilization and their own outcomes of care in the inpatient setting. J Grad Med Educ 2010; 2:175–180.
- Smith CD, Alguire PC. Is cardiac stress testing appropriate in asymptomatic adults at low risk? Cleve Clin J Med 2014; 81:405–406.
Health care costs in the United States are rising at an unsustainable rate, currently approaching 20% of the nation’s gross domestic product.1 The reasons for the rapidly increasing costs are many and complex and include new devices and drugs, greater intensity of care in the last years of life, and most perniciously, wasted care.
In its 2010 report The Healthcare Imperative: Lowering costs and Improving Outcomes, the Institute of Medicine estimated that we spend $765 billion annually on wasted care, defined as care that provides no value to the patient.2 Identified causes of wasted care include inefficiently delivered services, excessive pricing, and missed opportunities for prevention. Unnecessary services provided by physicians account for $210 billion annually, accounting for 30% of “wasted care.” Chief culprits are unnecessary imaging procedures and diagnostic tests. These two categories of physician-provided services have skyrocketed, with a cumulative increase of approximately 90% from 2000 to 2009.3
Despite our extensive use of diagnostic imaging and other testing, the US population does not benefit from better health or longer life than other industrialized nations. For example, US male life expectancy from birth is the lowest of 21 high-income countries despite greater use of health care resources, such as an 84% higher rate of magnetic resonance imaging testing per 1,000 population.4
These costs are generated directly by physicians. As aptly put by Walt Kelly’s cartoon character Pogo, “We have met the enemy, and he is us.”
COST AND VALUE
This economic crisis is not all about cost, but about value. The distinction between cost and value is important and provides a framework for physicians striving to be good shepherds of health care resources.
An expensive imaging procedure or diagnostic test may be a good value if its net benefit outweighs or at least justifies the cost. A computed tomographic angiogram provides good value for patients with an intermediate probability of pulmonary embolism in its ability to identify those who may benefit from potentially life-saving therapy.
Conversely, inexpensive tests may provide little value if they provide no patient benefit or even lead to downstream harm such as unnecessary additional testing or therapy. An example might be preoperative electrocardiography in a patient at low risk and without symptoms. Not uncommonly, unexpected electrocardiographic abnormalities are pursued with additional diagnostic tests, even though there is no evidence that patients without symptoms and at low risk benefit from this additional diagnostic scrutiny.
Because some high-cost interventions provide benefit and low-cost interventions may not, efforts to control cost should focus on value, not just cost.
REASONS FOR EXCESSIVE TESTING
Many reasons are offered for excessive testing, including assuaging concerns about diagnostic uncertainty, lack of confidence in diagnostic skills, meeting patient expectations, and lack of time to educate patients about the appropriate use of imaging and diagnostic testing.5 Both attending physicians and residents have knowledge gaps that contribute to overuse of testing.6 Physicians also report deliberate overtesting in a misguided attempt to prevent malpractice claims,5 an unproven defensive strategy that may be associated with more harm than benefit.
EDUCATIONAL INITIATIVES TO CONTROL COSTS
To meet this growing need for clinical guidance and education, regulatory agencies, professional societies, consumer groups, and foundations have prioritized high-value care as an important strategic objective. For example, cost-effective care has been incorporated into the training milestones reported to the Accreditation Council for Graduate Medical Education by internal medicine residency programs. The American College of Physicians (ACP) and the Alliance of Academic Internal Medicine have developed a curriculum to teach high-value care to internal medicine residents, and the ACP has released an interactive online curriculum for practicing physicians. The American Board of Internal Medicine Foundation launched its Choosing Wisely campaign, which asks professional societies to create lists of “things physicians and patients should question” to help make wise decisions about appropriate care. Consumer Reports has joined both the ACP and the American Board of Internal Medicine Foundation to promote high-value care to its consumer audience.
‘SMART TESTING’: THE JOURNAL’S CONTRIBUTION TO CONTROLLING COST
In this issue, Cleveland Clinic Journal of Medicine initiates its contribution to high-value care with a new series—“Smart Testing.”7 The series offers short, clinically engaging vignettes and discussions on the appropriate use of imaging procedures and other diagnostic tests. The vignettes depict common situations in clinical practice, and the discussions focus on identifying and incorporating evidence-based recommendations most likely to provide optimal patient outcome and value. This laudable goal of the Journal is reminiscent of the exhortation by Samuel Clemens (Mark Twain): “Always do right. This will gratify some people and astonish the rest.”
Physicians want to do the right thing, and with the help of the Journal, we can gratify ourselves and society with our efforts to deliver high-value care.
Health care costs in the United States are rising at an unsustainable rate, currently approaching 20% of the nation’s gross domestic product.1 The reasons for the rapidly increasing costs are many and complex and include new devices and drugs, greater intensity of care in the last years of life, and most perniciously, wasted care.
In its 2010 report The Healthcare Imperative: Lowering costs and Improving Outcomes, the Institute of Medicine estimated that we spend $765 billion annually on wasted care, defined as care that provides no value to the patient.2 Identified causes of wasted care include inefficiently delivered services, excessive pricing, and missed opportunities for prevention. Unnecessary services provided by physicians account for $210 billion annually, accounting for 30% of “wasted care.” Chief culprits are unnecessary imaging procedures and diagnostic tests. These two categories of physician-provided services have skyrocketed, with a cumulative increase of approximately 90% from 2000 to 2009.3
Despite our extensive use of diagnostic imaging and other testing, the US population does not benefit from better health or longer life than other industrialized nations. For example, US male life expectancy from birth is the lowest of 21 high-income countries despite greater use of health care resources, such as an 84% higher rate of magnetic resonance imaging testing per 1,000 population.4
These costs are generated directly by physicians. As aptly put by Walt Kelly’s cartoon character Pogo, “We have met the enemy, and he is us.”
COST AND VALUE
This economic crisis is not all about cost, but about value. The distinction between cost and value is important and provides a framework for physicians striving to be good shepherds of health care resources.
An expensive imaging procedure or diagnostic test may be a good value if its net benefit outweighs or at least justifies the cost. A computed tomographic angiogram provides good value for patients with an intermediate probability of pulmonary embolism in its ability to identify those who may benefit from potentially life-saving therapy.
Conversely, inexpensive tests may provide little value if they provide no patient benefit or even lead to downstream harm such as unnecessary additional testing or therapy. An example might be preoperative electrocardiography in a patient at low risk and without symptoms. Not uncommonly, unexpected electrocardiographic abnormalities are pursued with additional diagnostic tests, even though there is no evidence that patients without symptoms and at low risk benefit from this additional diagnostic scrutiny.
Because some high-cost interventions provide benefit and low-cost interventions may not, efforts to control cost should focus on value, not just cost.
REASONS FOR EXCESSIVE TESTING
Many reasons are offered for excessive testing, including assuaging concerns about diagnostic uncertainty, lack of confidence in diagnostic skills, meeting patient expectations, and lack of time to educate patients about the appropriate use of imaging and diagnostic testing.5 Both attending physicians and residents have knowledge gaps that contribute to overuse of testing.6 Physicians also report deliberate overtesting in a misguided attempt to prevent malpractice claims,5 an unproven defensive strategy that may be associated with more harm than benefit.
EDUCATIONAL INITIATIVES TO CONTROL COSTS
To meet this growing need for clinical guidance and education, regulatory agencies, professional societies, consumer groups, and foundations have prioritized high-value care as an important strategic objective. For example, cost-effective care has been incorporated into the training milestones reported to the Accreditation Council for Graduate Medical Education by internal medicine residency programs. The American College of Physicians (ACP) and the Alliance of Academic Internal Medicine have developed a curriculum to teach high-value care to internal medicine residents, and the ACP has released an interactive online curriculum for practicing physicians. The American Board of Internal Medicine Foundation launched its Choosing Wisely campaign, which asks professional societies to create lists of “things physicians and patients should question” to help make wise decisions about appropriate care. Consumer Reports has joined both the ACP and the American Board of Internal Medicine Foundation to promote high-value care to its consumer audience.
‘SMART TESTING’: THE JOURNAL’S CONTRIBUTION TO CONTROLLING COST
In this issue, Cleveland Clinic Journal of Medicine initiates its contribution to high-value care with a new series—“Smart Testing.”7 The series offers short, clinically engaging vignettes and discussions on the appropriate use of imaging procedures and other diagnostic tests. The vignettes depict common situations in clinical practice, and the discussions focus on identifying and incorporating evidence-based recommendations most likely to provide optimal patient outcome and value. This laudable goal of the Journal is reminiscent of the exhortation by Samuel Clemens (Mark Twain): “Always do right. This will gratify some people and astonish the rest.”
Physicians want to do the right thing, and with the help of the Journal, we can gratify ourselves and society with our efforts to deliver high-value care.
- Centers for Medicare & Medicaid Services, Office of the Actuary, National Health Statistics Group. www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthEx-pendData/downloads/tables.pdf. Accessed June 2, 2014.
- Institute of Medicine (US) Roundtable on Evidence-Based Medicine; Yong PL, Saunders RS, Olsen LA, editors. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Washington, DC: National Academies Press (US); 2010. www.ncbi.nlm.nih.gov/books/NBK53920/. Accessed June 2, 2014.
- Reinhardt UE. Fees, volume, and spending at Medicare. Economix. December 24, 2010. http://economix.blogs.nytimes.com/2010/12/24/fees-volume-and-spending-at-medicare/?_php=true&_type=blogs&_r=0. Accessed June 2, 2014.
- National Research Council (US); Institute of Medicine (US); Woolf SH, Aron L, eds. US Health in International Perspective: Shorter Lives, Poorer Health. Washington, DC: National Academies Press (US); 2013. www.ncbi.nlm.nih.gov/books/NBK115854/. Accessed June 2, 2014.
- Sirovich BE, Woloshin S, Schwartz LM. Too little? Too much? Primary care physicians’ views on US health care: a brief report. Arch Intern Med 2011; 171:1582–1585.
- Dine CJ, Miller J, Fuld A, Bellini LM, Iwashyna TJ. Educating physicians-in-training about resource utilization and their own outcomes of care in the inpatient setting. J Grad Med Educ 2010; 2:175–180.
- Smith CD, Alguire PC. Is cardiac stress testing appropriate in asymptomatic adults at low risk? Cleve Clin J Med 2014; 81:405–406.
- Centers for Medicare & Medicaid Services, Office of the Actuary, National Health Statistics Group. www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthEx-pendData/downloads/tables.pdf. Accessed June 2, 2014.
- Institute of Medicine (US) Roundtable on Evidence-Based Medicine; Yong PL, Saunders RS, Olsen LA, editors. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Washington, DC: National Academies Press (US); 2010. www.ncbi.nlm.nih.gov/books/NBK53920/. Accessed June 2, 2014.
- Reinhardt UE. Fees, volume, and spending at Medicare. Economix. December 24, 2010. http://economix.blogs.nytimes.com/2010/12/24/fees-volume-and-spending-at-medicare/?_php=true&_type=blogs&_r=0. Accessed June 2, 2014.
- National Research Council (US); Institute of Medicine (US); Woolf SH, Aron L, eds. US Health in International Perspective: Shorter Lives, Poorer Health. Washington, DC: National Academies Press (US); 2013. www.ncbi.nlm.nih.gov/books/NBK115854/. Accessed June 2, 2014.
- Sirovich BE, Woloshin S, Schwartz LM. Too little? Too much? Primary care physicians’ views on US health care: a brief report. Arch Intern Med 2011; 171:1582–1585.
- Dine CJ, Miller J, Fuld A, Bellini LM, Iwashyna TJ. Educating physicians-in-training about resource utilization and their own outcomes of care in the inpatient setting. J Grad Med Educ 2010; 2:175–180.
- Smith CD, Alguire PC. Is cardiac stress testing appropriate in asymptomatic adults at low risk? Cleve Clin J Med 2014; 81:405–406.
Is cardiac stress testing appropriate in asymptomatic adults at low risk?
A 48-year-old insurance executive is offered the option of several health insurance packages at the time of a promotion. He is healthy and a non-smoker; both his parents are alive and well; and he takes only vitamins and fish oil supplements on a regular basis. His levels of total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol are all in the normal range, as is his blood pressure. He plans to purchase the lowest price policy, but wants to know if he should also get a stress test to best guide his care.
GUIDELINES RECOMMEND AGAINST TESTING
Patients who are at low risk of disease and without symptoms should not undergo cardiac stress testing. The test is unlikely to be helpful in these patients and may expose them to harm unnecessarily. Cardiac stress testing such as exercise electrocardiography is most useful in patients who have chest pain and shortness of breath on exertion, to look for underlying cardiovascular disease. Despite this, the test is often used inappropriately as part of a routine health evaluation in low-risk, asymptomatic people, such as this patient.
Recent high-quality guidelines address exercise electrocardiography as a screening test for cardiovascular disease in asymptomatic, low-risk adults.
The US Preventive Services Task Force 2012 guideline1 recommends against screening with exercise electrocardiography for predicting coronary heart disease events in adults with no symptoms and at low risk of these events. A systematic review found no data from randomized controlled trials or prospective cohort studies of this test to screen asymptomatic adults compared with no screening.2
The American Academy of Family Physicians (AAFP) 2012 guideline3 recommends against routine screening with exercise electrocardiography either for the presence of severe coronary artery stenosis or for predicting coronary events in adults at low risk. The AAFP guideline notes that there is moderate or high certainty of no net benefit or that the harms outweigh the benefits of exercise electrocardiography in adults at low risk and without symptoms.
The 2010 joint guideline of the American College of Cardiology and the American Heart Association4 does not comment on the role of screening exercise electrocardiography in low-risk asymptomatic adults, but states that a physician may consider ordering exercise electrocardiography in asymptomatic adults at intermediate risk of coronary heart disease. The guideline recommends that the individual physician decide whether screening exercise electrocardiography is warranted in a patient at intermediate risk.
The Choosing Wisely initiative
As part of the Choosing Wisely initiative of the American Board of Internal Medicine Foundation, a number of medical specialty societies have published lists of recommendations and issues that physicians and patients should question and discuss. Cardiac stress testing in low-risk asymptomatic patients is on the list of a number of organizations, including the American College of Physicians, the American College of Cardiology, the AAFP, and the American Society of Nuclear Cardiology. These lists can be found at www.choosingwisely.org.
POSSIBLE HARM ASSOCIATED WITH CARDIAC STRESS TESTING
The overall risk of sudden cardiac death or an event that requires hospitalization during exercise electrocardiography is very small, estimated to be 1 per 10,000 tests, and the risk is probably even less in patients at low risk.5 But the risk of potential downstream harm from additional testing or interventions may be greater than direct harm. Still, no study has yet assessed harm associated with follow-up testing or interventions after screening with exercise electrocardiography.
On the basis of large, population-based registries that include symptomatic persons, the risk of any serious adverse event as a result of angiography is about 1.7%; this includes a 0.1% risk of death, a 0.05% risk of myocardial infarction, a 0.07% risk of stroke, and a 0.4% risk of arrhythmia.6 In addition, coronary angiography is associated with an average effective radiation dose of 7 mSv and myocardial perfusion imaging with a dose of 15.6 mSv.7 These are approximately two times and five times the amount of radiation an average person in the United States receives per year from exposure to ambient radiation (3 mSv).
Several studies that included symptomatic and asymptomatic patients who had undergone angiography reported that between 39% and 85% of patients had no coronary artery disease. This means that many patients were subjected to the risks of invasive testing and treatment without the possibility of benefit. Patients who receive lipid-lowering therapy or aspirin because of an abnormal exercise electrocardiogram are also exposed to the risks related to those interventions.
THE CLINICAL BOTTOM LINE
On the basis of current data, the insurance executive should not get a stress test because the results of the test are unlikely to have an impact on his medical management, are unlikely to improve his clinical outcome, and carry a small risk of harm. Low-risk, asymptomatic people with a positive stress test have the same mortality rate as those who have a negative stress test, and its usefulness beyond traditional risk-factor assessment in motivating patients and guiding therapy has not been established.8 In addition, the rate of false-positive results with exercise stress testing is as high as 71%.9 Although the risk of an adverse event from the initial stress test is low, ie, 1 serious event in 10,000 tests, the risk of subsequent cardiac catheterization after a positive test is significantly higher, ie, 170 serious events in 10,000 tests. For these reasons, the potential harm of exercise electrocardiography outweighs the benefits in this patient.
- Moyer VAUS Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 157:512–518.
- Chou R, Arora B, Dana T, Fu R, Walker M, Humphrey L. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2011; 155:375–385.
- Leawood KS; American Academy of Family Physicians (AAFP). Summary of recommendations for clinical preventive services. American Academy of Family Physicians (AAFP); 2012. http://www.guideline.gov/content.aspx?id=47554. Accessed May 12, 2014.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation; American Heart Association. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2010; 56:e50–e103.
- Myers J, Arena R, Franklin B, et al; American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention of the Council on Clinical Cardiology, the Council on Nutrition, Physical Activity, and Metabolism, and the Council on Cardiovascular Nursing. Recommendations for clinical exercise laboratories: a scientific statement from the American Heart Association. Circulation 2009; 119:3144–3161.
- Noto TJ, Johnson LW, Krone R, et al. Cardiac catheterization 1990: a report of the Registry of the Society for Cardiac Angiography and Interventions (SCA&I). Cathet Cardiovasc Diagn 1991; 24:75–83.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Pilote L, Pashkow F, Thomas JD, et al. Clinical yield and cost of exercise treadmill testing to screen for coronary artery disease in asymptomatic adults. Am J Cardiol 1998; 81:219–224.
- Hopkirk JA, Uhl GS, Hickman JR, Fischer J, Medina A. Discriminant value of clinical and exercise variables in detecting significant coronary artery disease in asymptomatic men. J Am Coll Cardiol 1984; 3:887–894.
A 48-year-old insurance executive is offered the option of several health insurance packages at the time of a promotion. He is healthy and a non-smoker; both his parents are alive and well; and he takes only vitamins and fish oil supplements on a regular basis. His levels of total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol are all in the normal range, as is his blood pressure. He plans to purchase the lowest price policy, but wants to know if he should also get a stress test to best guide his care.
GUIDELINES RECOMMEND AGAINST TESTING
Patients who are at low risk of disease and without symptoms should not undergo cardiac stress testing. The test is unlikely to be helpful in these patients and may expose them to harm unnecessarily. Cardiac stress testing such as exercise electrocardiography is most useful in patients who have chest pain and shortness of breath on exertion, to look for underlying cardiovascular disease. Despite this, the test is often used inappropriately as part of a routine health evaluation in low-risk, asymptomatic people, such as this patient.
Recent high-quality guidelines address exercise electrocardiography as a screening test for cardiovascular disease in asymptomatic, low-risk adults.
The US Preventive Services Task Force 2012 guideline1 recommends against screening with exercise electrocardiography for predicting coronary heart disease events in adults with no symptoms and at low risk of these events. A systematic review found no data from randomized controlled trials or prospective cohort studies of this test to screen asymptomatic adults compared with no screening.2
The American Academy of Family Physicians (AAFP) 2012 guideline3 recommends against routine screening with exercise electrocardiography either for the presence of severe coronary artery stenosis or for predicting coronary events in adults at low risk. The AAFP guideline notes that there is moderate or high certainty of no net benefit or that the harms outweigh the benefits of exercise electrocardiography in adults at low risk and without symptoms.
The 2010 joint guideline of the American College of Cardiology and the American Heart Association4 does not comment on the role of screening exercise electrocardiography in low-risk asymptomatic adults, but states that a physician may consider ordering exercise electrocardiography in asymptomatic adults at intermediate risk of coronary heart disease. The guideline recommends that the individual physician decide whether screening exercise electrocardiography is warranted in a patient at intermediate risk.
The Choosing Wisely initiative
As part of the Choosing Wisely initiative of the American Board of Internal Medicine Foundation, a number of medical specialty societies have published lists of recommendations and issues that physicians and patients should question and discuss. Cardiac stress testing in low-risk asymptomatic patients is on the list of a number of organizations, including the American College of Physicians, the American College of Cardiology, the AAFP, and the American Society of Nuclear Cardiology. These lists can be found at www.choosingwisely.org.
POSSIBLE HARM ASSOCIATED WITH CARDIAC STRESS TESTING
The overall risk of sudden cardiac death or an event that requires hospitalization during exercise electrocardiography is very small, estimated to be 1 per 10,000 tests, and the risk is probably even less in patients at low risk.5 But the risk of potential downstream harm from additional testing or interventions may be greater than direct harm. Still, no study has yet assessed harm associated with follow-up testing or interventions after screening with exercise electrocardiography.
On the basis of large, population-based registries that include symptomatic persons, the risk of any serious adverse event as a result of angiography is about 1.7%; this includes a 0.1% risk of death, a 0.05% risk of myocardial infarction, a 0.07% risk of stroke, and a 0.4% risk of arrhythmia.6 In addition, coronary angiography is associated with an average effective radiation dose of 7 mSv and myocardial perfusion imaging with a dose of 15.6 mSv.7 These are approximately two times and five times the amount of radiation an average person in the United States receives per year from exposure to ambient radiation (3 mSv).
Several studies that included symptomatic and asymptomatic patients who had undergone angiography reported that between 39% and 85% of patients had no coronary artery disease. This means that many patients were subjected to the risks of invasive testing and treatment without the possibility of benefit. Patients who receive lipid-lowering therapy or aspirin because of an abnormal exercise electrocardiogram are also exposed to the risks related to those interventions.
THE CLINICAL BOTTOM LINE
On the basis of current data, the insurance executive should not get a stress test because the results of the test are unlikely to have an impact on his medical management, are unlikely to improve his clinical outcome, and carry a small risk of harm. Low-risk, asymptomatic people with a positive stress test have the same mortality rate as those who have a negative stress test, and its usefulness beyond traditional risk-factor assessment in motivating patients and guiding therapy has not been established.8 In addition, the rate of false-positive results with exercise stress testing is as high as 71%.9 Although the risk of an adverse event from the initial stress test is low, ie, 1 serious event in 10,000 tests, the risk of subsequent cardiac catheterization after a positive test is significantly higher, ie, 170 serious events in 10,000 tests. For these reasons, the potential harm of exercise electrocardiography outweighs the benefits in this patient.
A 48-year-old insurance executive is offered the option of several health insurance packages at the time of a promotion. He is healthy and a non-smoker; both his parents are alive and well; and he takes only vitamins and fish oil supplements on a regular basis. His levels of total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol are all in the normal range, as is his blood pressure. He plans to purchase the lowest price policy, but wants to know if he should also get a stress test to best guide his care.
GUIDELINES RECOMMEND AGAINST TESTING
Patients who are at low risk of disease and without symptoms should not undergo cardiac stress testing. The test is unlikely to be helpful in these patients and may expose them to harm unnecessarily. Cardiac stress testing such as exercise electrocardiography is most useful in patients who have chest pain and shortness of breath on exertion, to look for underlying cardiovascular disease. Despite this, the test is often used inappropriately as part of a routine health evaluation in low-risk, asymptomatic people, such as this patient.
Recent high-quality guidelines address exercise electrocardiography as a screening test for cardiovascular disease in asymptomatic, low-risk adults.
The US Preventive Services Task Force 2012 guideline1 recommends against screening with exercise electrocardiography for predicting coronary heart disease events in adults with no symptoms and at low risk of these events. A systematic review found no data from randomized controlled trials or prospective cohort studies of this test to screen asymptomatic adults compared with no screening.2
The American Academy of Family Physicians (AAFP) 2012 guideline3 recommends against routine screening with exercise electrocardiography either for the presence of severe coronary artery stenosis or for predicting coronary events in adults at low risk. The AAFP guideline notes that there is moderate or high certainty of no net benefit or that the harms outweigh the benefits of exercise electrocardiography in adults at low risk and without symptoms.
The 2010 joint guideline of the American College of Cardiology and the American Heart Association4 does not comment on the role of screening exercise electrocardiography in low-risk asymptomatic adults, but states that a physician may consider ordering exercise electrocardiography in asymptomatic adults at intermediate risk of coronary heart disease. The guideline recommends that the individual physician decide whether screening exercise electrocardiography is warranted in a patient at intermediate risk.
The Choosing Wisely initiative
As part of the Choosing Wisely initiative of the American Board of Internal Medicine Foundation, a number of medical specialty societies have published lists of recommendations and issues that physicians and patients should question and discuss. Cardiac stress testing in low-risk asymptomatic patients is on the list of a number of organizations, including the American College of Physicians, the American College of Cardiology, the AAFP, and the American Society of Nuclear Cardiology. These lists can be found at www.choosingwisely.org.
POSSIBLE HARM ASSOCIATED WITH CARDIAC STRESS TESTING
The overall risk of sudden cardiac death or an event that requires hospitalization during exercise electrocardiography is very small, estimated to be 1 per 10,000 tests, and the risk is probably even less in patients at low risk.5 But the risk of potential downstream harm from additional testing or interventions may be greater than direct harm. Still, no study has yet assessed harm associated with follow-up testing or interventions after screening with exercise electrocardiography.
On the basis of large, population-based registries that include symptomatic persons, the risk of any serious adverse event as a result of angiography is about 1.7%; this includes a 0.1% risk of death, a 0.05% risk of myocardial infarction, a 0.07% risk of stroke, and a 0.4% risk of arrhythmia.6 In addition, coronary angiography is associated with an average effective radiation dose of 7 mSv and myocardial perfusion imaging with a dose of 15.6 mSv.7 These are approximately two times and five times the amount of radiation an average person in the United States receives per year from exposure to ambient radiation (3 mSv).
Several studies that included symptomatic and asymptomatic patients who had undergone angiography reported that between 39% and 85% of patients had no coronary artery disease. This means that many patients were subjected to the risks of invasive testing and treatment without the possibility of benefit. Patients who receive lipid-lowering therapy or aspirin because of an abnormal exercise electrocardiogram are also exposed to the risks related to those interventions.
THE CLINICAL BOTTOM LINE
On the basis of current data, the insurance executive should not get a stress test because the results of the test are unlikely to have an impact on his medical management, are unlikely to improve his clinical outcome, and carry a small risk of harm. Low-risk, asymptomatic people with a positive stress test have the same mortality rate as those who have a negative stress test, and its usefulness beyond traditional risk-factor assessment in motivating patients and guiding therapy has not been established.8 In addition, the rate of false-positive results with exercise stress testing is as high as 71%.9 Although the risk of an adverse event from the initial stress test is low, ie, 1 serious event in 10,000 tests, the risk of subsequent cardiac catheterization after a positive test is significantly higher, ie, 170 serious events in 10,000 tests. For these reasons, the potential harm of exercise electrocardiography outweighs the benefits in this patient.
- Moyer VAUS Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 157:512–518.
- Chou R, Arora B, Dana T, Fu R, Walker M, Humphrey L. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2011; 155:375–385.
- Leawood KS; American Academy of Family Physicians (AAFP). Summary of recommendations for clinical preventive services. American Academy of Family Physicians (AAFP); 2012. http://www.guideline.gov/content.aspx?id=47554. Accessed May 12, 2014.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation; American Heart Association. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2010; 56:e50–e103.
- Myers J, Arena R, Franklin B, et al; American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention of the Council on Clinical Cardiology, the Council on Nutrition, Physical Activity, and Metabolism, and the Council on Cardiovascular Nursing. Recommendations for clinical exercise laboratories: a scientific statement from the American Heart Association. Circulation 2009; 119:3144–3161.
- Noto TJ, Johnson LW, Krone R, et al. Cardiac catheterization 1990: a report of the Registry of the Society for Cardiac Angiography and Interventions (SCA&I). Cathet Cardiovasc Diagn 1991; 24:75–83.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Pilote L, Pashkow F, Thomas JD, et al. Clinical yield and cost of exercise treadmill testing to screen for coronary artery disease in asymptomatic adults. Am J Cardiol 1998; 81:219–224.
- Hopkirk JA, Uhl GS, Hickman JR, Fischer J, Medina A. Discriminant value of clinical and exercise variables in detecting significant coronary artery disease in asymptomatic men. J Am Coll Cardiol 1984; 3:887–894.
- Moyer VAUS Preventive Services Task Force. Screening for coronary heart disease with electrocardiography: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 157:512–518.
- Chou R, Arora B, Dana T, Fu R, Walker M, Humphrey L. Screening asymptomatic adults with resting or exercise electrocardiography: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med 2011; 155:375–385.
- Leawood KS; American Academy of Family Physicians (AAFP). Summary of recommendations for clinical preventive services. American Academy of Family Physicians (AAFP); 2012. http://www.guideline.gov/content.aspx?id=47554. Accessed May 12, 2014.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation; American Heart Association. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2010; 56:e50–e103.
- Myers J, Arena R, Franklin B, et al; American Heart Association Committee on Exercise, Cardiac Rehabilitation, and Prevention of the Council on Clinical Cardiology, the Council on Nutrition, Physical Activity, and Metabolism, and the Council on Cardiovascular Nursing. Recommendations for clinical exercise laboratories: a scientific statement from the American Heart Association. Circulation 2009; 119:3144–3161.
- Noto TJ, Johnson LW, Krone R, et al. Cardiac catheterization 1990: a report of the Registry of the Society for Cardiac Angiography and Interventions (SCA&I). Cathet Cardiovasc Diagn 1991; 24:75–83.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Pilote L, Pashkow F, Thomas JD, et al. Clinical yield and cost of exercise treadmill testing to screen for coronary artery disease in asymptomatic adults. Am J Cardiol 1998; 81:219–224.
- Hopkirk JA, Uhl GS, Hickman JR, Fischer J, Medina A. Discriminant value of clinical and exercise variables in detecting significant coronary artery disease in asymptomatic men. J Am Coll Cardiol 1984; 3:887–894.